 Agnieszka A. Kusiak1
Agnieszka A. Kusiak1 Monika A. Jakubowska
Monika A. Jakubowska Pawel E. Ferdek
Pawel E. Ferdek
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 14 February 2020
Sec. Gastrointestinal Sciences
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00078
This article is part of the Research TopicSpotlight on the Background Actors - Physiology and Pathophysiology of Supporting, Accessory and Less Common Cell Types in the Gastrointestinal TractView all 10 articles
The interest in pancreatic stellate cells (PSCs) has been steadily growing over the past two decades due mainly to the central role these cells have in the desmoplastic reaction associated with diseases of the pancreas, such as pancreatitis or pancreatic cancer. In recent years, the scientific community has devoted substantial efforts to understanding the signaling pathways that govern PSC activation and interactions with neoplastic cells. This mini review aims to summarize some very recent findings on signaling in PSCs and highlight their impact to the field.
Since their discovery, pancreatic stellate cells (PSCs) have often been overlooked in favor of other cellular components of the pancreas – pancreatic acinar cells (PACs), ductal cells, and pancreatic islets – that all are present in evident abundance in the tissue architecture and perform obvious exocrine or endocrine functions. Only relatively recently have PSCs gained substantial attention from the scientific community, and this was once it became clear that these cells play important roles in pancreatic pathophysiology. PSCs occupy periacinar space and form a dynamic network in between pancreatic acini. In health, PSCs predominantly occur in a quiescent phenotype and are a minority among cellular components of the pancreas, comprising merely 4–7% of all cells in the organ (Apte et al., 1998). One characteristic propensity of quiescent PSCs is the presence of retinoid droplets in the cytosol (Apte et al., 1998). Upon activation triggered by tissue injury, PSCs undergo a series of morphological alterations, which include the loss of retinoid droplets, increased prominence of the ER network, and elongation of the cellular processes; they also start expressing alpha smooth muscle actin (α-SMA) as well as collagen types I and III, laminin, and fibronectin (Apte et al., 1998; Bachem et al., 1998). As a result, activated PSCs increase in numbers, and their products – extracellular matrix (ECM) components – may become a significant part of the organ. If unbalanced, this mechanism underlies the development of pancreatic fibrosis. This mini review aims to summarize the most recent studies on signaling in PSCs relevant in physiology and pathophysiology of the pancreas.
Physiologically, the transition of quiescent PSCs into a proliferative, fibrogenic phenotype is an autonomous repair reaction to tissue injury. Since the damage to enzyme-storing PACs is particularly threatening to the integrity of the tissue, there is an obvious need for an efficient and well-balanced system orchestrating regeneration and containment of the injury with PSC activation at its center. This mechanism is expected to have cross-talks between numerous signaling pathways (Figure 1), given that PSC activation may be triggered by various stimuli, such as inflammatory mediators (Mews et al., 2002), alcohol metabolites (Apte et al., 2000), and growth factors, including transforming growth factors TGF-α and TGF-β, platelet-derived growth factor (PDGF) (Apte et al., 1999), or connective tissue growth factor (CTGF) (Gao and Brigstock, 2005). These activating factors are secreted not only by infiltrating immune cells but might also be coming from other sources e.g., acinar cells (PACs) (Masamune and Shimosegawa, 2009). Other pathophysiologically relevant factors associated with PSC activation include hyperglycemia (Nomiyama et al., 2007), hypoxia (Masamune et al., 2008), and oxidative stress (Casini et al., 2000). It is widely accepted that signaling pathways, such as the MAPK/ERK (Jaster et al., 2002; Yoshida et al., 2004), PI3K (McCarroll et al., 2004), Smad (Ohnishi et al., 2004), Jak/STAT (Komar et al., 2017), PKC (Nomiyama et al., 2007), and Hedgehog (Li et al., 2014), play a crucial role in PSC physiology and have been subjects of previous reviews (Masamune and Shimosegawa, 2009). However, recent evidence has indicated that the Hippo and Wnt pathways as well as authophagy and calcium signaling might be equally important players in PSCs. These new findings are briefly described below.
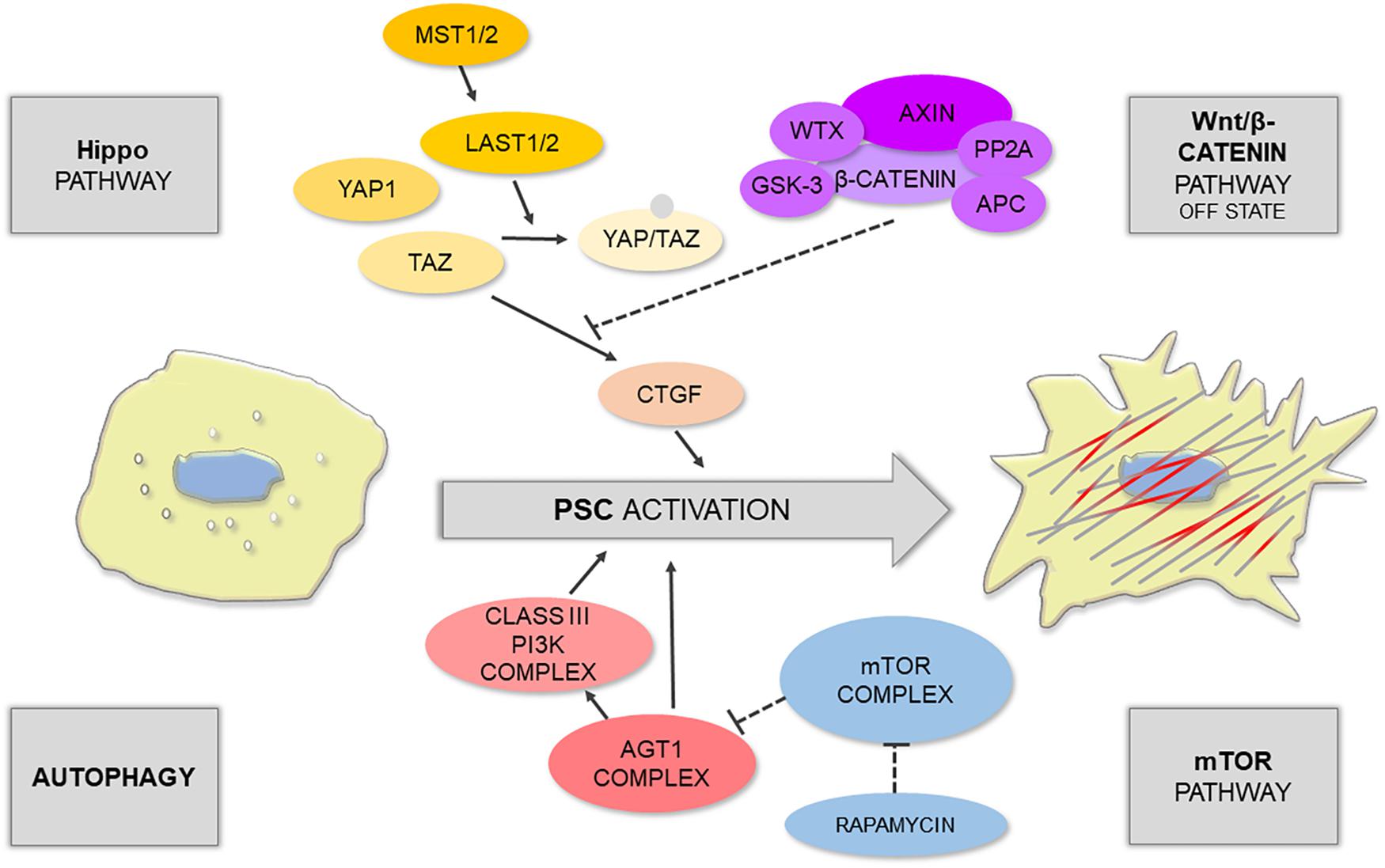
Figure 1. Schematic illustration shows that PSC activation is controlled by different signaling pathways.
