 Kinya Seo
Kinya Seo Victoria N. Parikh1
Victoria N. Parikh1- 1Division of Cardiovascular Medicine, Department of Medicine, Stanford University, Stanford, CA, United States
- 2Department of Genetics, Stanford University, Stanford, CA, United States
The myocardium has an intrinsic ability to sense and respond to mechanical load in order to adapt to physiological demands. Primary examples are the augmentation of myocardial contractility in response to increased ventricular filling caused by either increased venous return (Frank–Starling law) or aortic resistance to ejection (the Anrep effect). Sustained mechanical overload, however, can induce pathological hypertrophy and dysfunction, resulting in heart failure and arrhythmias. It has been proposed that angiotensin II type 1 receptor (AT1R) and apelin receptor (APJ) are primary upstream actors in this acute myocardial autoregulation as well as the chronic maladaptive signaling program. These receptors are thought to have mechanosensing capacity through activation of intracellular signaling via G proteins and/or the multifunctional transducer protein, β-arrestin. Importantly, ligand and mechanical stimuli can selectively activate different downstream signaling pathways to promote inotropic, cardioprotective or cardiotoxic signaling. Studies to understand how AT1R and APJ integrate ligand and mechanical stimuli to bias downstream signaling are an important and novel area for the discovery of new therapeutics for heart failure. In this review, we provide an up-to-date understanding of AT1R and APJ signaling pathways activated by ligand versus mechanical stimuli, and their effects on inotropy and adaptive/maladaptive hypertrophy. We also discuss the possibility of targeting these signaling pathways for the development of novel heart failure therapeutics.
Introduction
The working heart adjusts cardiac output to changes in hemodynamic load in order to adapt to physiological demand. The first adaptation occurs immediately, on a beat-to-beat basis, after the ventricle is dilated by increased inflow. The relationship between end-diastolic volume and cardiac output, described by Ernest Henry Starling in a series of papers between 1912 and 1914, has been called “Starling’s Law of the Heart” or the “Frank–Starling relationship” which explains how the heart adapts to change in mechanical load by adjusting its contractile function (Katz Arnold, 2002). The main cellular mechanism that underlies the Frank–Starling relationship is enhanced myofilament sensitivity to Ca2+ at a longer sarcomere length, commonly referred to as length-dependent activation (Allen et al., 1974; de Tombe, 2003) (Figure 1, LDA). This length-dependent activation is modulated by posttranslational modification of myofilament proteins, such as cardiac troponin I (cTnI) (Tachampa et al., 2007; Wijnker et al., 2014), myosin-binding protein C (MyBPC) (Kumar et al., 2015), myosin regulatory light chain (Toepfer et al., 2016; Breithaupt et al., 2019) and titin (Hamdani et al., 2017). While the upstream molecular mechanisms that induce myofilament modifications and length-dependent activation had been poorly understood, recent studies suggest the roles of G protein-coupled receptors (GPCRs): angiotensin II type 1 receptor (AT1R) (Abraham et al., 2016) and apelin receptor APJ (Peyronnet et al., 2017; Parikh et al., 2018).
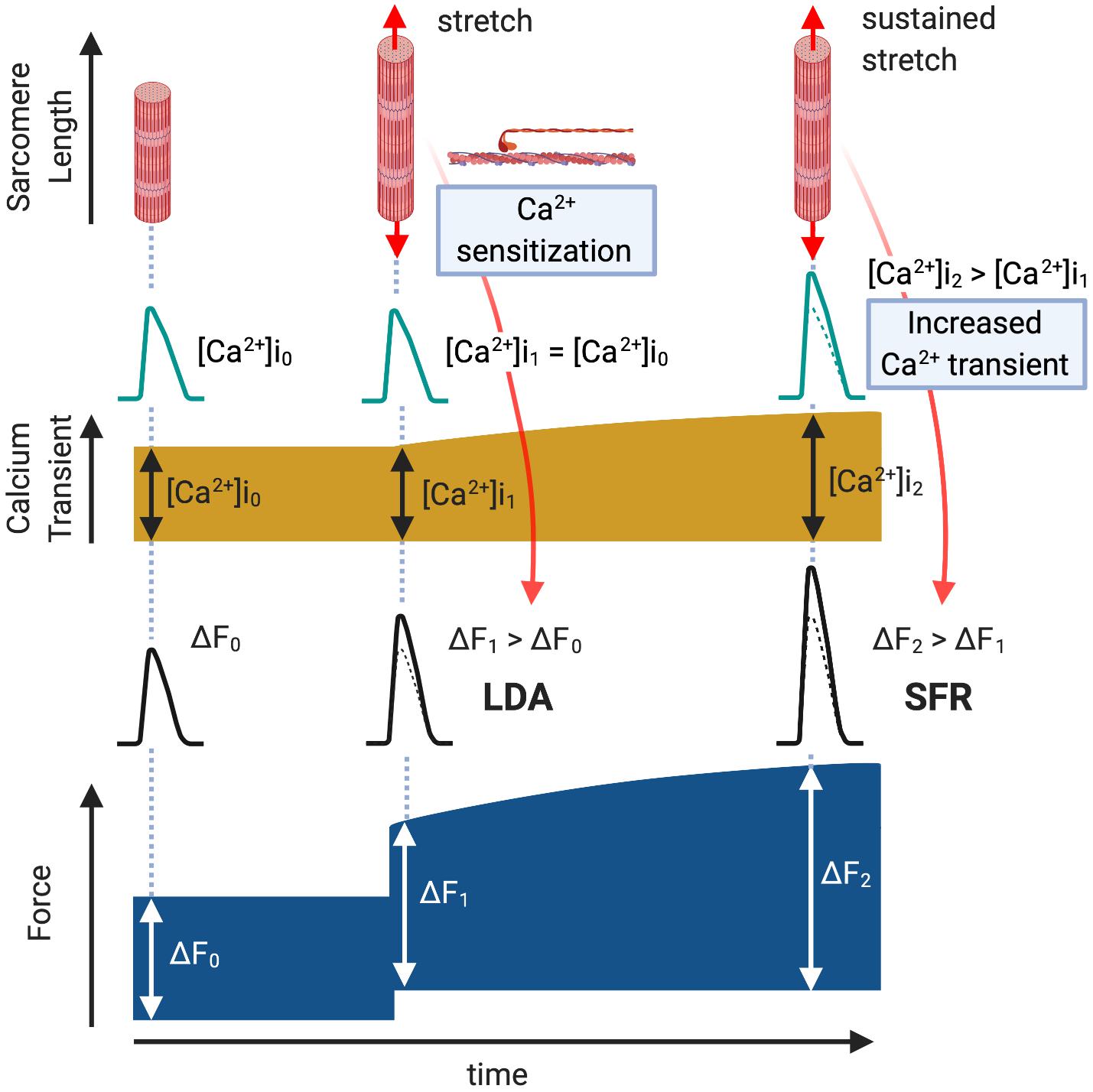
Figure 1. Length-Dependent Activation (LDA) and Slow Force Response (SFR). Immediately after a cardiac muscle is stretched, there is a rapid Ca2+-independent rise in developed force (ΔF1 > ΔF0, LDA) followed by a more gradual rise that is associated with an intracellular Ca2+ increase (ΔF2 > ΔF1, SFR).
The myocardium has another autoregulatory mechanism to gradually increase its contractility in the setting of increased systemic resistance. Gleb Von Anrep first described in 1912 that the heart exhibits progressive increases in contractility in response to left ventricular dilation induced by aortic clamping in vivo (von Anrep, 1912). This “Anrep effect” had been interpreted as secondary to a neurohormonal effect or increased oxygen consumption due to the change in coronary perfusion [known as the “Gregg phenomenon” (Gregg and Shipley, 1944)] until Stanley Sarnoff and his colleagues reproduced this phenomenon in pressure/flow controlled isolated hearts and defined it as an autoregulation of myocardium (Sarnoff et al., 1960; Sarnoff and Mitchell, 1961). Subsequently, in isolated ventricular muscle strips exposed to sudden myocardial stretch, a gradual secondary increase in isometric/isotonic force was observed to follow the initial rise in contractility induced by the Frank–Starling mechanism (Parmley and Chuck, 1973). It has since been proposed that this “Slow Force Response (SFR)” (Figure 1, SFR) is the in vitro equivalent of the Anrep effect (Alvarez et al., 1999). Unlike the Frank–Starling mechanism, SFR is induced by a gradual increase in Ca2+ transient amplitude (Allen and Kurihara, 1982; Kentish and Wrzosek, 1998) through the activation of multiple intracellular components and ion transporters (Cingolani et al., 2013). Notably, AT1R may control this signaling pathway (Cingolani et al., 2013).
In response to sustained mechanical stress, the heart undergoes hypertrophic enlargement characterized by an increase in the size of individual cardiac myocytes. Although cardiac hypertrophy can initially be a compensatory response that temporarily augments and maintains cardiac output along with the Frank–Starling mechanism and the Anrep effect, prolonged hypertrophic stimuli can eventually lead to decompensation, heart failure, and arrhythmia (Levy et al., 1990; Ho et al., 1993). This pathological hypertrophy is induced by the activation of GPCRs by ligand or stretch stimulation, which in turn activates downstream signaling pathways, including mitogen-activated protein kinase (MAPK), protein kinase C (PKC), and calcineurin–nuclear factor of activated T cells (NFAT), leading to myocyte hypertrophy (Heineke and Molkentin, 2006). Candidates for control of this mechano-transduction of hypertrophic signaling include AT1R (Zou et al., 2004) and APJ (Scimia et al., 2012).
The GPCR family is critical both at the bench and bedside, because the majority of current therapeutic drugs for heart failure target GPCRs (Lefkowitz, 2004). An expanding area of GPCR research is focused on the differential activation of G protein or β-arrestin signaling pathway in a “biased” manner to selectively promote cardiac beneficial pathways while preventing stimulation of cardiotoxic pathways. This biased agonism is achieved by ligands or mechanical stretch that can induce distinct active receptor conformations that in turn selectively activate only specific subsets of a given receptor (Figure 2) (Rakesh et al., 2010; Wisler et al., 2014). β-arrestin is a multifunctional scaffolding protein that desensitizes ligand-stimulated GPCRs but also can stimulate other signaling pathways distinct from G protein-dependent signaling (Reiter et al., 2012). Downstream of AT1R, chronic G protein-dependent signaling is associated with adverse outcomes, while β-arrestin-dependent signaling is considered beneficial for heart failure (Kim et al., 2012). Importantly, mechanical stress has been proposed to activate both G protein- and β-arrestin-dependent AT1R signaling pathways (Zou et al., 2004; Rakesh et al., 2010). In the APJ signaling system, in contrast, stretch stimulation selectively activates β-arrestin-dependent pathological pathway (Scimia et al., 2012), while apelin–APJ binding preferentially promotes G protein-dependent cardioprotective and prosurvival signaling. Because β-arrestins work as scaffolds that form complexes by binding to other proteins, it is conceivable that β-arrestins in AT1R and APJ show different functions due to their different binding partners. Thus, the role of these interacting pathways downstream of GPCRs in myocardial physiology appears to be receptor-dependent, and further investigation of how AT1R and APJ integrate ligand and mechanical stimuli to bias G protein or β-arrestin signaling, thus controlling cardioprotective versus cardiotoxic programs is important for the discovery of new therapeutics for heart failure.
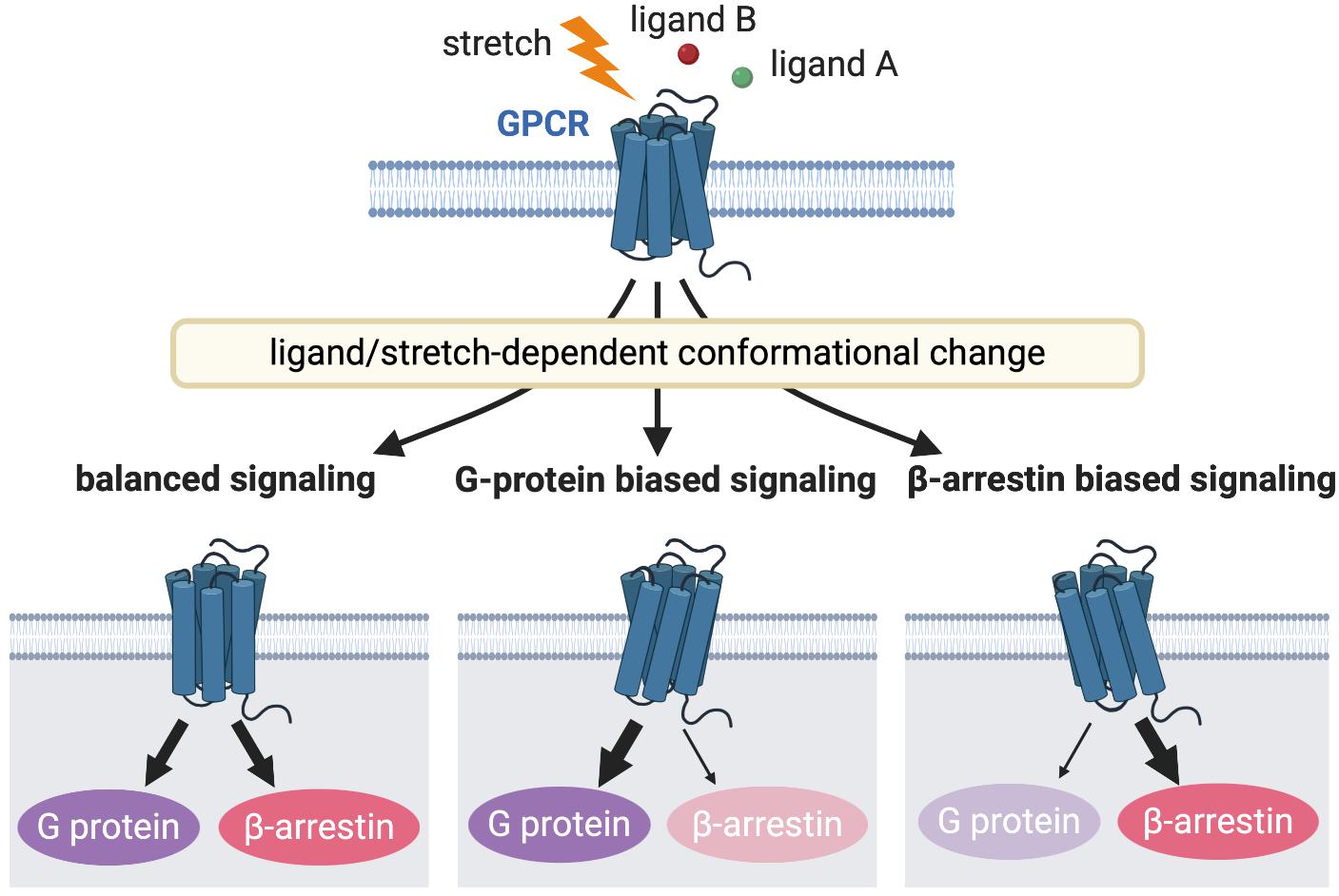
Figure 2. GPCR signaling induced by different receptor activation. Different stimulations stabilize the receptors into distinct active conformational states that couples to a particular G protein or β-arrestin or both to activate diverse arrays of downstream signaling, resulting in different functional outcomes.
