 Shuang Yu1†
Shuang Yu1† Ping Chen
Ping Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 18 June 2020
Sec. Striated Muscle Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00539
This article is part of the Research Topic Pathogenic Mechanisms in Cardiac and Skeletal Muscle Diseases View all 15 articles
The roles of the histone demethylase JMJD1C in cardiac hypertrophy remain unknown. JMJD1C was overexpressed in hypertrophic hearts of humans and mice, whereas the histone methylation was reduced. Jmjd1c knockdown repressed the angiotensin II (Ang II)-mediated increase in cardiomyocyte size and overexpression of hypertrophic genes in cardiomyocytes. By contrast, JMJD1C overexpression promoted the hypertrophic response of cardiomyocytes. Our further molecular mechanism study revealed that JMJD1C regulated AMP-dependent kinase (AMPK) in cardiomyocytes. JMJD1C did not influence LKB1 but repressed Camkk2 expression in cardiomyocytes. Inhibition of CAMKK2 with STO609 blocked the effects of JMJD1C on AMPK. AMPK knockdown blocked the inhibitory functions of JMJD1C knockdown on Ang II-induced hypertrophic response, whereas metformin reduced the functions of JMJD1C and repressed the hypertrophic response in cardiomyocytes.
Currently, cardiovascular disease has become the leading cause of death all around the world. Cardiac hypertrophy is a pathological foundation of diverse cardiovascular diseases, including heart failure and hypertension (Veselka et al., 2017). The heart of mature mammals shows the low potential of cardiomyocyte proliferation. When the heart is injured and cardiomyocyte apoptosis occurs, the number of cardiomyocytes decreases (Nakamura and Sadoshima, 2018). The cardiomyocytes are unable to proliferate to support the increased demand of the heart. Instead, the cardiomyocytes undergo growth or hypertrophy to satisfy the demand. Pathological cardiac hypertrophy is a fundamental mechanism underlying diverse cardiovascular diseases, and sustained hypertrophy leads to arrhythmia and heart failure (Hou and Kang, 2012).
Metabolic dysfunction is a core mechanism underlying cardiac hypertrophy. Under physiological conditions, the cardiomyocytes use fatty acids to support the energy demand of the heart (Noordali et al., 2018). However, when pathological cardiac hypertrophy occurs, the cardiomyocytes undergo a metabolic switch (Riehle and Abel, 2016; Noordali et al., 2018). In hypertrophic cardiomyocytes, the cells prefer to utilize glucose as the energy source (Riehle and Abel, 2016). The metabolism of cardiomyocytes is regulated by several core regulators, including Sirtuins, AMPK, FoxO, and insulin signaling (Doenst et al., 2013; Riehle and Abel, 2016). For example, AMPK signaling is repressed during cardiac hypertrophy, and cardiac-specific knockout of AMPK promoted stress-induced cardiac hypertrophy (Salt and Hardie, 2017). By contrast, metformin-mediated activation of AMPK prevented metabolic dysfunction and cardiac hypertrophy as well as heart failure (Daskalopoulos et al., 2016). Generally, AMPK activation is induced by the calcium/calmodulin–dependent protein kinase (CAMKK2) or the AMP-dependent liver kinase B1 (LKB1) (Steinberg and Carling, 2019). Inactivation of either LKB1 or CAMKK2 could facilitate the development of metabolic dysfunction and cardiac hypertrophy (Dolinsky et al., 2009; Watanabe et al., 2014). However, the mechanism by which the AMPK signaling is regulated during cardiac hypertrophy is not fully understood.
In general, H3K4 (lysine 4 at histone H3) methylation is correlated with gene activation, whereas the methylation of H3K9 (H3K9me) and H3K27 (H3K27me) is related to the repression of gene transcription (Greer and Shi, 2012). The demethylation of H3K9 is generally controlled by the Jumonji C domain-containing proteins (JMJD), which include the PHF8, JMJD1 family, and JMJD2 family (Jambhekar et al., 2019). The histone demethylase JMJD1C can remove the methylation motif from H3K9me1, H3K9me2, and H3K9me3 (Michalak et al., 2019). JMJD1C and WHISTLE mediate the balance of histone methylation by WHISTLE and demethylation by JMJD1C with WHISTLE acting as a transcriptional repressor during testis development in mice (Kim et al., 2010). During DNA break repair, the histone demethylase JMJD1C targets MDC1 to regulate the response of chromatin to DNA breaks mediated by RNF8 and BRCA1 (Watanabe et al., 2013). JMJD1C also regulates the expression of miR-302 to inhibit the differentiation of human embryonic stem cells to neurons (Wang et al., 2014). In addition, JMJD1C ensures cellular self-renewal and reprogramming by controlling the expression of microRNAs in mouse embryonic stem cells (Xiao et al., 2017). JMJD1C is also required for the maintenance of leukemia. JMJD1C depletion induces a growth defect that is primarily attributed to the increase in apoptosis of leukemia cells from either mouse or human (Sroczynska et al., 2014). JMJD1C is critically involved in the survival of acute myeloid leukemia cells by serving as the coactivator of pivotal transcription factors (Chen et al., 2015). Furthermore, JMJD1C essentially contributes to MLL-AF9/HOXA9-mediated self-renewal of leukemia stem cells (Zhu et al., 2016). Partially, JMJD1C-induced metabolic dysfunction contributes to HOXA9-dependent leukemogenesis (Lynch et al., 2019), which implies that JMJD1C may be a regulator of cellular metabolism. The role of JMJD1C in the cardiovascular system is not known.
Here, we investigate the roles of JMJD1C during cardiac hypertrophy in humans and mice. JMJD1C was significantly overexpressed in hypertrophic hearts of humans and mice, which was coupled with the downregulation of the methylation of H3K9. JMJD1C promoted cardiomyocyte hypertrophy induced by Ang II, which relied on the AMPK signaling in a CAMKK2-dependent manner.
Ten tissue samples of patients with hypertrophic cardiomyopathy and 10 from control donors were enrolled in this study. The hypertrophic heart tissues were obtained from patients who were diagnosed with hypertrophic cardiomyopathy (HCM). The control heart tissues were obtained from vehicle accident victims. The samples were obtained and stored in liquid nitrogen after informed consent was signed by the patients or the families of the donors. The study was approved by the institutional review boards for a clinical study at Jiamusi University.
Cardiac hypertrophy in 8-week-old male C57BL/6N mice was induced by chronic infusion of angiotensin II (Sigma, 1.5 mg/kg/day) for a continuous 4 weeks as described previously (Tang et al., 2017). In brief, an Ang II-contained minipump (ALZET 2004) was placed into the mouse subcutaneously. Mice and SD rats were purchased from Charles River. The protocol for the animal study was approved by the ethics review boards at Jiamusi University.
