 Federico Carbone1,2
Federico Carbone1,2 Aldo Bonaventura
Aldo Bonaventura Fabrizio Montecucco
Fabrizio Montecucco
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 04 February 2020
Sec. Redox Physiology
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.01587
This article is part of the Research TopicOxidative Stress in Myocardial and Neural RemodelingView all 10 articles
The inflammatory response associated with myocardial and brain ischemia/reperfusion injury (IRI) is a critical determinant of tissue necrosis, functional organ recovery, and long-term clinical outcomes. In the post-ischemic period, reactive oxygen species (ROS) are involved in tissue repair through the clearance of dead cells and cellular debris. Neutrophils play a critical role in redox signaling due to their early recruitment and the large variety of released ROS. Noteworthy, ROS generated during IRI have a relevant role in both myocardial healing and activation of neuroprotective pathways. Anatomical and functional differences contribute to the responses in the myocardial and brain tissue despite a significant gene overlap. The exaggerated activation of this signaling system can result in adverse consequences, such as cell apoptosis and extracellular matrix degradation. In light of that, blocking the ROS cascade might have a therapeutic implication for cardiomyocyte and neuronal loss after acute ischemic events. The translation of these findings from preclinical models to clinical trials has so far failed because of differences between humans and animals, difficulty of agents to penetrate into specific cellular organs, and specifically unravel oxidant and antioxidant pathways. Here, we update knowledge on ROS cascade in IRI, focusing on the role of neutrophils. We discuss evidence of ROS blockade as a therapeutic approach for myocardial infarction and ischemic stroke.
Neutrophil (PMN) activation is strongly implicated in the pathogenesis of cardiovascular (CV) disease (Bonaventura et al., 2019). In addition to favoring atherosclerotic plaque vulnerability and rupture (Carbone et al., 2015a, 2018b), PMNs enhance thrombosis through different mechanisms, which include generation of neutrophil extracellular traps (NETs) (Bonaventura et al., 2018), release of proteases, and direct PMN–platelet interactions (Lisman, 2018). PMNs also have a critical role in ischemia/reperfusion injury (IRI) (Carbone et al., 2013; Garcia-Culebras et al., 2018) and tissue repair (Montecucco et al., 2017) (e.g., in the myocardium and the brain). Oxidative burst characterizes PMN activation and generates several classes of reactive oxygen species (ROS). Among the different subtypes of NADPH oxidases (NOX), NOX2 is prevalent but not specific of PMNs, being commonly expressed in cardiomyocytes, endothelial cells, fibroblasts, and neurons (El-Benna et al., 2009). Components of NOX2 enzymatic complex are located either in the cytosol (p47phox/p67phox/p40phox and the GTPase Rac1/Rac2) or in the plasma membrane (flavocytochrome subunits gp91phox and p22phox). Once assembled, the glycosylated gp91phox subunit undergoes a conformational change which allows catalytic activity. Activated NOX2 then generates high concentration of superoxide anion (), which exerts a prevalent local effect due to the short life span. In addition, dismutation may generate hydrogen peroxide (H2O2), which, in turn, reacts to produce the hydroxyl radical (⋅OH). The phagocyte-specific enzyme myeloperoxidase (MPO) catalyzes the formation of hypochlorous acid (HClO) and promotes the generation of chloramines, aldehydes, 1O2, ozone (O3), and ⋅OH (Prokopowicz et al., 2012). Nitric oxide synthase (NOS) is another ROS-generating enzyme active in PMNs. Through the conversion of the L-arginine to L-citrulline, NOS produces nitric oxide (NO), which may generate peroxynitrite by interacting with (Szabo et al., 2007). On this basis, it is not surprising that oxidative stress largely contributes to IRI. Conversely, less is known about the potential involvement in tissue repair. In the next paragraphs, we will focus on both myocardial and brain remodeling, also discussing the potential therapeutic implication of oxidative stress modulation.
Neutrophil-derived ROS show a specific diffusion range, determined by their life span and reactivity, whereas has a short life span. The non-radical compound H2O2 generated by its dismutation readily diffuses across membranes. Therefore, ROS may differently oxidize DNA, RNA, protein, and lipids. Nucleic acids undergo direct oxidative processes (e.g., nitrosative deamination, oxidation, and halogenation) or alternatively generate adducts with oxidized polyunsaturated fatty acids, protein, carbohydrates, and even nucleic acids themselves (Lonkar and Dedon, 2011). Posttranslational modification of proteins may occur through a direct oxidation of amino acids or other cellular components. Endoplasmic reticulum is extremely sensitive to the redox stress, which may determine disruption of the protein folding mechanism and the production of misfolded proteins (Cao and Kaufman, 2014). Also, catabolic processes are under the control of oxidative stress, which modulates protein degradation and autophagy (Pajares et al., 2015). By targeting polyunsaturated fatty acids, ROS may also determine membrane permeability, cytosol efflux, loss of membrane protein activities, and even biomembrane disruption with loss of cell viability (Jaganjac et al., 2016).
Finally, ROS may themselves act as second messengers and then transduce signals. Mitogen-activated protein kinases (MAPKs) are regulated by oxidative stress via different signaling cascades, involving Jun, p38 or extracellular signal-regulated kinase (ERK) 1/2 pathways, protein kinase C (PKC) and phosphoinositide 3-kinase (PI3K) activation (Hotamisligil and Davis, 2016). By adding an additional level of control, oxidative stress regulates a large amount of transcription factors (e.g., hypoxia-inducible factor [HIF]-1, activator protein [AP]-1, nuclear factor κ-light-chain-enhancer of activated B cells [NF-kB], and p53). It is then not surprising that ROS strongly influence in both autocrine and paracrine manner different PMN functions including phagocytosis, cytokine secretion, and apoptosis. Noteworthy, PMN-derived ROS also drive tissue response to IRI by modulating pathophysiological processes of resident cells (e.g., cardiomyocytes, endothelial and microglial cells, and neurons). The following paragraph will focus on this complex interaction between PMNs and the surrounding environment (Figure 1).
