
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 08 May 2020
Sec. Membrane Physiology and Membrane Biophysics
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00332
This article is part of the Research TopicInvolvements of TRP Channels, Oxidative Stress and Apoptosis in Neurodegenerative DiseasesView all 12 articles
 Daniele Maria-Ferreira1,2
Daniele Maria-Ferreira1,2 Natalia Mulinari Turin de Oliveira1,2
Natalia Mulinari Turin de Oliveira1,2 Liziane Cristine Malaquias da Silva1,2
Liziane Cristine Malaquias da Silva1,2 Elizabeth Soares Fernandes1,2*
Elizabeth Soares Fernandes1,2*Parkinson’s disease (PD) represents one of the most common multifactorial neurodegenerative disorders affecting the elderly population. It is associated with the aggregation of α-synuclein protein and the loss of dopaminergic neurons in the substantia nigra pars compacta of the brain. The disease is mainly represented by motor symptoms, such as resting tremors, postural instability, rigidity, and bradykinesia, that develop slowly over time. Parkinson’s disease can also manifest as disturbances in non-motor functions. Although the pathology of PD has not yet been fully understood, it has been suggested that the disruption of the cellular redox status may contribute to cellular oxidative stress and, thus, to cell death. The generation of reactive oxygen species and reactive nitrogen intermediates, as well as the dysfunction of dopamine metabolism, play important roles in the degeneration of dopaminergic neurons. In this context, the transient receptor potential channel canonical (TRPC) sub-family plays an important role in neuronal degeneration. Additionally, PD gene products, including DJ-1, SNCA, UCH-L1, PINK-1, and Parkin, also interfere with mitochondrial function leading to reactive oxygen species production and dopaminergic neuronal vulnerability to oxidative stress. Herein, we discuss the interplay between these various biochemical and molecular events that ultimately lead to dopaminergic signaling disruption, highlighting the recently identified roles of TRPC in PD.
Neurological disorders continue to increase in tandem with longer lifespans in populations, with aging remaining the biggest risk factor for developing neurodegenerative diseases. Parkinson’s disease (PD) is one of the most common multifactorial neurodegenerative disorders. Indeed, it affects approximately 2% of the elderly population and 4% of individuals aged over 80 years (Berman and Nichols, 2019).
Disease onset usually occurs at the age of 65–70 years (Marino et al., 2019). However, its pathological changes can be observed as early as 20 years prior to the appearance of motor symptoms and include unspecific signs such as fatigue, hyposmia, and constipation (Hawkes et al., 2010). Motor symptoms develop slowly over time and are the main clinical characteristics of PD. These include dysfunctions of the somatomotor system such as resting tremors, rigidity, bradykinesia, and postural instability (Schapira et al., 2017). In turn, there is a progressive physical limitation, in addition to impairments in non-motor functions such as neuropsychiatric (sleep disorders, depression, and dementia) and autonomic symptoms (bladder and gastrointestinal alterations) (Sakakibara et al., 2012; Fasano et al., 2015).
The pathogenesis of PD is not completely understood. However, different studies have contributed to the dissection and determination of some of the mechanisms involved in its establishment and progression. Classically, the progressive neurodegeneration in PD is associated with the aggregation of α-synuclein, a small lipid-binding protein, into structures called Lewy bodies in the substantia nigra pars compacta (SNpc).
Accumulation of dopamine (DA) and DA products has also been pointed as a potential mechanism involved in neuronal death (Mullin and Schapira, 2015). Indeed, the neurotoxic effects of the endogenous DA derivative N-methyl-(R)-salsolinol (NMSAL) (Naoia et al., 2002) was shown to induce oxidative stress and decrease the levels of reduced glutathione (GSH) in dopaminergic SH-SY5Y cells (Wanpen et al., 2004). The progressive loss of DA neurons leads to a subsequent reduction of DA levels. All these alterations contribute to an abnormal neuronal functioning, and thus, to motor deficiency and worsening of the quality of life of patients at advanced stages of PD (Magrinelli et al., 2016).
For instance, many studies have provided substantial evidence of the role of neuroinflammation (Tansey and Goldberg, 2010), mitochondrial dysfunction (Park et al., 2018, 2019), and oxidative and nitrosative stresses in PD (Puspita et al., 2017). In this context, disruption of neuronal calcium ion (Ca2+) homeostasis in the central nervous system plays a critical role in the cascade of events that culminates in the degeneration of dopaminergic neurons (Zaichick et al., 2017). Also, a correlation between reactive oxygen species (ROS) production and Ca2+ channel activation has already been explored (Görlach et al., 2015).
Recent studies have focused in the identification of a link between Ca2+-mediated signaling and neuroinflammation (Sama and Norris, 2013). It observed an association between neurodegeneration, mitochondrial dysfunction, and, oxidative and nitrosative stresses (Celsia et al., 2009). This evidence points to a role for transient receptor potential channels (TRP) in PD (Takahashi and Mori, 2011).
First discovered in Drosophila melanogaster as key molecules in phototransduction, the TRP channels comprise a family of non-selective cation channels that are widely expressed on mammalian cells, including neurons and different types of non-neuronal cells. They are distributed in six different subfamilies: ankyrin (TRPA1), canonical (TRPC1-7), melastatin (TRPM1-8), mucolipin (TRPML1-3), polycystin (TRPP1-3), and vanilloid (TRPV1-6). Their broad tissue expression confers them the ability to influence different pathologies and physiological states. In this context, it is now known that these channels participate in the development and maintenance of inflammation and pain, are important sensors of molecules such as lipids and ROS, and are involved in thermoregulation, tissue remodeling, and neuronal plasticity, among other responses.
Reactive oxygen species and reactive nitrogen intermediates (RNIs) are natural byproducts necessary for cellular homeostasis (Liguori et al., 2018) (Figure 1). ROS are formed during metabolic redox reactions and include hydrogen peroxide (H2O2), singlet oxygen (1O2), hydroxyl (∙OH), and superoxide (O2∙−) radicals (Sies et al., 2017). RNIs are produced in neuronal cells from arginine by the neuronal nitric oxide synthase (nNOS) and include nitric oxide (NO∙), nitrite (NO2), and S-nitrosothiols and peroxynitrite (OONO−) (Adams et al., 2015).