Cardiomyocytes were isolated from the heart ventricles of 1- to 3-day neonatal SD rats as described previously (Tang et al., 2016). Neonatal rat cardiomyocytes (NRCMs) were then cultured in DMEM medium supplied with 10% fetal bovine serum (FBS, Gibco), 1% antibiotic-antimycotic (ThermoFisher) for 48 h. The NRCMs were cultured in DMEM with 1% fetal bovine serum for 24 h and then the hypertrophy of NRCMs was induced by angiotensin II (1 μM) treatment for 48 h as reffed from previous publications (Luo et al., 2017; Tang et al., 2017). Cardiomyocyte size was measured with Image J software. For each group at each experiment, >100 cardiomyocytes were quantified for average cardiomyocyte size, and the results of three independent experiments were shown.
For gene knockdown or overexpression, the adenovirus system was applied. Adenoviral vectors expressing rat Jmjd1c (Ad-Jmjd1c), control construct (Ad-Ctrl), shJmjd1c (Ad-shJmjd1c, shRNA sequence: GCGCTGACCTTCAAACCATTG), or shPrkaa1 (Ad-shPrkaa1, shRNA sequence: GCACGAGTTGACTGGACATAA) and negative control (Ad-shCtrl) were obtained using the AdEasy Vector kit (Jia et al., 2014).
The cultured cells and heart tissues were washed with cold PBS and then subjected to RNA extraction with TRIZOL reagent (ThermoFisher). Next, the SuperScript III first-strand synthesis system kit (Invitrogen) was applied for cDNA synthesis with 1 ug of total RNA. Finally, SYBR green real-time PCR master mixes (Invitrogen) were applied to analyze the expression of target genes with the cDNA. The primers are shown in Table 1. The double delta CT method was applied for the quantification of mRNA expression.

Table 1. Primers used for quantitative real-time PCR.
The cultured cells and heart tissues were washed with cold PBS and then subjected to protein extraction with RIPA buffer (Millipore) supplemented with a phosphatase inhibitor cocktail (Roche) and a protease inhibitor cocktail (Roche). Protein concentration was measured with a BCA kit (ThermoFisher). SDS-PAGE was applied to separate the proteins in gel with 30 ug of total protein; then the proteins were transferred to PVDF membranes (Beyotime) followed by blockade with 5% fat-free milk in TBST for 1 h. Then, the membranes were washed with TBST three times and incubated with primary antibodies at 4°C overnight. The next day, the membranes were washed and incubated with HRP-conjected secondary antibodies. Finally, the expression of proteins was analyzed with the chemiluminescent Western blot detection kit (Millipore). The following primary antibodies were used: anti-Tubulin antibody (Cell Signaling Technology, #2144), anti-JMJD1C antibody (ThermoFisher, #PA5-20804), anti-Histone H3 antibody (Abcam, #ab1791), anti-H3K9me1 antibody (Active Motif, #39249), anti-H3K9me2 antibody (Active Motif, #39683), anti-H3K9me3 antibody (Active Motif, #61013), anti-AMPK antibody (Proteintech, #66536-1-Ig), anti-pAMPK antibody (Cell Signaling Technology, #2535), anti-ACC antibody (Proteintech, #67373-1-Ig), anti-pACC antibody (Cell Signaling Technology, #11818), anti-LKB1 antibody (Santa Cruz Biotechnology, #sc-32245), anti-pLKB1 antibody (Santa Cruz Biotechnology, #sc-271924), anti-CAMKK2 antibody (ThermoFisher, #PA5-69921), anti-PGC1a antibody (Cell Signaling Technology, #2178), anti-O-GlcNAcylation antibody (Abcam, #ab2739), anti-p-p70S6K1 antibody (Cell Signaling Technology, #9205), anti-p70S6K1 antibody (Cell Signaling Technology, #2708), anti-p-eEF2 antibody (Cell Signaling Technology, #2331), anti-eEF2 antibody (Cell Signaling Technology, #2332), anti-H3K4me1 antibody (Abcam, #ab8895), anti-H3K4me3 antibody (Abcam, #ab8580), anti-H3K9ac antibody (Active Motif, #61663).
A ChIP assay was performed with the chromatin immunoprecipitation (ChIP) assay kit (17-295) with ChIP-grade primary antibodies. Immunoprecipitated chromatin fragments were quantified by SYBR-based qRT-PCR, normalized using the input.
The protein synthesis assay was performed by analyzing the incorporation of [14C]-phenylalanine into proteins of cultured NRVMs as described previously (Sundaresan et al., 2009).
The statistical analysis was performed with SPSS software, and all the values are expressed as mean ± SD. Student's t-test was applied for the analysis of the difference between the two groups. One-way ANOVA followed by Tukey's post hoc test was used when more than two groups exist.
To understand the functions of JMJD1C in cardiac hypertrophy, JMJD1C expression was examined in hypertrophic hearts of humans and mice. We collected heart tissues from 10 cases of hypertrophic cardiomyopathy (HCM) and from 10 control donors. We confirmed the upregulation of hypertrophy-related fetal genes (e.g., ANP, BNP, and MYH7) in HCM tissues compared with the control donors (Figure 1A). Compared with control tissues, the expression levels of JMJD1C mRNA and protein were significantly increased in HCM tissues (Figures 1B,C). Next, we observed that the levels of H3K9me1, H3Kme2, and H3K9me3 were downregulated in HCM tissues, which was associated with the upregulation of JMJD1C (Figure 1C). In addition, cardiac hypertrophy was induced in mice by chronic infusion of Ang II for 4 weeks. The overexpression of hypertrophy-related fetal genes was confirmed in hypertrophic hearts (Figure 1D). In hypertrophic hearts, the expression of JMJD1C was remarkably increased (Figures 1E,F), and the levels of H3K9me1, H3K9me2, and H3K9me3 were decreased (Figure 1F). In addition, we observed the increase in methylation of H3K4 (H3K4me1 and H3K4me3) as well as acetylation of H3K9 in hypertrophic mouse hearts (Supplementary Figure 1). Collectively, these findings demonstrated that JMJD1C was overexpressed in cardiac hypertrophy, which was associated with the downregulation of methylation of H3K9.