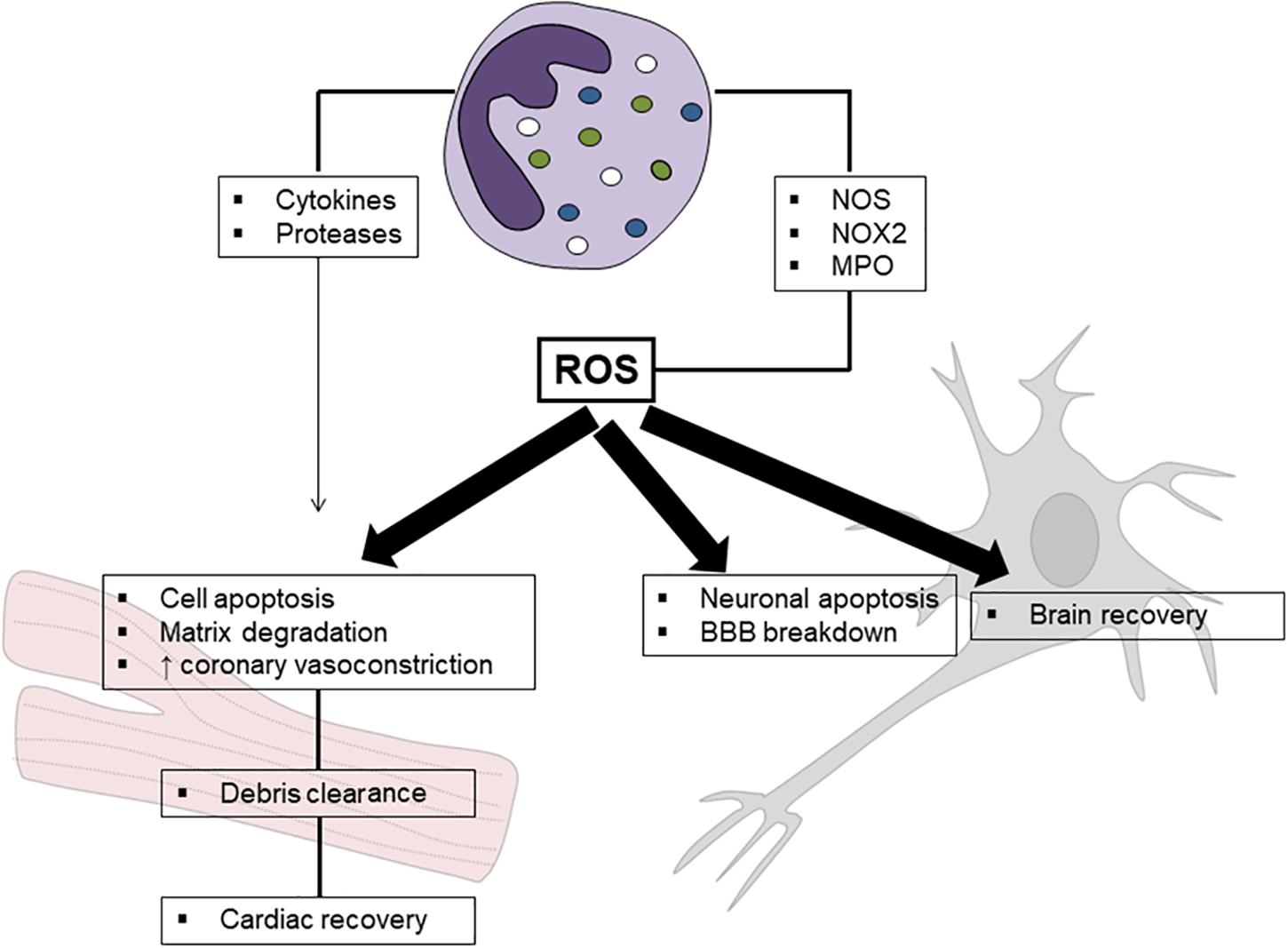
Figure 1. Schematic mechanism of neutrophil-related oxidative stress in ischemia/reperfusion injury. Reactive oxygen species (ROS) released by neutrophils are mainly produced by nitric oxide synthase (NOS), NADPH oxidase type 2 (NOX2), and myeloperoxidase (MPO). Although their detrimental role in ischemia reperfusion injury has been clearly established, a potential effect in promoting tissue healing has been suggested, especially in myocardial injury.
Myocardial reperfusion after an acute myocardial infarction (AMI) is recommended to save as much myocardium as possible from necrosis and dysfunction. Anyway, when the coronary flux is re-established, and the myocardium reperfused after ischemia, the hypercontracture of cardiac myocytes and their cytolysis may be paradoxically increased in response to reoxygenation. This phenomenon, known as “oxygen paradox,” forms a complementary dyad with the oxidative stress (Davies, 2016). The term “myocardial reperfusion injury” (MRI) describes myocardial injury and cardiomyocyte death usually occurring between 6 and 24 h after reperfusion of an ischemic area (Hausenloy and Yellon, 2016). Importantly, MRI is the main cause of death for stunned cardiomyocytes and ultimately accounts for more than half of the final size of myocardial ischemia. As widely reported in clinical and experimental studies, cardiomyocyte exposure to ROS causes apoptosis through different mechanisms. The leading one is likely the cytosolic and mitochondrial calcium overload that determines rapid alteration of intracellular pH. This, in turn, triggers the activation of MAPKs (Ong et al., 2015). Of interest, recent studies hypothesized oxidative stress as a promoter of myocardial fibrosis after ischemic injury, and both angiotensin 1 and the KCa3.1 channel have been suggested as involved pathways (Somanna et al., 2016; Wang et al., 2017). PMNs are recruited very early to the infarcted area (nearly after 30 min), and their actions are mediated by adhesive interactions with vascular endothelial cells, such as selectins, integrins, and molecules belonging to the immunoglobulin superfamily (Bonaventura et al., 2016). Selectin-dependent adhesion of PMNs mainly involves L-selectin (CD62L), E-selectin (CD62E), and P-selectin (CD62P). They play a critical role in neutrophil rolling so that their blocking is effective in reducing PMN recruitment and infarct size (Weyrich et al., 1993; Palazzo et al., 1998). However, selectins alone do not allow transmigration until the integrins start to play. PMNs express a combination of the β-chain CD18 with the α-chains CD11a [LFA-1 (lymphocyte function-associated antigen-1)], CD11b (macrophage-1 antigen), or CD11c (p150,95). PMN adhesion occurs with the binding of CD11/CD18 to the ligand intercellular adhesion molecule (ICAM)-1 expressed on the endothelial surface (Jordan et al., 1999). Hence, PMNs change shape to motile cells, and transendothelial migration takes place (Smith, 2000). The latter occurs via paracellular route through junctions between adjacent endothelial cells and involves various molecules (PECAM-1, CD99, ICAM-2, endothelial cell-selective adhesion molecule, and members of the junctional adhesion molecule family). Once the infarcted zone is infiltrated, PMNs release ROS alongside cytokines and proteolytic enzymes, ultimately feeding a vicious circle through a positive feedback loop (Panth et al., 2016; Montecucco et al., 2017). MPO is detected within the infarcted myocardium, including the luminal thrombi on eroded plaques (Ferrante et al., 2010), and MPO-generated oxidants are likely to have a negligible impact on infarct size and impact largely on the adverse left ventricle remodeling and function (Vasilyev et al., 2005). In experimental models, the mechanism of neutrophil-dependent MRI was shown to be further dependent on CD18 integrin activation and ICAM-1 expression by injured cardiac cells (Albelda et al., 1994). In vivo, ROS are mainly released by adherent PMNs, thus suggesting the crucial role of the PMN ligand-specific adhesion to cardiomyocytes in MRI (Frangogiannis, 2015). The generation of ROS peaks within 2–10 min from coronary artery reperfusion and acts as a trigger for immune cell chemotaxis – particularly PMN – through complement activation and upregulation of cytokines and chemokines via the NF-κB pathway (Hensley et al., 2000). The role of ROS is of utmost importance in cardiac healing after AMI as it promotes the clearance of dead cells and cellular debris. However, exaggerated oxidative stress may result in detrimental consequences, such as cell apoptosis and degradation of the extracellular matrix. This occurs when antioxidant systems (i.e., catalase, glutathione peroxidase, and superoxide dismutase) and intracellular antioxidants are overcome and the catastrophic actions of ROS may take place (Frangogiannis, 2015).
