
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 17 June 2020
Sec. Mitochondrial Research
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00595
This article is part of the Research TopicMitochondria in Health and DiseaseView all 23 articles
 Georgios Amanakis*
Georgios Amanakis* Elizabeth Murphy
Elizabeth MurphyCyclophilin D (CypD) is a mitochondrial peptidyl-prolyl cis-trans isomerase, well-known for regulating the mitochondrial permeability transition pore (PTP), a nonspecific large conductance pore whose opening leads to cell death and has been implicated in ischemia/reperfusion injury in multiple organs, in neurodegenerative disorders, and in muscular dystrophies. While the main target of CypD is a matter of ongoing research, inhibiting CypD protects in models of those diseases making it an interesting therapeutic target. The present review focuses on post-translational modifications of CypD that have been identified by recent studies, which can alter the regulation of the PTP and contribute to understanding the mechanisms of action of CypD.
Cyclophilin D (CypD) is a highly conserved peptidyl-prolyl cis-trans isomerase (PPIase) that plays an important role in mitochondrial biology. It is encoded by the genomic Ppif gene and contains a mitochondrial targeting sequence which is cleaved upon entering the mitochondrial matrix, reducing its size from 22 to 19 kDa. It was named, like all cyclophilins, after its ability to bind the drug cyclosporine A (CsA) (Walsh et al., 1992). Cyclophilins, a family with more than 15 members, have been shown to act as chaperones accelerating protein folding and maturation, as well as playing a critical role in signal transduction and the immune response. Although the physiological role of CypD remains elusive, CypD has been shown to be a sensitizer of the permeability transition pore (PTP), a nonspecific large conductance pore whose opening leads to dissipation of the inner mitochondrial membrane (IMM) potential, loss of ATP production, and eventually cell death (Bauer and Murphy, 2020). The PTP has been implicated in ischemia/reperfusion (I/R) injury in the heart (Lim et al., 2011; Bibli et al., 2019), brain (Uchino et al., 2002; Schinzel et al., 2005), and kidney (Park et al., 2011; Yang et al., 2019), in neurodegenerative disorders (Warne et al., 2016), and in muscular dystrophies (Pato et al., 1983; Dubinin et al., 2020). CypD has been reported to interact with the F1F0-ATP synthase (Giorgio et al., 2009; Bonora et al., 2017), the phosphate carrier (PiC) (Leung et al., 2008), and the adenine nucleotide translocator (ANT) (Kokoszka et al., 2004; Karch et al., 2019), all of which have been proposed as potential components of the PTP. While irreversible opening of the PTP is associated with cell death, short transient openings may have a role in modulating matrix calcium (Petronilli et al., 1999; Bernardi and von Stockum, 2012; Lu et al., 2016; Agarwal et al., 2017), which in turn can regulate mitochondrial bioenergetics (Elrod et al., 2010; Glancy and Balaban, 2012; Tarasov et al., 2012). Thus, CypD as a sensitizer of the PTP may affect mitochondrial bioenergetics, and this may provide insight into its physiological function. Interestingly, the ATP synthase can oligomerize with ANT and PiC to form a synthasome to increase the efficiency of ATP production and its translocation to the cytosol, and CypD has been reported to regulate its assembly (Beutner et al., 2017). The initial studies to elucidate the role of CypD involved inhibition, deletion, and overexpression of its encoding gene (Baines et al., 2005; Nakagawa et al., 2005; Devalaraja-Narashimha et al., 2009; Elrod et al., 2010; Menazza et al., 2013). Recent work revealed CypD can undergo many different post-translational modifications (Linard et al., 2009; Hafner et al., 2010; Kohr et al., 2011b; Nguyen et al., 2011; Sanchez et al., 2011; Bochaton et al., 2015; Parks et al., 2018; Amanakis et al., 2020), including oxidation, S-nitrosylation, S-palmitoylation, S-glutathionylation, phosphorylation, and acetylation. The present review will focus on CypD as an integrator of mitochondrial physiology and pathophysiology through its post-translational modifications (Figures 1, 2).
