
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 29 November 2019
Sec. Respiratory Physiology and Pathophysiology
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.01453
This article is part of the Research TopicTime Domains of Hypoxia Adaptation: Evolutionary Insights and ApplicationsView all 24 articles
 Kristiana Rood1
Kristiana Rood1 Vanessa Lopez1
Vanessa Lopez1 Michael R. La Frano2,3
Michael R. La Frano2,3 Oliver Fiehn4,5
Oliver Fiehn4,5 Lubo Zhang1
Lubo Zhang1 Arlin B. Blood1
Arlin B. Blood1 Sean M. Wilson1*
Sean M. Wilson1*Gestational hypoxia is a risk factor in the development of pulmonary hypertension in the newborn and other sequela, however, the mechanisms associated with the disease remain poorly understood. This review highlights disruption of metabolism by antenatal high altitude hypoxia and the impact this has on pulmonary hypertension in the newborn with discussion of model organisms and human populations. There is particular emphasis on modifications in glucose and lipid metabolism along with alterations in mitochondrial function. Additional focus is placed on increases in oxidative stress and the progression of pulmonary vascular disease in the newborn and on the need for further exploration using a combination of contemporary and classical approaches.
A mother’s womb provides a nurturing environment for her unborn child, helping to maintain physiological homeostasis when presented with various prenatal stressors. Such maternal compensation allows the fetus to develop fully and thrive under less than ideal conditions. Exposure to excessive gestational hypoxia or other intrauterine stress, however, may cause fetal abnormalities or death. Stress-related physiological aberrations that begin in utero can also cause fetal organ systems to become compromised or fail even before birth. Intrauterine stress can result in a myriad of newborn morbidities and also program the infant to have diseases later in life (Longo and Pearce, 2005; Pearce, 2014; Ducsay et al., 2018). This review is centered on the disruption of metabolism by antenatal high altitude hypoxia and the impact this has on pulmonary hypertension in the newborn with discussion of issues that arise in human populations and the use of model organisms.
Antenatal hypoxic exposure places a significant stress on the fetus, which can cause growth restriction that is dependent on the degree of exposure. Stress-related fetal growth restriction increases the risk of infant morbidity and mortality as well as enhances the possibility of developing diseases that occur later in life. Young children with growth restriction can have lower cognitive scores and worse academic performance compared with similar preterm infants who do not suffer growth restriction or a failure to thrive (Cole and Lanham, 2011; Homan, 2016). Complications and morbidities associated with stress-related developmental abnormalities and growth restriction are costly and present significant financial burdens on families of sick infants as well as our public health system. Gestational hypoxia can be a consequence of placental insufficiency, placental infarcts, high altitude residence, maternal smoking, congestive heart failure, heart valvar diseases, pulmonary diseases, acute/chronic respiratory tract infections, anemia, preeclampsia, and other conditions.
Worldwide many women live or sojourn to high altitude during pregnancy, which causes fetal hypoxia and places the fetus at risk of developing disease. Studies on humans have unmasked critical phenotypic changes associated with gestational hypoxia in native and non-native populations (Niermeyer et al., 1995; Weissmann et al., 2003; Scherrer et al., 2010), including congenital heart defects in Tibetan children with a prevalence that stratifies based on their altitude of residence (Chun et al., 2019). Others have begun to define a number of genetic modifications in native populations (Simonson et al., 2010; Eichstaedt et al., 2014; Nanduri et al., 2017; Gnecchi-Ruscone et al., 2018). Even still, animal models are vitally important to our understanding of the mechanistic underpinnings of how long term exposure to high altitude leads to disease. When using animal models the oxygen tensions can be titrated to induce stress on the mother and unborn infant that ranges from being relatively mild to extreme. The magnitude of stress can be adjusted because it is based on the altitude and duration of exposure as opposed to other multifactorial stress models in animals, such as uterine artery ligation and consequent placental insufficiency.
We have used a gestational long-term high altitude sheep (LTH) model for over 20 years to understand the relevant mechanisms that underlie functional and structural adaptations in the lung and other organ systems (Ducsay et al., 2018). Pregnant sheep are placed at the White Mountain Research Station at 3,801 m for the latter majority of pregnancy. This altitude is significant as it is similar to that of Lhasa Tibet and La Paz Bolivia, which are home to millions of people. Being at this altitude results in an inspired PO2 of approximately 90 Torr for pregnant mothers and animals, which is a ∼35% reduction from sea level. In turn, in the pregnant ewe the fetal arterial PO2 decreases by roughly 20% (Ducsay et al., 2018). Studying the effects of this antenatal hypoxic exposure in fetal and newborn sheep has enabled us to better understand the etiology of hypoxia-related dysregulation in fetal, newborn and adult physiology. Our group chose sheep for exploration of prenatal programing of disease because fetal sheep have a similar developmental profile, are of similar size to human infants, and because we have the ability to perform invasive studies. Findings from our group illustrate that LTH leads to many respiratory, cardiovascular, endocrine, adipocyte, and neural impairments in fetal and newborn lambs and can disrupt normal physiological function in pregnant and non-pregnant ewes (Lewis et al., 1999; Garcia et al., 2000; Arakawa et al., 2004; Longo and Pearce, 2005; Ducsay et al., 2007, 2018; Gao et al., 2007; Xue et al., 2008; Hubbell et al., 2012; Adeoye et al., 2014, 2015; Myers et al., 2015; Newby et al., 2015; Blum-Johnston et al., 2016).
The lung is particularly vulnerable to hypoxemic damage during the prenatal and neonatal periods in various species including humans, in part because the organ experiences marked developmental plasticity both before and after birth (Papamatheakis et al., 2013). Long term prenatal high altitude stress in particular places infants at risk of developing pulmonary hypertension (Niermeyer et al., 1995; Weissmann et al., 2003; Scherrer et al., 2010), high altitude pulmonary edema (Niermeyer et al., 2009), and idiopathic pulmonary hypertension later in life (Grunig et al., 2005). Human infants exposed to gestational hypoxia adapt poorly to breathing air because the low oxygen exposure impairs lung development. Lungs of these infants manifest with structural and functional defects that program them to be susceptible to disease throughout life (Goldberg et al., 1971).
We and others have found that hypoxia-induced pulmonary vascular disease, which occurs in human infants, can be recapitulated in fetal and newborn sheep from pregnant ewes living at high altitude. Fetal sheep that gestate at 3,801 m have thickened resistive pulmonary arteries, similar to afflicted human newborns (Bixby et al., 2007; Xue et al., 2008; Sheng et al., 2009), effects that persist in newborn lambs that remain at high altitude (Herrera et al., 2007, 2010). Fetuses and lambs born at high altitude have other abnormalities that parallel infants with hypoxia-induced pulmonary hypertension (Berger and Konduri, 2006; Abman, 2007; Konduri and Kim, 2009), including elevated pulmonary pressures, exacerbated hypoxic-induced pulmonary vasoconstriction (Herrera et al., 2007, 2010; Blood et al., 2013), impaired vasodilation (Blum-Johnston et al., 2016), arterial remodeling (Bixby et al., 2007), and right ventricular hypertrophy (Herrera et al., 2007). Studies further demonstrate that Ca2+ signals are disrupted in endothelium and smooth muscle and this involves modifications in the expression and function of multiple receptor signaling systems and ion channels (Herrera et al., 2007, 2010; Goyal et al., 2011; Hadley et al., 2012; Blood et al., 2013; Blum-Johnston et al., 2016; Shen et al., 2018).