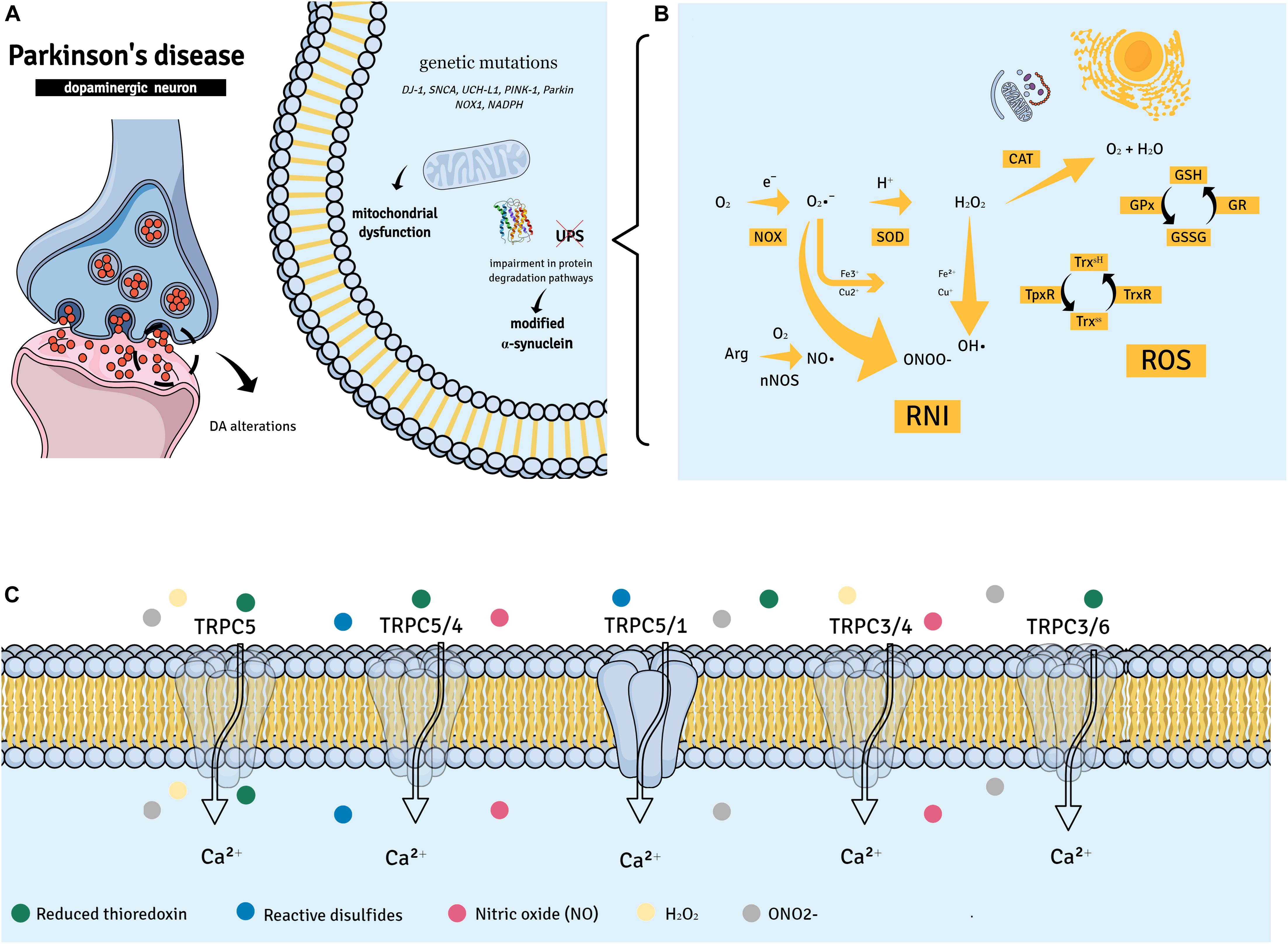
Figure 1. Parkinson’s disease (PD) suggested pathways. (A,B) PD has been associated with the aggregation of α-synuclein into Lewy bodies in dopaminergic neurons of the substantia nigra pars compacta. Other factors such as gene mutations (DJ-1, SNCA, UCH-L1, PINK-1, and Parkin) may contribute to mitochondrial dysfunction and neuronal death in PD. The accumulation of dopamine (DA) and its products in DA neurons may also be a causative factor of neuronal death. This may lead to mitochondrial dysfunction, changes in protein degradation [by impairing the ubiquitin-proteasome system (UPS) function], and increased generation of reactive oxygen species (ROS) and reactive nitrogen intermediates (RNIs). (C) Members of the transient receptor potential canonical (TRPC) subfamily of non-selective Ca2+ channels are able to recognize ROS and RNIs and have been implicated in neuronal survival; in fact, different oxidative/nitrosative stress products can directly activate TRPC complexes.
Excessive ROS and RNI formation during oxidative and nitrosative stresses results in a variety of detrimental effects in the cell, thus, contributing to organelle and membrane structural damages and cellular apoptosis (Guo et al., 2018). This cytotoxic environment has been recognized as a common underlying phenomenon in the dopaminergic neurodegenerative process (Dias et al., 2013). Indeed, an irregular oxidation of macromolecules, such as lipids, proteins, and nucleic acids, was observed in the brain tissues of PD patients (Bosco et al., 2006; Nakabeppu et al., 2007). Also, higher levels of the oxidative stress markers 8-OhdG (8-Oxo-2′-deoxyguanosine) and malondialdehyde, in addition to NO2, were detected in the peripheral blood of PD patients in comparison with healthy individuals (Wei et al., 2018). The same patients presented systemic down-regulation of the antioxidant proteins glutathione and catalase (CAT).
In addition, major genetic insights indicate that specific mutations in a series of primary genes that are responsible for PD-related synucleopathy and the regulation of mitochondrial and ROS equilibrium can disrupt cellular homeostasis (Cacabelos, 2017). For instance, an elevated expression of the wα-synuclein protein and oxidative stress genes [HSPB1, Heat Shock Protein Family B (Small) Member 1; NOX1, NADPH oxidase 1; and MAOB, Monoamine oxidase B] was observed in induced pluripotent stem cell (iPSC)-derived dopaminergic neurons (Nguyen et al., 2011). Similarly, iPSC midbrain dopaminergic neurons from patients with PTEN-induced putative kinase 1 (PINK1) or Parkin mutations presented abnormal mitochondria (Chung et al., 2016) (Figure 1).
Accordingly, evidence suggests that in PD, the mitochondrion represents the primary source of ROS, contributing to intracellular oxidative stress and therefore, to the vulnerability of dopaminergic neurons to apoptosis (Beal, 2005). Moreover, knockout mice for Dynamin-1-like protein (Drp1), a guanosine triphosphate (GTP)ase that regulates mitochondrial fission, exhibited degeneration of nigrostriatal dopaminergic neurons (Berthet et al., 2014). This response was associated with a reduced mitochondrial mass in axons, which was associated with impaired mitochondrial dynamics denoted by the loss of coordination of mitochondrial movements.
Additionally, disruption of respiratory chain complexes, especially the mitochondrial complex I (NADH-quinone oxidoreductase), was implicated in the enhanced production of ROS in PD (Ryan et al., 2015). Human studies also indicated that the dysfunction of this specific complex occurs in the SNpc of PD patients (Schapira et al., 1990). Of note, mitochondrial integrity in SNpc neurons was found to be dependent on Parkin expression (Park et al., 2006; Stichel et al., 2007).
In regard to RNIs, the excessive or inappropriate generation of NO and O2∙−-derived reactive species, plays a critical role in mediating the neurotoxicity associated with mitochondrial damage (Kaludercic and Giorgio, 2016). The reaction between NO and O2∙− represents an important source of OONO−, a highly reactive molecule for a broad range of chemical targets that potently inhibits mitochondrial proteins. OONO− overproduction was found to enhance the levels of oxidized lipids and DNA in the dopaminergic neurons of PD patients (Ebadi and Sharma, 2003). Depletion of antioxidant defenses, including GSH, was also observed in the same samples (Franco and Cidlowski, 2009). Interestingly, nNOS- and inducible NO synthase (iNOS)-dependent NO levels were increased in the SNpc of PD patients (Hancock et al., 2008). Also, high levels of NO and OONO− correlated with a worse prognosis in PD (Kouti et al., 2013), corroborating the hypothesis that both RNI and ROS generation may strongly contribute to neurodegeneration in PD.
Antioxidant proteins such as superoxide dismutase (SOD), CAT, glutathione peroxidase (GPx), and GSH counteract excessive ROS production. Therefore, reductions in their activities and/or expression may favor lipid peroxidation or promote neuronal excitotoxicity with subsequent protein modifications and eventual neuronal death (Deponte, 2013; Patlevič et al., 2016). Interestingly, evident differences were found in the levels of GSH of post-mortem brain samples of PD patients in comparison with other brain regions (Perry et al., 1982; Sian et al., 1994). Also, animal studies revealed that down-regulation of GSH synthesis results in a progressive degeneration of nigrostriatal dopaminergic neurons (Garrido et al., 2011).
By using agonists and antagonists, knockout mice and cells, and a diverse range of molecular biology techniques, several roles have been suggested for the TRPC subfamily. These include their importance as sensors of molecules involved in oxidative and nitrosative stresses (Figure 1) known to influence neuronal survival and function (Chen et al., 2009; Delgado-Camprubi et al., 2017).
In humans, the TRPC subfamily is formed by six channels (TRPC1 and TRPC3-7), which are considered the mammalian TRPs most closely related to those of D. melanogaster. TRPC channels are formed by four subunits and each subunit has six transmembrane domains and a pore region between the fifth and the sixth transmembrane domain (Feng, 2017).
TRPCs assemble into tetramers to form functional channels. Each monomer consists of a transmembrane domain and a cytosolic domain (Li et al., 2019). The cytosolic domain contains the N- and C-terminal subdomains. The N-terminal is composed of four ankyrin repeats and linker helices, whilst the C-terminal is formed by a connecting helix and a coiled-coil domain (Li et al., 2019). All TRPC channels contain the calmodulin and inositol trisphosphate (IP3) receptor-binding motif, which is able to interact with phosphoinositides, inositol polyphosphates, Gαi/o proteins, and SEC14 domain and spectrin repeat-containing protein 1 (SESTD1), a Ca2+-dependent phospholipid/cytoskeleton-binding protein (Wang et al., 2020). These different interacting pathways may influence TRPC functions.