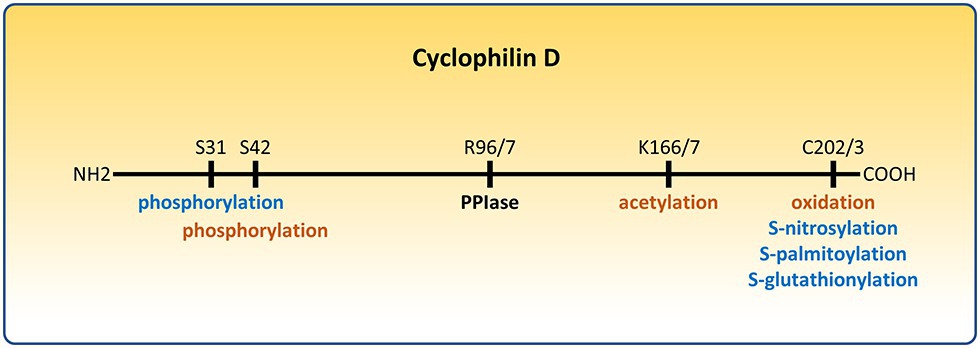
Figure 1. Post-translational modifications of cyclophilin D (CypD) that affect the permeability transition pore (PTP). Modifications that sensitize the PTP are depicted in orange. Modifications that desensitize the PTP are depicted in blue. R96/7, K166/7, and C202/3 represent homologous arginine, lysine, and cysteine residues in the mouse/human form of CypD, respectively.

Figure 2. Surface representation of the secondary structure of human CypD with key residues related to PTP activation and catalytic activity. S1 and S2 represent the PPIase catalytic pockets (Davis et al., 2010). The model was retrieved from the Protein Data Bank (PDB ID: 3QYU) and rendered using PyMol (Schrodinger LLC, 2015). R96/7, K166/7, and C202/3 represent homologous arginine, lysine, and cysteine residues in the mouse/human form of CypD, respectively. Serine residues 31 and 42 (identical in mouse/human CypD) are not included in any currently available CypD model.
Mitochondria are the power house of the cell; they create energy in the form of ATP through oxidative phosphorylation. The electron transport chain (ETC, complexes I–IV) uses reducing equivalents from fatty acid oxidation, glycolysis, and the Krebs cycle to transfer electrons ultimately to oxygen, pump protons from the matrix to the intermembrane space, and thus generate an electrochemical gradient across the inner mitochondrial membrane. The ATP synthase exploits this gradient to create ATP from ADP and inorganic phosphate. Inorganic phosphate is imported from the cytosol by the PiC. The newly produced ATP however is localized in the matrix and needs to be transferred to the cytosol for the cell to use it. This transport is mediated by the ANT, which also imports ADP from the cytosol. To increase the efficiency of ATP production and transportation to the cytosol, the ATP synthase can oligomerize with the ANT and PiC, forming synthasomes (Beutner et al., 2017). Furthermore, the ETC can form super complexes to increase the efficiency of proton gradient generation (Acin-Perez and Enriquez, 2014; Milenkovic et al., 2017). Transport of electrons by the ETC and consumption of oxygen by cytochrome oxidase, the final electron acceptor, are coupled to ATP production by the ATP synthase. In well coupled mitochondria, electrons are not transported and oxygen is not consumed unless ADP is present. Mitochondrial coupling is greatly dependent on the permeability of the IMM. If there are leaks across the IMM (uncoupled mitochondria), the electrochemical gradient is dissipated which leads to less efficient ATP production. The PTP (Bauer and Murphy, 2020) provides a means to directly regulate the conductance of the IMM and mitochondrial coupling as its opening dissipates the proton gradient and uncouples electron transport from ATP production.
CypD is a regulator of the PTP, and thus of mitochondrial coupling. It sensitizes PTP to calcium and oxidative stress. It can also directly bind to the ATP synthase and reduce ATP production while its displacement from the inner mitochondrial membrane by CsA increases ATP synthesis (Giorgio et al., 2009). In heart, brain, and liver mitochondria, CypD also regulates the assembly of synthasomes (Beutner et al., 2017). Their formation is stimulated by respiration while their disassembly favors PTP opening. Interestingly inhibiting or deleting CypD increases the stability of the synthasomes, suggesting a model in which CypD binds to and limits their assembly (Beutner et al., 2017). However, from the data provided it is unclear whether this is a direct effect of CypD or if matrix calcium levels, redox state, or inner membrane potential are also involved.
The PPIase activity of CypD may also have a regulatory role on the PTP. In a study employing CsA analogues, inhibition of PTP correlated well with inhibition of PPIase activity of CypD (Nicolli et al., 1996). The arginine at residue 96 (R96, homologous to R97 in human CypD) is important for PPIase activity and mutating it to a glycine abolishes the PPIase activity of CypD. CypD−/− fibroblasts are protected against oxidative stress; however, consistent with the previous study, the protection is lost in cells expressing a wild type (WT) CypD but not in cells expressing the R96G CypD mutant (Baines et al., 2005). These findings suggest that the PPIase activity of CypD facilitates PTP activation. However, the exact mechanism is unknown. Cis-trans prolyl isomerization of PTP components may expose binding sites to calcium which would affect the sensitivity of PTP to it. It has also been suggested that CypD activates the PTP independent of its PPIase activity (Scorrano et al., 1997). Future studies will be needed to address the role of the PPIase activity in regulating the PTP. Interestingly, the PPIase activity normalized to the expression level of the ATP synthase was associated with changes in synthasome assembly, leading to the conclusion that increased synthasome assembly with the ATP synthase in high order oligomers decreases the probability of PTP formation from ATP synthase monomers or dimers (Beutner et al., 2017). However, differentiating between independent effects of CypD and those resulting from uncoupling due to CypD-mediated sensitization of the PTP is challenging. Furthermore, it is unclear how post-translational modifications of CypD affect those functions.