The Hippo signaling pathway is one of the main restrictors of cell proliferation that tend to promote apoptosis; it can be triggered by numerous microenvironmental cues, such as cell–cell contact, mechanotransduction, and cellular stress (Meng et al., 2016). During tissue repair, which normally requires cell proliferation, the Hippo pathway is often downregulated by phosphorylation of its main effectors: yes-associated protein 1 (YAP1) and transcriptional co-activator with PDZ-binding motif (TAZ) (Furth et al., 2018). This results in the switched off expression of YAP1/TAZ target genes, particularly CTGF, responsible for PSC activation. Liu J. et al. (2019) have recently demonstrated that acinar-specific deletion of central kinases in Hippo signaling – large tumor suppressor 1 and 2 (LATS1/2) – contributed to inflammation and severe fibrosis in vivo, even without the initiation of acinar cell rupture; whereas removal of CTGF with a neutralizing antibody attenuated this effect. The authors have also shown that in the mouse model of caerulein-induced pancreatitis there was an upregulation of secreted phosphoprotein 1 (SPP1) (Liu J. et al., 2019), a previously reported target of YAP1/TAZ involved in fibroinflammatory responses in other organs (Pardo et al., 2005). Interestingly, YAP1 and TAZ activity can be further modulated by the Wnt/β-catenin pathway. For instance, Wnt signaling can activate YAP1/TAZ by preventing their degradation by the β-catenin destruction complex (Hansen et al., 2015). Since the Wnt/β-catenin pathway plays a role in embryonic development as well as in homeostasis of the developed tissues, Wnt signaling becomes particularly important in the pathogenesis of proliferative diseases, including cancer.
The main factors associated with the Wnt signaling pathway were found both in the endocrine and exocrine pancreas; this includes β-catenin, Wnt2, Wnt5a, and Wnt inhibitors e.g., the secreted frizzled-related protein (SFRP) family (Heller et al., 2003). The distribution of these factors is dependent on the condition of the tissue – that is it changes upon tissue damage and in fibrosis. An analysis of human samples revealed that in the non-fibrotic pancreatic tissue, β-catenin is mainly located at the cell membrane of PACs, inhibitory SFRP4 is present in these cells, and Wnt2 is expressed at a low level (Blauer et al., 2019). In moderate fibrosis, the expression of Wnt2 in PACs increases, and β-catenin can now be found in the acinar nuclei indicating transcriptional activation; whereas in advanced fibrosis, β-catenin mostly localizes to PSCs (Blauer et al., 2019). Interestingly, this distribution pattern can be modeled in vitro. PSCs express β-catenin in monocultures, but when co-cultured with PACs, the levels of SFRP4 increase compared to monoculture, and thus PSCs do not exhibit nuclear distribution of β-catenin (Blauer et al., 2019). The above indicates that, in terms of Wnt signaling, a monoculture of PSCs reflects advanced stages of fibrosis, whereas a co-culture of PACs-PSCs has the expression pattern seen in moderate fibrosis. In another study, reversion of activated PSCs to quiescence was associated with upregulation of retinoic acid receptor β (RARβ), an increase in SFRP4 expression, and a reduction in nuclear β-catenin content in these cells (Carapuca et al., 2016). However, other reports suggest that both Wnt2 and SFRP4 might be elevated upon PSC activation (Hu et al., 2014). It is likely that PSCs could utilize multiple mechanisms regulating SFRP4, and thus its role in the interplay between PSCs and PACs, as well as in the process of PSC phenotype transition, is yet to be fully elucidated.
Autophagy is traditionally viewed either as a means for nutrient seeking under stress or as a physiological process for a cell quality check and removal of dysfunctional cellular components (Glick et al., 2010; Bootman et al., 2018). While it is normally pro-survival, disruption of autophagy has been associated with non-apoptotic cell death via the interaction of its main effector Beclin-1 with anti-apoptotic Bcl-2 protein (Pattingre et al., 2005). Just like other autophagy-related proteins (Atg), including Atg1 or Atg5, Beclin-1 acts as an initiator of autophagosome formation (Shibutani et al., 2015). The relationship between PSC activation, pancreatic fibrosis, and autophagy has recently been well documented in the literature; autophagy has been suggested to be a source of energy and molecular material for PSC phenotype transition and ECM deposition (Endo et al., 2017). In highly fibrotic pancreatic solid tumors, cancer cells can stimulate autophagy of PSCs. This leads to the release of non-essential amino acids (NEAA), mainly alanine, from PSCs; NEAA are then used by cancer cells as an alternative (to glucose) energy source (Sousa et al., 2016). However, mouse models revealed that deregulation of Atg5 in the pancreas results in a similar phenotype to that seen in human chronic pancreatitis, indicating that the development of this disease appears to involve defects in autophagy (Diakopoulos et al., 2015). Mammalian target of rapamycin (mTOR) is a very well-known suppressor of autophagy that inhibits this process by regulating the activity of the initiator complex Atg1/ULK1 (Mizushima, 2010). In turn, mTOR can be upregulated by sphingosine-1-phosphate (S1P) or its analog, the immunomodulator fingolimod (FTY720), both of which inhibit autophagy (Thangada et al., 2014). FTY720 has been shown to suppress PSC activation and autophagy via the mTOR pathway; it can also increase the Bax/Bcl-2 ratio with a simultaneous decrease in the mitochondrial membrane potential (Cui et al., 2019). The mTOR pathway also links autophagy with intracellular calcium signals (Bootman et al., 2018).
