 Guoyuan Qi
Guoyuan Qi Yashi Mi
Yashi Mi Fei Yin
Fei Yin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 08 January 2020
Sec. Redox Physiology
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.01531
This article is part of the Research TopicAdvances in Metabolic Mechanisms of Aging and Its Related DiseasesView all 11 articles
As an organ with a highly heterogenous cellular composition, the brain has a bioenergetic system that is more complex than peripheral tissues. Such complexities are not only due to the diverse bioenergetic phenotypes of a variety of cell types that differentially contribute to the metabolic profile of the brain, but also originate from the bidirectional metabolic communications and coupling across cell types. While brain energy metabolism and mitochondrial function have been extensively investigated in aging and age-associated neurodegenerative disorders, the role of various cell types and their inter-cellular communications in regulating brain metabolic and synaptic functions remains elusive. In this review, we summarize recent advances in differentiating bioenergetic phenotypes of neurons, astrocytes, and microglia in the context of their functional specificity, and their metabolic shifts upon aging and pathological conditions. Moreover, the metabolic coordination between the two most abundant cell populations in brain, neurons and astrocytes, is discussed regarding how they jointly establish a dynamic and responsive system to maintain brain bioenergetic homeostasis and to combat against threats such as oxidative stress, lipid toxicity, and neuroinflammation. Elucidating the mechanisms by which brain cells with distinctive bioenergetic phenotypes individually and collectively shape the bioenergetic system of the brain will provide rationale for spatiotemporally precise interventions to sustain a metabolic equilibrium that is resilient against synaptic dysfunction in aging and neurodegeneration.
Active metabolism is fundamental in maintaining the life and activity of organisms by mediating the exchange of material, energy, and information with the environment. Most organisms rely on the catabolism of organic molecules to obtain energy. The acquirement of mitochondria by eukaryotic cells via endosymbiosis is proposed to remarkably expand their genomic capacity, which energetically drives and enables the evolution from single- to multi-cell systems (Lane and Martin, 2010; Lane, 2014). In higher organisms like the mammals, the production, storage, and utilization of fuels have been compartmentalized to, and distributed among, different organs. Organs such as liver, muscle, heart, and adipose tissue have adapted to possess specialized metabolic capacity and flexibility in using different fuels to meet their diverse functional needs.
Among all organs, brain is unique, not only because it consumes 20% of total glucose with its 2% of body weight (Bélanger et al., 2011), but also due to its unique metabolic profile and metabolite pool that are separated from the rest of the body by the blood–brain barrier (BBB). Although brain ultimately uses substrates from the periphery, its capacity to uptake and metabolize them varies from one fuel to another, with glucose being the predominant preference (Lundgaard et al., 2015). Within the brain, the highly heterogeneous cellular composition comes with diverse energetic capacity and fuel preference across cell types. While previous studies have established the vital role of bioenergetics system in brain aging and neurodegenerative diseases, the physiological and pathological roles of different cell types and their metabolic coordination are still being actively investigated. Accordingly, most strategies to restore brain bioenergetic homeostasis in neurodegenerative diseases either indistinguishably targeted all cell types or focused on neurons with limited consideration of the bioenergetic contributions from non-neuronal cells. In this review, we sought to discuss recent advances in revealing cell-type-specific bioenergetic role of brain cells with connection to their specialized functions. Moreover, inter-cellular metabolic coordination between neurons and astrocytes is reviewed in the context of brain aging and age-associated neurodegenerative disorders.
The mammalian brain is composed of diverse, specialized cell populations, including neurons, astrocytes, oligodendrocytes, microglia, and others. All of them together not only enable the highly refined electrophysiological activities, but also fulfill the organ’s nutritional needs and its defense against pathogens (Saunders et al., 2018). Recently, high-throughput single-cell RNA sequencing has achieved an unprecedented resolution in distinguishing and clustering cell types and sub-cell types in both rodent and human brains (Zeisel et al., 2015; Lake et al., 2016; Saunders et al., 2018). Moreover, studies in aging and degenerating brains also suggested that bioenergetic genes in different cell types are differentially altered in aging or diseases. Comparative analysis of bulk tissue- and single-cell RNA-seq of Alzheimer’s brains revealed distinct transcriptional changes across cell types, and a decline in mitochondrial genes at tissue level is only seen in neurons but not in glial cells from the same region of brain (Mathys et al., 2019).
Consistent with their transcriptomic profiles, different brain cell types have their unique metabolic phenotypes (Magistretti and Allaman, 2015; Figure 1). Both glia and neurons have the capacity to fully metabolize glucose, but their functional diversity differentiates their metabolic preference. Neurons, which generate and consume most brain ATPs, primarily rely on mitochondrial oxidative phosphorylation (OXPHOS) for energy transduction and possess low glycolytic capacity due to the suppression of a key glycolysis enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB) (Schönfeld and Reiser, 2013; Zheng et al., 2016; Figure 1). For non-glucose substrates, neurons have a low capacity to utilize fatty acids (Schönfeld and Reiser, 2013) but can metabolize ketone bodies generated in non-neuronal cells from fatty acid β-oxidation.