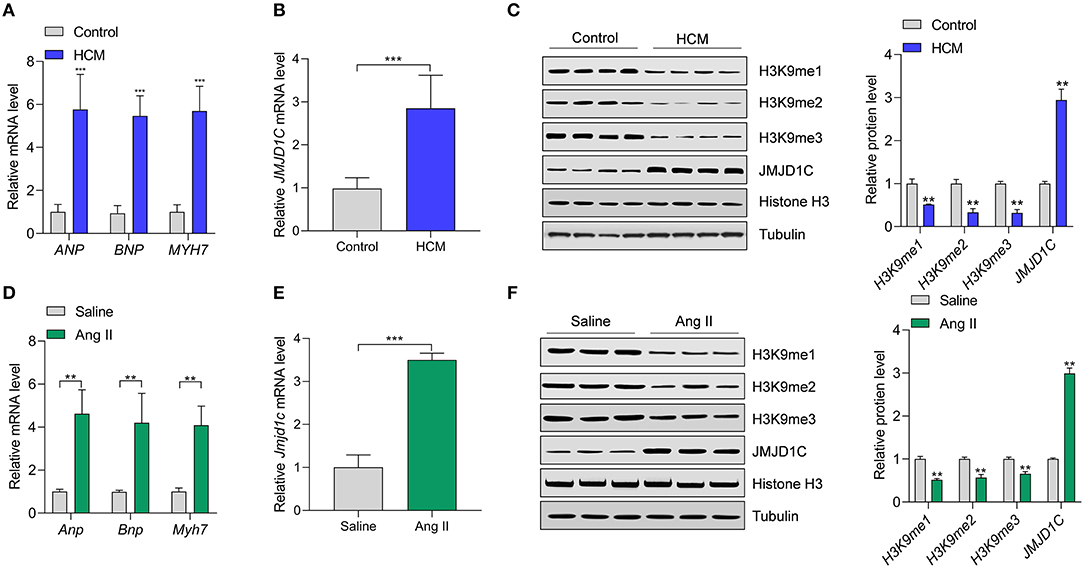
Figure 1. JMJD1C expression is increased in hypertrophic hearts. (A) Expression of hypertrophy-related fetal genes (ANP, BNP, MYH7) in hypertrophic hearts (HCM) of human patients (n = 10) and in control donors (n = 10). ***p < 0.001 vs. control. (B) JMJD1C mRNA expression in hypertrophic hearts of human patients (n = 10) and in control donors (n = 10). ***p < 0.001 vs. control. (C) JMJD1C protein and levels of related histone methylation marks (H3K9me1, H3K9me2, H3K9me3) in hypertrophic hearts of human patients and in control donors. **p < 0.01 vs. control. n = 4 in each group. (D) Hypertrophy-related fetal gene levels in the hearts of mice undergoing saline or Angiotensin II (Ang II) infusion for 4 weeks. **p < 0.01 vs. saline. n = 5 in each group. (E) Jmjd1c mRNA expression in control and hypertrophic hearts of mice treated as in (D). ***p < 0.001 vs. saline. n = 5 in each group. (F) Levels of JMJD1C protein and related histone methylation marks (H3K9me1, H3K9me2, H3K9me3) in the control and hypertrophic hearts of mice treated as in (D). In (C,F), a representative tubulin blot is shown as control, and all the loading controls were very similar. **p < 0.01 vs. saline. n = 4 in each group.
Next, we investigated the roles of JMJD1C in the regulation of cardiac hypertrophy using neonatal rat cardiomyocyte (NRCMs). We knocked down the expression of Jmjd1c in NRCMs with adenovirus-mediated shRNA (Figure 2A) and induced cardiomyocyte hypertrophy with Ang II. Treatment with Ang II increased the size of cardiomyocytes and led to overexpression of the hypertrophy-related fetal genes Anp, Bnp, and Myh7. Knockdown of Jmjd1c repressed hypertrophy of cardiomyocytes induced by Ang II treatment (Figures 2B,C). We also overexpressed Jmjd1c in NRCMs with adenovirus-mediated overexpression (Figure 2D). Jmjd1c overexpression promoted the Ang II-mediated increase in the size of cardiomyocytes and the overexpression of hypertrophy-related fetal markers (Figures 2E,F). Protein synthesis is a hallmark of cardiac hypertrophy (Tang et al., 2020a); thus, we tested the effects of JMJD1C on protein synthesis by performing [14C]-phenylalanine incorporation assay. We observed that JMJD1C knockdown reduced Ang II-induced protein synthesis (Figure 3A), whereas JMJD1C overexpression promoted Ang II-induced protein synthesis (Figure 3B). In addition, we tested the effects of JMJD1C on proteins that critically participate in protein synthesis. We analyzed the phosphorylations of p70S6K1 and eEF2. The results showed that JMJD1C knockdown inhibited the phosphorylations of p70S6K1 and Eef2, whereas the opposite effects were observed in JMJD1C-overexpressed cardiomyocytes (Figures 3C,D). Taken together, these findings revealed that JMJD1C promotes cardiomyocyte hypertrophy partially via regulating protein synthesis.
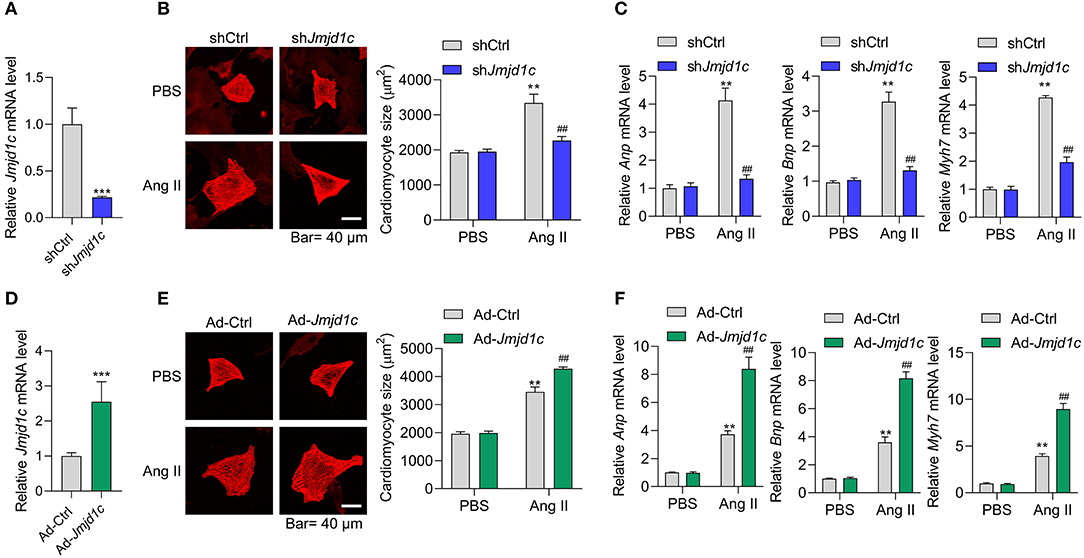
Figure 2. JMJD1C promotes cardiomyocyte hypertrophy. (A) Jmjd1c knockdown in neonatal rat cardiomyocytes (NRCMs). The cells were infected with adenovirus expressing shJmjd1c or shCtrl for 48 h. ***p < 0.001 vs. shCtrl. (B,C) Jmjd1c knockdown reduces the Ang II-mediated increase in the size of cardiomyocytes (B) and the expression of fetal genes (C). Cardiomyocytes infected with adenovirus expressing shJmjd1c or shCtrl were treated with Ang II (1 μM) for 48 h to induce hypertrophy. Then α-actinin staining was performed and cardiomyocyte size was analyzed. **p < 0.01 vs. shCtrl+PBS, ##p < 0.01 vs. shCtrl+Ang II. (D) Exogenous Jmjd1c overexpression in NRCMs. The cells were infected with adenovirus overexpressing Jmjd1c for 48 h. ***p < 0.001 vs. Ad-Ctrl. (E,F) Exogenous Jmjd1c overexpression enhances the increase in cardiomyocyte size (E) and hypertrophic fetal gene expression (F) induced by Ang II. NRCMs with/without Jmjd1c overexpression were treated with Ang II for 48 h to induce hypertrophy. Then cardiomyocyte size was analyzed. **p < 0.01 vs. Ad-Ctrl+PBS, ##p < 0.01 vs. Ad-Ctrl+Ang II. All results are from three independent experiments.
