 Mariapaola Nitti
Mariapaola Nitti Anna Lisa Furfaro
Anna Lisa Furfaro Giovanni E. Mann
Giovanni E. Mann
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 29 January 2020
Sec. Redox Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00023
This article is part of the Research TopicAdvances in Metabolic Mechanisms of Aging and Its Related DiseasesView all 11 articles
Among antioxidants in the human body, bilirubin has been recognized over the past 20 years to afford protection against different chronic conditions, including inflammation and cardiovascular disease. Moderate increases in plasma concentration and cellular bilirubin generation from metabolism of heme via heme oxygenase (HMOX) in virtually all tissues can modulate endothelial and vascular function and exert antioxidant and anti-inflammatory roles. This review aims to provide an up-to-date and critical overview of the molecular mechanisms by which bilirubin derived from plasma or from HMOX1 activation in vascular cells affects endothelial function. Understanding the molecular actions of bilirubin may critically improve the management not only of key cardiovascular diseases, but also provide insights into a broad spectrum of pathologies driven by endothelial dysfunction. In this context, therapeutic interventions aimed at mildly increasing serum bilirubin as well as bilirubin generated endogenously by endothelial HMOX1 should be considered.
Bilirubin is widely known as an end-product of the degradation of heme which occurs in the spleen primarily as part of red blood cell removal. Senescent or damaged red blood cells are degraded and the heme groups derived from hemoglobin are converted into biliverdin by the activity of heme oxygenases (HMOX1 and HMOX2) (Maines, 1988), and then converted into bilirubin via the activity of BLVRA. This form of bilirubin, known as UCB, is bound to albumin to enable its plasma transport to the liver where it is conjugated with glucuronic acid and excreted as main component of bile (Kapitulnik, 2004).
The elevation of bilirubin concentration in plasma is well known as a marker of hemolytic conditions, liver damage or bile-duct impairment. When the concentration of UCB exceeds the binding capability of albumin, the fraction of free bilirubin (unbound UCB) increases and can cause toxicity in the nervous system in particular, a serious condition known as bilirubin encephalopathy which has been the focus of excellent reviews (Ostrow et al., 2003; Shapiro, 2003; Kapitulnik, 2004). It is important to note that neuronal cells are particularly sensitive to UCB toxicity compared to other cell types (Silva et al., 2002). Indeed, neuronal toxicity is observed in vitro even at relatively low concentrations, e.g., 0.5 μM UCB is toxic to primary cells and acts to enhance glutamate-induced toxicity (Grojean et al., 2000). Moreover, mitochondrial damage is critically involved in neuronal damage induced by UCB especially in developing neurons (Rodrigues et al., 2002).
Nonetheless, different beneficial aspects linked to mild elevations of plasma bilirubin (up to 12 mg/dl) have been recognized. The anti-inflammatory effect of jaundice was already observed by Hench (1938) in patients affected by rheumatoid arthritis and by Engerman and Meyer (1959) in hyperbilirubinemic rats. More recent evidence comes from individuals with GS, where bilirubin affords cardiovascular protection (Vítek et al., 2002; Novotný and Vítek, 2003; Maruhashi et al., 2012; Gazzin et al., 2016). Furthermore, the degradation pathway of heme occurs in virtually all tissues, since heme-containing proteins are ubiquitously distributed and undergo physiological turnover. Notably, also the generation of bilirubin in peripheral tissues has been proposed to be protective (Sedlak et al., 2009; Takeda et al., 2015; Nam et al., 2018), and bilirubin derived from vascular cells could modulate tissue microenvironment playing both antioxidant and anti-inflammatory roles (Siow et al., 1999; He et al., 2015). We pointed out this aspect in the different contexts analyzed in the present review. Importantly, studies of cytoprotective pathways in endothelial cells is receiving considerable attention in view of potential therapeutic targets to maintain vascular health or to delay or treat vasculopathies (Kim Y.-M. et al., 2011; Calay and Mason, 2014; Mason, 2016).
Heme oxygenase (HMOX) is the first, rate-limiting enzyme in heme degradation pathway, with two major isoforms of HMOX identified. HMOX1 is inducible, expressed only under oxidative stress or when heme concentration increases; HMOX2 is constitutively present mainly in testis and neuronal cells (Maines et al., 1986) but has also been detected in vascular cells (Chen et al., 2014). HMOX catalyzes the opening of the prothoporphyrinic ring of heme, generating biliverdin, free iron (Fe2+) and CO (Maines, 1988). The activity of BLVRA converts biliverdin into UCB. Heme degradation occurs in all cell types in order to complete the turnover of heme-containing proteins. Furthermore, HMOX1 induction crucially drives cell adaptive responses to stressors and its metabolic products exert potent biological activities (Siow et al., 1999; Otterbein et al., 2016; Kishimoto et al., 2019). CO has anti-apoptotic and anti-inflammatory activities (Ryter et al., 2002), both intracellularly and in the microenvironment (Morita and Kourembanas, 1995) of blood vessels by increasing cGMP concentration and activating the MAPK pathway (Siow et al., 1999; Ryter et al., 2002). The generation of free iron is potentially highly toxic but, under physiological conditions, a parallel induction of the heavy chain of ferritin, and the activation of membrane Fe-ATPase transporters occurs (Dulak et al., 2002; Loboda et al., 2008). This adaptation is critical in decreasing intracellular Fe2+ content and prevents the generation of ROS via the Fenton reaction, and thereby maintains endothelial function (Balla et al., 2007). Finally, bilirubin is a potent antioxidant, as highlighted by the pioneering studies of Stocker et al. (1987). It has been well demonstrated that bilirubin efficiently scavenges ROS, peroxynitrite and peroxyradicals (Neuzil and Stocker, 1993). Moreover, as it is lipophilic, bilirubin is able to prevent lipid peroxidation (Stocker et al., 1987). Indeed, in HEK293 cells, the ability of UCB to prevent lipid peroxidation has been well documented (Sedlak et al., 2009), e.g., in hepatoblastoma cells and plasma samples exposed to oxidative stressors and LPS (Zelenka et al., 2012). Notably, UCB was shown to prevent LDL oxidation, 20 times more efficiently than vitamin E (Wu et al., 1994).
In addition to HMOX1, modulation of other steps involved in bilirubin production, from the synthesis of heme rings to the reduction of biliverdin catalyzed by BLVRA, plays a role in cellular protection (Figure 1). Indeed, recent evidence from different cell lines suggests that a continuous de novo synthesis of heme occurs with the specific purpose of bilirubin generation in order to provide cytoprotection (Takeda et al., 2015). Indeed, by inhibiting the synthesis of ALA the synthesis of bilirubin is reduced, leading to increased cell damage in response to oxidative stress and demonstrating that endogenous bilirubin is highly efficient in preventing cell damage.
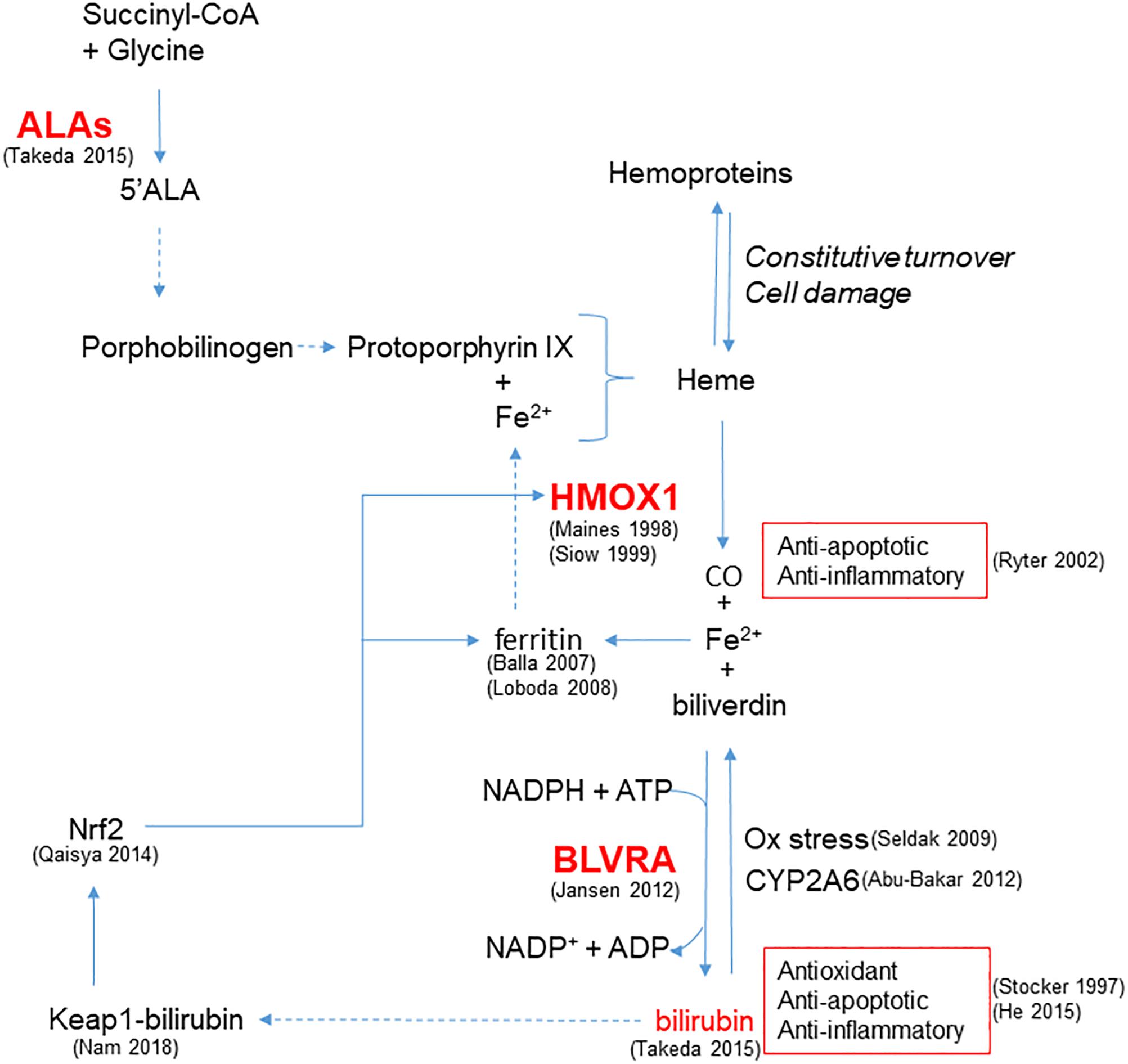
Figure 1. Enzymatic reactions modulated to increase bilirubin generation and cytoprotection. HMOX1 catalyzes the degradation of heme groups to CO, Fe2+ and biliverdin, the latter subsequently converted to bilirubin by BLVRA. By reaction with oxidant species, bilirubin is oxidized back to biliverdin, amplifying the antioxidant effect. Bilirubin is also the substrate of CYP2A6 responsible for its oxidation to biliverdin. Bilirubin and CO exert anti-apoptotic and anti-inflammatory activity. Fe2+ is quenched by the heavy chain of ferritin, and further released to form heme. In addition to iron availability, the synthesis of heme groups depends on the activity of ALAs that catalyzes the reaction between succinyl-CoA and glycine to form 5’ALA; this is then converted to porphobilinogen and protoporphyrin IX that forms heme. Heme groups can also derived from the constitutive turnover of hemoproteins that can be amplified by cell damage. A positive feedback of cytoprotection can be generated by the ability of bilirubin to bind nucleophiles such as thiol reactive cysteines on Keap1, favoring Nrf2-dependent HMOX1 gene transcription. References in brackets.
Furthermore, BLVRA contributes to the antioxidant properties of the HMOX1/bilirubin system, as pointed out by Jansen and Daiber (2012). Importantly, Nam et al. (2018) have recently discussed the physiological relevance of the conversion of biliverdin to bilirubin through an energy consuming reaction, highlighting that the reaction carried out by BLVRA dramatically increases the electrophilicity of bilirubin in comparison to biliverdin. This enables its binding to thiol reactive cysteines on Keap1, favoring Nrf2-dependent gene induction and thereby generating a positive feedback of cytoprotection. Moreover, the up-regulation of Nrf2 induced by bilirubin treatment has already been demonstrated in neuronal-like cells (Qaisiya et al., 2014) and is of relevance in the context of maintaining vascular health. Notably, bilirubin can be oxidized back to biliverdin and then be regenerated by the activity of BLVRA. The biliverdin-bilirubin cycle has been postulated to be highly active under reducing oxidative stress (Baranano et al., 2002) and, by cooperating with GSH (Sedlak et al., 2009), is able to counteract a 10,000-fold excess of H2O2 (Doré et al., 1999), affording protection in endothelial cells (Jansen et al., 2010). Moreover, mitochondrial CYP2A6 is able to perform the same oxidation of UCB back to biliverdin, highlighting the biological importance of biliverdin-bilirubin redox cycle to maintain cell homeostasis (Abu-Bakar et al., 2012).
UCB is able to bind different proteins to facilitate crossing plasma or intracellular membranes. As widely reviewed (Bellarosa et al., 2009), it can bind ABC transporters (ABCB1, ABCC1,2,3, ABCG2 and the OATP). In plasma, UCB binds to albumin and to enable uptake in the liver, it binds to transport proteins initially identified as ligandins (Litwack et al., 1971) and subsequently recognized as members of GSH transferases (Habig et al., 1974). Especially in the liver, UCB is transformed to conjugated bilirubin by the activity of uridine diphospho-glucoronosyl transferase 1A1 (UGT1A1) in order to increase its water solubility. Hepatic bilirubin metabolism and transport have been widely investigated (Kamisako et al., 2000).
Interestingly, as far as the intracellular transport is concerned, it was postulated as early as 1969 (Levi et al., 1969) that UCB could bind FABPs which are involved in the intracellular transport of organic anions. In this context, the discovery of UnaG protein from freshwater eel (Kumagai et al., 2013), a protein belonging to FABPs that selectively binds bilirubin and generates a fluorescent signal, improved the detection of endogenously generated bilirubin enabling studies of its subcellular localization and transport. Endogenous UCB seems to be mainly located at ER cytosolic membranes, associated with HMOX1/BLVRA activities and has also been detected in the nucleus and mitochondria (Park et al., 2016). However, intracellular transport mechanisms of endogenous UCB and its role in subcellular compartments remain to be explored.
