 Neoma T. Boardman
Neoma T. Boardman Line Rossvoll
Line Rossvoll Anne D. Hafstad
Anne D. Hafstad
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 02 October 2019
Sec. Exercise Physiology
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.01274
This article is part of the Research TopicExploration of the Physiological Effects of Exercise in Cardiovascular DiseasesView all 10 articles
Physical activity is an efficient strategy to delay development of obesity and insulin resistance, and thus the progression of obesity/diabetes-related cardiomyopathy. In support of this, experimental studies using animal models of obesity show that chronic exercise prevents the development of obesity-induced cardiac dysfunction (cardiomyopathy). Whether exercise also improves the tolerance to ischemia-reperfusion in these models is less clear, and may depend on the type of exercise procedure as well as time of initiation. We have previously shown a reduction in ischemic-injury in diet-induced obese mice, when the exercise was started prior to the development of cardiac dysfunction in this model. In the present study, we aimed to explore the effect of exercise on ischemic-tolerance when exercise was initiated after the development obesity-mediated. Male C57BL/6J mice were fed a high-fat diet (HFD) for 20–22 weeks, where they were subjected to high-intensity interval training (HIT) during the last 3 weeks of the feeding period. Sedentary HFD fed and chow fed mice served as controls. Left-ventricular (LV) post-ischemic functional recovery and infarct size were measured in isolated perfused hearts. We also assessed the effect of 3-week HIT on mitochondrial function and myocardial oxygen consumption (MVO2). Sedentary HFD fed mice developed marked obesity and insulin resistance, and demonstrated reduced post-ischemic cardiac functional recovery and increased infarct size. Three weeks of HIT did not induce cardiac hypertrophy and only had a mild effect on obesity and insulin resistance. Despite this, HIT improved post-ischemic LV functional recovery and reduced infarct size. This increase in ischemic-tolerance was accompanied by an improved mitochondrial function as well as reduced MVO2. The present study highlights the beneficial effects of exercise training with regard to improving the ischemic-tolerance in hearts with cardiomyopathy following obesity and insulin resistance. This study also emphasizes the exercise-induced improvement of cardiac energetics and mitochondrial function in obesity/diabetes.
Cardiovascular disease is a major cause of morbidity and mortality in type 2 diabetic patients (Kannel and McGee, 1979). Obese and diabetic patients are also at risk of developing a specific cardiomyopathy. The pathogenesis of this cardiomyopathy is multifactorial and complex, including fibrosis, inflammation, mitochondrial dysfunction, altered substrate utilization, oxidative stress and altered Ca2+ handling (Bugger and Abel, 2014; Jia et al., 2018). An early hallmark is also elevated myocardial oxygen consumption (MVO2), leading to cardiac inefficiency (Boudina et al., 2007; Wright et al., 2009; Cole et al., 2011; Hafstad et al., 2013; Lund et al., 2015). Diabetes also increases the risk of acute myocardial infarction as well as death following infarction (Haffner et al., 1998). In accordance, experimental studies using animal models of obesity, insulin resistance and/or diabetes generally show less tolerance to ischemic-reperfusion injury. Although the pathophysiological mechanisms contributing to ischemic injury in normal hearts has been subject to a comprehensive investigation, the underlying mechanisms leading to the higher ischemic susceptibility in the diabetic heart are not well known. As increased oxygen consumption will be particularly disadvantageous under conditions of limited oxygen availability, there is reason to suggest that the obesity/diabetes-induced increase in MVO2 contributes to a higher susceptibility to ischemic-reperfusion injury.
Exercise training is considered a key element in the management of type 2 diabetes (Colberg et al., 2010; Zanuso et al., 2010) as well as in the prevention and treatment of cardiovascular diseases (Myers et al., 2002). In support of this, experimental studies have shown that chronic exercise not only reduces obesity and insulin resistance (Zanuso et al., 2010) but also prevents or ameliorates the development of cardiac dysfunction (Bidasee et al., 2008; Lu et al., 2017). Accordingly, we have previously found reduced obesity and insulin resistance, accompanied by preserved cardiac function and decreased infarct size after 10 weeks of high-intensity exercise training (HIT) in mice fed a high-fat diet (HFD) for 18 weeks (Lund et al., 2015). As the exercise procedure was initiated prior to the development of cardiac dysfunction, its effect on the heart could have been due to delayed progression and severity of obesity/diabetes-related cardiomyopathy. In the present study, we aimed to explore the cardiac effect of exercise training with regard to improving ischemic-tolerance in a model with obesity-mediated cardiomyopathy.
C57BL/6J male mice (5–6 weeks) were purchased from Charles River Laboratories (Germany). Obesity and insulin resistance were induced by feeding the mice a (HFD 58V8, TestDiet, United Kingdom, 60% of calories from fat) for 20–22 weeks. All mice received chow and drinking water ad libitum and were housed at 23°C on a reversed light-dark cycle. Due to the nocturnal nature of mice, all exercise training occurred during their dark cycle. During the last 3 weeks of the feeding regime, the mice were assigned to maintain a sedentary lifestyle (HFDSED) or high-intensity interval training (HFDHIT) by treadmill running 5 days/week as previously described (Hafstad et al., 2011, 2013). The exercise protocol consisted of 10 bouts of 4-min, high-intensity, treadmill running at 25° inclination, corresponding to 85–90% of VO2max, interspersed by 2 min active rest (Hafstad et al., 2011, 2013). Aerobic capacity, determined as VO2max, was assessed before and after the 3–wk exercise protocol, using a treadmill in a metabolic chamber (Columbus Instruments, Columbus, OH) (Hafstad et al., 2011). Blood was collected from the saphenous vein following a 4-h fasting period. Blood glucose concentration was measured with a glucometer (FreeStyle Lite, Alameda, CA, United States), and plasma insulin was analyzed using commercial kits from DRG Diagnostics (Marburg, Germany). Animal experiments were approved by the Norwegian National Animal Research Authority (FDU ID 3698), which conforms to the National Institute of Health guidelines (NIH publication No. 85-23, revised 1996) and European Directive 2010/63/EU.