CypD can regulate the expression of mitochondrial genes, thus affecting cell proliferation and differentiation (Radhakrishnan et al., 2015). It interacts with mitochondrial transcription factor and regulates mitochondrial RNA synthesis of subunits of the NADH dehydrogenase (ND1), cytochrome c oxidase (COX1), and ATP synthase (ATP6). Furthermore, deletion of CypD has been shown to activate mitochondrial retrograde signaling leading to transcriptional changes in gene expression, modulation of a chemokine/chemokine receptor signature, and activation of the inflammatory mediator STAT3 that affects cell motility and proliferation (Tavecchio et al., 2013).
Loss or inhibition of CypD has been shown to increase mitochondrial calcium which activates the mitochondrial dehydrogenases and shifts the metabolism from fatty acids to glucose (Elrod et al., 2010). Additionally, the increase in NADH/NAD+ ratio results in inhibition of deacetylases and increases acetylation of proteins (Nguyen et al., 2013) comprising pathways involved in branched chain amino acid metabolism, pyruvate metabolism, and the Krebs cycle (Menazza et al., 2013). Interestingly, CypD has been reported to be essential for the activity of the trifunctional protein, a central enzyme in β-oxidation of fatty acids (Tavecchio et al., 2015). Consistent with the metabolic shift to glucose, CypD−/− mice were more susceptible to heart failure compared to WT. These results indicate that CypD has a central role in controlling mitochondrial bioenergetics by regulating matrix calcium. It is widely known that failing hearts exhibit a metabolic shift from fatty-acid to glucose oxidation (Karwi et al., 2018). However, the role of CypD in heart failure is unclear and future research should focus on how to exploit it as a therapeutic target in this disease.
Oxidative stress can modify structural and functional properties of proteins, and cysteine residues are of particular interest due to their reactive thiol group. Oxidation of cysteines of CypD influences the conformation of the enzyme and its activity (Linard et al., 2009). Site-directed mutagenesis allowed the identification of cysteine 203 of human CypD (homologous to cysteine 202 in mice, abbreviated as C202/3) as an important redox-sensitive residue. When C202/3 is oxidized it can form a disulfide bond with C157 which decreases its PPIase activity by 20% (Linard et al., 2009). As discussed, it is unclear whether PPIase activity is essential for PTP activation. Additional experiments are needed to address this issue. Previous studies have also shown that C202/3 undergoes S-nitrosylation in ischemic preconditioning, a cardioprotective strategy consisting of repetitive non-lethal I/R cycles (Kohr et al., 2011a,b). The mutation of C202/3 to a serine or treatment with a nitric oxide donor in mouse embryonic fibroblasts desensitized the PTP in oxidative cell death (Nguyen et al., 2011). In addition, tachycardic preconditioning desensitizes the PTP and increases S-glutathionylation of CypD (Sanchez et al., 2011). These findings suggest that S-nitrosylation and S-glutathionylation shield C202/3 from further oxidative damage and are thus cardio-protective. C202/3 also matches an S-palmitoylation motif common in soluble proteins (Collins et al., 2017). Recent data from our lab show that CypD is abundantly S-palmitoylated at C202/3, and that increased matrix calcium during ischemia leads to de-palmitoylation of C202/3, allowing it to undergo oxidation and to interact with the PTP (Amanakis et al., 2020). Of note, S-palmitoylation, S-nitrosylation, and the mutation of C202/3 to a serine do not affect the PPIase activity of CypD (Nguyen et al., 2011; Amanakis et al., 2020). In summary, oxidation on C202/3 sensitizes the PTP while S-nitrosylation, S-glutathionylation, and S-palmitoylation confer protection.