This review aims to provide an up-to-date understanding of AT1R and APJ signaling pathways activated by mechanical stimuli on cardiac function and pathological hypertrophy, with special emphasis on biased stretch-mediated engagement of both AT1R and APJ and their potential roles in initiation or amelioration of heart diseases. The possibilities of targeting these pathways for the development of novel heart failure therapeutics will be discussed. Mechanistic insight will be provided through review of cell-based and animal models, and the areas of need for continued investigation will be highlighted.
Angiotensin II Versus Stretch-Induced AT1R Signaling
Angiotensin II type 1 receptor plays pivotal roles in the regulation of cardiovascular function, and is one of the major targets for the therapeutic treatment of heart failure (Violin et al., 2013). Angiotensin II (Ang II), the endogenous ligand for AT1R, is well known for its action on vasoconstriction and aldosterone release, while its local action in the heart is also known to augment myocardial contraction (Petroff et al., 2000) and to activate hypertrophic signaling (Sadoshima and Izumo, 1993). It is widely accepted that myocytes respond to mechanical stretch to release Ang II in an autocrine fashion to activate AT1R and its intracellular signaling (Sadoshima et al., 1993). However, the receptor can also directly sense mechanical stress to activate its downstream signaling, even without Ang II binding (Zou et al., 2004; Yasuda et al., 2008; Rakesh et al., 2010). Komuro’s group showed for the first time that the mechanical activation of AT1R is agonist-independent in angiotensinogen-deficient mice (Zou et al., 2004). They also demonstrated that stretch stimulation of AT1R induces a conformational change of the receptor that is distinct from the ligand-activated receptor conformation (Yasuda et al., 2008). Despite some commonalities between stretch- and ligand-induced AT1R signaling pathways, recent studies have suggested that mechanical stress differentially activates β-arrestin-dependent AT1R signaling, which is distinct from ligand-activated pathways (Rakesh et al., 2010; Wang et al., 2018). This section summarizes the current understanding of how AT1R signaling regulates cardiac contractility and adaptive/maladaptive signaling when the heart is subjected to mechanical stress. In addition, the therapeutic possibility of targeting AT1R signaling for mechanical stress-relevant heart diseases is discussed.
The Role of Ang II and AT1R in Ca2+-Independent Inotropic Response and the Frank–Starling Mechanism
Angiotensin II has a positive inotropic effect in cardiomyocytes both by myofilament Ca2+ sensitization and intracellular Ca2+ rise (Mattiazzi, 1997; Watanabe and Endoh, 1998). Experiments in isolated rabbit myocytes demonstrated that Ang II induced a positive inotropic effect without associated increases in either Ca2+ inward current or Ca2+ transients, but was accompanied by intracellular alkalosis that could potentially increase myofilament sensitivity to Ca2+ (Ikenouchi et al., 1994). It has also been shown that Ang II increases phosphorylation of myosin light chain 2 (MLC2) by the MLC–PKC pathway, thereby increasing myofilament Ca2+ responsiveness (Morano et al., 1988; Clement et al., 1992). This Ang II–AT1R activation of myofilaments could, therefore, form the basis for length-dependent activation (Frank–Starling mechanism). Indeed, Rockman’s group recently revealed that gene deletion or selective inhibition of AT1R in mouse hearts abrogates the Frank–Starling relationship (Abraham et al., 2016).
Using β-arrestin1/2 deficient mice, they also found that the loss of β-arrestin proteins abrogates Frank–Starling relationship without activating PKC. It has been demonstrated that β-arrestin-biased AT1R activation enhances myocyte contractility without increasing intracellular Ca2+ concentration (Rajagopal et al., 2006), and myofilament Ca2+ sensitivity associated with reduced TnI and MyBPC phosphorylation and enhanced tropomyosin phosphorylation (Monasky et al., 2013; Ryba et al., 2017). However, protein phosphorylation in the β-arrestin1/2 deficient mice does not differ compared to wildtype controls. This may be due to distinct β-arrestin signaling pathways downstream of stretch–AT1R compared to ligand–AT1R stimulation (Wang et al., 2018). Further detailed investigation of posttranslational modifications of myofilament proteins by proteomic analyses will identify novel proteins critical to the AT1R-dependent modulation of the Frank–Starling relationship.
The Role of AT1R in Ca2+-Dependent Inotropic Response and Slow Force Response
In addition to the positive inotropic effect caused by myofilament Ca2+ sensitization, Ang II enhances contractility by a Ca2+ dependent mechanism. It has been proposed that Ang II–AT1R binding triggers endothelin-1 (ET-1) production/release, which in turn activates endothelin type A receptor (ETAR) to induce transactivation of epidermal growth factor receptor (EGFR) through ROS-induced ROS release (Cingolani et al., 2006; Yeves et al., 2015; Zhang et al., 2015). This results in NHE-1 activation to induce Na+ influx that in turn triggers Ca2+ entry via reverse mode NCX, thereby enhancing contractility (Petroff et al., 2000; Pérez et al., 2003) (Figure 3, red arrows). It has been suggested that these mechanisms could be the basis for the SFR (the in vitro equivalent of the Anrep effect). Cingolani’s group has described a similar complex signaling pathway upon stretch-induced Ang II production, involving autocrine/paracrine activation of AT1R and ETAR (Cingolani et al., 2011), ROS-induced ROS release (Caldiz et al., 2007), the transactivation of EGFR (Brea et al., 2016), ERK1/2 activation (Pérez et al., 2011), and NHE-1 and NCX activation (Pérez et al., 2001), thus increasing myocyte contraction (Figure 3, red arrows).
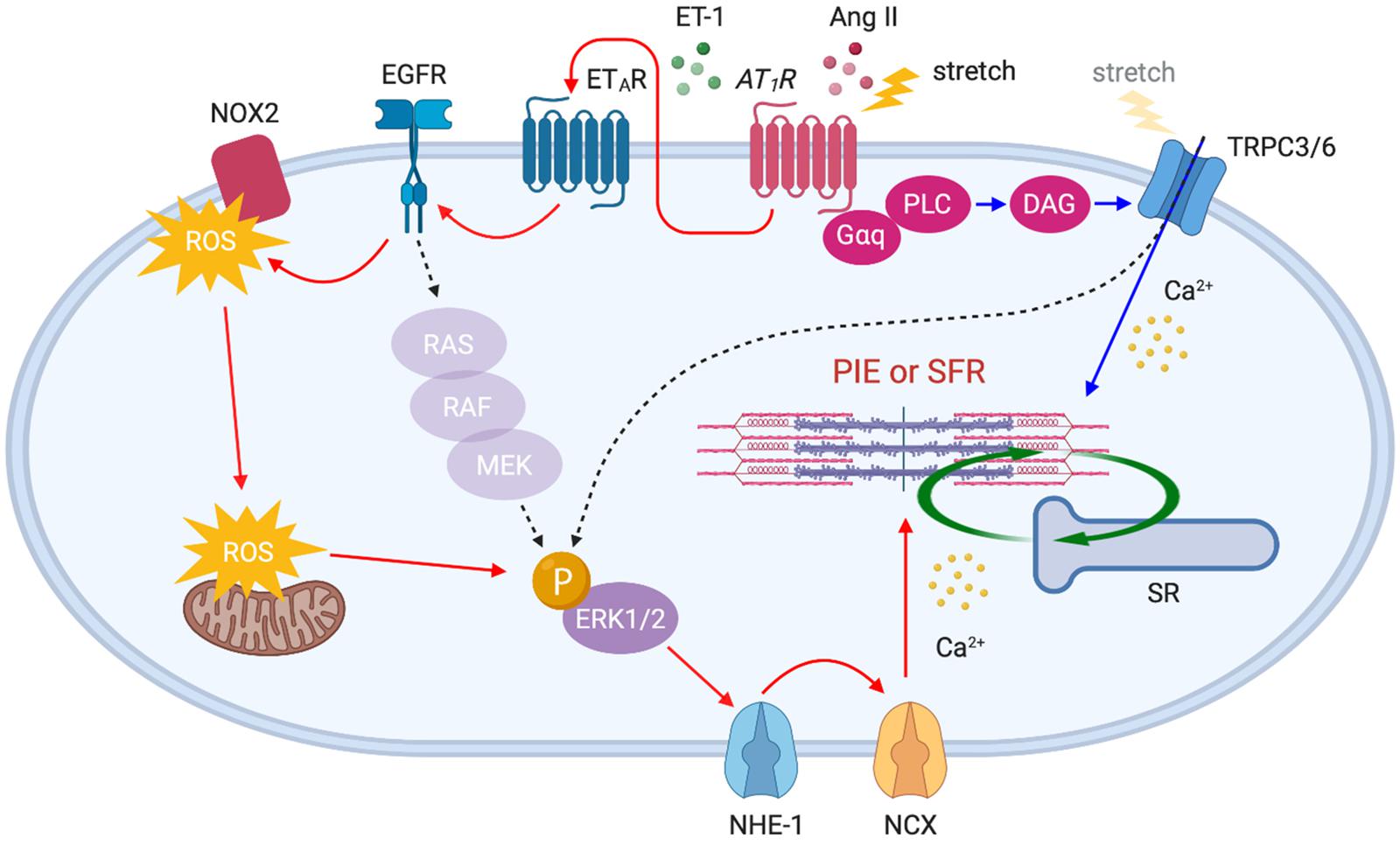
Figure 3. AT1R downstream signaling pathways activated by acute stretch or Ang II. Membrane stretch directly or indirectly (by the autocrine release of Ang II) stimulates AT1R which in turn activates distinct downstream signaling pathways: (1) ETAR–EGFR–ERK1/2–NHE-1–NCX axis (red arrows) and (2) PLC–DAG–TRPC3/6 axis (blue arrows). The gradual influx of Ca2+ through NCX and TRPC3/6 channels enhances myocardial contractility, thereby initiating SFR. Ang II-triggered positive inotropic effect (PIE) shares the same pathways.
Recently, we demonstrated that transient receptor potential canonical (TRPC) 6 channel is an additional important upstream determinant of SFR. Our study demonstrated that gene deletion and selective blockade of TRPC6 channel abrogates SFR and the Anrep effect in physiologically loaded myocytes, muscle strips, and intact hearts (Seo et al., 2014a). More recently, it was found that TRPC3 is an equivalent contributor to this process (Yamaguchi et al., 2018) as supported by evidence that TRPC3 and 6 can form functional heterotetramers (Hofmann et al., 2002). The mechanosensitivity of TRPC6 had been demonstrated in smooth muscle cells (Spassova et al., 2006) and adult cardiomyocytes (Dyachenko et al., 2009) exposed to membrane stretch or shear stress. Some, however, have questioned the mechanosensing capacity of TRPC6 and attributed it to its upstream stretch-activated GPCRs and/or artifacts from TRPC6 overexpression in heterologous systems (Gottlieb et al., 2008). Importantly, TRPC3/6 is a downstream component of AT1R, and Ang II stimulation is known to activate these channels via Gαq protein, PLC and diacylglycerol (DAG) signaling pathway (Onohara et al., 2006). Indeed, stretch-induced slow increase in Ca2+ was suppressed by either TRPC3/6 or AT1R inhibition (Yamaguchi et al., 2018). Ca2+ influx through TRPC3/6 may directly stimulate SFR, while it is also conceivable that TRPC3/6 activates ERK1/2 (Yao et al., 2009; Chiluiza et al., 2013) upstream of NHE-1, thus inducing an inward Na+ current that in turn triggers Ca2+ entry via NCX (Poburko et al., 2007, 2008; Louhivuori et al., 2010) (Figure 3, blue arrows). While many of these studies have suggested the involvement of AT1R as an upstream component of SFR signaling pathways, there are several opposing reports describing that Ang II and AT1R are not involved in the process because SFR was not suppressed by Ang II receptor blockers (ARBs) (Calaghan and White, 2001; von Lewinski et al., 2003; Shen et al., 2013). Despite the inverse agonistic effect of ARBs (Sato et al., 2016), mechanical stretch may differentially activate AT1R even in the presence of ARBs which should normally suppress the ligand-stimulated signaling pathway. More direct evidence that links AT1R to SFR, using AT1R-deficient models, will be needed to clarify this mechanism.