Distributed in two subgroups, diacylglycerol (DAG)-activated (TRPC3/6/7) and non-DAG-activated receptors (TRPC1/4/5), TRPC channels can form homo- and heterotetramers (Strübing et al., 2001; Zagranichnaya et al., 2005; Poteser et al., 2006; Woo et al., 2014; Myeong et al., 2016; Bröker-Lai et al., 2017; Sunggip et al., 2018; Ko et al., 2019). Their assembly in these complexes may vary with their expression sites and functions. Additionally, members of the TRPC subfamily, such as TRPC1, can also form heterotetramers with channels of other TRP subfamilies, including TRPV4 and TRPP2 (Kobori et al., 2009; Greenberg et al., 2017). Despite the advances in elucidating the structure and assembly of TRPCs, the definite functions of their homo- and heterotetramers remain unclear and represent a whole new avenue of knowledge to be pursued.
So far, different roles have been identified for TRPC channels including in cardiovascular, lung, kidney and neurological diseases, inflammation, and cancer, among others. Of importance to our review, TRPCs are involved in neurotransmission, neural development, excitotoxicity, and neurodegeneration (Wang et al., 2020). Interestingly, TRPC channels, especially TRPC1, have topped the list of molecules involved in store-operated Ca2+ entry. However, it is now well-established that their importance goes beyond the endoplasmic reticulum Ca2+ store (Wang et al., 2020). Herein, we will focus on the importance of TRPC channels as oxidative and nitrosative sensors in PD.
In regard to oxidative stress, TRPC5 is perhaps the most well investigated member of the TRPC subfamily. It can be activated by both oxidant and antioxidant molecules such as H2O2 and reduced thioredoxin, respectively (Yoshida et al., 2006; Xu et al., 2008; Naylor et al., 2011). TRPC5 can be also activated by NO and reactive disulfides (Yoshida et al., 2006; Maddox et al., 2018). However, TRPC5 sensitivity to NO has been argued by other studies (Xu et al., 2008; Wong et al., 2010), indicating this response may vary with cell type, generated NO concentrations, and other experimental conditions. Interestingly, TRPC5/TRPC4 complexes were found to be involved in the regulation of Ca2+-dependent production of NO by endothelial cells (Yoshida et al., 2006). TRPC5-dependent NO generation via endothelial NOS (eNOS) activation was later confirmed (Sunggip et al., 2018).
Another interesting finding is the ability of oxidant products such as OONO− to up-regulate both the mRNA and protein expressions of TRPC6 and TRPC3 in monocytes. Of note, OONO−-induced Ca2+ influx in these cells is reversed by the TRPC channel blocker 2-APB (Wuensch et al., 2010). Additionally, TRPC3/TRPC4 assembly forms redox-sensitive complexes on endothelial cells (Poteser et al., 2006). Adding another layer of complexity to TRPC roles in oxidative/nitrosative stresses, it is important to highlight that these channels do not only form complexes but are also able to down-regulate each other’s’ responses. Indeed, TRPC3/TRPC6-mediated Ca2+ influx can be down-regulated by activation of the TRPC5-NO axis (Sunggip et al., 2018).
Evidence also indicates that TRPC1 negatively regulates TRPC5-mediated Ca2+ influx in striatal neurons undergoing oxidative stress (Hong et al., 2015). Interestingly, TRPC1/TRPC5 complexes have been shown to mediate the protective effects of reduced thioredoxin in inflammation, therefore acting as a target for this antioxidant molecule (Xu et al., 2008).
Importantly, TRPCs are highly expressed in various regions of the brain in which they play different roles (Table 1). Thus, due to their ability to sense and modulate oxidative/nitrosative stress responses, they should be considered as potential mediators of neuroinflammation. Therefore, the importance of TRPC channels in PD will now be discussed.
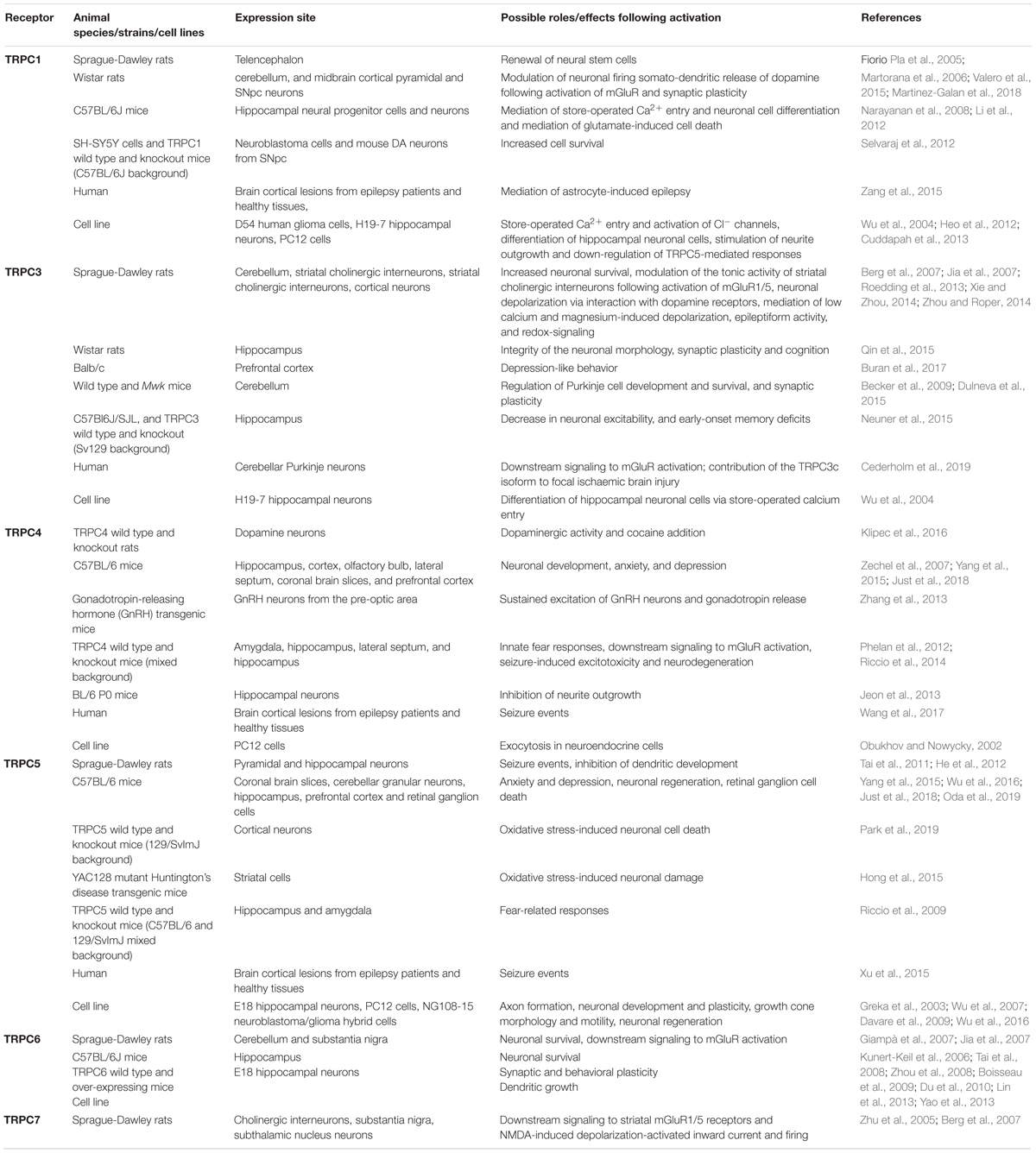
Table 1. Neuronal expression and functions of TRPC channels.
Reports of the contribution of TRPC channels in PD are relatively new and we have not yet uncovered their definite roles in disease progression and maintenance. Also, few studies have attempted to link their expression and/or activation with the ongoing oxidative and nitrosative stresses that occur in PD.