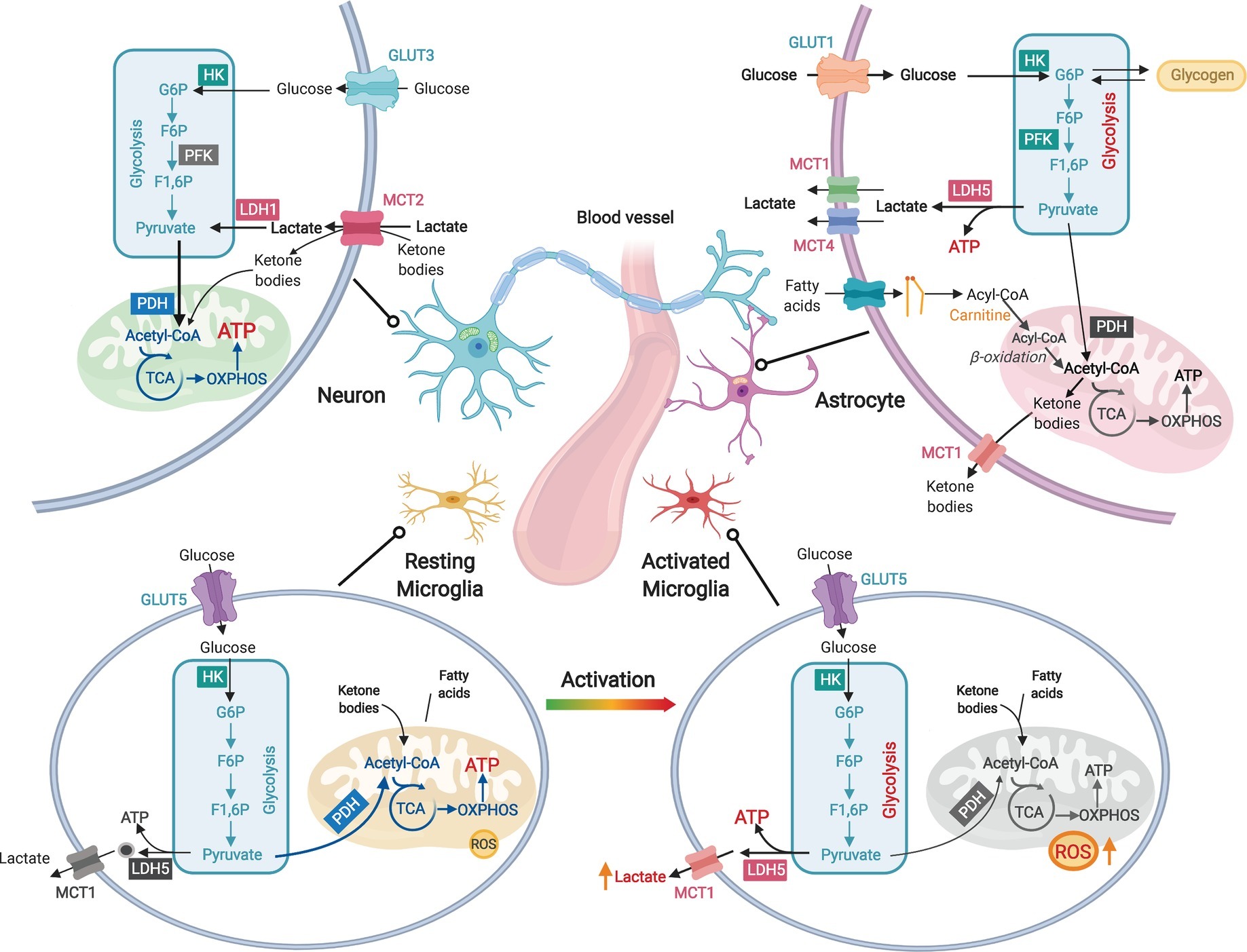
Figure 1. Diverse metabolic phenotypes of brain cells. Neurons produce ATP primarily via mitochondrial oxidative phosphorylation (OXPHOS), but their glycolytic rate is less active than that of astrocytes due to suppressed PFK activity. Both lactate and ketone bodies can enter neurons via monocarboxylate transporter 2 (MCT2). Astrocytes are highly active in glycolysis to produce lactate but less dependent on OXPHOS due to low pyruvate dehydrogenase (PDH) activity. Lactate is generated by lactate dehydrogenase 5 (LDH5) in astrocytes and exported by MCT1 and MCT4. Astrocytes are capable to metabolize fatty acids through β-oxidation. Fatty acids transport into mitochondria is facilitated by carnitine and carnitine palmitoyltransferases (CPTs). Ketone bodies can be generated in astrocytic mitochondria via ketogenesis. Excessive astrocytic glucose can be stored as glycogen, which can be converted back to glucose. Resting microglia have high rate of OXPHOS and low production of reactive oxygen species (ROS), whereas activated microglia produce large amount of ROS and undergo a metabolic reprogramming from OXPHOS toward glycolysis and lactate production.
As the most abundant cell type in the brain, astrocytes provide critical metabolic and structural support to neurons, including modulation of ion homeostasis, supply of nutrients, and control of BBB permeability (Suzuki et al., 2011; Bolaños, 2016; Boison and Steinhäuser, 2018). In contrast to neurons, astrocytes maintain lower OXPHOS activity but higher glycolysis rate (Bélanger et al., 2011; Bouzier-Sore and Pellerin, 2013; Figure 1). Large amount of glucose metabolized by glycolysis in astrocytes is released as lactate, which serves as important metabolic fuel for neurons (Bolaños, 2016). Astrocytes also store excessive fuels in glycogen that can rapidly regenerate glucose for glycolysis or glutamate synthesis (Suzuki et al., 2011). Multiple lines of evidence suggest that astrocytes can metabolize non-glucose substrates including fatty acids (Ioannou et al., 2019), glutamate (Schousboe et al., 2014), and ketone bodies (McNair et al., 2017).
Microglia are resident immune cells in the brain (Viader et al., 2015), and are closely related to host defense against pathogens and CNS disorders (Perry et al., 2010). The survival and activation of microglia depend on sufficient energy supply. Cell-type-specific RNA-seq analysis reveals that microglia express the full set of genes required for both glycolysis and OXPHOS (Zhang et al., 2014), but their bioenergetic phenotype is activation-state-dependent (Aldana, 2019). Resting microglia depend mainly on OXPHOS for ATP production, whereas activated microglia favor glycolysis as manifested by increased lactate production and decreased mitochondrial oxygen consumption (Ghosh et al., 2018; Aldana, 2019; Figure 1). Additionally, transcriptome data suggest that microglia express key enzymes for fatty acids mobilization and β-oxidation, which may alternatively meet their elevated energy demand upon activation (Zhang et al., 2014).
A byproduct of mitochondrial OXPHOS is the electron leak from respiratory chain to generate superoxide and other reactive oxygen species (ROS; Murphy, 2009). Increased ROS production and oxidized redox status characterizes brain aging and neurodegeneration (Markesbery, 1997; Cadenas and Davies, 2000; Lin and Beal, 2006; Yin et al., 2012a, 2014; Hwang, 2013). Of all brain cell types, neurons are particularly susceptible to redox changes due to their high metabolic rate and limited antioxidant capacity (Baxter and Hardingham, 2016). Astrocytes, in contrast, have greater antioxidative potential (Gupta et al., 2012). Multiple studies have demonstrated that the astrocytic support of neuronal antioxidant system is a key neuroprotective mechanism against oxidative damage (Magistretti and Allaman, 2013). Since the electrons used to reduce ROS are ultimately from NADPH, which is generated by the pentose phosphate pathways (PPP) from glucose or by mitochondrial enzymes including the transhydrogenase (Yin et al., 2012b), elevated oxidative stress could cause a switch in re-routing fuels toward NADPH production rather than ATP production (Agarwal et al., 2013). Accordingly, astrocytic oxidative stress dampens glucose uptake and diverts glucose into PPP for NADPH production, which subsequently decreases lactate release from astrocytes to neurons and compromises the neuronal redox homeostasis (Steele and Robinson, 2012). The low capacity to metabolize fatty acids of neurons is another cause for its sensitivity to oxidative challenges: a recent study revealed that excess ROS trigger lipids synthesis in neuron and give rise to neurodegeneration if managed improperly (Liu et al., 2017).
Glucose is brain’s dominating fuel under physiological conditions. Upon limited glucose availability, the brain undergoes a metabolic shift to use ketone bodies – including acetate, acetoacetate, and β-hydroxybutyrate – as its alternative energy source (Cunnane et al., 2011). Upon fasting, starvation, extended exercise, pregnancy, or development, fatty acids are mobilized from adipocytes and transported to liver for conversion to ketone bodies, which are subsequently transported to the brain to generate acetyl-CoA and eventually ATP in mitochondria (Drenick et al., 1972; Puchalska and Crawford, 2017; Mattson et al., 2018). Ketogenic interventions were reported with beneficial effect on cases of inborn metabolic disorder involving genetic deficiency of GLUT1 or pyruvate dehydrogenase, suggesting ketones as brain fuels to bypass glucose hypometabolism (Falk et al., 1976; Klepper, 2004; Wang et al., 2005). Although liver is believed to be the predominant supplier of ketone bodies, it was found that ketone bodies could also be generated in astrocytes from fatty acids (Martinez-Outschoorn et al., 2012). In brain aging and a variety of neurodegenerative diseases, a decline in glucose metabolism has been well-established (Ott et al., 1999; Fabre et al., 2002; Cunnane et al., 2011; Ding et al., 2013; Goyal et al., 2017). Under this situation, the brain may adapt a similar bioenergetic shift from exclusively relying on glucose toward using ketone bodies in response to energetic deficits (Ding et al., 2013; Klosinski et al., 2015; Camandola and Mattson, 2017). Beneficial effect of ketogenic diet on cognitive and cardiovascular outcomes has been observed in aging mice (Newman et al., 2017) and patients with mild cognitive impairment, a transition state toward AD (Krikorian et al., 2012).