Pathological AT1R–TRPC3/6 and Cardioprotective AT1R–β-Arrestin Signaling in Pressure-Overload Cardiac Hypertrophy
It is well established that Ang II can induce cardiomyocyte hypertrophy through the activation of multiple intracellular signaling pathways such as the mitogen-activated protein kinase (MAPK) signaling cascade (Thorburn et al., 1994; Zechner et al., 1997; Wang et al., 1998a), c-Jun N-terminal kinase (Sadoshima and Izumo, 1993; Ramirez et al., 1997; Wang et al., 1998b), Akt–mammalian target of rapamycin (mTOR) (Gorin et al., 2001; Diniz et al., 2009) and calcium–calmodulin-activated phosphatase calcineurin (Molkentin et al., 1998; Wilkins and Molkentin, 2004). In particular, calcineurin is a pivotal regulator of pathological cardiac hypertrophy preferentially activated by mechanical stress on GPCRs (Molkentin, 2004; Heineke and Molkentin, 2006). Once activated by increases in Ca2+, calcineurin mediates the hypertrophic response through its downstream transcriptional effector NFAT (Crabtree and Olson, 2002). It has been accepted that a major source of Ca2+ for activation of calcineurin is Ca2+ influx through TPRC3 and 6 channels. Indeed, Ang II–AT1R activation promotes calcineurin–NFAT signaling that requires the DAG-induced Ca2+ signaling pathway through TRPC3 and 6 (Onohara et al., 2006). Importantly, this signaling pathway can be activated by mechanical stress. TRPC3 overexpressed transgenic mice exhibit an increase in calcineurin–NFAT activation in vivo, and increased hypertrophy after Ang II/Phenylephrine and pressure-overload stimulation (Nakayama et al., 2006). In addition, TRPC6 transgenic mice also resulted in enhanced sensitivity to mechanical stress, with an increase in calcineurin–NFAT signaling, and severe cardiac hypertrophy and failure (Kuwahara et al., 2006). Although TRPC3/6 channels are linked to other GαqPCRs such as ETAR and α-adrenergic receptors, AT1R may be the putative central actor in stress-induced hypertrophy, considering the mechanosensing capacity of the receptor.
AT1R activation by either Ang II or mechanical stress not only induces pathological signaling but also promotes physiological hypertrophy and prosurvival signaling. In embryonic, neonatal, and adult cardiomyocytes, Ang II–AT1R activation promotes transactivation of EGFR, which in turn activates MAPK and Akt–mTOR pathways (Thomas et al., 2002; Diniz et al., 2009). Rockman’s group recently demonstrated that mechanical stress in cells and the hearts activates AT1R-induced prosurvival signaling in a β-arrestin-dependent manner that does not require Ang II release (Rakesh et al., 2010). The formation of an AT1R–β-arrestin complex by mechanical stress induces EGFR transactivation and subsequent ERK and Akt signaling pathways, which suppresses cardiomyocyte injury (Rakesh et al., 2010). They later found that membrane stretch uniquely promotes the coupling of the inhibitory G protein (Gαi) that is required for the recruitment of β-arrestin2 and activation of downstream signaling pathways, such as EGFR transactivation and ERK phosphorylation (Wang et al., 2018). Gαi proteins primarily inhibit the cAMP-dependent pathway by inhibiting adenylyl cyclase (AC) activity. Although previous studies have shown that Ang II may promote AT1R–Gαi coupling to inhibit AC and to regulate Ca2+ channels in certain tissues or cell types (Hescheler et al., 1988; Maturana et al., 1999), it is not yet clear if the Gαi–β-arrestin complex has the same function.
Taken together, chronic mechanical stress can induce both Gαq–TRPC3/6 dependent and Gαi–β-arrestin dependent signaling pathways to differentially promote pathological and prosurvival signaling (Figure 4). Although selective Gαq–TRPC3/6 signaling initially works as an adaptive response to mechanical stress by enhancing myocardial contractility through SFR, prolonged signaling eventually worsens cardiac function. On the other hand, β-arrestin-dependent AT1R signaling is proposed to enhance cardiac contractility by a Ca2+-independent mechanism, and chronically activates prosurvival signaling, making it a pathway of high clinical potential to ameliorate acute and chronic heart failure.
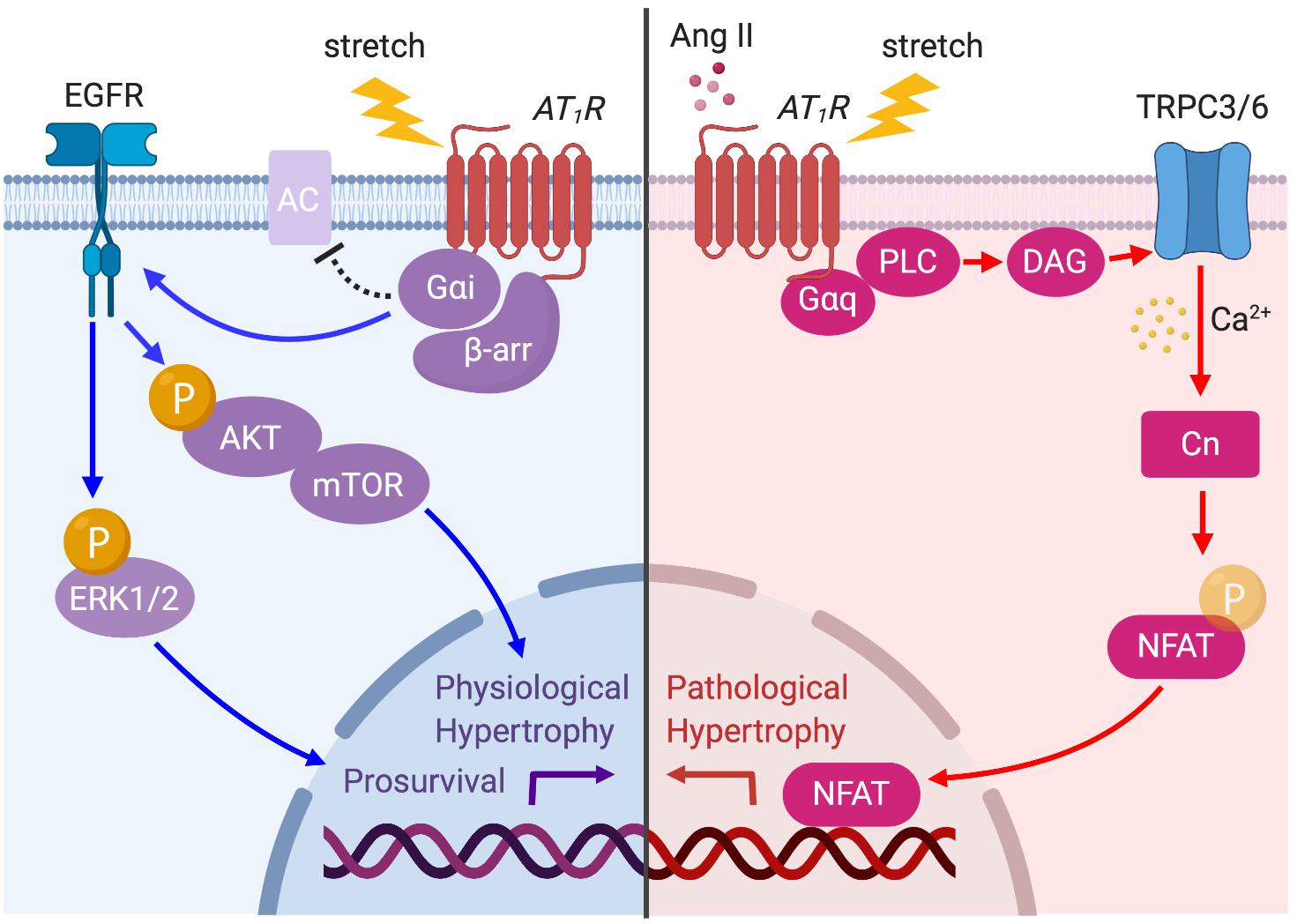
Figure 4. AT1R signaling pathways activated by prolonged mechanical load or Ang II. The AT1R is principally coupled to Gαq protein but is also coupled to Gαi protein through β-arrestin (β-arr) recruitment when the myocyte is subjected to mechanical load. Ang II- or stretch-induced Gαq signaling activates TRPC3/6 to initiate Ca2+ influx, which in turn activates calcineurin (CN)–NFAT pathway to promote pathological cardiac hypertrophy (red arrows). AT1R–Gαi coupling induces EGFR transactivation and Akt/ERK phosphorylation, which promotes physiological hypertrophy and prosurvival signaling (blue arrows).
AT1R Targeted Therapeutics for Mechanical Stress-Associated Heart Diseases (Table 1)
Pathological hypertrophy induced by the overstimulation of AT1R by Ang II or mechanical stress can eventually lead to heart failure and sudden death associated with arrhythmia. One of the current therapeutics for these conditions is AT1R blocking drugs, known as ARBs. Several ARBs are known to have inverse agonistic action which can inactivate the GPCR state, and thereby suppress the constitutive activity of receptors. Such drugs can suppress mechanical stretch-induced signals through AT1R and may exhibit enhanced therapeutic effects for these disease states (Zou et al., 2004; Wei et al., 2011).

Table 1. Emerging drugs for heart failure therapeutics targeting AT1R or APJ.
While endogenous Ang II activates both G protein and β-arrestin signaling pathways, several AT1R ligands, such as [Sar1, Ile4, Ile8]-Ang II (SII), TRV120023, TRV120027, and TRV120067 have been shown to selectively activate β-arrestin-mediated pathways and therefore termed as β-arrestin-biased agonists (Rajagopal et al., 2010; Strachan et al., 2014). These β-arrestin-biased AT1R agonists have been demonstrated to have beneficial effects on the disease condition caused by mechanical stress. For example, TRV120027 causes cardiac unloading action while preserving renal function in a canine model of acute heart failure (Boerrigter et al., 2011). However, a Phase 2B trial of TRV120027 in acute heart failure (BLAST-AHF) resulted in no benefit over placebo (De Vecchis et al., 2017). On the other hand, TRV120023 diminishes myocyte apoptosis caused by mechanical stress by selectively activating ERK1/2 cardioprotective signaling pathways in isolated mouse hearts (Kim et al., 2012). Long-term treatment with TRV120067 in the mouse model of dilated cardiomyopathy not only prevented maladaptive signaling but also improved cardiac function by altering the myofilament response to Ca2+ via β-arrestin signaling pathways (Ryba et al., 2017). Notably, TRV120023 and TRV120067 have shown better efficacy in cardiac function and cardioprotection compared to ARBs, suggesting their possibility to become novel therapeutic drugs for heart failure.
Targeting AT1R downstream signals, such as protein kinase G (PKG) and TRPC3/6, could be another therapeutic strategy. Our study previously revealed that adverse myocardial responses induced by mechanical stimulation to these channels are suppressed by post-transcriptional modification of TRPC6 channel by activation of the cGMP–PKG pathway (Seo et al., 2014a). Indeed, there is growing evidence that stimulation of the cGMP–PKG pathway within cardiac myocytes dampens cardiac stress responses, and its activation can attenuate pathological hypertrophy, protect against ischemic injury and enhance cell survival (Zhang and Kass, 2011). One means to activate the cGMP–PKG pathway is to inhibit the degradation of cGMP by phosphodiesterase-5 (PDE5). PDE5 inhibitors [e.g., sildenafil (Viagra)] have proven their efficacy in treating pressure-overload cardiac hypertrophy and failure in animal models (Takimoto et al., 2005; Nagayama et al., 2009). Although the RELAX trial, a large clinical trial of PDE5 inhibition in heart failure with preserved ejection fraction (HFpEF), failed to show robust beneficial effects (Redfield et al., 2013), single-center trials in patients with heart failure with reduced ejection fraction (HFrEF) reported improved exercise capacity and quality of life (Lewis et al., 2007). Indeed, a meta-analysis of randomized controlled trials in heart failure shows statistically significant improvement of clinical outcomes in patients with HFrEF rather than HFpEF (De Vecchis et al., 2017). Our study demonstrated that sildenafil attenuates pathological hypertrophy by promoting TRPC6 phosphorylation by PKG in the mouse model of muscular dystrophy in which the heart is susceptible to mechanical load (Seo et al., 2014a). Direct antagonism of TRPC3/6 channels is also proven effective for preventing pathological hypertrophy in experiment levels (Seo et al., 2014b). Recently, the orally bioavailable selective TRPC6 inhibitor (BI 749327) was tested in mice, providing in vivo evidence of therapeutic efficacy for cardiac and renal stress-induced disease with fibrosis (Lin et al., 2019). Thus, direct inhibition of TRPC6 could be an alternative strategy to effectively suppress pathological cardiac hypertrophy and failure induced by adverse mechanical stress.