The effects of phosphorylation of CypD differ according to the phosphorylated residue. Glycogen synthase kinase-3 (GSK-3) has been reported to phosphorylate CypD and to enhance PTP opening and, consistent with this concept, GSK-3 inhibition (which would decrease CypD phosphorylation) protects from PTP opening in human cancer cell models (Rasola et al., 2010). Similarly in a rat model, treatment with an inhibitor of GSK-3 decreased CypD phosphorylation and its association with ANT and prevented PTP opening which decreased I/R injury (Teodoro et al., 2018). The site of phosphorylation by GSK-3 was not identified in these studies, and it is unclear how GSK-3 localizes to the mitochondrial matrix since it lacks a mitochondrial targeting sequence (Rasola et al., 2010). In contrast to these studies, activation of the PI3K pathway resulted in Akt2 mediated phosphorylation of serine 31 (S31) of CypD in tumor cells. In this study the mutant S31A CypD, which cannot be phosphorylated, when expressed in CypD−/− cells lacked PPIase activity, exhibited defective mitochondrial bioenergetics with reduced glucose utilization, impaired oxygen consumption, and decreased ATP production and showed findings in accordance with sensitization of the PTP such as loss of membrane potential, discharge of cytochrome c, and reduced cell viability (Ghosh et al., 2015). Finally, it was reported recently that the germline deletion of the mitochondrial calcium uniporter results in increased CypD phosphorylation at serine 42 (S42), increased association of CypD with the ATP synthase, a proposed component of the PTP, and PTP sensitization to calcium (Parks et al., 2018). The kinase responsible for phosphorylation of S42 was not identified. Summarizing phosphorylation on S31 has been shown to be required for normal mitochondrial bioenergetics and cell viability while phosphorylation on S42 has been associated with PTP sensitization. Notably, since the site of phosphorylation was not identified in studies involving GSK-3 mediated phosphorylation (Rasola et al., 2010; Teodoro et al., 2018), there could be additional serine residues whose phosphorylation regulates the PTP. Thus, depending upon the site of phosphorylation, it appears to have different effects on CypD stimulation of PTP. Interestingly, PI3K increases phosphorylation of AKT, leading to phosphorylation and inhibition of GSK-3, which both lead to inhibition of the PTP. These findings suggest that AKT and GSK-3 phosphorylate different residues on CypD and are consistent with cardioprotection conferred by activation of the PI3K pathway (Tong et al., 2000, 2002).
CypD alters the cardiac mitochondrial acetylome and has been shown to be a substrate of the mitochondrial deacetylase sirtuin 3 (SIRT3) (Nguyen et al., 2013). Lysine 166 (K166, homologous to K167 in human CypD) has been reported to be deacetylated by SIRT3 while in SIRT3−/− mice CypD is hyperacetylated at this residue, leading to sensitization of the PTP and increased cell death in a model of transaortic constriction (Hafner et al., 2010) In addition, heart failure induced by coronary artery ligation in a rat model decreased the expression of SIRT3 and increased CypD acetylation. In line with these findings, another report showed that hypoxia in cardiac myoblasts also increased CypD acetylation while overexpression of SIRT3 attenuated this effect and was protective against cell death (Bochaton et al., 2015). Of interest, expressing the CypD acetylation mimic K166Q decreased calcium retention capacity and increased cell death in mouse embryonic fibroblasts, while the non-acetylated CypD mimic K166R had the opposite effects (Bochaton et al., 2015). In the same study, in vivo I/R injury increased CypD acetylation while ischemic postconditioning decreased infarct size and cardiac CypD acetylation but failed to protect in SIRT3−/− hearts. Similar effects of I/R injury in the heart and brain were attenuated by hypothermia and CypD inhibition in a rabbit model (Jahandiez et al., 2017). These data suggest that acetylation of CypD on K166 promotes cell death by sensitizing the PTP and that the protection with ischemic postconditioning is dependent on SIRT3 deacetylation of this residue. Notably, the acetylation of mitochondrial proteins is dependent on acetyl CoA abundance (Baeza et al., 2015; Weinert et al., 2015) and this may alter CypD acetylation, thereby altering PTP and perhaps regulating the synthasome assembly.
There are a large number of studies showing that CypD is a regulator of the PTP, an activator of cell death in I/R injury and that cyclosporine A desensitizes PTP and confers cardioprotection (Weinbrenner et al., 1998; Hausenloy et al., 2002; Argaud et al., 2005; Lim et al., 2007). However, the translation of these findings to the clinic has failed (Cung et al., 2015; Ottani et al., 2016). The reasons proposed for the lack of translation include the incomplete understanding of the cell-death pathway involving the PTP, the possibility that cyclosporine A did not reach the mitochondria in the first minutes of reperfusion, and the differences in comorbidities and co-medications between our patients and the experimental models (Heusch, 2017; Botker et al., 2020). Another possibility is that post-translational modifications of CypD might occur with comorbidities, co-medications, or aging, thus altering the regulation of PTP. Interestingly, there is evidence of increased CypD binding to the ATP synthase and decreased ATP synthase activity in the brain of aging mice (Gauba et al., 2017). The cause for the increased interaction might be a post-translational modification on CypD. In this case, it is unclear whether CsA would interact with CypD already bound to the ATP synthase and, thus, if it would protect in these mitochondria. In addition, the failure to translate may be due to the inhibition of a physiological function of CypD. As discussed, chronic genetic loss of CypD results in cardiac failure in response to both physiological and pathological stimuli, possibly suggesting a necessary function beyond cell death regulation (Elrod et al., 2010). A more complete understanding of the physiologic function of CypD and PTP mediated cell death is mandatory before pursuing CypD dependent PTP’s as a therapeutic target.