TRPC1 is the most well investigated member of the canonical subfamily in PD. A study in SH-SY5Y cells demonstrated that TRPC1 protein expression becomes down-regulated in these cells following incubation with salsolinol (Bollimuntha et al., 2006), a neurotoxin endogenously found in the nigrostriatal cells and cerebrospinal fluid samples of patients with PD (Moser et al., 1995; Maruyama et al., 1996). Despite its low expression on the cell membrane, the TRPC1 protein was detected in the cytosol (Bollimuntha et al., 2006). This result suggests that salsolinol may cause TRPC1 translocation from the neuronal cell membrane to the cytoplasm.
Interestingly, the endogenous salsolinol derivative NMSAL was detected in the nigrostriatum and intraventricular fluid samples of patients with PD (Maruyama et al., 1996). NMSAL induces neuronal apoptosis via mitochondrial and caspase-3-dependent pathways (Akao et al., 1999; Maruyama et al., 2001; Arshad et al., 2014) and it is considered to be far more toxic to neurons than salsolinol (Maruyama et al., 1996). NMSAL exhibited similar effects to those of salsolinol in neuronal TRPC1 expression and localization (Arshad et al., 2014). All this evidence indicates a protective role for TRPC1 in PD.
Ca2+-induced ROS generation in cultured rat DA neurons treated with the neurotoxin 1-methyl-4-phenylpyridinium ion (MPP+) was also linked to TRPC1 (Chen et al., 2013). Another study by Selvaraj et al. (2009) showed that 1-methyl- 4-phenyl-1, 2,3,6-tetrahyrdro-pyridine (MPTP), a compound known to cause PD in mice by inducing mitochondrial dysfunction and neuronal apoptosis, reduces the expression of TRPC1 in the SNpc. A similar result was observed in PC12 cells incubated with MPP+. The same study also found that TRPC1 over-expression increases the survival of PC12 cells incubated with MPP+ by preserving mitochondrial membrane potential and regulating the expression of the anti-apoptotic genes Bcl2 and Bcl-xl (Selvaraj et al., 2009). Of note, the authors highlighted in their study that TRPC1 over-expression only partly restores mitochondrial membrane potential and neuronal survival.
The contribution of other TRPCs to PD has also been investigated. Analysis of TRPC3 expression patterns revealed that the TRPC3 protein is increased in the SNpc following exposure to MPTP (Selvaraj et al., 2009). On the other hand, no alterations in TRPC3 levels were noted in DA neurons from PD patients (Sun et al., 2017). Of note, these controversial data on TRPC3 expression have been obtained in different experimental settings. Therefore, TRPC3’s role in PD cannot be overruled. Also, it is possible that other TRPC channels and their complexes may contribute to changes in neuronal survival in PD.
In this context, it is important to highlight the complexes formed by TRPC1 with TRPC5. Although no studies have yet investigated these complexes in PD, they have been pointed as mediators of other neurodegenerative diseases such as Huntington’s. In a recent report, it was demonstrated that intracellular oxidized glutathione activates TRPC5 in striatal cells of Huntington’s disease (Q111 cells). The same study showed that upon oxidative stress, TRPC5-mediated Ca2+ influx leads to increased cytosolic Ca2+ levels and activation of the calpain-caspase pathway, leading to apoptosis of striatal neurons (Hong et al., 2015). In parallel, as observed for PD, TRPC1 protein and mRNA expression is down-regulated in Huntington’s striatal cells favoring the formation of TRPC5 heterotetramers in these cells (Hong et al., 2015). These results reinforce the protective role of TRPC1 in neurodegenerative diseases and shed light on the deleterious importance of TRPC5 in neuronal survival.
From the best of our knowledge, no studies have yet investigated the association between TRPC channels and RNI in PD, highlighting the need for further studies to fill this gap of information.
Herein, we presented evidence and discussed the importance of TRPC channels in the recognition and regulation of oxidative and nitrosative stress responses, as well as their contributions to PD. The recent advances in the field of TRPC channels, in particular the protective functions of TRPC1 and the deleterious role of TRPC5 in PD, highlight their importance as pharmacological targets in treating neurodegenerative diseases. Considering the ability of TRPC channels to assemble as homo- and heterotetramers with channels of the same subfamily and also as members of other subfamilies of TRPs, and the lack of antagonists and agonists capable of selectively differentiating the individual actions of each one of these channels, their targeting of PD may become a difficult task. Therefore, efforts need to be made in order to develop effective and more selective pharmacological tools to investigate TRPC channels. This will be an essential step to achieve a broader knowledge of the pathophysiological roles of their different assembly modes and establish their definite importance in PD.
DM-F, NO, LS, and EF contributed to the conception and design and drafted and critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of the work.
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; 305676/2019-9 and 408053/2018-6), Instituto de Pesquisa Pelé Pequeno Príncipe, and INCT-INOVAMED. NO is a Ph.D. student receiving a grant from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and LS is a MSc student receiving a grant from Instituto de Pesquisa Pelé Pequeno Príncipe.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adams, L., Franco, M. C., and Estevez, A. G. (2015). Reactive nitrogen species in cellular signaling experimental biology and medicine. Exp. Biol. Med. 240, 711–717. doi: 10.1177/1535370215581314
Akao, Y., Nakagawa, Y., Maruyama, W., Takahashi, T., and Naoi, M. (1999). Apoptosis induced by an endogenous neurotoxin, N-methyl(R)salsolinol, is mediated by activation of caspase 3. Neurosci. Lett. 267, 153–156. doi: 10.1016/S0304-3940(99)00361-4
Arshad, A., Chen, X., Cong, Z., Qing, H., and Deng, Y. (2014). TRPC1 protects dopaminergic SH-SY5Y cells from MPP+, salsolinol, and N-methyl-(R)-salsolinol-induced cytotoxicity. Acta Biochim Biophys. Sin. 46, 22–30. doi: 10.1093/abbs/gmt127
Beal, M. F. (2005). Mitochondria take center stage in aging and neurodegeneration. Neurol. Progr. 58, 495–505. doi: 10.1002/ana.20624
Becker, E. B., Oliver, P. L., Glitsch, M. D., Banks, G. T., Achilli, F., Hardy, A., et al. (2009). A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc. Nal. Acad. Sci. U.S.A. 106, 6706–6711. doi: 10.1073/pnas.0810599106
Berg, A. P., Sen, N., and Bayliss, D. A. (2007). TrpC3/C7 and Slo2.1 are molecular targets for metabotropic glutamate receptor signaling in rat striatal cholinergic interneurons. J. Neurosci. 27, 8845–8856. doi: 10.1523/JNEUROSCI.0551-07.2007
Berman, M. H., and Nichols, T. W. (2019). Treatment of neurodegeneration: integrating photobiomodulation and neurofeedback in Alzheimer’s Dementia and Parkinson’s: a review. Photobiom.Photomed. Laser Surg 37, 623–634. doi: 10.1089/photob.2019.4685
Berthet, A., Margolis, E. B., Zhang, J., Hsieh, I., Zhang, J., Hnasko, T. S., et al. (2014). Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J. Neurosci. 34, 14304–14317. doi: 10.1523/JNEUROSCI.0930-14.2014
Boisseau, S., Kunert-Keil, C., Lucke, S., and Bouron, A. (2009). Heterogeneous distribution of TRPC proteins in the embryonic cortex. Histochem. Cell. Biol. 131, 355–363. doi: 10.1007/s00418-008-0532-6