Cytochrome P-450 (CYP), xanthine oxidase (XO), NOX, monoamine oxidase (MAO), and the mitochondrial electron transport chain (METC) are the most common enzymes involved in ROS production. CYP is likely the most known source of ROS in reperfused infarcted heart (Vinten-Johansen, 2004). CYP-derived ROS increase in an oxygen-dependent way following increased uncoupling reaction and oxygen supply (Hernandez-Resendiz et al., 2018). In particular, the release of O–2 and H2O2 by CYP 2C9 activates the NF-κB pathway which in turn upregulates the production of proinflammatory cytokines and the expression of adhesion molecules (Fleming et al., 2001). Similarly, the overexpression of CYP 2C8 was reported to favor ROS generation, finally exacerbating coronary vasoconstriction and increasing infarct size (Edin et al., 2011). XO derives from xanthine dehydrogenase upon myocardial reperfusion and reacts with purine substrates and O2 to finally generate and H2O2. XO is usually abundant within the vascular endothelium of normal hearts and is referred to as a primary source of ROS (Hernandez-Resendiz et al., 2018). This finding is largely based on the evidence that allopurinol, a known xanthine oxidoreductase, is effective in limiting the injury occurring during MRI and reducing the infarcted size (Pisarenko et al., 1994). Furthermore, XO was described to have a role in leukocyte recruitment and neutrophil adhesion in hypoxic conditions (Matsumura et al., 1998). Concerning NOX, the subtype NOX2 plays a central role and is typically overexpressed during MRI. Apart from the role in ROS generation, NOX can also indirectly provide damages via the activation of PKC-mediated phosphorylation of the cytosolic p47phox (Patterson et al., 1999). MAOs can catalyze oxidative deamination of several monoamines leading to a great production of ROS. Moreover, MAOs have a role in the production of H2O2 in the very early reperfusion period (Kunduzova et al., 2002). Finally, METC complexes are very important sources and targets of ROS arising during MRI, particularly complexes I and III (Hernandez-Resendiz et al., 2018). Mitochondrial complex I is formed by an active A-form and a deactivated D-form, the latter being the most abundant during MRI and producing and H2O2 (Gorenkova et al., 2013). Mitochondrial complex III is another fundamental source of ROS during reperfusion as demonstrated by the reduced activity in ischemic hearts compared to healthy ones. Interestingly, ROS burst from the mitochondria can induce the oxidation of cholesterol and the production of oxysterols, which induce interleukin-1β release in the vascular endothelial cells and the following expression of adhesion molecules for the recruitment of immune cells (Lemaire et al., 1998; Poli et al., 2013).
Apart from the known detrimental effects of PMNs in the ischemic area, current evidence suggests that PMNs can also display reparative functions by recruiting and activating mononuclear cells (Alard et al., 2015). Recently, neutrophil depletion by a specific monoclonal antibody has been reported to not affect infarct size, but rather favor the progressive deterioration of the post-AMI cardiac function (Horckmans et al., 2017). Accordingly, the positive influence of PMNs in this setting may be due to the polarization of macrophages toward a reparative phenotype mediated, at least partially, by neutrophil gelatinase-associated lipocalin.
In line with these findings, many antioxidants were studied, although clinical trials reported some controversial results, especially with mitochondria-targeted antioxidants (coenzyme Q10, mitoQ, and MTP-131) (Table 1; Argaud et al., 2005; Karlsson et al., 2010; Skyschally et al., 2010; Chiari et al., 2014; Hausenloy et al., 2014; Cung et al., 2015; Dare et al., 2015; Eleawa et al., 2015; Hernandez-Resendiz et al., 2015; Gibson et al., 2016; Ottani et al., 2016). Coenzyme Q10 was shown to decrease the infarcted area, the inflammatory burden, and the oxidative stress and to normalize left ventricle function following AMI (Eleawa et al., 2015). MitoQ was studied in IRI after heart transplantation to reduce ROS production showing to block graft oxidative damage and blunt the early proinflammatory response in the recipient. This effect would be dependent on reducing mitochondrial DNA damage and H2O2 (Dare et al., 2015). On the contrary, no reduction in myocardial infarct size was found in the EMBRACE STEMI trial with MTP-131 (also known as Szeto-Schiller-31 or elamipretide), a cell-permeable mitochondria-targeting peptide selectively binding to cardiolipin and optimizing mitochondrial electron transport by reducing ROS generation (Gibson et al., 2016). The EMBRACE STEMI was a multicenter, randomized, double-blind phase IIa trial evaluating the efficacy and safety of MTP-131 vs. placebo infused at a rate of 0.05 mg/kg/h for 1 h among first-time anterior STEMI subjects undergoing primary percutaneous coronary intervention for a proximal or mid left anterior descending artery occlusion. No reduction of the primary endpoint (infarct size by creatine kinase–myocardial band and area under the curve over 72 h) was reached as well as no improvement in prespecified magnetic resonance imaging, angiographic, electrocardiographic, or clinical outcomes was shown (Gibson et al., 2016). In a similar manner, cyclosporine A, a potent inhibitor of the mitochondrial permeability transition pore, showed to significantly reduce myocardial infarction size in most, but not all, experimental studies (Argaud et al., 2005; Karlsson et al., 2010; Skyschally et al., 2010). As well, in some phase II clinical trials, cyclosporine A was likely to protect the heart following an AMI (Chiari et al., 2014; Hausenloy et al., 2014). On the contrary, two large clinical trials reported disappointing results, although a clear explanation is still lacking. The CYCLE (CYCLosporinE A in Reperfused Acute Myocardial Infarction) trial of 410 patients ST elevation AMI did not find any benefit with cyclosporine A administered prior to primary percutaneous coronary intervention in terms of ST-segment resolution and enzymatic myocardial infarct size (Ottani et al., 2016). In the CIRCUS (Does Cyclosporine Improve Clinical Outcome in ST Elevation Myocardial Infarction Patients) trial conducted among 970 patients with anterior ST elevation AMI, cyclosporine A immediately before primary percutaneous coronary intervention failed to improve clinical outcomes (all-cause death, heart failure hospitalization, and adverse left ventricle remodeling) at 1 year (Cung et al., 2015). For a wider explanation on this topic, readers can be referred to the review by Hausenloy et al. (2017). Among natural molecules, curcumin was largely studied as an antioxidant molecule, with promising direct and indirect effects on ROS scavenging and myocardial remodeling. An extensive discussion on this topic can be found elsewhere (Hernandez-Resendiz et al., 2015).