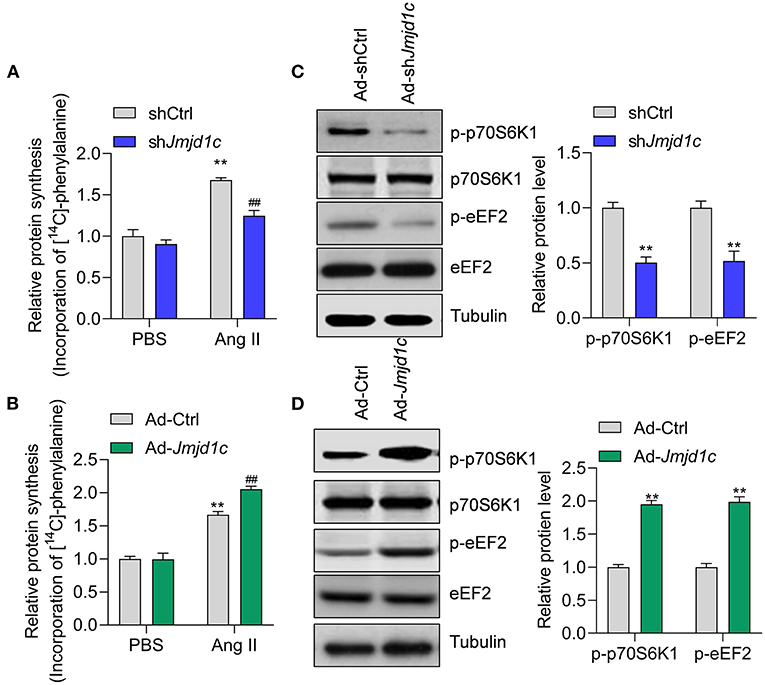
Figure 3. JMJD1C regulates protein synthesis in cardiomyocytes. (A) JMJD1C knockdown reduces Ang II-induced protein synthesis. **p < 0.01 vs. Ad-shCtrl+PBS; ##p < 0.01 vs. Ad-shCtrl+Ang II. n = 3 in each group. (B) JMJD1C overexpression promotes Ang II-induced protein synthesis. **p < 0.01 vs. Ad-Ctrl+PBS; ##p < 0.01 vs. Ad-Ctrl+Ang II. n = 3 in each group. (C) JMJD1C knockdown reduces phosphorylation of p70S6K1 and eEF2 in Ang II-treated cardiomyocytes. **p < 0.01 vs. Ad-shCtrl. n = 3 in each group. (D) JMJD1C overexpression increases phosphorylation of p70S6K1 and eEF2 in Ang II-treated cardiomyocytes. **p < 0.01 vs. Ad-Ctrl. n = 3 in each group.
We next explored the mechanism of JMJD1C function in hypertrophy of cardiomyocytes. Metabolism is critically involved in the physiological and pathological progress of the heart (Tang et al., 2020b). The metabolic sensor AMPK serves as one of the key regulators of glucose and fatty acid metabolism of cardiomyocytes, and the deregulation of AMPK signaling participates in hypertrophic cardiomyopathy and other cardiac diseases. In this study, we observed that the phosphorylation levels of the kinase AMPK and its downstream factor ACC were significantly downregulated in hypertrophic hearts in humans and mice (Figures 4A,B). Additionally, we observed that Jmjd1c knockdown promoted the activation of AMPK signaling, whereas Jmjd1c overexpression repressed AMPK signaling activation in Ang II-treated NRCMs (Figures 4C,D). AMPK was reported to regulate cardiac hypertrophy by inducing PGC1a (Peroxisome proliferator–activated receptor gamma coactivator 1-alpha) expression and reducing protein O-GlcNAcylation (Watanabe et al., 2014; Gelinas et al., 2018). Indeed, we observed that Jmjd1c knockdown increased the PGC1a protein level and reduced total protein O-GlcNAcylation, which was blocked by AMPK knockdown (Supplementary Figure 2). Therefore, these findings support the notion that JMJD1C represses AMPK signaling during cardiac hypertrophy.
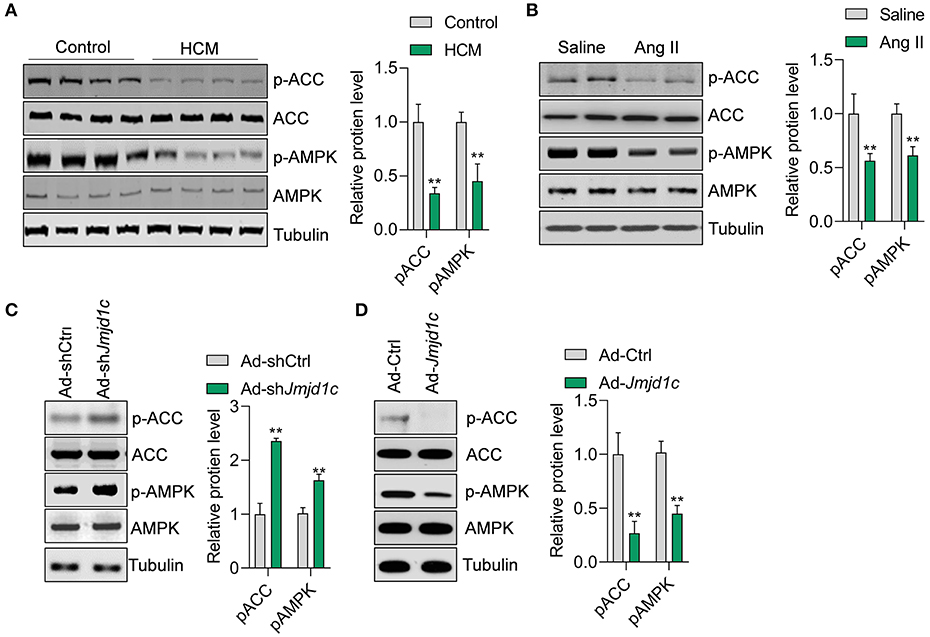
Figure 4. JMJD1C represses AMPK activation in cardiomyocytes. (A,B) The AMPK signaling pathway is repressed in hypertrophic hearts in humans and mice. **p < 0.01 vs. control or saline. n = 4 in each group. (C) Jmjd1c knockdown activates AMPK signaling. NRCMs were infected with adenovirus expressing shJmjd1c or shCtrl for 24 h, followed by Ang II treatment for an additional 24 h. **p < 0.01 vs. Ad-shCtrl. n = 3 in each group. (D) JMJD1C overexpression represses AMPK activation. NRCMs were infected with adenovirus expressing Jmjd1c for 24 h, followed by Ang II treatment for an additional 24 h. **p < 0.01 vs. Ad-Ctrl. n = 3 in each group.