Bollimuntha, S., Ebadi, M., and Singh, B. B. (2006). TRPC1 protects human SH-SY5Y cells against salsolinol-induced cytotoxicity by inhibiting apoptosis. Brain Res. 1099, 141–149. doi: 10.1016/j.brainres.2006.04.104
Bosco, D. A., Fowler, D. M., Zhang, Q., Nieva, J., Powers, E. T., Wentworth, P., et al. (2006). Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate α-synuclein fibrilization. Nat. Chem. Biol. 2, 249–253. doi: 10.1038/nchembio782
Bröker-Lai, J., Kollewe, A., Schindeldecker, B., Pohle, J., Nguyen Chi, V., Mathar, I., et al. (2017). Heteromeric channels formed by TRPC 1, TRPC 4 and TRPC 5 define hippocampal synaptic transmission and working memory. EMBO J. 36, 2770–2789. doi: 10.15252/embj.201696369
Buran, Ý, Etem, E. Ö, Tektemur, A., and Elyas, H. (2017). Treatment with TREK1 and TRPC3/6 ion channel inhibitors upregulates microRNA expression in a mouse model of chronic mild stress. Neurosci Lett. 656, 51–57. doi: 10.1016/j.neulet.2017.07.017
Cacabelos, R. (2017). Parkinson’s disease: from pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 18:551. doi: 10.3390/ijms18030551
Cederholm, J. M. E., Kim, Y., von Jonquieres, G., and Housley, G. D. (2019). Human brain region-specific alternative splicing of TRPC3, the Type 3 canonical transient receptor potential non-selective cation channel. Cerebellum 18, 536–543. doi: 10.1007/s12311-019-01026-4
Celsia, F., Pizzoc, P., Brinid, M., Leoa, S., Fotinoa, C., Pintona, P., et al. (2009). Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim. Biophys. Acta 1787, 335–344. doi: 10.1016/j.bbabio.2009.02.021
Chen, L., Liu, L., Yin, J., Luo, Y., and Huang, S. (2009). Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int. J. Biochem. Cell Biol. 41, 1284–1295. doi: 10.1016/j.biocel.2008.10.029
Chen, T., Yang, Y.-F., Luo, P., Liu, W., Dai, S.-H., Zheng, X.-R., et al. (2013). Homer1 knockdown protects dopamine neurons through regulating calcium homeostasis in an in vitro model of Parkinson’s disease. Cell. Signal. 25, 2863–2870. doi: 10.1016/j.cellsig.2013.09.004
Chung, S. Y., Kishinevsky, S., Mazzulli, J. R., Graziotto, J., Mrejeru, A., Mosharov, E. V., et al. (2016). Parkin and PINK1 Patient iPSC-Derived midbrain dopamine neurons exhibit mitochondrial dysfunction and α-Synuclein accumulation. Stem Cell Repo. 7, 664–677. doi: 10.1016/j.stemcr.2016.08.012
Cuddapah, V. A., Turner, K. L., and Sontheimer, H. (2013). Calcium entry via TRPC1 channels activates chloride currents in human glioma cells. Cell Calcium 53, 187–194. doi: 10.1016/j.ceca.2012.11.013
Davare, M. A., Fortin, D. A., Saneyoshi, T., Nygaard, S., Kaech, S., Banker, G., et al. (2009). Transient receptor potential Canonical 5 Channels Activate Ca2+/Calmodulin Kinase Iγ to promote axon formation in hippocampal neurons. J. Neurosci. 29, 9794–9808. doi: 10.1523/JNEUROSCI.1544-09.2009
Delgado-Camprubi, M., Esteras, N., Soutar, M. P., Plun-Favreau, H., and Abramov, A. Y. (2017). Deficiency of Parkinson’s disease-related gene Fbxo7 is associated with impaired mitochondrial metabolism by PARP activation. Cell Death Differ. 24, 120–131. doi: 10.1038/cdd.2016.104
Deponte, M. (2013). Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta Gen. Subjects 1830, 3217–3266. doi: 10.1016/j.bbagen.2012.09.018
Dias, V., Junn, E., and Mouradian, M. M. (2013). The role of oxidative stress in Parkinson’s disease. J. Parkinson’s Di. 3, 461–491. doi: 10.3233/JPD-130230
Du, W., Huang, J., Yao, H., Zhou, K., Duan, B., and Wang, Y. (2010). Inhibition of TRPC6 degradation suppresses ischemic brain damage in rats J. Clin. Invest. 120, 3480–3492. doi: 10.1172/JCI43165
Dulneva, A., Lee, S., Oliver, P. L., Di Gleria, K., Kessler, B. M., Davies, K. E., et al. (2015). The mutant Moonwalker TRPC3 channel links calcium signaling to lipid metabolism in the developing cerebellum. Hum. Mol. Genet. 24, 4114–4125. doi: 10.1093/hmg/ddv150
Ebadi, M., and Sharma, S. K. (2003). Peroxynitrite and mitochondrial dysfunction in the pathogenesis of Parkinson’s Disease. Antiox. Redox Signal. 5, 319–335. doi: 10.1089/152308603322110896
Fasano, A., Visanji, N. P., Liu, L. W. C., Lang, A. E., and Pfeiffer, R. F. (2015). Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol 14, 625–639. doi: 10.1016/S1474-4422(15)00007-1
Feng, S. (2017). TRPC channel structure and properties. Adv. Exp. Med. Biol. 976, 9–23. doi: 10.1007/978-94-024-1088-4_2
Franco, R., and Cidlowski, J. A. (2009). Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ 16, 1303–1314. doi: 10.1038/cdd.2009.107
Garrido, M., Tereshchenko, Y., Zhevtsova, Z., Taschenberger, G., Bähr, M., and Kügler, S. (2011). Glutathione depletion and overproduction both initiate degeneration of nigral dopaminergic neurons. Acta Neuropathol. 121, 475–485. doi: 10.1007/s00401-010-0791-x
Giampà, C., DeMarch, Z., Patassini, S., Bernardi, G., and Fusco, F. R. (2007). Immunohistochemical localization of TRPC6 in the rat substantia nigra. Neurosci. Lett. 424, 170–174. doi: 10.1016/j.neulet.2007.07.049
Görlach, A., Bertram, K., Hudecova, S., and Krizanovac, O. (2015). Calcium and ROS: a mutual interplay. Redox Biol. 6, 260–271. doi: 10.1016/j.redox.2015.08.010
Greenberg, H. Z. E., Carlton-Carew, S. R. E., Khan, D. M., Khan, D. M., Zargaran, A. K., Jahan, K. S., et al. (2017). Heteromeric TRPV4/TRPC1 channels mediate calcium-sensing receptor-induced nitric oxide production and vasorelaxation in rabbit mesenteric arteries. Vascul. Pharmacol. 96-98, 53–62. doi: 10.1016/j.vph.2017.08.005
Greka, A., Navarro, B., Oancea, E., Duggan, A., and Clapham, D. E. (2003). TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat. Neurosci. 6, 837–845. doi: 10.1038/nn1092
Guo, J. D., Zhao, X., Li, Y., Li, G. R., and Liu, X. L. (2018). Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (Review). Int. J. Mol. Med. 41, 1817–1825. doi: 10.3892/ijmm.2018.3406
Hancock, D. B., Martin, E. R., Vance, J. M., and Scott, W. K. (2008). Nitric oxide synthase genes and their interactions with environmental factors in Parkinson’s disease. Neurogenetics 9, 249–262. doi: 10.1007/s10048-008-0137-1
Hawkes, C. H., Del Tredici, K., and Braak, H. (2010). A timeline for Parkinson’s disease. Parkinsonism Related Disord. 16, 79–84. doi: 10.1016/j.parkreldis.2009.08.007
He, Z., Jia, C., Feng, S., Zhou, K., Bai, X., and Wang, Y. (2012). TRPC5 channel Is the mediator of Neurotrophin-3 in regulating dendritic growth via CaMKIIα in rat hippocampal neurons. J. Neurosci. 32, 9383–9395. doi: 10.1523/JNEUROSCI.6363-11.2012
Heo, D. K., Chung, W. Y., Park, H. W., Yuan, J. P., Lee, M. G., and Kim, J. Y. (2012). Opposite regulatory effects of TRPC1 and TRPC5 on neurite outgrowth in PC12 cells. Cell. Signal. 24, 899–906. doi: 10.1016/j.cellsig.2011.12.011
Hong, C., Seo, H., Kwak, M., Jeon, J., Jang, J., Jeong, E. M., et al. (2015). Increased TRPC5 glutathionylation contributes to striatal neuron loss in Huntington’s disease. Brain 138, 3030–3047. doi: 10.1093/brain/awv188
Jeon, J. P., Roh, S. E., Wie, J., Kim, J., Kim, H., Lee, K. P., et al. (2013). Activation of TRPC4β by Gαi subunit increases Ca2+ selectivity and controls neurite morphogenesis in cultured hippocampal neuron. Cell Calcium 54, 307–319. doi: 10.1016/j.ceca.2013.07.006
Jia, Y., Zhou, J., Tai, Y., and Wang, Y. (2007). TRPC channels promote cerebellar granule neuron survival. Nat. Neurosci. 10, 559–567.
