 Yu-Han Hung1
Yu-Han Hung1 Matt Kanke1Catherine Lisa Kurtz2Rebecca L. Cubitt1Rodica P. Bunaciu1Liye Zhou3
Matt Kanke1Catherine Lisa Kurtz2Rebecca L. Cubitt1Rodica P. Bunaciu1Liye Zhou3 Phillip J. White4Kasey C. Vickers5Mohammed Mahmood Hussain3Xiaoling Li6
Phillip J. White4Kasey C. Vickers5Mohammed Mahmood Hussain3Xiaoling Li6 Praveen Sethupathy1*
Praveen Sethupathy1*- 1Department of Biomedical Sciences, Cornell University, Ithaca, NY, United States
- 2Department of Genetics, The University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 3Diabetes and Obesity Research Center, NYU Winthrop Hospital, Mineola, NY, United States
- 4Duke Molecular Physiology Institute, Duke University, Durham, NC, United States
- 5Department of Medicine, Vanderbilt University, Nashville, TN, United States
- 6Laboratory of Signal Transduction, National Institute of Environmental Health Sciences, Durham, NC, United States
MicroRNAs (miRNAs) are known regulators of lipid homeostasis. We recently demonstrated that miR-29 controls the levels of circulating cholesterol and triglycerides, but the mechanisms remained unknown. In the present study, we demonstrated that systemic delivery of locked nucleic acid inhibitor of miR-29 (LNA29) through subcutaneous injection effectively suppresses hepatic expression of miR-29 and dampens de novo lipogenesis (DNL) in the liver of chow-fed mice. Next, we used mice with liver-specific deletion of Sirtuin 1 (L-Sirt1 KO), a validated target of miR-29, and demonstrated that the LNA29-induced reduction of circulating triglycerides, but not cholesterol, is dependent on hepatic Sirt1. Moreover, lipidomics analysis revealed that LNA29 suppresses hepatic triglyceride levels in a liver-Sirt1 dependent manner. A comparative transcriptomic study of liver tissue from LNA29-treated wild-type/floxed and L-Sirt1 KO mice identified the top candidate lipogenic genes and hepatokines through which LNA29 may confer its effects on triglyceride levels. The transcriptomic analysis also showed that fatty acid oxidation (FAO) genes respond differently to LNA29 depending on the presence of hepatic Sirt1. Overall, this study demonstrates the beneficial effects of LNA29 on DNL and circulating lipid levels. In addition, it provides mechanistic insight that decouples the effect of LNA29 on circulating triglycerides from that of circulating cholesterol.
Introduction
The liver is the primary site of lipid synthesis and is a major source of circulating lipids (Nguyen et al., 2008; Tiwari and Siddiqi, 2012). Dysregulation of hepatic lipid metabolism is involved in the pathogenesis of many metabolic disorders. For example, abnormal lipid accumulation in the liver is the hallmark of fatty liver disease (Matsuzaka et al., 2007; Dorn et al., 2010) and exacerbates insulin resistance (Banerji et al., 1995; Bajaj et al., 2003). Elevated lipid synthesis and secretion by the liver can cause hyperlipidemia, which is a significant risk factor for atherosclerosis and cardiovascular disease (Zoltowska et al., 2001; Adiels et al., 2008). Given the critical contribution of hepatic lipid metabolism to various metabolic diseases and related complications, there have been numerous efforts to identify key regulators of lipid levels (Singh et al., 2017; Townsend and Newsome, 2017), as these represent candidate therapeutic targets.
Accumulating evidence suggests that microRNAs (miRNAs) are key post-transcriptional regulators of lipid homeostasis (Moore, 2013; Vickers et al., 2013a). We and others have demonstrated that miRNAs play an important role in regulating lipid metabolism in the liver. For example, we identified that hepatic miR-27b acts as a master regulator of lipid balance through modulation of genes encoding key lipogenic enzymes such as Gpam (Vickers et al., 2013b). Another miRNA in the liver, miR-30c, is involved in the control of lipoprotein secretion by targeting Mttp (Soh et al., 2013). In addition, we and others demonstrated that hepatic miR-223 coordinates cholesterol biosynthesis, uptake and efflux in the liver in part through regulation of Abca1 (Vickers et al., 2014). Additional studies have identified other miRNAs that are involved in different aspects of lipid regulation in the liver, including but not limited to miR-34a (Choi et al., 2013), miR-33a/b (Najafi-Shoushtari et al., 2010; Rayner et al., 2011), miR-122 (Esau et al., 2006; Cheung et al., 2008), miR-192 (Lin et al., 2017), and miR-148a (Cheng et al., 2017; Heo et al., 2019), several of which have been shown to be dysregulated in fatty liver disease and hyperlipidemia (Jeon and Osborne, 2016).
Recently, we uncovered the role of another miRNA, miR-29, in regulating lipid homeostasis. First we demonstrated that miR-29 is significantly elevated in the livers of mice fed an obesogenic high-fat diet as well as in Zucker Diabetic Fatty (fa/fa) rats, and may be involved in fine-tuning the liver FOXA2 transcriptional network (Kurtz et al., 2014). Later we showed using locked nucleic acid (LNA) inhibitors (Veedu and Wengel, 2010) that 1-week, acute suppression of miR-29 (LNA29) in vivo effectively reduces hepatic miR-29 levels and leads to significant reduction of circulating triglyceride and cholesterol in adult chow-fed mice (Kurtz et al., 2015). We postulated in that study that the lipid-lowering effect induced by LNA29 may be in part due to elevated Sirtuin 1 (Sirt1), which is a direct target of miR-29 (Kurtz et al., 2015). However, the mechanisms underpinning the effects of LNA29 were not fully understood. In the present study, we demonstrate that acute suppression of miR-29 dampens hepatic de novo lipogenesis (DNL) in the mouse liver and show that Sirt1 mediates the effects of miR-29 inhibition on circulating triglycerides but not cholesterol.
Materials and Methods
Animals
Mouse studies – The mice enrolled in the in vivo LNA29 studies were maintained on a 12 h light/dark cycle with access to diet and water ad libitum. To evaluate the acute effects of LNA29 on lipid homeostasis, C57BL/6J mice (female 9–11 weeks old) on chow-diet (Lab Diet 5047) were used. To evaluate the role of hepatic Sirt1 in mediating the effects of LNA29 on lipid homeostasis, the mouse model of liver-specific knockout of Sirt1 and floxed control littermates (female 15–20 week old mice) with C57BL/6J background were used (Purushotham et al., 2009). In these studies, the blood and the liver tissues collected during the fed state were used for plasma metabolite analysis, RNA-seq analysis, and lipidomics. In addition, liver tissue from a subset of mice were collected from the fasted state as the standard procedure for de novo lipid synthesis assay, in which the tissues are incubated with radiolabeled acetate substrates ex vivo.
Rat studies – The rats included in the studies were maintained on a 12 h light/dark cycle with access to diet and water ad libitum. To evaluate the association of hepatic miR-29 levels and expressions of lipogenesis genes, 5.5 month-old male fa/fa Zucker Fatty rats (ZFRs) and control Zucker Lean rats (ZLRs) fed on chow diet were used. To study the hepatic genes in response to 3,6-dichlorobenzo[b]thiophene-2-carboxylic acid (BT2) in the ZFR model, 3 month-old male ZFR received daily intraperitoneal injections of BT2 (20 mg/kg; dissolved in sterile dimethylsulfoxide) or vehicle (equal volume of sterile dimethylsulfoxide) for 1 week. The injections were performed daily at Zeitgeber time (ZT) 3. More details of BT2 treatments are described in White et al. (2018). Liver tissue was collected during fed state for RNA-seq analysis.
All animal procedures were performed with the approval and authorization of the Institutional Animal Care and Use Committee (IACUC) at each participating institution.
In vivo LNA Study
Animals received one subcutaneous injection of RNase-free sterile saline (Bio Scientific, Austin, TX, United States) or LNAs against mmu-miR-29a-3p (5′-ATTTCAGATGGTGCT-3′) and mmu-miR-29bc-3p (5′-ATTTCAAATGGTGCT-3′) at 20 mg/kg each (Qiagen) at ZT 3. We have demonstrated previously that one subcutaneous injection of LNA against miR-29 with this concentration effectively suppresses hepatic miR-29 expressions (Kurtz et al., 2015). We have also demonstrated in a previously published study that saline and LNA-scramble sequence (LNA-scr) exert similar effects on our physiological parameters of interest and therefore both can serve as control treatment for the LNA-29 in vivo study (Hung et al., 2019). We therefore used saline as control treatment in the present study. For radiolabeled assays of de novo lipid synthesis, fresh liver was collected from overnight fasted animals at day 7 post-dose of LNA. For plasma metabolite and transcriptome analysis, blood and liver were collected from non-fasted animals at day 7 post-dose of LNA. The in vivo LNA study was performed at National Health Institutes as well as at NYU Winthrop Hospital. No significant differences were observed in body weights at day 7 post-dose between saline and LNA29 treatment. All animal procedures were performed with the approval and authorization of the IACUC at each participating institution.
Plasma Metabolite Analysis
Plasma was prepared by spinning whole blood for 10 min at 10,000 × g, 4°C. Plasma was used to measure circulating cholesterol and triglyceride at the UNC Nutrition Obesity Research Center (NORC) Nutritional Biochemistry and Molecular Biology core facility.