Isolated perfused hearts were subjected to ischemia-reperfusion, and post-ischemic recovery of left ventricular (LV) function was assessed using an intra-ventricular fluid-filled balloon where a vent was also inserted into the LV, through the apex. The volume of the balloon was adjusted to give an end-diastolic pressure of 5–10 mmHg. The hearts were perfused in a recirculating mode, with a modified Krebs-Henseleit bicarbonate buffer supplemented with 5 mM glucose and 0.4 mM palmitate prebound to 3% BSA. After 20 min stabilization and 25 min global ischemia, post-ischemic functional recovery was measured over a 60 min period. Reperfusion was continued for an additional 40 min to allow for determination of infarcted tissue. At the end of reperfusion, hearts were frozen at −20°C, prior to slicing and staining using a 1% 2,3,5-triphenyl-2H-tetrazolium chloride solution. Infarct size was determined using ImageJ software (National Institutes of Health, Bethesda, MD, United States). Fiber-optic oxygen probes (FOXY-AL300; Ocean Optics, Duiven, Netherlands) were used to assess PO2 in the perfusion buffer above the aortic cannula and in the pulmonary artery. Coronary flow and the arterial-venous difference of PO2 was used to determine myocardial oxygen consumption (MVO2) as previously described (Lund et al., 2015; Boardman et al., 2017). In order to assess a work-independent (unloaded) MVO2, the hearts were subjected to an unloaded perfusion condition to minimize the workload (Boardman et al., 2009).
Cardiac mitochondria were isolated from hearts harvested prior to the ischemic insult. Briefly, tissue from the left ventricle was homogenized and trypsinized (5 mg/mL) in isolation buffer (250 mM sucrose, 0.5 mM EDTA, 10 mM Tris; pH 7.4). After a further homogenization, and differential centrifugation, mitochondrial pellets were suspended in respiration buffer containing 0.5 mM EGTA, 3 mM MgCl2, 60 mM K-lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, 1 g/L BSA, 280 U/mL catalase, 20 mM histidine, 20 μM vitamin E succinate, 3 mM glutathione, 1 μM leupeptin, 2 mM glutamate, 2 mM malate, and 2 mM ATP; pH 7.1. Mitochondria were kept on ice for 1 h prior to mitochondrial respiration experiments. Oxygen consumption was measured using an oxygraph (O2-k, Oroboros Instruments, Austria), where pyruvate (5 mM) and malate (2 mM) or palmitoyl-CoA (25 μM), L-carnitine (5 mM), and malate (2 mM) served as substrates. V0 was defined as the respiration in the presence of substrates before ADP was added. An oxidative phosphorylation state (Vmax) was defined as the respiration peak after adding 100 μmol/L ADP. Respiration rates were adjusted to protein content (Bradford Protein Assay Kit). The respiratory coupling ratio (RCR) was calculated as Vmax/V0.
All data are presented as mean ± standard error of means. Numbers of observations are presented as “n.” Differences between three groups were analyzed using one-way ANOVA with multiple comparisons (Holm-Sidak method as post hoc test). Where the normality test failed (Shapiro–Wilk test), a Mann–Whitney rank-sum test was performed.
Sedentary mice fed an obesogenic diet for 20–22 weeks (HFDSED) developed obesity as indicated by elevated bodyweight and perirenal fat mass when compared to chow fed control (CON) mice (Table 1). Diet-induced obesity was accompanied by a marked insulin resistance (HOMA-IR), as well as reduced aerobic capacity (VO2max).
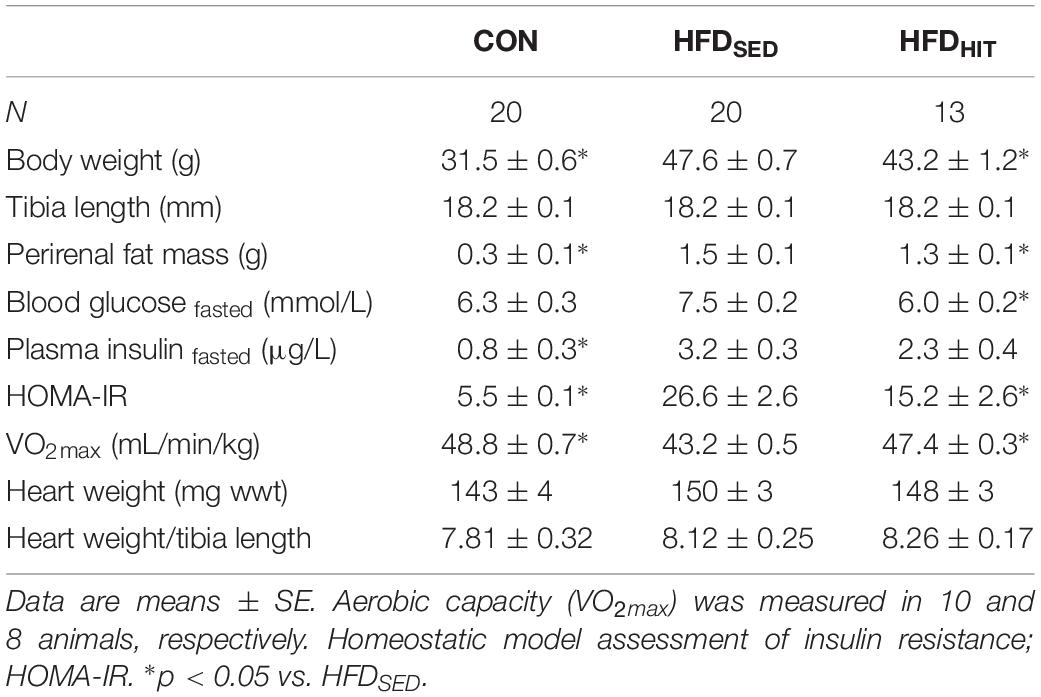
Table 1. Animal characteristics of control mice (CON) and HFD-fed obese mice subjected to 3 weeks of high-intensity (HFDHIT) exercise training or a sedentary (HFDSED) lifestyle.
As expected, subjecting obese mice to 3-week HIT (HFDHIT) resulted in increased aerobic capacity and a small but significant lowering of body weight and perirenal fat mass (Table 1). HIT also reduced insulin resistance (HOMA-IR), mainly due to a reduction in circulating insulin levels. It should be noted that 3-week of HIT did not induce cardiac hypertrophy, which was supported by unaltered gene expression of hypertrophic markers (data not shown).