Among the different cell types, neuronal cells are highly sensitive to UCB toxicity especially during development, and undergo apoptosis at relatively low concentrations (0.25–5 μM) of UCB (Grojean et al., 2000; Rodrigues et al., 2002). In addition, as far as endothelial cells are concerned, different sensitivities to UCB have been postulated depending on the different tissues from which endothelial cells were derived. Moreover, immortalized endothelial cell lines, such as bEnd.3 cells used as a model of BBB, undergo oxidative stress and apoptosis when treated with 20–40 μM UCB, while no significant damage occurs in endothelial cells derived from pancreatic islets of Langerhans (Kapitulnik et al., 2012). The peculiar sensitivity of BBB-like endothelial cells has been confirmed by other authors (Palmela et al., 2012), underlining the marked sensitivity of brain endothelial cells and neurons to UCB, as previously reviewed (Ostrow et al., 2003; Brito et al., 2014).
However, regardless of the endothelial cell type, the protective, pro-survival activity of low concentrations of UCB (0.1–5 μM) has been widely documented and ascribed to its antioxidant properties (Kapitulnik et al., 2012). Thus, although the different cell types exhibit varying degrees of sensitivity toward bilirubin toxicity, they all benefit from bilirubin cytoprotection at low concentrations. This has clinical implications, since recent research has shown that BRT prevents ROS generation in ex vivo platelets, opening the possibility of screening the actions of acute BRT treatment to improve platelets storage and function (Pennell et al., 2019).
The molecular mechanisms underlying the antioxidant activity of UCB in endothelial cells have been investigated extensively in the context of various pathophysiological conditions and diseases. Cytoprotection afforded by UCB has been attributed to quenching of superoxide anions derived from NOXs (Datla et al., 2007). Indeed, HMOX1 induction by hemin prevents NOX2/4 activation in apolipoprotein E-deficient mice characterized by vascular oxidative stress, and the same result is observed following bilirubin treatment in vitro or HMOX1 overexpression via gene transfer in VSMC (Datla et al., 2007). Notably, UCB derived from HMOX1 in endothelial cells has been shown to be able to scavenge NADPH oxidase derived ROS, impairing leukocyte transmigration and inflammatory responses (Vogel and Zucker, 2016), as discussed in Section “Effects on Adhesion Molecule Expression: Implications for Obesity, Atherosclerosis and Chronic Inflammatory Diseases.”
Protection afforded by bilirubin in endothelial cells has recently been reported Ziberna et al. (2016), who demonstrated that HMOX1 induction in response to CoPPIX or cell supplementation with a very low concentration of bilirubin (≤50 nM) significantly elevates intracellular bilirubin levels, leading to increased antioxidant defenses. It is important to note that bilirubin derived from HMOX2 can also protect vascular cells. Notably, based on the co-localization of HMOX2 with NOX4 in cerebral microvascular endothelial cells, it has been hypothesized that bilirubin may scavenge ROS derived from TNFα induced NOX4 activation (Basuroy et al., 2009). Thus, there is accumulating evidence that low concentration of UCB, both plasma and endothelial cell derived, can affect endothelial redox balance and thereby attenuate cell damage in oxidative stress.
Slight increases in plasma total bilirubin concentrations (1.53 ± 0.48 mg/dl) have been reported to preserve flow-mediated vasodilation compared to subjects with low levels of plasma bilirubin (0.40 ± 0.08 mg/dl) (Erdogan et al., 2006). As recently reviewed (Mortada, 2017), a negative correlation between serum level of bilirubin levels and hypertension has been established in different studies (Mortada, 2017). The antihypertensive activity of a moderate hyperbilirubinemia is related to an increase in NO bioavailability due to the reduction of AngII-dependent superoxide generation by endothelial cells (Pflueger et al., 2005; Vera et al., 2009). Indeed, bilirubin quenches superoxide anion derived from NOX in cultured vascular endothelial cells exposed to AngII and increases the bioavailability of NO, leading to vascular relaxation and lowering of blood pressure (Pflueger et al., 2005; Fujii et al., 2010).
Furthermore, HMOX1 derived UCB protects endothelial cells against nitrosative stress, induced by hemin pre-treatment, which is able to further increase HMOX1 dependent bilirubin generation to reduce endothelial apoptosis induced by peroxynitrite (Foresti et al., 1999). Indeed, NO and peroxynitrite are able to induce HMOX1 in endothelial cells, and bilirubin may directly scavenge NO to prevent excessive accumulation (Kaur et al., 2003) and the formation of peroxynitrite-dependent protein modification in human plasma (Minetti et al., 1998). Thus, a positive loop of cytoprotection seems to exist between NO and HMOX1-derived bilirubin (Figure 2).
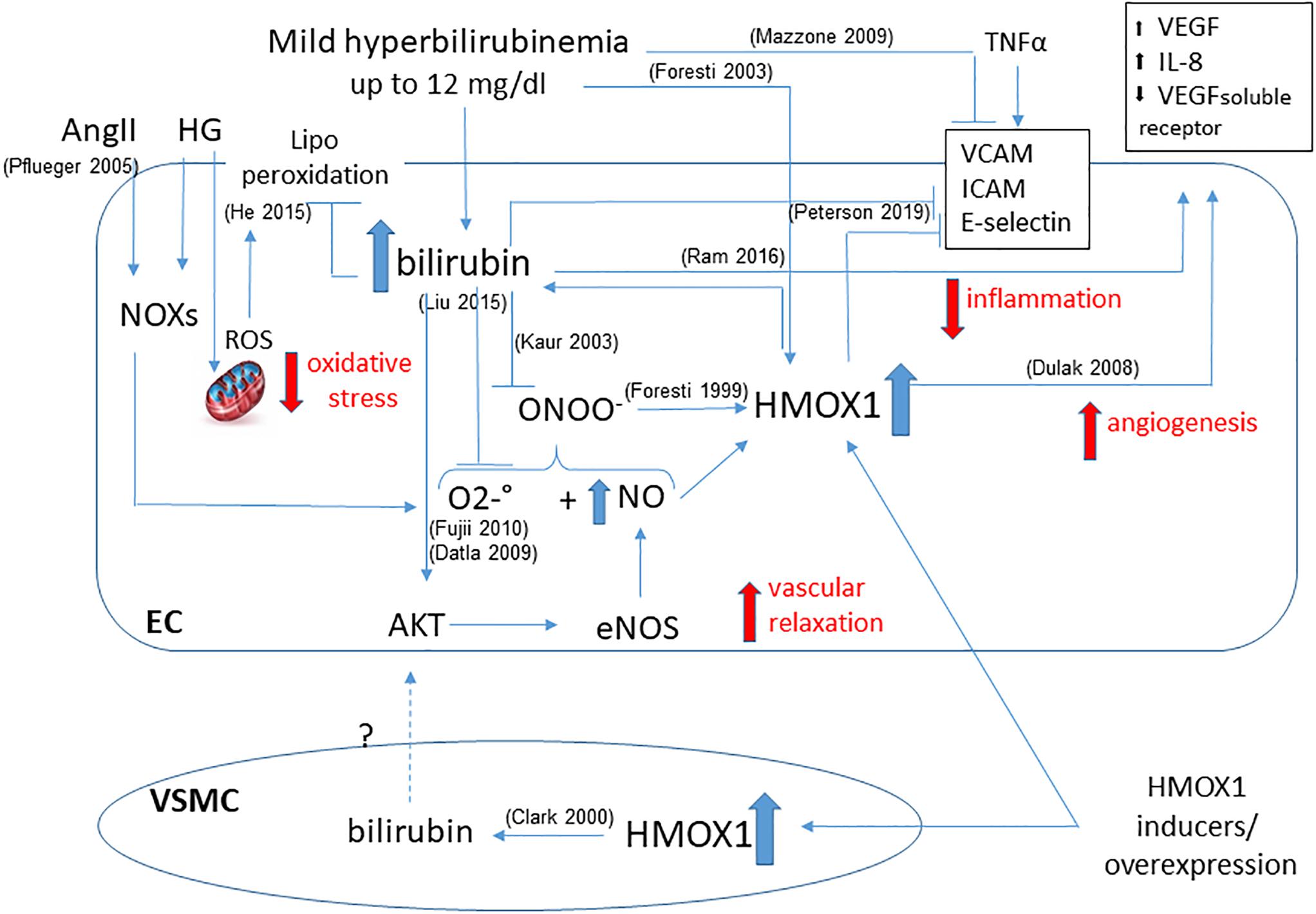
Figure 2. Functional effects of bilirubin on endothelial cells. Positive loop between plasma- and HMOX1 derived bilirubin is shown highlighting the main activities in maintaining endothelial function. The ability to reduce oxidative stress by quenching both O2–. and ONOO– prevents the dysfunction due to HG or AngII. The increased bioavailability of NO favors vascular relaxation and further activates HMOX1. The direct activity of bilirubin and HMOX1 in preventing the expression of adhesion molecules (VCAM, ICAM, E-selectin) is showed as well as the generation of angiogenetic factors (VEGF, IL-8, VEGF soluble receptor). A further contribution from VSMC-derived bilirubin has been hypothesized. References in brackets.
Moreover, the increased availability of NO due to UCB, positively affects the rate of glomerular filtration improving renal hemodynamics (Vera and Stec, 2010). However, it has also been hypothesized that other mechanisms in addition to superoxide scavenging can be involved in the ability of bilirubin to lower blood pressure and decrease hypertension, for instance the modulation of calcium and potassium channels (Stec et al., 2013). In fact, it has been shown that the induction of HMOX1, through the generation of bilirubin, favors vessel relaxation by acting on the transport of calcium and potassium, and thereby improving EDH in a rat model of spontaneous hypertension (Li et al., 2013).
The role played by UCB in preventing endothelial dysfunction in chronic sequaele of diabetes has been widely demonstrated (Vítek, 2012). Indeed, the incidence of retinopathy, coronary artery disease, and cerebrovascular diseases is low in Gilbert subjects (i.e., bilirubin level 1.2 mg/dl) with diabetes (Inoguchi et al., 2007). Moreover, the Hisayama Study revealed that increased bilirubin levels counteract diabetic retinopathy (Yasuda et al., 2011), with total bilirubin concentration negatively correlated with the severity of retinopathy (Sekioka et al., 2015).
Furthermore, in diabetic rats, up-regulation of HMOX1 increases serum bilirubin, reduces superoxide anion and endothelial sloughing induced by hyperglycemia (Quan et al., 2004). Moreover, we have reported that activation of the HMOX1/bilirubin pathway protects endothelial cells against HG-induced damage by reducing HNE production and lipid peroxidation (He et al., 2015). In addition, as mentioned before, bilirubin acts by inhibiting NOX. This action, at the level of endothelial cells and mesangial cells in diabetic kidney, has been shown to ameliorate diabetic nephropathy (Fujii et al., 2010). An elegant study by Liu et al. (2015), in different animal models of diabetes, established that HMOX1 overexpression improves vascular function, but that BLVRA activity is also required, underlining that conversion of biliverdin into bilirubin is fundamental. These authors also established that administration of exogenous bilirubin affords vascular protection by restoring AKT-dependent signal transduction (Liu et al., 2015). AKT seems to be a key target in bilirubin activity in endothelial cells, and the ability to restore the AKT-eNOS pathway is involved in not only maintaining endothelial redox balance and cell survival but also pro-angiogenic signals (see section “Effects on Angiogenesis: Implications for Wound Healing, Vascular Dysfunction in Pregnancy and Tumor Growth”). In addition, in a mouse model of obesity, bilirubin has been shown to activate AKT, reducing insulin resistance in skeletal muscle (Dong et al., 2014) and improving glucose tolerance. The role of HMOX1 and BLVRA in inflammation associated with diabetes has recently been reviewed (Rochette et al., 2018).
Bilirubin has been shown to be protective against the development of ischemic damage associated with ischemic/reperfusion injury and endothelial activation (Yang et al., 2016). A recent cross-sectional study performed on 1121 healthy Japanese demonstrated that low levels of plasma bilirubin are associated with a high prevalence of ischemic modifications in brain white matter (Higuchi et al., 2018). In this context, a recent review form Thakkar et al. (2019) rigorously analyzed clinical studies highlighting a relationship between bilirubin levels and cerebral ischemic stroke. These authors provided evidence that in the large majority of the studies, a negative relationship exists between the level of serum bilirubin and prevalence of stroke. However, they also discussed limitations of some of these studies that only considered total bilirubin levels and ischemic stroke without considering hemorrhagic stroke or traumatic brain injury. Interestingly, recent evidence in a rat model of MCAO established that intraperitoneal administration of biliverdin significantly reduces the infarct size by modulating microRNA-mRNA network (Zou et al., 2019). It is important to note that at the level of the nervous system, up-regulation of HMOX1 can be both protective and damaging as reviewed recently (Nitti et al., 2018), but the role played by HMOX1 derived bilirubin in neurons and non-neuronal cells in neuroprotection has been highlighted (Hung et al., 2010).
In different models of cardiac ischemia/reperfusion damage, bilirubin administration protects against tissue damage. Indeed, ex vivo heart perfusion with BRT improves post-ischemic outcomes by reducing oxidative damage (Bakrania et al., 2016). Moreover, intraperitoneal administration of bilirubin has been shown to prevent cardiolipin oxidation and to reduce the infarct size in a rat model of coronary ischemia/reperfusion injury (Ben-Amotz et al., 2014). More recently, preconditioning performed using nanoparticles of pegylated bilirubin was highly effective in preventing hepatic ischemic damage (Kim et al., 2017). Furthermore, the critical importance of the endogenous HMOX1/bilirubin pathway has been demonstrated following supplementation cell culture media not only with UCB but also with hemin. Both reduce ROS generation induced by re-oxygenation of cardiomyocytes subjected to hypoxia, preventing cell injury (Foresti et al., 2001). Furthermore, hemin efficiently prevents oxidative damage in VSMCs by increasing bilirubin generation (Clark et al., 2000). Even though the majority studies performed in vivo or ex vivo evaluate whole tissue without considering the specific role played by endothelial cells, it is conceivable that bilirubin supplementation can directly protect the endothelium. It is important to note that UCB induces HMOX1 up-regulation in aortic endothelial cells, generating a positive loop that increases endothelial resistance to oxidative stress (Foresti et al., 2003). The specific anti-inflammatory activity exerted by bilirubin on endothelial cells plays a role in preventing ischemia/reperfusion damage by reducing leukocyte infiltration, as discussed in section “Effects on Adhesion Molecule Expression: Implications for Obesity, Atherosclerosis and Chronic Inflammatory Diseases.”
Moreover, protection afforded by bilirubin against rejection of kidney transplantation has been demonstrated, and its antioxidant activity in preventing ischemia/reperfusion damage as well as its anti-inflammatory action that favors organ acceptance has been reviewed, highlighting the positive impact on renal hemodynamics (Sundararaghavan et al., 2018).