De novo Lipid Synthesis Assay
3H-acetate was purchased from NEN Life Science Products. Mice were fasted overnight (16 h) before anesthesia. 1 ml of serum free DMEM containing 100 uCi of 3H-acetate was added to each well in 12-well plate and pre-warmed at 37°C incubator for 20 min. About 50 mg of fresh liver pieces were weighed and incubated with isotope containing medium for 3 h. The wet weight was used for normalization later. After incubation, tissues were washed three times with PBS, chopped into fine pieces, and placed into 750 μl of chloroform: methanol (1:2) solution overnight. The next day, 250 μl of chloroform was added to each tube and incubated in room temperature for 15 min. 250 μl of distilled water was added to each tube and the tubes were centrifuged for 10 min at 12,000 × g. The bottom layer was taken and evaporated overnight. Total lipids were resuspended in 100 μl of chloroform. Different lipids were separated on silica-60 thin layer chromatography (TLC) plates (Hexane: diethyl ether: glacial acetic acid = 82:17:2). Pure TG, cholesterol, and phospholipid were used as markers on TLC plate. The plates were exposed to iodine to visualize bands. The bands containing TG, cholesterol, or phospholipid were scraped off from the plates and counted in a scintillation counter.
RNA Extraction and Real-Time qPCR
Total RNA was isolated using the Total RNA Purification kit (Norgen Biotek, Thorold, ON, Canada). High Capacity RNA to cDNA kit (Life Technologies, Grand Island, NY, United States) was used for reverse transcription of RNA. TaqMan microRNA Reverse Transcription kit (Life Technologies) was used for reverse transcription of miRNA. Both miRNA and gene expression qPCR were performed using TaqMan assays (Life Technologies) with either TaqMan Universal PCR Master Mix (miRNA qPCR) or TaqMan Gene Expression Master Mix (mRNA qPCR) per the manufacturer’s protocol, on a BioRad CFX96 Touch Real Time PCR Detection System (Bio-Rad Laboratories, Richmond, CA, United States). Reactions were performed in triplicate using either U6 (miRNA qPCR) and Rps9 (mRNA qPCR) as the normalizer.
RNA Library Preparation and Sequencing
RNA-sequencing libraries of the liver were prepared using the Illumina TruSeq polyA + Sample Prep Kit. Sequencing of mouse livers harvested during fed state was carried out on the Illumina HiSeq2000 platform with single-end 100 bp sequencing depth at the UNC High Throughput Sequencing Core Facility. Sequencing of ZFR livers harvested during fed state was performed using the same sequencing platform and parameters at the Cornell RNA-sequencing core facility. Raw sequencing data as well as normalized counts are available through GEO accession GSE129539.
Bioinformatics Analysis
The sequencing reads were aligned to mouse genome (mm10) for mouse samples and rat genome (rn6) for rat samples. RNA-sequencing reads were mapped to genome release using STAR (v2.5.3a) (Dobin et al., 2013) and transcript quantification was performed using Salmon (v0.6.0) (Patro et al., 2017). Differential gene expression analysis was accomplished using DESeq2 (Love et al., 2014). The Benjamini–Hochberg method was used for multiple testing correction. One sample in the group of floxed mice receiving saline treatment was considered as an outlier based on the PCA analysis and thereby excluded from the entire RNA-seq analyses. MiRNA binding site enrichment among differentially expressed genes was determined using miRhub (Baran-Gale et al., 2013). Gene pathway analyses with specific subsets of genes were performed using Enrichr (Chen et al., 2013; Kuleshov et al., 2016) and the results were shown using WikiPathways’s cell signaling pathway database.
Hepatic Lipid Extraction for Lipidomics
Snap-frozen liver tissues (10 mg) were homogenized in 300 μL chilled 50% methanol (OptimaTM LC/MS Grade, A456-500, Fisher Chemical) diluted in HPLC grade water (Fisher Chemical, W5-1). 600 μL of methylene chloride (OptimaTM LC/MS Grade, D151-1, Fisher Chemical) was added to the homogenates and vortexed for 10 s. 300 μL HPLC grade water was then added and vortexed for 10 s. The tissue homogenates were centrifuged at 13,000 rpm for 15 min at 4°C. The bottom organic layer was collected into silica tubes and the supernatants were air dried by speed vacuum. Samples were redissolved in 150 μL of isopropanol (IPA)/acetonitrile (ACN)/H2O (65:30:5) prior to analysis. 25 μg/mL of TG (15:15:15) were added as internal standard prior to analysis and used for normalization.
Liquid Chromatography and Mass Spectrometry
Chromatographic separation was performed on a Vanquish UHPLC system with an Accucore C30, 2.6 μm column (2.1 mm id × 150 mm) coupled to a Q ExactiveTM Hybrid Quadrupole-Orbitrap High Resolution Mass Spectrometer (Thermo Fisher Scientific, San Jose, CA, United States). The mobile phase consisted of solvent A containing 60% ACN, 40% H2O, 10 mM Ammonium Formate with 0.1% Formic Acid and solvent B containing 90% IPA, 10% ACN, 10 mM Ammonium Formate with 0.1% Formic Acid. The gradient was as follows: 0–1.5 min, 32% solvent B; 1.5–4 min, 32–45% solvent B; 4–5 min, 45–52% solvent B; 5–8 min 52–58% solvent B; 8–11 min, 58–66% solvent B; 11–14 min, 66–70% solvent B; 14–18 min, 70–75% solvent B; 21–25 min, isocratic 97% solvent B, 25–25.1 min 97–32% solvent B; followed by 4 min of re-equilibration of the column before the next run. The flow rate was 260 μl/min and the Injection volumes were set to 2 μL. To avoid possible bias, the sequence of injections was randomized. All of the samples were analyzed by negative electrospray ionization in data-dependent MS-MS mode, Nitrogen as sheath, auxiliary, and sweep gas was set at 50, 5, and 1 U, respectively. Other conditions included the following: resolution, 1,20,000 full width at half maximum; automatic gain control target, 3e6 ions; maximum injection time, 100 ms; scan range, 67–1000 m/z; spray voltage, 3.50 kV; and capillary temperature, 275°C. Data-dependent MS-MS spectra were generated through the use of the following conditions: resolution, 15,000 full width at half maximum; automatic gain control target, 1e5 ions; maximum injection time, 50 ms; isolation window, 0.4 m/z.
Lipidomics Data Analysis
The acquired data set was processed using Thermo Scientific LipidSearchTM software version 4.1 with the following workflow. First, the individual data files were searched for product ion MS/MS spectra of lipid precursor ions. MS/MS fragment ions were predicted for all precursor adduct ions measured within ±5 ppm. The product ions that matched the predicted fragment ions within a ±5 ppm mass tolerance were used to calculate a match-score, and those candidates providing the highest quality match were determined. Next, the search results from the individual positive ion files from each sample group were aligned within a retention time window (±0.1 min) and the data were merged for each annotated lipid. The annotated lipids that were not ionized by NH4+ were then filtered to reduce false positives. The resulting data of TG (with [M + NH4]+ ion) was analyzed using MetaboAnalyst (Chong and Xia, 2018; Chong et al., 2018). Specifically, the data was normalized by tissue weight followed by log transformation and Pareto scaling. Two-tailed Student’s t tests were performed to calculate raw P-values and adjusted P-values (FDR).
Statistics
The quantitative data are reported as an average of biological replicates ± standard error (SE) of the mean. Statistical differences were assessed by two-tailed Student’s t-test with threshold P-value <0.05. For the experiments that were performed previously and repeated in the present study, the statistical differences were assessed by one-tailed Student’s t-test with threshold P-value <0.05. Given that we only had n = 2 for the group of floxed mice receiving saline treatment and that there is no significant difference between WT and floxed mice in circulating cholesterol (WT 95 ± 10 mg/dL; floxed 105 ± 5 mg/dL) and TG (WT 86 ± 15 mg/dL; floxed 92 ± 19 mg/dL), we pooled WT and floxed mice into one group for meaningful statistical comparisons.
Results
Acute Systemic Suppression of miR-29 Dampens de novo Lipid Synthesis in the Liver and Lowers Circulating Lipid Levels
We have previously demonstrated that one dose of LNA29 injection (20 mg/kg for LNA29a and LNA29b/c each) in chow-fed C57BL/6J mice significantly lowers circulating triglyceride (TG) and cholesterol levels compared to saline treatment (Kurtz et al., 2015). We have also previously reported that LNA-mediated suppression of the miR-29 family leads to hepatic transcriptome profiles that indicate suppression of cholesterol and fatty acid synthesis pathways (Kurtz et al., 2015). However, whether LNA29 treatment in mice suppresses hepatic lipid synthesis in vivo has not yet been examined. To address this, we first repeated the LNA29 experiment, in which the chow-fed C57BL/6J mice received one subcutaneous injection of either LNA29 or saline control. Fresh liver tissues were collected from fasted mice at day 7 post-dose for DNL assays. We found that LNA29 treatment significantly suppresses the levels of hepatic miR-29 family members (miR-29a, b and c exhibit 444-, 126-, and 614-fold suppression, respectively) and significantly elevates a validated miR-29 target gene Col3a1 compared to saline treatment as determined by qPCR (Figures 1A,B). We showed that LNA29 moderately decreases 3H-acetate incorporation into cholesterol as well as TG but has no effects on phospholipids compared to saline control (Figures 1C–E). Also, as shown previously (Kurtz et al., 2015), non-fasted circulating levels of TG and cholesterol were significantly lowered by LNA29 compared to saline treatment (Figures 1F,G). These findings overall suggest that the moderate reduction of hepatic DNL may partially contribute to the significant reduction of circulating lipids upon treatment with LNA29. Note that treatment with LNA inhibitors of another liver-expressed miRNA, miR-455, has little effects on liver lipids (Supplementary Figures S1A–C), which demonstrates that the effects of LNA29 on circulating lipids and DNL does not represent a generic effect of LNAs but rather a specific effect of suppressing miR-29.