Previous reports from our group, have demonstrated delayed LV relaxation (increased Tau), increased end-diastolic pressure and an elevated end-diastolic pressure-volume relationship (Pedersen et al., 2018) in this mouse model following LV pressure-volume analysis. In the present study, intraventricular pressure was measured using a fluid-filled balloon in Langendorff perfused hearts (Table 2). However, this perfusion mode does not allow for the reliable determination of diastolic function (Pedersen et al., 2018). Although this represents a clear limitation of the present study, it should be noted that we have, in this model, repeatedly documented a diastolic dysfunction at identical age/feeding duration that is relevant for this study (18–20 week on obesogenic diets).
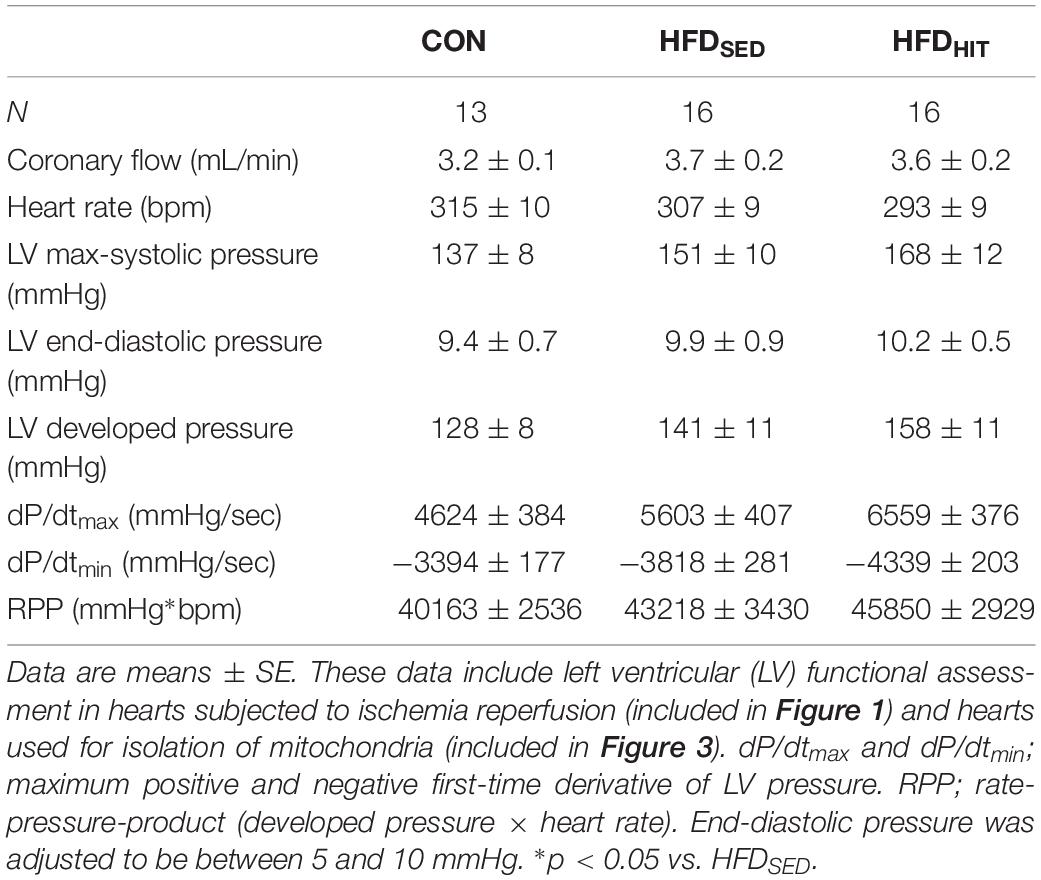
Table 2. Steady-state parameters of LV function obtained in isolated Langendorff perfused hearts from control mice (CON) and HFD-fed obese mice subjected to 3 weeks of high-intensity exercise training (HFDHIT) or a sedentary lifestyle (HFDSED).
The Langendorff perfusion mode is well suited to examine changes in tolerance to ischemia-reperfusion. Functional recovery was followed during the first 60 min of reperfusion after a period of global ischemia. Hearts from HFDSED mice showed impaired recovery of the rate-pressure-product (RPP, given as % of the pre-ischemic value) when compared to hearts from CON mice (Figure 1). As heart rate was not different between the groups, the reduced recovery of RPP was primarily due to the reduced recovery of LV developed pressure (Figure 1). Likewise, we also found a significantly impaired recovery of both the maximum and minimum of the pressure derivative (dP/dtmax and dP/dtmin) indicating increased post-ischemic stunning in HFDSED hearts (Figure 1). Three weeks of exercise training was found to significantly improve functional recovery not only of RPP, but also LV developed pressure, as well as dP/dtmax and dP/dtmin (Figure 1). HR was not altered by exercise, which emphasized that exercise increased LV contraction by improving both contractility and relaxation.