While mitochondrial function in neurons is known to be impaired during aging, astrocytes exhibit elevated mitochondrial respiration in aging rat brains (Jiang and Cadenas, 2014), which is consistent with human studies showing increased astrocytic TCA activity with age (Boumezbeur et al., 2010). In parallel with these bioenergetic changes, rodent and human studies also demonstrated that the expression of glial fibrillary acidic protein (GFAP), a marker protein for activated astrocytes, increases with age (Nichols et al., 1993; Wu et al., 2005). Overall, age-associated activation of astrocytic OXPHOS could restrict their capacity to supply neurons with substrates such as lactate or glucose (Yan et al., 2013; Rodríguez-Arellano et al., 2016), and is connected with a switch from being neurotrophic to neurotoxic and to neuroinflammation (Jiang and Cadenas, 2014).
Similar to their peripheral counterparts macrophages, the bioenergetics of microglia also undergo a switch from OXPHOS toward glycolysis upon activation (Orihuela et al., 2016). LPS-activation of microglia-like BV-2 cell blocks mitochondrial oxygen consumption while activating anaerobic glycolysis and PPP (Voloboueva et al., 2013; Gimeno-Bayón et al., 2014). Consistently, glucose transporters GLUT1 and GLUT4, and hexokinase 2 (HK2), are upregulated in activated microglia (Gimeno-Bayón et al., 2014). Such a metabolic switch is critical to enable the fast response of activated microglia to energy-demanding processes such as proliferation, migration, cytokine secretion, and phagocytosis, because of the much faster rate in generating ATP by glycolysis than OXPHOS (Orihuela et al., 2016). Activation of microglia and chronic neuroinflammation are key features of aging and neurodegeneration. Upon LPS challenge, microglia from aged animals produce more ROS, compared to those isolated from young mouse brains (Tichauer et al., 2014; Figure 1). In a familial AD mouse model, exposure to amyloid-β triggers acute microglial activation in parallel with a metabolic shift from OXPHOS to glycolysis (Baik et al., 2019). A similar shift was also found in microglia from multiple sclerosis patients (Van Der Poel et al., 2019).
The diverse ability to perform OXPHOS and glycolysis between neurons and astrocytes is proposed to be related to inter-cellular coupling of glucose metabolism by the astrocyte-neuron lactate shuttle (Pellerin and Magistretti, 1994; Díaz-García et al., 2017; Figure 2). Glutamate released from active neurons activates astrocytic glycolysis and the production of lactate, which is used by neurons for synaptic activities (Mächler et al., 2016). Astrocytes take up more glucose than their energetic needs, which suggests their role in maintaining an extracellular lactate pool for neuronal use (Chuquet et al., 2010). In Drosophila, glial cells express glycolytic enzymes to produce lactate and alanine, which are then used to derive pyruvate for neuronal OXPHOS (Brooks et al., 1999; Volkenhoff et al., 2015). Lactate, pyruvate, and ketone bodies cross cell membranes through proton-linked monocarboxylate transporters (MCTs) (Halestrap, 2011; Figure 2). Knocking down astrocytic lactate transporter MCT4 in mouse hippocampus leads to memory retention, supporting the role of astrocytic lactate for long-term synaptic plasticity and memory (Suzuki et al., 2011). A recent in vivo study using a genetically encoded FRET sensor Laconic revealed a lactate gradient from astrocytes to neurons (Mächler et al., 2016). Astrocyte-to-neuron lactate shuttle was also found to be more functional in young hippocampi whereas aged neurons exhibit reduced dependence on astrocytic lactate with disrupted metabolic crosstalk (Drulis-Fajdasz et al., 2018). Perturbations to neuron-astrocyte metabolic coupling are seen in multiple neurodegenerative diseases (Rama Rao and Kielian, 2015). Decreased expression of astrocytic MCTs and defective neuron-astrocyte coupling of glucose metabolism was found in mouse models of AD and amyotrophic lateral sclerosis (ALS) (Ferraiuolo et al., 2011; Ding et al., 2013). A decline in levels of lactate, glucose, and other glycolytic intermediates was seen in the cerebrospinal fluid of Parkinson’s patients (Ohman and Forsgren, 2015).
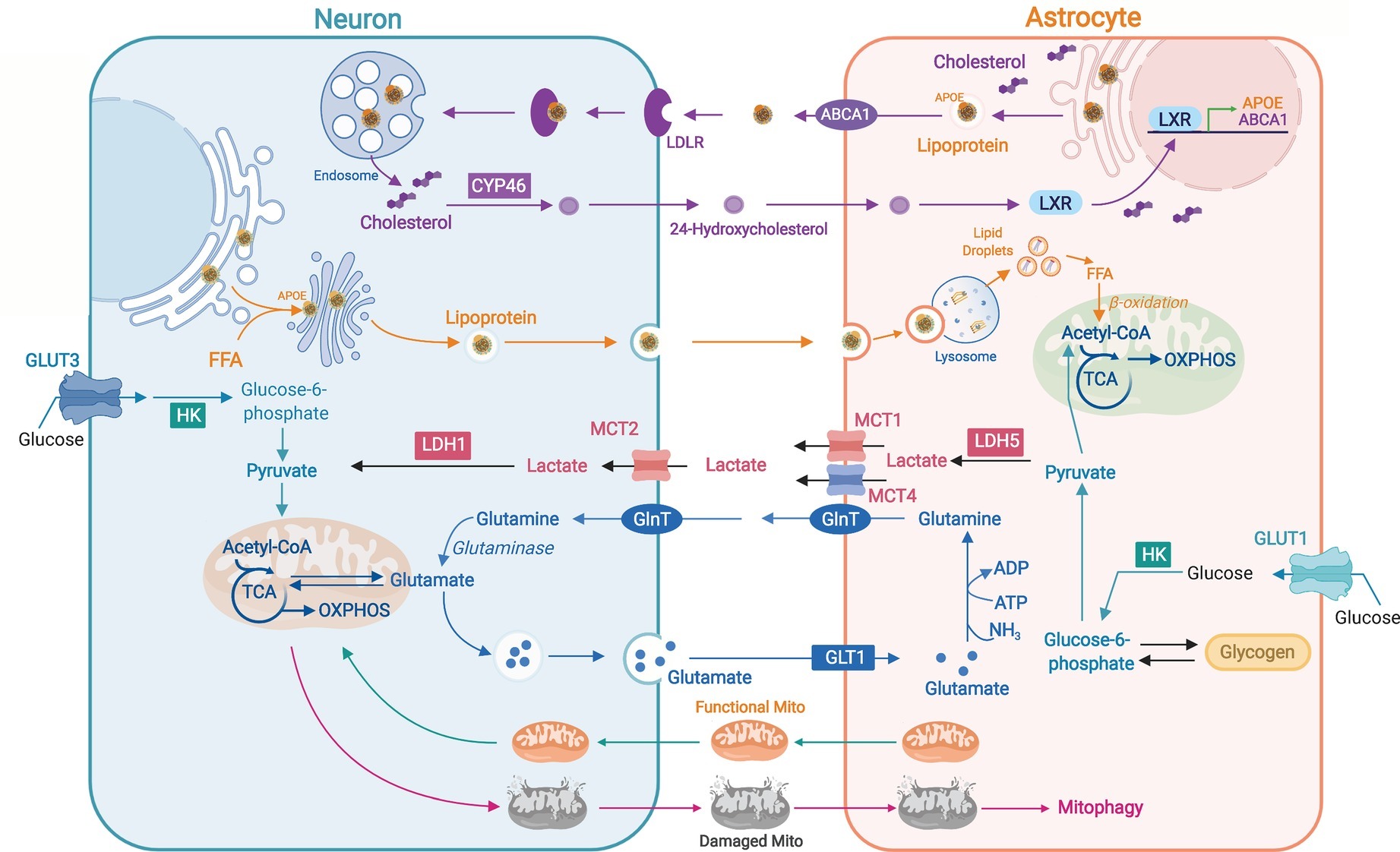