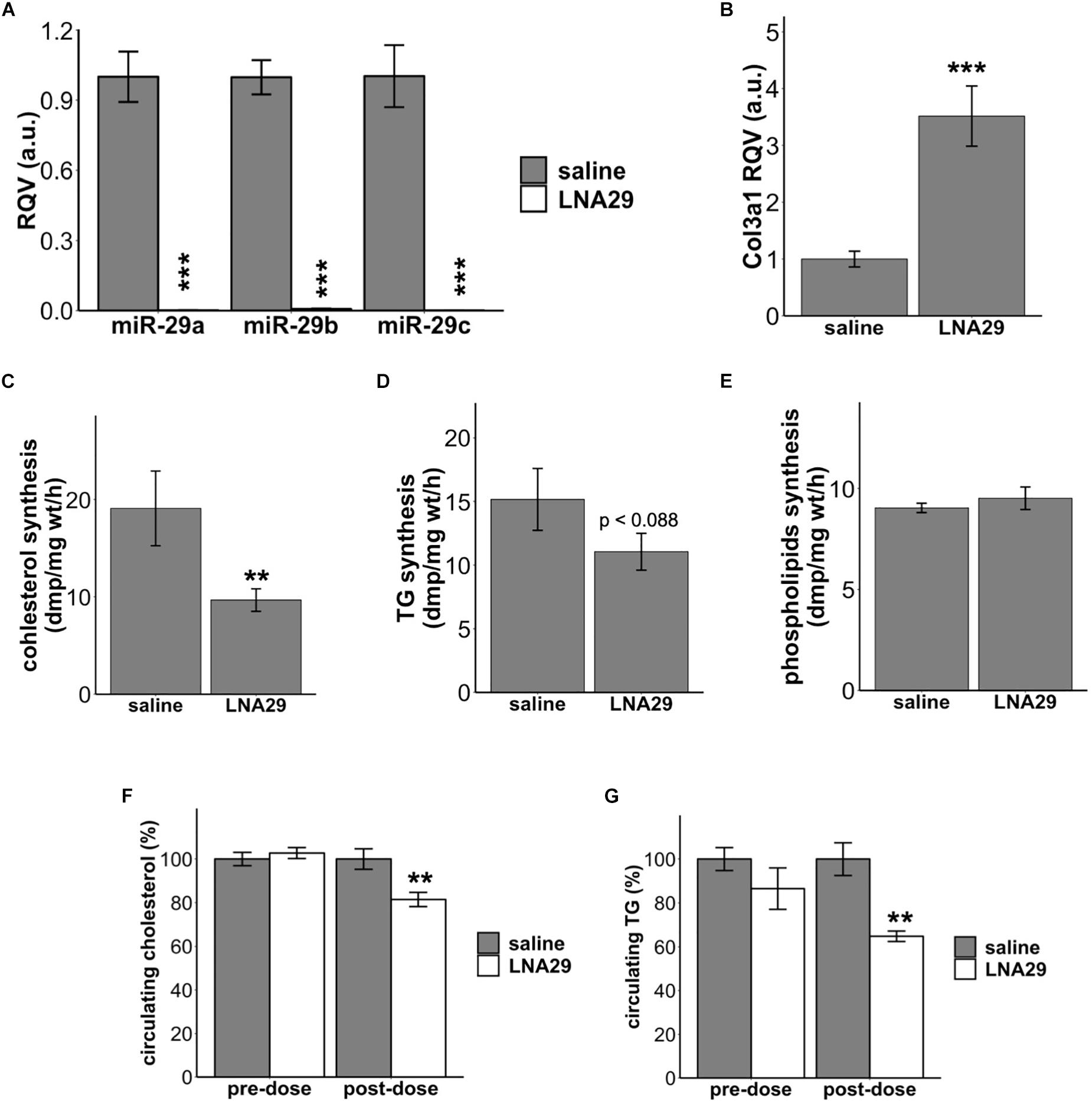
Figure 1. Acute treatment of LNA29 inhibits de novo lipid synthesis in the liver and lowers circulating cholesterol and triglyceride (TG) in chow-fed wild type (WT) mice. (A) RT-qPCR of hepatic miR-29 family of chow-fed WT mice with saline or LNA29 treatment. LNA29 leads to 444, 126, 614 fold suppression in miR-29a, b, and c, respectively. (B) qPCR of Col3a1, a validated miR-29 target gene, of chow-fed WT mice with saline or LNA29 treatment. (C) 3H-acetate incorporation into cholesterol with liver samples harvested from saline- or LNA29-treated WT mice. (D) 3H-acetate incorporation into TG with liver samples harvested from saline- or LNA29-treated WT mice. (E) 3H-acetate incorporation into phospholipids with liver samples harvested from saline- or LNA29-treated WT mice. (F) Non-fasted circulating cholesterol of mice with saline or LNA29 treatment. (G) Non-fasted circulating TG of mice with saline or LNA29 treatment. Given that the circulating data presented in this figure is a repeat of our previous findings, the one-tailed Student’s t-test was used. ∗∗P < 0.01, ∗∗∗P < 0.001 by one-tailed Students’ t test (saline, n = 6; LNA29, n = 6).
The Reduction of Circulating TG by LNA29 Is Abolished in the Absence of Liver Sirt1
We have previously validated that Sirtuin-1 (Sirt1) is a miR-29 target gene and that protein levels of hepatic Sirt1 are increased by LNA29 treatment in mice (Kurtz et al., 2015). Given that Sirt1 is known to be involved in the regulation of lipid metabolism in the liver (Purushotham et al., 2009; Yin et al., 2014) and that its expression is post-transcriptionally regulated by miR-29 (Bae et al., 2014), we hypothesized that the LNA29-induced lipid lowering effects are in part mediated by loss of miR-29 regulation of Sirt1 in the liver. To test this hypothesis, we employed a liver-specific Sirt1 knockout (L-Sirt1 KO) mouse model (Purushotham et al., 2009). A set of L-Sirt1 KO mice and control mice, including both C57BL/6J wild type and floxed (WT/Floxed) mice, were treated with LNA29 or saline and examined at day 7 post-dose (Supplementary Figure S2A). In the absence of hepatic Sirt1, LNA29 was still able to effectively suppress the miR-29 family and increase its target gene Col1a1 (Supplementary Figures S2B,C). We found that non-fasted circulating levels of cholesterol are significantly lowered by LNA29 in both L-Sirt1 KO and WT/Floxed mice (Figures 2A,B; by Student’s t-test). Notably, however, we found that non-fasted circulating levels of TG are significantly lowered by LNA29 only in WT/Floxed mice, and not in L-Sirt1 KO mice (Figures 2C,D; by Student’s t-test). A two-way ANOVA test was also applied to examine the effect of treatment type (saline vs. LNA29), genotype (WT/floxed vs. KO), and the interaction on the circulating cholesterol and TG. Consistent with the conclusions made by Student’s t-test, while the treatment effect is significant (P = 1.78 × 10–5; type II test), the treatment:genotype interaction does not exert significant impact (P = 0.343; type II test) in the context of LNA29 lowering blood cholesterol. In contrast, the treatment:genotype interaction is suggested to be a non-negligible factor (P = 0.068; type II test) in the context of LNA29 lowering blood TG. Taken together, these observations suggest that the LNA29-induced lowering of circulating cholesterol is hepatic Sirt1-independent, whereas the LNA29-induced lowering of circulating TG is hepatic Sirt1-dependent.
