
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 04 September 2019
Sec. Clinical and Translational Physiology
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.01108
This article is part of the Research Topic Heart Failure with Reduced, Mid-Range and Preserved Ejection Fraction: Mechanisms, Diagnosis and Therapy View all 8 articles
 Jens van de Wouw1†
Jens van de Wouw1† Michelle Broekhuizen1,2,3†
Michelle Broekhuizen1,2,3† Oana Sorop1
Oana Sorop1 Jaap A. Joles4
Jaap A. Joles4 Marianne C. Verhaar4
Marianne C. Verhaar4 Dirk J. Duncker1
Dirk J. Duncker1 A. H. Jan Danser2
A. H. Jan Danser2 Daphne Merkus1*
Daphne Merkus1*Heart failure (HF) and chronic kidney disease (CKD) co-exist, and it is estimated that about 50% of HF patients suffer from CKD. Although studies have been performed on the association between CKD and HF with reduced ejection fraction (HFrEF), less is known about the link between CKD and heart failure with preserved ejection fraction (HFpEF). Approximately, 50% of all patients with HF suffer from HFpEF, and this percentage is projected to rise in the coming years. Therapies for HFrEF are long established and considered quite successful. In contrast, clinical trials for treatment of HFpEF have all shown negative or disputable results. This is likely due to the multifactorial character and the lack of pathophysiological knowledge of HFpEF. The typical co-existence of HFpEF and CKD is partially due to common underlying comorbidities, such as hypertension, dyslipidemia and diabetes. Macrovascular changes accompanying CKD, such as hypertension and arterial stiffening, have been described to contribute to HFpEF development. Furthermore, several renal factors have a direct impact on the heart and/or coronary microvasculature and may underlie the association between CKD and HFpEF. These factors include: (1) activation of the renin-angiotensin-aldosterone system, (2) anemia, (3) hypercalcemia, hyperphosphatemia and increased levels of FGF-23, and (4) uremic toxins. This review critically discusses the above factors, focusing on their potential contribution to coronary dysfunction, left ventricular stiffening, and delayed left ventricular relaxation. We further summarize the directions of novel treatment options for HFpEF based on the contribution of these renal drivers.
Heart failure with preserved ejection fraction (HFpEF) is characterized by impaired relaxation of the left ventricle (LV) during diastole and accounts for over 50% of all patients with heart failure (HF) (Redfield et al., 2003; Yancy et al., 2006). Both the proportion of HFpEF-patients and morbidity, mortality, and healthcare costs associated with this disease are rising (Bhatia et al., 2006; Liao et al., 2006; Owan et al., 2006; Steinberg et al., 2012). Multiple processes including cardiomyocyte hypertrophy, interstitial fibrosis, impaired calcium handling, and increased passive cardiomyocyte stiffness contribute to the left ventricular stiffening characteristic for HFpEF (Borlaug, 2014; Gladden et al., 2014; Sharma and Kass, 2014). Although ejection fraction is still normal, systolic dysfunction is present in HFpEF, as measured by tissue Doppler or strain imaging (Borlaug, 2014; Tadic et al., 2017). In large population studies, the majority of the HFpEF patients are women (Masoudi et al., 2003). Whereas men have more coronary artery disease indicative of macrovascular disease, women typically present with obesity, left ventricular hypertrophy, diastolic dysfunction and more often have microvascular angina (Gori et al., 2014a; Crea et al., 2017).
The current paradigm for HFpEF proposes that commonly present comorbidities such as diabetes mellitus (DM), obesity, and hypertension lead to a systemic pro-inflammatory state. This pro-inflammatory state causes coronary microvascular dysfunction, evidenced by an imbalance between nitric oxide (NO) and reactive oxygen species (ROS) leading to stiffening of the LV (Paulus and Tschope, 2013; Gladden et al., 2014). Excessive ROS-production in the endothelium of the coronary microvasculature lowers NO bioavailability through scavenging of NO. Loss of NO reduces soluble guanylate cyclase (sGC) activity in the cardiomyocytes, thereby lowering cGMP levels and decreasing PKG activity. The latter results in hypophosphorylation of titin and induces cardiomyocyte hypertrophy (Paulus and Tschope, 2013; Franssen et al., 2016). Given the proposed central role for disruption of the NO pathway in pathogenesis of HFpEF, it is rather surprising that all large clinical trials, which targeted the NO-cGMP-PKG pathway failed to date. Organic and inorganic nitrates are therapeutic agents that can be metabolized to NO systemically and thus act as NO-donors. However, the NEAT-HFPEF trial showed that isosorbide mononitrate, a long working organic nitrate, tended to reduce physical activity and did not improve quality of life and exercise capacity (Redfield et al., 2015). Inhaled nebulized inorganic nitrate, also did not improve exercise capacity, as recently shown in the INDIE-HFpEF trial (Borlaug et al., 2018). The phase 2b SOCRATES-PRESERVED trial showed no reduction of NT-pro-BNP or left atrial dimensions at 12 weeks after treatment with the sGC stimulator Vericiguat. However, Vericiguat was well tolerated and increased quality of life, warranting further research (Pieske et al., 2017). Inhibition of the cGMP-degrading enzyme phosphodiesterase 5 with Sildenafil did not improve clinical status rank score or exercise capacity (Redfield et al., 2013), and failed to improve vascular and cardiac function (Borlaug et al., 2015). Therefore, new therapeutic targets need to be identified that can interfere with the development and progression of HFpEF.
It is important to note that the impact of microvascular dysfunction on cardiac structure and function is not limited to dysfunction of the NO-cGMP-PKG pathway. Indeed, upregulation of VCAM-1 and E-selectin on the coronary microvascular endothelium induces transendothelial leucocyte migration and activation, increased transforming growth factor β (TGF-β) levels, thereby promoting pro-fibrotic pathways and differentiation of fibroblast to myofibroblasts (Westermann et al., 2011; Paulus and Tschope, 2013) and increasing interstitial fibrosis (van Heerebeek et al., 2012; Sharma and Kass, 2014). Secretion of autocrine and paracrine factors, such as apelin, TGF-β, and endothelin-1, by dysfunctional coronary microvascular endothelial cells can also directly induce left ventricular hypertrophy (Kamo et al., 2015). Finally, capillary rarefaction and inadequate angiogenesis could contribute to a decreased oxygen supply and subsequent left ventricular myocardial stiffening (Gladden et al., 2014).