Calcium signaling is one of the most universal pathways, regulating virtually every cellular process from excitability and motility to apoptosis (Clapham, 2007). Sophisticated machinery consisting of pumps, ion channels, and active transporters controls Ca2+ homeostasis; while spatiotemporal changes in Ca2+ concentration encode signals that exert cell responses. In the pancreas, Ca2+ signals play a particularly important role in regulating secretion of digestive enzymes by PACs, which has been a relatively frequent subject of research in the past decades (Petersen and Tepikin, 2008). In contrast, Ca2+ signaling in PSCs has so far been investigated only by a handful of studies. Nevertheless, recent reports indicate that Ca2+ signals are also important in the regulation of PSC physiology, showing a clear cross-talk with other pathways, such as NO signaling (Jakubowska et al., 2016). We already know that PSCs express bradykinin receptor type 2 (BDKRB2) (Ferdek et al., 2016), and pharmacological studies revealed that these cells produce Ca2+ responses even to very low doses of bradykinin (Won et al., 2011; Gryshchenko et al., 2016a). Since bradykinin is a well-known pro-inflammatory mediator, BDKRB2 signaling may well play a role in activation of PSCs in diseases of the pancreas (Gryshchenko et al., 2016b). PSCs also express a number of Ca2+ channels, particularly those from the family of transient receptor potential (TRP). For example, TRPC6 has been associated with autocrine stimulation of PSCs in hypoxic conditions (Nielsen et al., 2017); whereas another TRP member, TRPC1, was proposed to contribute to pressure-induced activation of these cells (Fels et al., 2016). Deregulated Ca2+ signals directly underlie the pathophysiology of acute pancreatitis, and thus it is somewhat surprising that so little is known about the effects that common inducers of pancreatic pathology, such as bile acids and ethanol metabolites, have on Ca2+ signaling in PSCs. Given that ethanol induces the expression of TRPV4 in these cells (Zhang et al., 2013), whereas sodium cholate and taurocholate generate noxious and sustained Ca2+ signals (Ferdek et al., 2016), the contribution of PSCs in the development of acute pancreatitis might still be underrated by the current dogma.
Despite being a minority in the normal pancreas, the role of activated PSCs becomes apparent in pathophysiological conditions. It is well established that persistent activation of PSCs is the main contributor to fibrosis in pancreatic diseases, with pancreatic cancer and pancreatitis being the most prevalent. In health, PSCs regulate ECM turnover not only by producing its components but also by secreting key enzymes engaged in EMC remodeling – matrix metalloproteinases (MMP) as well as their inhibitors – tissue inhibitors of metalloproteinases (TIMPs) (Phillips et al., 2003). PSCs have been shown to activate in response to cytokines upregulated in acute pancreatitis (Mews et al., 2002). Upon activation, the fate of PSCs largely depends on the external stimuli and can follow different scenarios. If the injury is transient, activated PSCs revert to quiescence or become senescent (McCarroll et al., 2003). However, a subpopulation of PSCs may expand excessively when activating factors accumulate or occur in a persistent manner e.g., in chronic pancreatitis (Haber et al., 1999; Masamune et al., 2009). Activated PSCs secrete increased amounts of MMP-2 (Phillips et al., 2003), and this enzyme breaks down normal basement membrane, which then may hasten its replacement by fibril-forming collagen (Friedman, 2000).
Pancreatic cancer – mainly pancreatic ductal adenocarcinoma (PDAC) – remains one of the most serious problems of our modern society. According to the Global Cancer Statistics 2018 (GLOBOCAN 2018), PDAC has been ranked the 11th most frequent cancer worldwide and the 7th most deadly cancer, accounting for 4.5% of all cancer-related deaths (Bray et al., 2018). The hallmark of pancreatic tumorigenesis is a desmoplastic reaction that leads to the formation of fibrotic stroma. The stroma not only provides a hypoxic microenvironment for the neoplastic cells, but it also constitutes a physical barrier that limits the efficacy of drug delivery to the tumor. Despite significant advances in the treatment regimens and surgical procedures, the collagen-rich fibrotic microenvironment of pancreatic cancer is the main reason why chemotherapeutics are largely ineffective and the clinical outcome remains very poor (Dangi-Garimella et al., 2011; Shields et al., 2012). It is becoming increasingly clear that a successful therapy should not only be focused on cancer cells but also target the tumor-associated fibrotic stroma.
Pancreatic stellate cells are the main cellular contributors to the desmoplastic reaction in PDAC. They become progressively activated in the process of tumorigenesis and deposit collagen fibers that embed and protect cancer cells. This collagen-rich microenvironment becomes an integral part of the developing tumor, and the interactions between the stroma and cancer cells are essential practically at every stage of tumorigenesis from its initiation, through progression, and to metastasis. As an example, PSCs have been shown to increase the viability and proliferative capacity of cancer cells as well as reduce gemcitabine-induced apoptosis of these cells. This was attenuated by pharmacological blockade of TGF-β receptor I (TGF-βRI, ALK5) (Liu S.L. et al., 2019). The inhibition of stroma-cancer cell interactions is currently attracting a lot of interest as a promising strategy that may sensitize PDAC to therapy and increase the 5-year survival rate of patients suffering from this disease (Cannon et al., 2018).
In light of the above, a number of recent studies have been devoted to investigating signaling pathways in tumor–stroma interactions. Yet again, the Hippo pathway stands out as an important player in pancreatic tumorigenesis. YAP1 is expressed by cancer cells as well as is present in the nuclei of PDAC-derived PSCs, correlating with their activated phenotype. Xiao et al. (2019) have recently demonstrated in vitro that depletion of YAP1 in PSCs (via silencing its expression) results in a shift of PSC phenotype toward quiescence, evidenced by a downregulation of markers such as α-SMA and collagen I as well as a decrease in PSC contractility and proliferation. While secreted protein acidic and cysteine rich (SPARC) has previously been associated with the poor outcomes of pancreatic cancer patients (Erkan et al., 2008; Whatcott et al., 2015), very recently SPARC has been identified as a downstream target of YAP1 that mediates its effects on cancer cell proliferation via paracrine signaling (Xiao et al., 2019). Since YAP1 expression shows a strong correlation with the degree of tissue fibrosis in patient samples, YAP1 was suggested as a feasible target in PDAC therapy that, in principle, could inactivate PSCs and limit the development of the tumor-associated stroma (Xiao et al., 2019).
Another recently revealed paracrine factor involved in stroma–cancer cell interactions is leukemia inhibitory factor (LIF) (Ohlund et al., 2017; Bressy et al., 2018; Shi et al., 2019). This cytokine is secreted by activated PSCs in PDAC lesions and acts on the neighboring cancer cells via its receptor LIFR, activating the STAT3 pathway, driving tumor progression and increasing chemoresistance (Shi et al., 2019). Since its circulating and tissue levels increase in PDAC patients, LIF was postulated to be both a novel biomarker and a feasible therapeutic target.