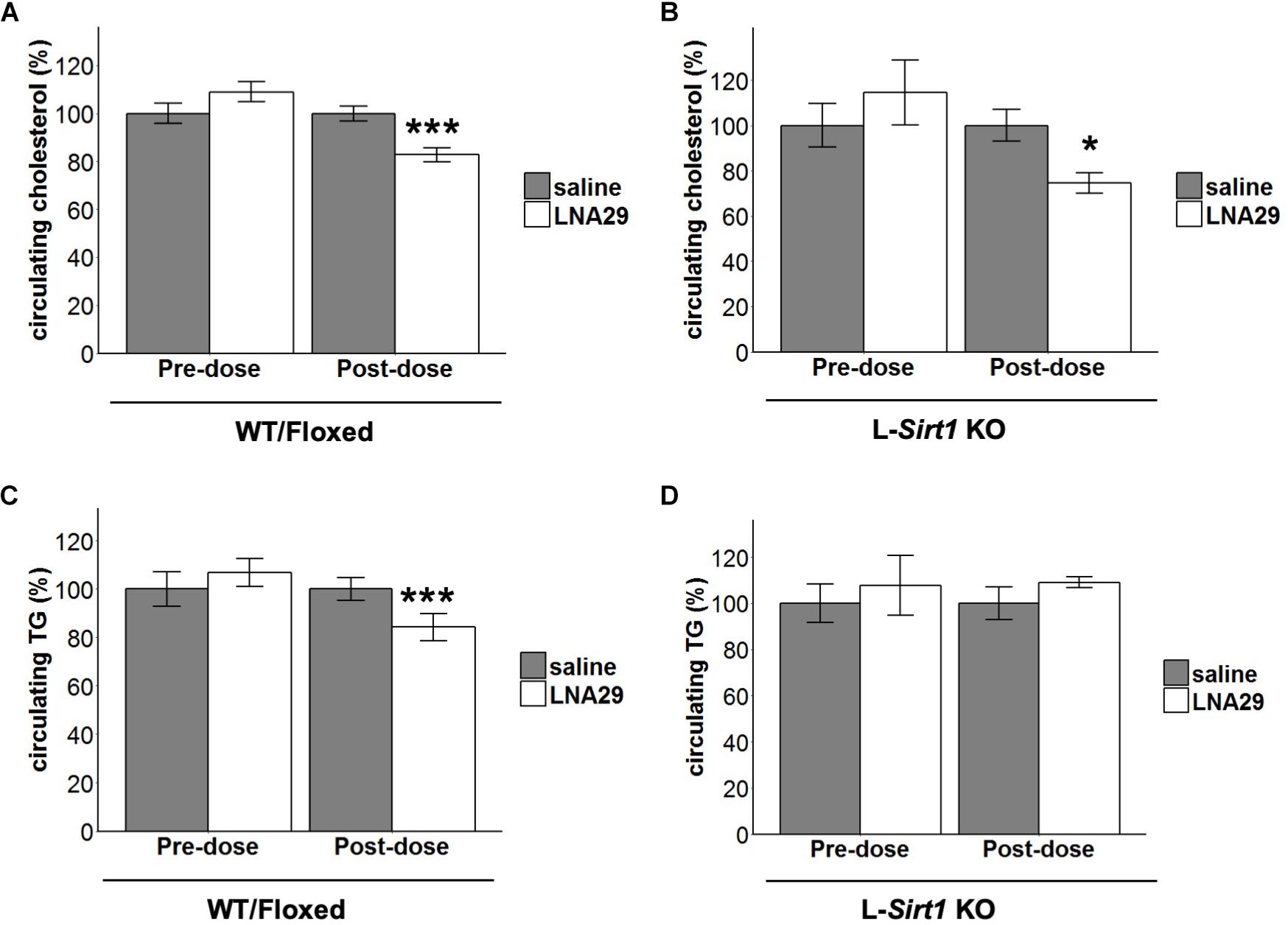
Figure 2. The absence of hepatic Sirt1 blunts LNA29-induced reduction of circulating triglyceride (TG). (A) Non-fasted circulating cholesterol of WT/Floxed mice with saline or LNA29 treatment. (B) Non-fasted circulating cholesterol of L-Sirt1 KO mice with saline or LNA29 treatment. (C) Non-fasted circulating TG of WT/Floxed mice with saline or LNA29 treatment. (D) Non-fasted circulating TG of L-Sirt1 KO mice with saline or LNA29 treatment. Data of WT/Floxed mice in response to LNA29 was assessed by one-tailed Student’s t-test (Floxed/WT, n = 11–12/treatment group) as the data is a validation of our previous findings. The data of L-Sirt1 KO mice in response to LNA29 was assessed by two-tailed Student’s t-test (L-Sirt1 KO, n = 5–6/treatment group). ∗P < 0.05, ∗∗∗P < 0.001.
Identification of Sirt1-Independent and Sirt1-Dependent Gene/Pathway Changes in the Liver After LNA29 Treatment
In order to characterize the hepatic Sirt1-independent mechanisms that mediate in part the beneficial effects of LNA29, we performed RNA-seq analysis on the liver of L-Sirt1 KO and control WT/Floxed mice treated with either LNA29 or saline. In WT/Floxed mice, we identified a total of 2397 genes (1254 up; 1143 down) that are altered by LNA29 (Supplementary Figures S2D,E; P < 0.05). In L-Sirt1 KO mice, we identified a total of 1987 genes (1098 up; 889 down) that are altered by LNA29 (Supplementary Figures S2H,I; P < 0.05). In theory, miR-29 target genes should be upregulated by LNA29 treatment. By performing miRhub analysis (Baran-Gale et al., 2013), we confirmed that the genes that are upregulated by LNA29 are significantly enriched for predicted target sites of miR-29 in both WT/Floxed (Supplementary Figure S2F) and L-Sirt1 KO mice (Supplementary Figure S2J), whereas the genes that are downregulated by LNA29 are not in both groups (Supplementary Figures S2G,K).
The genes that are altered by LNA29 in both WT/Floxed and L-Sirt1 KO mice are by definition those that are regulated in a Sirt1-independent manner. By comparing the set of genes that are altered in LNA29-treated WT/Floxed and L-Sirt1 KO mice (P < 0.05, adjusted P < 0.2, base mean >50, fold change >1.2 compared to saline) (Figures 3A,C), we identified subsets of genes that are unique to WT/Floxed mice (gene list i in Figure 3A and iv in Figure 3C), unique to L-Sirt1 KO mice (gene list iii in Figure 3A and vi in Figure 3C), or shared (gene list ii in Figure 3A and v in Figure 3C). The shared genes that are upregulated by LNA29 (gene list ii in Figure 3A) are most enriched in the extracellular matrix (ECM) pathway (Supplementary Figure S3A), which is consistent with the published evidence of a well-established role for miR-29 in regulating ECM genes (Luna et al., 2009; Roderburg et al., 2011). We identified several other shared upregulated genes, such as Enho, Igfbp2, Lpl, and Sparc (gene list ii in Figure 3A), that are involved in regulating energy balance and/or metabolism (Figure 3B). In addition, among the small subset of downregulated genes shared between WT/Floxed and L-Sirt1 KO mice in response to LNA29 (gene list v in Figure 3C and Supplementary Figure S3D) were Ldhd, Fitm1 and Onecut2 (Figure 3D), which are involved in the process of lipogenesis.
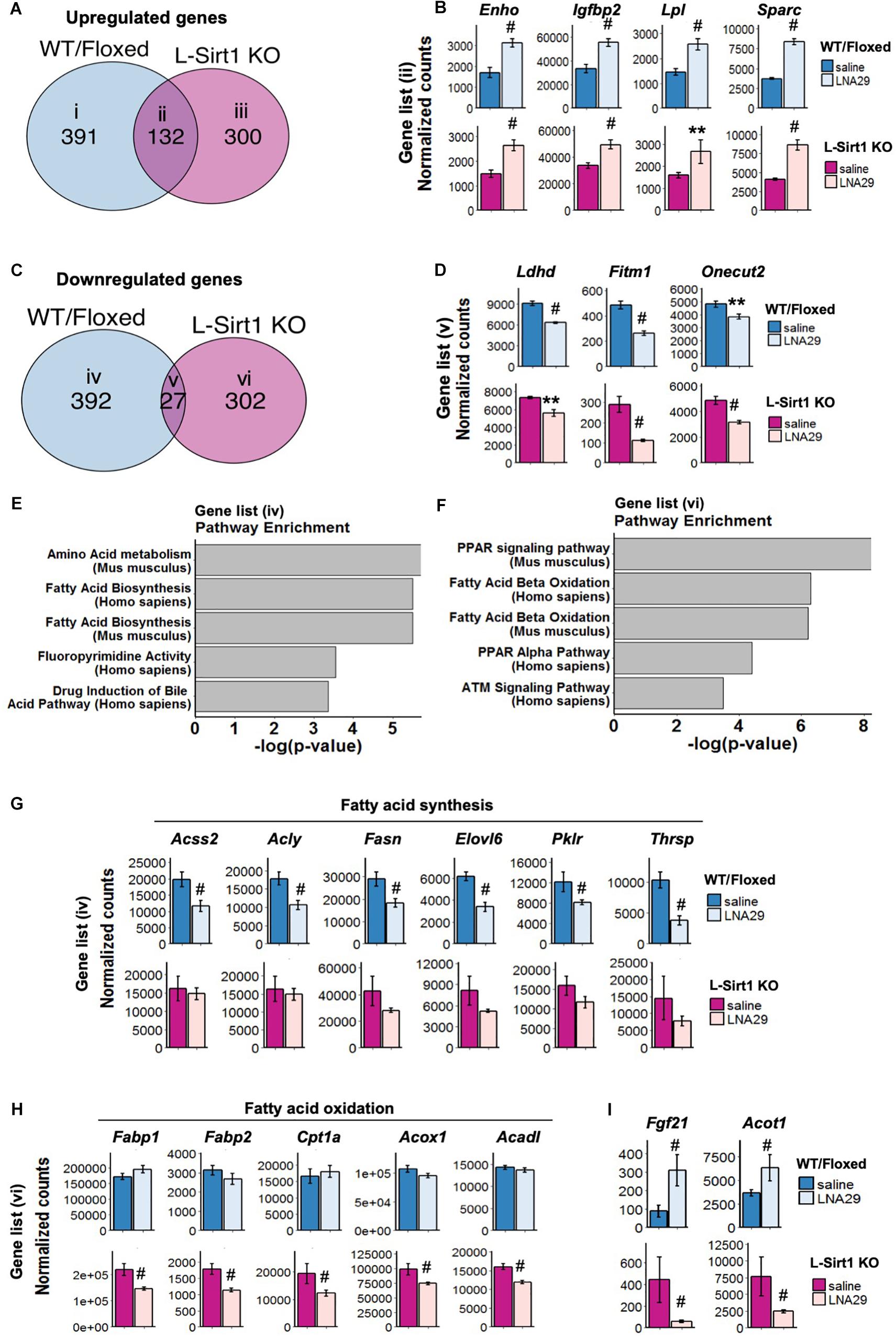
Figure 3. The absence of hepatic Sirt1 blunts the suppression of lipogenesis genes and downregulates fatty acid oxidation (FAO) genes upon LNA29 treatment. (A) Venn diagram of upregulated genes that are shared or unique in WT/Floxed and L-Sirt1 KO mice in response to LNA29 (P < 0.05, adjusted P < 0.2, base mean >50, fold change>1.2). (B) Expressions of upregulated genes shared between WT/Floxed and L-Sirt1 KO mice in response to LNA29. (C) Venn diagram of downregulated genes that are shared or unique in WT/Floxed and L-Sirt1 KO mice in response to LNA29 (P < 0.05, adjusted P < 0.2, base mean>50, fold change 1.2). (D) Expressions of downregulated genes shared between WT/Floxed and L-Sirt1 KO mice in response to LNA29. (E) Pathway analysis of the downregulated genes unique to WT/Floxed mice in response to LNA29. (F) Pathway analysis of the downregulated genes unique to L-Sirt1 KO mice in response to LNA29. (G) Lipogenesis genes are downregulated in WT/Floxed but blunted in L-Sirt1 KO mice in response to LNA29. (H) FAO genes are unchanged in WT/Floxed but downregulated in L-Sirt1 KO mice in response to LNA29. (I) Genes that are changed in opposite direction in WT/Floxed and L-Sirt1KO mice in response to LNA29. ∗∗P < 0.01, #P < 0.001 by Wald test (DESeq2) (WT/Floxed-saline, n = 5; WT/Floxed-LNA29, n = 8; L-Sirt1 KO-saline, n = 4; L-Sirt1 KO-LNA29, n = 4).