Figure 2. Neuron-astrocyte metabolic coordination. Astrocyte-synthesized cholesterols are packaged in APOE-associated lipoproteins and exported by ATP-binding cassette transporter A1 (ABCA1); released lipoproteins bind to neuronal LDL receptors and are internalized for neuronal use or conversion to 24-hydroxycholesterol (24-OHC) by cholesterol 24-hydroxylase (CYP46). 24-OHC produced by neurons can activate astrocytic live X receptor (LXR) to induce the expression of APOE and ABCA1. Hyperactive neurons release excessive free fatty acids (FFAs) in APOE-associated lipid particles to astrocytes where FFAs are targeted to lipid droplets for subsequent degradation by mitochondrial β-oxidation. High glycolytic rate in astrocytes produces lactate that can be transported to neurons by MCTs and used for ATP production. Neurotransmitter glutamate released by neurons is uptaken by astrocytes through glutamate transporter 1 (GLT1) and converted to glutamine for recycle to neurons and the re-generation of glutamate. Neurons can transfer damaged axonal mitochondria to astrocytes for autophagic degradation (mitophagy) whereas astrocytes can transfer healthy mitochondria to adjacent neurons as a neuroprotective and neurorecovery mechanism after stroke.
Another metabolic coupling between neurons and astrocytes that was described recently is the coordination of fatty acid metabolism (Liu et al., 2015, 2017; Rambold et al., 2015; Ioannou et al., 2019; Figure 2). Lipids constitute 50% of the brain dry weight, mainly as fundamental component of membrane structures (Bruce et al., 2017). Since neurons are known to have minimum capacity to catabolize fatty acids (Schonfeld and Reiser, 2017), unused or recycled neuronal lipids are transported to astrocytes and stored in lipid droplets (LDs) before the metabolism in mitochondria (Liu et al., 2017; Ioannou et al., 2019). Sequestering of excessive fatty acids in LD could prevent lipotoxicity and mitochondrial dysfunction (Listenberger et al., 2003). In Drosophila, LD formation in niche glia under oxidative stress dampens intracellular ROS generation and oxidation of polyunsaturated fatty acids, which protects glia and neuroblasts from the peroxidation chain (Bailey et al., 2015). Upon nutrient depletion, LDs can deliver FAs to mitochondria for β-oxidation (Greenberg et al., 2011; Khor et al., 2013), and LD accumulation was found in olfactory bulb and vestibular nucleus of mice with dysfunctional mitochondria (Liu et al., 2015). Notably, apolipoprotein E-ε4 (APOE4), the strongest genetic risk factor for AD (Corder et al., 1993), reduces neuron-to-astrocyte transfer of fatty acids, which could be the underlying mechanism of the lipid dyshomeostasis seen in the disease (Liu et al., 2017). Additionally, LDs were also found to buildup in microglia of aged mouse or human brains, which then trigger ROS production and secretion of pro-inflammatory cytokines, and contribute to neurodegeneration (Marschallinger et al., 2019).
Brain contains ~20% cholesterol of the body (Zhang and Liu, 2015), which is essential for membrane fluidity, vesicle formation, and synaptic transmission (Björkhem and Meaney, 2004). Brain synthesizes its own cholesterol due to the block of cholesterol-carrying lipoproteins across the BBB. Since neurons acquire extra cholesterol for synaptogenesis from astrocytes (Mauch, 2001), and brain cholesterol needs to be hydroxylated to 24-hydroxycholesterol in neurons before excretion (Bjorkhem et al., 1998; Meaney et al., 2002), cholesterol transport from astrocytes to neurons is vital for synaptic function and sterol homeostasis in the brain. Upon co-culture, astrocytes stimulate neuronal neurite outgrowth, which is impaired when astrocytes without SREBP2 (a cholesterol synthesis regulatory protein) are used. In vivo, SREBP2 knockout in astrocytes alters brain development and impairs behavioral and motor functions in mice (Ferris et al., 2017), suggesting astrocyte-to-neuron cholesterol trafficking is key for brain development and synaptic function. APOE is the primary apolipoprotein that transports cholesterol in the brain. APOE lipoproteins carrying cholesterol and phospholipids are effluxed of astrocytes and uptaken into neurons by binding to lipoprotein receptors (Pfrieger and Ungerer, 2011; Figure 2). APOE-deficient mice exhibit markedly reduced cholesterol levels in hippocampus, and AD-like behavioral and synaptic impairments (Champagne et al., 2002; Levi et al., 2005). Astrocytes and neurons expressing APOE4 have reduced capacity in secreting or binding to cholesterol and phospholipids (Mahley, 2016). LDL receptor and LRP1 from LDL receptor family are main receptors to uptake APOE-containing lipoprotein particles to neurons. LRP1 knockout in forebrain neurons disrupts lipid metabolism, and leads to neuroinflammation and synapse loss (Liu et al., 2010). During aging, mRNA levels of the rate-limiting enzyme of cholesterol synthesis HMGCR decrease in astrocytes (Boisvert et al., 2018), which could contribute to dendrite atrophy and synaptic dysfunction seen in the aging brain. A study in AD-patient iPSC-derived neurons suggests that neuron-specific activation of cholesterol degradation could be a potential therapeutic target to alleviate Aβ and Tau pathology in AD (Van Der Kant et al., 2019).