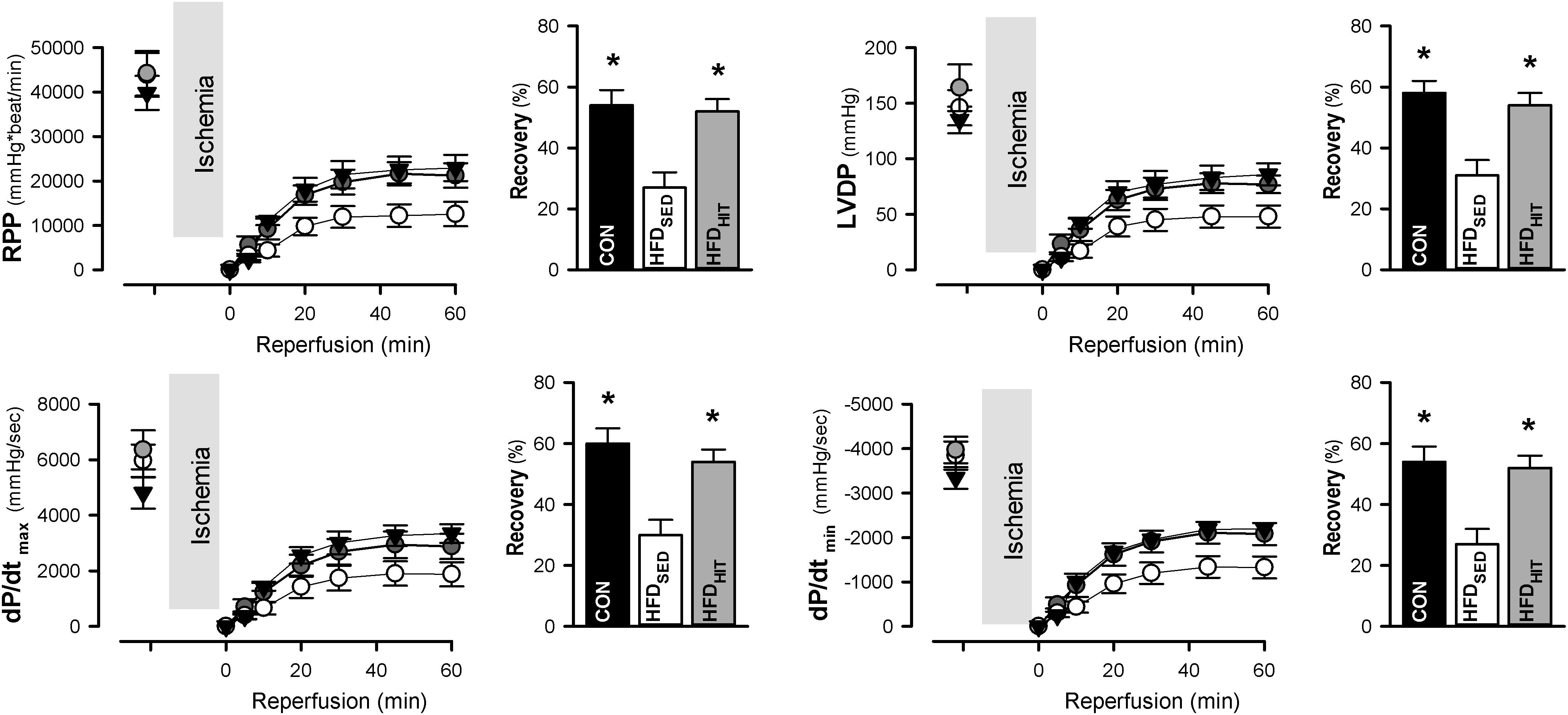
Figure 1. Rate-pressure product (RPP), LV developed pressure (LVDP), and the maximum and minimum of the pressure derivative (dP/dtmax, dP/dtmin) in isolated perfused hearts from control mice (CON, n = 8) and HFD fed mice subjected to a sedentary lifestyle (HFDSED, n = 10) or to 3 weeks of high-intensity exercise training (HFDHIT, n = 6). Bars indicate the calculated post-ischemic recovery (% of pre-ischemic values) of these parameters. Data are means ± SE. ∗p < 0.05 vs. HFDSED.
Impaired functional recovery of post-ischemic function in HFDSED hearts was accompanied by increased infarct size when compared to CON (Figure 2). In addition, 3-week HIT was found to reduce ischemia-reperfusion induced cell death (i.e., infarct size) in hearts from HFD mice.
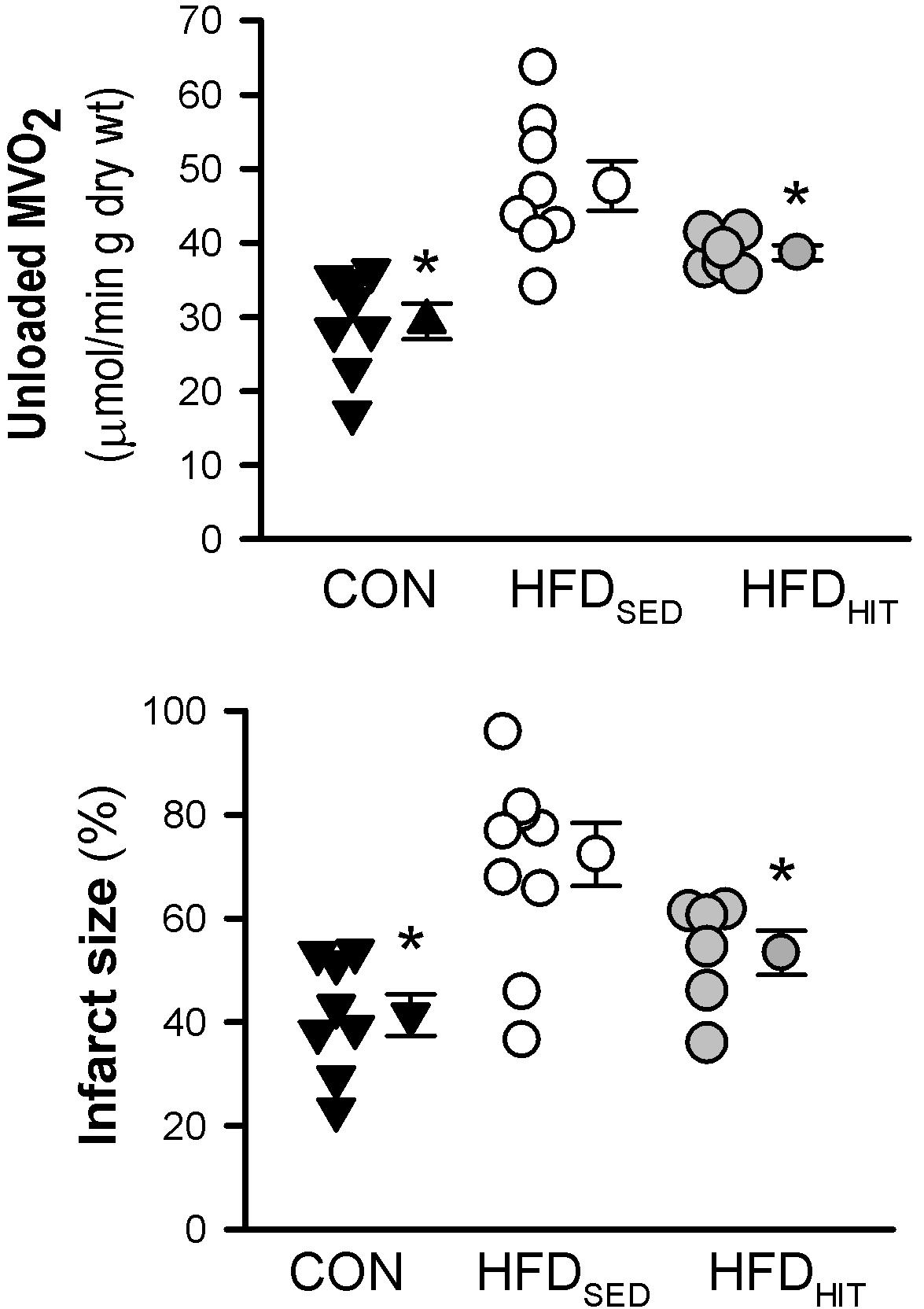
Figure 2. Unloaded MVO2 and infarct size was measured in isolated perfused hearts from control mice (CON, n = 8) and HFD fed mice subjected to a sedentary lifestyle (HFDSED, n = 10) or 3 weeks of high-intensity exercise training (HFDHIT, n = 6). The unloaded MVO2 was obtained by deflating the balloon in the LV for a short period during baseline conditions and pacing the hearts at 7 Hz. Data are means ± SE. ∗p < 0.05 vs. HFDSED.