To investigate how the absence of hepatic Sirt1 blunts the LNA29-induced TG lowering effect (Figure 2D), we turned our attention to the genes that are altered by LNA29 only in WT/Floxed or in L-Sirt1 KO mice but not in both (Figures 3E,F and Supplementary Figures S3B,C,E,F). We found that the genes that are downregulated by LNA29 only in WT/Floxed mice (gene list iv in Figure 3C) are significantly enriched in the fatty acid synthesis pathway (Figure 3E). Several key lipogenic genes, including Acss2, Acly, Fasn, Elovl6, Pklr, and Thrsp (though especially the first two), are significantly downregulated in WT/Floxed but not in L-Sirt1 KO mice (Figure 3G). In addition, the genes that downregulated by LNA29 only in L-Sirt1 KO mice (gene list vi in Figure 3C) are significantly enriched in fatty acid oxidation (FAO) pathways (Figure 3F), including Fabp1, Fabp2, Cpt1a, Acox1, and Acadl (Figure 3H). Taken together, these data show that in the absence of hepatic Sirt1, the inhibition of lipogenic genes is blunted and instead FAO genes are significantly downregulated in response to LNA29 treatment. These findings are consistent with the importance of hepatic Sirt1 in mediating the LNA29-induced TG lowering effect (Figure 2D). Interestingly, we also identified genes that are oppositely affected by LNA29 treatment depending on the Sirt1 genotype (Supplementary Figures S3E,F), which may also contribute to the difference in TG phenotype between LNA29-treated WT/Floxed and L-Sirt1 KO mice.
The Effect of LNA29 on Lipogenesis Genes Are Shared With That of BT2 Compound Known to Reduce Liver Fat Partly Through the Suppression of miR-29
The aforementioned RNA-seq analysis of the liver from LNA29-treated L-Sirt1 KO and WT/Floxed mice suggests that changes in lipogenesis and FAO pathways together determine the LNA29-induced TG lowering phenotype through either Sirt1-dependent or Sirt1-independent manner (Figure 3). We next examined the genes involved in these pathways in an animal model of fatty liver, the ZFR. A recent study described that ZFRs, which develop obesity and NAFLD, have reduced hepatic TG levels upon 1 week treatment with the BT2 compound (inhibitor of branched chain α-keto acid dehydrogenase kinase) (White et al., 2018). We observed elevated miR-29 levels in the liver of ZFRs compared to control ZLRs and a rescue of miR-29 levels in the liver of ZFRs upon BT2 treatment (Supplementary Figures S4A,B), indicating that the reduction of hepatic miR-29 levels is associated with the TG-lowering phenotype of BT2 treatment. We also showed that the same lipogenic genes that are downregulated by LNA29 treatment in a Sirt1-dependent manner (Figure 3G) are significantly upregulated in ZFRs compared to ZLRs and significantly downregulated in ZFRs after BT2 treatment (Figure 4A). Among these lipogenic genes are Acly, Fasn, Thrsp, and Pklr, for which protein levels were also significantly reduced as determined by unbiased proteomics analysis (Supplementary Figure S4C). It is worth noting that we found the FAO genes, which are altered by LNA29 only when Sirt1 is absent in the liver (Figure 3H), are not changed much in ZFRs compared to ZLRs or in response to BT2 treatment, with the singular exception of Fabp1 (Figure 4B). Taken together, these findings suggest that the effects of BT2, a compound shown to ameliorate fatty liver (White et al., 2018), on lipogenic and FAO genes in the ZF rat liver are consistent with the effects of LNA29 on the same pathways in the chow-fed mouse liver. This provides further support to the notion that the TG-improving effect of LNA29 is mediated by the suppression of lipogenic genes.
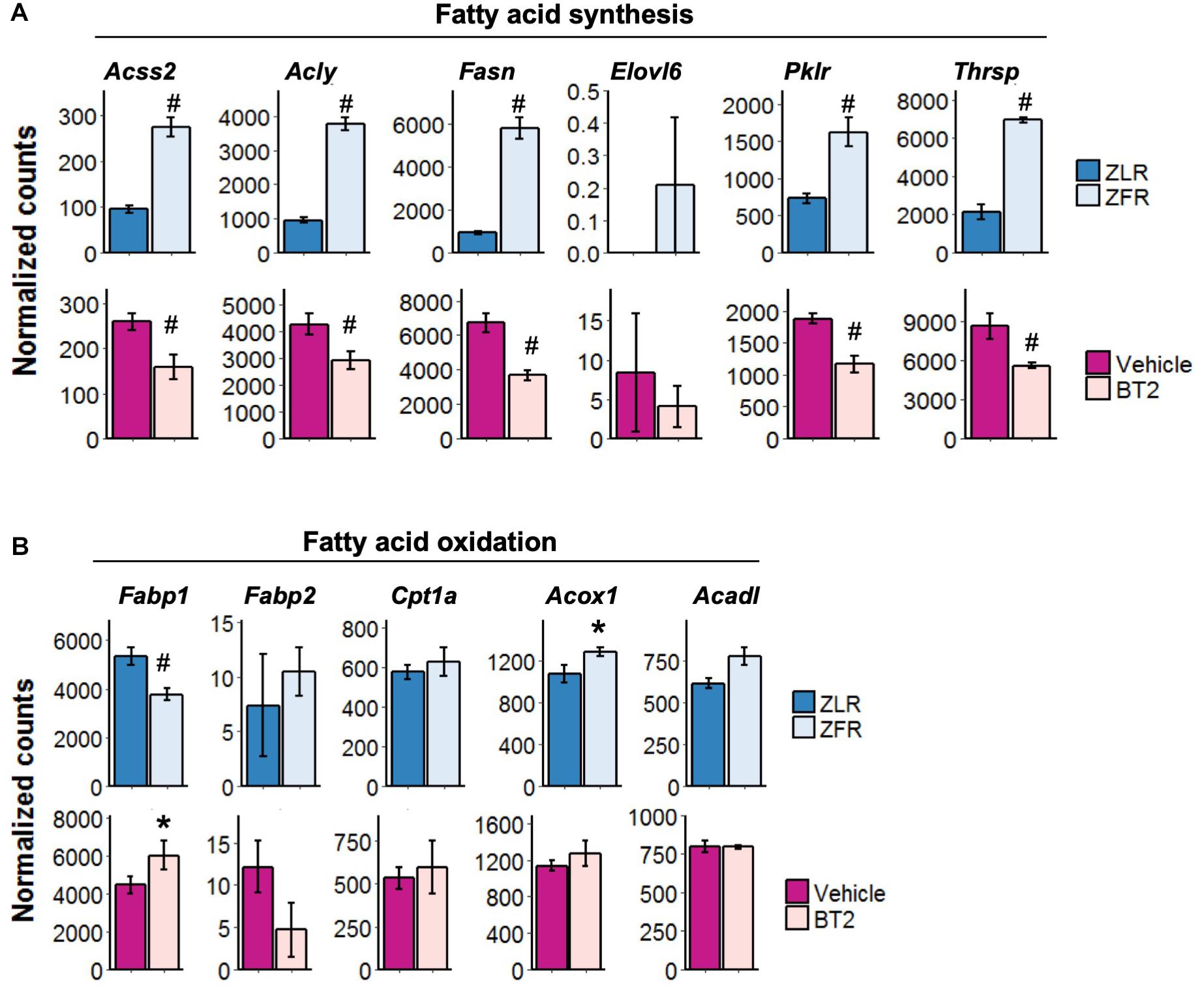
Figure 4. The effect of LNA29 on lipogenesis genes are shared with that of BT2 compound known to reduce liver fat partly through the suppression of miR-29. (A) Expressions of lipogenesis genes which are downregulated in LNA29-treated WT/Floxed mice in ZFR compared to ZLR (upper panel) and in ZFR upon BT2 treatment (lower panel). (B) Expressions of FAO genes which are downregulated in LNA29-treated L-Sirt1 KO mice are in ZFR compared to ZLR (upper panel) and in ZFR upon BT2 treatment (lower panel). FAO, fatty acid oxidation, ZFR, Zucker Fatty rat, ZLR, Zucker Lean rat. ∗P < 0.05 and #P < 0.001 by two tailed Students’ t test after Bonferroni correction (ZFR, n = 4; ZLR, n = 4; Vehicle, n = 4; BT2, n = 4).