Apelin Versus Stretch-Induced APJ Signaling
Apelin receptor is a GPCR that binds the endogenous peptide apelin (Tatemoto et al., 1998; Lee et al., 2000). This receptor is widely expressed in the cardiovascular system and is emerging as an important mediator of both cardiac and vascular function (Chandrasekaran et al., 2008). In the heart, apelin has been shown to increase myocardial contraction (Szokodi et al., 2002), reduce cardiac load (Ashley et al., 2005) and promote cardioprotective effects (Zhang et al., 2009). Recently, a peptide named ELABELA (Ryba et al., 2017) or Toddler (Pauli et al., 2014) was found to bind APJ, exhibiting a cardiac protective role comparable to apelin (Sato et al., 2017). In addition to this new ligand, it recently turned out that mechanical stretch can also directly activate this receptor. It has been proposed that APJ acts as a bifunctional receptor for both mechanical stress and apelin to activate separate signaling pathways directed to inotropic, cardiotoxic, and cardioprotective effects (Scimia et al., 2012). Intriguingly, unlike AT1R, stretch-induced activation of APJ triggers pathological hypertrophy through β-arrestin, while β-arrestin-dependent signaling in AT1R activates prosurvival signaling. This section summarizes the up-to-date findings of APJ signaling pathways stimulated by apelin or mechanical stress, and introduces how each stimulation can bring different cardiac outcomes. Therapeutic possibilities of biased agonists for heart failure targeting G protein versus β-arrestin-dependent signaling pathways are discussed.
The Role of Apelin–APJ in Ca2+-Independent Positive Inotropic Effect and Frank–Starling Relationship
A potent inotropic effect of apelin has been demonstrated in cardiomyocytes (Farkasfalvi et al., 2007; Wang et al., 2008; Peyronnet et al., 2017), muscle strips (Dai et al., 2006), isolated hearts (Szokodi et al., 2002; Perjés et al., 2014) and in vivo heart disease models (Berry et al., 2004; Charo et al., 2009). It has been proposed that the increase in myocardial contractility is attributed to both Ca2+-dependent (Dai et al., 2006; Wang et al., 2008) and Ca2+-independent mechanisms (Farkasfalvi et al., 2007; Charo et al., 2009; Parikh et al., 2018). The latter is considered to rely on apelin’s action on myofilament sensitivity to Ca2+. It was first demonstrated by Farkasfalvi et al. (2007) in studies with normal and failing cardiomyocytes that displayed increased sarcomere shortening in the absence of increased Ca2+ transient amplitude after apelin administration. They suggested one of the mechanisms involves increased myofilament sensitivity to Ca2+ as apelin activates NHE-1 with consequent intracellular alkalinization. Subsequently, a study using isolated single left ventricular myocytes from apelin deficient and APJ deficient mice revealed that loss of apelin or APJ causes impaired contraction with no difference in intracellular Ca2+ kinetics, suggesting apelin and APJ affect either myofilament Ca2+ sensitivity or cross-bridge cycling kinetics (Charo et al., 2009). This may be attributable to the activation of MLC kinase through parallel and independent activation of PKCε and ERK1/2 signaling stimulated by apelin (Perjés et al., 2014).
The phosphorylation level of myofilament proteins can affect length-dependent activation, which positively regulates the Frank–Starling relationship. Recently, the direct effect of apelin on length-dependent activation was examined in mechanically preloaded cardiomyocytes; apelin increased compliance of the myocytes as indicated by the negative regulation of end-diastolic force-length relationship, which in turn enhanced contractility as indicated by increased Frank–Starling gain (dimensionless index for contractility) (Peyronnet et al., 2017). Increased cardiomyocyte compliance is presumably related to titin phosphorylation. However, the observed positive regulation of the Frank–Starling relationship by apelin must be dependent on other contractile proteins such as cTnI and MLC or alkalosis, because decreased titin-based stiffness is associated with reduced length-dependent activation of myocardium (Methawasin et al., 2014; Ait-Mou et al., 2016; Beqqali et al., 2016). Recently, we demonstrated that APJ-deficient cardiomyocytes showed negative regulation of the Frank–Starling relationship with no increase in Ca2+ transients in response to stretch (Parikh et al., 2018). Our study also provided mechanistic insights for apelin’s positive inotropic and Frank–Starling effects. We demonstrated reduced protein kinase A (PKA) phosphorylation of cTnI at Ser22/Ser23 in response to apelin. This is known to increase myofilament Ca2+ sensitivity, and is consistent with apelin-dependent AC–cAMP–PKA inhibition through Gαi activation (Szokodi et al., 2002; Scimia et al., 2012) (Figure 5).
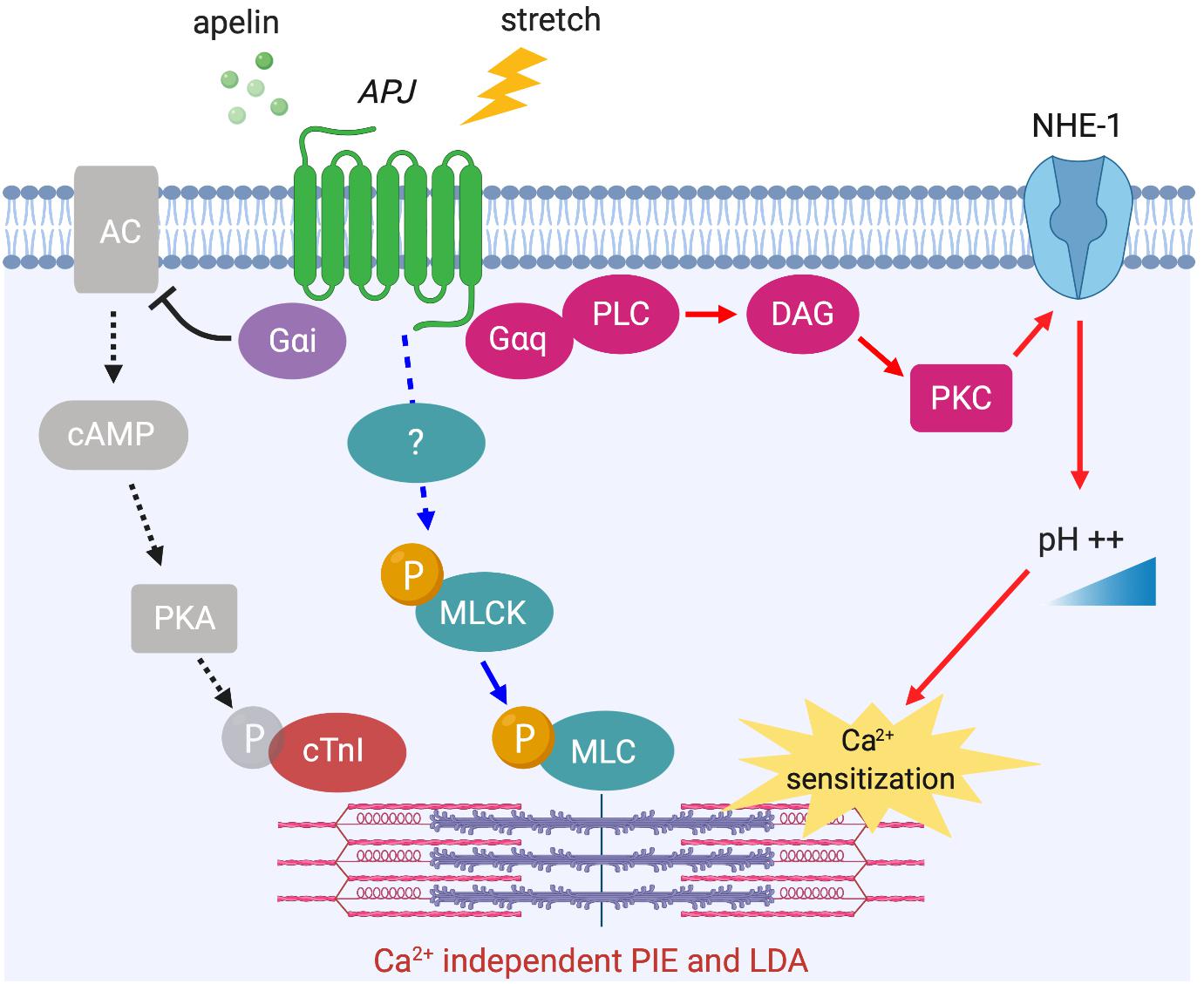
Figure 5. APJ downstream signaling pathways for Ca2+ independent positive inotropic effect (PIE) and length-dependent activation (LDA). Apelin-induced activation of APJ enhances myofilament sensitivity to Ca2+, which leads to the positive regulation of LDA. The mechanisms include intracellular alkalinization by Gαq-dependent NHE-1 activation (red arrows), Gαi-dependent dephosphorylation of cTnI (black dashed arrows), and the phosphorylation of MLC (blue arrows, mechanism unknown).
The Role of Apelin–APJ in Ca2+-Dependent Positive Inotropy and the Anrep Effect
In addition to this Ca2+-independent mechanism, apelin is thought to exert its inotropic action by increasing the availability of intracellular Ca2+. Apelin’s inotropic effect in isolated hearts is dependent on PLC, PKC, NHE-1, and NCX activation (Szokodi et al., 2002). Notably, this inotropic response develops slowly to reach a plateau within 10–30 min and is then sustained, which is different from classical β-adrenergic receptor activation that develops rapidly over a matter of seconds. Although Ca2+ dynamics were not examined in this study, a later study proposed that apelin-induced increased contractility is the result of increased Ca2+ transients rather than changes in myofilament Ca2+ responsiveness (Dai et al., 2006). Most recently, these observations were consolidated in a report showing that apelin has positive inotropic and lusitropic actions on isolated myocytes with enhanced calcium-induced calcium release. This enhanced Ca2+ release is achieved through increased Ca2+ influx through NCX and increased rate of Ca2+ uptake to Ca2+ storage by sarcoplasmic reticulum Ca2+-ATPase (SERCA), as controlled by PKC-directed phosphorylation (Wang et al., 2008). It is intriguing that the time course and the signaling pathways underlying the effect of apelin show some similarities to the mechanism of SFR (Figure 6 compared to Figure 3), in which NHE-1 and NCX are the primary downstream actors. Although the role of APJ in SFR has yet to be fully explored, it is conceivable that this signaling pathway modulates this physiological phenomenon both through apelin–APJ binding and APJ’s mechanosensing ability.
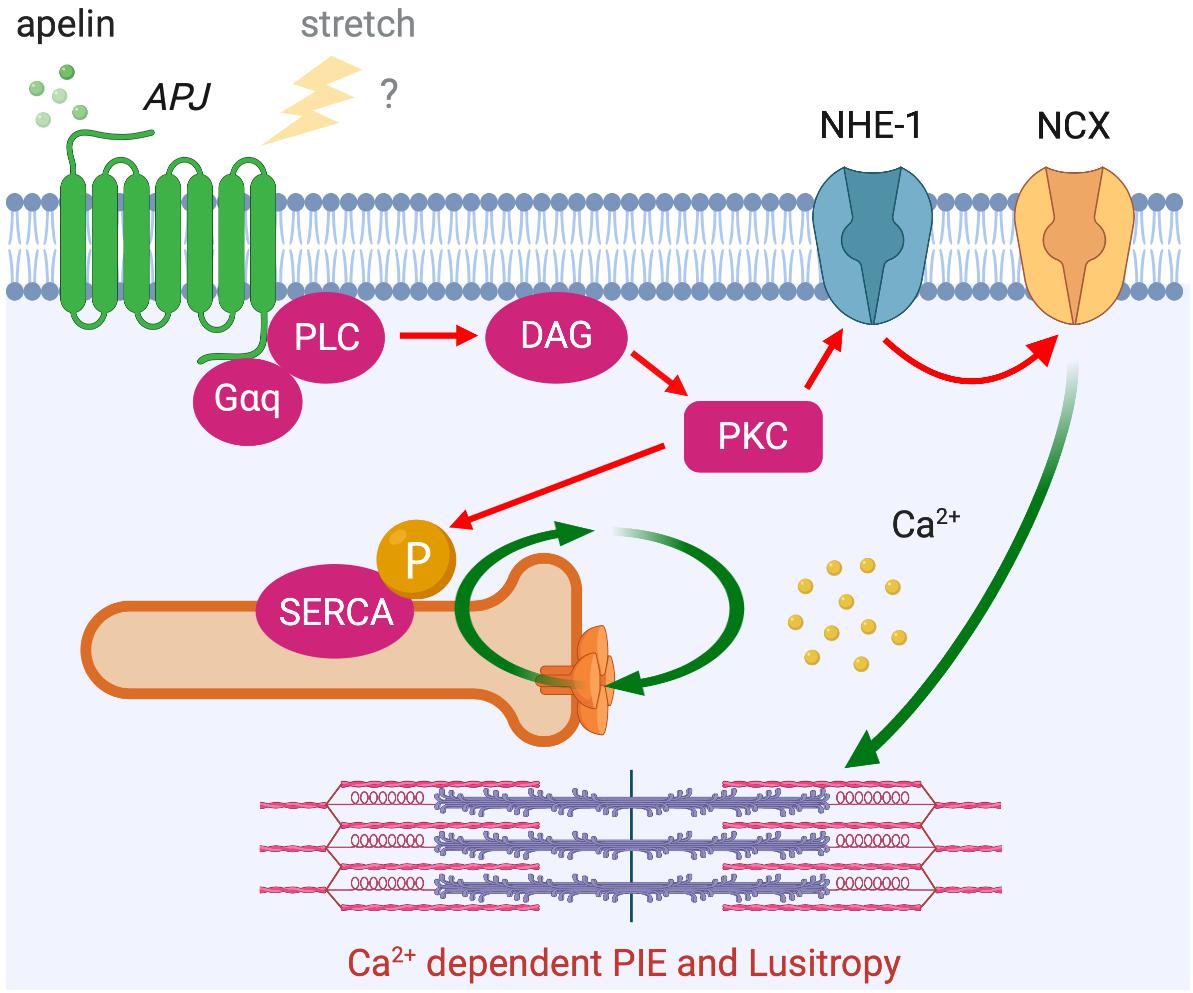
Figure 6. APJ downstream signaling pathway for Ca2+ dependent positive inotropic effect (PIE). APJ activation by apelin also induces PIE that accompanies increased Ca2+ transient through Gαq-dependent NCX activation (red arrows). Positive lusitropic effect (increased velocity of myocardial relaxation) is also induced via PKC-directed phosphorylation of SERCA.