This study confirmed that HFDSED hearts demonstrate higher oxygen consumption (MVO2) compared to CON hearts when perfused in an unloaded state (Lund et al., 2015). Further, we examined mitochondrial respiration in isolated mitochondria from hearts perfused for 30 min, but not subjected to ischemia-reperfusion. The ADP-dependent oxidative phosphorylation state (Vmax) in mitochondria from HFDSED hearts was reduced when compared to CON hearts, both when using pyruvate as well as palmitoyl-CoA as respiratory substrates (Figure 3). As ADP-independent respiration (V0) was also lower in these hearts, the rate-dependent RCR remained the same in CON and HFDSED (pyruvate: 5.7 ± 0.3 and 5.6 ± 0.3, palmitoyl-CoA: 5.3 ± 0.1 and 5.2 ± 0.2, respectively).
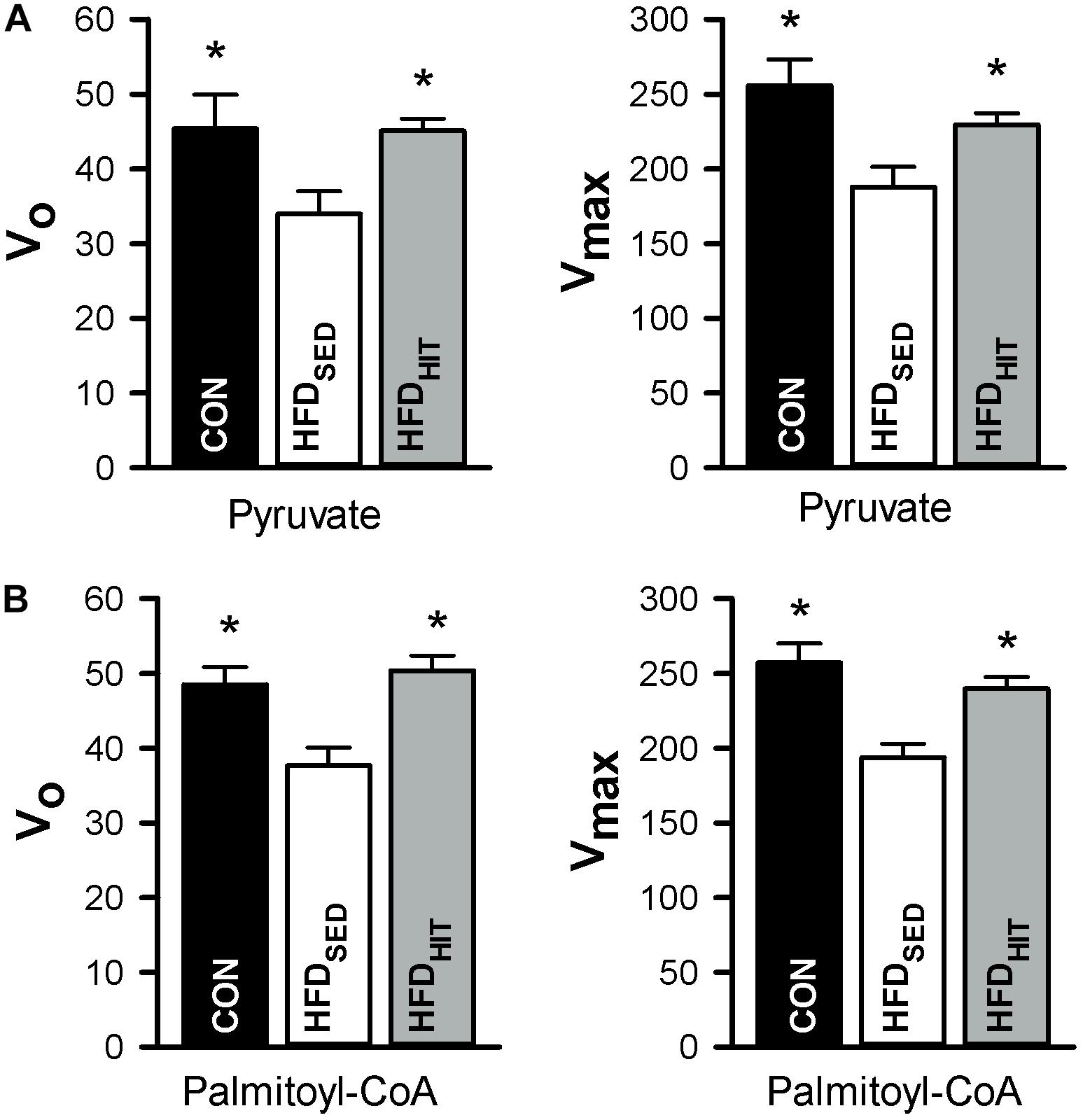
Figure 3. Respiration in isolated mitochondria (nmol O2/min/mg protein) from hearts of control mice (CON, n = 5) mice and HFD-fed obese mice subjected to a sedentary lifestyle (HFDSED, n = 7) or 3 weeks of high-intensity exercise training (HFDHIT, n = 7). The respiratory medium contained pyruvate (5 mM) and malate (2 mM) (A,B) or palmitoyl-CoA (25 μM), L-carnitine (5 mM) and malate (2 mM). V0 respiration, the respiratory state before ADP is added. Vmax, the respiration peak after adding 100 μmol/L ADP. Data are means ± SE. ∗p < 0.05 vs. HFDSED.