The first evidence for the anti-inflammatory properties of bilirubin stems from Philip Hench who in 1938 described the remission of rheumatoid arthritis in the 94% of patients who developed jaundice (Hench, 1938). Even though Hench tried to correlate this phenomenon with an increased level of cortisol (Hench, 1949), this has never been demonstrated, and the anti-inflammatory effect of jaundice has been then proved to be due to UCB itself (Jangi et al., 2013). In this context, a negative correlation between bilirubin plasma concentration and clinical manifestations of other immune-mediated diseases such as MS (Peng et al., 2011) and SLE (Vítek et al., 2010) has been demonstrated. Moreover, an elevation of UCB between 2 and 12 mg/dl has been shown to reduce experimental autoimmune encephalomyelitis in an animal model of MS (Liu et al., 2008). However, it is important to note that UCB concentrations higher than 15–20 mg/dl always induce cytotoxicity.
The ability of UCB to modulate immunoglobulin production, native or acquired immunity as well as complement activity has been reviewed by others (Basiglio et al., 2009; Jangi et al., 2013), highlighting how UCB can impact neutrophil and macrophage phagocytosis, as well as antigen presenting functions to lymphocytes and the generation of pro-inflammatory cytokines. In this way, UCB exerts beneficial effects in different chronic inflammatory and autoimmune diseases, as discussed in section “Bilirubin and Chronic Inflammatory Diseases.” However, we here focus on the role played by endothelial cells, which are closely involved in the onset and development of inflammation.
Endothelial dysfunction and inflammation characterize the development and progression of obesity, and mild hyperbilirubinemia contributes to a reduction of obesity especially during aging. These findings were particularly significant when abdominal obesity, triglyceridemia and hip and waist circumference were considered (Seyed Khoei et al., 2018). The same authors have shown that in Gilbert individuals expression level of p-AMPK, PPAR-α and γ, and PGC1-α in PBMCs were significantly higher in comparison to age- and gender-matched control subjects (Mölzer et al., 2016), whilst others have shown that UCB limits lipid deposition in adipose cells by binding PPAR-α nuclear receptors (Stec et al., 2016). The effect of UCB toward PPARα is discussed subsequently with a focus on atherosclerotic lesions (see section “Bilirubin and Atherosclerosis”).
Furthermore, a positive effect of CoPPIX-induced HMOX1 activation on reduction of body weight has been reported by Abraham et al. (2016). The same group, more recently focused on long-term endothelial HMOX1 activation in mice fed a HFD demonstrating the gene therapy to increase expression of HMOX1, reduced ICAM and VCAM expression, decreased serum markers of inflammation, such as IL-1 and TNFα, reduced the size of adipocytes and down-regulated PPARγ. Furthermore, these effects were prevented by using a specific inhibitor of HMOX1 activity, indirectly suggesting that the ability to restore a proper crosstalk between the vasculature and adipocytes depends on the metabolic products of endothelial HMOX1 activity (Peterson et al., 2019). Together these findings demonstrate that endothelial activation of HMOX1 could play an important role in the treatment/prevention of obesity, and further highlight the importance of AMPK-dependent signaling in mediating the anti-obesogenic effects of bilirubin.
Endothelial activation and recruitment of inflammatory cells are two pivotal steps in the development of atherosclerotic lesions (Moore et al., 2013). The negative correlation between plasma concentrations of bilirubin and onset and progression of CVD is clearly demonstrated by numerous studies and is evident in individuals with GS (Hopkins et al., 1996; Vítek et al., 2002; Novotný and Vítek, 2003; Maruhashi et al., 2012; Vítek, 2017).
UCB has been shown to impair leukocyte migration by affecting endothelial adhesion molecules. The activation of endothelial cells induced by pro-inflammatory cytokines such as TNFα leads to the expression of adhesion molecules (Young et al., 2002). In vitro studies demonstrated that UCB prevents TNFα-induced leukocyte adhesion to endothelial cells by reducing the expression of E-selectin, VCAM and ICAM (Mazzone et al., 2009a) by impairing NF-kB nuclear translocation (Mazzone et al., 2009b). However, others maintain that bilirubin does not modulate expression of adhesion molecules but instead impairs adhesion molecule-dependent intracellular signals. Indeed, the signaling cascade activated by the binding of leukocyte integrins to VCAM and ICAM is known to generate ROS from the activity of NOX and XO, favoring leukocyte transmigration following activation of MMP2 (Wang and Doerschuk, 2000, 2002). Vogel et al. (2017) have reported that the intraperitoneal administration of bilirubin in LdL–/– mice prevents atherosclerotic plaque formation by inhibiting leukocyte infiltration in vessel wall. These authors also confirmed in vitro that both exogenous bilirubin and HMOX1 derived bilirubin are highly efficient in reducing VCAM and ICAM dependent signals by quenching NOX4- and XO-derived ROS.
It has clearly been shown that UCB mimics the hypolipidemic activity of fenofibrate (Hinds and Stec, 2018). In addition, its ability to act as Selective PPAR Modulator (SPPARM) toward PPARα has been pointed out, confirming that, by enhancing lipid metabolism, bilirubin is able to reduce cholesterol deposition, decreasing plaque formation (Hinds and Stec, 2018). However, it has been also reported that UCB decreases the export of cholesterol from macrophages through the degradation of the ABCA1 (Wang et al., 2017). These data show that, in the vessel wall, the activity of bilirubin on lipid metabolism is complex and merits further investigation. However, serum UCB does exert a strong antioxidant activity at the level of atherosclerotic plaque by reducing the content of lipoperoxides in the lesions (Lapenna et al., 2018).
As recently reviewed, bilirubin prevent platelets aggregation due its ability to interfere with the surface expression of adhesion molecules and its antioxidant activity, thereby supporting a role played in the prevention of hypercoagulability and thrombosis (Kundur et al., 2015). Even though at high concentration (50–200 μM) UCB activates p38/MAPK favoring platelet apoptosis (NaveenKumar et al., 2015), the protective role of bilirubin against platelet aggregation has been confirmed in Gilbert individuals (Kundur et al., 2017), providing evidence for the inhibition of collagen-induced activation (Kundur et al., 2014).
As mentioned before, the beneficial role of UCB in autoimmune and chronic inflammatory diseases is widely accepted. Indeed, there is clinical evidence that higher total serum bilirubin levels reduce the risk of rheumatoid arthritis (Fischman et al., 2010), Gilbert’s subjects are less likely to develop Crohn’s disease (de Vries et al., 2012), and patients with MS have low levels of total, conjugated and UCB (Peng et al., 2011, 2012), as highlighted in a previous extensive review (Jangi et al., 2013).
Focusing on the role played by endothelial cells in the context of chronic inflammation, it has been shown that bilirubin impairs the expression and the activity of adhesion molecules as documented in animal models of airways inflammation and inflammatory colitis. Indeed, intraperitoneal administration of bilirubin blocks the influx of leukocytes into the lungs of mice with OVA-induced asthma (Keshavan et al., 2005), and bilirubin is able to counteract iNOS expression and activity in colonic tissue by preventing leukocytes infiltration (Zucker et al., 2015). In both these studies, the molecular mechanism involves the ability of bilirubin to disrupt VCAM1-dependent signaling by quenching intracellular ROS.
Pro-angiogenetic activity of bilirubin has been demonstrated and related both to vascular activation of HMOX1 and to an increased plasma concentration of UCB. Favorable or detrimental actions can be associated with increased angiogenesis, depending on the different pathological conditions involved. Indeed, pro-angiogenetic activity can be desirable in repairing tissue damage or in preventing placental dysfunction (Chen and Zheng, 2014) but becomes deleterious when is linked to retinal degeneration in diabetes (Wang and Lo, 2018) or in the context of tumor growth (Viallard and Larrivée, 2017).
At a molecular level, pro-angiogenetic activity of bilirubin is achieved by the modulation of different pathways, some of which are not directly related to its antioxidant activity. In vivo and in vitro studies have shown that exogenous bilirubin promotes angiogenesis in response to ischemia via direct modulation of the PI3K/AKT pathway, which favors eNOS activation (Ikeda et al., 2015). The activation of AKT pathway in response to bilirubin exposure has been also been confirmed in other studies (Liu et al., 2015).
Furthermore, vascular HMOX1 induction favors the production of angiogenic factors, such as VEGF and IL-8, and decreases antiangiogenic factors such as VEGF soluble receptor (Dulak et al., 2008; Kim Y.-M. et al., 2011), with all HMOX1 metabolic products seeming to mediate these proangiogenic activities (Dulak et al., 2008; Kim Y.-M. et al., 2011). Moreover, the angiogenic potential of endothelial cells exposed to intermittent HG is restored by CoPPIX-induced HMOX1 activation to reduce ER stress (Maamoun et al., 2017). In this context, the protective role of HMOX1 in maintaining endothelial function by preventing both oxidative and ER stress has recently been reviewed (Maamoun et al., 2019). VEGF, in addition, is able to activate endothelial HMOX1 favoring angiogenesis, leading to positive feedback. However, the role played by HMOX1 in the vasculature is more complex, since it has been shown in VSMC that HMOX1-derived CO is able to increase VEGF production, whilst HMOX1-derived iron can inhibit VEGF, thus affecting endothelial responses in different ways (Dulak et al., 2002) which are not completely understood.
Furthermore, constitutive HMOX2 has been proposed to play an important role in angiogenesis. In HMOX2 null mice, an increase in corneal angiogenesis has been observed, which is paralleled by a down-regulation of HMOX1 (Seta et al., 2006). However, in another study HMOX2 deletion favors angiogenesis but, in this case, a parallel up-regulation of HMOX1 was observed (Bellner et al., 2009). Thus, the interplay between HMOX1 and HMOX2 seems to be more complex and merits further investigation.
The process of wound healing is strictly dependent on tightly regulated angiogenesis. The role played by both HMOX1 (Lundvig et al., 2012) and HMOX2 (Halilovic et al., 2011) in wound healing has been well documented and related to the generation of CO and bilirubin. In particular, HMOX1 deficient mice exhibit less capability for wound healing compared to wild type mice due to a reduced ability to recruit endothelial progenitor cells (EPC) for capillary formation (Deshane et al., 2007). Interestingly, gene therapy performed using an hypoxia-regulated vector encoding for HMOX1 protects against oxidative damage and favors tissue regeneration by improving angiogenesis both in vitro and in vivo, avoiding the potential toxic effects due to iron overload (Jazwa et al., 2013). In this context, it has recently been reported that hyperbilirubinemic infants have an increased number of circulating endothelial progenitor cells (cEPC) which also exhibit an increased ability to proliferate, migrate and repair wounds. Indeed, tissues treated with conditioned medium derived from these cells up-regulate the expression of VEGF and IL-10 and reduce TNFα (Jabarpour et al., 2018). The involvement of bilirubin has also been documented in a model of cutaneous wounds, where i.p. administration of bilirubin (30 mg/kg) increases contraction and extracellular matrix deposition, with a down regulation of pro-inflammatory markers such as TNFα (Ahanger et al., 2016). Further evidence for a vasoactive role of bilirubin was reported by Ram and colleagues, who demonstrated that the topical treatment with 0.3% bilirubin ointment reduces oxidative stress in the granulation tissue of wounds in diabetic rats to favor wound healing (Ram et al., 2014, 2016). Other studies have underlined that, in chronic inflammation, HMOX1 could also be involved in the inhibition of leukocyte infiltration thereby favoring angiogenesis to complete tissue repair (Bussolati and Mason, 2006).
The involvement of HMOX1 in placental development and pathology has been reviewed by Levytska et al. (2013) and also highlighted by others (George et al., 2011, 2013). HMOX1 deficiency is associated with the pathogenesis of pregnancy complications and PE (Zhao et al., 2008). In this context, studies with human umbilical vein endothelial cells and trophoblasts highlight the importance of bilirubin in angiogenesis, particularly in the regulation of spiral artery remodeling (Ha et al., 2015). Moreover, in choriocarcinoma cells, it has been shown that the up-regulation of HMOX1 or exposure to bilirubin prevents advanced glycation end-product induced generation of sFTL-1, the soluble decoy receptor for VEGF, known to play a critical role in the pathogenesis of PE (Jeong et al., 2014). However, it is worth noting that placenta-derived soluble factors involved in maternal hypertension act by increasing endothelin-1 production from endothelial cells. In this context, it has been shown that HMOX1 expression prevents endothelin-1 production in glomerular endothelial cells (Bakrania et al., 2018) and pharmacological activation of HMOX1 or treatment with bilirubin has been proposed to attenuate PE (Ramma and Ahmed, 2014).
In general, bilirubin is associated with anti-proliferative and anti-neoplastic activity. In fact, specific polymorphisms in UGT1A1 gene and the consequent hyperbilirubinemia are correlated positively with an increased overall survival in cancer patients (Vitek et al., 2019b), particularly considering colorectal (Jirásková et al., 2012), lung and breast cancers (Wagner et al., 2015). Moreover, reduced bilirubin levels are associated with increased cancer risk (Ching et al., 2002; Zucker et al., 2004). There are a few exceptions, for instance, a reported increase in the incidence of breast cancer in Gilbert individuals, which is related to specific polymorphisms of UGT1A1 promoter, as reviewed by Wagner et al. (2015).
At molecular level, an antigenotoxic activity of bilirubin has been hypothesized and a negative correlation between bilirubin concentration and DNA damage in epithelial cells has been documented (Wallner et al., 2012) even though, this has not been confirmed from an analysis of 8oxodGuo in PBMCs from GS individuals or Gunn rats (Wallner et al., 2013).
Thus, bilirubin not unlike other antioxidants, can counteract cancer onset and development by preventing DNA damage and the oxidative stress which plays a central role in carcinogenesis (Marengo et al., 2016).
Moreover, the anti-cancer proprieties of bilirubin have been documented, with bilirubin administration i.p. in mice increasing plasma bilirubin up to 40μM and drastically reducing colon cancer growth via activation of ERK1/2 (Ollinger et al., 2007). These findings have been confirmed by in vitro experiments showing that bilirubin concentrations >25μM exert pro-apoptotic activity whereas concentrations <25μM have no effect on cell viability or proliferation.