High altitude-induced changes in the structure and function of the cardio-respiratory system that result in pulmonary hypertension in humans and animal models may be related to hypoxia-mediated changes in cellular metabolism, oxidative stress, and inflammatory processes, elements that have not been thoroughly examined in humans or animal models. The influence of high altitude gestation on metabolism is important to consider as the ensuing chronic hypoxia reduces oxygen uptake and delivery to immature fetal tissues (Cosse and Michiels, 2008). Varying degrees and durations of high altitude exposure are also likely to cause differential impact on metabolic adaptations in the fetus. Long-term multigenerational living or relocation to high altitude can lead to the selection of genetic and epigenetic traits (Simonson et al., 2010; Eichstaedt et al., 2014; Nanduri et al., 2017; Gnecchi-Ruscone et al., 2018). The phenotypic and genetic changes can be unique to the human populations examined, as illustrated by studies on Andean and Tibetan populations (Simonson et al., 2010; Eichstaedt et al., 2014; Heinrich et al., 2019). Native Tibetans have modifications to the EGLN1 and EPAS genes that result in modifications to hypoxia inducible factors (HIF1α) and HIF2α, respectably that prevent these transcription factors from being activated normally. Given the role of these genes in activating erythropoietin and erythrogenesis these modifications have caused Tibetans to adapt such that high altitude does not increase hematocrit levels (Simonson et al., 2010; Tashi et al., 2017; Heinrich et al., 2019). This differs from Andean populations who can have extremely high hematocrit values, and who do not have the changes in EGLN1 that restrict erythrogenesis (Heinrich et al., 2019). Epigenetic changes can result in modifications in DNA methylation, histone modifications, as well as changes in non-coding RNAs all of which regulate gene transcription and translation allowing for developmental plasticity (Ducsay et al., 2018). Associated with these epigenetic modifications, short term sojourns to high altitude can induce developmental adaptations as well as abnormalities (Jones et al., 2019). While we do not know all of the changes in the pulmonary vasculature due to prenatal hypoxic exposure and high altitude living, the genetic and epigenetic changes due to high altitude living are likely to have great impact on cellular metabolism (Woolcott et al., 2015; Murray, 2016; Murray and Horscroft, 2016; Stenmark et al., 2018).
The general influence of hypoxia on cellular metabolism is known, however, less is understood regarding the effects in the lung and even less is known about the influences on the fetus or infant as compared to that of the adult. The impact of gestational hypoxia has been mostly investigated in animal models including rodents, sheep, and other species (Bixby et al., 2007; Herrera et al., 2007, 2010; Al-Hasan et al., 2013; Breckenridge et al., 2013; Neary et al., 2014; Allison et al., 2016; Mcgillick et al., 2017; Blum-Johnston et al., 2018). In humans, there have also been examinations of short-term high altitude adaptation in native adult Tibetans and non-native lowlanders (Ge et al., 2012; Woolcott et al., 2015; Murray, 2016; Murray and Horscroft, 2016). There are distinctions between the metabolic adaptations to high altitude that occur in these native and non-native adult populations, though individuals from both populations exhibit an increase in cellular glycolysis along with a decrease in beta-oxidation of fatty acids as well as citric acid intermediates (Table 1; Murray, 2009, 2016; Murray and Horscroft, 2016). Further, in a fetal sheep model, exposure to a low oxygen environment can elicit a glycolytic shift in glucose metabolism with an increase in lactic acid production and appearance in the plasma (Allison et al., 2016).
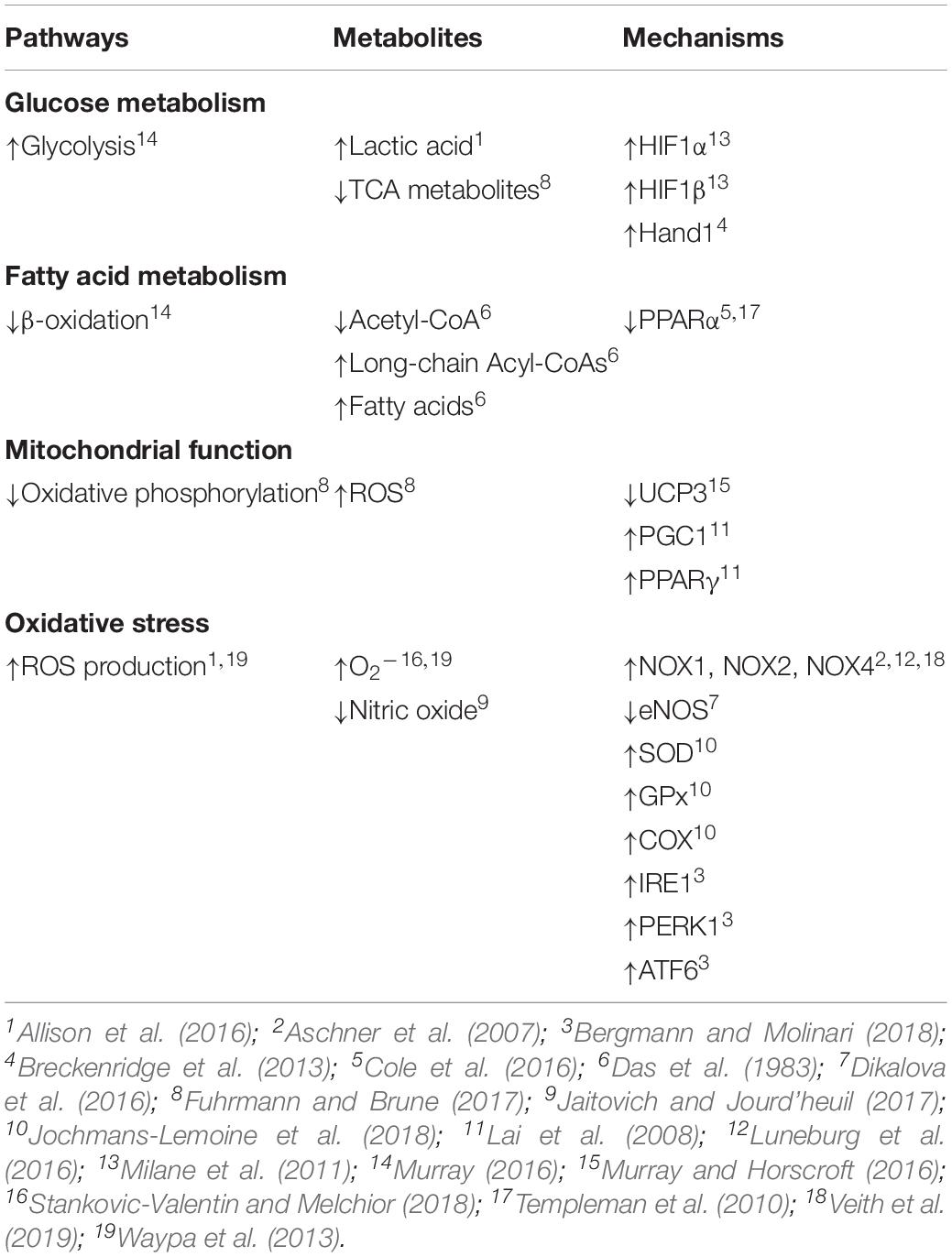
Table 1. Collective evidence of metabolic changes induced by hypoxia.
Even though many of the effects of high altitude on glycolysis have been observed with transition and residency in high altitude environments, a majority of our knowledge regarding the effect of hypoxia on glycolytic metabolism is derived from cancer biology. In the case of cancer, tumors often have limited nutrient supply and oxygenation as cellular growth outpaces vascularization. Even when cancer cells do have sufficient oxygen they still metabolize more glucose than normal cells and produce greater lactic acid than non-cancerous tissues in what is known as “aerobic glycolysis” or the “Warburg effect” (Huang et al., 2014). Cancer cells reduce their reliance on mitochondrial oxygen-dependent ATP production in favor of cytoplasmic glycolysis, which is far less efficient at generating energy. The result is that cancer cells consume more glucose to produce the ATP required for cell growth and survival (Huang et al., 2014).