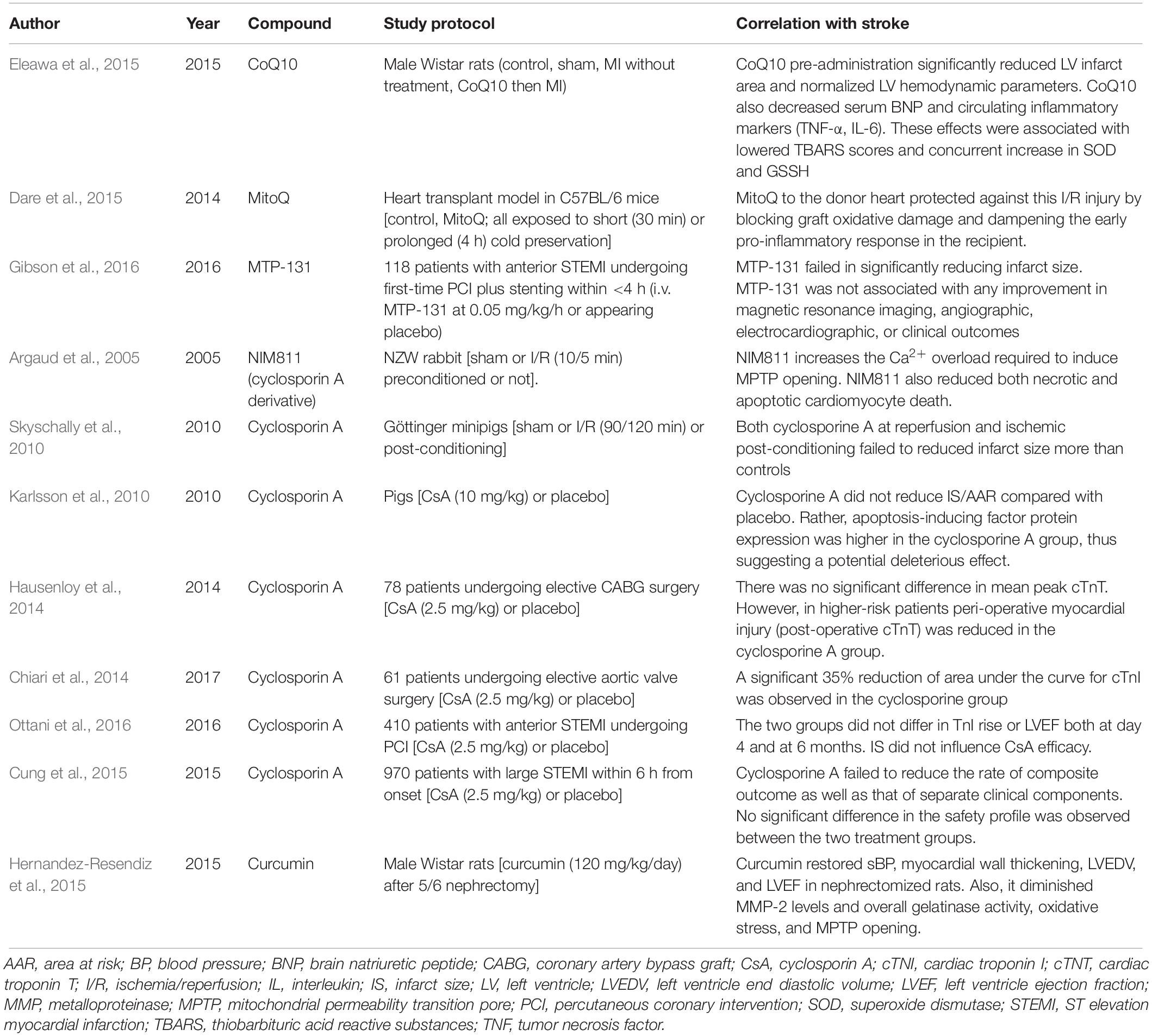
Table 1. Efficacy of antioxidant compounds in myocardial ischemia/reperfusion injury.
Differently from other peripheral organs, the brain parenchyma does not elicit stereotypic immune responses. This is largely due to the unique anatomical composition, as endothelial, epithelial, and glial barriers tightly regulate the accessibility of immune cells. The anatomical and functional characteristics of this immune-privileged organ then imply a different response to IRI despite a significant gene overlapping (Zhang et al., 2014). The low levels of antioxidants associated with high polyunsaturated fatty acids in cellular membranes make the brain more susceptible to oxidative damage (Adibhatla and Hatcher, 2010). In the early phase of ischemic IRI, mitochondrial depolarization and activation of XO determine a first oxidative burst with ROS generation (mainly and H2O2). They trigger PMN recruitment and activation, further precipitating IRI, even more when thrombolytic drugs are administered. Indeed, we have previously demonstrated how recombinant tissue-type plasminogen activator induces an early neutrophil degranulation via PI3K/Akt, which contributes to the increased risk of hemorrhagic transformation (Carbone et al., 2015b, c). PMNs then represent the leading source of ROS in the subacute phase of stroke. Experimental models largely emphasized the detrimental effect of NOX2 activation in the ischemic brain, being brain swelling and infarct size significantly reduced in NOX2–/– mice. In this context, especially ⋅OH has been described as typical phagocyte ROS and amplifier of ischemic injury on neuronal cells. ⋅OH is generated via the Fenton reaction and has great affinity for unsaturated fatty acids, ultimately leading to peroxyl radical (ROOS) generation. In turn, ROOS trigger a cycle of lipid peroxidation that destroys cellular membranes. Products of lipid peroxidation further sustain oxidative stress by generating aldehydes, dienals, or alkanes (e.g., malondialdehyde and 4-hydroxynonenal). The consequent redox unbalance leads to neuronal apoptosis and blood–brain barrier (BBB) breakdown.
More recently, NO has been suggested as an additional mediator of IRI. At low oxygen concentrations, NO accumulates and reacts with to generate peroxynitrite. Nitrosative stress may lead to BBB breakdown, inflammation, and caspase activation, which ultimately lead to cell apoptosis through interacting with different cellular signaling pathways including matrix metalloproteinase, high-mobility group box 1, toll-like receptors 2 and 4, poly(ADP-ribose) polymerase, Src, Rho-associated protein kinase (ROCK), and glycogen synthase kinase (GSK)-3β (Radi et al., 2015). Oxidative stress may also influence epigenetic mechanisms (i.e., DNA methylation, histone modification, microRNAs) (Zhao et al., 2016; Narne et al., 2017), well known to be implicated in neuroprotection (Felling and Song, 2015; Simon, 2016; Chandran et al., 2017). Therefore, it has been suggested that ROS may contribute themselves to neuronal recovery after ischemic stroke. In line with this hypothesis, ROS-induced activation of HIF-1α/β-catenin pathway has been associated with neuronal recovery in rats (Hu et al., 2014), whereas a biphasic role of ROS has been recently suggested also in human beings (Yang et al., 2018). Furthermore, oxidative stress (mainly H2O2 and NOX signaling) regulates neural stem and progenitor cell proliferation, self-renewal, and neurogenesis (Le Belle et al., 2011). As compared with ⋅OH, ROS are generated constantly as part of a normal aerobic life and are active players of several metabolic pathways ranging from cell adhesion to lipid metabolism (Roy et al., 2017). Nevertheless, the extent to which the redox status potentially contributes to brain recovery has not been established yet. Rather, a large body of evidence indicates the suppression of oxidative stress as a promising strategy to reduce brain injury. Improving reperfusion is certainly the best approach to reduce ROS generation (Carbone et al., 2018a; Chamorro, 2018; Taskiran-Sag et al., 2018). Furthermore, it is likely that antioxidant compounds tested in previous neuroprotection trials might be more effective if reperfusion therapies are co-administered (Chamorro, 2018). Among different antioxidant compounds, edaravone has already been used for years in the Far East countries. As a free radical scavenger with inhibitory effects on lipid peroxidation (Yamamoto, 2017), edaravone promotes neuroprotection when combined with thrombolysis (Logallo et al., 2011; Kono et al., 2013; Wada et al., 2014; Liu et al., 2015; Llull et al., 2015; Chamorro et al., 2017; Yamaguchi et al., 2017; Lee and Xiang, 2018; Table 2). Surprisingly, free radical scavenging properties have also been described for uric acid (UA). Serum levels of UA are classically associated with increased CV risk, but this association may rather be a compensatory mechanism (Li et al., 2015). Furthermore, when administered with recombinant tissue-type plasminogen activator, UA significantly improves the efficacy of thrombolysis (Logallo et al., 2011; Liu et al., 2015; Llull et al., 2015; Chamorro et al., 2017; Table 2). Other classes of compounds are currently under evaluation in preclinical studies and include a synthetic analog of vitamin E and specific inhibitors of NOX and NOS (Sun et al., 2018). Finally, some advances in nanomedicine are expected to improve drug delivery (Zhang et al., 2017; Shen et al., 2018) or even provide new nanoparticles with antioxidant potential (Hosoo et al., 2017).