Stroma–cellular interaction may also occur via cell surface receptors such as integrins (Schnittert et al., 2018). Integrin α-11 (ITGA11), a collagen type I–binding receptor, is an interesting example: essentially absent in the healthy pancreas, it becomes expressed within the stromal fraction of PDAC tissues. It has been found that approximately 80% of α-SMA-positive cells are also ITGA11-positive, and knockdown of ITGA11 in PSCs results in attenuation of differentiation, migration, and secretion of ECM components by these cells (Schnittert et al., 2019). Another cell surface protein, integrin α-5 (ITGA5), was also shown to be present in as many as 72% of α-SMA-positive cells in human PDAC tissues, and its high expression was associated with poor prognosis (Kuninty et al., 2019). ITGA5 was demonstrated to play a role in TGF-β–mediated activation of PSCs via Smad2 and FAK pathways; and its knockdown inhibited both PSC-induced cancer cell proliferation in vitro and tumor growth in vivo (Kuninty et al., 2019). The authors went even one step further and developed a novel ITGA5-antagonizing peptidomimetic (AV3) that could inhibit PSC activation and enhance the cytotoxic effects of gemcitabine in spheroid co-cultures of cancer cell lines with PSCs (Kuninty et al., 2019).
Bcl2-associated athanogene 3 (BAG3) is expressed by multiple cancer types, including PDAC, and correlates with poor prognosis. Recent studies show that this marker is also present in activated PSCs and may promote, via IL−6, TGF−β2, and insulin-like growth factor-binding protein 2 (IGFBP2) signaling, autocrine-driven maintenance of the activated phenotype in these cells (Yuan et al., 2019). Vice versa, BAG3−positive PSCs also increase migration and invasion capacity of nearby cancer cells via soluble factors such as IL−8, monocyte chemoattractant protein-1 (MCP-1), TGF−β2, and IGFBP2 (Yuan et al., 2019). A recent report shows that plasminogen activator inhibitor-1 (PAI-1), a common regulator of blood coagulation, cell apoptosis, and migration, is secreted by pancreatic cancer cells and activates PSCs LRP-1/ERK/c-JUN signaling (Wang et al., 2019). Interestingly, knockdown of PAI-1 in cancer cells abolished activation in co-cultured PSCs, indicating that PAI-1 might be one of the key players in PSC – cancer cell interaction (Fang et al., 2012).
While cancer-associated PSCs secrete hepatocyte growth factor (HGF), its receptor c-MET is present on pancreatic cancer cells. Simultaneous inhibition of HGF and c-MET combined with gemcitabine is more effective in reduction of tumor volume in an orthotopic model of pancreatic cancer than chemotherapy alone, indicating a critical role of the HGF/c-MET pathway in PSC–cancer cell interactions (Pothula et al., 2017). What is more, inhibition of HGF, c-MET, and urokinase-type plasminogen activator (uPA) has been shown to decrease the angiogenic properties of endothelial cells, suggesting the role of PSCs and the HGF/c-MET pathway in neoangionesis (Patel et al., 2014). The authors envision that a novel antiangiogenic approach that targets the HGF/c-MET and uPA pathways could be used against pancreatic cancer (Patel et al., 2014).
The human pancreas is an organ that weights only approximately 100 g (Innes and Carey, 1994), but it produces daily as much as 1 L (or 1 kg) of pancreatic juice, comprising water, bicarbonate, and a variety of digestive enzymes (Pallagi et al., 2015). This substantial difference between the juice mass and the mass of the tissue is associated with mechanical stress in the organ exerted by the fluid pressure on pancreatic duct walls and pancreatic acini. PSCs have recently been attributed to sensing the mechanical properties of pancreatic microenvironment (Cortes et al., 2019b). PSCs control mechanostasis (mechanical homeostasis of the organ in response to various types of forces) not only in the normal tissue but also under pathophysiological conditions like that present in the development of fibrosis (Ferdek and Jakubowska, 2017). Deposition of collagen fibers by activated PSCs, as well as the presence of cross-linking in these fibers, affects the mechanical properties of the pancreatic microenvironment, making it dense and rigid (Cortes et al., 2019a, b). In light of the recent findings, activated PSCs might be able to sense fiber topology, adhesiveness of their surroundings, and viscoelasticity of the stroma (Papalazarou et al., 2018). Stress, strain, and forces that stretch the fibrillary proteins are also mechanosensed by myofibroblast-like cells (Chen et al., 2017). Importantly, the increased matrix stiffness may further promote activation of PSCs, supporting a positive feedback loop that perpetuates formation of the dense fibrotic tumor stroma (Bachem et al., 2005). Therefore, modulation of the mechanical properties of the desmoplastic pancreatic tissue, in order to decrease its density and overcome problems with drug delivery, may become one of key strategies in successful therapy of pancreatic cancer (Papalazarou et al., 2018). Only very recently has tamoxifen, a drug used successfully against breast cancer cells, been demonstrated to inhibit differentiation of quiescent PSCs into myofibroblasts via the G protein-coupled estrogen receptor (GPER)- and hypoxia-inducible factor-1 alpha (HIF-1α)-mediated mechanism and suppress matrix remodeling (Cortes et al., 2019a, b). Another plausible anti-fibrotic and mechanomodulatory strategy against pancreatic diseases could be targeting YAP and TAZ signal transduction of the Hippo pathway: YAP/TAZ, via their interaction with the cell cytoskeleton, promote cell “stemness,” tissue regeneration, and remodeling of the stroma; whereas YAP can modulate fibroinflammatory responses (Martinez et al., 2019).
What is more, activated PSCs express hyaluronan synthase 2 (HAS2) as well as hyaluronidase 1 (HYAL1) and have been identified as an important source of stromal hyaluronic acid (HA) (Junliang et al., 2019). In turn, HA has been attributed to high interstitial fluid pressure and vascular collapse in PDAC desmoplastic reaction (Provenzano et al., 2012). Enzymatic degradation of HA has been shown to reduce the interstitial pressure, restore the microvasculature and enhance the efficacy of chemotherapy (Kultti et al., 2012; Provenzano et al., 2012; Jacobetz et al., 2013). Of note is that HA binds to CD44, commonly expressed by many types of cancer cells, and this interaction promotes tumor-driving signaling and transport activities (Slomiany et al., 2009; Toole, 2009). Since CD44 has also been found on the surface of PDAC cells (Zhao et al., 2016), the full spectrum of HA roles in pancreatic cancer might be even more complex; but this notion requires further investigation.