Stimulation of hypoxia inducible factors (HIFs) by the low oxygen environment is mechanistically important to the metabolic adaptations that occur at high altitude. HIF-1α and HIF-1β are the primary responders, although HIF-2 and other isoforms may also serve important roles (Milane et al., 2011). Mechanistically, the low oxygen environment decreases HIF-1α subunit degradation through depression of prolyl hydroxylase (PHD) activity, which enhances HIF-1α stabilization. Stabilization of HIF-1α favors formation of an activated complex with the beta subunit, retention in the nucleus, and binding to hypoxia responsive elements (HRE) on target genes and induction of transcription (Milane et al., 2011). In the mouse heart, HIF-1α activation due to hypoxia increases transcription of Hand1, which is vital to cardiac development and causes a fundamental shift toward an increase in glycolysis and a decrease in oxidative phosphorylation (Table 1; Breckenridge et al., 2013; Wong et al., 2017). Activation of HIF-1α is also important to the glycolytic shift in cancer cells, fibroblasts and other cell types (Yang et al., 2014). However, whether or not hypoxia induced HIF-1α activation causes an analogous shift toward glycolytic metabolism in cells within the fetal lung remains unresolved; although chronic hypoxia induced HIF-1α activation is important to medial wall thickening associated with the development of pulmonary hypertension in various species (Yang et al., 2014), and delays surfactant protein production in the fetal sheep lung (Orgeig et al., 2015).
Similar to glucose metabolism, fatty acid metabolism is also affected by reduced oxygenation. Oxidation of fatty acids usually occurs in the mitochondria where an acyl-CoA is catalyzed to produce acetyl-CoA, NADH+ H+ and FADH2 (Huang et al., 2014). Acute ischemia of the rabbit fetus reduces fatty acid metabolism in the lung with decreased levels of acetyl-CoA and a buildup of long chain acyl-CoA derivatives (Table 1; Das et al., 1983). Anoxia to the myocardium of rats causes similar reductions in acetyl-CoA and increases in long chain acyl-CoA products (Whitmer et al., 1978). Hypoxia induced activation of HIF-1 contributes to the buildup of fatty acid metabolites in myocardium of mice as it suppresses activity of peroxisome proliferator activator receptor alpha (PPARα) (Templeman et al., 2010; Cole et al., 2016). The reduction in PPARα activity restricts fatty acid uptake and metabolism. These decreases in fatty acid metabolism and changes in CoA and acyl derivatives with low oxygen exposure further illustrate that beta oxidation is a limiting step in fatty acid metabolism (Whitmer et al., 1978). Related with this, under aerobic conditions in the adult between 60 and 90% of the total oxygen consumption may be used to oxidize fatty acids (Whitmer et al., 1978). The fetal heart, however, is far more reliant on glycolytic pathways than the adult and thus already is not very dependent on oxygen, and chronic hypoxia causes a further reduction in fatty acid oxidation (Thompson, 2003). These adaptations to hypoxia that lessen fatty acid oxidation and oxygen utilization, generally, can be energetically advantageous in rarified environments (Ge et al., 2012). Whether hypoxia due to high altitude gestation modifies fatty acid metabolism and increases glycolytic metabolism in the developing lung remains to be determined and is important to resolve as the changes in bioenergetics may be linked to disease.
High altitude gestation and the accompanying low oxygen environment alter mitochondrial function in an altitude dependent manner (Murray, 2016; Chicco et al., 2018). The density of skeletal muscle mitochondria may be reduced in humans who remain in particularly rarified environments (>5500 m) for long periods (Hoppeler et al., 1990; Levett et al., 2012; Murray, 2016). Electron transport chain complexes are downregulated along with uncoupling protein 3 in these extreme high altitude environments, while there is a decrease in fatty acid oxidation capacity and creatine kinase expression (Table 1; Murray and Horscroft, 2016). The reduction in proton leak results in changes in the coupling efficiency of the electron transport chain and contributes to modifications in fuel utilization at altitude.
Evidence suggests that carotid arteries of newborn sheep experience mitochondrial stress when subjected to LTH due to a build-up of compounds related to glycolysis, the pentose phosphate pathway, and the mitochondrial citric acid cycle (Goyal and Longo, 2015). Furthermore in rat heart, long term hypoxia decreases fatty acid oxidation, respiratory capacity, and pyruvate oxidation (Essop et al., 2004; Adrogue et al., 2005; Murray, 2016). At moderately high altitudes, decreased mitochondrial respiratory capacity of humans may occur without mitochondrial volume density being affected, but mitochondrial volume density decreases at extreme altitudes (>5500 m), a process that may be governed by hypoxia-signaling pathways (Murray, 2016). Studies using cultured cells and genetic mouse models have explained a number of adaptations that allow cells with a diminished mitochondrial density to function effectively in hypoxic situations (Murray, 2009).
High altitude hypoxia generally results in changes in citric acid cycle metabolism and oxidative phosphorylation (OXPHOS), as well as alterations in mitochondrial morphology, mass, fusion, fission, and mitophagy, reviewed recently (Fuhrmann and Brune, 2017). Mitochondria are one of the main consumers of oxygen through OXPHOS and when oxygen levels decrease they become a more prominent contributor of reactive oxygen species (ROS) (Fuhrmann and Brune, 2017). Notably, the fetal pulmonary vasculature offers a dramatic example of the differences between fetal and neonatal oxygen tensions. During fetal life, the unventilated lung is perfused predominantly with less-oxygenated blood returned from the upper body with a PO2 in the low 20 s (Vali and Lakshminrusimha, 2017). With the initiation of pulmonary ventilation at birth, the PO2 of the pulmonary vasculature increases to ∼40 Torr in the arteries and >90 Torr in the pulmonary veins. Thus, the mitochondria of the fetal pulmonary vasculature experiences a two to fivefold increase in PO2 during the transition from fetus to newborn. Little is known regarding how the fetal lung adapts to these changes in O2 concentrations and studies using both classic and modern approaches are needed to resolve the adaptive processes during the birth transition. In the hearts of mice, the substantial increase in O2 tensions after birth decreases cardiomyocyte HIF-signaling, a process that leads to mitochondrial fusion and biogenesis (Neary et al., 2014). The upregulation of mitochondrial biogenesis after birth in mice is due, in part, to increases in peroxisome proliferator-activated receptor gamma coactivator-1 (Pgc1alpha/beta) expression (Lai et al., 2008). PPARγ activation is also important for upregulation of lipid uptake along with enhanced expression of citrate synthase, a key mitochondrial enzyme involved in the citric acid cycle. Interestingly, fetal mitochondria of mice and rabbits have a “fragmented” appearance while postnatal mitochondria are elongated in appearance, a process due to the metabolic changes that occur with birth (Lopaschuk et al., 1991; Neary et al., 2014); a process that may be related to the increase in PGC1 expression.
The low oxygen environment associated with high altitude exposure is well regarded for enhancing oxidative stress in tissues from adults and in cultured cells. Although maternal chronic hypoxia increases oxidative stress in intact fetal lamb (Table 1; Allison et al., 2016), far less is known about the impact of gestational hypoxia on oxidative stress in the fetal and newborn lung. What is more, hypoxia associated with other acute and chronic lung diseases also increases oxidative stress (Van Der Vliet et al., 2018), and thus gestational hypoxia may increase oxidative stress in fetal lung. Reactive oxygen species are not just harmful byproducts of cellular metabolism, but rather are complex signaling molecules that regulate cell function. Under normal conditions free radicals are produced by cells in a highly controlled way by various enzymatic systems, but most prominently by NADPH oxidases (NOX) that produce superoxide (O2–) (Stankovic-Valentin and Melchior, 2018). There are seven members of the NOX family, which have varied tissue and subcellular distributions. NOX1 is well expressed in epithelial and endothelial cells (Veith et al., 2019). NOX2 plays a prominent role in phagocytic cells and the innate immune response (Veith et al., 2019). NOX4 has been implicated in fibrotic diseases including those of the liver, skin, kidney, heart, and lung (Veith et al., 2019).