Integration of Apelin and Mechanical Stimuli in APJ to Differentially Activate Cardiac Prosurvival and Hypertrophic Signaling
We previously demonstrated a beneficial effect of chronic apelin supplementation on cardiac performance with reduced left ventricular preload and afterload, and increased contractile function without evidence of hypertrophy (Ashley et al., 2005). Chronic stimulation of APJ by apelin not only increases cardiac performance but also attenuates the development of pressure-overload heart failure through the inhibition of TGF-β-driven profibrotic activity (Pchejetski et al., 2012) and NOX2-derived ROS production (Koguchi et al., 2012). In apelin-deficient mice, hypertrophic response to pressure overload was unchanged, but the progressive impairment of systolic function was observed (Kuba et al., 2007). Myocardial infarcted hearts in apelin deficient mice exhibit exacerbated postinfarction remodeling and impaired functional recovery with a significant reduction of prosurvival phospho-Akt and ERK1/2 signals in the infarct and peri-infarct regions (Wang et al., 2003). These studies clearly demonstrate the simultaneous inotropic and antihypertrophic effects of apelin in ischemia and pressure overload.
Conversely, cardiomyocyte-specific overexpression of APJ causes cardiac hypertrophy and contractile dysfunction in mice (Murata et al., 2016). This indicates that APJ has a capacity to activate multiple downstream signaling pathways such as Gαi- and Gαq-dependent pathways (Chapman et al., 2014), some of which may be independent of apelin’s protective effects. It has also been shown that APJ can integrate chemical (apelin) and mechanical (stretch) stimuli and translates these into opposite cardiac outcomes by differentially activating downstream pathways (Scimia et al., 2012). Specifically, apelin activates APJ through Gαi protein to exert its cardioprotective effect, while stretch stimulates APJ to recruit β-arrestins, which promote pathological hypertrophy (Figure 7). At the cellular level, the mechanosensing capacity of APJ has been confirmed in H9c2 cardiomyocytes in which the increase in diameter, volume and protein content of cardiomyocytes under static pressure was ameliorated by APJ shRNA (Xie et al., 2014). Recently, our study provided supporting evidence that myocyte-specific deletion of APJ is protective against pressure-overload heart failure, showing the abrogation of mechanosensing capacity, reduced Ca2+ transient, and remarkable suppression of cellular hypertrophy and fibrosis (Parikh et al., 2018). These studies suggest that APJ integrates apelin and stretch stimuli, biasing the level of G protein versus β-arrestin signaling to attenuate or stimulate hypertrophy. While the downstream mechanisms of stretch-induced β-arrestin signaling remain undefined, the function of β-arrestins has also been observed to block the interaction of APJ with Gαi proteins (i.e., desensitization), which may contribute to the pro-hypertrophic program (Scimia et al., 2012). These studies have implications for the consideration of APJ as a drug target, because the greatest benefit may be obtained not simply by apelin stimulation, but rather by selectively activating the Gαi-dependent signaling pathway or by inhibiting the ability of APJ to respond to mechanical stretch. For this purpose, further efforts are critical to clarify the intricacies of downstream integration of ligand and mechanosensitive signaling by APJ.
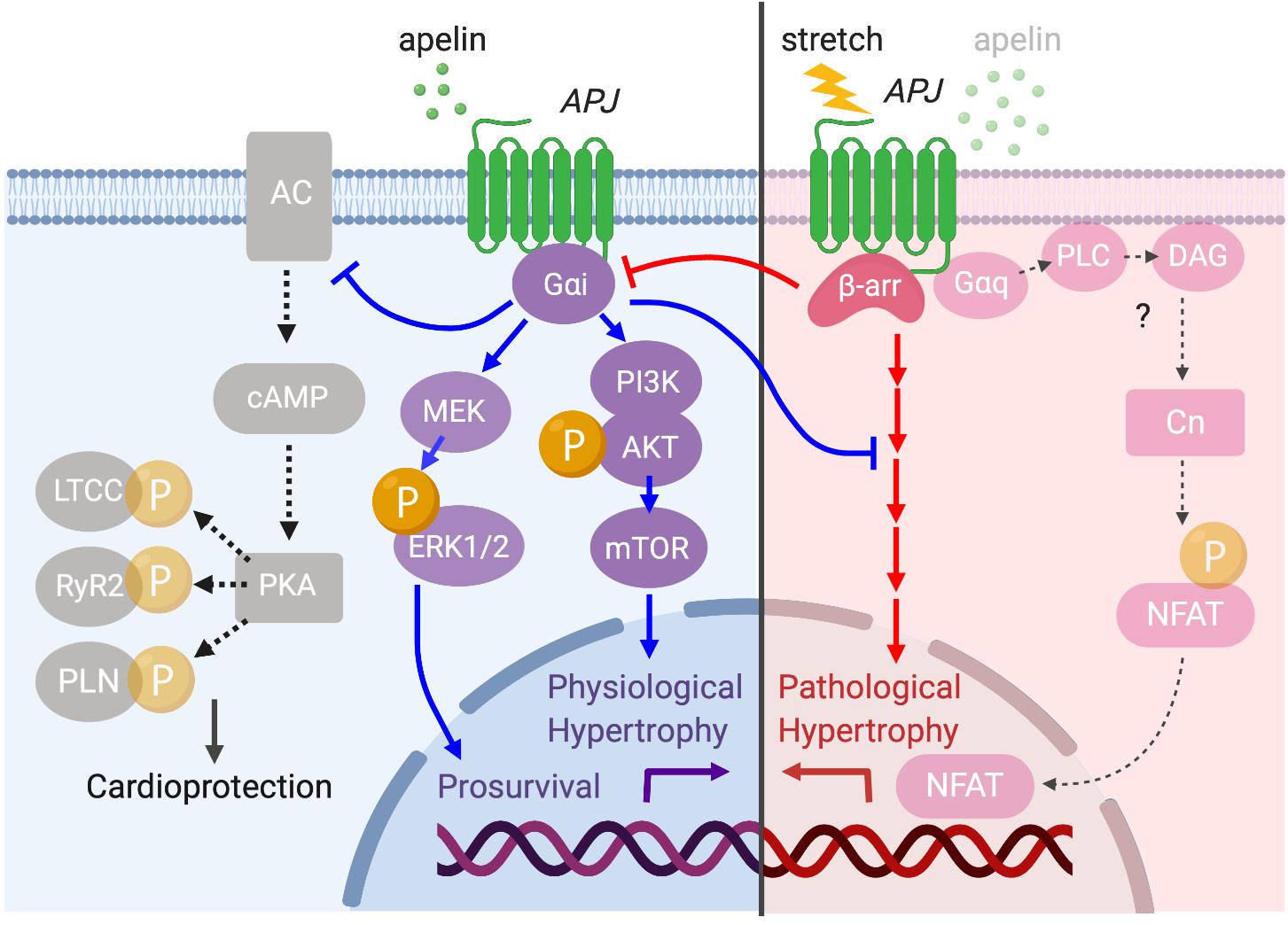
Figure 7. Diverse APJ signaling pathways activated by prolonged mechanical load or apelin. APJ translates apelin and stretch stimuli into distinct downstream signaling pathways. Apelin stimulation induces Gαi-dependent signaling to promote physiological hypertrophy and prosurvival signaling through the activation of Akt–mTOR and ERK1/2 (blue arrows). Gαi also inhibits deleterious cAMP–PKA pathway to serve a cardioprotective role (black dashed arrows). On the other hand, membrane stretch activates a β-arrestin-dependent program that results in pathological hypertrophy although the detailed downstream mechanisms are unresolved (red arrows). Importantly, there is a functional interplay between apelin- and stretch-dependent APJ signaling, in which apelin–APJ signaling blunts stretch-induced pathological signaling while stretch–APJ signaling interferes with apelin-induced Gαi activation. Although the apelin–APJ system involves Gαq-dependent signaling pathway (Figure 6), the relevance of this pathway to pathological hypertrophy has not been examined.
APJ Targeted Therapeutics for Mechanical Stress-Associated Heart Diseases (Table 1)
Apelin’s positive inotropy and anti-hypertrophic effects support its therapeutic potential in preventing and treating cardiovascular disease. The majority of current heart failure therapies are targeted at the inhibition of deleterious neurohormonal axes that are upregulated in the later stages of the disease. In this aspect, the apelin–APJ system is attractive because it appears to be downregulated in heart failure (Japp and Newby, 2008), and stimulation of this pathway may have additive or even synergistic efficacy to current therapy by targeting complementary but separate pathways. Indeed, intravenous administration of [Pyr1] apelin-13, an active fragment of apelin, in heart failure patients showed efficacy with peripheral and coronary vasodilatation and increases in cardiac output (Japp et al., 2010).
Nonetheless, the therapeutic application of apelin is limited because of its extremely short biological half-life and parenteral administration. This is attributed to the degradation by endogenous proteases circulating in the blood (Japp et al., 2008; Zhen et al., 2013). Furthermore, apelin is hydrolyzed and partially deactivated by angiotensin I converting enzyme 2 (ACE2) (Vickers et al., 2002; Wang et al., 2016). Thus, many efforts have been directed to the development of apelin analogs or novel agonists of APJ (Yamaleyeva et al., 2016). In particular, combinatorial screening of the NIH small molecule library identified a full non-peptide APJ agonist, ML233, at activating both Gαi- and β-arrestin-dependent pathways (Khan et al., 2011). In addition, using molecular dynamics simulations, Brame et al. (2015) designed cyclic analogs and identified a biased agonist, MM07, which activates APJ by preferentially stimulating Gαi-dependent pathways but not β-arrestin. Because stimulation of β-arrestin pathway could be deleterious, G protein-biased APJ ligand MM07 represents a potential new therapeutic for heart failure.
Stretch-Induced Conformational Change in AT1R and APJ
G protein-coupled receptors adopt distinct conformations to selectively activate different arrays of downstream signaling (Rosenbaum et al., 2009; Vaidehi and Kenakin, 2010). Detailed analysis of the conformational changes is often performed by crystal structure analysis, but this only allows a static view of a given receptor conformation. Bioluminescence resonance energy transfer (BRET) and fluorescence resonance energy transfer (FRET) techniques allow insight into the kinetics and amplitudes of agonist- and stretch-induced receptor conformations. Using these techniques, stretch-induced conformational changes of AT1R have been studied. BRET revealed that membrane stretch induces an active AT1R conformation allowing for G protein activation and subsequent β-arrestin recruitment (y Schnitzler et al., 2008). More recently, a study using BRET and FRET demonstrated that G protein activation is not necessary for β-arrestin recruitment and that mechanical stretch induces a particular β-arrestin conformation that is distinct from the agonist-stimulated conformation (Rakesh et al., 2010). Another approach was also taken to examine the conformational change of AT1R in response to stretch. Using the substituted cysteine accessibility method (SCAM) and molecular modeling approach, Yasuda et al. (2008) showed that stretch stimulation of AT1R induces a dislocation and a counterclockwise rotation of transmembrane domain 7 toward the ligand-binding pocket, a conformation that is distinct from the ligand-activated receptor conformation. These fascinating data suggest that conformational change is responsible for biased signaling in AT1R. Such studies of APJ and examination of the differential effect of novel AT1R ligands on its conformation could eventually lead to structure-based drug design focused on selective inhibition of adverse remodeling via stretch-dependent pathways.