We have previously demonstrated that 10 weeks HIT reduced unloaded MVO2 (Lund et al., 2015). The present study shows that 3 weeks of HIT was sufficient to induce this effect (Figure 2). In isolated cardiac mitochondria, 3-week HIT was also shown to increase oxidative phosphorylation (Vmax) under both substrate conditions (Figure 3). Interestingly, exercise also significantly increased V0, indicating an exercise-mediated induction of a mitochondrial proton-leak. Due to a concomitant increase in both the ADP-dependent and -independent O2 consumption, we did not find exercise to alter RCR (pyruvate: 5.1 ± 0.3, palmitoyl-CoA: 4.9 ± 0.2). Finally, although 10 weeks of HIT was previously shown to increase CS activity in cardiac tissue in normal hearts (Hafstad et al., 2013), 3-week HIT was not sufficient to alter CS activity in cardiac tissue from HFD mice (10.6 ± 1.0 vs. 12.3 ± 0.4 IU/g wwt, HFDSED vs. HFDHIT, respectively).
We have previously shown in diet-induced obese mice, that exercise training initiated prior to changes in cardiac function, prevented the subsequent development of dysfunction (Hafstad et al., 2013; Lund et al., 2015) as well as exacerbated infarction (Lund et al., 2015). As this effect could have been due to delayed progression of the cardiomyopathy (because of reduced obesity and insulin resistance), we have now explored the effect of exercise on cardiac ischemic-tolerance in mice with obesity-mediated cardiomyopathy. We found that a shorter duration of exercise, initiated at the end of the feeding protocol, improved ischemic-tolerance, despite having only mild effects on co-morbidities. We also found that this effect was associated with an exercise-mediated reduction in myocardial oxygen consumption (MVO2) and improved cardiac mitochondrial respiration.
In accordance with previous studies (Lund et al., 2015; Pedersen et al., 2018), feeding C57BL/6J mice an HFD for more than 18 weeks led to obesity, insulin resistance and low aerobic capacity (VO2max). These mice also develop impaired LV function, primarily manifested as a diastolic dysfunction (Lund et al., 2015; Pedersen et al., 2018). A hallmark of obesity/diabetes related cardiomyopathy is mechanical inefficiency (i.e., the ratio between cardiac work and MVO2) (Cole et al., 2011; Hafstad et al., 2013; Lund et al., 2015). Cardiac inefficiency occurs prior to development of dysfunction (Wright et al., 2009; Hafstad et al., 2013), and is due to a higher oxygen cost for non-mechanical processes (Hafstad et al., 2013; Lund et al., 2015). Accordingly, in the present study, hearts from the sedentary HFD fed mice, showed a higher unloaded MVO2. As a high O2 consumption is particularly detrimental under conditions of limited O2 availability, this O2 wasting can contribute to render these hearts more susceptible to an ischemic-injury. In support of this, the present study, as well as a range of other studies (Yi et al., 2011; Pons et al., 2013; Littlejohns et al., 2014; Lund et al., 2015), have reported a decreased tolerance to myocardial ischemia-reperfusion in models of obesity/diabetes.
Chronic exercise training improves obesity and insulin resistance (Colberg et al., 2010; Qiu et al., 2014), and may thus delay or ameliorate the severity of diabetic cardiomyopathy. In accordance, experimental studies from our lab have demonstrated that 10 weeks of exercise training of diet-induced obese mice delayed the progression of obesity, reduced insulin resistance (Hafstad et al., 2013; Lund et al., 2015), prevented the development of LV dysfunction (Hafstad et al., 2013; Lund et al., 2015) and decreased cardiac susceptibility to ischemic-injury (Lund et al., 2015). Notably, in these studies, exercise was started prior to the development of dysfunction (i.e., halfway through the 18-week feeding protocol). The cardiac effects could therefore be due to a postponed development of cardiomyopathy. In the present study, we wanted to examine the effect of shorter exercise period, which did not prevent the development of cardiomyopathy. Therefore, mice were fed HFD for 20–22 weeks, and subjected to HIT during the final 3 weeks of the feeding period.
Compared to the longer exercise protocol (10-week HIT) (Lund et al., 2015), the systemic effects of 3-week HIT were, as expected, less marked. Accordingly, while 10-week HIT reduced perirenal fat by approximately 36–44% (Lund et al., 2015; Boardman et al., 2017), the corresponding reduction by 3-week HIT was only 11%. Similarly, 3-week HIT also resulted in a lesser decrease in HOMA (30 vs. 60%), and a smaller increase in VO2max (7 vs. 20%). Finally, in contrast to 10-week, 3-week HIT was not sufficient to develop cardiac hypertrophy.
We have assessed ischemic susceptibility in hearts perfused in Langendorff perfusion mode with an intraventricular balloon. As this perfusion mode cannot detect changes in diastolic function in mouse hearts (Lund et al., 2015; Pedersen et al., 2018), the present study could not confirm the diastolic dysfunction that has been shown to develop in this model (Lund et al., 2015; Pedersen et al., 2018). Nor can the present study determine whether or not 3-week HIT ameliorated LV diastolic dysfunction. This study, however, clearly demonstrated that 3-week HIT improved post-ischemic functional recovery as well as reduced infarct size. Thus, despite a less marked modification of the associated co-morbidities, these hearts show improved ischemic-tolerance. This is also supported by Pons et al. (2013), who demonstrated that 4 weeks of exercise in ob/ob mice, reduced infarct size without altering hyperglycemia, hypercholesterolemia, hyperinsulinemia, fat mass or body weight.