Just, S., Chenard, B. L., Ceci, A., Strassmaier, T., Chong, J. A., Blair, N. T., et al. (2018). Treatment with HC-070, a potent inhibitor of TRPC4 and TRPC5, leads to anxiolytic and antidepressant effects in mice. PLoS One 13:e0191225. doi: 10.1371/journal.pone.0191225
Kaludercic, N., and Giorgio, V. (2016). The dual function of reactive oxygen/nitrogen species in bioenergetics and cell death: the role of ATP synthase. Oxidative Med. Cell. Longevity 2016:3869610. doi: 10.1155/2016/3869610
Klipec, W. D., Burrow, K. R., O’Neill, C., Cao, J. L., Lawyer, C. R., Ostertag, E., et al. (2016). Loss of the trpc4 gene is associated with a reduction in cocaine self-administration and reduced spontaneous ventral tegmental area dopamine neuronal activity, without deficits in learning for natural rewards. Behav. Brain Res. 306, 117–127. doi: 10.1016/j.bbr.2016.03.027
Ko, J., Myeong, J., Shin, Y. C., and So, I. (2019). Differential PI(4,5)P 2 sensitivities of TRPC4, C5 homomeric and TRPC1/4, C1/5 heteromeric channels. Scie. Rep. 9:1849. doi: 10.1038/s41598-018-38443-0
Kobori, T., Smith, G. D., Sandford, R., and Edwardson, J. M. (2009). The transient receptor potential channels TRPP2 and TRPC1 form a heterotetramer with a 2:2 stoichiometry and an alternating subunit arrangement. J. Biol. Chem. 284, 35507–35513. doi: 10.1074/jbc.M109.060228
Kouti, L., Noroozian, M., Akhondzadeh, S., Abdollahi, M., Javadi, M. R., Faramarzi, M. A., et al. (2013). Nitric oxide and peroxynitrite serum levels in Parkinson’s disease: correlastion of oxidative stress and the severity of the disease. Eur. Rev. Med. Pharmacol. Sci. 17, 964–970.
Kunert-Keil, C., Bisping, F., Krüger, J., and Brinkmeier, H. (2006). Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genomics 7:159. doi: 10.1186/1471-2164-7-159
Li, J., Zhang, X., Song, X., Liu, R., Zhang, J., and Lid, Z. (2019). The structure of TRPC ion channels. Cell Calcium 80, 25–28. doi: 10.1016/j.ceca.2019.03.005
Li, M., Chen, C., Zhou, Z., Xu, S., and Yu, Z. (2012). TRPC1-mediated increase in store-operated Ca2+ entry is required for the proliferation of adult hippocampal neural progenitor cells. Cell Calcium 51, 486–496. doi: 10.1016/j.ceca.2012.04.014
Liguori, I., Russo, G., Curcio, F., Bulli, G., Aran, L., Della-Morte, D., et al. (2018). Oxidative stress, aging, and diseases. Clin. Interv. Aging 13, 757–772. doi: 10.2147/CIA.S158513
Lin, Y., Zhang, J.-C., Fu, J., Chen, F., Wanget, J., Wu, Z.-L., et al. (2013). Hyperforin attenuates brain damage induced by transient middle cerebral artery occlusion (MCAO) in rats via inhibition of TRPC6 channels degradation. J Cereb. Blood. Flow. Metab. 33, 253–262. doi: 10.1038/jcbfm.2012.164
Maddox, J. W., Khorsandi, N., and Gleason, E. (2018). TRPC5 is required for the NO-dependent increase in dendritic Ca2+ and GABA release from chick retinal amacrine cells. J. Neurophysiol. 119, 262–273. doi: 10.1152/jn.00500.2017
Magrinelli, F., Picelli, A., Tocco, P., Federico, A., Roncari, L., Smania, N., et al. (2016). Pathophysiology of motor dysfunction in Parkinson’s Disease as the rationale for drug treatment and rehabilitation. Parkinson’s Dis. 2016:9832839. doi: 10.1155/2016/9832839
Marino, B. L. B., Souza, L. R., Sousa, K. P. A., Ferreira, J. V., Padilha, E. C., Silva, C. H. T. P., et al. (2019). Parkinson’s disease: a review from the pathophysiology to diagnosis, new perspectives for pharmacological TReatment. Mini Rev. Med. Chemi. doi: 10.2174/1389557519666191104110908
Martinez-Galan, J. R., Verdejo, A., and Caminos, E. (2018). TRPC1 channels are expressed in pyramidal neurons and in a subset of somatostatin interneurons in the rat neocortex. Front. Neuroanat. 12:15. doi: 10.3389/fnana.2018.00015
Martorana, A., Giampà, C., DeMarch, Z., Viscomi, M. T., Patassini, S., Sancesario, G., et al. (2006). Distribution of TRPC1 receptors in dendrites of rat substantia nigra: a confocal and electron microscopy study. Eur. J. Neurosci. 24, 732–738. doi: 10.1111/j.1460-9568.2006.04932.x
Maruyama, W., Boulton, A. A., Davis, B. A., Dostert, P., and Naoi, M. (2001). Enantio-specific induction of apoptosis by an endogenous neurotoxin, N-methyl(R)salsolinol, in dopaminergic SH-SY5Y cells: suppression of apoptosis by N-(2-heptyl)-N-methylpropargylamine. J. Neural. Transm. 108, 11–24. doi: 10.1007/s007020170093
Maruyama, W., Narabayashi, H., Dostert, P., and Naoi, M. (1996). Stereospecific occurrence of a parkinsonism-inducing catechol isoquinoline, N-methyl(R)salsolinol, in the human intraventricular fluid. J. Neural. Transm. 103, 1069–1076. doi: 10.1007/BF01291791
Moser, A., Scholz, J., Nobbe, F., Vieregge, P., Böhme, V., and Bamberg, H. (1995). Presence of N-methyl-norsalsolinol in the CSF: correlations with dopamine metabolites of patients with Parkinson’s disease. J. Neurol. Sci. 131, 183–189. doi: 10.1016/0022-510X(95)00110-N
Mullin, S., and Schapira, A. H. V. (2015). Pathogenic mechanisms of neurodegeneration in Parkinson Disease. Neurol. Clin. 33, 1–17. doi: 10.1016/j.ncl.2014.09.010
Myeong, J., Ko, J., Hong, C., Yang, D., Lee, K. P., Jeon, J. H., et al. (2016). The interaction domains of transient receptor potential canonical (TRPC)1/4 and TRPC1/5 heteromultimeric channels. Biochem. Biophys. Res. Commun. 474, 476–481. doi: 10.1016/j.bbrc.2016.04.138
Nakabeppu, Y., Tsuchimoto, D., Yamaguchi, H., and Sakumi, K. (2007). Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 85, 919–934. doi: 10.1002/jnr.21191
Naoia, M., Maruyamab, W., Akaoc, Y., and Yic, H. (2002). Dopamine-derived endogenous N-methyl-(R)-salsolinol: its role in Parkinson’s disease. Neurotoxicol. Teratol. 24, 579–591. doi: 10.1016/S0892-0362(02)00211-8
Narayanan, K. L., Irmady, K., Subramaniam, S., Unsicker, K., Von Bohlen und Halbach, O., and Von Bohlen und Halbach, O. (2008). Evidence that TRPC1 is involved in hippocampal glutamate-induced cell death. Neurosci. Lett. 446, 117–122. doi: 10.1016/j.neulet.2008.09.034
Naylor, J., Al-Shawaf, E., McKeown, L., Manna, P. T., Porter, K. E., O’Regan, D., et al. (2011). TRPC5 channel sensitivities to antioxidants and hydroxylated stilbenes. J. Biol. Chem. 286, 5078–5086. doi: 10.1074/jbc.M110.196956
Neuner, S. M., Wilmott, L. A., Hope, K. A., Hoffmann, B., Chong, J. A., Abramowitz, J., et al. (2015). TRPC3 channels critically regulate hippocampal excitability and contextual fear memory. Behav. Brain Res. 281, 69–77. doi: 10.1016/j.bbr.2014.12.018
Nguyen, H. N., Byers, B., Cord, B., Shcheglovitov, A., Byrne, J., Gujar, P., et al. (2011). LRRK2 mutant iPSC-derived da neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280. doi: 10.1016/j.stem.2011.01.013