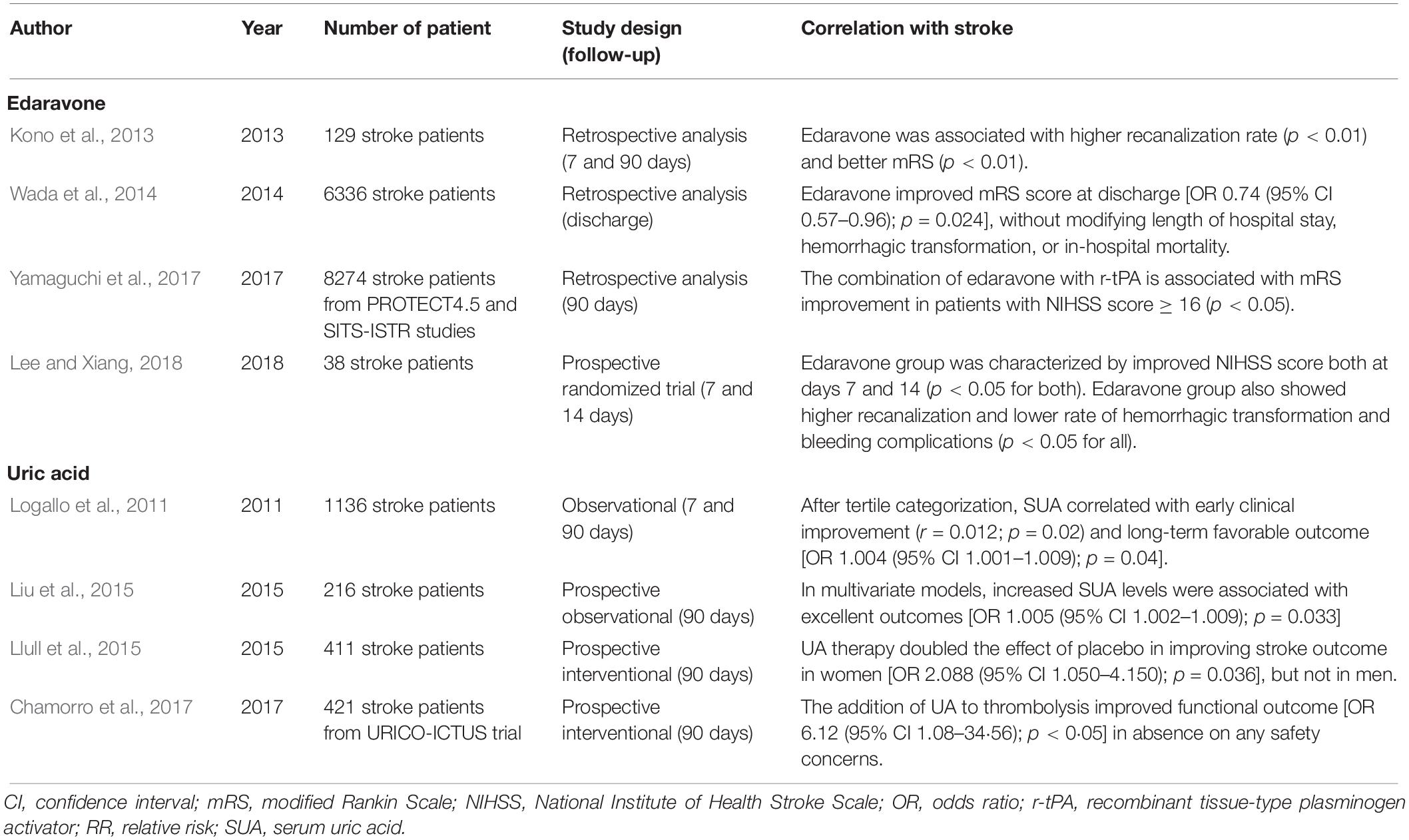
Table 2. Efficacy of antioxidant compounds in stroke.
Free radicals have strong oxidative properties in ischemic tissues. When reperfusion occurs, the massive generation of ROS and reactive nitrogen species leads to cell death via DNA damage, protein dysfunction, and lipid peroxidation. However, signaling pathways activated by oxidative stress are also likely to be involved in the healing processes. Appropriate consideration of the role of PMN-related oxidative stress in IRI might potentially improve current therapeutic strategies. However, several critical points should be taken into account. PMN-generated ROS may have both detrimental and beneficial roles in different models of IRI. This dual role might depend on the cell and tissue types and the timing of response (Montecucco et al., 2017). ROS activity is not limited to nearby cells/tissues. Rather there is variability in the diffusion range due to the different life span and reactivity. Finally, the side effects induced by immune suppression should be considered (Horckmans et al., 2017). Addressing these issues may explain how the results of clinical trials have so far failed to reproduce the success of preclinical studies.
All the authors wrote, made substantial corrections and contribution, and approved the final version of the manuscript to be published.
This study was supported by a grant from the Italian Ministry of Health to the Italian Cardiovascular Network (#2754291) and a grant from the Fondazione CARIGE to FM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adibhatla, R. M., and Hatcher, J. F. (2010). Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox. Signal. 12, 125–169. doi: 10.1089/ARS.2009.2668
Alard, J. E., Ortega-Gomez, A., Wichapong, K., Bongiovanni, D., Horckmans, M., Megens, R. T., et al. (2015). Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci. Transl. Med. 7:ra196. doi: 10.1126/scitranslmed.aad5330
Albelda, S. M., Smith, C. W., and Ward, P. A. (1994). Adhesion molecules and inflammatory injury. FASEB J. 8, 504–512.