Hypoxia is known to uncouple endothelial nitric oxide synthase (eNOS) (Jaitovich and Jourd’heuil, 2017). This uncoupling of eNOS impairs nitric oxide (NO) signaling and increases generation of superoxide. Such eNOS uncoupling affects nitric oxide (NO) signaling in a variety of cardiopulmonary disorders, including pulmonary hypertension (Dikalova et al., 2016). The contribution of eNOS uncoupling to cardiopulmonary disease is mediated through a number of mechanisms related to the superoxide production. When eNOS becomes uncoupled, electrons travel to molecular oxygen producing superoxide instead of NO (Dikalova et al., 2016). For example, eNOS becomes uncoupled following exposure of newborn piglets to hypoxia for as few as 3 days and is associated with increases in generation of O2– and decreases in both eNOS dimer formation and NO production (Dikalova et al., 2016). Changes in the cellular redox status are biologically relevant as the reactive oxygen molecules elicit reversible or irreversible oxidative protein or DNA modifications, mitochondrial dysfunction, as well as changes in the expression or activity of NOX enzymes and antioxidant enzyme systems (Van Der Vliet et al., 2018). When considering protein folding, cells work to compensate for the misfolded or oxidized proteins by marking them for degradation using posttranslational modifications such as ubiquitination and SUMOylation (Stankovic-Valentin and Melchior, 2018).
Acute alveolar hypoxia is well known to trigger constriction of resistance pulmonary arteries (Waypa et al., 2013). Accompanying this, acute hypoxia also leads to superoxide generation in smooth muscle cells (Table 1; Waypa et al., 2013). These cytosolic oxidant signals are thought to be important to the increases in [Ca2+]i that cause acute hypoxic pulmonary vasoconstriction (HPV) (Waypa et al., 2013). We find that newborn sheep born at high altitude have exacerbated HPV responses (Blood et al., 2013). Potentially this enhanced responsiveness may be due to alterations in the pro and anti-oxidant systems. Long-term high-altitude exposure of rats as compared to mice provides some insight about the diversity in inter-species responses. In response to high altitude living at 3,600 m rats, but not mice, have a decrease in gas exchange across the lung epithelia that is associated with loss of alveolar surface area (Jochmans-Lemoine et al., 2018). While the authors did not determine causation, rats had elevated oxidative stress and mitochondrial compensatory pathways, while mice were less effected. Associated with the elevation in oxidative stress, mitochondrial superoxide dismutase (SOD), glutathione peroxidase (GPx) and cytochrome oxidase-c (COX) activities were 2–3 times higher in high altitude rats while cytosolic enzymatic activities for NOX, xanthine oxidase (XO), SOD, and GPx were not as greatly effected in high altitude mice (Table 1; Jochmans-Lemoine et al., 2018).
Low oxygen tensions generally reduce mitochondrial function, as previously discussed. However, in the fetus, where oxygen tension is already low the additional impact of the high altitude environment on free radicals is largely unresolved. In most tissues, ATP production predominantly happens via mitochondrial oxidative phosphorylation of reduced intermediates of the citric acid cycle from substrates, the majority of which come from glucose and fatty acids (Murray, 2009). Mitochondria need a constant supply of fuels and oxygen to maintain ATP production (Murray, 2009). In high altitude environments, tissue oxygen levels fall and cells must work to limit oxidative stress (Murray, 2009). Exposure to the rarified environment in tissues of the adult increases superoxide production from mitochondrial complexes of the respiratory chain (Guzy et al., 2005; Guzy and Schumacker, 2006; Murray, 2009; Murray and Horscroft, 2016). Fetal lungs of guinea pigs exposed to 10.5% O2 for the last 14 days of gestation had reduced cytochrome oxidase activity and expression of COX4, illustrative of perturbations to the generation of free radicals by mitochondria (Al-Hasan et al., 2013). While there is a fair amount of knowledge regarding the importance of oxidative stress to the regulation of vascular function further exploration is needed to fully understand the underlying cellular and molecular mechanisms.
The endoplasmic reticulum (ER) is a critical organelle that is important for protein processing, lipid synthesis, as well as for intracellular Ca2+ signaling and homeostasis. Protein translation and folding functions of the organelle are strongly regulated by reactive oxygen species. Although the ER can respond to ROS generated anywhere throughout the cell, NOX4 is known to be closely associated with the ER and nucleus in rats, and regulates ER function through superoxide generation (Camargo et al., 2018). Elevated levels of oxidative stress due to superoxide and other free radicals is part of the normal signaling process. However, abnormally high oxidative stress can cause ER dysfunction. Heightened oxidative stress due to hypoxia and other stresses can elicit protein misfolding and unfolding responses in the ER. The unfolding protein response is a prototypical marker of ER stress and induces cellular responses that act to preserve homeostasis. The unfolding protein responses are graded and highly conserved across phylogeny suggesting reactive oxygen species are critical regulators of organelle function (Bergmann and Molinari, 2018).
Modest levels of ER stress leads to activation of signaling pathways that increase protein synthesis, enhance protein trafficking through the ER, increase protein folding and augment ER-associated protein degradation processes, all of which allows for maintenance of organelle function. However, elevated ROS and ER stress levels cause greater organelle dysregulation and magnified UPR responses and activation of IRE1, PERK1, and ATF6 (Table 1; Bergmann and Molinari, 2018). High levels of stress and coordinated activation of these pathways then leads to cell autophagy. While the exact role of NOX4 in the fetal or newborn lung is unresolved, recent studies show that NOX4 expression is elevated in pulmonary arteries of adult rats exposed to chronic hypoxia (Luneburg et al., 2016) and in systemic arteries of spontaneous hypertensive rats (Camargo et al., 2018). These findings have focused attention on the potential that ER stress is important in the development of systemic as well as pulmonary hypertension. SHR rats have increased ER stress that is associated with an upregulation of NOX4. Suppression of oxidative stress as well as NOX4 expression blunts the hypertension response. These effects in the systemic vasculature of SHR rats are similar to piglets exposed to chronic hypoxia, which have increased NOX dependent pulmonary hypertension (Aschner et al., 2007). Similarly, fetal sheep lungs of ewes exposed to 10.5% O2 from 105 to 138 days of gestation had increased expression of the antioxidant catalase but decreased pro-oxidant NOX4 expression, illustrative of changes in oxidative stress in the neonatal lung (Mcgillick et al., 2017).
Chronic hypoxia is closely associated with the induction of ER stress, disruption of mitochondrial function and the development of pulmonary hypertension. Chronic hypoxia-induced pulmonary hypertension in mice is associated with increases in ER stress and the uncoupling protein response (Dromparis et al., 2013a). The hypoxic stress is further linked to loss of the mitochondrial membrane potential as well as cellular proliferation and medial wall thickening. Reducing ER stress with the chemical chaperones 4-phenylbutyrate or tauroursodeoxycholic acid in these mice provides additional evidence for these interactions as the chemicals were able to mitigate the hypoxia related impacts on mitochondrial calcium and membrane potential, activation of ER stress pathways, as well as the proliferative responses (Dromparis et al., 2013a). Downregulation of uncoupling protein 2 in mice, which is normally expressed in the mitochondria, causes dysregulation of mitochondrial Ca2+ signaling and induces pulmonary hypertension (Dromparis et al., 2013b). The close association between the mitochondria and ER also appear to be important to the development of PH in mice (Sutendra et al., 2011). ER stress due to hypoxia may cause a breakdown in the close interaction and Ca2+ movement between the ER and mitochondria, which further dysregulates the function of both organelles (Sutendra et al., 2011; Raffaello et al., 2016). Based on this evidence, it will be important to pursue the impact of gestational hypoxia on ER stress and its relevance to the development of pulmonary hypertension in the newborn.
A number of human populations have lived at high altitudes for many generations, including citizens of Tibet, Ethiopia, and the Andes. Among these, Tibetans are the best studied high altitude population. Interestingly, they have a variety of adaptations that improve their capacity to develop and thrive at high altitude (Niermeyer et al., 1995; Ge et al., 2012). However, beyond only a handful of studies our knowledge remains limited concerning the developmental progression of cellular metabolism and the impact of the low oxygen environment on these populations. This includes functional compensatory mechanisms consisting of increased ventilation and greater pulmonary diffusion capacity relative to non-native populations (Bianba et al., 2014). The lungs of Tibetan infants are better adapted to provide enhanced blood oxygenation through developmental alterations in cardiorespiratory structure and function (Niermeyer et al., 1995). This combined with the decreased oxygen utilization by the mitochondria and lessened fatty acid oxidation in peripheral tissues help to enhance physiological performance at high altitude (Murray, 2016).