Nonetheless, it is important to note that once a tumor mass has developed, cancer cells can take advantage of antioxidants, especially when they derived from the activation of intrinsic adaptive pathways, and can be used for their growth and survival, favoring resistance to therapies and disease progression (Gorrini et al., 2013; Gill et al., 2016). This has been well documented for other antioxidants such as GSH (Traverso et al., 2013) and may apply to bilirubin. Indeed, role of HMOX1 in cancer progression has been highlighted and, even though some tissue specificity need to be considered, in many cancer types its expression correlates with tumor growth, aggressiveness, metastatic and angiogenetic potential, resistance to therapy, tumor escape, and poor prognosis (Furfaro et al., 2016; Nitti et al., 2017; Piras et al., 2017). It is important to note that HMOX1 and its metabolic byproducts can be involved in the generation of a permissive microenvironment, which is fundamental for cancer progression. In this context, a role for HMOX1 derived bilirubin has been implicated by Di Biase et al. (2016) and our group (Furfaro et al., 2019) in the progression of melanoma. Considering the role HMOX1 in reducing immune-surveillance, in favoring angiogenesis and invasiveness, it seems likely that some of these properties can be ascribed to the in loco generation of bilirubin.
Moreover, the involvement of HMOX1 in physiological angiogenesis in pregnancy and pathological angiogenesis in cancer has been proposed, since both share a necessity for a permissive microenvironment in which cell invasion, cytoprotection, angiogenesis and immune-escape are favored (Zhao et al., 2015). However, the specific role of bilirubin in this context has yet to be elucidated, providing a new field for investigation.
Different therapeutic approaches aimed at elevating plasma bilirubin concentration as well as modulating cellular HMOX1 activity have been proposed for the treatment of many disorders, as widely reviewed by others (McCarty, 2007; Vitek et al., 2019b). Here, we focused on that ones in which the modulation of endothelial cell function is pivotal.
Elevating bilirubin plasma concentration needs to be considered carefully due to bilirubin’s well-known toxic effects. As discussed, bilirubin supplementation significantly decreases infarct size in animal models of ischemia damage (Ben-Amotz et al., 2014) as does BRT (Bakrania et al., 2016). Notably, in a mouse model of islet transplantation, bilirubin supplementation i.p. to a donor mouse as well as in vitro preconditioning of cells significantly suppresses the immune response and favors tolerance improving the outcomes (Adin et al., 2017). Furthermore, the use of nanoparticle of PEGylated bilirubin has been reported to protect liver tissue from ischemia-reperfusion damage (Kim et al., 2017) and to reduce lung inflammation (Kim et al., 2017). Notably, the use of bilirubin-coated stents reduces inflammation and endothelial activation preventing restenosis in porcine carotid arteries (Bae et al., 2018).
In humans, elevated plasma bilirubin levels, as side effects of drugs such as HIV PIs, have been proposed to improve endothelial function. For instance, 3-day atazanavir treatment in type II diabetes patients significantly ameliorates endothelial dysfunction (Dekker et al., 2011). Notably, three different HIV PIs, ritonavir, atazanavir and lopinavir, have been shown to be inducers of endothelial HMOX1. Moreover, HMOX1 induction is able to attenuate PI-dependent antiproliferative and inflammatory activity on endothelial cells primarily through the generation of bilirubin (Liu et al., 2016).
Importantly, therapeutic approaches leading to induce iatrogenic GS and to improve bioavailability of bilirubin might represent a valid strategy to prevent and ameliorate oxidative stress-related diseases, and McCarty (2007) already highlighted endothelial cells as crucially targeted by these therapies. More recently, the efficacy of different natural compounds which act by inhibiting UGT1A1 and elevate serum bilirubin has been confirmed, for instance silymarin, a seed extract of milk thistle (Silybum marianum), is able to protect against lipoperoxidation (Šuk et al., 2019) as are other natural compounds and herbal extracts (Vitek et al., 2019a). In addition, it is important to note that many natural compounds can increase bilirubin generation by activating HMOX1, as demonstrated that curcumin inducing HMOX1 expression and elevating bilirubin serum level, leading to protection against acute vascular inflammation both in vivo and in vitro (Xiao et al., 2018).
Furthermore, bilirubin analogs or related compounds can exert anti-proliferative effects as discussed earlier. In this context it is important to note that administration of natural tetrapyrrolic compounds structurally related to bilirubin, such as molecules extracted from Spirulina platensis, exert in vitro and in vivo actions including antiproliferative effects in cancer cells (Konícková et al., 2014). In addition, nanoparticles of both biotinylated and PEGylated bilirubin have recently been developed as drug delivery system improving the anti-angiogenetic efficacy of target tumor therapy (Lee et al., 2018; Yu et al., 2019).
The role of HMOX1 activity in protection of endothelial cells against oxidative stress has been already highlighted (Kim Y.-M. et al., 2011) as has the opportunity to target HMOX1 to improve endothelial function and prevent CVD. However, induction of HMOX1 activity can be potentially harmful, mainly due to the generation of toxic amounts of iron. Thus, even though hemin is commonly used in experimental settings both in vitro and in animals to activate HMOX1, the use of the heme analogs such as heme arginate or hematin remains potentially toxic at the vascular level (Balla et al., 2000). However, other drugs such as niacin (Wu et al., 2012), statins (Grosser et al., 2004) and fenofibrates (Krönke et al., 2007) have been shown to be vasculoprotective via activation of HMOX1. Interestingly, valsartan induces HMOX1 expression in the aortic wall and increases serum bilirubin levels via down-regulation of AT1, leading to the reduction of intimal thickening (Li et al., 2014).
Finally, to achieve a more efficient up-regulation of HMOX1 and to avoid its pathological overexpression a molecular approach has been proposed: the use a hypoxia-regulated plasmid coding for HMOX1 to enhance vascular cell resistance in vitro and improve recovery from ischemia/reperfusion injury in vivo (Jazwa et al., 2013).
As far as tumor therapy is concerned, it has been reported that chlorophyll is able to inhibit HMOX1 expression and activity, exerting antiproliferative and antioxidative effects toward pancreatic cancer and inducing a significant reduction of pancreatic tumor size (Vanková et al., 2018). However, the efficacy of HMOX1 modulation specifically toward tumor angiogenesis has yet to be investigated.
Bilirubin, far from being just a waste product highly toxic for neurons, has been recognized as a powerful antioxidant and anti-inflammatory molecule. The potential application of bilirubin for the treatment of human diseases is driven by its ability to prevent endothelial dysfunction, thus affecting not only the treatment and prevention of CVDs (Ayer et al., 2016; Bulmer et al., 2018; Maruhashi et al., 2019), but also many other pathologies in which the alterations of endothelial cells has been proved to play a role (Figure 3 and Tables 1, 2).
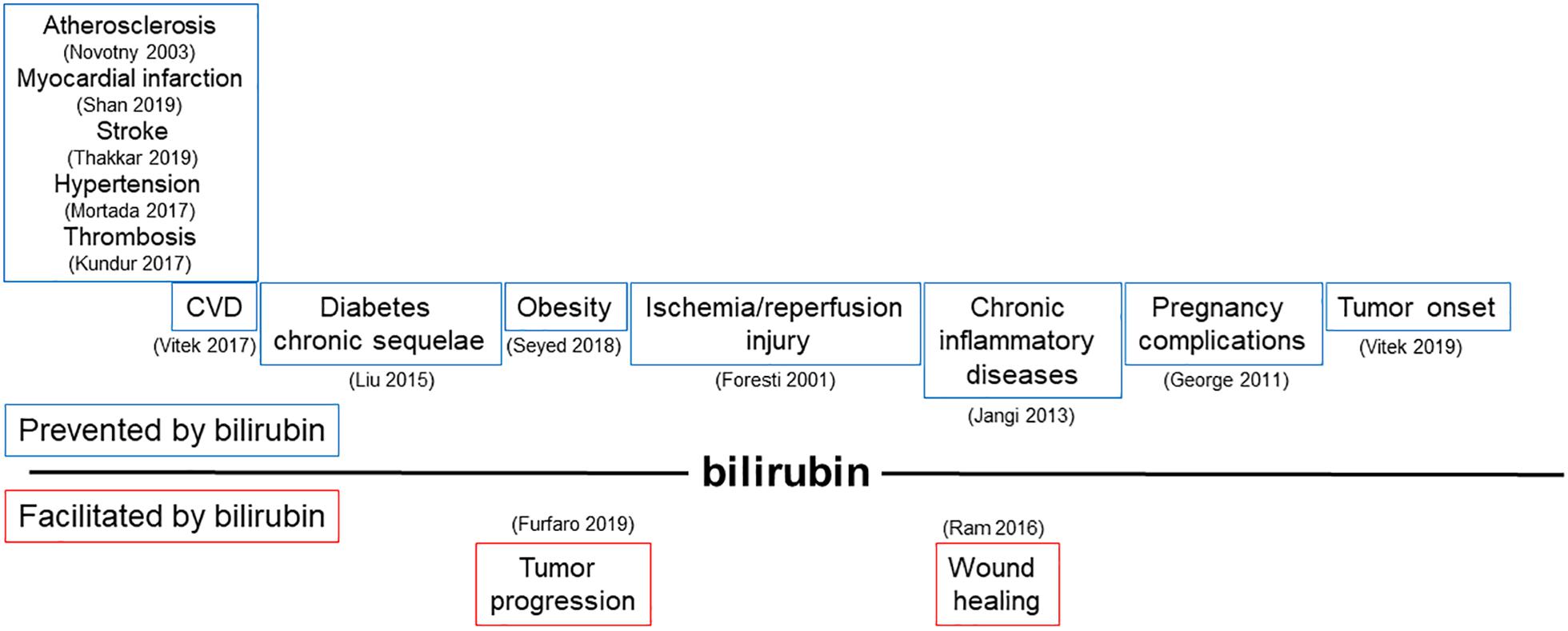
Figure 3. Modulation of bilirubin production can affect a broad range of pathologies driven by endothelial dysfunction. Schematic representation of pathophysiological processes influenced by the ability of bilirubin to modulate endothelial function. In the upper part, diseases prevented by bilirubin are shown; in the lower part, conditions favored by locally generated bilirubin are indicated. References in brackets.
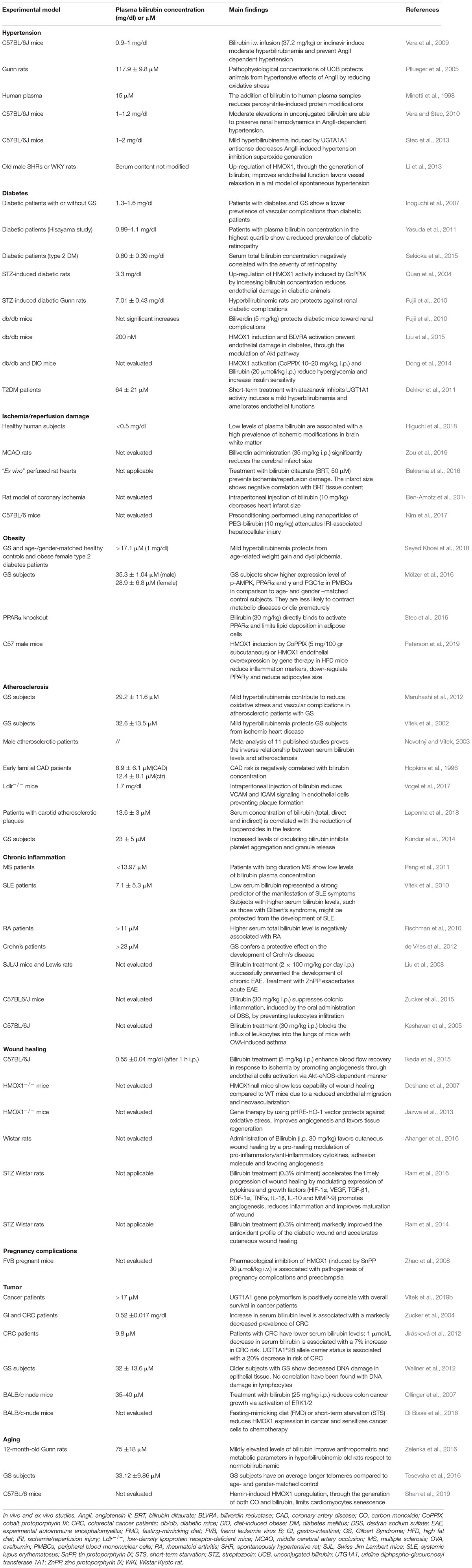
Table 1. Bilirubin affects vascular cell functions: In vivo and ex vivo studies.
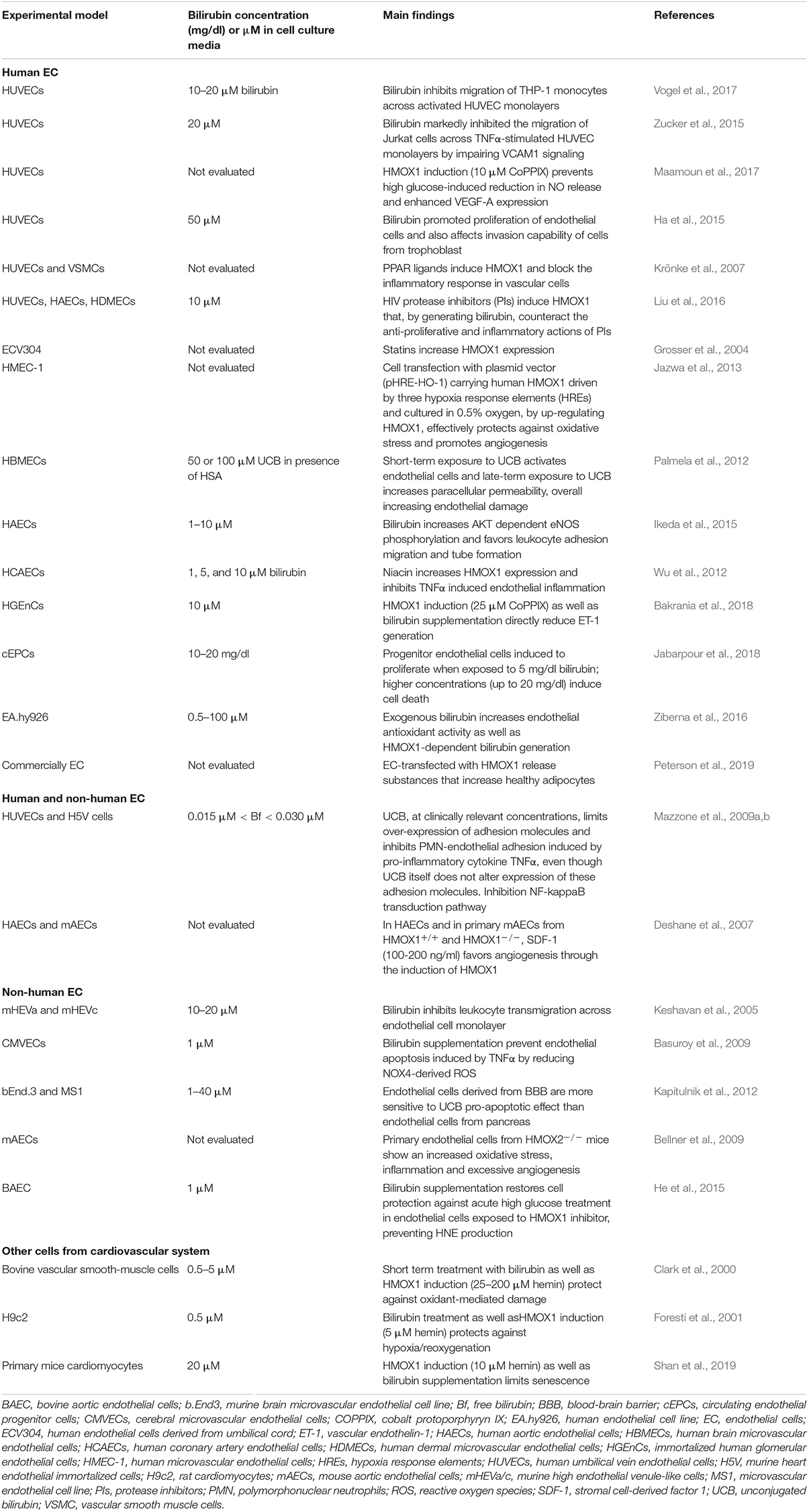
Table 2. Bilirubin affects vascular cell functions: In vitro studies.