Argaud, L., Gateau-Roesch, O., Muntean, D., Chalabreysse, L., Loufouat, J., Robert, D., et al. (2005). Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J. Mol. Cell Cardiol. 38, 367–374. doi: 10.1016/j.yjmcc.2004.12.001
Bonaventura, A., Liberale, L., Carbone, F., Vecchie, A., Diaz-Canestro, C., Camici, G. G., et al. (2018). The pathophysiological role of neutrophil extracellular traps in inflammatory diseases. Thromb. Haemost. 118, 6–27. doi: 10.1160/TH17-09-0630
Bonaventura, A., Montecucco, F., and Dallegri, F. (2016). Cellular recruitment in myocardial ischaemia/reperfusion injury. Eur. J. Clin. Invest. 46, 590–601. doi: 10.1111/eci.12633
Bonaventura, A., Montecucco, F., Dallegri, F., Carbone, F., Lüscher, T. F., Camici, G. G., et al. (2019). Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc. Res. 115, 1266–1285. doi: 10.1093/cvr/cvz084
Cao, S. S., and Kaufman, R. J. (2014). Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox. Signal. 21, 396–413. doi: 10.1089/ars.2014.5851
Carbone, F., Busto, G., Padroni, M., Bernardoni, A., Colagrande, S., Dallegri, F., et al. (2018a). Radiologic cerebral reperfusion at 24 h predicts good clinical outcome. Transl. Stroke Res. 10, 178–188. doi: 10.1007/s12975-018-0637-8
Carbone, F., Mach, F., and Montecucco, F. (2015a). Update on the role of neutrophils in atherosclerotic plaque vulnerability. Curr. Drug Targets 16, 321–333. doi: 10.2174/1389450115666141110093013
Carbone, F., Nencioni, A., Mach, F., Vuilleumier, N., and Montecucco, F. (2013). Pathophysiological role of neutrophils in acute myocardial infarction. Thromb. Haemost. 110, 501–514. doi: 10.1160/TH13-03-0211
Carbone, F., Rigamonti, F., Burger, F., Roth, A., Bertolotto, M., Spinella, G., et al. (2018b). Serum levels of osteopontin predict major adverse cardiovascular events in patients with severe carotid artery stenosis. Int. J. Cardiol. 255, 195–199. doi: 10.1016/j.ijcard.2018.01.008
Carbone, F., Vuilleumier, N., Bertolotto, M., Burger, F., Galan, K., Roversi, G., et al. (2015b). Treatment with recombinant tissue plasminogen activator (r-TPA) induces neutrophil degranulation in vitro via defined pathways. Vascul. Pharmacol. 64, 16–27. doi: 10.1016/j.vph.2014.11.007
Carbone, F., Vuilleumier, N., Burger, F., Roversi, G., Tamborino, C., Casetta, I., et al. (2015c). Serum osteopontin levels are upregulated and predict disability after an ischaemic stroke. Eur. J. Clin. Invest. 45, 579–586. doi: 10.1111/eci.12446
Chamorro, A. (2018). Neuroprotectants in the era of reperfusion therapy. J. Stroke 20, 197–207. doi: 10.5853/jos.2017.02901
Chamorro, A., Amaro, S., Castellanos, M., Gomis, M., Urra, X., Blasco, J., et al. (2017). Uric acid therapy improves the outcomes of stroke patients treated with intravenous tissue plasminogen activator and mechanical thrombectomy. Int. J. Stroke 12, 377–382. doi: 10.1177/1747493016684354
Chandran, R., Mehta, S. L., and Vemuganti, R. (2017). Non-coding RNAs and neuroprotection after acute CNS injuries. Neurochem. Int. 111, 12–22. doi: 10.1016/j.neuint.2017.01.015
Chiari, P., Angoulvant, D., Mewton, N., Desebbe, O., Obadia, J. F., Robin, J., et al. (2014). Cyclosporine protects the heart during aortic valve surgery. Anesthesiology 121, 232–238. doi: 10.1097/ALN.0000000000000331
Cung, T. T., Morel, O., Cayla, G., Rioufol, G., Garcia-Dorado, D., Angoulvant, D., et al. (2015). Cyclosporine before PCI in patients with acute myocardial infarction. N. Engl. J. Med. 373, 1021–1031. doi: 10.1056/NEJMoa1505489
Dare, A. J., Logan, A., Prime, T. A., Rogatti, S., Goddard, M., Bolton, E. M., et al. (2015). The mitochondria-targeted anti-oxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. J. Heart Lung Transplant. 34, 1471–1480. doi: 10.1016/j.healun.2015.05.007
Davies, K. J. (2016). The oxygen paradox, oxidative stress, and ageing. Arch. Biochem. Biophys. 595, 28–32. doi: 10.1016/j.abb.2015.11.015
Edin, M. L., Wang, Z., Bradbury, J. A., Graves, J. P., Lih, F. B., DeGraff, L. M., et al. (2011). Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J. 25, 3436–3447. doi: 10.1096/fj.11-188300
El-Benna, J., Dang, P. M., Gougerot-Pocidalo, M. A., Marie, J. C., and Braut-Boucher, F. (2009). p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp. Mol. Med. 41, 217–225. doi: 10.3858/emm.2009.41.4.058
Eleawa, S. M., Alkhateeb, M., Ghosh, S., Al-Hashem, F., Shatoor, A. S., Alhejaily, A., et al. (2015). Coenzyme Q10 protects against acute consequences of experimental myocardial infarction in rats. Int. J. Physiol. Pathophysiol. Pharmacol. 7, 1–13.
Felling, R. J., and Song, H. (2015). Epigenetic mechanisms of neuroplasticity and the implications for stroke recovery. Exp. Neurol. 268, 37–45. doi: 10.1016/j.expneurol.2014.09.017
Ferrante, G., Nakano, M., Prati, F., Niccoli, G., Mallus, M. T., and Ramazzotti, V. (2010). High levels of systemic myeloperoxidase are associated with coronary plaque erosion in patients with acute coronary syndromes: a clinicopathological study. Circulation 122, 2505–2513. doi: 10.1161/CIRCULATIONAHA.110.955302
Fleming, I., Michaelis, U. R., Bredenkotter, D., Fisslthaler, B., Dehghani, F., Brandes, R. P., et al. (2001). Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ. Res. 88, 44–51. doi: 10.1161/01.res.88.1.44
Frangogiannis, N. G. (2015). Pathophysiology of myocardial infarction. Compr. Physiol. 5, 1841–1875. doi: 10.1002/cphy.c150006
Garcia-Culebras, A., Duran-Laforet, V., Pena-Martinez, C., Ballesteros, I., Pradillo, J. M., Diaz-Guzman, J., et al. (2018). cells as therapeutic targets in neuroinflammation after stroke: specific roles of neutrophils and neutrophil-platelet interactions. J. Cereb. Blood Flow Metab. 38, 2150–2164. doi: 10.1177/0271678X18795789
Gibson, C. M., Giugliano, R. P., Kloner, R. A., Bode, C., Tendera, M., Janosi, A., et al. (2016). EMBRACE STEMI study: a Phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur. Heart J. 37, 1296–1303. doi: 10.1093/eurheartj/ehv597
Gorenkova, N., Robinson, E., Grieve, D. J., and Galkin, A. (2013). Conformational change of mitochondrial complex I increases ROS sensitivity during ischemia. Antioxid. Redox. Signal. 19, 1459–1468. doi: 10.1089/ars.2012.4698
Hausenloy, D., Kunst, G., Boston-Griffiths, E., Kolvekar, S., Chaubey, S., John, L., et al. (2014). The effect of cyclosporin-A on peri-operative myocardial injury in adult patients undergoing coronary artery bypass graft surgery: a randomised controlled clinical trial. Heart 100, 544–549. doi: 10.1136/heartjnl-2013-304845
Hausenloy, D. J., Botker, H. E., Engstrom, T., Erlinge, D., Heusch, G., Ibanez, B., et al. (2017). Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: trials and tribulations. Eur. Heart J. 38, 935–941.