CypD can undergo multiple post-translational modifications and regulate mitochondrial bioenergetics and the PTP. However, most post-translational modifications of CypD have been studied in relation to the PTP. It is unclear how the majority of those post-translational modifications affect mitochondrial bioenergetics. In addition, although CypD sensitizes the opening of the PTP to stressful stimuli and possibly acts as an uncoupler by interacting directly with the ETC and the ATP synthase, its physiological role remains elusive.
GA wrote initial draft of the manuscript. EM edited the manuscript.
This work was supported by intramural funding from the National Heart Lung and Blood Institute at the National Institutes of Health (ZIA HL002066 and ZIA HL006059 to EM) and by the Leducq Foundation (6CVD04 to EM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acin-Perez, R., and Enriquez, J. A. (2014). The function of the respiratory supercomplexes: the plasticity model. Biochim. Biophys. Acta 1837, 444–450. doi: 10.1016/j.bbabio.2013.12.009
Agarwal, A., Wu, P. H., Hughes, E. G., Fukaya, M., Tischfield, M. A., Langseth, A. J., et al. (2017). Transient opening of the mitochondrial permeability transition pore induces microdomain calcium transients in astrocyte processes. Neuron 93, 587–605. doi: 10.1016/j.neuron.2016.12.034
Amanakis, G., Sun, J., Fergusson, M. M., Mcginty, S., Liu, C., Molkentin, J. D., et al. (2020). Cysteine 202 of cyclophilin D is a site of multiple post-translational modifications and plays a role in cardioprotection. Cardiovasc. Res. cvaa053. doi: 10.1093/cvr/cvaa053
Argaud, L., Gateau-Roesch, O., Muntean, D., Chalabreysse, L., Loufouat, J., Robert, D., et al. (2005). Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J. Mol. Cell. Cardiol. 38, 367–374. doi: 10.1016/j.yjmcc.2004.12.001
Baeza, J., Smallegan, M. J., and Denu, J. M. (2015). Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem. Biol. 10, 122–128. doi: 10.1021/cb500848p
Baines, C. P., Kaiser, R. A., Purcell, N. H., Blair, N. S., Osinska, H., Hambleton, M. A., et al. (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662. doi: 10.1038/nature03434
Bauer, T. M., and Murphy, E. (2020). Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ. Res. 126, 280–293. doi: 10.1161/CIRCRESAHA.119.316306
Bernardi, P., and von Stockum, S. (2012). The permeability transition pore as a Ca (2+) release channel: new answers to an old question. Cell Calcium 52, 22–27. doi: 10.1016/j.ceca.2012.03.004
Beutner, G., Alanzalon, R. E., and Porter, G. A. Jr. (2017). Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes. Sci. Rep. 7:14488. doi: 10.1038/s41598-017-14795-x
Bibli, S. I., Papapetropoulos, A., Iliodromitis, E. K., Daiber, A., Randriamboavonjy, V., Steven, S., et al. (2019). Nitroglycerine limits infarct size through S-nitrosation of cyclophilin D: a novel mechanism for an old drug. Cardiovasc. Res. 115, 625–636. doi: 10.1093/cvr/cvy222
Bochaton, T., Crola-Da-Silva, C., Pillot, B., Villedieu, C., Ferreras, L., Alam, M. R., et al. (2015). Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3-mediated deacetylation of cyclophilin D. J. Mol. Cell. Cardiol. 84, 61–69. doi: 10.1016/j.yjmcc.2015.03.017
Bonora, M., Morganti, C., Morciano, G., Pedriali, G., Lebiedzinska-Arciszewska, M., Aquila, G., et al. (2017). Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 18, 1077–1089. doi: 10.15252/embr.201643602
Botker, H. E., Cabrera-Fuentes, H. A., Ruiz-Meana, M., Heusch, G., and Ovize, M. (2020). Translational issues for mitoprotective agents as adjunct to reperfusion therapy in patients with ST-segment elevation myocardial infarction. J. Cell. Mol. Med. 24, 2717–2729. doi: 10.1111/jcmm.14953
Collins, M. O., Woodley, K. T., and Choudhary, J. S. (2017). Global, site-specific analysis of neuronal protein S-acylation. Sci. Rep. 7:4683. doi: 10.1038/s41598-017-04580-1