The so-called cardio-renal syndrome describes the co-existence of HF and chronic kidney disease (CKD). Approximately 50% of the patients with HFpEF also suffer from CKD (Ter Maaten et al., 2016). Although this co-existence is partially due to shared risk factors, such as hypertension, DM and obesity, it has also been proposed that HF directly impacts kidney function, and vice versa, CKD worsens cardiac function (Brouwers et al., 2013). Interdependence of the heart and kidneys, similarities between their microvascular networks, and the coexistence of CKD and HF further imply a role for microvascular dysfunction in development and progression of both diseases (Ter Maaten et al., 2016).
Given the co-incidence of HFpEF and CKD, the present review aims to provide a mechanistic link between CKD and HFpEF, by describing potential pathways through which CKD can induce or aggravate coronary microvascular dysfunction and thereby contribute to the development and progression of left ventricular hypertrophy and diastolic dysfunction. These include mechanical effects, neurohumoral activation, systemic inflammation, anemia and changes in mineral metabolism as induced by CKD (Figure 1). As some of these CKD-induced effects may induce HFpEF and contribute to cardiovascular disease in general, they may provide targets to intervene with the development of diastolic dysfunction and/or its progression towards HFpEF. Hence, this review will also describe the (potential) druggable therapeutic targets within these pathways, and where applicable, clinical trials intervening with these pathways.
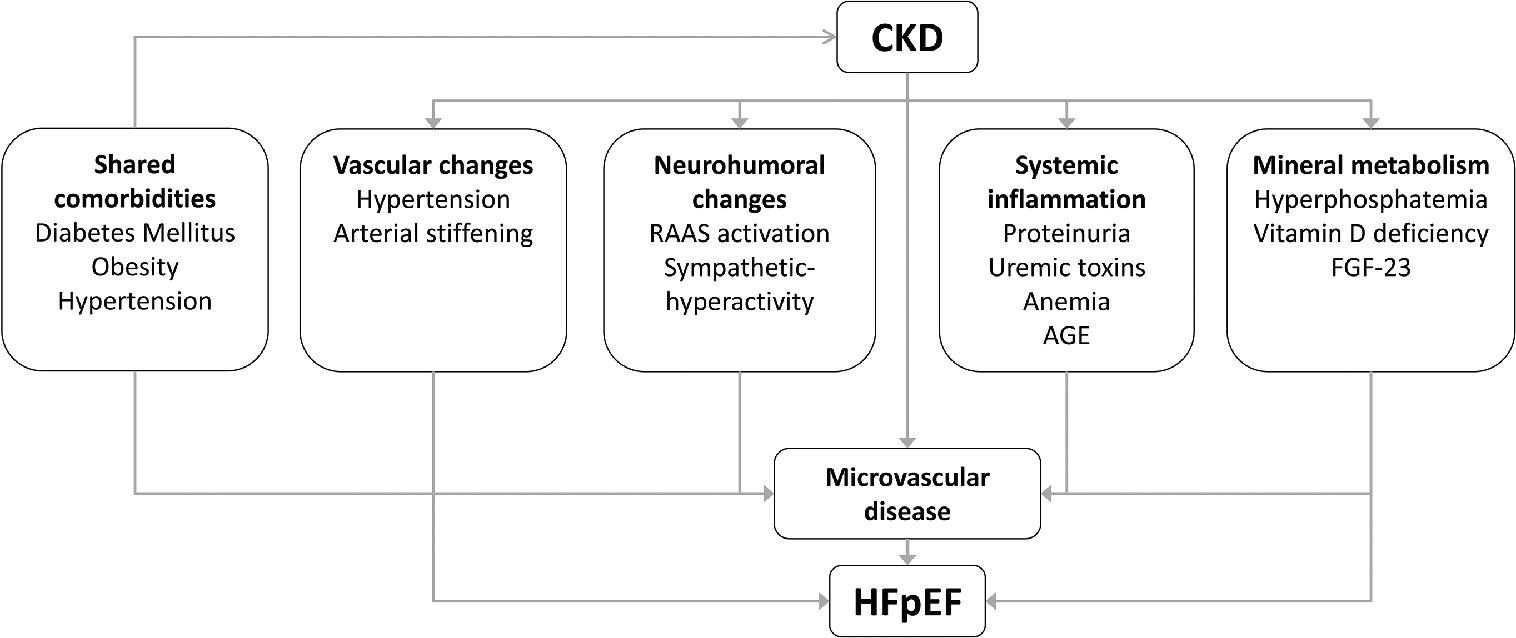
Figure 1. Schematic overview of the risk factors that can contribute to the development of heart failure with preserved ejection fraction (HFpEF) in patients with chronic kidney disease (CKD).