Other than the coupling of glucose and lipid metabolism, additional metabolic interactions between neurons and astrocytes are essential for brain functions. Neuronal levels of reduced glutathione (GSH), the most abundant redox modulator eliminating ROS in the brain, are found to be synthesized using precursors amino acids, including glycine and cysteine, from astrocytes. GSH released from astrocytes is cleaved by γ-glutamyl transpeptidase on astrocyte surface to glycine and cysteine before being uptaken by neurons for GSH synthesis (Dringen, 2000). Inhibition of GSH biosynthesis in astrocytes triggers neuronal toxicity, and depletion of GSH during aging leads to microglia activation and increased neuronal susceptibility to cell death (Hirrlinger et al., 2002; Lee et al., 2010).
Intra-cellularly, damaged mitochondria are removed via mitophagy. Inter-cellularly, neurons release damaged axonal mitochondria to adjacent astrocytes for autophagic degradation (Chung-Ha et al., 2014). Conversely, astrocytes can transfer healthy mitochondria to adjoining neurons for ATP production and recovery from stroke (Hayakawa et al., 2016; Figure 2). When extracellular mitochondria are removed from astrocyte-conditional media, their neuroprotective effect is abolished (Hayakawa et al., 2016). At the molecular level, astrocyte-generated ATP, together with its degradation product adenosine, is involved in supporting synaptic transmission and neuronal excitability (Chen et al., 2013; Tan et al., 2017).
The high energy demand of the brain renders it sensitive to changes in bioenergetic capacity. Disruptions to substrate availability and/or mitochondrial function are well-known hallmarks of brain aging and a variety of age-associated neurodegenerative disorders. Distinguishing cell type-specific bioenergetic contributions to brain aging is particularly of significance because of previous findings linking aging with diverse metabolic changes between different brain cells. Elucidating the cellular specificity and inter-cellular coupling within the brain bioenergetic system is also critical to understand the mechanisms underlying selective cellular vulnerability in neurodegeneration. Mitochondrial enhancers targeting all brain cells, such as creatine, coenzyme-Q, and Mito-Q, have thus far exhibited limited efficacy for neurodegenerative diseases (Moreira et al., 2010; Chaturvedi and Flint Beal, 2013; Murphy and Hartley, 2018). While such a lack of success may be ascribed to multiple reasons including window of intervention and translatability of existing animal models (Murphy and Hartley, 2018), for neurodegenerative diseases that typically harbor complex and dynamic etiologies, precision strategies targeting specific cells or inter-cellular metabolic interactions may be needed at different disease stages. For stages when reactive astrocytes and microglia are playing a central role, interventions that facilitate their metabolic reprogramming could be more effective than direct OXPHOS enhancers (Baik et al., 2019; Polyzos et al., 2019). Recent methodological advances have started to enable targeted delivery of therapeutics to different brain cells via nanoparticles or viral carriers (Zhang et al., 2016; Sharma et al., 2018). Furthermore, emerging intra- and inter-cellular strategies targeting non-energetic facets of the mitochondria have manifested encouraging efficacy (Fang et al., 2019; Joshi et al., 2019), which suggest an necessity of combinational therapies for these currently incurable diseases.
All authors contributed to the discussion of content, generation of figures, and writing, review, editing of the manuscript.
This work was supported by the University of Arizona Center for Innovation in Brain Science Startup fund to FY, National Institute on Aging P01AG026572 to Roberta Diaz Brinton (Analytic Core to FY), and Arizona Alzheimer’s Consortium Pilot Project grant to FY.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Agarwal, A. R., Yin, F., and Cadenas, E. (2013). Metabolic shift in lung alveolar cell mitochondria following acrolein exposure. Am. J. Phys. Lung Cell. Mol. Phys. 305, L764–L773. doi: 10.1152/ajplung.00165.2013
Aldana, B. I. (2019). Microglia specific metabolic changes in neurodegeneration. J. Mol. Biol. 431, 1830–1842. doi: 10.1016/j.jmb.2019.03.006
Baik, S. H., Kang, S., Lee, W., Choi, H., Chung, S., Kim, J. I., et al. (2019). A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 30, 493–507.e6. doi: 10.1016/j.cmet.2019.06.005
Bailey, A. P., Koster, G., Guillermier, C., Hirst, E. M., Macrae, J. I., Lechene, C. P., et al. (2015). Antioxidant role for lipid droplets in a stem cell niche of drosophila. Cell 163, 340–353. doi: 10.1016/j.cell.2015.09.020
Baxter, P. S., and Hardingham, G. E. (2016). Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 100, 147–152. doi: 10.1016/j.freeradbiomed.2016.06.027
Bélanger, M., Allaman, I., and Magistretti, P. J. (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738. doi: 10.1016/j.cmet.2011.08.016
Bjorkhem, I., Lutjohann, D., Diczfalusy, U., Stahle, L., Ahlborg, G., and Wahren, J. (1998). Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 39, 1594–1600.
Björkhem, I., and Meaney, S. (2004). Brain cholesterol: long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 24, 806–815. doi: 10.1161/01.ATV.0000120374.59826.1b
Boison, D., and Steinhäuser, C. (2018). Epilepsy and astrocyte energy metabolism. Glia 66, 1235–1243. doi: 10.1002/glia.23247
Boisvert, M. M., Erikson, G. A., Shokhirev, M. N., and Allen, N. J. (2018). The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 22, 269–285. doi: 10.1016/j.celrep.2017.12.039
Bolaños, J. P. (2016). Bioenergetics and redox adaptations of astrocytes to neuronal activity. J. Neurochem. 139, 115–125. doi: 10.1111/jnc.13486
Boumezbeur, F., Mason, G. F., De Graaf, R. A., Behar, K. L., Cline, G. W., Shulman, G. I., et al. (2010). Altered brain mitochondrial metabolism in healthy aging as assessed by in vivo magnetic resonance spectroscopy. J. Cereb. Blood Flow Metab. 30, 211–221. doi: 10.1038/jcbfm.2009.197
Bouzier-Sore, A.-K., and Pellerin, L. (2013). Unraveling the complex metabolic nature of astrocytes. Front. Cell. Neurosci. 7:179. doi: 10.3389/fncel.2013.00179
Brooks, G. A., Dubouchaud, H., Brown, M., Sicurello, J. P., and Butz, C. E. (1999). Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc. Natl. Acad. Sci. USA 96, 1129–1134. doi: 10.1073/pnas.96.3.1129
Bruce, K. D., Zsombok, A., and Eckel, R. H. (2017). Lipid processing in the brain: a key regulator of systemic metabolism. Front. Endocrinol. 8:60. doi: 10.3389/fendo.2017.00060
Cadenas, E., and Davies, K. J. A. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222–230. doi: 10.1016/S0891-5849(00)00317-8
Camandola, S., and Mattson, M. P. (2017). Brain metabolism in health, aging, and neurodegeneration. EMBO J. 36, 1474–1492. doi: 10.15252/embj.201695810