Hausenloy, D. J., and Yellon, D. M. (2016). Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol. 13, 193–209. doi: 10.1038/nrcardio.2016.5
Hensley, K., Robinson, K. A., Gabbita, S. P., Salsman, S., and Floyd, R. A. (2000). Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 28, 1456–1462. doi: 10.1016/s0891-5849(00)00252-5
Hernandez-Resendiz, S., Chinda, K., Ong, S. B., Cabrera-Fuentes, H., Zazueta, C., and Hausenloy, D. J. (2018). The role of redox dysregulation in the inflammatory response to acute myocardial ischaemia-reperfusion injury - adding fuel to the fire. Curr. Med. Chem. 25, 1275–1293. doi: 10.2174/0929867324666170329100619
Hernandez-Resendiz, S., Correa, F., Garcia-Nino, W. R., Buelna-Chontal, M., Roldan, F. J., Ramirez-Camacho, I., et al. (2015). Cardioprotection by curcumin post-treatment in rats with established chronic kidney disease. Cardiovasc. Drugs Ther. 29, 111–120. doi: 10.1007/s10557-015-6581-x
Horckmans, M., Ring, L., Duchene, J., Santovito, D., Schloss, M. J., Drechsler, M., et al. (2017). Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 38, 187–197. doi: 10.1093/eurheartj/ehw002
Hosoo, H., Marushima, A., Nagasaki, Y., Hirayama, A., Ito, H., Puentes, S., et al. (2017). Neurovascular unit protection from cerebral ischemia-reperfusion injury by radical-containing nanoparticles in mice. Stroke 48, 2238–2247. doi: 10.1161/STROKEAHA.116.016356
Hotamisligil, G. S., and Davis, R. J. (2016). Cell signaling and stress responses. Cold Spring Harb. Perspect. Biol. 8:a006072. doi: 10.1101/cshperspect.a006072
Hu, Q., Liang, X., Chen, D., Chen, Y., Doycheva, D., Tang, J., et al. (2014). Delayed hyperbaric oxygen therapy promotes neurogenesis through reactive oxygen species/hypoxia-inducible factor-1alpha/beta-catenin pathway in middle cerebral artery occlusion rats. Stroke 45, 1807–1814. doi: 10.1161/STROKEAHA.114.005116
Jaganjac, M., Cipak, A., Schaur, R. J., and Zarkovic, N. (2016). Pathophysiology of neutrophil-mediated extracellular redox reactions. Front. Biosci. 21, 839–855. doi: 10.2741/4423
Jordan, J. E., Zhao, Z. Q., and Vinten-Johansen, J. (1999). The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc. Res. 43, 860–878. doi: 10.1016/s0008-6363(99)00187-x
Karlsson, L. O., Zhou, A. X., Larsson, E., Astrom-Olsson, K., Mansson, C., Akyurek, L. M., et al. (2010). Cyclosporine does not reduce myocardial infarct size in a porcine ischemia-reperfusion model. J. Cardiovasc. Pharmacol. Ther. 15, 182–189. doi: 10.1177/1074248410362074
Kono, S., Deguchi, K., Morimoto, N., Kurata, T., Yamashita, T., Ikeda, Y., et al. (2013). Intravenous thrombolysis with neuroprotective therapy by edaravone for ischemic stroke patients older than 80 years of age. J. Stroke Cerebrovasc. Dis. 22, 1175–1183. doi: 10.1016/j.jstrokecerebrovasdis.2013.02.010
Kunduzova, O. R., Bianchi, P., Parini, A., and Cambon, C. (2002). Hydrogen peroxide production by monoamine oxidase during ischemia/reperfusion. Eur. J. Pharmacol. 448, 225–230. doi: 10.1016/s0014-2999(02)01913-1
Le Belle, J. E., Orozco, N. M., Paucar, A. A., Saxe, J. P., Mottahedeh, J., Pyle, A. D., et al. (2011). Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 8, 59–71. doi: 10.1016/j.stem.2010.11.028
Lee, X. R., and Xiang, G. L. (2018). Effects of edaravone, the free radical scavenger, on outcomes in acute cerebral infarction patients treated with ultra-early thrombolysis of recombinant tissue plasminogen activator. Clin. Neurol. Neurosurg. 167, 157–161. doi: 10.1016/j.clineuro.2018.02.026
Lemaire, S., Lizard, G., Monier, S., Miguet, C., Gueldry, S., Volot, F., et al. (1998). Different patterns of IL-1beta secretion, adhesion molecule expression and apoptosis induction in human endothelial cells treated with 7alpha-, 7beta-hydroxycholesterol, or 7-ketocholesterol. FEBS Lett. 440, 434–439. doi: 10.1016/s0014-5793(98)01496-3
Li, R., Huang, C., Chen, J., Guo, Y., and Tan, S. (2015). The role of uric acid as a potential neuroprotectant in acute ischemic stroke: a review of literature. Neurol. Sci. 36, 1097–1103. doi: 10.1007/s10072-015-2151-z
Lisman, T. (2018). Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. 371, 567–576. doi: 10.1007/s00441-017-2727-4
Liu, X., Liu, M., Chen, M., Ge, M. Q., and Pan, S. M. (2015). Serum uric acid is neuroprotective in Chinese patients with acute ischemic stroke treated with intravenous recombinant tissue plasminogen activator. J. Stroke Cerebrovasc. Dis. 24, 1080–1086. doi: 10.1016/j.jstrokecerebrovasdis.2015.01.011
Llull, L., Laredo, C., Renu, A., Perez, B., Vila, E., Obach, V., et al. (2015). Uric acid therapy improves clinical outcome in women with acute ischemic stroke. Stroke 46, 2162–2167. doi: 10.1161/STROKEAHA.115.009960
Logallo, N., Naess, H., Idicula, T., Brogger, J., Waje-Andreassen, U., and Thomassen, L. (2011). Serum uri acid: neuroprotection in thrombolysis. The bergen NORSTROKE study. BMC Neurol. 11:114. doi: 10.1186/1471-2377-11-114
Lonkar, P., and Dedon, P. C. (2011). Reactive species and DNA damage in chronic inflammation: reconciling chemical mechanisms and biological fates. Int. J. Cancer 128, 1999–2009. doi: 10.1002/ijc.25815
Matsumura, F., Yamaguchi, Y., Goto, M., Ichiguchi, O., Akizuki, E., Matsuda, T., et al. (1998). Xanthine oxidase inhibition attenuates kupffer cell production of neutrophil chemoattractant following ischemia-reperfusion in rat liver. Hepatology 28, 1578–1587. doi: 10.1002/hep.510280618
Montecucco, F., Liberale, L., Bonaventura, A., Vecchie, A., Dallegri, F., and Carbone, F. (2017). The Role of inflammation in cardiovascular outcome. Curr. Atheroscler. Rep. 19:11. doi: 10.1007/s11883-017-0646-1
Narne, P., Pandey, V., and Phanithi, P. B. (2017). Interplay between mitochondrial metabolism and oxidative stress in ischemic stroke: an epigenetic connection. Mol. Cell. Neurosci. 82, 176–194. doi: 10.1016/j.mcn.2017.05.008
Ong, S. B., Samangouei, P., Kalkhoran, S. B., and Hausenloy, D. J. (2015). The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell Cardiol. 78, 23–34. doi: 10.1016/j.yjmcc.2014.11.005
Ottani, F., Latini, R., Staszewsky, L., La Vecchia, L., Locuratolo, N., Sicuro, M., et al. (2016). Cyclosporine a in reperfused myocardial infarction: the multicenter. Controlled, open-label CYCLE trial. J. Am. Coll. Cardiol. 67, 365–374. doi: 10.1016/j.jacc.2015.10.081
Pajares, M., Jimenez-Moreno, N., Dias, I. H., Debelec, B., Vucetic, M., Fladmark, K. E., et al. (2015). Redox control of protein degradation. Redox Biol. 6, 409–420. doi: 10.1016/j.redox.2015.07.003
Palazzo, A. J., Jones, S. P., Anderson, D. C., Granger, D. N., and Lefer, D. J. (1998). Coronary endothelial P-selectin in pathogenesis of myocardial ischemia-reperfusion injury. Am. J. Physiol. 275, H1865–H1872. doi: 10.1152/ajpheart.1998.275.5.H1865
Panth, N., Paudel, K. R., and Parajuli, K. (2016). Reactive oxygen species: a key hallmark of cardiovascular disease. Adv. Med. 2016:9152732.