Cung, T. T., Morel, O., Cayla, G., Rioufol, G., Garcia-Dorado, D., Angoulvant, D., et al. (2015). Cyclosporine before PCI in patients with acute myocardial infarction. N. Engl. J. Med. 373, 1021–1031. doi: 10.1056/NEJMoa1505489
Davis, T. L., Walker, J. R., Campagna-Slater, V., Finerty, P. J., Paramanathan, R., Bernstein, G., et al. (2010). Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 8:e1000439. doi: 10.1371/journal.pbio.1000439
Devalaraja-Narashimha, K., Diener, A. M., and Padanilam, B. J. (2009). Cyclophilin D gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Renal Physiol. 297, F749–F759. doi: 10.1152/ajprenal.00239.2009
Dubinin, M. V., Talanov, E. Y., Tenkov, K. S., Starinets, V. S., Mikheeva, I. B., Sharapov, M. G., et al. (2020). Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis. Dis. 1866:165674. doi: 10.1016/j.bbadis.2020.165674
Elrod, J. W., Wong, R., Mishra, S., Vagnozzi, R. J., Sakthievel, B., Goonasekera, S. A., et al. (2010). Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120, 3680–3687. doi: 10.1172/JCI43171
Gauba, E., Guo, L., and Du, H. (2017). Cyclophilin D promotes brain mitochondrial F1FO ATP synthase dysfunction in aging mice. J. Alzheimers Dis. 55, 1351–1362. doi: 10.3233/JAD-160822
Ghosh, J. C., Siegelin, M. D., Vaira, V., Faversani, A., Tavecchio, M., Chae, Y. C., et al. (2015). Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J. Natl. Cancer Inst. 107:dju502. doi: 10.1093/jnci/dju502
Giorgio, V., Bisetto, E., Soriano, M. E., Dabbeni-Sala, F., Basso, E., Petronilli, V., et al. (2009). Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 284, 33982–33988. doi: 10.1074/jbc.M109.020115
Glancy, B., and Balaban, R. S. (2012). Role of mitochondrial Ca(2+) in the regulation of cellular energetics. Biochemistry 51, 2959–2973. doi: 10.1021/bi2018909
Hafner, A. V., Dai, J., Gomes, A. P., Xiao, C. Y., Palmeira, C. M., Rosenzweig, A., et al. (2010). Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2, 914–923. doi: 10.18632/aging.100252
Hausenloy, D. J., Maddock, H. L., Baxter, G. F., and Yellon, D. M. (2002). Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc. Res. 55, 534–543. doi: 10.1016/S0008-6363(02)00455-8
Heusch, G. (2017). Critical issues for the translation of cardioprotection. Circ. Res. 120, 1477–1486. doi: 10.1161/CIRCRESAHA.117.310820
Jahandiez, V., Cour, M., Bochaton, T., Abrial, M., Loufouat, J., Gharib, A., et al. (2017). Fast therapeutic hypothermia prevents post-cardiac arrest syndrome through cyclophilin D-mediated mitochondrial permeability transition inhibition. Basic Res. Cardiol. 112:35. doi: 10.1007/s00395-017-0624-3
Karch, J., Bround, M. J., Khalil, H., Sargent, M. A., Latchman, N., Terada, N., et al. (2019). Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 5:eaaw4597. doi: 10.1126/sciadv.aaw4597
Karwi, Q. G., Uddin, G. M., Ho, K. L., and Lopaschuk, G. D. (2018). Loss of metabolic flexibility in the failing heart. Front. Cardiovasc. Med. 5:68. doi: 10.3389/fcvm.2018.00068
Kohr, M. J., Aponte, A. M., Sun, J., Wang, G., Murphy, E., Gucek, M., et al. (2011a). Characterization of potential S-nitrosylation sites in the myocardium. Am. J. Physiol. Heart Circ. Physiol. 300, H1327–H1335. doi: 10.1152/ajpheart.00997.2010
Kohr, M. J., Sun, J., Aponte, A., Wang, G., Gucek, M., Murphy, E., et al. (2011b). Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ. Res. 108, 418–426. doi: 10.1161/CIRCRESAHA.110.232173
Kokoszka, J. E., Waymire, K. G., Levy, S. E., Sligh, J. E., Cai, J., Jones, D. P., et al. (2004). The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465. doi: 10.1038/nature02229
Leung, A. W., Varanyuwatana, P., and Halestrap, A. P. (2008). The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 283, 26312–26323. doi: 10.1074/jbc.M805235200