The excessive oxidative stress that occurs with chronic hypoxia has physiological consequences (Jochmans-Lemoine et al., 2015, 2018). Oxygen sensing is essential for stimulating gene expression and transcription for growth processes and angiogenesis (Kurlak et al., 2016). However, this is best examined in animal models that share critical features of human disease because of the ability to perform invasive examinations regarding the phenotypic impacts of high altitude, the associated cellular and molecular mechanisms, and evaluation of new therapeutics. For example, in Bolivia at 3,600 m above sea level, rats exhibit a chronic mountain sickness phenotype that is similar to humans with elevated hematocrit, lower ventilation, signs of severe pulmonary hypertension, and lower arterial oxygen saturations when breathing either high or low levels of oxygen (Jochmans-Lemoine et al., 2018). From a therapeutic perspective, some of these effects can be partially reversed by exposing affected rats to enriched oxygen during the first 2 weeks after birth (Jochmans-Lemoine et al., 2018).
One key adaptation in adults with exposure to even moderately high altitudes above 1,500 m is that there are dramatic changes in glucose tolerance, which may be linked to oxidative stress (Woolcott et al., 2015). Initial exposure to the rarified environment causes a person’s glycemic index to increase with an increase in anaerobic glucose metabolism (Kelly et al., 2010). Related to this, healthy people exposed to high-altitude hypoxia may become insulin-resistant (Siervo et al., 2014). Mechanistically, the insulin resistance may be related to cellular inflammation and ROS production (Trayhurn, 2013). Long-term residence at high altitude in comparison may lower a person’s glycemic index and improve glucose uptake and utilization (Woolcott et al., 2015). Recent work in the sheep fetus exposed to gestational hypoxia for 9 days may provide some insight as the data shows there is an increase in hepatic expression of G6PC and PCK1 without any change in plasma glucose (Jones et al., 2019). Overall, the relationships between ROS, cellular metabolism, and the functional consequences remain to be elucidated in humans and animal models. Whether or not many of the changes outlined in fetal and adult organs also occur in the lung are unresolved but are important to address as changes in ROS and cellular metabolism are projected to have major impact on lung development and function.
High altitude gestation and birth places a significant stress on both the mother and fetus and results in metabolic reprogramming of the lung and other organ systems, which give rise to functional defects including pulmonary hypertension as well as diseases in other systems. Focused evaluation of individual signaling pathways, related genes and proteins has provided some insights into the mechanistic basis for stress related diseases due to high altitude gestation. Even still, our comprehension of the impact of low oxygen on fetal development is far from complete. Better understanding of the etiology as well as treatment of disease will require integration of information from various sources. Studies that use contemporary omics approaches including metabolomics, proteomics, transcriptomics and genomics along with traditional functional studies using manipulated systems hold great promise for providing a deeper understanding of the mechanisms associated with hypoxia-related disease in the fetus as well as the development of novel treatments.
KR and SW provided the initial draft of the manuscript with VL, ML, OF, LZ, and AB providing further input and edits.
This work was supported in part by USPHS Grants NIH R03HD098477 and WCMC Pilot project (SW), P01HD083132 (LZ), U24DK097154 (OF). VL was an APS STRIDE Research Fellow supported by R25HL115473.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abman, S. H. (2007). Recent advances in the pathogenesis and treatment of persistent pulmonary hypertension of the newborn. Neonatology 91, 283–290. doi: 10.1159/000101343
Adeoye, O. O., Bouthors, V., Hubbell, M. C., Williams, J. M., and Pearce, W. J. (2014). VEGF receptors mediate hypoxic remodeling of adult ovine carotid arteries. J. Appl. Physiol. 117, 777–787. doi: 10.1152/japplphysiol.00012.2014
Adeoye, O. O., Silpanisong, J., Williams, J. M., and Pearce, W. J. (2015). Role of the sympathetic autonomic nervous system in hypoxic remodeling of the fetal cerebral vasculature. J. Cardiovasc. Pharmacol. 65, 308–316. doi: 10.1097/FJC.0000000000000192
Adrogue, J. V., Sharma, S., Ngumbela, K., Essop, M. F., and Taegtmeyer, H. (2005). Acclimatization to chronic hypobaric hypoxia is associated with a differential transcriptional profile between the right and left ventricle. Mol. Cell Biochem. 278, 71–78. doi: 10.1007/s11010-005-6629-5
Al-Hasan, Y. M., Evans, L. C., Pinkas, G. A., Dabkowski, E. R., Stanley, W. C., and Thompson, L. P. (2013). Chronic hypoxia impairs cytochrome oxidase activity via oxidative stress in selected fetal guinea pig organs. Reprod. Sci. 20, 299–307. doi: 10.1177/1933719112453509
Allison, B. J., Brain, K. L., Niu, Y., Kane, A. D., Herrera, E. A., Thakor, A. S., et al. (2016). Fetal in vivo continuous cardiovascular function during chronic hypoxia. J. Physiol. 594, 1247–1264. doi: 10.1113/JP271091
Arakawa, T. K., Mlynarczyk, M., Kaushal, K. M., Zhang, L., and Ducsay, C. A. (2004). Long-term hypoxia alters calcium regulation in near-term ovine myometrium. Biol. Reprod. 71, 156–162. doi: 10.1095/biolreprod.103.026708
Aschner, J. L., Foster, S. L., Kaplowitz, M., Zhang, Y., Zeng, H., and Fike, C. D. (2007). Heat shock protein 90 modulates endothelial nitric oxide synthase activity and vascular reactivity in the newborn piglet pulmonary circulation. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L1515–L1525.
Berger, S., and Konduri, G. G. (2006). Pulmonary hypertension in children: the twenty-first century. Pediatr. Clin. North Am. 53, 961–987. doi: 10.1016/j.pcl.2006.08.001
Bergmann, T. J., and Molinari, M. (2018). Three branches to rule them all? UPR signalling in response to chemically versus misfolded proteins-induced ER stress. Biol. Cell 110, 197–204. doi: 10.1111/boc.201800029
Bianba, Berntsen, S., Andersen, L. B., Stigum, H., Ouzhuluobu, Nafstad, P., et al. (2014). Exercise capacity and selected physiological factors by ancestry and residential altitude: cross-sectional studies of 9-10-year-old children in Tibet. High Alt. Med. Biol. 15, 162–169. doi: 10.1089/ham.2013.1084
Bixby, C. E., Ibe, B. O., Abdallah, M. F., Zhou, W., Hislop, A. A., Longo, L. D., et al. (2007). Role of platelet-activating factor in pulmonary vascular remodeling associated with chronic high altitude hypoxia in ovine fetal lambs. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L1475–L1482.