Champagne, D., Dupuy, J.-B., Rochford, J., and Poirier, J. (2002). Apolipoprotein E knockout mice display procedural deficits in the Morris water maze: analysis of learning strategies in three versions of the task. Neuroscience 114, 641–654. doi: 10.1016/S0306-4522(02)00313-5
Chaturvedi, R. K., and Flint Beal, M. (2013). Mitochondrial diseases of the brain. Free Radic. Biol. Med. 63, 1–29. doi: 10.1016/j.freeradbiomed.2013.03.018
Chen, J., Tan, Z., Zeng, L., Zhang, X., He, Y., Gao, W., et al. (2013). Heterosynaptic long-term depression mediated by ATP released from astrocytes. Glia 61, 178–191. doi: 10.1002/glia.22425
Chung-Ha, O. D., Kim, K.-Y., Bushong, E. A., Mills, E. A., Boassa, D., Shih, T., et al. (2014). Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 111, 9633–9638. doi: 10.1073/pnas.1404651111
Chuquet, J., Quilichini, P., Nimchinsky, E. A., and Buzsáki, G. (2010). Predominant enhancement of glucose uptake in astrocytes versus neurons during activation of the somatosensory cortex. J. Neurosci. 30, 15298–15303. doi: 10.1523/JNEUROSCI.0762-10.2010
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Cunnane, S., Nugent, S., Roy, M., Courchesne-Loyer, A., Croteau, E., Tremblay, S., et al. (2011). Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 27, 3–20. doi: 10.1016/j.nut.2010.07.021
Díaz-García, C. M., Mongeon, R., Lahmann, C., Koveal, D., Zucker, H., and Yellen, G. (2017). Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab. 26, 361–374.e4. doi: 10.1016/j.cmet.2017.06.021
Ding, F., Yao, J., Rettberg, J. R., Chen, S., and Brinton, R. D. (2013). Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One 8:e79977. doi: 10.1371/journal.pone.0079977
Drenick, E. J., Alvarez, L. C., Tamasi, G. C., and Brickman, A. S. (1972). Resistance to symptomatic insulin reactions after fasting. J. Clin. Invest. 51, 2757–2762. doi: 10.1172/JCI107095
Dringen, R. (2000). Metabolism and functions of glutathione in brain. Prog. Neurobiol. 62, 649–671. doi: 10.1016/S0301-0082(99)00060-X
Drulis-Fajdasz, D., Gizak, A., Wojtowicz, T., Wisniewski, J. R., and Rakus, D. (2018). Aging-associated changes in hippocampal glycogen metabolism in mice. Evidence for and against astrocyte-to-neuron lactate shuttle. Glia 66, 1481–1495. doi: 10.1002/glia.23319
Fabre, C., Chamari, K., Mucci, P., Masse-Biron, J., and Prefaut, C. (2002). Improvement of cognitive function by mental and/or individualized aerobic training in healthy elderly subjects. Int. J. Sports Med. 23, 415–421. doi: 10.1055/s-2002-33735
Falk, R. E., Cederbaum, S. D., Blass, J. P., Gibson, G. E., Kark, R. P., and Carrel, R. E. (1976). Ketonic diet in the management of pyruvate dehydrogenase deficiency. Pediatrics 58, 713–721.
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Ferraiuolo, L., Higginbottom, A., Heath, P. R., Barber, S., Greenald, D., Kirby, J., et al. (2011). Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134, 2627–2641. doi: 10.1093/brain/awr193
Ferris, H. A., Perry, R. J., Moreira, G. V., Shulman, G. I., Horton, J. D., and Kahn, C. R. (2017). Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc. Natl. Acad. Sci. U. S. A. 114, 1189–1194. doi: 10.1073/pnas.1620506114
Ghosh, S., Castillo, E., Frias, E. S., and Swanson, R. A. (2018). Bioenergetic regulation of microglia. Glia 66, 1200–1212. doi: 10.1002/glia.23271
Gimeno-Bayón, J., López-López, A., Rodríguez, M., and Mahy, N. (2014). Glucose pathways adaptation supports acquisition of activated microglia phenotype. J. Neurosci. Res. 92, 723–731. doi: 10.1002/jnr.23356
Goyal, M. S., Vlassenko, A. G., Blazey, T. M., Su, Y., Couture, L. E., Durbin, T. J., et al. (2017). Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 26, 353–360.e353. doi: 10.1016/j.cmet.2017.07.010
Greenberg, A. S., Coleman, R. A., Kraemer, F. B., Mcmanaman, J. L., Obin, M. S., Puri, V., et al. (2011). The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Invest. 121, 2102–2110. doi: 10.1172/JCI46069
Gupta, K., Patani, R., Baxter, P., Serio, A., Story, D., Tsujita, T., et al. (2012). Human embryonic stem cell derived astrocytes mediate non-cell-autonomous neuroprotection through endogenous and drug-induced mechanisms. Cell Death Differ. 19, 779–787. doi: 10.1038/cdd.2011.154
Halestrap, A. P. (2011). Monocarboxylic acid transport. Compr. Physiol. 3, 1611–1643. doi: 10.1002/cphy.c130008
Hayakawa, K., Esposito, E., Wang, X., Terasaki, Y., Liu, Y., Xing, C., et al. (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. doi: 10.1038/nature18928
Hirrlinger, J., Schulz, J. B., and Dringen, R. (2002). Glutathione release from cultured brain cells: multidrug resistance protein 1 mediates the release of GSH from rat astroglial cells. J. Neurosci. Res. 69, 318–326. doi: 10.1002/jnr.10308
Hwang, O. (2013). Role of oxidative stress in Parkinson’s disease. Exp. Neurobiol. 22, 11–17. doi: 10.5607/en.2013.22.1.11
Ioannou, M. S., Jackson, J., Sheu, S. H., Chang, C. L., Weigel, A. V., Liu, H., et al. (2019). Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell 177, 1522–1535.e14. doi: 10.1016/j.cell.2019.04.001
Jiang, T., and Cadenas, E. (2014). Astrocytic metabolic and inflammatory changes as a function of age. Aging Cell 13, 1059–1067. doi: 10.1111/acel.12268
Joshi, A. U., Minhas, P. S., Liddelow, S. A., Haileselassie, B., Andreasson, K. I., Dorn, G. W. 2nd., et al. (2019). Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 22, 1635–1648. doi: 10.1038/s41593-019-0486-0
Khor, V. K., Shen, W.-J., and Kraemer, F. B. (2013). Lipid droplet metabolism. Curr. Opin. Clin. Nutr. Metab. Care 16, 632–637. doi: 10.1097/MCO.0b013e3283651106
Klepper, J. (2004). Impaired glucose transport into the brain: the expanding spectrum of glucose transporter type 1 deficiency syndrome. Curr. Opin. Neurol. 17, 193–196. doi: 10.1097/00019052-200404000-00018
Klosinski, L. P., Yao, J., Yin, F., Fonteh, A. N., Harrington, M. G., Christensen, T. A., et al. (2015). White matter lipids as a Ketogenic fuel supply in aging female brain: implications for Alzheimer’s disease. EBioMed. 2, 1888–1904. doi: 10.1016/j.ebiom.2015.11.002
Krikorian, R., Shidler, M. D., Dangelo, K., Couch, S. C., Benoit, S. C., and Clegg, D. J. (2012). Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 33, 425.e419–425.e427. doi: 10.1016/j.neurobiolaging.2010.10.006
Lake, B. B., Ai, R., Kaeser, G. E., Salathia, N. S., Yung, Y. C., Liu, R., et al. (2016). Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590. doi: 10.1126/science.aaf1204
Lane, N. (2014). Bioenergetic constraints on the evolution of complex life. Cold Spring Harb. Perspect. Biol. 6:a015982. doi: 10.1101/cshperspect.a015982
Lane, N., and Martin, W. (2010). The energetics of genome complexity. Nature 467, 929–934. doi: 10.1038/nature09486
Lee, M., Cho, T., Jantaratnotai, N., Wang, Y. T., Mcgeer, E., and Mcgeer, P. L. (2010). Depletion of GSH in glial cells induces neurotoxicity: relevance to aging and degenerative neurological diseases. FASEB J. 24, 2533–2545. doi: 10.1096/fj.09-149997
Levi, O., Lütjohann, D., Devir, A., Von Bergmann, K., Hartmann, T., and Michaelson, D. M. (2005). Regulation of hippocampal cholesterol metabolism by apoE and environmental stimulation. J. Neurochem. 95, 987–997. doi: 10.1111/j.1471-4159.2005.03441.x