CKD is defined as a progressive decline of renal function and is associated with hypertension, proteinuria, and the loss of nephron mass (Noone and Licht, 2014). CKD is an independent risk factor for the development of HF, with increasing cardiovascular risk and mortality as renal function declines (Tonelli et al., 2006; Ronco et al., 2008). Additionally, HF is the major cause of death among patients with CKD (Kottgen et al., 2007; Bansal et al., 2017). Although renal dysfunction is present in about half of the patients with HF in general (Hillege et al., 2006; Smith et al., 2013), and is an important prognostic marker for adverse outcomes (Yancy et al., 2006; Ahmed et al., 2007; McAlister et al., 2012), particularly the association between HFpEF and CKD is very strong. In a cohort comparing patients with heart failure with reduced ejection fraction (HFrEF), HF with mid-range ejection fraction and HFpEF, renal dysfunction was associated with increased mortality in all HF subtypes, but was most prevalent in HFpEF (Streng et al., 2018). Gori et al. showed that 62% of the patients with HFpEF display abnormalities in at least one marker of renal insufficiency, with different markers correlating with different HFpEF phenotypes (Gori et al., 2014b). Further evidence for a causal relationship between CKD and HFpEF comes from a rat model, in which CKD was mimicked by nephrectomy of one whole kidney and two-third of the remaining kidney. Loss of nephron mass in these rats resulted in a cardiac HFpEF-like phenotype, with LV hypertrophy and diastolic dysfunction, but critical HFpEF features such as lung congestion and exercise intolerance were not reported (Sarkozy et al., 2019). In accordance with CKD as a causative factor for HFpEF, the majority of patients on hemodialysis display diastolic dysfunction and left ventricular hypertrophy, whereas overt systolic dysfunction and HFrEF are visible in only a minority of these patients (Hickson et al., 2016; Antlanger et al., 2017). In a prospective cohort study, 74% of the patients admitted for dialysis displayed left ventricular hypertrophy. In contrast, systolic dysfunction and left ventricular dilatation were present in only 15% and 32% of the patients, respectively (Foley et al., 2010). Left ventricular hypertrophy is not restricted to end stage CKD, but is already highly prevalent in the general CKD population (Collins, 2003). Indeed, the first visible myocardial alteration in patients with CKD is left ventricular hypertrophy (London, 2002), developing early in the progression of kidney dysfunction (Levin et al., 1996; Pecoits-Filho et al., 2012) and often co-occurring with myocardial fibrosis and diastolic dysfunction (Silberberg et al., 1989). Hypertension is an important predictor for development of left ventricular hypertrophy and HFpEF in patients with CKD (Levin et al., 1996; Thomas et al., 2008), while blood pressure reduction is associated with a lower cardiovascular risk (Blood Pressure Lowering Treatment Trialists Collaboration et al., 2013).
It should be noted however, that in addition to decreased diastolic function, both hemodialysis and pre-dialysis CKD patients show impaired regional systolic function measured by longitudinal, circumferential, and radial strain while ejection fraction was preserved (Yan et al., 2011). Similarly, patients with HFpEF can also display signs of systolic dysfunction defined by decreased global longitudinal strain and S′ velocity measured with tissue Doppler. Unger et al. showed in a large group of HFpEF patients that not only diastolic dysfunction, but also the severity of systolic dysfunction and mortality increased in parallel with CKD stage (Unger et al., 2016).
Arterial remodeling in CKD patients is characterized by arterial stiffening, increasing pulse pressure, as a consequence of premature aging, and atherosclerosis of the arteries (Laurent et al., 2006; Briet et al., 2012). Premature vascular aging is common in both CKD and HFpEF. Increased aortic stiffness has been strongly associated with both left ventricular dysfunction, and markers of renal dysfunction (Bortolotto et al., 1999; Borlaug and Kass, 2011), which precede and increase cardiovascular risk in patients with CKD (Kendrick et al., 2010; Middleton and Pun, 2010). Stiffer arteries result in an increased pulse pressure, as well as an increased pulse wave velocity, which cause the increased pulsatility to be transmitted into the microvasculature (Mitchell, 2008). Renal and coronary microvascular networks are very vulnerable to pulsatile pressure and flow, thus failure in decreasing pulsatility can result in damage of the capillary networks (Mitchell, 2008; Safar et al., 2015), and thereby contribute to coronary microvascular dysfunction. Fukushima et al. showed an impaired global myocardial flow reserve in CKD patients, even with a normal regional perfusion and function of the LV (Fukushima et al., 2012). Furthermore, coronary microvascular dysfunction was shown to be present in patients with end stage CKD (Bozbas et al., 2009), and was associated with an increased risk of cardiac death in patients with renal failure (Murthy et al., 2012).
Hypertension in CKD is thought to be mainly a consequence of volume overload due to increased sodium reabsorption by the kidneys (Charra and Chazot, 2003; Judd and Calhoun, 2015). Increased sodium loading might also contribute to HFpEF development independent of hypertension, through inducing a systemic pro-inflammatory state, which is detrimental to the coronary microvasculature (Yilmaz et al., 2012). Indeed, empagliflozin, a sodium glucose co-transporter-2 (SGLT2) inhibitor, initially developed as an anti-diabetic drug, resulted in decreased cardiovascular mortality in an initial type 2 diabetes cohort (Zinman et al., 2015). Interestingly, these effects seem to, at least for some part, be specific for empagliflozin as canagliflozin protected less against cardiovascular death (Neal et al., 2017). Although the mechanisms of action have not completely been elucidated yet, multiple pre-clinical studies are being conducted to investigate the myocardial effects of SGLT2-inhibitors (Uthman et al., 2018a,b). Currently, three mechanisms have been proposed to contribute to reduced cardiovascular mortality in patients receiving SGLT2-inhibitors in general and/or empagliflozin in particular (Bertero et al., 2018); (1) osmotic diuresis and natriuresis lower blood pressure and subsequently reduce left ventricular afterload; (2) empagliflozin may instigate a shift to cardiac ketone body oxidation, increasing mitochondrial respiratory efficiency and reducing ROS production; (3) empagliflozin can lower intracellular Na+ by inhibition of the cardiac Na+/H+ exchanger (NHE) and induce coronary vasodilation (Uthman et al., 2018a). The latter effect is especially promising as increased intracellular Na+, as present in failing cardiomyocytes, results in altered mitochondrial Ca2+ handling and subsequent ROS production, which may be ameliorated by SGLT2-inhibitors (Bertero et al., 2018). SGLT2-inhibitors, therefore, seem promising in the cardiorenal field as they are both cardio- and reno-protective (Butler et al., 2017). The effect of empagliflozin on cardiovascular mortality in HFpEF specifically, regardless of diabetic status, is being investigated in the ongoing EMPEROR-Preserved trial (ClinicalTrials.gov NCT03057951).