Blood, A. B., Terry, M. H., Merritt, T. A., Papamatheakis, D. G., Blood, Q., Ross, J. M., et al. (2013). Effect of chronic perinatal hypoxia on the role of rho-kinase in pulmonary artery contraction in newborn lambs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R136–R146. doi: 10.1152/ajpregu.00126.2012
Blum-Johnston, C., Thorpe, R. B., Wee, C., Opsahl, R., Romero, M., Murray, S., et al. (2018). Long-term hypoxia uncouples Ca(2+) and eNOS in bradykinin-mediated pulmonary arterial relaxation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 314, R870–R882. doi: 10.1152/ajpregu.00311.2017
Blum-Johnston, C., Thorpe, R. B., Wee, C., Romero, M., Brunelle, A., Blood, Q., et al. (2016). Developmental acceleration of bradykinin-dependent relaxation by prenatal chronic hypoxia impedes normal development after birth. Am. J. Physiol. Lung. Cell Mol. Physiol. 310, L271–L286. doi: 10.1152/ajplung.00340.2015
Breckenridge, R. A., Piotrowska, I., Ng, K. E., Ragan, T. J., West, J. A., Kotecha, S., et al. (2013). Hypoxic regulation of hand1 controls the fetal-neonatal switch in cardiac metabolism. PLoS Biol. 11:e1001666. doi: 10.1371/journal.pbio.1001666
Camargo, L. L., Harvey, A. P., Rios, F. J., Tsiropoulou, S., Da Silva, R. N. O., Cao, Z., et al. (2018). Vascular nox (NADPH Oxidase) compartmentalization, protein hyperoxidation, and endoplasmic reticulum stress response in hypertension. Hypertension 72, 235–246. doi: 10.1161/HYPERTENSIONAHA.118.10824
Chicco, A. J., Le, C. H., Gnaiger, E., Dreyer, H. C., Muyskens, J. B., D’alessandro, A., et al. (2018). Adaptive remodeling of skeletal muscle energy metabolism in high-altitude hypoxia: lessons from AltitudeOmics. J. Biol. Chem. 293, 6659–6671. doi: 10.1074/jbc.RA117.000470
Chun, H., Yue, Y., Wang, Y., Dawa, Z., Zhen, P., La, Q., et al. (2019). High prevalence of congenital heart disease at high altitudes in Tibet. Eur. J. Prev. Cardiol. 26, 756–759. doi: 10.1177/2047487318812502
Cole, M. A., Abd Jamil, A. H., Heather, L. C., Murray, A. J., Sutton, E. R., Slingo, M., et al. (2016). On the pivotal role of PPARalpha in adaptation of the heart to hypoxia and why fat in the diet increases hypoxic injury. Faseb J. 30, 2684–2697. doi: 10.1096/fj.201500094R
Cole, S. Z., and Lanham, J. S. (2011). Failure to thrive: an update. Am. Fam. Physician 83, 829–834.
Cosse, J. P., and Michiels, C. (2008). Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med. Chem. 8, 790–797. doi: 10.2174/187152008785914798
Das, D. K., Ayromlooi, J., and Neogi, A. (1983). Effect of ischemia on fatty acid metabolism in fetal lung. Life Sci. 33, 569–576. doi: 10.1016/0024-3205(83)90132-7
Dikalova, A., Aschner, J. L., Kaplowitz, M. R., Summar, M., and Fike, C. D. (2016). Tetrahydrobiopterin oral therapy recouples eNOS and ameliorates chronic hypoxia-induced pulmonary hypertension in newborn pigs. Am. J. Physiol. Lung. Cell Mol. Physiol. 311, L743–L753. doi: 10.1152/ajplung.00238.2016
Dromparis, P., Paulin, R., Stenson, T. H., Haromy, A., Sutendra, G., and Michelakis, E. D. (2013a). Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation 127, 115–125. doi: 10.1161/circulationaha.112.133413
Dromparis, P., Paulin, R., Sutendra, G., Qi, A. C., Bonnet, S., and Michelakis, E. D. (2013b). Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ. Res. 113, 126–136. doi: 10.1161/CIRCRESAHA.112.300699
Ducsay, C. A., Goyal, R., Pearce, W. J., Wilson, S., Hu, X. Q., and Zhang, L. (2018). Gestational hypoxia and developmental plasticity. Physiol. Rev. 98, 1241–1334. doi: 10.1152/physrev.00043.2017
Ducsay, C. A., Hyatt, K., Mlynarczyk, M., Root, B. K., Kaushal, K. M., and Myers, D. A. (2007). Long-term hypoxia modulates expression of key genes regulating adrenomedullary function in the late gestation ovine fetus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1997–R2005.
Eichstaedt, C. A., Antão, T., Pagani, L., Cardona, A., Kivisild, T., and Mormina, M. (2014). The Andean adaptive toolkit to counteract high altitude maladaptation: genome-wide and phenotypic analysis of the Collas. PloS One 9:e93314. doi: 10.1371/journal.pone.0093314
Essop, M. F., Razeghi, P., Mcleod, C., Young, M. E., Taegtmeyer, H., and Sack, M. N. (2004). Hypoxia-induced decrease of UCP3 gene expression in rat heart parallels metabolic gene switching but fails to affect mitochondrial respiratory coupling. Biochem. Biophys. Res. Commun. 314, 561–564. doi: 10.1016/j.bbrc.2003.12.121
Fuhrmann, D. C., and Brune, B. (2017). Mitochondrial composition and function under the control of hypoxia. Redox Biol. 12, 208–215. doi: 10.1016/j.redox.2017.02.012
Gao, Y., Portugal, A. D., Negash, S., Zhou, W., Longo, L. D., and Raj, J. U. (2007). Role of Rho kinases in PKG-mediated relaxation of pulmonary arteries of fetal lambs exposed to chronic high altitude hypoxia. Am. J. Physiol. Lung. Cell Mol. Physiol. 292, L678–L684.
Garcia, F. C., Stiffel, V. M., and Gilbert, R. D. (2000). Effects of long-term high-altitude hypoxia on isolated fetal ovine coronary arteries. J. Soc. Gynecol. Investig. 7, 211–217.
Ge, R. L., Simonson, T. S., Cooksey, R. C., Tanna, U., Qin, G., Huff, C. D., et al. (2012). Metabolic insight into mechanisms of high-altitude adaptation in Tibetans. Mol. Genet. Metab. 106, 244–247. doi: 10.1016/j.ymgme.2012.03.003
Gnecchi-Ruscone, G. A., Abondio, P., De Fanti, S., Sarno, S., Sherpa, M. G., Sherpa, P. T., et al. (2018). Evidence of polygenic adaptation to high altitude from tibetan and sherpa genomes. Genome Biol. Evol. 10, 2919–2930. doi: 10.1093/gbe/evy233
Goldberg, S. J., Levy, R. A., Siassi, B., and Betten, J. (1971). The effects of maternal hypoxia and hyperoxia upon the neonatal pulmonary vasculature. Pediatrics 48, 528–533.
Goyal, R., and Longo, L. D. (2015). Metabolic profiles in ovine carotid arteries with developmental maturation and long-term Hypoxia. PLoS One 10:e0130739. doi: 10.1371/journal.pone.0130739
Goyal, R., Papamatheakis, D. G., Loftin, M., Vrancken, K., Dawson, A. S., Osman, N. J., et al. (2011). Long-term maternal hypoxia: the role of extracellular Ca2+ entry during serotonin-mediated contractility in fetal ovine pulmonary arteries. Reprod. Sci. 18, 948–962. doi: 10.1177/1933719111401660
Grunig, E., Dehnert, C., Mereles, D., Koehler, R., Olschewski, H., Bartsch, P., et al. (2005). Enhanced hypoxic pulmonary vasoconstriction in families of adults or children with idiopathic pulmonary arterial hypertension. Chest 128, 630S–633S. doi: 10.1378/chest.128.6_suppl.630s-a
Guzy, R. D., Hoyos, B., Robin, E., Chen, H., Liu, L., Mansfield, K. D., et al. (2005). Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1, 401–408. doi: 10.1016/j.cmet.2005.05.001
Guzy, R. D., and Schumacker, P. T. (2006). Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 91, 807–819. doi: 10.1113/expphysiol.2006.033506
Hadley, S. R., Blood, Q., Rubalcava, M., Waskel, E., Lumbard, B., Le, P., et al. (2012). Maternal high-altitude hypoxia and suppression of ryanodine receptor-mediated Ca2+ sparks in fetal sheep pulmonary arterial myocytes. Am. J. Physiol. Lung. Cell Mol. Physiol. 303, L799–L813. doi: 10.1152/ajplung.00009.2012
Heinrich, E. C., Wu, L., Lawrence, E. S., Cole, A. M., Anza-Ramirez, C., Villafuerte, F. C., et al. (2019). Genetic variants at the EGLN1 locus associated with high-altitude adaptation in tibetans are absent or found at low frequency in highland Andeans. Ann. Hum. Genet. 83, 171–176. doi: 10.1111/ahg.12299
Herrera, E. A., Pulgar, V. M., Riquelme, R. A., Sanhueza, E. M., Reyes, R. V., Ebensperger, G., et al. (2007). High-altitude chronic hypoxia during gestation and after birth modifies cardiovascular responses in newborn sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R2234–R2240.