We attempted to investigate the molecular mechanism underlying JMJD1C-mediated AMPK inactivation. AMPK is generally activated by two kinases, LKB1 and CAMKK2. We observed that LKB1 expression and activation and its interaction partners STRAD and MO25 were not affected by Jmjd1c knockdown or overexpression (Figures 5A,B). Interestingly, we observed that Jmjd1c knockdown increased the protein levels of CAMKK2, whereas Jmjc1c overexpression downregulated the protein levels of CAMKK2 in NRCMs (Figure 5C). Importantly, we showed that JMJD1C controlled the expression of CAMKK2 at the transcriptional level because JMJD1C overexpression reduced and knockdown increased the levels of CAMKK2 mRNA (Figure 5E). Next, we analyzed the mechanism by which JMJD1C regulated CAMKK2 expression. JMJD2A and JMJD3 were reported to regulate cardiac hypertrophy by epigenetically targeting the FHL1 (four-and-a-half LIM domains 1) and the β-MHC promoter, respectively (Zhang et al., 2011; Guo et al., 2018). Our chromatin immunoprecipitation assay did not reveal the enrichment of JMJD2A and JMJD3 at the CAMKK2 promoter nor of JMJD1C at the FHL1 or MHC promoters. By contrast, JMJD1C selectively bound the CAMKK2 promoter (Figure 5F). In addition, overexpression of JMJD1C reduced H3K9me1/2/3 levels at the CAMKK2 promoter (Figure 5G, Supplementary Figure 3). Finally, we treated NRCMs with the CAMKK2 inhibitor STO-609 and observed that STO-609 treatment blocked the roles of JMJD1C on the activation of the AMPK pathway (Figure 5H). These results demonstrated that JMJD1C controls AMPK signaling via a CAMKK2-dependent mechanism.
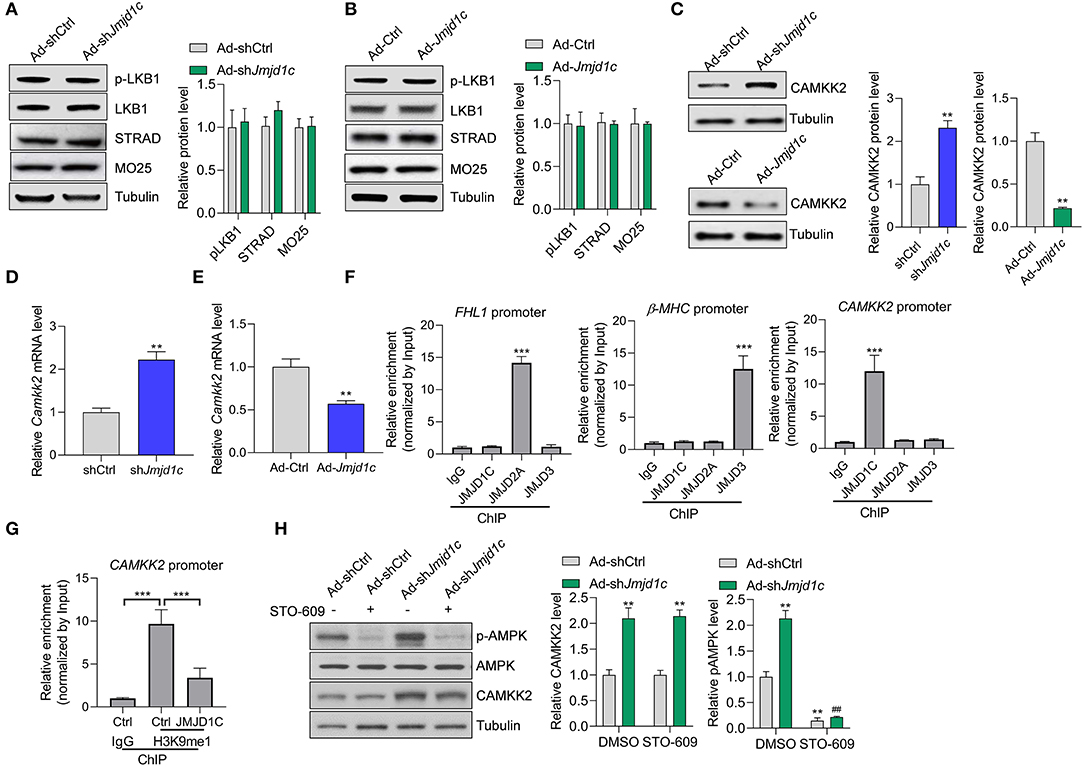
Figure 5. JMJD1C regulates AMPK in a CAMKK2-dependent manner. (A,B) JMJD1C does not affect activation of LKB1 or levels of its activation partners STRAD and MO25 in NRCMs. The cells were infected with adenovirus expressing Jmjd1c, shJmjd1c, or the control constructs for 24 h, followed by Ang II treatment for an additional 24 h. n = 3 in each group. (C) Effects of JMJD1C on CAMKK2 expression. NRCMs were treated as in (A). **p < 0.01 vs. Ad-Ctrl or shCtrl. n = 3 in each group. (D,E) Effects of JMJD1C on CAMKK2 mRNA expression. NRCMs were treated as in (A). **p < 0.01 vs. Ad-Ctrl or shCtrl. (F) Chromatin immunoprecipitation (ChIP) assay showing the binding of JMJD1C, JMJD2A, and JMJD3 on the FHL1, β-MHC, and CAMKK2 promoters. IgG was used for negative control, and the enrichment was normalized to the input. ***p < 0.001 vs. IgG. (G) ChIP assay showing that JMJD1C overexpression reduced H3K9me1 enrichment at the CAMKK2 promoter in cardiomyocytes. ***p < 0.001 (H). The inhibition of CAMKK2 blocks the effects of JMJD1C on AMPK activation in NRCMs. The cells were treated with adenovirus expressing shJmjd1c or shCtrl; then the cells were treated with Ang II and the CAMKK2 inhibitor STO-609 (20 μM) for an additional 24 h. All results are from three independent experiments. **p < 0.01 vs. Ad-shCtrl+PBS; ##p < 0.01 vs. Ad-shCtrl+STO-609. n = 3 in each group.