While the mechanisms leading to exercise-induced cardioprotection in normal hearts have been widely studied, fewer studies have examined this in models of insulin resistance and/or type 2 diabetes. The present study supports that exercise can decrease myocardial O2 wasting found in diabetic cardiomyopathy. The increased O2-consumption and decreased efficiency are an early and consistent finding in these hearts (Wright et al., 2009; Hafstad et al., 2013). Although the mechanisms for O2 wasting are not fully understood, several of the reported pathophysiological changes in these hearts can impair mechanoenergetics, by altering ATP production and/or utilization. These changes include elevated fatty acid supply and/or utilization (Belke et al., 2000; Aasum et al., 2003), structural remodeling (i.e., myocardial stiffness) (How et al., 2006), impaired mitochondrial function and oxidative stress (Fauconnier et al., 2007; Anderson et al., 2009), as well as altered Ca2+ handling (Stolen et al., 2009; Epp et al., 2013). Importantly, several of these processes, can also be altered by exercise training (Hafstad et al., 2015). As a major part of the work-independent O2 consumption is linked to maintainance of Ca2+ homeostasis, exercise-mediated effects on myocardial Ca2+ transport (Stolen et al., 2009; Epp et al., 2013) can lead to decreased MVO2 (Hafstad et al., 2013). Exercise can also reduce oxidative stress [by increasing antioxidant capacity (Muthusamy et al., 2012)], and improve the redox state (Fisher-Wellman et al., 2013). As we found an exercise-induced mitochondrial proton leak [in the present study, and previously (Hafstad et al., 2013)], it could be suggested that obesity-mediated redox modification of the Ca2+ handling proteins is prevented by exercise due to leak-induced decrease in mitochondrial membrane potential.
The present study highlights the beneficial effects of exercise training with regard to improving the ischemic-tolerance in hearts with obesity-induced cardiomyopathy. This study also emphasizes exercise-induced improvement of cardiac energetics and mitochondrial function.
The raw data supporting the conclusions of this manuscript will be made available by the corresponding author, without undue reservation, to any qualified researcher.
This study was carried out in accordance with the recommendation of the National Institute of Health guidelines (NID publication No. 85–23, revised 1996) and European Directive 2010/63/EU. All protocols involving animals in the present study were approved by the Norwegian National Animal Research Authority.
NB, AH, and EA conceived and designed the research, and edited and revised the manuscript. NB, AH, JL, and LR performed the experiments. NB, LR, and EA analyzed the data. NB and EA interpreted the results of the experiments, prepared figures, and drafted the manuscript. NB, AH, JL, LR, and EA approved the final version of the manuscript.
This work was supported by grants from the Northern Norway Regional Health Authority (Helse Nord RHF, UNIKARD; NB) and UiT – The Arctic University of Norway (JL and LR). This publication also arises from the research funded by The Norwegian Health Association. The publication charges for this article have been funded by a grant from the publication fund of UiT – The Arctic University of Norway.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge Trine Lund and Tina Myhre Pedersen for technical assistance.
Aasum, E., Hafstad, A. D., Severson, D. L., and Larsen, T. S. (2003). Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes Metab. Res. Rev. 52, 434–441. doi: 10.2337/diabetes.52.2.434
Anderson, E. J., Kypson, A. P., Rodriguez, E., Anderson, C. A., Lehr, E. J., and Neufer, P. D. (2009). Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 54, 1891–1898. doi: 10.1016/j.jacc.2009.07.031
Belke, D. D., Larsen, T. S., Gibbs, E. M., and Severson, D. L. (2000). Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am. J. Physiol. Endocrinol. Metab. 279, E1104–E1113. doi: 10.1152/ajpendo.2000.279.5.E1104
Bidasee, K. R., Zheng, H., Shao, C. H., Parbhu, S. K., Rozanski, G. J., and Patel, K. P. (2008). Exercise training initiated after the onset of diabetes preserves myocardial function: effects on expression of beta-adrenoceptors. J. Appl. Physiol. 105, 907–914. doi: 10.1152/japplphysiol.00103.2008
Boardman, N., Hafstad, A. D., Larsen, T. S., Severson, D. L., and Aasum, E. (2009). Increased O2 cost of basal metabolism and excitation-contraction coupling in hearts from type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 296, H1373–H1379. doi: 10.1152/ajpheart.01264.2008
Boardman, N. T., Hafstad, A. D., Lund, J., Rossvoll, L., and Aasum, E. (2017). Exercise of obese mice induces cardioprotection and oxygen sparing in hearts exposed to high-fat load. Am. J. Physiol. Heart Circ. Physiol. 313, H1054–H1062. doi: 10.1152/ajpheart.00382.2017
Boudina, S., Sena, S., Theobald, H., Sheng, X., Wright, J. J., Hu, X. X., et al. (2007). Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes Metab. Res. Rev. 56, 2457–2466. doi: 10.2337/db07-0481
Bugger, H., and Abel, E. D. (2014). Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57, 660–671. doi: 10.1007/s00125-014-3171-3176
Colberg, S. R., Sigal, R. J., Fernhall, B., Regensteiner, J. G., Blissmer, B. J., Rubin, R. R., et al. (2010). Exercise and type 2 diabetes: the american college of sports medicine and the american diabetes association: joint position statement executive summary. Diabetes Care 33, 2692–2696. doi: 10.2337/dc10-1548
Cole, M. A., Murray, A. J., Cochlin, L. E., Heather, L. C., McAleese, S., Knight, N. S., et al. (2011). A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res. Cardiol. 106, 447–457. doi: 10.1007/s00395-011-0156-151