Obukhov, A. G., and Nowycky, M. C. (2002). TRPC4 can be activated by G-protein-coupled receptors and provides sufficient Ca2+ to trigger exocytosis in neuroendocrine cells. J. Biol. Chem. 277, 16172–16178. doi: 10.1074/jbc.M111664200
Oda, M., Yamamoto, H., Matsumoto, H., Ishizaki, Y., and Shibasaki, K. (2019). TRPC5 regulates axonal outgrowth in developing retinal ganglion cells. Lab. Investig. 100, 1–14. doi: 10.1038/s41374-019-0347-1
Park, J., Lee, S. B., Lee, S., Kim, Y., Song, S., Kim, S., et al. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161. doi: 10.1038/nature04788
Park, J. S., Davis, R. L., and Sue, C. M. (2018). Mitochondrial dysfunction in parkinson’s disease: new mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep. 18:21. doi: 10.1007/s11910-018-0829-3
Park, S. E., Song, J. H., Hong, C., Kim, D. E., Sul, J.-W., Kim, T.-Y., et al. (2019). Contribution of zinc-dependent delayed calcium influx via TRPC5 in oxidative neuronal death and its prevention by Novel TRPC antagonist. Mol. Neurobiol. 56, 2822–2835. doi: 10.1007/s12035-018-1258-7
Patlevič, P., Vašková, J., Švorc, P., Vaško, L., and Švorc, P. (2016). Reactive oxygen species and antioxidant defense in human gastrointestinal diseases. Integ. Med. Res. 5, 250–258. doi: 10.1016/j.imr.2016.07.004
Perry, T. L., Godin, D. V., and Hansen, S. (1982). Parkinson’s disease: a disorder due to nigral glutathione deficiency? Neurosci. Lett. 33, 305–310. doi: 10.1016/0304-3940(82)90390-1
Phelan, K. D., Mock, M. M., Kretz, O., Shwe, U. T., Kozhemyakin, M., Greenfield, L. J., et al. (2012). Heteromeric canonical transient receptor potential 1 and 4 channels play a critical role in epileptiform burst firing and seizure-induced neurodegeneration. Mol. Pharmacol. 81, 384–392. doi: 10.1124/mol.111.075341
Pla, A. F., Maric, D., Brazer, S. C., Giacobini, P., Liu, X., Chang, Y. H., et al. (2005). Canonical transient receptor potential 1 plays a role in basic fibroblast growth factor (bFGF)/FGF receptor-1-induced Ca2+ entry and embryonic rat neural stem cell proliferation. J. Neurosci. 25, 2687–2701. doi: 10.1523/JNEUROSCI.0951-04.2005
Poteser, M., Graziani, A., Rosker, C., Eder, P., Derler, I., Kahr, H., et al. (2006). TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. J. Biol. Chem. 281, 13588–13595. doi: 10.1074/jbc.m512205200
Puspita, L., Chung, S. Y., and Shim, J. W. (2017). Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 10:53. doi: 10.1186/s13041-017-0340-9
Qin, X., Liu, Y., Zhu, M., and Yang, Z. (2015). The possible relationship between expressions of TRPC3/5 channels and cognitive changes in rat model of chronic unpredictable stress. Behav. Brain Res. 290, 180–186. doi: 10.1016/j.bbr.2015.04.054
Riccio, A., Li, Y., Moon, J., Kim, K. S., Smith, K. S., Rudolph, U., et al. (2009). Essential Role for TRPC5 in amygdala function and fear-related behavior. Cell 137, 761–772. doi: 10.1016/j.cell.2009.03.039
Riccio, A., Li, Y., Tsvetkov, E., Gapon, S., Yao, G. L., Smith, K. S., et al. (2014). Decreased anxiety-like behavior and Gαq/11-dependent responses in the amygdala of mice lacking TRPC4 channels. J. Neurosci. 34, 3653–3667. doi: 10.1523/JNEUROSCI.2274-13.2014
Roedding, A. S., Tong, S. Y., Au-Yeung, W., Li, P. P., and Warsh, J. J. (2013). Chronic oxidative stress modulates TRPC3 and TRPM2 channel expression and function in rat primary cortical neurons: relevance to the pathophysiology of bipolar disorder. Brain Res. 1517, 16–27. doi: 10.1016/j.brainres.2013.04.025
Ryan, B. J., Hoek, S., Fon, E. A., and Wade-Martins, R. (2015). Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem. Sci. 40, 200–210. doi: 10.1016/j.tibs.2015.02.003
Sakakibara, R., Tateno, F., Kishi, M., Tsuyuzaki, Y., Uchiyama, T., and Yamamoto, T. (2012). Pathophysiology of bladder dysfunction in Parkinson’s disease. Neurobiol. Dis. 46, 565–571. doi: 10.1016/j.nbd.2011.10.002
Sama, D. M., and Norris, C. M. (2013). Calcium dysregulation and neuroinflammation: discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev. 12, 982–995. doi: 10.1016/j.arr.2013.05.008
Schapira, A. H., Cooper, J. M., Dexter, D., Clark, J. B., Jenner, P., and Marsden, C. D. (1990). Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 54, 823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x
Schapira, A. H. V., Chaudhuri, K. R., and Jenner, P. (2017). Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 18, 435–450. doi: 10.1038/nrn.2017.62
Selvaraj, S., Sun, Y., Watt, J. A., Wang, S., Lei, S., Birnbaumer, L., et al. (2012). Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Investig. 122, 1354–1367. doi: 10.1172/JCI61332
Selvaraj, S., Watt, J. A., and Singh, B. B. (2009). TRPC1 inhibits apoptotic cell degeneration induced by dopaminergic neurotoxin MPTP/MPP+. Cell Calcium 46, 209–218. doi: 10.1016/j.ceca.2009.07.008
Sian, J., Dexter, D. T., Lees, A. J., Daniel, S., Agid, Y., Javoy-Agid, F., et al. (1994). Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 36, 348–355. doi: 10.1002/ana.410360305
Sies, H., Berndt, C., and Jones, D. P. (2017). Oxidative Stress. Annu. Rev. Biochem 86, 715–748. doi: 10.1146/annurev-biochem-061516-045037
Stichel, C. C., Zhu, X. R., Bader, V., Linnartz, B., Schmidt, S., and Lübbert, H. (2007). Mono- and double-mutant mouse models of Parkinson’s disease display severe mitochondrial damage. Hum. Mol. Genet. 16, 2377–2393. doi: 10.1093/hmg/ddm083
Strübing, C., Krapivinsky, G., Krapivinsky, L., and Clapham, D. E. (2001). TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 29, 645–655. doi: 10.1016/S0896-6273(01)00240-9
Sun, Y., Zhang, H., Selvaraj, S., Sukumaran, P., Lei, S., Birnbaumer, L., et al. (2017). Inhibition of L-type Ca2+ channels by TRPC1-STIM1 complex is essential for the protection of dopaminergic neurons. J. Neurosci. 37, 3364–3377. doi: 10.1523/JNEUROSCI.3010-16.2017
Sunggip, C., Shimoda, K., Oda, S., Tanaka, T., Nishiyama, K., Mangmool, S., et al. (2018). TRPC5-eNOS axis negatively regulates ATP-induced cardiomyocyte hypertrophy. Front. Pharmacol. 9:523. doi: 10.3389/fphar.2018.00523
Tai, C., Hines, D. J., Choi, H. B., and MacVicar, B. A. (2011). Plasma membrane insertion of TRPC5 channels contributes to the cholinergic plateau potential in hippocampal CA1 pyramidal neurons. Hippocampus 21, 958–967. doi: 10.1002/hipo.20807
Tai, Y., Feng, S., Ge, R., Du, W., Zhang, X., He, Z., et al. (2008). TRPC6 channels promote dendritic growth via the CaMKIV-CREB pathway. J. Cell Sci. 121, 2301–2307. doi: 10.1242/jcs.026906