Herrera, E. A., Riquelme, R. A., Ebensperger, G., Reyes, R. V., Ulloa, C. E., Cabello, G., et al. (2010). Long-term exposure to high-altitude chronic hypoxia during gestation induces neonatal pulmonary hypertension at sea level. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R1676–R1684. doi: 10.1152/ajpregu.00123.2010
Hoppeler, H., Kleinert, E., Schlegel, C., Claassen, H., Howald, H., Kayar, S. R., et al. (1990). Morphological adaptations of human skeletal muscle to chronic hypoxia. Int. J. Sports Med. 11(Suppl. 1), S3–S9.
Huang, D., Li, C., and Zhang, H. (2014). Hypoxia and cancer cell metabolism. Acta Biochim. Biophys. Sin. 46, 214–219. doi: 10.1093/abbs/gmt148
Hubbell, M. C., Semotiuk, A. J., Thorpe, R. B., Adeoye, O. O., Butler, S. M., Williams, J. M., et al. (2012). Chronic hypoxia and VEGF differentially modulate abundance and organization of myosin heavy chain isoforms in fetal and adult ovine arteries. Am. J. Physiol. Cell Physiol. 303, C1090–C1103. doi: 10.1152/ajpcell.00408.2011
Jaitovich, A., and Jourd’heuil, D. (2017). A brief overview of nitric oxide and reactive oxygen species signaling in hypoxia-induced pulmonary hypertension. Adv. Exp. Med. Biol. 967, 71–81. doi: 10.1007/978-3-319-63245-2_6
Jochmans-Lemoine, A., Revollo, S., Villalpando, G., Valverde, I., Gonzales, M., Laouafa, S., et al. (2018). Divergent mitochondrial antioxidant activities and lung alveolar architecture in the lungs of rats and mice at high altitude. Front. Physiol. 9:311. doi: 10.3389/fphys.2018.00311
Jochmans-Lemoine, A., Villalpando, G., Gonzales, M., Valverde, I., Soria, R., and Joseph, V. (2015). Divergent physiological responses in laboratory rats and mice raised at high altitude. J. Exp. Biol. 218, 1035–1043. doi: 10.1242/jeb.112862
Jones, A. K., Rozance, P. J., Brown, L. D., Goldstrohm, D. A., William, W., Hay, J., et al. (2019). Sustained hypoxemia in late gestation potentiates hepatic gluconeogenic gene expression but does not activate glucose production in the ovine fetus. Am. J. Phys. Endocrinol. Metab. 317, E1–E10. doi: 10.1152/ajpendo.00069.2019
Kelly, K. R., Williamson, D. L., Fealy, C. E., Kriz, D. A., Krishnan, R. K., Huang, H., et al. (2010). Acute altitude-induced hypoxia suppresses plasma glucose and leptin in healthy humans. Metabolism 59, 200–205. doi: 10.1016/j.metabol.2009.07.014
Konduri, G. G., and Kim, U. O. (2009). Advances in the diagnosis and management of persistent pulmonary hypertension of the newborn. Pediatr. Clin. North Am. 56, 579–600. doi: 10.1016/j.pcl.2009.04.004
Kurlak, L. O., Mistry, H. D., Cindrova-Davies, T., Burton, G. J., and Broughton Pipkin, F. (2016). Human placental renin-angiotensin system in normotensive and pre-eclamptic pregnancies at high altitude and after acute hypoxia-reoxygenation insult. J. Physiol. 594, 1327–1340. doi: 10.1113/JP271045
Lai, L., Leone, T. C., Zechner, C., Schaeffer, P. J., Kelly, S. M., Flanagan, D. P., et al. (2008). Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 22, 1948–1961. doi: 10.1101/gad.1661708
Levett, D. Z., Radford, E. J., Menassa, D. A., Graber, E. F., Morash, A. J., Hoppeler, H., et al. (2012). Acclimatization of skeletal muscle mitochondria to high-altitude hypoxia during an ascent of Everest. Faseb J. 26, 1431–1441. doi: 10.1096/fj.11-197772
Lewis, A. M., Mathieu-Costello, O., Mcmillan, P. J., and Gilbert, R. D. (1999). Quantitative electron microscopic study of the hypoxic fetal sheep heart. Anat. Rec. 256, 381–388. doi: 10.1002/(sici)1097-0185(19991201)256:4<381::aid-ar5>3.0.co;2-w
Longo, L. D., and Pearce, W. J. (2005). Fetal cerebrovascular acclimatization responses to high-altitude, long-term hypoxia: a model for prenatal programming of adult disease? Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R16–R24.
Lopaschuk, G. D., Spafford, M. A., and Marsh, D. R. (1991). Glycolysis is predominant source of myocardial ATP production immediately after birth. Am. J. Physiol. 261, H1698–H1705.
Luneburg, N., Siques, P., Brito, J., Arriaza, K., Pena, E., Klose, H., et al. (2016). Long-term chronic intermittent hypobaric hypoxia in rats causes an imbalance in the asymmetric dimethylarginine/nitric oxide pathway and ROS activity: a possible synergistic mechanism for altitude pulmonary hypertension? Pulm. Med. 2016:6578578. doi: 10.1155/2016/6578578
Mcgillick, E. V., Orgeig, S., Allison, B. J., Brain, K. L., Niu, Y., Itani, N., et al. (2017). Maternal chronic hypoxia increases expression of genes regulating lung liquid movement and surfactant maturation in male fetuses in late gestation. J. Physiol. 595, 4329–4350. doi: 10.1113/JP273842
Milane, L., Duan, Z., and Amiji, M. (2011). Role of hypoxia and glycolysis in the development of multi-drug resistance in human tumor cells and the establishment of an orthotopic multi-drug resistant tumor model in nude mice using hypoxic pre-conditioning. Cancer Cell Int. 11:3. doi: 10.1186/1475-2867-11-3
Murray, A. J. (2009). Metabolic adaptation of skeletal muscle to high altitude hypoxia: how new technologies could resolve the controversies. Genome Med. 1:117. doi: 10.1186/gm117
Murray, A. J. (2016). Energy metabolism and the high-altitude environment. Exp. Physiol. 101, 23–27. doi: 10.1113/EP085317
Murray, A. J., and Horscroft, J. A. (2016). Mitochondrial function at extreme high altitude. J. Physiol. 594, 1137–1149. doi: 10.1113/JP270079
Myers, D. A., Singleton, K., Hyatt, K., Mlynarczyk, M., Kaushal, K. M., and Ducsay, C. A. (2015). Long-term gestational hypoxia modulates expression of key genes governing mitochondrial function in the perirenal adipose of the late gestation sheep fetus. Reprod. Sci. 22, 654–663. doi: 10.1177/1933719114561554
Nanduri, J., Semenza, G. L., and Prabhakar, N. R. (2017). Epigenetic changes by DNA methylation in chronic and intermittent hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L1096–L1100. doi: 10.1152/ajplung.00325.2017
Neary, M. T., Ng, K. E., Ludtmann, M. H., Hall, A. R., Piotrowska, I., Ong, S. B., et al. (2014). Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J. Mol. Cell Cardiol. 74, 340–352. doi: 10.1016/j.yjmcc.2014.06.013
Newby, E. A., Myers, D. A., and Ducsay, C. A. (2015). Fetal endocrine and metabolic adaptations to hypoxia: the role of the hypothalamic-pituitary-adrenal axis. Am. J. Physiol. Endocrinol. Metab. 309, E429–E439. doi: 10.1152/ajpendo.00126.2015
Niermeyer, S., Andrade Mollinedo, P., and Huicho, L. (2009). Child health and living at high altitude. Arch. Dis. Child 94, 806–811. doi: 10.1136/adc.2008.141838