Since activation of PSCs can often become part of the pathophysiological process, a number of attempts have been made to either block the phenotype transition in these cells or force already activated PSCs back to quiescence. Treatment with retinol and retinoic acid (both ATRA and 9-RA) was used to inhibit cell proliferation, expression of activation markers and the MAPK signaling pathway in these cells; and retinol even blocked ethanol-induced activation of PSCs (McCarroll et al., 2006). Further, ligands of PPARγ, a nuclear receptor regulating lipid storage and glucose metabolism, inhibited PSC proliferation and decreased expression of α-SMA and MCP-1, suggesting a potential role of PPARγ in the development of pancreatic fibrosis and inflammation (Masamune et al., 2002). More recently, it was shown that vitamin D receptor (VDR) is present in the stroma of human pancreatic tumors, and its ligand, calcipotriol, reduces markers of inflammation and fibrosis in mouse models of pancreatitis and pancreatic cancer (Sherman et al., 2014). A different group demonstrated that while vitamin D2, vitamin D3 and calcipotriol inhibit activation of PSCs in vitro, they fail to reverse the phenotype transition after the cells have already been activated (Wallbaum et al., 2018).
For the last two decades, there has been a steady (linear) increase in the number of papers on PSCs published every year (Figure 2). This new field continues to expand, and increasingly more efforts are directed toward uncovering the signaling pathways that control PSC physiology. Since PSC activation underlies the pathogenesis of pancreatic disorders, the signals that induce phenotype transition in these cells are of particular interest. Targeted manipulation of these signals might prevent or even revert PSC activation and become a useful tool in the therapy of pancreatic diseases.
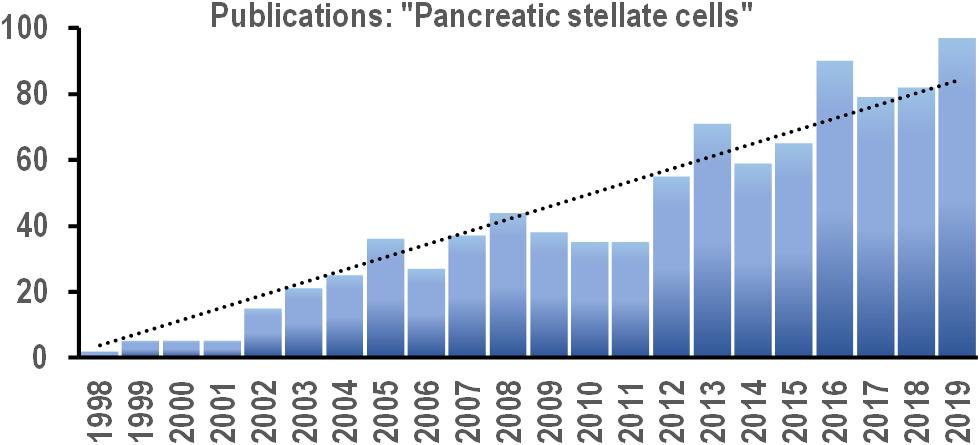
Figure 2. Bar chart shows the number of new scientific papers published each year between 2008 and 2019 with the phrase “pancreatic stellate cells” in the text (based on online search via PubMed.gov). Trend line (dotted): y = 3.8179x; R2 = 0.914.
AK, MS, MJ, and PF contributed to writing the manuscript. PF edited the final version of the manuscript.
The authors were supported by the HOMING/2017-4/31 (PF) and HOMING/2017-3/23 (MJ) project grants, both carried out within the HOMING program of the Foundation for Polish Science (Fundacja na rzecz Nauki Polskiej, FNP), co-financed by the European Union under the European Regional Development Fund. AK and MS are M.Sc. students supported by the HOMING/2017-4/31 project grant.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Apte, M. V., Haber, P. S., Applegate, T. L., Norton, I. D., Mccaughan, G. W., Korsten, M. A., et al. (1998). Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 43, 128–133. doi: 10.1136/gut.43.1.128
Apte, M. V., Haber, P. S., Darby, S. J., Rodgers, S. C., Mccaughan, G. W., Korsten, M. A., et al. (1999). Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut 44, 534–541. doi: 10.1136/gut.44.4.534
Apte, M. V., Phillips, P. A., Fahmy, R. G., Darby, S. J., Rodgers, S. C., Mccaughan, G. W., et al. (2000). Does alcohol directly stimulate pancreatic fibrogenesis? Studies with rat pancreatic stellate cells. Gastroenterology 118, 780–794. doi: 10.1016/s0016-5085(00)70148-x
Bachem, M. G., Schneider, E., Gross, H., Weidenbach, H., Schmid, R. M., Menke, A., et al. (1998). Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 115, 421–432. doi: 10.1016/s0016-5085(98)70209-4
Bachem, M. G., Schunemann, M., Ramadani, M., Siech, M., Beger, H., Buck, A., et al. (2005). Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 128, 907–921. doi: 10.1053/j.gastro.2004.12.036
Blauer, M., Laaninen, M., Sand, J., and Laukkarinen, J. (2019). Wnt/beta-catenin signalling plays diverse functions during the process of fibrotic remodelling in the exocrine pancreas. Pancreatology 19, 252–257. doi: 10.1016/j.pan.2019.02.003
Bootman, M. D., Chehab, T., Bultynck, G., Parys, J. B., and Rietdorf, K. (2018). The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium. 70, 32–46. doi: 10.1016/j.ceca.2017.08.005
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Bressy, C., Lac, S., Nigri, J., Leca, J., Roques, J., Lavaut, M. N., et al. (2018). LIF drives neural remodeling in pancreatic cancer and offers a new candidate biomarker. Cancer Res. 78, 909–921. doi: 10.1158/0008-5472.CAN-15-2790
Cannon, A., Thompson, C., Hall, B. R., Jain, M., Kumar, S., and Batra, S. K. (2018). Desmoplasia in pancreatic ductal adenocarcinoma: insight into pathological function and therapeutic potential. Genes Cancer 9, 78–86. doi: 10.18632/genesandcancer.171
Carapuca, E. F., Gemenetzidis, E., Feig, C., Bapiro, T. E., Williams, M. D., Wilson, A. S., et al. (2016). Anti-stromal treatment together with chemotherapy targets multiple signalling pathways in pancreatic adenocarcinoma. J. Pathol. 239, 286–296. doi: 10.1002/path.4727
Casini, A., Galli, A., Pignalosa, P., Frulloni, L., Grappone, C., Milani, S., et al. (2000). Collagen type I synthesized by pancreatic periacinar stellate cells (PSC) co-localizes with lipid peroxidation-derived aldehydes in chronic alcoholic pancreatitis. J. Pathol. 192, 81–89. doi: 10.1002/1096-9896(2000)9999:9999<::aid-path675>3.0.co;2-n
Chen, Y., Ju, L., Rushdi, M., Ge, C., and Zhu, C. (2017). Receptor-mediated cell mechanosensing. Mol. Biol. Cell 28, 3134–3155. doi: 10.1091/mbc.E17-04-0228
Cortes, E., Lachowski, D., Robinson, B., Sarper, M., Teppo, J. S., Thorpe, S. D., et al. (2019a). Tamoxifen mechanically reprograms the tumor microenvironment via HIF-1A and reduces cancer cell survival. EMBO Rep. 2:e46557. doi: 10.15252/embr.201846557
Cortes, E., Sarper, M., Robinson, B., Lachowski, D., Chronopoulos, A., Thorpe, S. D., et al. (2019b). GPER is a mechanoregulator of pancreatic stellate cells and the tumor microenvironment. EMBO Rep. 20:e46556. doi: 10.15252/embr.201846556
Cui, L., Li, C., Gao, G., Zhuo, Y., Yang, L., Cui, N., et al. (2019). FTY720 inhibits the activation of pancreatic stellate cells by promoting apoptosis and suppressing autophagy via the AMPK/mTOR pathway. Life Sci. 217, 243–250. doi: 10.1016/j.lfs.2018.12.019