Lin, M., and Beal, M. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Listenberger, L. L., Han, X., Lewis, S. E., Cases, S., Farese, R. V., Ory, D. S., et al. (2003). Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. U. S. A. 100, 3077–3082. doi: 10.1073/pnas.0630588100
Liu, L., Mackenzie, K. R., Putluri, N., Maletić-Savatić, M., and Bellen, H. J. (2017). The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab. 26, 719–737.e6. doi: 10.1016/j.cmet.2017.08.024
Liu, Q., Trotter, J., Zhang, J., Peters, M. M., Cheng, H., Bao, J., et al. (2010). Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J. Neurosci. 30, 17068–17078. doi: 10.1523/JNEUROSCI.4067-10.2010
Liu, L., Zhang, K., Sandoval, H., Yamamoto, S., Jaiswal, M., Sanz, E., et al. (2015). Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160, 177–190. doi: 10.1016/j.cell.2014.12.019
Lundgaard, I., Li, B., Xie, L., Kang, H., Sanggaard, S., Haswell, J. D., et al. (2015). Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat. Commun. 6:6807. doi: 10.1038/ncomms7807
Mächler, P., Wyss, M. T., Elsayed, M., Stobart, J., Gutierrez, R., Von Faber-Castell, A., et al. (2016). In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 23, 94–102. doi: 10.1016/j.cmet.2015.10.010
Magistretti, P. J., and Allaman, I. (2013). “Brain energy metabolism” in Neuroscience in the 21st century: From basic to clinical. ed. D. W. Pfaff (New York, NY: Springer), 1591–1620.
Magistretti, P. J., and Allaman, I. (2015). A cellular perspective on brain energy metabolism and functional imaging. Neuron 86, 883–901. doi: 10.1016/j.neuron.2015.03.035
Mahley, R. W. (2016). Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arterioscler. Thromb. Vasc. Biol. 36, 1305–1315. doi: 10.1161/ATVBAHA.116.307023
Markesbery, W. (1997). Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 23, 134–147. doi: 10.1016/S0891-5849(96)00629-6
Marschallinger, J., Iram, T., Zardeneta, M., Lee, S., Lehallier, B., Haney, M., et al. (2019). Lipid droplet accumulating microglia represent a dysfunctional and pro-inflammatory state in the aging brain. bioRxiv [Preprint]. doi: 10.1101/722827
Martinez-Outschoorn, U. E., Lin, Z., Whitaker-Menezes, D., Howell, A., Sotgia, F., and Lisanti, M. P. (2012). Ketone body utilization drives tumor growth and metastasis. Cell Cycle 11, 3964–3971. doi: 10.4161/cc.22137
Mathys, H., Davila-Velderrain, J., Peng, Z., Gao, F., Mohammadi, S., Young, J. Z., et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. doi: 10.1038/s41586-019-1195-2
Mattson, M. P., Moehl, K., Ghena, N., Schmaedick, M., and Cheng, A. (2018). Intermittent metabolic switching, neuroplasticity and brain health. Nat. Rev. Neurosci. 19, 81–94. doi: 10.1038/nrn.2017.156
Mauch, D. H. (2001). CNS synaptogenesis promoted by glia-derived cholesterol. Science 294, 1354–1357. doi: 10.1126/science.294.5545.1354
McNair, L. F., Kornfelt, R., Walls, A. B., Andersen, J. V., Aldana, B. I., Nissen, J. D., et al. (2017). Metabolic characterization of acutely isolated hippocampal and cerebral cortical slices using [U-13 C] glucose and [1, 2-13 C] acetate as substrates. Neurochem. Res. 42, 810–826. doi: 10.1007/s11064-016-2116-5
Meaney, S., Bodin, K., Diczfalusy, U., and Bjorkhem, I. (2002). On the rate of translocation in vitro and kinetics in vivo of the major oxysterols in human circulation: critical importance of the position of the oxygen function. J. Lipid Res. 43, 2130–2135. doi: 10.1194/jlr.M200293-JLR200
Moreira, P. I., Zhu, X., Wang, X., Lee, H. G., Nunomura, A., Petersen, R. B., et al. (2010). Mitochondria: a therapeutic target in neurodegeneration. Biochim. Biophys. Acta 1802, 212–220. doi: 10.1016/j.bbadis.2009.10.007
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Murphy, M. P., and Hartley, R. C. (2018). Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 17, 865–886. doi: 10.1038/nrd.2018.174
Newman, J. C., Covarrubias, A. J., Zhao, M., Yu, X., Gut, P., Ng, C.-P., et al. (2017). Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 26, 547–557.e8. doi: 10.1016/j.cmet.2017.08.004
Nichols, N. R., Day, J. R., Laping, N. J., Johnson, S. A., and Finch, C. E. (1993). GFAP mRNA increases with age in rat and human brain. Neurobiol. Aging 14, 421–429. doi: 10.1016/0197-4580(93)90100-P
Ohman, A., and Forsgren, L. (2015). NMR metabonomics of cerebrospinal fluid distinguishes between Parkinson’s disease and controls. Neurosci. Lett. 594, 36–39. doi: 10.1016/j.neulet.2015.03.051
Orihuela, R., Mcpherson, C. A., and Harry, G. J. (2016). Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 173, 649–665. doi: 10.1111/bph.13139
Ott, A., Stolk, R., Van Harskamp, F., Pols, H., Hofman, A., and Breteler, M. (1999). Diabetes mellitus and the risk of dementia: the Rotterdam study. Neurology 53, 1937–1937. doi: 10.1212/WNL.53.9.1937
Pellerin, L., and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 91, 10625–10629. doi: 10.1073/pnas.91.22.10625
Perry, V. H., Nicoll, J. A., and Holmes, C. (2010). Microglia in neurodegenerative disease. Nat. Rev. Neurol. 6, 193–201. doi: 10.1038/nrneurol.2010.17
Pfrieger, F. W., and Ungerer, N. (2011). Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 50, 357–371. doi: 10.1016/j.plipres.2011.06.002
Polyzos, A. A., Lee, D. Y., Datta, R., Hauser, M., Budworth, H., Holt, A., et al. (2019). Metabolic reprogramming in astrocytes distinguishes region-specific neuronal susceptibility in Huntington mice. Cell Metab. 29, 1258–1273.e11. doi: 10.1016/j.cmet.2019.03.004
Puchalska, P., and Crawford, P. A. (2017). Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 25, 262–284. doi: 10.1016/j.cmet.2016.12.022
Rama Rao, K. V., and Kielian, T. (2015). Neuron-astrocyte interactions in neurodegenerative diseases: role of neuroinflammation. Clin. Exp. Neuroimmunol. 6, 245–263. doi: 10.1111/cen3.12237
Rambold, A. S., Cohen, S., and Lippincott-Schwartz, J. (2015). Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692. doi: 10.1016/j.devcel.2015.01.029
Rodríguez-Arellano, J., Parpura, V., Zorec, R., and Verkhratsky, A. (2016). Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 323, 170–182. doi: 10.1016/j.neuroscience.2015.01.007
Saunders, A., Macosko, E. Z., Wysoker, A., Goldman, M., Krienen, F. M., De Rivera, H., et al. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16. doi: 10.1016/j.cell.2018.07.028
Schönfeld, P., and Reiser, G. (2013). Why does brain metabolism not favor burning of fatty acids to provide energy?-reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 33, 1493–1499. doi: 10.1038/jcbfm.2013.128
Schonfeld, P., and Reiser, G. (2017). Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem. Int. 109, 68–77. doi: 10.1016/j.neuint.2017.03.018
Schousboe, A., Scafidi, S., Bak, L. K., Waagepetersen, H. S., and Mckenna, M. C. (2014). “Glutamate metabolism in the brain focusing on astrocytes” in Glutamate and ATP at the interface of metabolism and signaling in the brain. eds. V. Parpura, A. Schousboe, and A. Verkhratsky (Switzerland: Springer), 13–30.