LNA29 Treatment Leads to a Global Reduction of TG Species in the Liver Through a Hepatic Sirt1-Dependent Manner
Our results suggest that the LNA29-induced changes in genes involved in lipogenesis and FAO depends on the presence of hepatic Sirt1 (Figure 3). Specifically, LNA29-treated WT/Floxed mice exhibit downregulation of genes involved in lipogenesis, whereas LNA29-treatment in L-Sirt1 KO mice leads to little-to-no changes in lipogenic genes and downregulation of FAO genes (Figure 3). To validate whether the differences we observed at the transcriptomic level between WT/Floxed and L-Sirt1 KO mice leads to differences in hepatic TG content, we performed lipidomic analysis to profile TG species in the liver of WT/Floxed and L-Sirt1 KO mice with either saline or LNA29 treatment. We showed that LNA29 treatment leads to a global reduction of TG species in the liver of WT/Floxed mice (Figure 5A, Supplementary Figure S5, and Supplementary Table S1). In contrast, this LNA29-mediated hepatic TG lowering effect is lost in the liver of L-Sirt1 KO mice (Figure 5B, Supplementary Figure S5, and Supplementary Table S2). Overall, the finding suggests the critical role of hepatic Sirt1 in contributing to the LNA29-mediated TG lowering effect in the liver as well as in the circulation (Figure 5C).
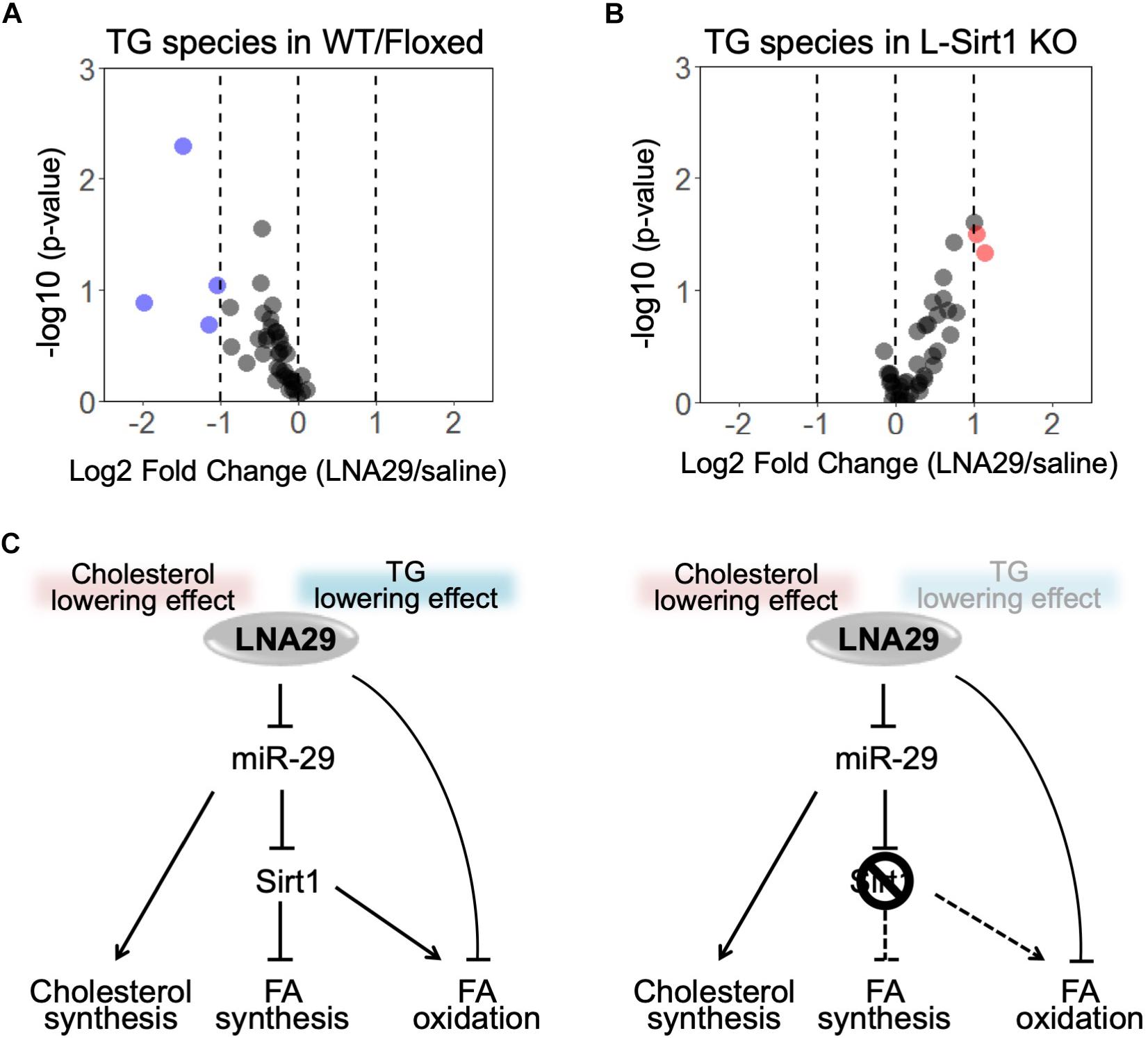
Figure 5. The absence of hepatic Sirt1 blunts LNA29-induced reduction of triglyceride (TG) in the liver. (A) Lipidomic analysis of changes in hepatic TG species in WT/Floxed mice with LNA29 compared to saline treatment (saline, n = 5; LNA29, n = 8). (B) Lipidomic analysis of changes in hepatic TG species in L-Sirt1 KO mice with LNA29 compared to saline treatment (saline, n = 5; LNA29, n = 5). (C) Proposed model of LNA29-mediated effects on hepatic lipid metabolism. The LNA29-mediated cholesterol lowering effect is hepatic Sirt1 independent. The LNA29-mediated TG lowering effect through the suppression of lipogenesis network is hepatic Sirt1 independent. LNA29 leads to downregulation in genes of fatty acid oxidation (FAO) only when hepatic Sirt1 is absent.
Discussion
Our previous studies demonstrated that LNA29 treatment improves lipid levels in circulation and highlighted hepatic Sirt1 as a candidate mediator of the effects of LNA29 (Kurtz et al., 2014, 2015). In the present study, we first performed radio-labeled assays to demonstrate that LNA29 treatment leads to suppression of DNL in mouse liver, which likely contributes to the reduction in circulating lipids observed in LNA29-treated mice. Secondly, we evaluated whether hepatic Sirt1 is critical for mediating the beneficial effects of LNA29 using liver-specific Sirt1-deficient (L-Sirt1 KO) mice. We showed that the LNA29-mediated reduction of liver and circulating TG, but not cholesterol, is in part dependent on hepatic Sirt1. Furthermore, lipidomics analysis revealed a systematic lowering of TG in the liver by LNA29 only in WT but not in L-Sirt1 KO mice, and comprehensive transcriptomic analysis identified the key fatty acid synthesis genes through which LNA29 may exert these effects.
By performing transcriptome analysis in L-Sirt1 KO mouse model, we were able to identify Sirt1-independent candidate genes that may mediate some LNA29 effects. For example, Enho, Igfbp2, Lpl, and Sparc are upregulated by LNA29 irrespective of the presence of Sirt1 (Figure 3B) and are known to be involved in improving energy homeostasis and/or lipid metabolism (Bradshaw et al., 2003; Wheatcroft et al., 2007; Kumar et al., 2008; Nie et al., 2011; Ganesh Kumar et al., 2012; Chen et al., 2017). Indeed, Enho, Lpl, and Sparc have been shown to be directly targeted by miR-29 (Yang et al., 2016; Song et al., 2018; Hung et al., 2019) and therefore the suppression of miR-29 would be expected to increase the expression and activity of these genes. Igfbp2, on the other hand, does not have predicted target sites for miR-29 and its upregulation by LNA29 is possibly through an indirect mechanism. These genes may contribute to some of the LNA29-induced beneficial effects through Sirt1-independent mechanisms. Recently, we reported that LNA29 treatment in WT mice exerts beneficial effects on insulin sensitivity in part through upregulation of hepatic Enho (Hung et al., 2019). Enho is also reported to improve cholesterol homeostasis (Ghoshal et al., 2018). In the present study we observed that upon LNA29 treatment L-Sirt1 KO mice exhibit elevated hepatic Enho and reduced circulating cholesterol. Whether Enho plays a critical role in contributing to the effects of LNA29 on cholesterol reduction through a Sirt1-independent manner merits detailed future investigation. In addition, Igfbp2 is known to be involved in the control of circulating cholesterol (Narayanan et al., 2014). Interrogating how this gene may contribute to the reduction of circulating cholesterol in LNA29-treated L-Sirt1 KO mice will also provide additional insights of Sirt1-independent LNA29 effects.