Epp, R. A., Susser, S. E., Morissette, M. P., Kehler, D. S., Jassal, D. S., and Duhamel, T. A. (2013). Exercise training prevents the development of cardiac dysfunction in the low-dose streptozotocin diabetic rats fed a high-fat diet. Can. J. Physiol. Pharmacol. 91, 80–89. doi: 10.1139/cjpp-2012-0294
Fauconnier, J., Andersson, D. C., Zhang, S. J., Lanner, J. T., Wibom, R., Katz, A., et al. (2007). Effects of palmitate on Ca(2+) handling in adult control and ob/ob cardiomyocytes: impact of mitochondrial reactive oxygen species. Diabetes Metab. Res. Rev. 56, 1136–1142. doi: 10.2337/db06-0739
Fisher-Wellman, K. H., Mattox, T. A., Thayne, K., Katunga, L. A., La Favor, J. D., Neufer, P. D., et al. (2013). Novel role for thioredoxin reductase-2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J. Physiol. 591, 3471–3486. doi: 10.1113/jphysiol.2013.254193
Haffner, S. M., Lehto, S., Ronnemaa, T., Pyorala, K., and Laakso, M. (1998). Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 339, 229–234. doi: 10.1056/nejm199807233390404
Hafstad, A. D., Boardman, N., and Aasum, E. (2015). How exercise may amend metabolic disturbances in diabetic cardiomyopathy. Antioxid. Redox. Signal. 22, 1587–1605. doi: 10.1089/ars.2015.6304
Hafstad, A. D., Boardman, N. T., Lund, J., Hagve, M., Khalid, A. M., Wisloff, U., et al. (2011). High intensity interval training alters substrate utilization and reduces oxygen consumption in the heart. J. Appl. Physiol. 111, 1235–1241. doi: 10.1152/japplphysiol.00594.2011
Hafstad, A. D., Lund, J., Hadler-Olsen, E., Hoper, A. C., Larsen, T. S., and Aasum, E. (2013). High- and moderate-intensity training normalizes ventricular function and mechanoenergetics in mice with diet-induced obesity. Diabetes Metab. Res. Rev. 62, 2287–2294. doi: 10.2337/db12-1580
How, O. J., Aasum, E., Severson, D. L., Chan, W. Y., Essop, M. F., and Larsen, T. S. (2006). Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes Metab. Res. Rev. 55, 466–473. doi: 10.2337/diabetes.55.02.06.db05-1164
Jia, G., Hill, M. A., and Sowers, J. R. (2018). Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ. Res. 122, 624–638. doi: 10.1161/CIRCRESAHA.117.311586
Kannel, W. B., and McGee, D. L. (1979). Diabetes and cardiovascular disease. Framingham study. JAMA 241, 2035–2038. doi: 10.1001/jama.241.19.2035
Littlejohns, B., Pasdois, P., Duggan, S., Bond, A. R., Heesom, K., Jackson, C. L., et al. (2014). Hearts from mice fed a non-obesogenic high-fat diet exhibit changes in their oxidative state, calcium and mitochondria in parallel with increased susceptibility to reperfusion injury. PLoS One 9:e100579. doi: 10.1371/journal.pone.0100579
Lu, G. Z. X., Sun, Z., Shi, X., Liu, T., and Xu, X. (2017). Effects of exercise training on systolic and diastolic function of mice with diabetic cardiomyopathy. Cardiol. Plus 2, 1–6. doi: 10.4103/cp.cp_1_18
Lund, J., Hafstad, A. D., Boardman, N. T., Rossvoll, L., Rolim, N. P., Ahmed, M. S., et al. (2015). Exercise training promotes cardioprotection through oxygen-sparing action in high fat-fed mice. Am. J. Physiol. Heart Circ. Physiol. 308, H823–H829. doi: 10.1152/ajpheart.00734.2014
Muthusamy, V. R., Kannan, S., Sadhaasivam, K., Gounder, S. S., Davidson, C. J., Boeheme, C., et al. (2012). Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic Biol. Med. 52, 366–376. doi: 10.1016/j.freeradbiomed.2011.10.440
Myers, J., Prakash, M., Froelicher, V., Do, D., Partington, S., and Atwood, J. E. (2002). Exercise capacity and mortality among men referred for exercise testing. N. Engl. J. Med. 346, 793–801. doi: 10.1056/nejmoa011858
Pedersen, T. M., Boardman, N. T., Hafstad, A. D., and Aasum, E. (2018). Isolated perfused working hearts provide valuable additional information during phenotypic assessment of the diabetic mouse heart. PLoS One 13:e0204843. doi: 10.1371/journal.pone.0204843
Pons, S., Martin, V., Portal, L., Zini, R., Morin, D., Berdeaux, A., et al. (2013). Regular treadmill exercise restores cardioprotective signaling pathways in obese mice independently from improvement in associated co-morbidities. J. Mol. Cell Cardiol. 54, 82–89. doi: 10.1016/j.yjmcc.2012.11.010
Qiu, S., Mintz, J. D., Salet, C. D., Han, W., Giannis, A., Chen, F., et al. (2014). Increasing muscle mass improves vascular function in obese (db/db) mice. J. Am. Heart Assoc. 3:e000854. doi: 10.1161/JAHA.114.000854
Stolen, T. O., Hoydal, M. A., Kemi, O. J., Catalucci, D., Ceci, M., Aasum, E., et al. (2009). Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ. Res. 105, 527–536. doi: 10.1161/CIRCRESAHA.109.199810
Wright, J. J., Kim, J., Buchanan, J., Boudina, S., Sena, S., Bakirtzi, K., et al. (2009). Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding. Cardiovasc. Res. 82, 351–360. doi: 10.1093/cvr/cvp017
Yi, W., Sun, Y., Gao, E., Wei, X., Lau, W. B., Zheng, Q., et al. (2011). Reduced cardioprotective action of adiponectin in high-fat diet-induced type II diabetic mice and its underlying mechanisms. Antioxid. Redox. Signal. 15, 1779–1788. doi: 10.1089/ars.2010.3722
Keywords: obesity, exercise, mice, Langendorff, mitochondria, MVO2, infarct size, functional recovery
Citation: Boardman NT, Rossvoll L, Lund J, Hafstad AD and Aasum E (2019) 3-Weeks of Exercise Training Increases Ischemic-Tolerance in Hearts From High-Fat Diet Fed Mice. Front. Physiol. 10:1274. doi: 10.3389/fphys.2019.01274
Received: 05 March 2019; Accepted: 19 September 2019;
Published: 02 October 2019.
Edited by:
Ines Drenjančević, Josip Juraj Strossmayer University of Osijek, CroatiaReviewed by:
Robert Blazekovic, Clinical Hospital Dubrava, CroatiaCopyright © 2019 Boardman, Rossvoll, Lund, Hafstad and Aasum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ellen Aasum, ZWxsZW4uYWFzdW1AdWl0Lm5v
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.