Interaction Between Ang-II–AT1R and Apelin–APJ
Neurohormonal interaction between AT1R and APJ systems has been well-studied. Infusion of Ang II in rats causes an acute reduction of apelin and APJ levels in the heart, which is abolished by treatment with an ARB (Iwanaga et al., 2006). The left ventricular dysfunction observed in apelin deficient mice is restored to normal levels either by the treatment with an ARB or by AT1R gene deletion (Sato et al., 2013). Furthermore, apelin overexpression abolishes Ang II-induced cardiac hypertrophy in cultured myocytes (Ye et al., 2015). While a growing literature suggests the antagonistic interplay between AT1R and APJ systems through neurohormonal interactions, direct physical interaction between AT1R and APJ has also been reported. Co-immunoprecipitation and FRET analysis revealed heterodimerization of AT1R and APJ, in which the interaction appears to induce the inhibition of the AT1R signaling pathway (Chun et al., 2008). More recently, it was revealed that AT1R–APJ heterodimerization is induced by apelin, but is not affected by Ang II (Siddiquee et al., 2013). To date, the downstream effects of ligand and stretch stimulation on heterodimeric AT1R–APJ have not been examined. It is conceivable that AT1R–APJ heterodimers activate distinct signaling pathways compared to monomeric receptors, or that the multiple reported downstream pathways of AT1R and APJ may be partially dependent on the activation of the heterodimeric receptor. Understanding of this may lead to the identification of novel ligands with optimal effects on heart failure pathobiology.
Conclusion
Angiotensin II type 1 receptor and APJ are mechanosensitive GPCRs in the heart, playing vital roles in cardiac physiological adaptation to changes in mechanical load. However, when a mechanical load is excessive and sustained, it can induce maladaptive hypertrophic signaling. Enormous effort has been invested in understanding the physiological and pathophysiological roles of AT1R and APJ signaling to identify novel therapeutic strategies. Selective activation or inhibition of mechanically stimulated signaling components by biased agonists may yield more precise molecular enhancers of desired inotropic or cardioprotective effects while avoiding detrimental signaling. In addition, by gaining an improved understanding of the signaling mechanisms of these receptors in the heart, it is likely that complementary downstream targets will be identified to modulate cardiac function and ameliorate disease.
Author Contributions
KS took the lead in writing the manuscript with input from VP and EA.
Funding
This work was supported by the American Heart Association (Career Development Award 18CDA34020088 to KS), NIH (K08-HL143185 to VP), and the Sarnoff Cardiovascular Research and John Taylor Babbitt Foundation (VP).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abraham, D. M., Davis, R. T. III, Warren, C. M., Mao, L., Wolska, B. M., Solaro, R. J., et al. (2016). β-Arrestin mediates the Frank-Starling mechanism of cardiac contractility. Proc. Natl. Acad. Sci. U.S.A. 113, 14426–14431. doi: 10.1073/pnas.1609308113
Ait-Mou, Y., Hsu, K., Farman, G. P., Kumar, M., Greaser, M. L., Irving, T. C., et al. (2016). Titin strain contributes to the Frank-Starling law of the heart by structural rearrangements of both thin- and thick-filament proteins. Proc. Natl. Acad. Sci. U.S.A. 113, 2306–2311. doi: 10.1073/pnas.1516732113
Allen, D. G., Jewell, B. R., and Murray, J. W. (1974). The contribution of activation processes to the length–tension relation of cardiac muscle. Nature 248, 606–607. doi: 10.1038/248606a0
Allen, D. G., and Kurihara, S. (1982). The effects of muscle length on intracellular calcium transients in mammalian cardiac muscle. J. Physiol. 327, 79–94. doi: 10.1113/jphysiol.1982.sp014221
Alvarez, B. V., Pérez, N. G., Ennis, I. L., Camilión de Hurtado, M. C., and Cingolani, H. E. (1999). Mechanisms underlying the increase in force and Ca(2+) transient that follow stretch of cardiac muscle: a possible explanation of the Anrep effect. Circ. Res. 85, 716–722. doi: 10.1161/01.res.85.8.716
Ashley, E. A., Powers, J., Chen, M., Kundu, R., Finsterbach, T., Caffarelli, A., et al. (2005). The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc. Res. 65, 73–82. doi: 10.1016/j.cardiores.2004.08.018
Beqqali, A., Bollen, I. A. E., Rasmussen, T. B., van den Hoogenhof, M. M., van Deutekom, H. W. M., Schafer, S., et al. (2016). A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank–Starling mechanism. Cardiovasc. Res. 112, 452–463. doi: 10.1093/cvr/cvw192
Berry, M. F., Pirolli, T. J., Jayasankar, V., Burdick, J., Morine, K. J., Gardner, T. J., et al. (2004). Apelin has in vivo inotropic effects on normal and failing hearts. Circulation 110, II187–II193.
Boerrigter, G., Lark, M. W., Whalen, E. J., Soergel, D. G., Violin, J. D., and Burnett, J. C. Jr. (2011). Cardiorenal actions of Trv120027, a Novel ß-arrestin–biased ligand at the angiotensin Ii TYPE I RECEPTOR, in healthy and heart failure canines: a novel therapeutic strategy for acute heart failure. Circ. Heart Fail. 4, 770–778. doi: 10.1161/circheartfailure.111.962571
Brame, A. L., Maguire, J. J., Yang, P., Dyson, A., Torella, R., Cheriyan, J., et al. (2015). Design, characterization, and first-in-human study of the vascular actions of a novel biased apelin receptor agonist. Hypertension 65, 834–840. doi: 10.1161/hypertensionaha.114.05099
Brea, M. S., Díaz, R. G., Escudero, D. S., Caldiz, C. I., Portiansky, E. L., Morgan, P. E., et al. (2016). Epidermal growth factor receptor silencing blunts the slow force response to myocardial stretch. J. Am. Heart Assoc. 5:e004017. doi: 10.1161/JAHA.116.004017
Breithaupt, J. J., Pulcastro, H. C., Awinda, P. O., DeWitt, D. C., and Tanner, B. C. W. (2019). Regulatory light chain phosphorylation augments length-dependent contraction in PTU-treated rats. J. Gen. Physiol. 151, 66–76. doi: 10.1085/jgp.201812158
Calaghan, S. C., and White, E. (2001). Contribution of angiotensin II, endothelin 1 and the endothelium to the slow inotropic response to stretch in ferret papillary muscle. Pflugers Arch. 441, 514–520. doi: 10.1007/s004240000458
Caldiz, C. I., Garciarena, C. D., Dulce, R. A., Novaretto, L. P., Yeves, A. M., Ennis, I. L., et al. (2007). Mitochondrial reactive oxygen species activate the slow force response to stretch in feline myocardium. J. Physiol. 584, 895–905. doi: 10.1113/jphysiol.2007.141689
Chandrasekaran, B., Dar, O., and McDonagh, T. (2008). The role of apelin in cardiovascular function and heart failure. Eur. J. Heart Fail. 10, 725–732. doi: 10.1016/j.ejheart.2008.06.002
Chapman, N. A., Dupré, D. J., and Rainey, J. K. (2014). The apelin receptor: physiology, pathology, cell signalling, and ligand modulation of a peptide-activated class A GPCR. Biochem. Cell Biol. 92, 431–440. doi: 10.1139/bcb-2014-0072
Charo, D. N., Ho, M., Fajardo, G., Kawana, M., Kundu, R. K., Sheikh, A. Y., et al. (2009). Endogenous regulation of cardiovascular function by apelin-APJ. Am. J. Physiol. Heart Circ. Physiol. 297, H1904–H1913.
Chiluiza, D., Krishna, S., Schumacher, V. A., and Schlöndorff, J. (2013). Gain-of-function mutations in transient receptor potential C6 (TRPC6) activate extracellular signal-regulated kinases 1/2 (ERK1/2). J. Biol. Chem. 288, 18407–18420. doi: 10.1074/jbc.m113.463059
Chun, H. J., Ali, Z. A., Kojima, Y., Kundu, R. K., Sheikh, A. Y., Agrawal, R., et al. (2008). Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J. Clin. Invest. 118, 3343–3354.
Cingolani, H. E., Ennis, I. L., Aiello, E. A., and Pérez, N. G. (2011). Role of autocrine/paracrine mechanisms in response to myocardial strain. Pflugers Arch. 462, 29–38. doi: 10.1007/s00424-011-0930-9
Cingolani, H. E., Pérez, N. G., Cingolani, O. H., and Ennis, I. L. (2013). The Anrep effect: 100 years later. Am. J. Physiol. Heart Circ. Physiol. 304, H175–H182.
Cingolani, H. E., Villa-Abrille, M. C., Cornelli, M., Nolly, A., Ennis, I. L., Garciarena, C., et al. (2006). The positive inotropic effect of angiotensin II: role of endothelin-1 and reactive oxygen species. Hypertension 47, 727–734. doi: 10.1161/01.hyp.0000208302.62399.68
Clement, O., Puceat, M., Walsh, M. P., and Vassort, G. (1992). Protein kinase C enhances myosin light-chain kinase effects on force development and ATPase activity in rat single skinned cardiac cells. Biochem. J. 285(Pt 1), 311–317. doi: 10.1042/bj2850311
Crabtree, G. R., and Olson, E. N. (2002). NFAT signaling: choreographing the social lives of cells. Cell 109(Suppl.), S67–S79.
Dai, T., Ramirez-Correa, G., and Gao, W. D. (2006). Apelin increases contractility in failing cardiac muscle. Eur. J. Pharmacol. 553, 222–228. doi: 10.1016/j.ejphar.2006.09.034
de Tombe, P. P. (2003). Cardiac myofilaments: mechanics and regulation. J. Biomech. 36, 721–730. doi: 10.1016/s0021-9290(02)00450-5
De Vecchis, R., Cesaro, A., Ariano, C., Giasi, A., and Cioppa, C. (2017). Phosphodiesterase-5 inhibitors improve clinical outcomes, exercise capacity and pulmonary hemodynamics in patients with heart failure with reduced left ventricular ejection fraction: a meta-analysis. J. Clin. Med. Res. 9, 488–498. doi: 10.14740/jocmr3008w
Diniz, G. P., Carneiro-Ramos, M. S., and Barreto-Chaves, M. L. M. (2009). Angiotensin type 1 receptor mediates thyroid hormone-induced cardiomyocyte hypertrophy through the Akt/GSK-3beta/mTOR signaling pathway. Basic Res. Cardiol. 104, 653–667. doi: 10.1007/s00395-009-0043-1
Dyachenko, V., Husse, B., Rueckschloss, U., and Isenberg, G. (2009). Mechanical deformation of ventricular myocytes modulates both TRPC6 and Kir2.3 channels. Cell Calcium 45, 38–54. doi: 10.1016/j.ceca.2008.06.003
Farkasfalvi, K., Stagg, M. A., Coppen, S. R., Siedlecka, U., Lee, J., Soppa, G. K., et al. (2007). Direct effects of apelin on cardiomyocyte contractility and electrophysiology. Biochem. Biophys. Res. Commun. 357, 889–895. doi: 10.1016/j.bbrc.2007.04.017
Gorin, Y., Kim, N. H., Feliers, D., Bhandari, B., Choudhury, G. G., and Abboud, H. E. (2001). Angiotensin II activates Akt/protein kinase B by an arachidonic acid/redox-dependent pathway and independent of phosphoinositide 3-kinase. FASEB J. 15, 1909–1920. doi: 10.1096/fj..01-0165com
Gottlieb, P., Folgering, J., Maroto, R., Raso, A., Wood, T. G., Kurosky, A., et al. (2008). Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch. 455, 1097–1103. doi: 10.1007/s00424-007-0359-3
Gregg, D. E., and Shipley, R. E. (1944). Augmentation of left coronary inflow with elevation of left ventricular pressure and observations on the mechanism for increased coronary inflow with increased cardiac load. Am. J. Physiol. 142:44 doi: 10.1152/ajplegacy.1944.142.1.44
Hamdani, N., Herwig, M., and Linke, W. A. (2017). Tampering with springs: phosphorylation of titin affecting the mechanical function of cardiomyocytes. Biophys. Rev. 9, 225–237. doi: 10.1007/s12551-017-0263-9
Heineke, J., and Molkentin, J. D. (2006). Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 7, 589–600. doi: 10.1038/nrm1983
Hescheler, J., Rosenthal, W., Hinsch, K. D., Wulfern, M., Trautwein, W., and Schultz, G. (1988). Angiotensin II-induced stimulation of voltage-dependent Ca2+ currents in an adrenal cortical cell line. EMBO J. 7, 619–624. doi: 10.1002/j.1460-2075.1988.tb02855.x
Ho, K. K., Pinsky, J. L., Kannel, W. B., and Levy, D. (1993). The epidemiology of heart failure: the Framingham study. J. Am. Coll. Cardiol. 22, 6A–13A.