Dangi-Garimella, S., Krantz, S. B., Barron, M. R., Shields, M. A., Heiferman, M. J., Grippo, P. J., et al. (2011). Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res. 71, 1019–1028. doi: 10.1158/0008-5472.CAN-10-1855
Diakopoulos, K. N., Lesina, M., Wormann, S., Song, L., Aichler, M., Schild, L., et al. (2015). Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology 148, 626–638e617. doi: 10.1053/j.gastro.2014.12.003
Endo, S., Nakata, K., Ohuchida, K., Takesue, S., Nakayama, H., Abe, T., et al. (2017). Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology 152, 1492.e24–1506.e24. doi: 10.1053/j.gastro.2017.01.010
Erkan, M., Michalski, C. W., Rieder, S., Reiser-Erkan, C., Abiatari, I., Kolb, A., et al. (2008). The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 6, 1155–1161. doi: 10.1016/j.cgh.2008.05.006
Fang, H., Placencio, V. R., and Declerck, Y. A. (2012). Protumorigenic activity of plasminogen activator inhibitor-1 through an antiapoptotic function. J. Natl. Cancer Inst. 104, 1470–1484. doi: 10.1093/jnci/djs377
Fels, B., Nielsen, N., and Schwab, A. (2016). Role of TRPC1 channels in pressure-mediated activation of murine pancreatic stellate cells. Eur. Biophys. J. 45, 657–670. doi: 10.1007/s00249-016-1176-4
Ferdek, P. E., and Jakubowska, M. A. (2017). Biology of pancreatic stellate cells - more than pancreatic cancer. Pflugers Arch. 469, 1039–1050. doi: 10.1007/s00424-017-1968-0
Ferdek, P. E., Jakubowska, M. A., Gerasimenko, J. V., Gerasimenko, O. V., and Petersen, O. H. (2016). Bile acids induce necrosis in pancreatic stellate cells dependent on calcium entry and sodium-driven bile uptake. J. Physiol. 594, 6147–6164. doi: 10.1113/JP272774
Friedman, S. L. (2000). Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol Chem. 275, 2247–2250. doi: 10.1074/jbc.275.4.2247
Furth, N., Aylon, Y., and Oren, M. (2018). p53 shades of Hippo. Cell Death Differ. 25, 81–92. doi: 10.1038/cdd.2017.163
Gao, R., and Brigstock, D. R. (2005). Connective tissue growth factor (CCN2) in rat pancreatic stellate cell function: integrin alpha5beta1 as a novel CCN2 receptor. Gastroenterology 129, 1019–1030. doi: 10.1053/j.gastro.2005.06.067
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Gryshchenko, O., Gerasimenko, J. V., Gerasimenko, O. V., and Petersen, O. H. (2016a). Ca2+ signals mediated by bradykinin type 2 receptors in normal pancreatic stellate cells can be inhibited by specific Ca2+ channel blockade. J. Physiol. 594, 281–293. doi: 10.1113/jp271468
Gryshchenko, O., Gerasimenko, J. V., Gerasimenko, O. V., and Petersen, O. H. (2016b). Calcium signalling in pancreatic stellate cells: mechanisms and potential roles. Cell Calcium. 59, 140–144. doi: 10.1016/j.ceca.2016.02.003
Haber, P. S., Keogh, G. W., Apte, M. V., Moran, C. S., Stewart, N. L., Crawford, D. H., et al. (1999). Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am. J. Pathol. 155, 1087–1095. doi: 10.1016/s0002-9440(10)65211-x
Hansen, C. G., Moroishi, T., and Guan, K. L. (2015). YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 25, 499–513. doi: 10.1016/j.tcb.2015.05.002
Heller, R. S., Klein, T., Ling, Z., Heimberg, H., Katoh, M., Madsen, O. D., et al. (2003). Expression of Wnt, Frizzled, sFRP, and DKK genes in adult human pancreas. Gene Expr. 11, 141–147. doi: 10.3727/000000003108749035
Hu, Y., Wan, R., Yu, G., Shen, J., Ni, J., Yin, G., et al. (2014). Imbalance of Wnt/Dkk negative feedback promotes persistent activation of pancreatic stellate cells in chronic pancreatitis. PLoS One 9:e95145. doi: 10.1371/journal.pone.0095145
Innes, J. T., and Carey, L. C. (1994). Normal pancreatic dimensions in the adult human. Am. J. Surg. 167, 261–263. doi: 10.1016/0002-9610(94)90088-4
Jacobetz, M. A., Chan, D. S., Neesse, A., Bapiro, T. E., Cook, N., Frese, K. K., et al. (2013). Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62, 112–120. doi: 10.1136/gutjnl-2012-302529
Jakubowska, M. A., Ferdek, P. E., Gerasimenko, O. V., Gerasimenko, J. V., and Petersen, O. H. (2016). Nitric oxide signals are interlinked with calcium signals in normal pancreatic stellate cells upon oxidative stress and inflammation. Open Biol. 6:160149. doi: 10.1098/rsob.160149
Jaster, R., Sparmann, G., Emmrich, J., and Liebe, S. (2002). Extracellular signal regulated kinases are key mediators of mitogenic signals in rat pancreatic stellate cells. Gut 51, 579–584. doi: 10.1136/gut.51.4.579
Junliang, L., Lili, W., Xiaolong, L., Xuguang, L., Huanwen, W., and Zhiyong, L. (2019). High-molecular-weight hyaluronan produced by activated pancreatic stellate cells promotes pancreatic cancer cell migration via paracrine signaling. Biochem. Biophys. Res. Commun. 515, 493–498. doi: 10.1016/j.bbrc.2019.05.167
Komar, H. M., Serpa, G., Kerscher, C., Schwoegl, E., Mace, T. A., Jin, M., et al. (2017). Inhibition of Jak/STAT signaling reduces the activation of pancreatic stellate cells in vitro and limits caerulein-induced chronic pancreatitis in vivo. Sci. Rep. 7:1787. doi: 10.1038/s41598-017-01973-0
Kultti, A., Li, X., Jiang, P., Thompson, C. B., Frost, G. I., and Shepard, H. M. (2012). Therapeutic targeting of hyaluronan in the tumor stroma. Cancers 4, 873–903. doi: 10.3390/cancers4030873
Kuninty, P. R., Bansal, R., De Geus, S. W. L., Mardhian, D. F., Schnittert, J., Van Baarlen, J., et al. (2019). ITGA5 inhibition in pancreatic stellate cells attenuates desmoplasia and potentiates efficacy of chemotherapy in pancreatic cancer. Sci. Adv. 5:eaax2770. doi: 10.1126/sciadv.aax2770
Li, X., Wang, Z., Ma, Q., Xu, Q., Liu, H., Duan, W., et al. (2014). Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clin. Cancer Res. 20, 4326–4338. doi: 10.1158/1078-0432.CCR-13-3426
Liu, J., Gao, M., Nipper, M., Deng, J., Sharkey, F. E., Johnson, R. L., et al. (2019). Activation of the intrinsic fibroinflammatory program in adult pancreatic acinar cells triggered by Hippo signaling disruption. PLoS Biol. 17:e3000418. doi: 10.1371/journal.pbio.3000418
Liu, S. L., Cao, S. G., Li, Y., Sun, B., Chen, D., Wang, D. S., et al. (2019). Pancreatic stellate cells facilitate pancreatic cancer cell viability and invasion. Oncol. Lett. 17, 2057–2062.