CKD is associated with hyperactivation of the renin-angiotensin-aldosterone system (RAAS) in response to renal hypoxia resulting in volume overload (Nangaku and Fujita, 2008), which may contribute to the development and/or progression of HFpEF. Interestingly, testosterone can increase, whereas estrogen can lower renin concentrations (Fischer et al., 2002). Such protective effects of estrogen would especially be relevant in pre-menopausal women, and be lost in the typically older, post-menopausal female HFpEF population. Consistent with a detrimental effect of RAAS activation on HFpEF progression, RAAS activation can increase myocardial workload, by elevating systemic vascular resistance and left ventricular afterload, through vasoconstriction of systemic blood vessels in response to angiotensin II or by causing volume expansion due to increased sodium and water reabsorption in response to increased aldosterone levels (Brown, 2013; Forrester et al., 2018). It is not clear if angiotensin II can also induce myocardial cell hypertrophy and fibrosis independently of hypertension. Although in vitro studies have shown that there is a hypertension-independent effect of angiotensin-II on cardiomyocytes, multiple in vivo studies could not confirm these findings, suggesting that the effect of angiotensin II is blood pressure-dependent (Reudelhuber et al., 2007; Qi et al., 2011). Furthermore, RAAS-activation induces coronary microvascular endothelial dysfunction, through NADP(H)-oxidase activation and subsequent ROS formation (Bongartz et al., 2005; Wong et al., 2013). Myocardial perfusion might also be impaired by the vasoconstrictor effects of angiotensin II. During prolonged exercise, vasoconstriction occurs within metabolically less active tissues, mediated by angiotensin II and endothelin-1. Such response is inhibited in metabolically active tissues by NO and prostanoids, resulting in an efficient distribution of blood (Merkus et al., 2006). In a state of systemic inflammation, locally decreased NO bioavailability in the coronary microvasculature might result in disinhibition of angiotensin II-mediated vasoconstriction, resulting in reduced blood delivery to the heart.
Downstream from angiotensin II in the RAAS, aldosterone regulates blood pressure and sodium/potassium homeostasis through the mineralocorticoid receptor in the kidneys, by enhancing sodium reabsorption, thereby contributing to hypertension and high plasma sodium levels. Besides the renal effects, aldosterone has been shown to directly promote myocardial fibrosis, left ventricular hypertrophy, and coronary microvascular dysfunction, acting through endothelial and myocardial mineralocorticoid receptors, independently of angiotensin II (Brown, 2013).
RAAS inhibition is the preferred therapeutic strategy to slow down progression of renal failure and reduce proteinuria in CKD (Levin and Stevens, 2014). Despite the fact that most data show RAAS overactivation in HFpEF, clinical trials in HFpEF with drugs acting on the RAAS, have failed to improve (all-cause) mortality so far (Pitt et al., 2014; Zhang et al., 2016). It is, however, important to note that AT1-blockade with Irbesartan reduced mortality and improved outcome on cardiovascular endpoints in patients with natriuretic peptides below the median, but not in patients with higher natriuretic peptide levels (Anand et al., 2011), suggesting that RAAS inhibition may be beneficial in early HFpEF. Furthermore, post hoc analysis of the TOPCAT trial demonstrated geographically different effects of the mineralocorticoid receptor blocker spironolactone, with small clinical benefits in patients from America (Pfeffer et al., 2015). However, these patients were generally older, had a higher prevalence of atrial fibrillation and diabetes, were less likely to have experienced prior myocardial infarction, had a higher ejection fraction and had a worse renal function (Pfeffer et al., 2015), suggesting that a benefit of spironolactone was associated with a more HFpEF-like phenotype. A more recent post hoc analysis of this trial further showed that spironolactone did show an improvement in primary endpoints in patients with lower levels of natriuretic peptides and hence less advanced disease (Anand et al., 2017). Consistent with this suggestion, a recent meta-analysis showed that mineralocorticoid receptor antagonists do improve indices of diastolic function and cardiac structure in HFpEF patients (Kapelios et al., 2019). Interestingly, treatment of DM type 2 with mineralocorticoid receptor antagonists also improved coronary microvascular function (Garg et al., 2015). Altogether, these data suggest that intervening with the RAAS is beneficial in patients with less advanced HFpEF, whereas beneficial effects are lost in patients with more advanced disease. Therefore, clinical studies investigating HFpEF progression and clinical trials focusing on reducing or preventing progression of early HFpEF into advanced HFpEF need to be conducted.
Another approach intervening with the RAAS is the use of Entresto, an angiotensin receptor and a neprilysin inhibitor (ARNI), which is a combination of valsartan (AT1 receptor blocker) and sacubitril (neprilysin inhibitor). Neprilysin inhibition exerts its beneficial effects through inhibition of the breakdown of natriuretic peptides. Entresto was superior to the standard therapy, enalapril, in patients with HFrEF in reducing mortality and number of hospitalizations for HF (McMurray et al., 2014). In hypertensive rats with diabetes, ARNI reduced proteinuria, glomerulosclerosis, and heart weight more strongly than AT1 receptor blockade, and this occurred independently of blood pressure (Roksnoer et al., 2015, 2016). In a phase 2 double-blind randomized controlled trial in HFpEF patients, Entresto reduced NT-pro-BNP plasma levels and left atrial diameters to a greater extent than valsartan (Solomon et al., 2012). These findings led to the ongoing PARAGON-HF trial (ClinicalTrials.gov NCT01920711), which investigates the long-term effect (26 months) of Entresto compared to valsartan in HFpEF (Solomon et al., 2017).