Niermeyer, S., Yang, P., Shanmina, Drolkar, Zhuang, J., and Moore, L. G. (1995). Arterial oxygen saturation in tibetan and han infants born in Lhasa, Tibet. N. Engl. J. Med. 333, 1248–1252. doi: 10.1056/nejm199511093331903
Orgeig, S., Mcgillick, E. V., Botting, K. J., Zhang, S., Mcmillen, I. C., and Morrison, J. L. (2015). Increased lung prolyl hydroxylase and decreased glucocorticoid receptor are related to decreased surfactant protein in the growth-restricted sheep fetus. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L84–L97. doi: 10.1152/ajplung.00275.2014
Papamatheakis, D. G., Blood, A. B., Kim, J. H., and Wilson, S. M. (2013). Antenatal hypoxia and pulmonary vascular function and remodeling. Curr. Vasc. Pharmacol. 11, 616–640. doi: 10.2174/1570161111311050006
Pearce, W. J. (2014). The fetal cerebral circulation: three decades of exploration by the LLU center for perinatal biology. Adv. Exp. Med. Biol. 814, 177–191. doi: 10.1007/978-1-4939-1031-1_16
Raffaello, A., Mammucari, C., Gherardi, G., and Rizzuto, R. (2016). Calcium at the Center of Cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 41, 1035–1049. doi: 10.1016/j.tibs.2016.09.001
Scherrer, U., Allemann, Y., Jayet, P. Y., Rexhaj, E., and Sartori, C. (2010). High altitude, a natural research laboratory for the study of cardiovascular physiology and pathophysiology. Prog. Cardiovasc. Dis. 52, 451–455. doi: 10.1016/j.pcad.2010.02.002
Shen, C. P., Romero, M., Brunelle, A., Wolfe, C., Dobyns, A., Francis, M., et al. (2018). Long-term high-altitude hypoxia influences pulmonary arterial L-type calcium channel-mediated Ca(2+) signals and contraction in fetal and adult sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 314, R433–R446. doi: 10.1152/ajpregu.00154.2017
Sheng, L., Zhou, W., Hislop, A. A., Ibe, B. O., Longo, L. D., and Raj, J. U. (2009). Role of epidermal growth factor receptor in ovine fetal pulmonary vascular remodeling following exposure to high altitude long-term hypoxia. High Alt. Med. Biol. 10, 365–372. doi: 10.1089/ham.2008.1034
Siervo, M., Riley, H. L., Fernandez, B. O., Leckstrom, C. A., Martin, D. S., Mitchell, K., et al. (2014). Effects of prolonged exposure to hypobaric hypoxia on oxidative stress, inflammation and gluco-insular regulation: the not-so-sweet price for good regulation. PLoS One 9:e94915. doi: 10.1371/journal.pone.0094915
Simonson, T. S., Yang, Y., Huff, C. D., Yun, H., Qin, G., Witherspoon, D. J., et al. (2010). Genetic evidence for high-altitude adaptation in Tibet. Science 329, 72–75. doi: 10.1126/science.1189406
Stankovic-Valentin, N., and Melchior, F. (2018). Control of SUMO and Ubiquitin by ROS: signaling and disease implications. Mol. Aspects Med. 63, 3–17. doi: 10.1016/j.mam.2018.07.002
Stenmark, K. R., Frid, M. G., Graham, B. B., and Tuder, R. M. (2018). Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc. Res. 114, 551–564. doi: 10.1093/cvr/cvy004
Sutendra, G., Dromparis, P., Wright, P., Bonnet, S., Haromy, A., Hao, Z., et al. (2011). The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci. Transl. Med. 3:88ra55. doi: 10.1126/scitranslmed.3002194
Tashi, T., Scott Reading, N., Wuren, T., Zhang, X., Moore, L. G., Hu, H., et al. (2017). Gain-of-function EGLN1 prolyl hydroxylase (PHD2 D4E:C127S) in combination with EPAS1 (HIF-2alpha) polymorphism lowers hemoglobin concentration in Tibetan highlanders. J. Mol. Med. 95, 665–670. doi: 10.1007/s00109-017-1519-3
Templeman, N. M., Beaudry, J. L., Le Moine, C. M., and Mcclelland, G. B. (2010). Chronic hypoxia- and cold-induced changes in cardiac enzyme and gene expression in CD-1 mice. Biochim. Biophys. Acta 1800, 1248–1255. doi: 10.1016/j.bbagen.2010.08.004
Thompson, L. P. (2003). Effects of chronic hypoxia on fetal coronary responses. High Alt. Med. Biol. 4, 215–224. doi: 10.1089/152702903322022811
Trayhurn, P. (2013). Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 93, 1–21. doi: 10.1152/physrev.00017.2012
Vali, P., and Lakshminrusimha, S. (2017). The fetus can teach us: oxygen and the pulmonary vasculature. Children 4:E67. doi: 10.3390/children4080067
Van Der Vliet, A., Janssen-Heininger, Y. M. W., and Anathy, V. (2018). Oxidative stress in chronic lung disease: from mitochondrial dysfunction to dysregulated redox signaling. Mol. Aspects Med. 63, 59–69. doi: 10.1016/j.mam.2018.08.001
Veith, C., Boots, A., Idris, M., Van Schooten, F. J., and Van Der Vliet, A. (2019). Redox imbalance in idiopathic pulmonary fibrosis: a role for oxidant cross-talk between NOX enzymes and mitochondria. Antioxid. Redox Signal. 31, 1092–1115. doi: 10.1089/ars.2019.7742
Waypa, G. B., Marks, J. D., Guzy, R. D., Mungai, P. T., Schriewer, J. M., Dokic, D., et al. (2013). Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 187, 424–432. doi: 10.1164/rccm.201207-1294OC
Weissmann, N., Nollen, M., Gerigk, B., Ardeschir Ghofrani, H., Schermuly, R. T., Gunther, A., et al. (2003). Downregulation of hypoxic vasoconstriction by chronic hypoxia in rabbits: effects of nitric oxide. Am. J. Physiol. Heart Circ. Physiol. 284, H931–H938.
Whitmer, J. T., Idell-Wenger, J. A., Rovetto, M. J., and Neely, J. R. (1978). Control of fatty acid metabolism in ischemic and hypoxic hearts. J. Biol. Chem. 253, 4305–4309.
Wong, B. W., Marsch, E., Treps, L., Baes, M., and Carmeliet, P. (2017). Endothelial cell metabolism in health and disease: impact of hypoxia. EMBO J. 36, 2187–2203. doi: 10.15252/embj.201696150
Woolcott, O. O., Ader, M., and Bergman, R. N. (2015). Glucose homeostasis during short-term and prolonged exposure to high altitudes. Endocr. Rev. 36, 149–173. doi: 10.1210/er.2014-1063
Xue, Q., Ducsay, C. A., Longo, L. D., and Zhang, L. (2008). Effect of long-term high-altitude hypoxia on fetal pulmonary vascular contractility. J. Appl. Physiol. 104, 1786–1792. doi: 10.1152/japplphysiol.01314.2007
Keywords: pulmonary artery, hypoxia, metabolomics, fetus, sheep
Citation: Rood K, Lopez V, La Frano MR, Fiehn O, Zhang L, Blood AB and Wilson SM (2019) Gestational Hypoxia and Programing of Lung Metabolism. Front. Physiol. 10:1453. doi: 10.3389/fphys.2019.01453
Received: 25 July 2019; Accepted: 11 November 2019;
Published: 29 November 2019.
Edited by:
Francisco C. Villafuerte, Universidad Peruana Cayetano Heredia, PeruReviewed by:
Vincent Joseph, Laval University, CanadaCopyright © 2019 Rood, Lopez, La Frano, Fiehn, Zhang, Blood and Wilson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sean M. Wilson, c2VhbndpbHNvbkBsbHUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.