Martinez, B., Yang, Y., Harker, D. M. R., Farrar, C., Mukundan, H., Nath, P., et al. (2019). YAP/TAZ related biomechano signal transduction and cancer metastasis. Front. Cell Dev. Biol. 7:199. doi: 10.3389/fcell.2019.00199
Masamune, A., Kikuta, K., Satoh, M., Sakai, Y., Satoh, A., and Shimosegawa, T. (2002). Ligands of peroxisome proliferator-activated receptor-gamma block activation of pancreatic stellate cells. J. Biol. Chem. 277, 141–147. doi: 10.1074/jbc.m107582200
Masamune, A., Kikuta, K., Watanabe, T., Satoh, K., Hirota, M., and Shimosegawa, T. (2008). Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver. Physiol. 295, G709–G717. doi: 10.1152/ajpgi.90356.2008
Masamune, A., and Shimosegawa, T. (2009). Signal transduction in pancreatic stellate cells. J. Gastroenterol. 44, 249–260. doi: 10.1007/s00535-009-0013-2
Masamune, A., Watanabe, T., Kikuta, K., and Shimosegawa, T. (2009). Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin. Gastroenterol. Hepatol. 7, S48–S54. doi: 10.1016/j.cgh.2009.07.038
McCarroll, J. A., Phillips, P. A., Kumar, R. K., Park, S., Pirola, R. C., Wilson, J. S., et al. (2004). Pancreatic stellate cell migration: role of the phosphatidylinositol 3-kinase(PI3-kinase) pathway. Biochem. Pharmacol. 67, 1215–1225. doi: 10.1016/j.bcp.2003.11.013
McCarroll, J. A., Phillips, P. A., Park, S., Doherty, E., Pirola, R. C., Wilson, J. S., et al. (2003). Pancreatic stellate cell activation by ethanol and acetaldehyde: is it mediated by the mitogen-activated protein kinase signaling pathway? Pancreas 27, 150–160. doi: 10.1097/00006676-200308000-00008
McCarroll, J. A., Phillips, P. A., Santucci, N., Pirola, R. C., Wilson, J. S., and Apte, M. V. (2006). Vitamin A inhibits pancreatic stellate cell activation: implications for treatment of pancreatic fibrosis. Gut 55, 79–89. doi: 10.1136/gut.2005.064543
Meng, Z., Moroishi, T., and Guan, K. L. (2016). Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17. doi: 10.1101/gad.274027.115
Mews, P., Phillips, P., Fahmy, R., Korsten, M., Pirola, R., Wilson, J., et al. (2002). Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut 50, 535–541. doi: 10.1136/gut.50.4.535
Mizushima, N. (2010). The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 22, 132–139. doi: 10.1016/j.ceb.2009.12.004
Nielsen, N., Kondratska, K., Ruck, T., Hild, B., Kovalenko, I., Schimmelpfennig, S., et al. (2017). TRPC6 channels modulate the response of pancreatic stellate cells to hypoxia. Pflugers Arch. 469, 1567–1577. doi: 10.1007/s00424-017-2057-0
Nomiyama, Y., Tashiro, M., Yamaguchi, T., Watanabe, S., Taguchi, M., Asaumi, H., et al. (2007). High glucose activates rat pancreatic stellate cells through protein kinase C and p38 mitogen-activated protein kinase pathway. Pancreas 34, 364–372. doi: 10.1097/mpa.0b013e31802f0531
Ohlund, D., Handly-Santana, A., Biffi, G., Elyada, E., Almeida, A. S., Ponz-Sarvise, M., et al. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596. doi: 10.1084/jem.20162024
Ohnishi, H., Miyata, T., Yasuda, H., Satoh, Y., Hanatsuka, K., Kita, H., et al. (2004). Distinct roles of Smad2-, Smad3-, and ERK-dependent pathways in transforming growth factor-beta1 regulation of pancreatic stellate cellular functions. J. Biol. Chem. 279, 8873–8878. doi: 10.1074/jbc.m309698200
Pallagi, P., Hegyi, P., and Rakonczay, Z. Jr. (2015). The physiology and pathophysiology of pancreatic ductal secretion: the background for clinicians. Pancreas 44, 1211–1233. doi: 10.1097/MPA.0000000000000421
Papalazarou, V., Salmeron-Sanchez, M., and Machesky, L. M. (2018). Tissue engineering the cancer microenvironment-challenges and opportunities. Biophys. Rev. 10, 1695–1711. doi: 10.1007/s12551-018-0466-8
Pardo, A., Gibson, K., Cisneros, J., Richards, T. J., Yang, Y., Becerril, C., et al. (2005). Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2:e251. doi: 10.1371/journal.pmed.0020251
Patel, M. B., Pothula, S. P., Xu, Z., Lee, A. K., Goldstein, D., Pirola, R. C., et al. (2014). The role of the hepatocyte growth factor/c-MET pathway in pancreatic stellate cell-endothelial cell interactions: antiangiogenic implications in pancreatic cancer. Carcinogenesis 35, 1891–1900. doi: 10.1093/carcin/bgu122
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939. doi: 10.1016/j.cell.2005.07.002
Petersen, O. H., and Tepikin, A. V. (2008). Polarized calcium signaling in exocrine gland cells. Annu. Rev. Physiol. 70, 273–299. doi: 10.1146/annurev.physiol.70.113006.100618
Phillips, P. A., Mccarroll, J. A., Park, S., Wu, M. J., Pirola, R., Korsten, M., et al. (2003). Rat pancreatic stellate cells secrete matrix metalloproteinases: implications for extracellular matrix turnover. Gut 52, 275–282. doi: 10.1136/gut.52.2.275
Pothula, S. P., Xu, Z., Goldstein, D., Merrett, N., Pirola, R. C., Wilson, J. S., et al. (2017). Targeting the HGF/c-MET pathway: stromal remodelling in pancreatic cancer. Oncotarget 8, 76722–76739. doi: 10.18632/oncotarget.20822
Provenzano, P. P., Cuevas, C., Chang, A. E., Goel, V. K., Von Hoff, D. D., and Hingorani, S. R. (2012). Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429. doi: 10.1016/j.ccr.2012.01.007