Both CKD and HFpEF are accompanied by autonomic dysregulation (Salman, 2015). Sympathetic hyperactivity has a detrimental effect on both the heart and the kidney and aggravates hypertension and proteinuria. Furthermore, HFpEF patients show attenuated withdrawal of parasympathetic tone and excessive sympathoexcitation during exercise that cause β-adrenergic desensitization, chronotropic incompetence, and may thereby contribute to the limited exercise tolerance of these patients (Phan et al., 2010). A critical role for CKD in this process was suggested by Klein et al. (Klein et al., 2015), showing a clear correlation between CKD, decreased heart rate variability, chronotropic incompetence in HFpEF, and decreased peak VO2. Unfortunately, neither the SENIORS trial (van Veldhuisen et al., 2009), nor the OPTIMIZE-HF registry (Hernandez et al., 2009) showed a beneficial effect of beta-adrenoceptor blockade on all-cause mortality or cardiovascular hospitalizations. Furthermore, beta-adrenoceptor blockade failed to improve LV systolic or diastolic function in patients with ejection fraction >35%, as measured in the SENIORS echocardiography sub-study (Ghio et al., 2006). It should be noted that in the SENIORS trial ejection fraction cutoff was set at 35%, which is lower than current consensus about the cutoff of reduced and preserved ejection fraction. Additionally, in these studies, beta-adrenoceptor blockade was administered on top of existing medication, which often included RAAS-inhibitors. Conversely, in patients with treatment resistant hypertension, renal sympathetic denervation did improve diastolic function and reduce left ventricular hypertrophy, besides reducing blood pressure (Brandt et al., 2012), suggesting that there is indeed an interaction between CKD, sympathetic hyperactivity and diastolic cardiac function.
A pro-inflammatory state is already present in early stages of CKD (Stenvinkel et al., 2002), and is likely an important risk factor for cardiovascular morbidity and mortality on the long term (Ruggenenti et al., 2001; Sarnak et al., 2003). In HFpEF, a systemic pro-inflammatory state has been proposed to be a critical causal factor in coronary microvascular dysfunction as inflammatory cytokines can directly induce endothelial cell dysfunction, cause upregulation of adhesion molecules on coronary microvascular endothelial cells, and reduce NO bioavailability, resulting in impaired vasodilation and pro-fibrotic signaling (Figure 2; Rosner et al., 2012; Paulus and Tschope, 2013).
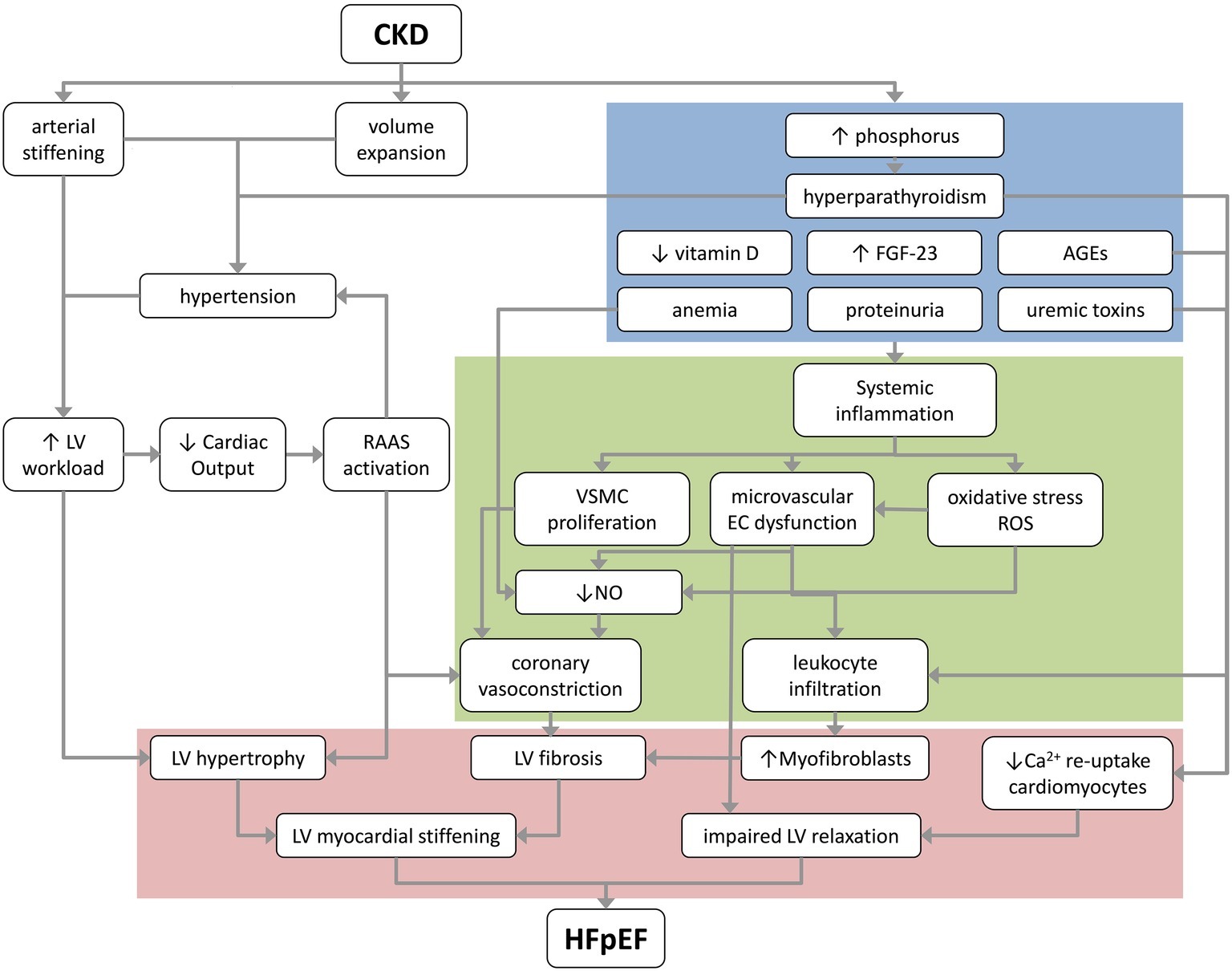
Figure 2. A proposed schematic overview of the pathological mechanisms that underlie the progression of CKD to HFpEF. Blue box depicts renal factors; green box depicts coronary microvascular factors; and red box depicts myocardial changes contributing to HFpEF. AGEs, advanced glycation products; CKD, chronic kidney disease; EC, endothelial cell; FGF-23, fibroblast growth factor 23; HFpEF, heart failure with preserved ejection fraction; LV, left ventricle; NO, nitric oxide; RAAS, renin-angiotensin-aldosterone system; ROS, reactive oxygen species; VSMC, vascular smooth muscle cell.