Lim, S. Y., Davidson, S. M., Hausenloy, D. J., and Yellon, D. M. (2007). Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovasc. Res. 75, 530–535. doi: 10.1016/j.cardiores.2007.04.022
Lim, S. Y., Hausenloy, D. J., Arjun, S., Price, A. N., Davidson, S. M., Lythgoe, M. F., et al. (2011). Mitochondrial cyclophilin-D as a potential therapeutic target for post-myocardial infarction heart failure. J. Cell. Mol. Med. 15, 2443–2451. doi: 10.1111/j.1582-4934.2010.01235.x
Linard, D., Kandlbinder, A., Degand, H., Morsomme, P., Dietz, K.-J., and Knoops, B. (2009). Redox characterization of human cyclophilin D: identification of a new mammalian mitochondrial redox sensor? Arch. Biochem. Biophys. 491, 39–45. doi: 10.1016/j.abb.2009.09.002
Lu, X., Kwong, J. Q., Molkentin, J. D., and Bers, D. M. (2016). Individual cardiac mitochondria undergo rare transient permeability transition pore openings. Circ. Res. 118, 834–841. doi: 10.1161/CIRCRESAHA.115.308093
Menazza, S., Wong, R., Nguyen, T., Wang, G., Gucek, M., and Murphy, E. (2013). CypD(−/−) hearts have altered levels of proteins involved in Krebs cycle, branch chain amino acid degradation and pyruvate metabolism. J. Mol. Cell. Cardiol. 56, 81–90. doi: 10.1016/j.yjmcc.2012.12.004
Milenkovic, D., Blaza, J. N., Larsson, N. G., and Hirst, J. (2017). The enigma of the respiratory chain supercomplex. Cell Metab. 25, 765–776. doi: 10.1016/j.cmet.2017.03.009
Nakagawa, T., Shimizu, S., Watanabe, T., Yamaguchi, O., Otsu, K., Yamagata, H., et al. (2005). Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658. doi: 10.1038/nature03317
Nguyen, T. T., Stevens, M. V., Kohr, M., Steenbergen, C., Sack, M. N., and Murphy, E. (2011). Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 286, 40184–40192. doi: 10.1074/jbc.M111.243469
Nguyen, T. T., Wong, R., Menazza, S., Sun, J., Chen, Y., Wang, G., et al. (2013). Cyclophilin D modulates mitochondrial acetylome. Circ. Res. 113, 1308–1319. doi: 10.1161/CIRCRESAHA.113.301867
Nicolli, A., Basso, E., Petronilli, V., Wenger, R. M., and Bernardi, P. (1996). Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, a cyclosporin A-sensitive channel. J. Biol. Chem. 271, 2185–2192. doi: 10.1074/jbc.271.4.2185
Ottani, F., Latini, R., Staszewsky, L., La Vecchia, L., Locuratolo, N., Sicuro, M., et al. (2016). Cyclosporine A in reperfused myocardial infarction: the multicenter, controlled, open-label CYCLE trial. J. Am. Coll. Cardiol. 67, 365–374. doi: 10.1016/j.jacc.2015.10.081
Park, J. S., Pasupulati, R., Feldkamp, T., Roeser, N. F., and Weinberg, J. M. (2011). Cyclophilin D and the mitochondrial permeability transition in kidney proximal tubules after hypoxic and ischemic injury. Am. J. Physiol. Renal Physiol. 301, F134–F150. doi: 10.1152/ajprenal.00033.2011
Parks, R. J., Menazza, S., Holmstrom, K. M., Amanakis, G., Fergusson, M., Ma, H., et al. (2018). Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc. Res. 115, 385–394. doi: 10.1093/cvr/cvy218
Pato, C. N., Davis, M. H., Doughty, M. J., Bryant, S. H., and Gruenstein, E. (1983). Increased membrane permeability to chloride in duchenne muscular dystrophy fibroblasts and its relationship to muscle function. Proc. Natl. Acad. Sci. U. S. A. 80, 4732–4736. doi: 10.1073/pnas.80.15.4732
Petronilli, V., Miotto, G., Canton, M., Brini, M., Colonna, R., Bernardi, P., et al. (1999). Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 76, 725–734. doi: 10.1016/S0006-3495(99)77239-5
Radhakrishnan, J., Bazarek, S., Chandran, B., and Gazmuri, R. J. (2015). Cyclophilin-D: a resident regulator of mitochondrial gene expression. FASEB J. 29, 2734–2748. doi: 10.1096/fj.14-263855
Rasola, A., Sciacovelli, M., Chiara, F., Pantic, B., Brusilow, W. S., and Bernardi, P. (2010). Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. U. S. A. 107, 726–731. doi: 10.1073/pnas.0912742107