Finally, we investigated whether JMJD1C-mediated AMPK inactivation was critically involved in the function of JMJD1C. We knocked down the expression of AMPK (Prkaa1) in NRCMs (Figure 6A) and induced cardiomyocyte hypertrophy with Ang II. We observed that AMPK knockdown promoted the hypertrophic response induced by Ang II and blocked the prevention of cardiomyocyte hypertrophy mediated by Jmjd1c knockdown (Figures 6B,C). In addition, we observed that metformin, an AMPK activator, rescued AMPK activation in NRCMs with Jmjd1c overexpression (Figure 6D). Significantly, metformin treatment repressed cardiomyocyte hypertrophy and blocked the function of JMJD1C overexpression (Figures 6E,F). Finally, we also tested whether JMJD1C promoted cardiomyocyte hypertrophy via CAMKK2. We inhibited CAMKK2 with STO-609 or knocked down Camkk2 expression and observed that either inhibition of CAMKK2 with STO-609 or Camkk2 knockdown blocked the inhibitory effects of Jmjd1c knockdown on cardiomyocyte hypertrophy (Figures 6G,H). Therefore, AMPK signaling critically participated in the roles of JMJD1C during the hypertrophic response of cardiomyocytes.
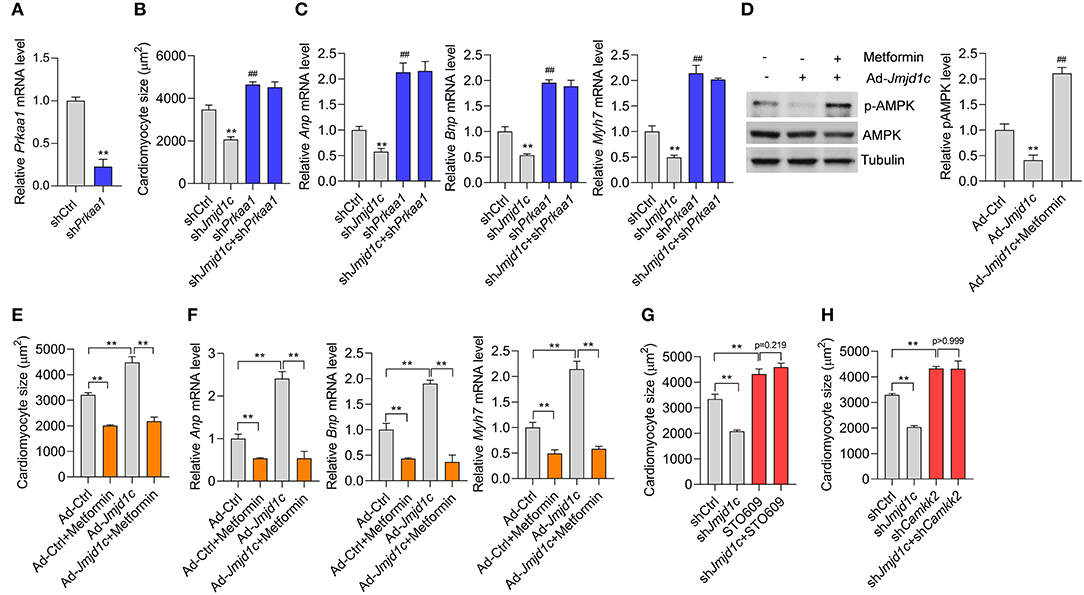
Figure 6. AMPK participates in the role of JMJD1C during cardiomyocyte hypertrophy. (A) Prkaa1 knockdown in cardiomyocytes. The cells were treated with adenovirus expressing shPrkaa1 or with a control virus for 48 h (B,C). Prkaa1 knockdown blocks the influence of Jmjd1c knockdown on Ang II-induced increase in cardiomyocytes. The cells with/without Prkaa1 or Jmjd1c knockdown were subjected to hypertrophy induction with Ang II for 48 h. Then cardiomyocyte size (B) and gene expression (C) were analyzed. **p < 0.01 vs. shCtrl. ##p < 0.01 vs. shCtrl (D). Metformin activates AMPK. The indicated cardiomyocytes were treated with metformin (1 mM) for 24 h. **p < 0.01 vs. Ad-Ctrl; ##p < 0.01 vs. Ad-Jmjd1c. n = 3 in each group (E,F). Metformin-mediated activation of AMPK blocks the influence of JMJD1C overexpression on cardiomyocyte hypertrophy induced by Ang II stimuli. NRCMs were infected with/without adenovirus carrying Jmjd1c for 24 h; then the cells were treated with Ang II (1 μM) metformin (1 mM) for an additional 48 h. Cardiomyocyte size (E) and gene expression (F) were analyzed **p < 0.01. (G) STO-609 blocks the influence of Jmjd1c knockdown on the Ang II-induced increase in cardiomyocytes. **p < 0.01. (H) Camkk2 knockdown blocks the influence of Jmjd1c knockdown on Ang II-induced increase in cardiomyocytes. **p < 0.01. All results are from three independent experiments.
Here we demonstrate that JMJD1C serves as an epigenetic regulator that is involved in pathological cardiac hypertrophy. The expression of JMJD1C mRNA and protein was increased during cardiac hypertrophy in humans and mice, which was associated with decreased methylation of H3K9. Further experimental evidence demonstrated that JMJD1C promoted the hypertrophic response of cardiomyocytes induced by Ang II. JMJD1C repressed AMPK through transcriptionally repressing the expression of CAMKK2 but not LKB1 in cardiomyocytes, which was involved in the roles of JMJD1C during cardiomyocyte hypertrophy.
Epigenetic regulators have been reported to be essential for the initiation and progress of cardiac hypertrophy. For instance, the histone deacetylase (HDACs) and Sirtuins participated in cardiac hypertrophy by targeting histone and non-histone targets (Li et al., 2019). The histone demethylase family is also critically involved in pathological cardiac hypertrophy. For instance, JMJD2A demethylated H3K9me3 and activated transcription of pro-hypertrophic genes. In animals, JMJD2A promoted cardiac hypertrophy (Zhang et al., 2011). KDM3A promoted pressure overload–induced cardiac hypertrophy and fibrosis. And KDM3A activated Timp1 expression with pro-fibrotic activity in cardiomyocytes. Interestingly, JIB-04, the pan-KDM inhibitor, repressed pathological cardiac hypertrophy and fibrosis (Zhang et al., 2018).
In this study, JMJD1C expression levels were increased in hypertrophic hearts in mice and humans. The upregulation of JMJD1C was associated with downregulated levels of H3K9me1, H3K9me2, and H3K9me3. We knocked down or overexpressed JMJD1C in cardiomyocytes and found that JMJD1C promoted the increase in the size of cardiomyocytes and expression of hypertrophy-related fetal genes, including Anp, Bnp, and Myh7, during Ang II-induced cardiomyocyte hypertrophy. Therefore, the JMJD family members are generally pro-hypertrophic epigenetic regulators (Zhang et al., 2011, 2018). However, our study was based on cardiomyocytes in vitro; further study is needed to explore the in vivo function of JMJD1C during cardiac hypertrophy with a cardiomyocyte-specific JMJD1C knockout mouse line.