Transcriptome analysis in the L-Sirt1 KO mouse model also reveals Sirt1-dependent candidate genes that mediate some of the effects of LNA29, especially as it pertains to TG levels. The data shows that FAO genes respond differently to LNA29 depending on the presence of hepatic Sirt1. Specifically, after LNA29 treatment, we observed little-to-no changes in FAO genes in WT/Floxed control mice but dramatic reduction of FAO genes in L-Sirt1 KO mice. One hypothesis is that in WT mice hepatic Sirt1 exerts opposing regulatory effects on FAO genes through direct and indirect means, thereby resulting in minimal net changes, and that this balance is disturbed in L-Sirt1 KO mice (Figure 5C). Furthermore, Fgf21 and Acot1 are significantly upregulated in WT/Floxed mice but significantly downregulated in L-Sirt1 KO mice in response to LNA29 treatment (Figure 3I). Fgf21 has recently emerged as a potential therapeutic target for treating obesity and type 2 diabetes (Coskun et al., 2008; Jimenez et al., 2018). Acot1 has been reported to regulate PPARα-mediated FAO (Dongol et al., 2007). Thus, the downregulation of Fgf21 and Acot1 in the absence of hepatic Sirt1 may also diminish the TG lowering effect of LNA29. It is important to note, however, that FAO usually occurs in the fasted state. In this study, we focused on the fed state because our primary focus was lipogenesis. Future work in the fasted state is necessary to dissect the effects of LNA29 on FAO.
It is tempting to consider the potential therapeutic value of LNA29 in the context of fatty liver disease or hyperlipidemia; however, despite the encouraging findings in this study, we issue at least four points of caution and suggestions for next steps. First, it is imperative that more testing of the efficacy of LNA29 in suppressing DNL is performed in animal models of disease. Second, while the delivery method of LNA29 (subcutaneous injection) in this study is very effective for suppressing miR-29 levels in the liver, it is possible that some of the LNA can be taken up by other tissue types as well, albeit likely with reduced efficacy (Kurtz et al., 2015). Thus, it is critical to develop and test tissue-specific LNA delivery mechanisms, and also to expand the examination of miR-29’s biological and molecular functions beyond the liver. Third, miR-29 is known to regulate genes encoding ECM proteins and LNA-mediated suppression of miR-29, especially in hepatic stellate cells, might contribute to the development of hepatic fibrosis (Wang et al., 2015). Careful evaluation of the effects of LNA29 on fibrotic phenotypes is required. Finally, all of the in vivo studies conducted thus far have involved single dose, one-week acute response experiments. While single doses of LNA29 appear to be non-toxic (Kurtz et al., 2015), it is unknown what the systemic responses would be to long-term dosing, and this needs to be tested in the future.
Overall, this study demonstrates the beneficial effects of LNA29 on DNL and circulating lipid levels. In addition, this study provides mechanistic insight, using liver-specific Sirt1-deficient mice, that decouples the effect of LNA29 on circulating TG from that of circulating cholesterol.
Data Availability Statement
The datasets generated for this study can be found in the GEO accession GSE129539.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of National Health Institutes, NYU Winthrop Hospital and Duke University.
Author Contributions
Y-HH contributed to the study design, experiments execution, data collection, and bioinformatic analyses. CK, RC, RB, and LZ carried out the experiments and involved in the data collection. MK performed the bioinformatic analyses. PW, KV, MH, and XL provided resources and animals for critical experiments. PS initiated the project, led the experimental designs, and supervised the study. Y-HH and PS wrote the manuscript. All authors made important intellectual contributions to the experiments and discussion.
Funding
This study was supported by the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (R01DK105965, awarded to PS), American Diabetes Association Pathway Program (1-16-ACE-47, awarded to PS; 1-16-INI-17, awarded to PW), and Empire State Stem Cell Fund through the New York State Department of Health (Contract #C30293GG, awarded to Y-HH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank members of the Sethupathy laboratory for helpful comments and feedback, Dr. Zhao Li and the Greehey Children’s Cancer Research Institute at University of Texas Health Science Center at San Antonio for small RNA library preparation and sequencing. We thank Sheng Zhang and Maria Elena Diaz Rubio (Cornell Metabolomics Facility) for lipidomics analysis. We also thank the UNC Nutrition Obesity Research Center (NORC) Nutritional Biochemistry and Molecular Biology core facility, UNC High Throughput Sequencing Core Facility for critical services, and the Cornell Transcriptional Regulation and Expression (TREx) facility.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.01367/full#supplementary-material
References
Adiels, M., Olofsson, S. O., Taskinen, M. R., and Boren, J. (2008). Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 28, 1225–1236. doi: 10.1161/ATVBAHA.107.160192
Bae, H. J., Noh, J. H., Kim, J. K., Eun, J. W., Jung, K. H., Kim, M. G., et al. (2014). MicroRNA-29c functions as a tumor suppressor by direct targeting oncogenic SIRT1 in hepatocellular carcinoma. Oncogene 33, 2557–2567. doi: 10.1038/onc.2013.216
Bajaj, M., Suraamornkul, S., Pratipanawatr, T., Hardies, L. J., Pratipanawatr, W., Glass, L., et al. (2003). Pioglitazone reduces hepatic fat content and augments splanchnic glucose uptake in patients with type 2 diabetes. Diabetes Metab. Res. Rev. 52, 1364–1370. doi: 10.2337/diabetes.52.6.1364
Banerji, M. A., Buckley, M. C., Chaiken, R. L., Gordon, D., Lebovitz, H. E., and Kral, J. G. (1995). Liver fat, serum triglycerides and visceral adipose tissue in insulin-sensitive and insulin-resistant black men with NIDDM. Int. J. Obes. Relat. Metab. Disord. 19, 846–850.
Baran-Gale, J., Fannin, E. E., Kurtz, C. L., and Sethupathy, P. (2013). Beta cell 5’-shifted isomiRs are candidate regulatory hubs in type 2 diabetes. PLoS One 8:e73240. doi: 10.1371/journal.pone.0073240
Bradshaw, A. D., Graves, D. C., Motamed, K., and Sage, E. H. (2003). SPARC-null mice exhibit increased adiposity without significant differences in overall body weight. Proc. Natl. Acad. Sci. U.S.A. 100, 6045–6050. doi: 10.1073/pnas.1030790100
Chen, E. Y., Tan, C. M., Kou, Y., Duan, Q., Wang, Z., Meirelles, G. V., et al. (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14:128. doi: 10.1186/1471-2105-14-128
Chen, S., Zeng, K., Liu, Q. C., Guo, Z., Zhang, S., Chen, X. R., et al. (2017). Adropin deficiency worsens HFD-induced metabolic defects. Cell Death Dis. 8:e3008. doi: 10.1038/cddis.2017.362
Cheng, L., Zhu, Y., Han, H., Zhang, Q., Cui, K., Shen, H., et al. (2017). MicroRNA-148a deficiency promotes hepatic lipid metabolism and hepatocarcinogenesis in mice. Cell Death Dis. 8:e2916. doi: 10.1038/cddis.2017.309
Cheung, O., Puri, P., Eicken, C., Contos, M. J., Mirshahi, F., Maher, J. W., et al. (2008). Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 48, 1810–1820. doi: 10.1002/hep.22569
Choi, S. E., Fu, T., Seok, S., Kim, D. H., Yu, E., Lee, K. W., et al. (2013). Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell 12, 1062–1072. doi: 10.1111/acel.12135
Chong, J., Soufan, O., Li, C., Caraus, I., Li, S., Bourque, G., et al. (2018). MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46, W486–W494. doi: 10.1093/nar/gky310
Chong, J., and Xia, J. (2018). MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 34, 4313–4314. doi: 10.1093/bioinformatics/bty528
Coskun, T., Bina, H. A., Schneider, M. A., Dunbar, J. D., Hu, C. C., Chen, Y., et al. (2008). Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 149, 6018–6027. doi: 10.1210/en.2008-0816
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Dongol, B., Shah, Y., Kim, I., Gonzalez, F. J., and Hunt, M. C. (2007). The acyl-CoA thioesterase I is regulated by PPARalpha and HNF4alpha via a distal response element in the promoter. J. Lipid Res. 48, 1781–1791. doi: 10.1194/jlr.m700119-jlr200
Dorn, C., Riener, M. O., Kirovski, G., Saugspier, M., Steib, K., Weiss, T. S., et al. (2010). Expression of fatty acid synthase in nonalcoholic fatty liver disease. Int. J. Clin. Exp. Pathol. 3, 505–514.