Patterson, C., Ruef, J., Madamanchi, N. R., Barry-Lane, P., Hu, Z., Horaist, C., et al. (1999). Stimulation of a vascular smooth muscle cell NAD(P)H oxidase by thrombin. Evidence that p47(phox) may participate in forming this oxidase in vitro and in vivo. J. Biol. Chem. 274, 19814–19822. doi: 10.1074/jbc.274.28.19814
Pisarenko, O. I., Lakomkin, V. L., Studneva, I. M., Timoshin, A. A., Kuzmin, A. I., Ruuge, E. K., et al. (1994). Allopurinol-enhanced postischemic recovery in the isolated rat heart involves repletion of high-energy phosphates. Biochem. Med. Metab. Biol. 51, 16–26. doi: 10.1006/bmmb.1994.1002
Poli, G., Biasi, F., and Leonarduzzi, G. (2013). Oxysterols in the pathogenesis of major chronic diseases. Redox Biol. 1, 125–130. doi: 10.1016/j.redox.2012.12.001
Prokopowicz, Z., Marcinkiewicz, J., Katz, D. R., and Chain, B. M. (2012). Neutrophil myeloperoxidase: soldier and statesman. Arch. Immunol. Ther. Exp. 60, 43–54. doi: 10.1007/s00005-011-0156-8
Radi, R., Beckman, J. S., Bush, K. M., and Freeman, B. A. (2015). When the good and the bad make the ugly: the discovery of peroxynitrite. J. Biol. Chem. 290, 30726–30727. doi: 10.1074/jbc.o115.000001
Roy, J., Galano, J. M., Durand, T., Le Guennec, J. Y., and Lee, J. C. (2017). Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J. 31, 3729–3745. doi: 10.1096/fj.201700170R
Shen, Y., Cao, B., Snyder, N. R., Woeppel, K. M., Eles, J. R., and Cui, X. T. (2018). ROS responsive resveratrol delivery from LDLR peptide conjugated PLA-coated mesoporous silica nanoparticles across the blood-brain barrier. J. Nanobiotechnol. 16:13. doi: 10.1186/s12951-018-0340-7
Simon, R. P. (2016). Epigenetic modulation of gene expression governs the brain’s response to injury. Neurosci. Lett. 625, 16–19. doi: 10.1016/j.neulet.2015.12.024
Skyschally, A., Schulz, R., and Heusch, G. (2010). Cyclosporine A at reperfusion reduces infarct size in pigs. Cardiovasc. Drugs. Ther. 24, 85–87. doi: 10.1007/s10557-010-6219-y
Smith, C. W. (2000). Introduction: functional polarity of motile neutrophils. Blood 95, 2459–2461. doi: 10.1182/blood.v95.8.2459.008a37_2459_2461
Somanna, N. K., Valente, A. J., Krenz, M., Fay, W. P., Delafontaine, P., and Chandrasekar, B. (2016). The Nox1/4 dual inhibitor GKT137831 or Nox4 knockdown inhibits angiotensin-II-induced adult mouse cardiac fibroblast proliferation and migration. AT1 physically associates with Nox4. J. Cell. Physiol. 231, 1130–1141. doi: 10.1002/jcp.25210
Sun, M. S., Jin, H., Sun, X., Huang, S., Zhang, F. L., Guo, Z. N., et al. (2018). Free radical damage in ischemia-reperfusion injury: an obstacle in acute ischemic stroke after revascularization therapy. Oxid. Med. Cell Longev. 2018:3804979. doi: 10.1155/2018/3804979
Szabo, C., Ischiropoulos, H., and Radi, R. (2007). Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 6, 662–680. doi: 10.1038/nrd2222
Taskiran-Sag, A., Yemisci, M., Gursoy-Ozdemir, Y., Erdener, S. E., Karatas, H., Yuce, D., et al. (2018). Improving microcirculatory reperfusion reduces parenchymal oxygen radical formation and provides neuroprotection. Stroke 49, 1267–1275. doi: 10.1161/STROKEAHA.118.020711
Vasilyev, N., Williams, T., Brennan, M. L., Unzek, S., Zhou, X., Heinecke, J. W., et al. (2005). Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation 112, 2812–2820. doi: 10.1161/circulationaha.105.542340
Vinten-Johansen, J. (2004). Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc. Res. 61, 481–497. doi: 10.1016/j.cardiores.2003.10.011
Wada, T., Yasunaga, H., Inokuchi, R., Horiguchi, H., Fushimi, K., Matsubara, T., et al. (2014). Effects of edaravone on early outcomes in acute ischemic stroke patients treated with recombinant tissue plasminogen activator. J. Neurol. Sci. 345, 106–111. doi: 10.1016/j.jns.2014.07.018
Wang, L. P., Fan, S. J., Li, S. M., Wang, X. J., Gao, J. L., and Yang, X. H. (2017). Oxidative stress promotes myocardial fibrosis by upregulating KCa3.1 channel expression in AGT-REN double transgenic hypertensive mice. Pflugers Archiv. 469, 1061–1071. doi: 10.1007/s00424-017-1984-0
Weyrich, A. S., Ma, X. Y., Lefer, D. J., Albertine, K. H., and Lefer, A. M. (1993). In vivo neutralization of P-selectin protects feline heart and endothelium in myocardial ischemia and reperfusion injury. J. Clin. Invest. 91, 2620–2629. doi: 10.1172/jci116501
Yamaguchi, T., Awano, H., Matsuda, H., Tanahashi, N., and Investigators, P. (2017). edaravone with and without .6 Mg/Kg Alteplase within 4.5 hours after ischemic stroke: a prospective cohort study (PROTECT4.5). J. Stroke Cerebrovasc. Dis. 26, 756–765. doi: 10.1016/j.jstrokecerebrovasdis.2016.10.011
Yamamoto, Y. (2017). Plasma marker of tissue oxidative damage and edaravone as a scavenger drug against peroxyl radicals and peroxynitrite. J. Clin. Biochem. Nutr. 60, 49–54. doi: 10.3164/jcbn.16-63
Yang, J., Qi, J., Xiu, B., Yang, B., Niu, C., and Yang, H. (2018). Reactive oxygen species play a biphasic role in brain ischemia. J. Invest. Surg. 32, 97–102. doi: 10.1080/08941939.2017.1376131
Zhang, C., Ling, C. L., Pang, L., Wang, Q., Liu, J. X., Wang, B. S., et al. (2017). Direct macromolecular drug delivery to cerebral ischemia area using neutrophil-mediated nanoparticles. Theranostics 7, 3260–3275. doi: 10.7150/thno.19979
Zhang, Y., Kong, P., Chen, Y., Yu, Y., Liu, J., Yang, L., et al. (2014). Significant overlapping modules and biological processes between stroke and coronary heart disease. CNS Neurol. Disord. Drug Targets 13, 652–660. doi: 10.2174/1871527312666131223115112
Keywords: inflammation, reactive oxygen species, neutrophils, acute myocardial infarction, stroke
Citation: Carbone F, Bonaventura A and Montecucco F (2020) Neutrophil-Related Oxidants Drive Heart and Brain Remodeling After Ischemia/Reperfusion Injury. Front. Physiol. 10:1587. doi: 10.3389/fphys.2019.01587
Received: 29 October 2018; Accepted: 18 December 2019;
Published: 04 February 2020.
Edited by:
Mansoureh Eghbali, University of California, Los Angeles, United StatesReviewed by:
Houzao Chen, Chinese Academy of Medical Sciences, ChinaCopyright © 2020 Carbone, Bonaventura and Montecucco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabrizio Montecucco, ZmFicml6aW8ubW9udGVjdWNjb0B1bmlnZS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.