Sharma, A., Liaw, K., Sharma, R., Zhang, Z., Kannan, S., and Kannan, R. M. (2018). Targeting mitochondrial dysfunction and oxidative stress in activated microglia using dendrimer-based therapeutics. Theranostics 8, 5529–5547. doi: 10.7150/thno.29039
Steele, M. L., and Robinson, S. R. (2012). Reactive astrocytes give neurons less support: implications for Alzheimer’s disease. Neurobiol. Aging 33, 423.e1–423.e13. doi: 10.1016/j.neurobiolaging.2010.09.018
Suzuki, A., Stern, S. A., Bozdagi, O., Huntley, G. W., Walker, R. H., Magistretti, P. J., et al. (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. doi: 10.1016/j.cell.2011.02.018
Tan, Z., Liu, Y., Xi, W., Lou, H.-F., Zhu, L., Guo, Z., et al. (2017). Glia-derived ATP inversely regulates excitability of pyramidal and CCK-positive neurons. Nat. Commun. 8:13772. doi: 10.1038/ncomms13772
Tichauer, J. E., Flores, B., Soler, B., Eugenín-Von Bernhardi, L., Ramírez, G., and Von Bernhardi, R. (2014). Age-dependent changes on TGFβ1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 37, 187–196. doi: 10.1016/j.bbi.2013.12.018
Van Der Kant, R., Langness, V. F., Herrera, C. M., Williams, D. A., Fong, L. K., Leestemaker, Y., et al. (2019). Cholesterol metabolism is a druggable axis that independently regulates tau and amyloid-β in iPSC-derived Alzheimer’s disease neurons. Cell Stem Cell 24, 363–375.e9. doi: 10.1016/j.stem.2018.12.013
Van Der Poel, M., Ulas, T., Mizee, M. R., Hsiao, C. C., Miedema, S. S. M., Adelia,, et al. (2019). Transcriptional profiling of human microglia reveals grey-white matter heterogeneity and multiple sclerosis-associated changes. Nat. Commun. 10:1139. doi: 10.1038/s41467-019-08976-7
Viader, A., Blankman, J. L., Zhong, P., Liu, X., Schlosburg, J. E., Joslyn, C. M., et al. (2015). Metabolic interplay between astrocytes and neurons regulates endocannabinoid action. Cell Rep. 12, 798–808. doi: 10.1016/j.celrep.2015.06.075
Volkenhoff, A., Weiler, A., Letzel, M., Stehling, M., Klämbt, C., and Schirmeier, S. (2015). Glial glycolysis is essential for neuronal survival in drosophila. Cell Metab. 22, 437–447. doi: 10.1016/j.cmet.2015.07.006
Voloboueva, L. A., Emery, J. F., Sun, X., and Giffard, R. G. (2013). Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett. 587, 756–762. doi: 10.1016/j.febslet.2013.01.067
Wang, D., Pascual, J. M., Yang, H., Engelstad, K., Jhung, S., Sun, R. P., et al. (2005). Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 57, 111–118. doi: 10.1002/ana.20331
Wu, Y., Zhang, A.-Q., and Yew, D. T. (2005). Age related changes of various markers of astrocytes in senescence-accelerated mice hippocampus. Neurochem. Int. 46, 565–574. doi: 10.1016/j.neuint.2005.01.002
Yan, L.-J., Xiao, M., Chen, R., and Cai, Z. (2013). Metabolic dysfunction of astrocyte: an initiating factor in beta-amyloid pathology? Aging Neurodegener. 1, 7–14.
Yin, F., Boveris, A., and Cadenas, E. (2014). Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. 20, 353–371. doi: 10.1089/ars.2012.4774
Yin, F., Sancheti, H., and Cadenas, E. (2012a). Mitochondrial thiols in the regulation of cell death pathways. Antioxid. Redox Signal. 17, 1714–1727. doi: 10.1089/ars.2012.4639
Yin, F., Sancheti, H., and Cadenas, E. (2012b). Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim. Biophys. Acta 1817, 401–409. doi: 10.1016/j.bbabio.2011.12.004
Zeisel, A., Muñoz-Manchado, A. B., Codeluppi, S., Lönnerberg, P., La Manno, G., Juréus, A., et al. (2015). Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. doi: 10.1126/science.aaa1934
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O’keeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014
Zhang, F., Lin, Y.-A., Kannan, S., and Kannan, R. M. (2016). Targeting specific cells in the brain with nanomedicines for CNS therapies. J. Control. Release 240, 212–226. doi: 10.1016/j.jconrel.2015.12.013
Zhang, J., and Liu, Q. (2015). Cholesterol metabolism and homeostasis in the brain. Protein Cell 6, 254–264. doi: 10.1007/s13238-014-0131-3
Keywords: mitochondria, neuron, astrocyte, microglia, metabolic shift, metabolic coupling, brain aging, neurodegenerative diseases
Citation: Qi G, Mi Y and Yin F (2020) Cellular Specificity and Inter-cellular Coordination in the Brain Bioenergetic System: Implications for Aging and Neurodegeneration. Front. Physiol. 10:1531. doi: 10.3389/fphys.2019.01531
Edited by:
Katia Aquilano, University of Rome Tor Vergata, ItalyReviewed by:
Elia Ranzato, University of Eastern Piedmont, ItalyCopyright © 2020 Qi, Mi and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Yin, ZmVpeWluQGVtYWlsLmFyaXpvbmEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.