Schnittert, J., Bansal, R., Mardhian, D. F., Van Baarlen, J., Ostman, A., and Prakash, J. (2019). Integrin alpha11 in pancreatic stellate cells regulates tumor stroma interaction in pancreatic cancer. FASEB J. 33, 6609–6621. doi: 10.1096/fj.201802336R
Schnittert, J., Bansal, R., Storm, G., and Prakash, J. (2018). Integrins in wound healing, fibrosis and tumor stroma: High potential targets for therapeutics and drug delivery. Adv. Drug Deliv. Rev. 129, 37–53. doi: 10.1016/j.addr.2018.01.020
Sherman, M. H., Yu, R. T., Engle, D. D., Ding, N., Atkins, A. R., Tiriac, H., et al. (2014). Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159, 80–93. doi: 10.1016/j.cell.2014.08.007
Shi, Y., Gao, W., Lytle, N. K., Huang, P., Yuan, X., Dann, A. M., et al. (2019). Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 569, 131–135. doi: 10.1038/s41586-019-1130-6
Shibutani, S. T., Saitoh, T., Nowag, H., Munz, C., and Yoshimori, T. (2015). Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 16, 1014–1024. doi: 10.1038/ni.3273
Shields, M. A., Dangi-Garimella, S., Redig, A. J., and Munshi, H. G. (2012). Biochemical role of the collagen-rich tumour microenvironment in pancreatic cancer progression. Biochem. J. 441, 541–552. doi: 10.1042/BJ20111240
Slomiany, M. G., Grass, G. D., Robertson, A. D., Yang, X. Y., Maria, B. L., Beeson, C., et al. (2009). Hyaluronan, CD44, and emmprin regulate lactate efflux and membrane localization of monocarboxylate transporters in human breast carcinoma cells. Cancer Res. 69, 1293–1301. doi: 10.1158/0008-5472.CAN-08-2491
Sousa, C. M., Biancur, D. E., Wang, X., Halbrook, C. J., Sherman, M. H., Zhang, L., et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. doi: 10.1038/nature19084
Thangada, S., Shapiro, L. H., Silva, C., Yamase, H., Hla, T., and Ferrer, F. A. (2014). Treatment with the immunomodulator FTY720 (fingolimod) significantly reduces renal inflammation in murine unilateral ureteral obstruction. J. Urol. 191, 1508–1516. doi: 10.1016/j.juro.2013.10.072
Toole, B. P. (2009). Hyaluronan-CD44 interactions in cancer: paradoxes and possibilities. Clin. Cancer Res. 15, 7462–7468. doi: 10.1158/1078-0432.ccr-09-0479
Wallbaum, P., Rohde, S., Ehlers, L., Lange, F., Hohn, A., Bergner, C., et al. (2018). Antifibrogenic effects of vitamin D derivatives on mouse pancreatic stellate cells. World J. Gastroenterol. 24, 170–178. doi: 10.3748/wjg.v24.i2.170
Wang, H. C., Lin, Y. L., Hsu, C. C., Chao, Y. J., Hou, Y. C., Chiu, T. J., et al. (2019). Pancreatic stellate cells activated by mutant KRAS-mediated PAI-1 upregulation foster pancreatic cancer progression via IL-8. Theranostics 9, 7168–7183. doi: 10.7150/thno.36830
Whatcott, C. J., Diep, C. H., Jiang, P., Watanabe, A., Lobello, J., Sima, C., et al. (2015). Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin. Cancer Res. 21, 3561–3568. doi: 10.1158/1078-0432.CCR-14-1051
Won, J. H., Zhang, Y., Ji, B., Logsdon, C. D., and Yule, D. I. (2011). Phenotypic changes in mouse pancreatic stellate cell Ca2+ signaling events following activation in culture and in a disease model of pancreatitis. Mol. Biol. Cell 22, 421–436. doi: 10.1091/mbc.E10-10-0807
Xiao, Y., Zhang, H., Ma, Q., Huang, R., Lu, J., Liang, X., et al. (2019). YAP1-mediated pancreatic stellate cell activation inhibits pancreatic cancer cell proliferation. Cancer Lett. 462, 51–60. doi: 10.1016/j.canlet.2019.07.015
Yoshida, S., Yokota, T., Ujiki, M., Ding, X. Z., Pelham, C., Adrian, T. E., et al. (2004). Pancreatic cancer stimulates pancreatic stellate cell proliferation and TIMP-1 production through the MAP kinase pathway. Biochem. Biophys. Res. Commun. 323, 1241–1245. doi: 10.1016/j.bbrc.2004.08.229
Yuan, Y., Jiang, J. Y., Wang, J. M., Sun, J., Li, C., Liu, B. Q., et al. (2019). BAG3-positive pancreatic stellate cells promote migration and invasion of pancreatic ductal adenocarcinoma. J. Cell. Mol. Med. 23, 5006–5016. doi: 10.1111/jcmm.14352
Zhang, L. P., Ma, F., Abshire, S. M., and Westlund, K. N. (2013). Prolonged high fat/alcohol exposure increases TRPV4 and its functional responses in pancreatic stellate cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R702–R711. doi: 10.1152/ajpregu.00296.2012
Keywords: calcium, Hippo, pancreatic cancer, pancreatic stellate cells, pancreas, PDAC, signaling, Wnt
Citation: Kusiak AA, Szopa MD, Jakubowska MA and Ferdek PE (2020) Signaling in the Physiology and Pathophysiology of Pancreatic Stellate Cells – a Brief Review of Recent Advances. Front. Physiol. 11:78. doi: 10.3389/fphys.2020.00078
Received: 06 December 2019; Accepted: 23 January 2020;
Published: 14 February 2020.
Edited by:
Peter Hegyi, University of Szeged, HungaryReviewed by:
Minoti Apte, University of New South Wales, AustraliaCopyright © 2020 Kusiak, Szopa, Jakubowska and Ferdek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pawel E. Ferdek, cC5lLmZlcmRla0BnbWFpbC5jb20=; cGF3ZWwuZmVyZGVrQHVqLmVkdS5wbA==
†ORCID: Monika A. Jakubowska, orcid.org/0000-0002-4899-2606; Pawel E. Ferdek, orcid.org/0000-0001-5582-6588
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.