As both increasing plasma levels of bilirubin and up-regulating HMOX1 could lead to toxic effects, the discovery of new strategies to reduce its toxic potential to maintain therapeutic effectiveness will be key for the treatment/prevention of endothelial dysfunction. Finally, it is worth noting that senescence and aging processes, strongly associated with accumulation of oxidative damage, are reduced/slowed by bilirubin. Notably, has been recently highlighted the negative relationship among bilirubin concentration and cancer mortality, even though the relationship is missing among bilirubin and CVD mortality (Vitek et al., 2019b). It is conceivable that larger studies focused on older population are needed to explain this lack of association. Indeed, the inverse correlation between plasma UCB levels and the prevalence of obesity has be proved to be stronger in older subjects (Seyed Khoei et al., 2018).
Nonetheless, it has been proved that in Gilbert subjects and in Gunn rats both mitochondrial and cytosolic ROS production is reduced leading to a decrease of chronic inflammation markers and better anthropometric parameters (Zelenka et al., 2016). Furthermore, telomere shortening is slower in patients with mild hyperbilirubinemia, implicating a correlation with reduced oxidative stress and blood markers of inflammation (Tosevska et al., 2016). Furthermore, in senescent fibroblasts the activity of BLVRA is significantly reduced in comparison to cells derived from younger donors, suggesting that a deficit of bilirubin may be responsible for increased oxidative stress and DNA damage (Kim S. Y. et al., 2011). Moreover, hemin-induced HMOX1 up-regulation limits cardiomyocyte senescence both in vitro and in aged mice via the generation of both CO and bilirubin (Shan et al., 2019). Thus, the careful modulation of HMOX1 activity and bilirubin generation offers a promising target in the context of prevention and treatment of endothelial dysfunction but also with an aim to ensure healthy aging.
All authors conceived and wrote the manuscript, and revised the literature. MN and GM provided financial support.
This study was supported by research funding from the University of Genoa (MN), British Heart Foundation (GM), and Heart Research UK (GM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
8-oxodGuo, 8-oxo-7,8-dihydro-2’-deoxyguanosine; ALA, 5-aminolevulinic acid; ABCA1, ATP-binding cassette transporter A; AngII, angiotensin II; AT1, angiotensin type 1 receptor; BBB, blood-brain barrier; BRT, bilirubin ditaurate; BLVRA, biliverdin reductase; CO, carbon monoxide; CoPPIX, cobalt protoporphyrin IX; CVD, cardiovascular disease; DM, diabetes mellitus; EDH, endothelial dependent hyperpolarization; EPC/cEPC, endothelial progenitor cells/circulating endothelial progenitor cells; FABPs, fatty acid binding proteins; GS, Gilbert syndrome; HFD, high fat diet; HG, high glucose; HMOX1, heme oxygenase 1; HMOX2, heme oxygenase 2; HNE, hydroxynonenal; MCAO, middle cerebral artery occlusion; MMP2, metalloproteinases; MS, multiple sclerosis; NOXs, NADPH oxidases; NSCLC, non-small cell lung cancer; OATP, organic anion transport binding protein; OVA, ovalbumin; PE, preeclampsia; PI, protease inhibitor; PMBCs, peripheral blood mononuclear cells; ROS, reactive oxygen species; SLE, systemic lupus erythematosus; UCB, unconjugated bilirubin; UTG1A1, uridine diphpspho-glucuronosyl transferase 1A1; VSMC, vascular smooth muscle cells; XO, xanthine oxidase
Abraham, N. G., Junge, J. M., and Drummond, G. S. (2016). Translational significance of heme oxygenase in obesity and metabolic syndrome. Trends Pharmacol. Sci. 37, 17–36. doi: 10.1016/j.tips.2015.09.003
Abu-Bakar, A., Arthur, D. M., Wikman, A. S., Rahnasto, M., Juvonen, R. O., Vepsäläinen, J., et al. (2012). Metabolism of bilirubin by human cytochrome P450 2A6. Toxicol. Appl. Pharmacol. 261, 50–58. doi: 10.1016/j.taap.2012.03.010
Adin, C. A., VanGundy, Z. C., Papenfuss, T. L., Xu, F., Ghanem, M., Lakey, J., et al. (2017). Physiologic doses of bilirubin contribute to tolerance of islet transplants by suppressing the innate immune response. Cell Transplant. 26, 11–21. doi: 10.3727/096368916X692096
Ahanger, A. A., Leo, M. D., Gopal, A., Kant, V., Tandan, S. K., and Kumar, D. (2016). Pro-healing effects of bilirubin in open excision wound model in rats. Int. Wound J. 13, 398–402. doi: 10.1111/iwj.12319
Ayer, A., Zarjou, A., Agarwal, A., and Stocker, R. (2016). Heme oxygenases in cardiovascular health and disease. Physiol. Rev. 96, 1449–1508. doi: 10.1152/physrev.00003.2016
Bae, I.-H., Park, D. S., Lee, S.-Y., Jang, E.-J., Shim, J.-W., Lim, K.-S., et al. (2018). Bilirubin coating attenuates the inflammatory response to everolimus-coated stents. J. Biomed Mater. Res. B Appl. Biomater. 106, 1486–1495. doi: 10.1002/jbm.b.33955
Bakrania, B., Du Toit, E. F., Wagner, K.-H., Headrick, J. P., and Bulmer, A. C. (2016). Pre- or post-ischemic bilirubin ditaurate treatment reduces oxidative tissue damage and improves cardiac function. Int. J. Cardiol. 202, 27–33. doi: 10.1016/j.ijcard.2015.08.192
Bakrania, B. A., Spradley, F. T., Satchell, S. C., Stec, D. E., Rimoldi, J. M., Gadepalli, R. S. V., et al. (2018). Heme oxygenase-1 is a potent inhibitor of placental ischemia-mediated endothelin-1 production in cultured human glomerular endothelial cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 314, R427–R432. doi: 10.1152/ajpregu.00370.2017
Balla, J., Balla, G., Jeney, V., Kakuk, G., Jacob, H. S., and Vercellotti, G. M. (2000). Ferriporphyrins and endothelium: a 2-edged sword-promotion of oxidation and induction of cytoprotectants. Blood 95, 3442–3450. doi: 10.1182/blood.v95.11.3442.011k51_3442_3450
Balla, J., Vercellotti, G. M., Jeney, V., Yachie, A., Varga, Z., Jacob, H. S., et al. (2007). Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid. Redox Signal. 9, 2119–2137. doi: 10.1089/ars.2007.1787
Baranano, D. E., Rao, M., Ferris, C. D., and Snyder, S. H. (2002). Biliverdin reductase: a major physiologic cytoprotectant. Proc. Natl. Acad. Sci. U.S.A 99, 16093–16098. doi: 10.1073/pnas.252626999
Basiglio, C. L., Arriaga, S. M., Pelusa, F., Almará, A. M., Kapitulnik, J., and Mottino, A. D. (2009). Complement activation and disease: protective effects of hyperbilirubinaemia. Clin. Sci. Lond. Engl. 1979, 99–113. doi: 10.1042/CS20080540
Basuroy, S., Bhattacharya, S., Leffler, C. W., and Parfenova, H. (2009). Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-alpha in cerebral vascular endothelial cells. Am. J. Physiol Cell Physiol. 296, C422–C432. doi: 10.1152/ajpcell.00381.2008
Bellarosa, C., Bortolussi, G., and Tiribelli, C. (2009). The role of ABC transporters in protecting cells from bilirubin toxicity. Curr. Pharm. Des. 15, 2884–2892. doi: 10.2174/138161209789058246
Bellner, L., Martinelli, L., Halilovic, A., Patil, K., Puri, N., Dunn, M. W., et al. (2009). Heme oxygenase-2 deletion causes endothelial cell activation marked by oxidative stress, inflammation, and angiogenesis. J. Pharmacol. Exp. Ther. 331, 925–932. doi: 10.1124/jpet.109.158352
Ben-Amotz, R., Bonagura, J., Velayutham, M., Hamlin, R., Burns, P., and Adin, C. (2014). Intraperitoneal bilirubin administration decreases infarct area in a rat coronary ischemia/reperfusion model. Front. Physiol. 5:53. doi: 10.3389/fphys.2014.00053
Brito, M. A., Palmela, I., Cardoso, F. L., Sá-Pereira, I., and Brites, D. (2014). Blood-brain barrier and bilirubin: clinical aspects and experimental data. Arch. Med. Res. 45, 660–676. doi: 10.1016/j.arcmed.2014.11.015
Bulmer, A. C., Bakrania, B., Du Toit, E. F., Boon, A.-C., Clark, P. J., Powell, L. W., et al. (2018). Bilirubin acts as a multipotent guardian of cardiovascular integrity: more than just a radical idea. Am. J. Physiol. Heart Circ. Physiol. 315, H429–H447. doi: 10.1152/ajpheart.00417.2017
Bussolati, B., and Mason, J. C. (2006). Dual role of VEGF-induced heme-oxygenase-1 in angiogenesis. Antioxid. Redox Signal. 8, 1153–1163. doi: 10.1089/ars.2006.8.1153
Calay, D., and Mason, J. C. (2014). The multifunctional role and therapeutic potential of HO-1 in the vascular endothelium. Antioxid. Redox Signal. 20, 1789–1809. doi: 10.1089/ars.2013.5659
Chen, D.-B., and Zheng, J. (2014). Regulation of placental angiogenesis. Microcirc. N. Y. N. 21, 15–25. doi: 10.1111/micc.12093
Chen, R.-J., Yuan, H.-H., Zhang, T.-Y., Wang, Z.-Z., Hu, A.-K., Wu, L.-L., et al. (2014). Heme oxygenase-2 suppress TNF-α and IL6 expression via TLR4/MyD88-dependent signaling pathway in mouse cerebral vascular endothelial cells. Mol. Neurobiol. 50, 971–978. doi: 10.1007/s12035-014-8693-x
Ching, S., Ingram, D., Hahnel, R., Beilby, J., and Rossi, E. (2002). Serum levels of micronutrients, antioxidants and total antioxidant status predict risk of breast cancer in a case control study. J. Nutr. 132, 303–306. doi: 10.1093/jn/132.2.303
Clark, J. E., Foresti, R., Green, C. J., and Motterlini, R. (2000). Dynamics of haem oxygenase-1 expression and bilirubin production in cellular protection against oxidative stress. Biochem. J. 348(Pt 3), 615–619. doi: 10.1042/bj3480615
Datla, S. R., Dusting, G. J., Mori, T. A., Taylor, C. J., Croft, K. D., and Jiang, F. (2007). Induction of heme oxygenase-1 in vivo suppresses NADPH oxidase derived oxidative stress. Hypertens Dallas Tex 1979, 636–642. doi: 10.1161/HYPERTENSIONAHA.107.092296
de Vries, H. S., Te Morsche, R. H. M., Jenniskens, K., Peters, W. H. M., and de Jong, D. J. (2012). A functional polymorphism in UGT1A1 related to hyperbilirubinemia is associated with a decreased risk for Crohn’s disease. J. Crohns Colitis 6, 597–602. doi: 10.1016/j.crohns.2011.11.010
Dekker, D., Dorresteijn, M. J., Pijnenburg, M., Heemskerk, S., Rasing-Hoogveld, A., Burger, D. M., et al. (2011). The bilirubin-increasing drug atazanavir improves endothelial function in patients with type 2 diabetes mellitus. Arter. Thromb. Vasc. Biol. 31, 458–463. doi: 10.1161/ATVBAHA.110.211789
Deshane, J., Chen, S., Caballero, S., Grochot-Przeczek, A., Was, H., Li Calzi, S., et al. (2007). Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J. Exp. Med. 204, 605–618. doi: 10.1084/jem.20061609
Di Biase, S., Lee, C., Brandhorst, S., Manes, B., Buono, R., Cheng, C. W., et al. (2016). Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell 30, 136–146. doi: 10.1016/j.ccell.2016.06.005
Dong, H., Huang, H., Yun, X., Kim, D., Yue, Y., Wu, H., et al. (2014). Bilirubin increases insulin sensitivity in leptin-receptor deficient and diet-induced obese mice through suppression of ER stress and chronic inflammation. Endocrinology 155, 818–828. doi: 10.1210/en.2013-1667
Doré, S., Takahashi, M., Ferris, C. D., Zakhary, R., Hester, L. D., Guastella, D., et al. (1999). Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. U.S.A. 96, 2445–2450. doi: 10.1073/pnas.96.5.2445
Dulak, J., Deshane, J., Jozkowicz, A., and Agarwal, A. (2008). Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation 117, 231–241. doi: 10.1161/CIRCULATIONAHA.107.698316
Dulak, J., Józkowicz, A., Foresti, R., Kasza, A., Frick, M., Huk, I., et al. (2002). Heme oxygenase activity modulates vascular endothelial growth factor synthesis in vascular smooth muscle cells. Antioxid. Redox Signal. 4, 229–240. doi: 10.1089/152308602753666280
Engerman, R. L., and Meyer, R. K. (1959). Anti-inflammatory effect of obstructive jaundice in rats. Ann. Rheum. Dis. 18, 309–312. doi: 10.1136/ard.18.4.309
Erdogan, D., Gullu, H., Yildirim, E., Tok, D., Kirbas, I., Ciftci, O., et al. (2006). Low serum bilirubin levels are independently and inversely related to impaired flow-mediated vasodilation and increased carotid intima-media thickness in both men and women. Atherosclerosis 184, 431–437. doi: 10.1016/j.atherosclerosis.2005.05.011
Fischman, D., Valluri, A., Gorrepati, V. S., Murphy, M. E., Peters, I., and Cheriyath, P. (2010). Bilirubin as a protective factor for rheumatoid arthritis: an NHANES study of 2003 - 2006 data. J. Clin. Med. Res. 2, 256–260. doi: 10.4021/jocmr444w
Foresti, R., Goatly, H., Green, C. J., and Motterlini, R. (2001). Role of heme oxygenase-1 in hypoxia-reoxygenation: requirement of substrate heme to promote cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 281, H1976–H1984.