Targeting this pro-inflammatory state with 14 days of treatment with the recombinant human IL-1 receptor antagonist Anakinra, increased peak VO2, which correlated with a reduction in C-reactive protein (CRP) in the D-HART trial including 12 patients (Van Tassell et al., 2014). Unfortunately, prolonged treatment (12 weeks) in the follow-up D-HART2 trial in 28 patients did not increase VO2, despite small improvements in exercise duration and quality of life, as well as reductions in CRP and NT-pro-BNP compared to baseline values (Van Tassell et al., 2018).
It is possible that targeting systemic inflammation in general to ameliorate HFpEF is too broad to be successful. In the subsequent paragraphs, the contribution of the individual systemic factors: anemia, proteinuria, and reduced excretion of so-called uremic toxins as consequences of renal dysfunction and possible contributors to systemic inflammation, development of microvascular dysfunction, and HFpEF will be considered in more detail.
Anemia is an independent risk factor for development of HFpEF (Foley et al., 2010; Gori et al., 2014b), and is strongly associated with CKD (Thomas et al., 2008). Although hemoglobin levels decreased with worsening of kidney function in both patients with HFpEF and HFrEF, hemoglobin levels were slightly lower in patients with HFpEF as compared to HFrEF (Lofman et al., 2017). The main causes for anemia are iron deficiency and deficient erythropoietin (EPO) production in the renal tubular cells. In addition, urinary loss of red blood cells through enlarged fenestrations of endothelial cells in diseased glomeruli, hemolysis, vitamin B12 deficiency, hyperparathyroidism, and hemodilution may contribute to anemia in CKD patients (Westenbrink et al., 2007; van der Putten et al., 2008). Furthermore, the bone marrow erythropoietic response to EPO is impaired in CKD patients (van der Putten et al., 2008). Finally, the pro-inflammatory cytokine Il-6 can impair erythroid development, by inducing production of the iron regulatory peptide hepcidin by hepatocytes, increasing degradation of iron exporter ferroportin, and decreasing iron delivery to developing erythrocytes (Fraenkel, 2015). Hence, the systemic inflammatory state in CKD, but also in HFpEF, can aggravate anemia.
It is unknown whether anemia, iron deficiency, and/or reduced EPO are causal factors in the development of HFpEF or mere markers of CKD. The most obvious effect of anemia is a general reduction in O2 transport. In 75% of the HFpEF patients, peripheral oxygen consumption was impaired due to impaired diffusive oxygen transport and utilization (Dhakal et al., 2015). Hence, cardiac output needs to be increased to maintain systemic oxygen delivery. Both the consequent increase in myocardial work, and the reduced oxygen-carrying capacity of the blood may contribute to an impaired myocardial O2 balance. Such a disbalance between myocardial oxygen demand and supply is also present in ischemia with no obstructive coronary artery disease (INOCA), in which myocardial oxygen supply is limited by coronary microvascular dysfunction. Indeed, INOCA is increasingly being recognized as a risk factor for development of HFpEF (Crea et al., 2017; Obokata et al., 2018).
Anemia can also directly affect microvascular function as red blood cells can modulate microvascular tone (Cosby et al., 2003; Singel and Stamler, 2005). Red blood cells release NO, which is produced, particularly at low oxygen tensions, from deoxygenated hemoglobin and nitrite, to stimulate vasodilation, cGMP formation in smooth muscle cells and cardiomyocytes, and to inhibit mitochondrial respiration (Crawford et al., 2006). Thus, low levels of red blood cells simulate a condition of coronary microvascular dysfunction, with increased ROS and reduced NO, thereby inducing true coronary microvascular dysfunction and cardiomyocyte damage, which eventually can contribute to progression of HFpEF (Figure 2).
CKD patients on EPO therapy have shown signs of cardiovascular improvement and reversal of left ventricular hypertrophy (Goldberg et al., 1992; Frank et al., 2004), suggesting that correction of anemia may prevent progression of HFpEF. In addition to promoting red blood cell formation and correction of anemia, EPO can protect cardiomyocytes against ischemic injury and induce NO production by endothelial cells, thereby improving microvascular function (van der Putten et al., 2008). EPO can also have tissue protective properties by activating the EPO receptor and β common receptor, which are found in multiple peripheral tissues and are present on endothelial cells. EPO sensitivity can be increased by hypoxia but is decreased by a pro-inflammatory state, which is considered a hallmark of HFpEF; therefore, lower eNOS expression due to lower EPO or lower EPO receptors on the endothelium can contribute to the lower NO-bioavailability in the coronary microcirculation (Congote et al., 2010). Interestingly, in patients, EPO resistance is shown to be present in early CKD prior to the decrease in EPO levels that occurs in later stages of CKD (Mercadal et al., 2012). However, in a randomized controlled trial conducted in older adults with HFpEF, EPO supplementation with epoetin alfa did not improve left ventricular geometry or exercise capacity despite increases in hemoglobin levels (Maurer et al., 2013). One potential explanation would be that the 1.5 g/dl increase in hemoglobin in the treatment group was insufficient, particularly since the placebo-treated patients also showed a 0.8 g/dl increase in hemoglobin. Alternatively, decreased endothelial and/or cardiomyocyte sensitivity to, rather than too low levels of EPO and/or anemia are important in the progression of HFpEF (van der Putten et al., 2008). If so, it would be more beneficial to restore EPO sensitivity of specific cells rather than changing its levels. Reducing the pro-inflammatory phenotype of endothelial cells could potentially be beneficial in increasing endothelial EPO sensitivity. Alternatively, although not specific an enhancer of EPO sensitivity, targeting the protective tissue-specific effects of EPO might prove a viable therapeutic target, although to date, this was mostly evaluated in neurological disorders (Leist et al., 2004).