Hofmann, T., Schaefer, M., Schultz, G., and Gudermann, T. (2002). Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. U.S.A. 99, 7461–7466. doi: 10.1073/pnas.102596199
Ikenouchi, H., Barry, W. H., Bridge, J. H., Weinberg, E. O., Apstein, C. S., and Lorell, B. H. (1994). Effects of angiotensin II on intracellular Ca2+ and pH in isolated beating rabbit hearts and myocytes loaded with the indicator indo-1. J. Physiol. 480(Pt 2), 203–215. doi: 10.1113/jphysiol.1994.sp020353
Iwanaga, Y., Kihara, Y., Takenaka, H., and Kita, T. (2006). Down-regulation of cardiac apelin system in hypertrophied and failing hearts: possible role of angiotensin II–angiotensin type 1 receptor system. J. Mol. Cell. Cardiol. 41, 798–806. doi: 10.1016/j.yjmcc.2006.07.004
Japp, A. G., Cruden, N. L., Amer, D. A. B., Li, V. K. Y., Goudie, E. B., Johnston, N. R., et al. (2008). Vascular effects of apelin in vivo in man. J. Am. Coll. Cardiol. 52, 908–913. doi: 10.1016/j.jacc.2008.06.013
Japp, A. G., Cruden, N. L., Barnes, G., van Gemeren, N., Mathews, J., Adamson, J., et al. (2010). Acute cardiovascular effects of apelin in humans: potential role in patients with chronic heart failure. Circulation 121, 1818–1827. doi: 10.1161/circulationaha.109.911339
Japp, A. G., and Newby, D. E. (2008). The apelin-APJ system in heart failure: pathophysiologic relevance and therapeutic potential. Biochem. Pharmacol. 75, 1882–1892. doi: 10.1016/j.bcp.2007.12.015
Katz Arnold, M. (2002). Ernest Henry starling, his predecessors, and the “Law of the HEART.”. Circulation 106, 2986–2992. doi: 10.1161/01.cir.0000040594.96123.55
Kentish, J. C., and Wrzosek, A. (1998). Changes in force and cytosolic Ca2+ concentration after length changes in isolated rat ventricular trabeculae. J. Physiol. 506(Pt 2), 431–444. doi: 10.1111/j.1469-7793.1998.431bw.x
Khan, P., Maloney, P. R., Hedrick, M., Gosalia, P., Milewski, M., Li, L., et al. (2011). “Functional agonists of the apelin (APJ) receptor. Probe Reports from the NIH Molecular Libraries Program, (Bethesda, MD: National Center for Biotechnology Information).
Kim, K.-S., Abraham, D., Williams, B., Violin, J. D., Mao, L., and Rockman, H. A. (2012). β-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am. J. Physiol. Heart Circ. Physiol. 303, H1001–H1010.
Koguchi, W., Kobayashi, N., Takeshima, H., Ishikawa, M., Sugiyama, F., and Ishimitsu, T. (2012). Cardioprotective effect of apelin-13 on cardiac performance and remodeling in end-stage heart failure. Circ. J. 76, 137–144. doi: 10.1253/circj.cj-11-0689
Kuba, K., Zhang, L., Imai, Y., Arab, S., Chen, M., Maekawa, Y., et al. (2007). Impaired heart contractility in Apelin gene-deficient mice associated with aging and pressure overload. Circ. Res. 101, e32–e42.
Kumar, M., Govindan, S., Zhang, M., Khairallah, R. J., Martin, J. L., Sadayappan, S., et al. (2015). Cardiac myosin-binding protein C and Troponin-I phosphorylation independently modulate myofilament length-dependent activation. J. Biol. Chem. 290, 29241–29249. doi: 10.1074/jbc.m115.686790
Kuwahara, K., Wang, Y., McAnally, J., Richardson, J. A., Bassel-Duby, R., Hill, J. A., et al. (2006). TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Invest. 116, 3114–3126. doi: 10.1172/jci27702
Lee, D. K., Cheng, R., Nguyen, T., Fan, T., Kariyawasam, A. P., Liu, Y., et al. (2000). Characterization of apelin, the ligand for the APJ receptor. J. Neurochem. 74, 34–41.
Lefkowitz, R. J. (2004). Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol. Sci. 25, 413–422. doi: 10.1016/j.tips.2004.06.006
Levy, D., Garrison, R. J., Savage, D. D., Kannel, W. B., and Castelli, W. P. (1990). Prognostic implications of echocardiographically determined left ventricular mass in the Framingham heart study. N. Engl. J. Med. 322, 1561–1566. doi: 10.1056/nejm199005313222203
Lewis, G. D., Shah, R., Shahzad, K., Camuso, J. M., Pappagianopoulos, P. P., Hung, J., et al. (2007). Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation 116, 1555–1562. doi: 10.1161/circulationaha.107.716373
Lin, B. L., Matera, D., Doerner, J. F., Zheng, N., del Camino, D., Mishra, S., et al. (2019). In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. 116, 10156–10161. doi: 10.1073/pnas.1815354116
Louhivuori, L. M., Jansson, L., Nordström, T., Bart, G., Näsman, J., and Akerman, K. E. O. (2010). Selective interference with TRPC3/6 channels disrupts OX1 receptor signalling via NCX and reveals a distinct calcium influx pathway. Cell Calcium 48, 114–123. doi: 10.1016/j.ceca.2010.07.005
Mattiazzi, A. (1997). Positive inotropic effect of angiotensin II. Increases in intracellular Ca2+ or changes in myofilament Ca2+ responsiveness? J. Pharmacol. Toxicol. Methods 37, 205–214. doi: 10.1016/s1056-8719(97)00020-8
Maturana, A. D., Casal, A. J., Demaurex, N., Vallotton, M. B., Capponi, A. M., and Rossier, M. F. (1999). Angiotensin II negatively modulates L-type calcium channels through a pertussis toxin-sensitive G protein in adrenal glomerulosa cells. J. Biol. Chem. 274, 19943–19948. doi: 10.1074/jbc.274.28.19943
Methawasin, M., Hutchinson, K. R., Lee, E.-J., Smith, J. E. III, Saripalli, C., Hidalgo, C. G., et al. (2014). Experimentally increasing titin compliance in a novel mouse model attenuates the Frank-Starling mechanism but has a beneficial effect on diastole. Circulation 129, 1924–1936. doi: 10.1161/circulationaha.113.005610
Molkentin, J. D. (2004). Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 63, 467–475. doi: 10.1016/j.cardiores.2004.01.021
Molkentin, J. D., Lu, J. R., Antos, C. L., Markham, B., Richardson, J., Robbins, J., et al. (1998). A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93, 215–228. doi: 10.1016/s0092-8674(00)81573-1
Monasky, M. M., Taglieri, D. M., Henze, M., Warren, C. M., Utter, M. S., Soergel, D. G., et al. (2013). The β-arrestin-biased ligand TRV120023 inhibits angiotensin II-induced cardiac hypertrophy while preserving enhanced myofilament response to calcium. Am. J. Physiol. Heart Circ. Physiol. 305, H856–H866.
Morano, I., Bächle-Stolz, C., Katus, A., and Rüegg, J. C. (1988). Increased calcium sensitivity of chemically skinned human atria by myosin light chain kinase. Basic Res. Cardiol. 83, 350–359. doi: 10.1007/bf02005820
Murata, K., Ishida, J., Ishimaru, T., Mizukami, H., Hamada, J., Saito, C., et al. (2016). Lactation is a risk factor of postpartum heart failure in mice with cardiomyocyte-specific apelin receptor (APJ) overexpression. J. Biol. Chem. 291, 11241–11251. doi: 10.1074/jbc.m115.699009
Nagayama, T., Hsu, S., Zhang, M., Koitabashi, N., Bedja, D., Gabrielson, K. L., et al. (2009). Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy caused by pressure overload. J. Am. Coll. Cardiol. 53, 207–215. doi: 10.1016/j.jacc.2008.08.069
Nakayama, H., Wilkin, B. J., Bodi, I., and Molkentin, J. D. (2006). Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 20, 1660–1670. doi: 10.1096/fj.05-5560com
Onohara, N., Nishida, M., Inoue, R., Kobayashi, H., Sumimoto, H., Sato, Y., et al. (2006). TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 25, 5305–5316. doi: 10.1038/sj.emboj.7601417
Parikh, V. N., Liu, J., Shang, C., Woods, C., Chang, A. C., Zhao, M., et al. (2018). Apelin and APJ orchestrate complex tissue-specific control of cardiomyocyte hypertrophy and contractility in the hypertrophy-heart failure transition. Am. J. Physiol. Heart Circ. Physiol. 315, H348–H356.
Parmley, W. W., and Chuck, L. (1973). Length-dependent changes in myocardial contractile state. Am. J. Physiol. 224, 1195–1199. doi: 10.1152/ajplegacy.1973.224.5.1195
Pauli, A., Norris, M. L., Valen, E., Chew, G.-L., Gagnon, J. A., Zimmerman, S., et al. (2014). Toddler: an embryonic signal that promotes cell movement via apelin receptors. Science 343:1248636. doi: 10.1126/science.1248636
Pchejetski, D., Foussal, C., Alfarano, C., Lairez, O., Calise, D., Guilbeau-Frugier, C., et al. (2012). Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur. Heart J. 33, 2360–2369. doi: 10.1093/eurheartj/ehr389
Pérez, N. G., de Hurtado, M. C., and Cingolani, H. E. (2001). Reverse mode of the Na+-Ca2+ exchange after myocardial stretch: underlying mechanism of the slow force response. Circ. Res. 88, 376–382. doi: 10.1161/01.res.88.4.376
Pérez, N. G., Nolly, M. B., Roldan, M. C., Villa-Abrille, M. C., Cingolani, E., Portiansky, E. L., et al. (2011). Silencing of NHE-1 blunts the slow force response to myocardial stretch. J. Appl. Physiol. 111, 874–880. doi: 10.1152/japplphysiol.01344.2010
Pérez, N. G., Villa-Abrille, M. C., Aiello, E. A., Dulce, R. A., Cingolani, H. E., and Camilión de Hurtado, M. C. (2003). A low dose of angiotensin II increases inotropism through activation of reverse Na+/Ca2+ exchange by endothelin release. Cardiovasc. Res. 60, 589–597. doi: 10.1016/j.cardiores.2003.09.004
Perjés, Á, Skoumal, R., Tenhunen, O., Kónyi, A., Simon, M., Horváth, I. G., et al. (2014). Apelin increases cardiac contractility via protein kinase Cε- and extracellular signal-regulated kinase-dependent mechanisms. PLoS One 9:e93473. doi: 10.1371/journal.pone.0093473
Petroff, M. G., Aiello, E. A., Palomeque, J., Salas, M. A., and Mattiazzi, A. (2000). Subcellular mechanisms of the positive inotropic effect of angiotensin II in cat myocardium. J. Physiol. 529(Pt 1), 189–203. doi: 10.1111/j.1469-7793.2000.00189.x
Peyronnet, R., Bollensdorff, C., Capel, R. A., Rog-Zielinska, E. A., Woods, C. E., Charo, D. N., et al. (2017). Load-dependent effects of apelin on murine cardiomyocytes. Prog. Biophys. Mol. Biol. 130, 333–343. doi: 10.1016/j.pbiomolbio.2017.09.013
Poburko, D., Fameli, N., Kuo, K.-H., and van Breemen, C. (2008). Ca2+ signaling in smooth muscle: TRPC6, NCX and LNats in nanodomains. Channels 2, 10–12. doi: 10.4161/chan.2.1.6053
Poburko, D., Liao, C.-H., Lemos, V. S., Lin, E., Maruyama, Y., Cole, W. C., et al. (2007). Transient receptor potential channel 6–mediated, localized cytosolic [Na+] transients drive Na+/Ca2+ exchanger–mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ. Res. 101, 1030–1038. doi: 10.1161/circresaha.107.155531
Rajagopal, K., Whalen, E. J., Violin, J. D., Stiber, J. A., Rosenberg, P. B., Premont, R. T., et al. (2006). β-Arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 103, 16284–16289. doi: 10.1073/pnas.0607583103
Rajagopal, S., Rajagopal, K., and Lefkowitz, R. J. (2010). Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 9, 373–386. doi: 10.1038/nrd3024
Rakesh, K., Yoo, B., Kim, I.-M., Salazar, N., Kim, K.-S., and Rockman, H. A. (2010). β-arrestin–biased agonism of the angiotensin receptor induced by mechanical stress. Sci. Signal. 3:ra46. doi: 10.1126/scisignal.2000769
Ramirez, M. T., Sah, V. P., Zhao, X.-L., Hunter, J. J., Chien, K. R., and Brown, J. H. (1997). The MEKK-JNK pathway is stimulated by α1-adrenergic receptor and ras activation and is associated with in Vitroand in vivo cardiac hypertrophy. J. Biol. Chem. 272, 14057–14061. doi: 10.1074/jbc.272.22.14057
Redfield, M. M., Chen, H. H., Borlaug, B. A., Semigran, M. J., Lee, K. L., Lewis, G., et al. (2013). Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 309, 1268–1277.
Reiter, E., Ahn, S., Shukla, A. K., and Lefkowitz, R. J. (2012). Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197. doi: 10.1146/annurev.pharmtox.010909.105800
Rosenbaum, D. M., Rasmussen, S. G. F., and Kobilka, B. K. (2009). The structure and function of G-protein-coupled receptors. Nature 459, 356–363.
Ryba, D. M., Li, J., Cowan, C. L., Russell, B., Wolska, B. M., and Solaro, R. J. (2017). Long-Term Biased β-arrestin signaling improves cardiac structure and function in dilated cardiomyopathy. Circulation 135, 1056–1070. doi: 10.1161/circulationaha.116.024482
Sadoshima, J., and Izumo, S. (1993). Molecular characterization of angiotensin II–induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ. Res. 73, 413–423. doi: 10.1161/01.res.73.3.413
Sadoshima, J., Xu, Y., Slayter, H. S., and Izumo, S. (1993). Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 75, 977–984. doi: 10.1016/0092-8674(93)90541-w
Sarnoff, S. J., and Mitchell, J. H. (1961). The regulation of the performance of the heart. Am. J. Med. 30, 747–771.