Esau, C., Davis, S., Murray, S. F., Yu, X. X., Pandey, S. K., Pear, M., et al. (2006). miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 3, 87–98. doi: 10.1016/j.cmet.2006.01.005
Ganesh Kumar, K., Zhang, J., Gao, S., Rossi, J., McGuinness, O. P., Halem, H. H., et al. (2012). Adropin deficiency is associated with increased adiposity and insulin resistance. Obesity 20, 1394–1402. doi: 10.1038/oby.2012.31
Ghoshal, S., Stevens, J. R., Billon, C., Girardet, C., Sitaula, S., Leon, A. S., et al. (2018). Adropin: an endocrine link between the biological clock and cholesterol homeostasis. Mol. Metab. 8, 51–64. doi: 10.1016/j.molmet.2017.12.002
Heo, M. J., Kim, T. H., You, J. S., Blaya, D., Sancho-Bru, P., and Kim, S. G. (2019). Alcohol dysregulates miR-148a in hepatocytes through FoxO1, facilitating pyroptosis via TXNIP overexpression. Gut 68, 708–720. doi: 10.1136/gutjnl-2017-315123
Hung, Y. H., Kanke, M., Kurtz, C. L., Cubitt, R., Bunaciu, R. P., Miao, J., et al. (2019). Acute suppression of insulin resistance-associated hepatic miR-29 in vivo improves glycemic control in adult mice. Physiol. Genomics 51, 379–389. doi: 10.1152/physiolgenomics.00037.2019
Jeon, T. I., and Osborne, T. F. (2016). miRNA and cholesterol homeostasis. Biochim. Biophys. Acta 1861, 2041–2046. doi: 10.1016/j.bbalip.2016.01.005
Jimenez, V., Jambrina, C., Casana, E., Sacristan, V., Munoz, S., Darriba, S., et al. (2018). FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 10:e8791. doi: 10.15252/emmm.201708791
Kuleshov, M. V., Jones, M. R., Rouillard, A. D., Fernandez, N. F., Duan, Q., Wang, Z., et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. doi: 10.1093/nar/gkw377
Kumar, K. G., Trevaskis, J. L., Lam, D. D., Sutton, G. M., Koza, R. A., Chouljenko, V. N., et al. (2008). Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab. 8, 468–481. doi: 10.1016/j.cmet.2008.10.011
Kurtz, C. L., Fannin, E. E., Toth, C. L., Pearson, D. S., Vickers, K. C., and Sethupathy, P. (2015). Inhibition of miR-29 has a significant lipid-lowering benefit through suppression of lipogenic programs in liver. Sci. Rep. 5:12911. doi: 10.1038/srep12911
Kurtz, C. L., Peck, B. C., Fannin, E. E., Beysen, C., Miao, J., Landstreet, S. R., et al. (2014). MicroRNA-29 fine-tunes the expression of key FOXA2-activated lipid metabolism genes and is dysregulated in animal models of insulin resistance and diabetes. Diabetes Metab. Res. Rev. 63, 3141–3148. doi: 10.2337/db13-1015
Lin, Y., Ding, D., Huang, Q., Liu, Q., Lu, H., Lu, Y., et al. (2017). Downregulation of miR-192 causes hepatic steatosis and lipid accumulation by inducing SREBF1: novel mechanism for bisphenol A-triggered non-alcoholic fatty liver disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 869–882. doi: 10.1016/j.bbalip.2017.05.001
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550.
Luna, C., Li, G., Qiu, J., Epstein, D. L., and Gonzalez, P. (2009). Role of miR-29b on the regulation of the extracellular matrix in human trabecular meshwork cells under chronic oxidative stress. Mol. Vis. 15, 2488–2497.
Matsuzaka, T., Shimano, H., Yahagi, N., Kato, T., Atsumi, A., Yamamoto, T., et al. (2007). Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat. Med. 13, 1193–1202. doi: 10.1038/nm1662
Moore, K. J. (2013). microRNAs: small regulators with a big impact on lipid metabolism. J. Lipid Res. 54, 1159–1160. doi: 10.1194/jlr.e036954
Najafi-Shoushtari, S. H., Kristo, F., Li, Y., Shioda, T., Cohen, D. E., Gerszten, R. E., et al. (2010). MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 328, 1566–1569. doi: 10.1126/science.1189123
Narayanan, R. P., Fu, B., Oliver, R. L., Siddals, K. W., Donn, R., Hudson, J. E., et al. (2014). Insulin-like growth factor-II and insulin-like growth factor binding protein-2 prospectively predict longitudinal elevation of HDL-cholesterol in type 2 diabetes. Ann. Clin. Biochem. 51, 468–475. doi: 10.1177/0004563213499145
Nguyen, P., Leray, V., Diez, M., Serisier, S., Le Bloc’h, J., Siliart, B., et al. (2008). Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 92, 272–283.
Nie, J., Bradshaw, A. D., Delany, A. M., and Sage, E. H. (2011). Inactivation of SPARC enhances high-fat diet-induced obesity in mice. Connect. Tissue Res. 52, 99–108. doi: 10.3109/03008207.2010.483747
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Purushotham, A., Schug, T. T., Xu, Q., Surapureddi, S., Guo, X., and Li, X. (2009). Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9, 327–338. doi: 10.1016/j.cmet.2009.02.006
Rayner, K. J., Esau, C. C., Hussain, F. N., McDaniel, A. L., Marshall, S. M., van Gils, J. M., et al. (2011). Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 478, 404–407. doi: 10.1038/nature10486
Roderburg, C., Urban, G. W., Bettermann, K., Vucur, M., Zimmermann, H., Schmidt, S., et al. (2011). Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53, 209–218. doi: 10.1002/hep.23922
Singh, S., Osna, N. A., and Kharbanda, K. K. (2017). Treatment options for alcoholic and non-alcoholic fatty liver disease: a review. World J. Gastroenterol. 23, 6549–6570. doi: 10.3748/wjg.v23.i36.6549
Soh, J., Iqbal, J., Queiroz, J., Fernandez-Hernando, C., and Hussain, M. M. (2013). MicroRNA-30c reduces hyperlipidemia and atherosclerosis in mice by decreasing lipid synthesis and lipoprotein secretion. Nat. Med. 19, 892–900. doi: 10.1038/nm.3200
Song, H., Ding, L., Zhang, S., and Wang, W. (2018). MiR-29 family members interact with SPARC to regulate glucose metabolism. Biochem. Biophys. Res. Commun. 497, 667–674. doi: 10.1016/j.bbrc.2018.02.129
Tiwari, S., and Siddiqi, S. A. (2012). Intracellular trafficking and secretion of VLDL. Arterioscler. Thromb. Vasc. Biol. 32, 1079–1086. doi: 10.1161/ATVBAHA.111.241471
Townsend, S. A., and Newsome, P. N. (2017). Review article: new treatments in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 46, 494–507. doi: 10.1111/apt.14210
Veedu, R. N., and Wengel, J. (2010). Locked nucleic acids: promising nucleic acid analogs for therapeutic applications. Chem. Biodivers. 7, 536–542. doi: 10.1002/cbdv.200900343
Vickers, K. C., Landstreet, S. R., Levin, M. G., Shoucri, B. M., Toth, C. L., Taylor, R. C., et al. (2014). MicroRNA-223 coordinates cholesterol homeostasis. Proc. Natl. Acad. Sci. U.S.A. 111, 14518–14523. doi: 10.1073/pnas.1215767111
Vickers, K. C., Sethupathy, P., Baran-Gale, J., and Remaley, A. T. (2013a). Complexity of microRNA function and the role of isomiRs in lipid homeostasis. J. Lipid Res. 54, 1182–1191. doi: 10.1194/jlr.R034801
Vickers, K. C., Shoucri, B. M., Levin, M. G., Wu, H., Pearson, D. S., Osei-Hwedieh, D., et al. (2013b). MicroRNA-27b is a regulatory hub in lipid metabolism and is altered in dyslipidemia. Hepatology 57, 533–542. doi: 10.1002/hep.25846
Wang, J., Chu, E. S., Chen, H. Y., Man, K., Go, M. Y., Huang, X. R., et al. (2015). microRNA-29b prevents liver fibrosis by attenuating hepatic stellate cell activation and inducing apoptosis through targeting PI3K/AKT pathway. Oncotarget 6, 7325–7338.
Wheatcroft, S. B., Kearney, M. T., Shah, A. M., Ezzat, V. A., Miell, J. R., Modo, M., et al. (2007). IGF-binding protein-2 protects against the development of obesity and insulin resistance. Diabetes Metab. Res. Rev. 56, 285–294. doi: 10.2337/db06-0436
White, P. J., McGarrah, R. W., Grimsrud, P. A., Tso, S. C., Yang, W. H., Haldeman, J. M., et al. (2018). The BCKDH Kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab. 27:e1287. doi: 10.1016/j.cmet.2018.04.015
Yang, Y., Pan, Q., Sun, B., Yang, R., Fang, X., Liu, X., et al. (2016). miR-29b Targets LPL and TDG genes and regulates apoptosis and triglyceride production in MECs. DNA Cell Biol. 35, 758–765. doi: 10.1089/dna.2016.3443
Yin, H., Hu, M., Liang, X., Ajmo, J. M., Li, X., Bataller, R., et al. (2014). Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 146, 801–811. doi: 10.1053/j.gastro.2013.11.008
Keywords: microRNA (miRNA), miR-29, liver, lipogenesis, Sirtuin 1 (Sirt1)
Citation: Hung Y-H, Kanke M, Kurtz CL, Cubitt RL, Bunaciu RP, Zhou L, White PJ, Vickers KC, Hussain MM, Li X and Sethupathy P (2019) MiR-29 Regulates de novo Lipogenesis in the Liver and Circulating Triglyceride Levels in a Sirt1-Dependent Manner. Front. Physiol. 10:1367. doi: 10.3389/fphys.2019.01367
Received: 08 August 2019; Accepted: 15 October 2019;
Published: 29 October 2019.
Edited by:
Kevin I. Watt, The University of Melbourne, AustraliaReviewed by:
Hervé Guillou, Institut National de la Recherche Agronomique de Toulouse, FranceRaffaella Crescenzo, University of Naples Federico II, Italy
Copyright © 2019 Hung, Kanke, Kurtz, Cubitt, Bunaciu, Zhou, White, Vickers, Hussain, Li and Sethupathy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Praveen Sethupathy, pr46@cornell.edu