Foresti, R., Hoque, M., Bains, S., Green, C. J., and Motterlini, R. (2003). Haem and nitric oxide: synergism in the modulation of the endothelial haem oxygenase-1 pathway. Biochem. J. 372, 381–390. doi: 10.1042/BJ20021516
Foresti, R., Sarathchandra, P., Clark, J. E., Green, C. J., and Motterlini, R. (1999). Peroxynitrite induces haem oxygenase-1 in vascular endothelial cells: a link to apoptosis. Biochem. J. 339(Pt 3), 729–736. doi: 10.1042/bj3390729
Fujii, M., Inoguchi, T., Sasaki, S., Maeda, Y., Zheng, J., Kobayashi, K., et al. (2010). Bilirubin and biliverdin protect rodents against diabetic nephropathy by downregulating NAD(P)H oxidase. Kidney Int. 78, 905–919. doi: 10.1038/ki.2010.265
Furfaro, A. L., Piras, S., Domenicotti, C., Fenoglio, D., De Luigi, A., Salmona, M., et al. (2016). Role of Nrf2, HO-1 and GSH in neuroblastoma cell resistance to bortezomib. Plos One 11:e0152465. doi: 10.1371/journal.pone.0152465
Furfaro, A. L., Ottonello, S., Loi, G., Cossu, I., Piras, S., Spagnolo, F., et al. (2019). HO-1 downregulation favors BRAFV600 melanoma cell death induced by Vemurafenib/PLX4032 and increases NK recognition. Int. J. Cancer doi: 10.1002/ijc.32611 [Epub ahead of print].
Gazzin, S., Vitek, L., Watchko, J., Shapiro, S. M., and Tiribelli, C. (2016). A novel perspective on the biology of bilirubin in health and disease. Trends Mol. Med. 22, 758–768. doi: 10.1016/j.molmed.2016.07.004
George, E. M., Cockrell, K., Aranay, M., Csongradi, E., Stec, D. E., and Granger, J. P. (2011). Induction of heme oxygenase 1 attenuates placental ischemia-induced hypertension. Hypertens Dallas Tex 1979, 941–948. doi: 10.1161/HYPERTENSIONAHA.111.169755
George, E. M., Hosick, P. A., Stec, D. E., and Granger, J. P. (2013). Heme oxygenase inhibition increases blood pressure in pregnant rats. Am. J. Hypertens 26, 924–930. doi: 10.1093/ajh/hpt045
Gill, J. G., Piskounova, E., and Morrison, S. J. (2016). Cancer, oxidative stress, and metastasis. Cold Spring Harb. Symp. Quant. Biol. 81, 163–175. doi: 10.1101/sqb.2016.81.030791
Gorrini, C., Harris, I. S., and Mak, T. W. (2013). Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947. doi: 10.1038/nrd4002
Grojean, S., Koziel, V., Vert, P., and Daval, J. L. (2000). Bilirubin induces apoptosis via activation of NMDA receptors in developing rat brain neurons. Exp. Neurol. 166, 334–341. doi: 10.1006/exnr.2000.7518
Grosser, N., Hemmerle, A., Berndt, G., Erdmann, K., Hinkelmann, U., Schürger, S., et al. (2004). The antioxidant defense protein heme oxygenase 1 is a novel target for statins in endothelial cells. Free Radic. Biol. Med. 37, 2064–2071. doi: 10.1016/j.freeradbiomed.2004.09.009
Ha, J. G., Li, L., Lee, D., Na, S., Ha, K. S., Kim, Y.-M., et al. (2015). The effects of heme oxygenase by-products on the proliferation and invasion of HUVECs, HTR-8/SVneo cells, 3A(tPA 30-1) cells, and HESCs under varying oxygen concentrations. Reprod. Sci. Thousand. Oaks Calif. 22, 1530–1538. doi: 10.1177/1933719115589415
Habig, W. H., Pabst, M. J., Fleischner, G., Gatmaitan, Z., Arias, I. M., and Jakoby, W. B. (1974). The identity of glutathione S-Transferase B with ligandin, a major binding protein of liver. Proc. Natl. Acad. Sci. U.S.A. 71, 3879–3882. doi: 10.1073/pnas.71.10.3879
Halilovic, A., Patil, K. A., Bellner, L., Marrazzo, G., Castellano, K., Cullaro, G., et al. (2011). Knockdown of heme oxygenase-2 impairs corneal epithelial cell wound healing. J. Cell Physiol. 226, 1732–1740. doi: 10.1002/jcp.22502
He, M., Nitti, M., Piras, S., Furfaro, A. L., Traverso, N., Pronzato, M. A., et al. (2015). Heme oxygenase-1-derived bilirubin protects endothelial cells against high glucose-induced damage. Free Radic. Biol. Med. 89, 91–98. doi: 10.1016/j.freeradbiomed.2015.07.151
Hench, P. S. (1938). Effect of jaundice on rheumatoid arthritis. Br. Med. J. 2, 394–398. doi: 10.1136/bmj.2.4050.394
Hench, P. S. (1949). Potential reversibility of rheumatoid arthritis. Ann. Rheum. Dis. 8, 90–96. doi: 10.1136/ard.8.2.90
Higuchi, S., Kabeya, Y., Uchida, J., Kato, K., and Tsukada, N. (2018). Low bilirubin levels indicate a high risk of cerebral deep white matter lesions in apparently healthy subjects. Sci. Rep. 8:6473. doi: 10.1038/s41598-018-24917-8
Hinds, T. D., and Stec, D. E. (2018). Bilirubin, a cardiometabolic signaling molecule. Hypertens Dallas Tex 72, 788–795. doi: 10.1161/HYPERTENSIONAHA.118.11130
Hopkins, P. N., Wu, L. L., Hunt, S. C., James, B. C., Vincent, G. M., and Williams, R. R. (1996). Higher serum bilirubin is associated with decreased risk for early familial coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 16, 250–255. doi: 10.1161/01.atv.16.2.250
Hung, S.-Y., Liou, H.-C., and Fu, W.-M. (2010). The mechanism of heme oxygenase-1 action involved in the enhancement of neurotrophic factor expression. Neuropharmacology 58, 321–329. doi: 10.1016/j.neuropharm.2009.11.003
Ikeda, Y., Hamano, H., Satoh, A., Horinouchi, Y., Izawa-Ishizawa, Y., Kihira, Y., et al. (2015). Bilirubin exerts pro-angiogenic property through Akt-eNOS-dependent pathway. Hypertens Res. Off. J. Jpn. Soc. Hypertens 38, 733–740. doi: 10.1038/hr.2015.74
Inoguchi, T., Sasaki, S., Kobayashi, K., Takayanagi, R., and Yamada, T. (2007). Relationship between gilbert syndrome and prevalence of vascular complications in patients with diabetes. JAMA 298, 1398–1400. doi: 10.1001/jama.298.12.1398-b
Jabarpour, M., Siavashi, V., Asadian, S., Babaei, H., Jafari, S. M., and Nassiri, S. M. (2018). Hyperbilirubinemia-induced pro-angiogenic activity of infantile endothelial progenitor cells. Microvasc Res 118, 49–56. doi: 10.1016/j.mvr.2018.02.005
Jangi, S., Otterbein, L., and Robson, S. (2013). The molecular basis for the immunomodulatory activities of unconjugated bilirubin. Int. J. Biochem. Cell Biol. 45, 2843–2851. doi: 10.1016/j.biocel.2013.09.014
Jansen, T., and Daiber, A. (2012). Direct antioxidant properties of bilirubin and biliverdin. is there a role for biliverdin reductase. Front. Pharmacol. 3:30. doi: 10.3389/fphar.2012.00030
Jansen, T., Hortmann, M., Oelze, M., Opitz, B., Steven, S., Schell, R., et al. (2010). Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of bilirubin. J. Mol. Cell Cardiol. 49, 186–195. doi: 10.1016/j.yjmcc.2010.04.011
Jazwa, A., Stepniewski, J., Zamykal, M., Jagodzinska, J., Meloni, M., Emanueli, C., et al. (2013). Pre-emptive hypoxia-regulated HO-1 gene therapy improves post-ischaemic limb perfusion and tissue regeneration in mice. Cardiovasc. Res. 97, 115–124. doi: 10.1093/cvr/cvs284
Jeong, J. H., Kim, H. G., and Choi, O. H. (2014). sildenafil inhibits advanced glycation end products-induced sFlt-1 release through upregulation of heme Oxygenase-1. J. Menopausal. Med. 20, 57–68. doi: 10.6118/jmm.2014.20.2.57
Jirásková, A., Novotný, J., Novotný, L., Vodicka, P., Pardini, B., Naccarati, A., et al. (2012). Association of serum bilirubin and promoter variations in HMOX1 and UGT1A1 genes with sporadic colorectal cancer. Int. J. Cancer 131, 1549–1555. doi: 10.1002/ijc.27412
Kamisako, T., Kobayashi, Y., Takeuchi, K., Ishihara, T., Higuchi, K., Tanaka, Y., et al. (2000). Recent advances in bilirubin metabolism research: the molecular mechanism of hepatocyte bilirubin transport and its clinical relevance. J. Gastroenterol. 35, 659–664. doi: 10.1007/s005350070044
Kapitulnik, J. (2004). Bilirubin: an endogenous product of heme degradation with both cytotoxic and cytoprotective properties. Mol. Pharmacol. 66, 773–779. doi: 10.1124/mol.104.002832
Kapitulnik, J., Benaim, C., and Sasson, S. (2012). Endothelial cells derived from the blood-brain barrier and islets of langerhans differ in their response to the effects of bilirubin on oxidative stress under hyperglycemic conditions. Front. Pharmacol. 3:131. doi: 10.3389/fphar.2012.00131
Kaur, H., Hughes, M. N., Green, C. J., Naughton, P., Foresti, R., and Motterlini, R. (2003). Interaction of bilirubin and biliverdin with reactive nitrogen species. FEBS Lett. 543, 113–119. doi: 10.1016/S0014-5793(03)00420-4
Keshavan, P., Deem, T. L., Schwemberger, S. J., Babcock, G. F., Cook-Mills, J. M., and Zucker, S. D. (2005). Unconjugated bilirubin inhibits VCAM-1-mediated transendothelial leukocyte migration. J. Immunol. Baltim. Md. 1950, 3709–3718. doi: 10.4049/jimmunol.174.6.3709
Kim, J. Y., Lee, D. Y., Kang, S., Miao, W., Kim, H., Lee, Y., et al. (2017). Bilirubin nanoparticle preconditioning protects against hepatic ischemia-reperfusion injury. Biomaterials 133, 1–10. doi: 10.1016/j.biomaterials.2017.04.011
Kim, S. Y., Kang, H. T., Choi, H. R., and Park, S. C. (2011). Biliverdin reductase A in the prevention of cellular senescence against oxidative stress. Exp. Mol. Med. 43, 15–23. doi: 10.3858/emm.2011.43.1.002
Kim, Y.-M., Pae, H.-O., Park, J. E., Lee, Y. C., Woo, J. M., Kim, N.-H., et al. (2011). Heme oxygenase in the regulation of vascular biology: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 14, 137–167. doi: 10.1089/ars.2010.3153
Kishimoto, Y., Kondo, K., and Momiyama, Y. (2019). The protective role of heme oxygenase-1 in atherosclerotic diseases. Int. J. Mol. Sci. 20:3628. doi: 10.3390/ijms20153628
Konícková, R., Vaòková, K., Vaňková, J., Váňová, K., Muchová, L., Subhanová, I., et al. (2014). Anti-cancer effects of blue-green alga spirulina platensis, a natural source of bilirubin-like tetrapyrrolic compounds. Ann. Hepatol. 13, 273–283. doi: 10.1016/s1665-2681(19)30891-9
Krönke, G., Kadl, A., Ikonomu, E., Blüml, S., Fürnkranz, A., Sarembock, I. J., et al. (2007). Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler. Thromb. Vasc. Biol. 27, 1276–1282. doi: 10.1161/ATVBAHA.107.142638
Kumagai, A., Ando, R., Miyatake, H., Greimel, P., Kobayashi, T., Hirabayashi, Y., et al. (2013). A bilirubin-inducible fluorescent protein from eel muscle. Cell 153, 1602–1611. doi: 10.1016/j.cell.2013.05.038
Kundur, A. R., Bulmer, A. C., and Singh, I. (2014). Unconjugated bilirubin inhibits collagen induced platelet activation. Platelets 25, 45–50. doi: 10.3109/09537104.2013.764405
Kundur, A. R., Santhakumar, A. B., Bulmer, A. C., and Singh, I. (2017). Mildly elevated unconjugated bilirubin is associated with reduced platelet activation-related thrombogenesis and inflammation in Gilbert’s syndrome. Platelets 28, 779–785. doi: 10.1080/09537104.2017.1280146
Kundur, A. R., Singh, I., and Bulmer, A. C. (2015). Bilirubin, platelet activation and heart disease: a missing link to cardiovascular protection in Gilbert’s syndrome? Atherosclerosis 239, 73–84. doi: 10.1016/j.atherosclerosis.2014.12.042
Lapenna, D., Ciofani, G., Pierdomenico, S. D., Giamberardino, M. A., Ucchino, S., and Davì, G. (2018). Association of serum bilirubin with oxidant damage of human atherosclerotic plaques and the severity of atherosclerosis. Clin. Exp. Med. 18, 119–124. doi: 10.1007/s10238-017-0470-5
Lee, Y., Lee, S., and Jon, S. (2018). Biotinylated bilirubin nanoparticles as a tumor microenvironment-responsive drug delivery system for targeted cancer therapy. Adv. Sci. Weinh. Baden- Wurtt. Ger. 5:800017. doi: 10.1002/advs.201800017
Levi, A. J., Gatmaitan, Z., and Arias, I. M. (1969). Two hepatic cytoplasmic protein fractions, Y and Z, and their possible role in the hepatic uptake of bilirubin, sulfobromophthalein, and other anions. J. Clin. Invest. 48, 2156–2167. doi: 10.1172/JCI106182
Levytska, K., Kingdom, J., Baczyk, D., and Drewlo, S. (2013). Heme oxygenase-1 in placental development and pathology. Placenta 34, 291–298. doi: 10.1016/j.placenta.2013.01.004
Li, Y., Wang, Q., Xu, Q., Cai, S., Zhou, J., Ren, B., et al. (2014). Valsartan decreases neointimal hyperplasia in balloon-injured rat aortic arteries by upregulating HO-1 and inhibiting angiotensin II type 1 receptor. Life Sci. 110, 70–76. doi: 10.1016/j.lfs.2014.06.021
Li, Z., Wang, Y., Man, R. Y. K., and Vanhoutte, P. M. (2013). Upregulation of heme oxygenase-1 potentiates EDH-type relaxations in the mesenteric artery of the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 305, H1471–H1483. doi: 10.1152/ajpheart.00962.2012
Litwack, G., Ketterer, B., and Arias, I. M. (1971). Ligandin: a hepatic protein which binds steroids, bilirubin, carcinogens and a number of exogenous organic anions. Nature 234, 466–467. doi: 10.1038/234466a0
Liu, J., Wang, L., Tian, X. Y., Liu, L., Wong, W. T., Zhang, Y., et al. (2015). Unconjugated bilirubin mediates heme oxygenase-1-induced vascular benefits in diabetic mice. Diabetes 64, 1564–1575. doi: 10.2337/db14-1391
Liu, X.-M., Durante, Z. E., Peyton, K. J., and Durante, W. (2016). Heme oxygenase-1-derived bilirubin counteracts HIV protease inhibitor-mediated endothelial cell dysfunction. Free Radic. Biol. Med. 94, 218–229. doi: 10.1016/j.freeradbiomed.2016.03.003
Liu, Y., Li, P., Lu, J., Xiong, W., Oger, J., Tetzlaff, W., et al. (2008). Bilirubin possesses powerful immunomodulatory activity and suppresses experimental autoimmune encephalomyelitis. J. Immunol. Baltim. Md. 1950, 1887–1897. doi: 10.4049/jimmunol.181.3.1887
Loboda, A., Jazwa, A., Grochot-Przeczek, A., Rutkowski, A. J., Cisowski, J., Agarwal, A., et al. (2008). Heme oxygenase-1 and the vascular bed: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 10, 1767–1812. doi: 10.1089/ars.2008.2043
Lundvig, D. M. S., Immenschuh, S., and Wagener, F. A. (2012). Heme oxygenase, inflammation, and fibrosis: the good, the bad, and the ugly? Front. Pharmacol. 3:81. doi: 10.3389/fphar.2012.00081
Maamoun, H., Benameur, T., Pintus, G., Munusamy, S., and Agouni, A. (2019). Crosstalk between oxidative stress and endoplasmic reticulum (ER) stress in endothelial dysfunction and aberrant angiogenesis associated with diabetes: a focus on the protective roles of heme oxygenase (HO)-1. Front. Physiol. 10:70. doi: 10.3389/fphys.2019.00070
Maamoun, H., Zachariah, M., McVey, J. H., Green, F. R., and Agouni, A. (2017). Heme oxygenase (HO)-1 induction prevents endoplasmic reticulum stress-mediated endothelial cell death and impaired angiogenic capacity. Biochem. Pharmacol. 127, 46–59. doi: 10.1016/j.bcp.2016.12.009
Maines, M. D. (1988). Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. Faseb. J. 2, 2557–2568. doi: 10.1096/fasebj.2.10.3290025
Maines, M. D., Trakshel, G. M., and Kutty, R. K. (1986). Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 261, 411–419.