The metabolic switch is a hallmark of hypertrophic cardiomyocytes. The metabolic sensor AMPK has critical functions in cardiac hypertrophy by diverse metabolism-dependent and independent mechanisms (Daskalopoulos et al., 2016). The previous study implicated the histone deacetylase SIRT2 in the activation of AMPK via LKB1 to repress cardiac hypertrophy (Tang et al., 2017). However, the negative epigenetic regulator of AMPK during cardiac hypertrophy remains unknown. Here we observed that JMJD1C repressed the activation of AMPK during cardiac hypertrophy. However, JMJD1C-mediated repression of AMPK signaling did not depend on LKB1, but rather on reducing CAMKK2 expression. Finally, our findings showed that AMPK activation with metformin blocked the effects of JMJD1C hyperexpression, whereas AMPK knockdown blockaded the effects of JMJD1C downregulation during cardiac hypertrophy. These findings demonstrated that the AMPK signaling pathway is essentially involved in JMJD1C-mediated function in pathological cardiac hypertrophy.
JMJD2A and JMJD3 were reported to regulate cardiac hypertrophy by epigenetically targeting FHL1 and MHC promoter, respectively (Zhang et al., 2011; Guo et al., 2018). We observed that JMJD2A was enriched at the FHL1 and JMJD3 at the beta-MHC promoter, respectively, but neither bound to the CaMKK2 promoter. In contrast, JMJD1C bound to the CaMKK2 promoter and regulated methylation of H3K9me1, implicating JMJD1C in the regulation of the expression of CaMKK2 via demethylating H3K9. In this study, we observed that JMJD1C bound the CaMKK2 promoter and reduced the H3K9me level, which was associated with CaMKK2 gene silence. This finding was contradictory to previous notions that H3K9me was a mark of gene repression and that JMJD1C was a transcriptional activator (Chen et al., 2015). Thus, the effects of JMJD1C on CaMKK2 expression may also rely on other mechanisms. For instance, the JMJD1C-mediated changes in the local chromatin 3-D structure at the CaMKK2 promoter may also recruit some transcriptional repressor. The transcriptional repressive roles were also observed in JMJD2A (Mallette and Richard, 2012; Li et al., 2014). In leukemia cells, Runx1 (Runt-related transcription factor 1) directly interacts with and recruits JMJD1C to target genes (Chen et al., 2015). In addition, JMJD1C regulates Runx1 target genes by maintaining low H3K9me1/2 levels (Chen et al., 2015). Interestingly, the promoter and 5'UTR region of the human CaMKK2 gene has consensus DNA-binding sequences for Runx1 (Racioppi and Means, 2012). Given that Runx1 also plays a critical role in the cardiovascular system (McCarroll et al., 2018), it will be very interesting to investigate whether Runx1 is also enriched at the CAMKK2 promoter. To this end, JMJD1C regulated CaMKK2 expression via a complex mechanism. In addition, we did not observe the effects of JMJD1C on total H3K27me3 (data not shown). As such, JMJD2A, JMJD3, and JMJD1C may show different enrichment at the genome and regulate the expression of different genes.
Taken together, here we identified JMJD1C as a new epigenetic regulator of cardiac hypertrophy by targeting the metabolic AMPK pathway.
The datasets generated for this study are available on request to the corresponding author.
The studies involving human participants were reviewed and approved by institutional review boards for a clinical study at Jiamusi University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by ethics review boards at Jiamusi University.
SY and PC designed the study. SY and YL performed most of the experiments. PC wrote the manuscript. QW performed cell culture and qPCR experiments. All authors contributed to the article and approved the submitted version.
This work was supported by Health Department of Heilongjiang Province (Grand No. 2018329).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.00539/full#supplementary-material
Chen, M., Zhu, N., Liu, X., Laurent, B., Tang, Z., Eng, R., et al. (2015). JMJD1C is required for the survival of acute myeloid leukemia by functioning as a coactivator for key transcription factors. Genes Dev. 29, 2123–2139. doi: 10.1101/gad.267278.115
Daskalopoulos, E. P., Dufeys, C., Bertrand, L., Beauloye, C., and Horman, S. (2016). AMPK in cardiac fibrosis and repair: actions beyond metabolic regulation. J. Mol. Cell. Cardiol. 91, 188–200. doi: 10.1016/j.yjmcc.2016.01.001
Doenst, T., Nguyen, T. D., and Abel, E. D. (2013). Cardiac metabolism in heart failure: implications beyond ATP production. Circ. Res. 113, 709–724. doi: 10.1161/CIRCRESAHA.113.300376
Dolinsky, V. W., Chan, A. Y., Robillard Frayne, I., Light, P. E., Des Rosiers, C., and Dyck, J. R. (2009). Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation 119, 1643–1652. doi: 10.1161/CIRCULATIONAHA.108.787440
Gelinas, R., Mailleux, F., Dontaine, J., Bultot, L., Demeulder, B., Ginion, A., et al. (2018). AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 9:374. doi: 10.1038/s41467-017-02795-4
Greer, E. L., and Shi, Y. (2012). Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357. doi: 10.1038/nrg3173
Guo, Z., Lu, J., Li, J., Wang, P., Li, Z., Zhong, Y., et al. (2018). JMJD3 inhibition protects against isoproterenol-induced cardiac hypertrophy by suppressing beta-MHC expression. Mol. Cell. Endocrinol. 477, 1–14. doi: 10.1016/j.mce.2018.05.009
Hou, J., and Kang, Y. J. (2012). Regression of pathological cardiac hypertrophy: signaling pathways and therapeutic targets. Pharmacol. Ther. 135, 337–354. doi: 10.1016/j.pharmthera.2012.06.006
Jambhekar, A., Dhall, A., and Shi, Y. (2019). Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 20, 625–641. doi: 10.1038/s41580-019-0151-1
Jia, Y.-Y., Lu, J., Huang, Y., Liu, G., Gao, P., Wan, Y.-Z., et al. (2014). The involvement of NFAT transcriptional activity suppression in SIRT1-mediated inhibition of COX-2 expression induced by PMA/Ionomycin. PLoS ONE 9:e97999. doi: 10.1371/journal.pone.0097999
Kim, S.-M., Kim, J.-Y., Choe, N.-W., Cho, I.-H., Kim, J.-R., Kim, D.-W., et al. (2010). Regulation of mouse steroidogenesis by WHISTLE and JMJD1C through histone methylation balance. Nucleic Acids Res. 38, 6389–6403. doi: 10.1093/nar/gkq491