Takahashi, N., and Mori, Y. (2011). TRP channels as sensors and signal integrators of redox status changes. Front. Pharmacol. 2:58. doi: 10.3389/fphar.2011.00058
Tansey, M. G., and Goldberg, M. S. (2010). Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 37, 510–518. doi: 10.1016/j.nbd.2009.11.004
Valero, M. L., Caminos, E., Juiz, J. M., and Martinez-Galan, J. R. (2015). TRPC1 and metabotropic glutamate receptor expression in rat auditory midbrain neurons. J. Neurosci. Res. 93, 964–972. doi: 10.1002/jnr.23557
Wang, H., Cheng, X., Tian, J., Xiao, Y., Tian, T., Xu, F., et al. (2020). TRPC channels: structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. 209:107497. doi: 10.1016/j.pharmthera.2020.107497
Wang, L.-K., Chen, X., Zhang, C.-Q., Liang, C., Wei, Y.-J., Yue, J., et al. (2017). Elevated expression of TRPC4 in cortical lesions of focal Cortical Dysplasia II and tuberous sclerosis complex. J. Mol. Neurosci. 62, 222–231. doi: 10.1007/s12031-017-0923-z
Wanpen, S., Govitrapong, S., Shavalia, S., Sangchota, P., and Ebadi, M. (2004). Salsolinol, a dopamine-derived tetrahydroisoquinoline, induces cell death by causing oxidative stress in dopaminergic SH-SY5Y cells, and the said effect is attenuated by metallothionein. Brain Res. 1005, 67–76. doi: 10.1016/j.brainres.2004.01.054
Wei, Z., Li, X., Li, X., Liu, Q., and Cheng, Y. (2018). Oxidative stress in Parkinson’s disease: a systematic review and meta-analysis. Front. Mol. Neurosci. 11:236. doi: 10.3389/fnmol.2018.00236
Wong, C. O., Sukumar, P., Beech, D. J., and Yao, X. (2010). Nitric oxide lacks direct effect on TRPC5 channels but suppresses endogenous TRPC5-containing channels in endothelial cells. Pflügers Arch. Eur. J. Physiol. 460, 121–130. doi: 10.1007/s00424-010-0823-3
Woo, J. S., Lee, K. J., Huang, M., Cho, C. H., and Lee, E. H. (2014). Heteromeric TRPC3 with TRPC1 formed via its ankyrin repeats regulates the resting cytosolic Ca2+ levels in skeletal muscle. Biochem. Biophys. Res. Commun. 446, 454–459. doi: 10.1016/j.bbrc.2014.02.127
Wu, G., Lu, Z.-H., André, S., Gabius, H.-J., and Ledeen, R. W. (2016). Functional interplay between ganglioside GM1 and cross-linking galectin-1 induces axon-like neuritogenesis via integrin-based signaling and TRPC5-dependent Ca2+ influx. J. Neurochem. 136, 550–563.
Wu, G., Lu, Z. H., Obukhov, A. G., Nowycky, M. C., and Ledeen, R. W. (2007). Induction of calcium influx through TRPC5 channels by cross-linking of GM1 ganglioside associated with α5β1 integrin initiates neurite outgrowth. J. Neurosci. 27, 7447–7458. doi: 10.1523/JNEUROSCI.4266-06.2007
Wu, X., Zagranichnaya, T. K., Gurda, G. T., Eves, E. M., and Villereal, M. L. (2004). A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19-7 hippocampal neuronal cells. J. Biol. Chem. 279, 43392–43402. doi: 10.1074/jbc.M408959200
Wuensch, T., Thilo, F., Krueger, K., Scholze, A., Ristow, M., and Tepel, M. (2010). High glucose-induced oxidative stress increases transient receptor potential channel expression in human monocytes. Diabetes Metab. Res. Rev. 59, 844–849. doi: 10.2337/db09-1100
Xie, Y.-F., and Zhou, F. (2014). TRPC3 channel mediates excitation of striatal cholinergic interneurons. Neurol. Sci. 35, 1757–1761. doi: 10.1007/s10072-014-1827-0
Xu, G.-Z., Shu, H.-F., Yue, H.-Y., Zheng, D.-H., Guo, W., and Yang, H. (2015). Increased Expression of TRPC5 in cortical lesions of the focal cortical dysplasia. J. Mol. Neuroci. 55, 561–569. doi: 10.1007/s12031-014-0390-8
Xu, S. Z., Sukumar, P., Zeng, F., Li, J., Jairaman, A., English, A., et al. (2008). TRPC channel activation by extracellular thioredoxin. Nature 451, 69–72. doi: 10.1038/nature06414
Yao, C., Zhang, J., Chen, F., and Lin, Y. (2013). Neuroprotectin D1 attenuates brain damage induced by transient middle cerebral artery occlusion in rats through TRPC6/CREB pathways. Mol. Med. Rep. 8, 543–550. doi: 10.3892/mmr.2013.1543
Yang, L. P., Jiang, F. J., Wu, G. S., Deng, K., Wen, M., Zhou, X., et al. (2015). Acute treatment with a Novel TRPC4/C5 channel inhibitor produces antidepressant and anxiolytic-like effects in mice. PLoS One 10:e013625. doi: 10.1371/journal.pone.0136255
Yoshida, T., Inoue, R., Morii, T., Takahashi, N., Yamamoto, S., Hara, Y., et al. (2006). Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2, 596–607. doi: 10.1038/nchembio821
Zagranichnaya, T. K., Wu, X., and Villereal, M. L. (2005). Endogenous TRPC1, TRPC3, and TRPC7 proteins combine to form native store-operated channels in HEK-293 cells. J. Biol. Chem. 280, 29559–29569. doi: 10.1074/jbc.M505842200
Zaichick, S. V., McGrath, K. M., and Caraveo, G. (2017). The role of Ca2+ signaling in Parkinson’s disease. Dis. Models Mech. 10, 519–535. doi: 10.1242/dmm.028738
Zang, Z., Li, S., Zhang, W., Chen, X., Zheng, D., Shu, H., et al. (2015). Expression patterns of TRPC1 in cortical lesions from patients with focal cortical dysplasia. J. Mol. Neurosci. 57, 265–272. doi: 10.1007/s12031-015-0615-5
Zechel, S., Werner, S., and Von Bohlen Und Halbach, O. (2007). Distribution of TRPC4 in developing and adult murine brain. Cell Tissue Res. 328, 651–656. doi: 10.1007/s00441-007-0388-4
Zhang, C., Bosch, M. A., Ronnekleiv, O. K., and Kelly, M. J. (2013). Kisspeptin activation of TRPC4 channels in female GnRH neurons requires PIP2 depletion and cSrc kinase activation. Endocrinology 154, 2772–2783. doi: 10.1210/en.2013-1180
Zhou, F.-W., and Roper, S. N. (2014). TRPC3 mediates hyperexcitability and epileptiform activity in immature cortex and experimental cortical dysplasia. J. Neurophysiol. 111, 1227–1237. doi: 10.1152/jn.00607.2013
Zhou, J., Du, W., Zhou, K., Tai, Y., Yao, H., Jia, Y., et al. (2008). Critical role of TRPC6 channels in the formation of excitatory synapses. Nat. Neurosci. 11, 741–743. doi: 10.1038/nn.2127
Keywords: TRPC channels, Parkinson’s disease, oxidative stress, dopamine release, neuronal apoptosis
Citation: Maria-Ferreira D, de Oliveira NMT, da Silva LCM and Fernandes ES (2020) Evidence of a Role for the TRPC Subfamily in Mediating Oxidative Stress in Parkinson’s Disease. Front. Physiol. 11:332. doi: 10.3389/fphys.2020.00332
Received: 23 December 2019; Accepted: 23 March 2020;
Published: 08 May 2020.
Edited by:
Nady Braidy, University of New South Wales, AustraliaReviewed by:
José Javier López Barba, University of Extremadura, SpainCopyright © 2020 Maria-Ferreira, de Oliveira, da Silva and Fernandes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth Soares Fernandes, ZWxpemFiZXRoLmZlcm5hbmRlc0BwZWxlcGVxdWVub3ByaW5jaXBlLm9yZy5icg==; bGl6YmV0aF9mZXJuYW5kZXNAeWFob28uY28udWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.