Iron deficiency, even without anemia, was also shown to be detrimental to the functional capacity of advanced HFpEF patients (Nunez et al., 2016), while diastolic dysfunction was not associated with functional iron deficiency (Kasner et al., 2013). Functional iron deficiency is detrimental to cardiomyocyte function as it reduces antioxidant capacity and limits oxidative phosphorylation thereby limiting energy production, potentially impairing energy-dependent Ca2+ reuptake during diastole (Anand and Gupta, 2018). Currently, iron supplementation with IV ferric carboxymaltose is being investigated in both anemic and non-anemic HFpEF patients in the FAIR-HFpEF trial (ClinicalTrial.org NCT03074591).
Proteinuria, an abnormal high protein concentration in urine, is present in up to 26% of CKD patients with an eGFR below 30 ml/min/1.73 m2 (Garg et al., 2002; Agrawal et al., 2009). Not only proteinuria, but also, more specifically, elevated urinary levels albumin, were associated with declining renal function (Klahr et al., 1994; GISEN Group, 1997; Brenner et al., 2001). Proteinuria is not just a marker of CKD, but also contributes to the exacerbation of CKD, by aggravating renal interstitial inflammatory cell influx resulting in interstitial fibrosis (Figure 2; Abbate et al., 2006; Ruggenenti et al., 2012).
In 1989, Deckert et al. already introduced the Steno hypothesis, which implies that albuminuria is not just reflecting local renal disease, but indicating more general endothelial microvascular dysfunction (Deckert et al., 1989). Indeed, large population based studies have shown that microalbuminuria correlates with a decrease in flow-mediated endothelium-dependent vasodilation in brachial arteries (Stehouwer et al., 2004), as well as in coronary arteries of diabetic patients (Cosson et al., 2006). In patients with essential hypertension, microalbuminuria was shown to correlate with levels of circulating von Willebrand factor, a marker for endothelial damage (Pedrinelli et al., 1994). Multiple studies have shown that (micro)albuminuria is highly prevalent in HFpEF, being associated with LV remodeling, and is a prognostic marker for further disease development (Miura et al., 2012; Brouwers et al., 2013; Katz et al., 2014; Gori et al., 2014b; Nayor et al., 2017). Consistent with a role for microalbuminuria as a prognostic marker for HFpEF, women with HFpEF are less likely to have albuminuria, while their eGFR is similar to that of men (Gori et al., 2014a), potentially explaining the better prognosis (±20% less likely to reach a MACE) in women with HFpEF (Lam et al., 2012). Furthermore, presence of CKD increased the risk for an all-cause event in women, to a similar risk present in men (Lam et al., 2012).
Currently, it is unclear, whether microalbuminuria simply reflects a more generalized microvascular endothelial dysfunction or may act as a causal contributing factor to HFpEF development by inducing coronary microvascular endothelial damage.
Insufficient glomerular filtration results in the retention of a variety of biologically active compounds in the blood, called uremic toxins. The accumulation of uremic toxins can have a deleterious effect on multiple organs, of which the cardiovascular system is most severely affected (Vanholder et al., 2008). Increased levels of uremic toxins are associated with an increased cardiovascular morbidity and mortality (Moradi et al., 2013). Moreover, blood urea nitrogen was shown to be an independent predictor for the progression from preclinical diastolic dysfunction to HFpEF, but not HFrEF (Zhang et al., 2017).
The mechanisms mediating the detrimental effects on the vascular system are multiple. The elevated uremia-associated pro-inflammatory cytokine levels, together with the associated chronic inflammatory state, can inhibit proliferation and enhance apoptosis of endothelial cells (Figure 2; Moradi et al., 2013). Furthermore, uremic toxins can increase von Willebrand factor levels, decrease NO bioavailability by inhibition of endothelial nitric oxide synthase (eNOS), and increase circulating endothelial microparticles (Brunet et al., 2011). Additionally, chronic low grade inflammation increases expression of adhesion molecules on endothelial cells and induces leukocyte activation with differentiation of fibroblasts to myofibroblasts, with subsequent production of collagen in the extracellular matrix, and migration and proliferation of vascular smooth muscle cells (Jourde-Chiche et al., 2011; Paulus and Tschope, 2013). Tryptophan-derived toxins can specifically activate the aryl hydrocarbon receptor pathway, and thereby induce endothelial dysfunction, and activate pro-fibrotic pathways in the myocardium, further enhancing inflammation and increasing vascular oxidative stress (Sallee et al., 2014). All these processes contribute to (coronary) microvascular dysfunction and remodeling.
Uremic toxins might also directly affect the left ventricular relaxation. Exposure of cardiomyocytes to uremic serum of CKD patients elicited inhibition of Na+/K+-ATPase, increased contractile force, impaired calcium re-uptake, and delayed relaxation (Figure 2; Periyasamy et al., 2001).
Elevated circulating and cellular levels of advanced glycation end products (AGEs) have been measured in patients with CKD (Stinghen et al., 2016). This is the result of impaired renal clearance of AGEs together with their increased formation resulting from oxidative stress and/or diabetes mellitus. Elevated circulating AGEs are linked to development and progression of both HFpEF and HFrEF (Hartog et al., 2006, 2007) and correlated positively with increased diastolic dysfunction in patients with diabetes mellitus type 1 (Berg et al., 1999).