Li, L.-L., Xue, A.-M., Li, B.-X., Shen, Y.-W., Li, Y.-H., Luo, C.-L., et al. (2014). JMJD2A contributes to breast cancer progression through transcriptional repression of the tumor suppressor ARHI. Breast Cancer Res. 16:R56. doi: 10.1186/bcr3667
Li, P., Ge, J., and Li, H. (2019). Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat. Rev. Cardiol. 17, 96–115. doi: 10.1038/s41569-019-0235-9
Luo, Y.-X., Tang, X., An, X.-Z., Xie, X.-M., Chen, X.-F., Zhao, X., et al. (2017). Sirt4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur. Heart J. 38:1389–1398. doi: 10.1093/eurheartj/ehw138
Lynch, J. R., Salik, B., Connerty, P., Vick, B., Leung, H., Pijning, A., et al. (2019). JMJD1C-mediated metabolic dysregulation contributes to HOXA9-dependent leukemogenesis. Leukemia 33, 1400–1410. doi: 10.1038/s41375-018-0354-z
Mallette, F. A., and Richard, S. (2012). JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5, Cell reports 2, 1233–1243. doi: 10.1016/j.celrep2012.09.033
McCarroll, C. S., He, W., Foote, K., Bradley, A., McGlynn, K., Vidler, F., et al. (2018). Runx1 deficiency protects against adverse cardiac remodeling after myocardial infarction. Circulation 137, 57–70. doi: 10.1161/CIRCULATIONAHA.117.028911
Michalak, E. M., Burr, M. L., Bannister, A. J., and Dawson, M. A. (2019). The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 20, 573–589. doi: 10.1038/s41580-019-0143-1
Nakamura, M., and Sadoshima, J. (2018). Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 15, 387–407. doi: 10.1038/s41569-018-0007-y
Noordali, H., Loudon, B. L., Frenneaux, M. P., and Madhani, M. (2018). Cardiac metabolism - A promising therapeutic target for heart failure. Pharmacol. Ther. 182, 95–114. doi: 10.1016/j.pharmthera.2017.08.001
Racioppi, L., and Means, A. R. (2012). Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J. Biol. Chem. 287, 31658–31665. doi: 10.1074/jbc.R112.356485
Riehle, C., and Abel, E. D. (2016). Insulin signaling and heart failure. Circ. Res. 118, 1151–1169. doi: 10.1161/CIRCRESAHA.116.306206
Salt, I. P., and Hardie, D. G. (2017). AMP-activated protein kinase: an ubiquitous signaling pathway with key roles in the cardiovascular system. Circ. Res. 120, 1825–1841. doi: 10.1161/CIRCRESAHA.117.309633
Sroczynska, P., Cruickshank, V. A., Bukowski, J.-P., Miyagi, S., Bagger, F. O., Walfridsson, J., et al. (2014). shRNA screening identifies JMJD1C as being required for leukemia maintenance. Blood 123, 1870–1882. doi: 10.1182/blood-2013-08-522094
Steinberg, G. R., and Carling, D. (2019). AMP-activated protein kinase: the current landscape for drug development. Nat. Rev. Drug Discov. 18, 527–551. doi: 10.1038/s41573-019-0019-2
Sundaresan, N. R., Gupta, M., Kim, G., Rajamohan, S. B., Isbatan, A., and Gupta, M. P. (2009). Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 119, 2758–2771. doi: 10.1172/JCI39162
Tang, X., Chen, X., and Chen, X.-F. (2020b). Short-chain fatty acid, acylation and cardiovascular diseases. Clin. Sci. 134, 657–676. doi: 10.1042/CS20200128
Tang, X., Chen, X.-F., Wang, N.-Y., Wang, X.-M., Liang, S.-T., Zheng, W., et al. (2017). SIRT2 acts as a cardioprotective deacetylase in pathological cardiac hypertrophy. Circulation 136, 2051–2067. doi: 10.1161/CIRCULATIONAHA.117.028728
Tang, X., Li, P.-H., and Chen, H.-Z. (2020a). Cardiomyocyte senescence and cellular communications within myocardial microenvironment. Front. Endocrinol. (Lausanne). 11:280. doi: 10.3389/fendo.2020.00280
Tang, X., Ma, H., Han, L., Zheng, W., Lu, Y.-B., Chen, X.-F., et al. (2016). SIRT1 deacetylates the cardiac transcription factor Nkx2.5 and inhibits its transcriptional activity. Sci. Rep. 6:36576. doi: 10.1038/srep36576
Veselka, J., Anavekar, N. S., and Charron, P. (2017). Hypertrophic obstructive cardiomyopathy. Lancet 389, 1253–1267. doi: 10.1016/S0140-6736(16)31321-6
Wang, J., Park, J. W., Drissi, H., Wang, X., and R.-,Xu, H. (2014). Epigenetic regulation of miR-302 by JMJD1C inhibits neural differentiation of human embryonic stem cells. J. Biol. Chem. 289, 2384–2395. doi: 10.1074/jbc.M113.535799
Watanabe, S., Horie, T., Nagao, K., Kuwabara, Y., Baba, O., Nishi, H., et al. (2014). Cardiac-specific inhibition of kinase activity in calcium/calmodulin-dependent protein kinase kinase-beta leads to accelerated left ventricular remodeling and heart failure after transverse aortic constriction in mice. PLoS ONE 9:e108201. doi: 10.1371/journal.pone.0108201
Watanabe, S., Watanabe, K., Akimov, V., Bartkova, J., Blagoev, B., Lukas, J., et al. (2013). JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1–mediated chromatin response to DNA breaks. Nat. Struct. Mol. Biol. 20, 1425–1433. doi: 10.1038/nsmb.2702
Xiao, F., Liao, B., Hu, J., Li, S., Zhao, H., Sun, M., et al. (2017). JMJD1C ensures mouse embryonic stem cell self-renewal and somatic cell reprogramming through controlling microRNA expression. Stem Cell Reports 9, 927–942. doi: 10.1016/j.stemcr.2017.07.013
Zhang, Q.-J., Chen, H.-Z., Wang, L., Liu, D.-P., Hill, J. A., and Liu, Z.-P. (2011). The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J. Clin. Invest. 121, 2447–2456. doi: 10.1172/JCI46277
Zhang, Q.-J., Tran, A. T., Wang, M., Ranek, M. J., Kokkonen-Simon, K. M., Gao, J., et al. (2018). Histone lysine dimethyl-demethylase KDM3A controls pathological cardiac hypertrophy and fibrosis. Nat. Commun. 9:5230. doi: 10.1038/s41467-018-07173-2
Keywords: cardiac hypertrophy, histone methylation, JMJD1C, AMPK, CAMKK2
Citation: Yu S, Li Y, Zhao H, Wang Q and Chen P (2020) The Histone Demethylase JMJD1C Regulates CAMKK2-AMPK Signaling to Participate in Cardiac Hypertrophy. Front. Physiol. 11:539. doi: 10.3389/fphys.2020.00539
Received: 02 March 2020; Accepted: 30 April 2020;
Published: 18 June 2020.
Edited by:
Xuejun Wang, University of South Dakota, United StatesReviewed by:
Manish Gupta, University of Central Florida, United StatesCopyright © 2020 Yu, Li, Zhao, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Chen, cGluZ2NoZW5qbXN1QHNvaHUuY29t; bWhuaHVhQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.