Marengo, B., Nitti, M., Furfaro, A. L., Colla, R., Ciucis, C. D., Marinari, U. M., et al. (2016). Redox homeostasis and cellular antioxidant systems: crucial players in cancer growth and therapy. Oxid. Med. Cell Longev 2016:6235641. doi: 10.1155/2016/6235641
Maruhashi, T., Kihara, Y., and Higashi, Y. (2019). Bilirubin and endothelial function. J. Atheroscler. Thromb. 26, 688–696. doi: 10.5551/jat.RV17035
Maruhashi, T., Soga, J., Fujimura, N., Idei, N., Mikami, S., Iwamoto, Y., et al. (2012). Hyperbilirubinemia, augmentation of endothelial function, and decrease in oxidative stress in Gilbert syndrome. Circulation 126, 598–603. doi: 10.1161/CIRCULATIONAHA.112.105775
Mason, J. C. (2016). Cytoprotective pathways in the vascular endothelium. Do they represent a viable therapeutic target? Vasc. Pharmacol. 86, 41–52. doi: 10.1016/j.vph.2016.08.002
Mazzone, G. L., Rigato, I., Ostrow, J. D., Bossi, F., Bortoluzzi, A., Sukowati, C. H., et al. (2009a). Bilirubin inhibits the TNFalpha-related induction of three endothelial adhesion molecules. Biochem. Biophys. Res. Commun. 386, 338–344. doi: 10.1016/j.bbrc.2009.06.029
Mazzone, G. L., Rigato, I., Ostrow, J. D., and Tiribelli, C. (2009b). Bilirubin effect on endothelial adhesion molecules expression is mediated by the NF-kappaB signaling pathway. Biosci. Trends. 3, 151–157.
McCarty, M. F. (2007). “Iatrogenic Gilbert syndrome” –a strategy for reducing vascular and cancer risk by increasing plasma unconjugated bilirubin. Med. Hypotheses 69, 974–994. doi: 10.1016/j.mehy.2006.12.069
Minetti, M., Mallozzi, C., Di Stasi, A. M., and Pietraforte, D. (1998). Bilirubin is an effective antioxidant of peroxynitrite-mediated protein oxidation in human blood plasma. Arch. Biochem. Biophys. 352, 165–174. doi: 10.1006/abbi.1998.0584
Mölzer, C., Wallner, M., Kern, C., Tosevska, A., Schwarz, U., Zadnikar, R., et al. (2016). Features of an altered AMPK metabolic pathway in Gilbert’s syndrome, and its role in metabolic health. Sci. Rep. 6:30051. doi: 10.1038/srep30051
Moore, K. J., Sheedy, F. J., and Fisher, E. A. (2013). Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 13, 709–721. doi: 10.1038/nri3520
Morita, T., and Kourembanas, S. (1995). Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J. Clin. Invest. 96, 2676–2682. doi: 10.1172/JCI118334
Mortada, I. (2017). Hyperbilirubinemia, hypertension, and CKD: the links. Curr. Hypertens Rep. 19:58. doi: 10.1007/s11906-017-0756-8
Nam, J., Lee, Y., Yang, Y., Jeong, S., Kim, W., Yoo, J.-W., et al. (2018). Is it worth expending energy to convert biliverdin into bilirubin? Free Radic. Biol. Med. 124, 232–240. doi: 10.1016/j.freeradbiomed.2018.06.010
NaveenKumar, S. K., Thushara, R. M., Sundaram, M. S., Hemshekhar, M., Paul, M., Thirunavukkarasu, C., et al. (2015). Unconjugated bilirubin exerts pro-apoptotic effect on platelets via p38-MAPK activation. Sci. Rep. 5:15045. doi: 10.1038/srep15045
Neuzil, J., and Stocker, R. (1993). Bilirubin attenuates radical-mediated damage to serum albumin. FEBS Lett. 331, 281–284. doi: 10.1016/0014-5793(93)80353-v
Nitti, M., Piras, S., Brondolo, L., Marinari, U. M., Pronzato, M. A., and Furfaro, A. L. (2018). Heme oxygenase 1 in the nervous system: does it favor neuronal cell survival or induce neurodegeneration? Int. J. Mol. Sci. 19:E2260. doi: 10.3390/ijms19082260
Nitti, M., Piras, S., Marinari, U. M., Moretta, L., Pronzato, M. A., and Furfaro, A. L. (2017). HO-1 induction in cancer progression: a matter of cell adaptation. Antioxid. Basel. Switz. 6, E29. doi: 10.3390/antiox6020029
Novotný, L., and Vítek, L. (2003). Inverse relationship between serum bilirubin and atherosclerosis in men: a meta-analysis of published studies. Exp. Biol. Med. Maywood. N. J. 228, 568–571. doi: 10.1177/15353702-0322805-29
Ollinger, R., Kogler, P., Troppmair, J., Hermann, M., Wurm, M., Drasche, A., et al. (2007). Bilirubin inhibits tumor cell growth via activation of ERK. Cell Cycle Georget Tex 6:3078–3085. doi: 10.4161/cc.6.24.5022
Ostrow, J. D., Pascolo, L., Shapiro, S. M., and Tiribelli, C. (2003). New concepts in bilirubin encephalopathy. Eur. J. Clin. Invest. 33, 988–997. doi: 10.1046/j.1365-2362.2003.01261.x
Otterbein, L. E., Foresti, R., and Motterlini, R. (2016). Heme oxygenase-1 and carbon monoxide in the heart: the balancing act between danger signaling and pro-survival. Circ. Res. 118, 1940–1959. doi: 10.1161/CIRCRESAHA.116.306588
Palmela, I., Sasaki, H., Cardoso, F. L., Moutinho, M., Kim, K. S., Brites, D., et al. (2012). Time-dependent dual effects of high levels of unconjugated bilirubin on the human blood-brain barrier lining. Front. Cell Neurosci. 6:22. doi: 10.3389/fncel.2012.00022
Park, J.-S., Nam, E., Lee, H.-K., Lim, M. H., and Rhee, H.-W. (2016). In cellulo mapping of subcellular localized bilirubin. ACS Chem. Biol. 11, 2177–2185. doi: 10.1021/acschembio.6b00017
Peng, F., Deng, X., Yu, Y., Chen, X., Shen, L., Zhong, X., et al. (2011). Serum bilirubin concentrations and multiple sclerosis. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 18, 1355–1359. doi: 10.1016/j.jocn.2011.02.023
Peng, F., Yang, Y., Liu, J., Jiang, Y., Zhu, C., Deng, X., et al. (2012). Low antioxidant status of serum uric acid, bilirubin and albumin in patients with neuromyelitis optica. Eur. J. Neurol. 19, 277–283. doi: 10.1111/j.1468-1331.2011.03488.x
Pennell, E. N., Wagner, K.-H., Mosawy, S., and Bulmer, A. C. (2019). Acute bilirubin ditaurate exposure attenuates ex vivo platelet reactive oxygen species production, granule exocytosis and activation. Redox. Biol. 26:101250. doi: 10.1016/j.redox.2019.101250
Peterson, S. J., Rubinstein, R., Faroqui, M., Raza, A., Boumaza, I., Zhang, Y., et al. (2019). Positive effects of heme oxygenase upregulation on adiposity and vascular dysfunction: gene targeting vs. Pharmacologic Therapy. Int. J. Mol. Sci. 20:E2514. doi: 10.3390/ijms20102514
Pflueger, A., Croatt, A. J., Peterson, T. E., Smith, L. A., d’Uscio, L. V., Katusic, Z. S., et al. (2005). The hyperbilirubinemic gunn rat is resistant to the pressor effects of angiotensin II. Am. J. Physiol. Renal. Physiol. 288, F552–F558. doi: 10.1152/ajprenal.00278.2004
Piras, S., Furfaro, A. L., Brondolo, L., Passalacqua, M., Marinari, U. M., Pronzato, M. A., et al. (2017). Differentiation impairs Bach1 dependent HO-1 activation and increases sensitivity to oxidative stress in SH-SY5Y neuroblastoma cells. Sci. Rep. 7:7568. doi: 10.1038/s41598-017-08095-7
Qaisiya, M., Coda Zabetta, C. D., Bellarosa, C., and Tiribelli, C. (2014). Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell Signal. 26, 512–520. doi: 10.1016/j.cellsig.2013.11.029
Quan, S., Kaminski, P. M., Yang, L., Morita, T., Inaba, M., Ikehara, S., et al. (2004). Heme oxygenase-1 prevents superoxide anion-associated endothelial cell sloughing in diabetic rats. Biochem. Biophys. Res. Commun. 315, 509–516. doi: 10.1016/j.bbrc.2004.01.086
Ram, M., Singh, V., Kumar, D., Kumawat, S., Gopalakrishnan, A., Lingaraju, M. C., et al. (2014). Antioxidant potential of bilirubin-accelerated wound healing in streptozotocin-induced diabetic rats. Naunyn Schmiedebergs Arch. Pharmacol. 387, 955–961. doi: 10.1007/s00210-014-1011-3
Ram, M., Singh, V., Kumawat, S., Kant, V., Tandan, S. K., and Kumar, D. (2016). Bilirubin modulated cytokines, growth factors and angiogenesis to improve cutaneous wound healing process in diabetic rats. Int. Immunopharmacol. 30, 137–149. doi: 10.1016/j.intimp.2015.11.037
Ramma, W., and Ahmed, A. (2014). Therapeutic potential of statins and the induction of heme oxygenase-1 in preeclampsia. J. Reprod. Immunol. 10, 153–160. doi: 10.1016/j.jri.2013.12.120
Rochette, L., Zeller, M., Cottin, Y., and Vergely, C. (2018). Redox functions of heme oxygenase-1 and biliverdin reductase in diabetes. Trends Endocrinol. Metab. 29, 74–85. doi: 10.1016/j.tem.2017.11.005
Rodrigues, C. M., Sola, S., Silva, R. F., and Brites, D. (2002). Aging confers different sensitivity to the neurotoxic properties of unconjugated bilirubin. Pediatr. Res. 51, 112–118. doi: 10.1203/00006450-200201000-00020
Ryter, S. W., Otterbein, L. E., Morse, D., and Choi, A. M. (2002). Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol. Cell Biochem. 23, 249–263. doi: 10.1007/978-1-4615-1087-1_29
Sedlak, T. W., Saleh, M., Higginson, D. S., Paul, B. D., Juluri, K. R., and Snyder, S. H. (2009). Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. U.S.A 106, 5171–5176. doi: 10.1073/pnas.0813132106
Sekioka, R., Tanaka, M., Nishimura, T., and Itoh, H. (2015). Serum total bilirubin concentration is negatively associated with increasing severity of retinopathy in patients with type 2 diabetes mellitus. J. Diabetes Complications 29, 218–221. doi: 10.1016/j.jdiacomp.2014.12.002
Seta, F., Bellner, L., Rezzani, R., Regan, R. F., Dunn, M. W., Abraham, N. G., et al. (2006). Heme oxygenase-2 is a critical determinant for execution of an acute inflammatory and reparative response. Am. J. Pathol. 169, 1612–1623. doi: 10.2353/ajpath.2006.060555
Seyed Khoei, N., Grindel, A., Wallner, M., Mölzer, C., Doberer, D., Marculescu, R., et al. (2018). Mild hyperbilirubinaemia as an endogenous mitigator of overweight and obesity: Implications for improved metabolic health. Atherosclerosis 269, 306–311. doi: 10.1016/j.atherosclerosis.2017.12.021
Shan, H., Li, T., Zhang, L., Yang, R., Li, Y., Zhang, M., et al. (2019). Heme oxygenase-1 prevents heart against myocardial infarction by attenuating ischemic injury-induced cardiomyocytes senescence. EBioMedicine 39, 59–68. doi: 10.1016/j.ebiom.2018.11.056
Shapiro, S. M. (2003). Bilirubin toxicity in the developing nervous system. Pediatr. Neurol. 29, 410–421. doi: 10.1016/j.pediatrneurol.2003.09.011
Silva, R. F., Rodrigues, C. M., and Brites, D. (2002). Rat cultured neuronal and glial cells respond differently to toxicity of unconjugated bilirubin. Pediatr. Res. 51, 535–541. doi: 10.1203/00006450-200204000-00022
Siow, R. C., Sato, H., and Mann, G. E. (1999). Heme oxygenase-carbon monoxide signalling pathway in atherosclerosis: anti-atherogenic actions of bilirubin and carbon monoxide? Cardiovasc. Res. 41, 385–394. doi: 10.1016/s0008-6363(98)00278-8
Stec, D. E., John, K., Trabbic, C. J., Luniwal, A., Hankins, M. W., Baum, J., et al. (2016). Bilirubin binding to PPARα inhibits lipid accumulation. PloS One 11:e0153427. doi: 10.1371/journal.pone.0153427