Sanchez, G., Fernandez, C., Montecinos, L., Domenech, R. J., and Donoso, P. (2011). Preconditioning tachycardia decreases the activity of the mitochondrial permeability transition pore in the dog heart. Biochem. Biophys. Res. Commun. 410, 916–921. doi: 10.1016/j.bbrc.2011.06.095
Schinzel, A. C., Takeuchi, O., Huang, Z., Fisher, J. K., Zhou, Z., Rubens, J., et al. (2005). Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. U. S. A. 102, 12005–12010. doi: 10.1073/pnas.0505294102
Scorrano, L., Nicolli, A., Basso, E., Petronilli, V., and Bernardi, P. (1997). Two modes of activation of the permeability transition pore: the role of mitochondrial cyclophilin. Mol. Cell. Biochem. 174, 181–184. doi: 10.1023/A:1006887921810
Tarasov, A. I., Griffiths, E. J., and Rutter, G. A. (2012). Regulation of ATP production by mitochondrial Ca (2+). Cell Calcium 52, 28–35. doi: 10.1016/j.ceca.2012.03.003
Tavecchio, M., Lisanti, S., Bennett, M. J., Languino, L. R., and Altieri, D. C. (2015). Deletion of cyclophilin D impairs beta-oxidation and promotes glucose metabolism. Sci. Rep. 5:15981. doi: 10.1038/srep15981
Tavecchio, M., Lisanti, S., Lam, A., Ghosh, J. C., Martin, N. M., O’connell, M., et al. (2013). Cyclophilin D extramitochondrial signaling controls cell cycle progression and chemokine-directed cell motility. J. Biol. Chem. 288, 5553–5561. doi: 10.1074/jbc.M112.433045
Teodoro, J. S., Varela, A. T., Duarte, F. V., Gomes, A. P., Palmeira, C. M., and Rolo, A. P. (2018). Indirubin and NAD(+) prevent mitochondrial ischaemia/reperfusion damage in fatty livers. Eur. J. Clin. Invest. 48:e12932. doi: 10.1111/eci.12932
Tong, H., Chen, W., Steenbergen, C., and Murphy, E. (2000). Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ. Res. 87, 309–315. doi: 10.1161/01.RES.87.4.309
Tong, H., Imahashi, K., Steenbergen, C., and Murphy, E. (2002). Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase--dependent pathway is cardioprotective. Circ. Res. 90, 377–379. doi: 10.1161/01.RES.0000012567.95445.55
Uchino, H., Minamikawa-Tachino, R., Kristian, T., Perkins, G., Narazaki, M., Siesjo, B. K., et al. (2002). Differential neuroprotection by cyclosporin A and FK506 following ischemia corresponds with differing abilities to inhibit calcineurin and the mitochondrial permeability transition. Neurobiol. Dis. 10, 219–233. doi: 10.1006/nbdi.2002.0514
Walsh, C. T., Zydowsky, L. D., and Mckeon, F. D. (1992). Cyclosporin A, the cyclophilin class of peptidylprolyl isomerases, and blockade of T cell signal transduction. J. Biol. Chem. 267, 13115–13118.
Warne, J., Pryce, G., Hill, J. M., Shi, X., Lenneras, F., Puentes, F., et al. (2016). Selective inhibition of the mitochondrial permeability transition pore protects against neurodegeneration in experimental multiple sclerosis. J. Biol. Chem. 291, 4356–4373. doi: 10.1074/jbc.M115.700385
Weinbrenner, C., Liu, G. S., Downey, J. M., and Cohen, M. V. (1998). Cyclosporine A limits myocardial infarct size even when administered after onset of ischemia. Cardiovasc. Res. 38, 676–684. doi: 10.1016/S0008-6363(98)00064-9
Weinert, B. T., Moustafa, T., Iesmantavicius, V., Zechner, R., and Choudhary, C. (2015). Analysis of acetylation stoichiometry suggests that SIRT3 repairs nonenzymatic acetylation lesions. EMBO J. 34, 2620–2632. doi: 10.15252/embj.201591271
Keywords: cyclophilin D, permeability transition pore, mitochondrial function, ATP synthase, cyclosporine A, peptidyl-prolyl cis-trans isomerase
Citation: Amanakis G and Murphy E (2020) Cyclophilin D: An Integrator of Mitochondrial Function. Front. Physiol. 11:595. doi: 10.3389/fphys.2020.00595
Edited by:
Orian S. Shirihai, University of California, Los Angeles, United StatesReviewed by:
John W. Elrod, Lewis Katz School of Medicine at Temple University, United StatesCopyright © 2020 Amanakis and Murphy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georgios Amanakis, Z2VvcmdlLmFtYW5ha2lzQG5paC5nb3Y=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.