Sarnoff, S. J., Mitchell, J. H., Gilmore, J. P., and Remensnyder, J. P. (1960). Homeometric autoregulation in the heart. Circ. Res. 8, 1077–1091. doi: 10.1161/01.res.8.5.1077
Sato, J., Makita, N., and Iiri, T. (2016). Inverse agonism: the classic concept of GPCRs revisited [Review]. Endocr. J. 63, 507–514. doi: 10.1507/endocrj.ej16-0084
Sato, T., Sato, C., Kadowaki, A., Watanabe, H., Ho, L., Ishida, J., et al. (2017). ELABELA-APJ axis protects from pressure overload heart failure and angiotensin II-induced cardiac damage. Cardiovasc. Res. 113, 760–769. doi: 10.1093/cvr/cvx061
Sato, T., Suzuki, T., Watanabe, H., Kadowaki, A., Fukamizu, A., Liu, P. P., et al. (2013). Apelin is a positive regulator of ACE2 in failing hearts. J. Clin. Invest. 123, 5203–5211. doi: 10.1172/jci69608
Scimia, M. C., Hurtado, C., Ray, S., Metzler, S., Wei, K., Wang, J., et al. (2012). APJ acts as a dual receptor in cardiac hypertrophy. Nature 488, 394–398. doi: 10.1038/nature11263
Seo, K., Rainer, P. P., Lee, D.-I., Hao, S., Bedja, D., Birnbaumer, L., et al. (2014a). Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cGMP-protein kinase G modulation. Circ. Res. 114, 823–832. doi: 10.1161/circresaha.114.302614
Seo, K., Rainer, P. P., Shalkey Hahn, V., Lee, D.-I., Jo, S.-H., Andersen, A., et al. (2014b). Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 111, 1551–1556. doi: 10.1073/pnas.1308963111
Shen, X., Cannell, M. B., and Ward, M.-L. (2013). Effect of SR load and pH regulatory mechanisms on stretch-dependent Ca(2+) entry during the slow force response. J. Mol. Cell. Cardiol. 63, 37–46. doi: 10.1016/j.yjmcc.2013.07.008
Siddiquee, K., Hampton, J., McAnally, D., May, L., and Smith, L. (2013). The apelin receptor inhibits the angiotensin II type 1 receptor via allosteric trans-inhibition. Br. J. Pharmacol. 168, 1104–1117. doi: 10.1111/j.1476-5381.2012.02192.x
Spassova, M. A., Hewavitharana, T., Xu, W., Soboloff, J., and Gill, D. L. (2006). A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl. Acad. Sci. U.S.A. 103, 16586–16591. doi: 10.1073/pnas.0606894103
Strachan, R. T., Sun, J.-P., Rominger, D. H., Violin, J. D., Ahn, S., Rojas Bie Thomsen, A., et al. (2014). Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J. Biol. Chem. 289, 14211–14224. doi: 10.1074/jbc.m114.548131
Szokodi, I., Tavi, P., Földes, G., Voutilainen-Myllylä, S., Ilves, M., Tokola, H., et al. (2002). Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ. Res. 91, 434–440. doi: 10.1161/01.res.0000033522.37861.69
Tachampa, K., Wang, H., Farman, G. P., and de Tombe, P. P. (2007). Cardiac troponin I threonine 144: role in myofilament length–dependent activation. Circ. Res. 101, 1081–1083. doi: 10.1161/circresaha.107.165258
Takimoto, E., Champion, H. C., Li, M., Belardi, D., Ren, S., Rodriguez, E. R., et al. (2005). Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 11, 214–222. doi: 10.1038/nm1175
Tatemoto, K., Hosoya, M., Habata, Y., Fujii, R., Kakegawa, T., Zou, M. X., et al. (1998). Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 251, 471–476. doi: 10.1006/bbrc.1998.9489
Thomas, W. G., Brandenburger, Y., Autelitano, D. J., Pham, T., Qian, H., and Hannan, R. D. (2002). Adenoviral-directed expression of the type 1A angiotensin receptor promotes cardiomyocyte hypertrophy via transactivation of the epidermal growth factor receptor. Circ. Res. 90, 135–142. doi: 10.1161/hh0202.104109
Thorburn, J., Frost, J. A., and Thorburn, A. (1994). Mitogen-activated protein kinases mediate changes in gene expression, but not cytoskeletal organization associated with cardiac muscle cell hypertrophy. J. Cell Biol. 126, 1565–1572. doi: 10.1083/jcb.126.6.1565
Toepfer, C. N., West, T. G., and Ferenczi, M. A. (2016). Revisiting Frank-Starling: regulatory light chain phosphorylation alters the rate of force redevelopment (ktr) in a length-dependent fashion. J. Physiol. 594, 5237–5254. doi: 10.1113/jp272441
Vaidehi, N., and Kenakin, T. (2010). The role of conformational ensembles of seven transmembrane receptors in functional selectivity. Curr. Opin. Pharmacol. 10, 775–781. doi: 10.1016/j.coph.2010.09.004
Vickers, C., Hales, P., Kaushik, V., Dick, L., Gavin, J., Tang, J., et al. (2002). Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 277, 14838–14843. doi: 10.1074/jbc.m200581200
Violin, J. D., Soergel, D. G., Boerrigter, G., Burnett, J. C. Jr., and Lark, M. W. (2013). GPCR biased ligands as novel heart failure therapeutics. Trends Cardiovasc. Med. 23, 242–249. doi: 10.1016/j.tcm.2013.01.002
von Anrep, G. (1912). On the part played by the suprarenals in the normal vascular reactions of the body. J. Physiol. 45, 307–317. doi: 10.1113/jphysiol.1912.sp001553
von Lewinski, D., Stumme, B., Maier, L. S., Luers, C., Bers, D. M., and Pieske, B. (2003). Stretch-dependent slow force response in isolated rabbit myocardium is Na+ dependent. Cardiovasc. Res. 57, 1052–1061. doi: 10.1016/s0008-6363(02)00830-1
Wang, C., Du, J.-F., Wu, F., and Wang, H.-C. (2008). Apelin decreases the SR Ca2+ content but enhances the amplitude of [Ca2+]i transient and contractions during twitches in isolated rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 294, H2540–H2546.
Wang, J., Hanada, K., Gareri, C., and Rockman, H. A. (2018). Mechanoactivation of the angiotensin II type 1 receptor induces β-arrestin-biased signaling through Gαi coupling. J. Cell. Biochem. 119, 3586–3597. doi: 10.1002/jcb.26552
Wang, W., McKinnie, S. M., Farhan, M., Paul, M., McDonald, T., McLean, B., et al. (2016). Angiotensin-converting enzyme 2 metabolizes and partially inactivates pyr-apelin-13 and apelin-17. Hypertension 68, 365–377. doi: 10.1161/hypertensionaha.115.06892
Wang, W., McKinnie, S. M., Patel, V. B., Haddad, G., Wang, Z., Zhabyeyev, P., et al. (2003). Loss of apelin exacerbates myocardial infarction adverse remodeling and ischemia-reperfusion injury: therapeutic potential of synthetic apelin analogues. J. Am. Heart Assoc. 2:e000249.
Wang, Y., Huang, S., Sah, V. P., Ross, J. Jr., Brown, J. H., Han, J., et al. (1998a). Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J. Biol. Chem. 273, 2161–2168. doi: 10.1074/jbc.273.4.2161
Wang, Y., Su, B., Sah, V. P., Brown, J. H., Han, J., and Chien, K. R. (1998b). Cardiac hypertrophy induced by mitogen-activated protein kinase kinase 7, a specific activator for c-Jun NH2-terminal kinase in ventricular muscle cells. J. Biol. Chem. 273, 5423–5426. doi: 10.1074/jbc.273.10.5423
Watanabe, A., and Endoh, M. (1998). Relationship between the increase in Ca2+ transient and contractile force induced by angiotensin II in aequorin-loaded rabbit ventricular myocardium. Cardiovasc. Res. 37, 524–531. doi: 10.1016/s0008-6363(97)00287-3
Wei, F., Jia, X.-J., Yu, S.-Q., Gu, Y., Wang, L., Guo, X.-M., et al. (2011). Candesartan versus imidapril in hypertension: a randomised study to assess effects of anti-AT1 receptor autoantibodies. Heart 97, 479–484. doi: 10.1136/hrt.2009.192104
Wijnker, P. J. M., Sequeira, V., Foster, D. B., Li, Y., Dos Remedios, C. G., Murphy, A. M., et al. (2014). Length-dependent activation is modulated by cardiac troponin I bisphosphorylation at Ser23 and Ser24 but not by Thr143 phosphorylation. Am. J. Physiol. Heart Circ. Physiol. 306, H1171–H1181.
Wilkins, B. J., and Molkentin, J. D. (2004). Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem. Biophys. Res. Commun. 322, 1178–1191. doi: 10.1016/j.bbrc.2004.07.121
Wisler, J. W., Xiao, K., Thomsen, A. R. B., and Lefkowitz, R. J. (2014). Recent developments in biased agonism. Curr. Opin. Cell Biol. 27, 18–24. doi: 10.1016/j.ceb.2013.10.008
Xie, F., Liu, W., Feng, F., Li, X., Yang, L., Lv, D., et al. (2014). A static pressure sensitive receptor APJ promote H9c2 cardiomyocyte hypertrophy via PI3K-autophagy pathway. Acta Biochim. Biophys. Sin. 46, 699–708. doi: 10.1093/abbs/gmu046
y Schnitzler, M. M., Storch, U., Meibers, S., Nurwakagari, P., Breit, A., Essin, K., et al. (2008). Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 27, 3092–3103. doi: 10.1038/emboj.2008.233
Yamaguchi, Y., Iribe, G., Kaneko, T., Takahashi, K., Numaga-Tomita, T., Nishida, M., et al. (2018). TRPC3 participates in angiotensin II type 1 receptor-dependent stress-induced slow increase in intracellular Ca2+ concentration in mouse cardiomyocytes. J. Physiol. Sci. 68, 153–164. doi: 10.1007/s12576-016-0519-3
Yamaleyeva, L. M., Shaltout, H. A., and Varagic, J. (2016). Apelin-13 in blood pressure regulation and cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 25, 396–403. doi: 10.1097/mnh.0000000000000241
Yao, H., Peng, F., Fan, Y., Zhu, X., Hu, G., and Buch, S. J. (2009). TRPC channel-mediated neuroprotection by PDGF involves Pyk2/ERK/CREB pathway. Cell Death Differ. 16, 1681–1693. doi: 10.1038/cdd.2009.108
Yasuda, N., Miura, S.-I., Akazawa, H., Tanaka, T., Qin, Y., Kiya, Y., et al. (2008). Conformational switch of angiotensin II type 1 receptor underlying mechanical stress-induced activation. EMBO Rep. 9, 179–186. doi: 10.1038/sj.embor.7401157
Ye, L., Ding, F., Zhang, L., Shen, A., Yao, H., Deng, L., et al. (2015). Serum apelin is associated with left ventricular hypertrophy in untreated hypertension patients. J. Transl. Med. 13:290.
Yeves, A. M., Caldiz, C. I., Aiello, E. A., Villa-Abrille, M. C., and Ennis, I. L. (2015). Reactive oxygen species partially mediate high dose angiotensin II-induced positive inotropic effect in cat ventricular myocytes. Cardiovasc. Pathol. 24, 236–240. doi: 10.1016/j.carpath.2015.01.002
Zechner, D., Thuerauf, D. J., Hanford, D. S., McDonough, P. M., and Glembotski, C. C. (1997). A role for the p38 mitogen-activated protein kinase pathway in myocardial cell growth, sarcomeric organization, and cardiac-specific gene expression. J. Cell Biol. 139, 115–127. doi: 10.1083/jcb.139.1.115
Zhang, M., and Kass, D. A. (2011). Phosphodiesterases and cardiac cGMP: evolving roles and controversies. Trends Pharmacol. Sci. 32, 360–365. doi: 10.1016/j.tips.2011.02.019
Zhang, M., Prosser, B. L., Bamboye, M. A., Gondim, A. N. S., Santos, C. X., Martin, D., et al. (2015). Contractile function during angiotensin-II activation: increased nox2 activity modulates cardiac calcium handling via phospholamban phosphorylation. J. Am. Coll. Cardiol. 66, 261–272.
Zhang, Z., Yu, B., and Tao, G.-Z. (2009). Apelin protects against cardiomyocyte apoptosis induced by glucose deprivation. Chin. Med. J. 122, 2360–2365.
Zhen, E. Y., Higgs, R. E., and Gutierrez, J. A. (2013). Pyroglutamyl apelin-13 identified as the major apelin isoform in human plasma. Anal. Biochem. 442, 1–9. doi: 10.1016/j.ab.2013.07.006
Keywords: angiotensin II, AT1R, apelin, APJ, β-arrestin, Frank–Starling law, Anrep effect, cardiac hypertrophy
Citation: Seo K, Parikh VN and Ashley EA (2020) Stretch-Induced Biased Signaling in Angiotensin II Type 1 and Apelin Receptors for the Mediation of Cardiac Contractility and Hypertrophy. Front. Physiol. 11:181. doi: 10.3389/fphys.2020.00181
Received: 29 November 2019; Accepted: 17 February 2020;
Published: 13 March 2020.
Edited by:
Remi Peyronnet, University of Freiburg, GermanyReviewed by:
Lewis J. Watson, University of Pikeville, United StatesSumanth D. Prabhu, The University of Alabama at Birmingham, United States
Copyright © 2020 Seo, Parikh and Ashley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kinya Seo, a3NlbzFAc3RhbmZvcmQuZWR1