Stec, D. E., Storm, M. V., Pruett, B. E., and Gousset, M. U. (2013). Antihypertensive actions of moderate hyperbilirubinemia: role of superoxide inhibition. Am. J. Hypertens 26, 918–923. doi: 10.1093/ajh/hpt038
Stocker, R., Yamamoto, Y., McDonagh, A. F., Glazer, A. N., and Ames, B. N. (1987). Bilirubin is an antioxidant of possible physiological importance. Science 235, 1043–1046. doi: 10.1126/science.3029864
Šuk, J., Jašprová, J., Biedermann, D., Petrásková, L., Valentová, K., Křen, V., et al. (2019). Isolated silymarin flavonoids increase systemic and hepatic bilirubin concentrations and lower lipoperoxidation in mice. Oxid. Med. Cell Longev 2019:6026902. doi: 10.1155/2019/6026902
Sundararaghavan, V. L., Binepal, S., Stec, D. E., Sindhwani, P., and Hinds, T. D. (2018). Bilirubin, a new therapeutic for kidney transplant? Transplant. Rev. Orlando. Fla 32, 234–240. doi: 10.1016/j.trre.2018.06.003
Takeda, T. A., Mu, A., Tai, T. T., Kitajima, S., and Taketani, S. (2015). Continuous de novo biosynthesis of haem and its rapid turnover to bilirubin are necessary for cytoprotection against cell damage. Sci. Rep. 5:10488. doi: 10.1038/srep10488
Thakkar, M., Edelenbos, J., and Doré, S. (2019). Bilirubin and ischemic stroke: rendering the current paradigm to better understand the protective effects of bilirubin. Mol. Neurobiol. 56, 5483–5496. doi: 10.1007/s12035-018-1440-y
Tosevska, A., Moelzer, C., Wallner, M., Janosec, M., Schwarz, U., Kern, C., et al. (2016). Longer telomeres in chronic, moderate, unconjugated hyperbilirubinaemia: insights from a human study on Gilbert’s syndrome. Sci. Rep. 6:22300. doi: 10.1038/srep22300
Traverso, N., Ricciarelli, R., Nitti, M., Marengo, B., Furfaro, A. L., Pronzato, M. A., et al. (2013). Role of glutathione in cancer progression and chemoresistance. Oxid. Med Cell Longev 2013:972913. doi: 10.1155/2013/972913
Vanková, K., Marková, I., Jašprová, J., Dvořák, A., Subhanová, I., Zelenka, J., et al. (2018). Chlorophyll-mediated changes in the redox status of pancreatic cancer cells are associated with its anticancer effects. Oxid. Med. Cell Longev 2018:4069167. doi: 10.1155/2018/4069167
Vera, T., Granger, J. P., and Stec, D. E. (2009). Inhibition of bilirubin metabolism induces moderate hyperbilirubinemia and attenuates ANG II-dependent hypertension in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R738–R743. doi: 10.1152/ajpregu.90889.2008
Vera, T., and Stec, D. E. (2010). Moderate hyperbilirubinemia improves renal hemodynamics in ANG II-dependent hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R1044–R1049. doi: 10.1152/ajpregu.00316.2010
Viallard, C., and Larrivée, B. (2017). Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis 20, 409–426. doi: 10.1007/s10456-017-9562-9
Vítek, L. (2012). The role of bilirubin in diabetes, metabolic syndrome, and cardiovascular diseases. Front. Pharmacol. 3:55. doi: 10.3389/fphar.2012.00055
Vitek, L., Bellarosa, C., and Tiribelli, C. (2019a). Induction of mild hyperbilirubinemia: hype or real therapeutic opportunity? Clin. Pharmacol. Ther. 106, 568–575. doi: 10.1002/cpt.1341
Vitek, L., Hubacek, J. A., Pajak, A., Doryńska, A., Kozela, M., Eremiasova, L., et al. (2019b). Association between plasma bilirubin and mortality. Ann. Hepatol. 18, 379–385. doi: 10.1016/j.aohep.2019.02.001
Vítek, L., Jirsa, M., Brodanová, M., Kalab, M., Marecek, Z., Danzig, V., et al. (2002). Gilbert syndrome and ischemic heart disease: a protective effect of elevated bilirubin levels. Atherosclerosis 160, 449–456. doi: 10.1016/s0021-9150(01)00601-3
Vítek, L., Muchová, L., Janèová, E., Pešičková, S., Tegzová, D., Peterová, V., et al. (2010). Association of systemic lupus erythematosus with low serum bilirubin levels. Scand. J. Rheumatol. 39, 480–484. doi: 10.3109/03009741003742748
Vogel, M. E., Idelman, G., Konaniah, E. S., and Zucker, S. D. (2017). Bilirubin prevents atherosclerotic lesion formation in low-density lipoprotein receptor-deficient mice by inhibiting endothelial VCAM-1 and ICAM-1 signaling. J. Am. Heart Assoc. 6:e004820. doi: 10.1161/JAHA.116.004820
Vogel, M. E., and Zucker, S. D. (2016). Bilirubin acts as an endogenous regulator of inflammation by disrupting adhesion molecule-mediated leukocyte migration. Inflamm. Cell Signal. 3:e1178. doi: 10.14800/ics.1178
Wagner, K.-H., Wallner, M., Mölzer, C., Gazzin, S., Bulmer, A. C., Tiribelli, C., et al. (2015). Looking to the horizon: the role of bilirubin in the development and prevention of age-related chronic diseases. Clin. Sci. Lond. Engl. 129, 1–25. doi: 10.1042/CS20140566
Wallner, M., Antl, N., Rittmannsberger, B., Schreidl, S., Najafi, K., Müllner, E., et al. (2013). Anti-genotoxic potential of bilirubin in vivo: damage to DNA in hyperbilirubinemic human and animal models. Cancer Prev. Res. Phila. Pa 6, 1056–1063. doi: 10.1158/1940-6207.CAPR-13-0125
Wallner, M., Blassnigg, S. M., Marisch, K., Pappenheim, M. T., Müllner, E., Mölzer, C., et al. (2012). Effects of unconjugated bilirubin on chromosomal damage in individuals with Gilbert’s syndrome measured with the micronucleus cytome assay. Mutagenesis 27, 731–735. doi: 10.1093/mutage/ges039
Wang, D., Tosevska, A., Heiß, E. H., Ladurner, A., Mölzer, C., Wallner, M., et al. (2017). Bilirubin decreases macrophage cholesterol efflux and ATP-binding cassette transporter a1 protein expression. J. Am. Heart Assoc. 6:e005520. doi: 10.1161/JAHA.117.005520
Wang, Q., and Doerschuk, C. M. (2000). Neutrophil-induced changes in the biomechanical properties of endothelial cells: roles of ICAM-1 and reactive oxygen species. J. Immunol. Baltim. Md. 1950, 6487–6494. doi: 10.4049/jimmunol.164.12.6487
Wang, Q., and Doerschuk, C. M. (2002). The signaling pathways induced by neutrophil-endothelial cell adhesion. Antioxid. Redox Signal. 4, 39–47. doi: 10.1089/152308602753625843
Wang, W., and Lo, A. C. Y. (2018). Diabetic retinopathy: pathophysiology and treatments. Int. J. Mol. Sci. 19, E1816. doi: 10.3390/ijms19061816
Wu, B. J., Chen, K., Barter, P. J., and Rye, K.-A. (2012). Niacin inhibits vascular inflammation via the induction of heme oxygenase-1. Circulation 125, 150–158. doi: 10.1161/CIRCULATIONAHA.111.053108
Wu, T. W., Fung, K. P., and Yang, C. C. (1994). Unconjugated bilirubin inhibits the oxidation of human low density lipoprotein better than Trolox. Life Sci. 54, 477–481.
Xiao, Y., Xia, J., Wu, S., Lv, Z., Huang, S., Huang, H., et al. (2018). curcumin inhibits acute vascular inflammation through the activation of heme Oxygenase-1. Oxid. Med. Cell Longev 2018:3295807. doi: 10.1155/2018/3295807
Yang, Q., He, G.-W., Underwood, M. J., and Yu, C.-M. (2016). Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: perspectives and implications for postischemic myocardial protection. Am. J. Transl. Res. 8, 765–777.
Yasuda, M., Kiyohara, Y., Wang, J. J., Arakawa, S., Yonemoto, K., Doi, Y., et al. (2011). High serum bilirubin levels and diabetic retinopathy: the Hisayama Study. Ophthalmology 118, 1423–1428. doi: 10.1016/j.ophtha.2010.12.009
Young, J. L., Libby, P., and Schönbeck, U. (2002). Cytokines in the pathogenesis of atherosclerosis. Thromb. Haemost. 88, 554–567. doi: 10.1055/s-0037-1613256
Yu, M., Su, D., Yang, Y., Qin, L., Hu, C., Liu, R., et al. (2019). D-T7 peptide-modified pegylated bilirubin nanoparticles loaded with cediranib and paclitaxel for antiangiogenesis and chemotherapy of glioma. ACS Appl. Mater. Interfaces 11, 176–186. doi: 10.1021/acsami.8b16219
Zelenka, J., Dvořák, A., Alán, L., Zadinová, M., Haluzík, M., and Vítek, L. (2016). Hyperbilirubinemia protects against aging-associated inflammation and metabolic deterioration. Oxid Med. Cell Longev 2016:6190609. doi: 10.1155/2016/6190609
Zelenka, J., Muchova, L., Zelenkova, M., Vanova, K., Vreman, H. J., Wong, R. J., et al. (2012). Intracellular accumulation of bilirubin as a defense mechanism against increased oxidative stress. Biochimie 94, 1821–1827. doi: 10.1016/j.biochi.2012.04.026
Zhao, H., Ozen, M., Wong, R. J., and Stevenson, D. K. (2015). Heme oxygenase-1 in pregnancy and cancer: similarities in cellular invasion, cytoprotection, angiogenesis, and immunomodulation. Front. Pharmacol. 5:295. doi: 10.3389/fphar.2014.00295
Zhao, H., Wong, R. J., Doyle, T. C., Nayak, N., Vreman, H. J., Contag, C. H., et al. (2008). Regulation of maternal and fetal hemodynamics by heme oxygenase in mice. Biol. Reprod. 78, 744–751. doi: 10.1095/biolreprod.107.064899
Ziberna, L., Martelanc, M., Franko, M., and Passamonti, S. (2016). Bilirubin is an endogenous antioxidant in human vascular endothelial cells. Sci. Rep. 6:29240. doi: 10.1038/srep29240
Zou, Z.-Y., Liu, J., Chang, C., Li, J.-J., Luo, J., Jin, Y., et al. (2019). Biliverdin administration regulates the microRNA-mRNA expressional network associated with neuroprotection in cerebral ischemia reperfusion injury in rats. Int. J. Mol. Med. 43, 1356–1372. doi: 10.3892/ijmm.2019.4064
Zucker, S. D., Horn, P. S., and Sherman, K. E. (2004). Serum bilirubin levels in the U.S. population: gender effect and inverse correlation with colorectal cancer. Hepatol. Baltim. Md. 40, 827–835. doi: 10.1002/hep.20407
Zucker, S. D., Vogel, M. E., Kindel, T. L., Smith, D. L. H., Idelman, G., Avissar, U., et al. (2015). Bilirubin prevents acute DSS-induced colitis by inhibiting leukocyte infiltration and suppressing upregulation of inducible nitric oxide synthase. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G841–G854. doi: 10.1152/ajpgi.00149.2014
Keywords: HMOX1, bilirubin, endothelial cells, diabetes, atherosclerosis, hypertension, obesity, tumor
Citation: Nitti M, Furfaro AL and Mann GE (2020) Heme Oxygenase Dependent Bilirubin Generation in Vascular Cells: A Role in Preventing Endothelial Dysfunction in Local Tissue Microenvironment? Front. Physiol. 11:23. doi: 10.3389/fphys.2020.00023
Received: 30 September 2019; Accepted: 13 January 2020;
Published: 29 January 2020.
Edited by:
Raffaella Faraonio, University of Naples Federico II, ItalyReviewed by:
William Durante, University of Missouri, United StatesCopyright © 2020 Nitti, Furfaro and Mann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariapaola Nitti, TWFyaWFwYW9sYS5OaXR0aUB1bmlnZS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.