In the LV, AGEs are particularly prominent in the coronary microvasculature, where their presence induces a pro-inflammatory phenotype (van Heerebeek et al., 2008), endothelial dysfunction by increasing oxidative stress and decreasing NO bioavailability and vascular stiffening by crosslinking of extracellular matrix (ECM) proteins (Smit and Lutgers, 2004; Hartog et al., 2007). In the myocardium, AGE-induced crosslinking of ECM proteins increases myocardial stiffness (Smit and Lutgers, 2004; Hartog et al., 2007). Furthermore, AGEs impair calcium handling in cardiomyocytes (Petrova et al., 2002). The latter is mediated by carbonylation of SERCA2a, which impairs its activity (Shao et al., 2011), as well as by enhancing calcium leakage from the sarcoplasmic reticulum through the ryanodine receptor (RyR2), thereby promoting mitochondrial damage and oxidative stress (Ruiz-Meana et al., 2019). Hence, reducing production and enhancing breakdown of AGEs could be a therapeutic option in HFpEF patients (Paulus and Dal Canto, 2018), particularly in patients with diabetes and CKD.
Besides glycemic control, there are three classes of drugs that can reduce AGEs: inhibitors of de novo AGE synthesis, drugs that break pre-existing AGE crosslinks and AGE receptor blockers (Zieman and Kass, 2004). Although, to our knowledge, none of these have been tested in HFpEF patients, treatment with aminoguanidine, a small hydrazine-like molecule capable of inhibiting AGE formation through interaction with and quenching of dicarbonyl compounds, resulted in a decrease of diabetes mellitus associated myocardial stiffening in rats, albeit without altering fibrosis (Norton et al., 1996). Furthermore, in DM type 2 patients, benfotiamine, a transketolase activator that blocks several hyperglycemia-induced pathways, prevented microvascular endothelial dysfunction and oxidative stress after an AGE rich meal (Stirban et al., 2006). Similarly, treatment with the AGE crosslink breaker alagebrium, improved endothelial function in patients with isolated systolic hypertension, which was associated with reduced vascular fibrosis and vascular inflammation (Zieman et al., 2007). For an overview of trials conducted with AGE-lowering therapies in CKD patients we refer to Stinghen et al. (2016). Some of these therapies which reduced AGEs in CKD patients might also be a viable chronic treatment option, to prevent or reverse AGE-associated microvascular dysfunction and subsequent diastolic dysfunction in HFpEF.
Lowering uremic toxin levels in general might also provide a viable, but challenging treatment option for HFpEF. The main challenges are to identify the specific uremic toxins that play a role in the pathogenesis of HFpEF, and to target a large variety of uremic toxins with just one class of drugs. Clinical trials with allopurinol, a therapy to decrease uric acid levels, resulted in slower disease progression and a decreased cardiovascular risk in patients with CKD (Goicoechea et al., 2010; Sezer et al., 2014). Even asymptomatic hyperuricemic patients may benefit from allopurinol treatment, as they showed improvements in endothelial function and eGFR (Kanbay et al., 2011).
Declining renal function results in a reduced capacity to perform 1α-hydroxylation and in progressive loss of active vitamin D (Schroeder and Cunningham, 2000). Loss of active vitamin D subsequently leads to increased parathyroid hormone (PTH) production, so-called secondary hyperparathyroidism, eventually contributing to increased calcium, phosphate, and FGF-23 levels. In patients on hemodialysis, an association was reported between low vitamin D levels, systemic inflammation, and myocardial hypertrophy (Bucharles et al., 2011). Furthermore, low levels of vitamin D in these patients were related to increased cardiovascular mortality (Wolf et al., 2007; Drechsler et al., 2010; Bucharles et al., 2011). In non-dialysis CKD patients, lower vitamin D levels were shown to be associated with decreased flow mediated dilatation in the brachial artery, reflecting systemic endothelial dysfunction (Chitalia et al., 2012). Low vitamin D correlates with reduced coronary flow reserve in patients with atypical chest pain, suggesting that vitamin D also affects coronary microvascular function (Capitanio et al., 2013). Recently, in a large cohort of patients with diastolic dysfunction or HFpEF, lower vitamin D levels were associated with increased cardiovascular hospitalizations but not with 5-year mortality (Nolte et al., 2019). Furthermore, in a univariate analysis, calcidiol, but not its active metabolite, calcitriol, was associated with new onset HFpEF in the PREVEND study, but the association disappeared after adjustment for confounding variables (Meems et al., 2016). However, in patients with established HFpEF, vitamin D levels were lower as compared to healthy, sex-, race-, and age-matched controls, and inversely correlated with exercise capacity (Pandey et al., 2018).
In a trial of vitamin D supplementation by cholecalciferol therapy, reductions were observed in the left ventricular mass, inflammatory markers and brain natriuretic peptide levels of CKD patients on hemodialysis (Matias et al., 2010). In contrast, in the PRIMO-trial, 48 weeks of treatment with paricalcitol in a CKD cohort with preserved systolic function neither resulted in improved diastolic function, nor reduced left ventricular mass (Thadhani et al., 2012). However, cardiac MRI unveiled that just a minority of the included patients had left ventricular hypertrophy at baseline, possibly explaining the lack of a beneficial effect. Although the administration of vitamin D has positive effects through inhibition of PTH secretion, it also results in increased serum phosphate levels, with opposing effects (see next paragraph for details). When modulating vitamin D status, one should consider the use of vitamin D analogues, such as paricalcitol, which inhibit PTH synthesis, without substantially inducing hyperphosphatemia, providing promising therapies for restoration of vitamin D levels (Cozzolino et al., 2012).
In large cohorts of patients on hemodialysis, strong associations were found between serum phosphate, calcium, hyperparathyroidism, and an increased risk for overall cardiac mortality, elevated levels of cardiac injury markers, and a worse systolic and diastolic cardiac function (Block et al., 2004; Wang et al., 2014). Additionally, in a cohort of hospitalized patients with CKD, serum phosphate was related to elevated left ventricular concentric remodeling and diastolic dysfunction (Zou et al., 2016). Furthermore, in late stage CKD patients—on peritoneal dialysis—phosphate was independently associated with impairment of left ventricular diastolic function (Ye et al., 2016). At the structural level, elevated levels of phosphate (hyperphosphatemia) and PTH have been associated with the presence of hypertrophy and fibrosis of the LV specifically (Rostand and Drueke, 1999; Block et al., 2004). In addition, in a small cohort of patients on chronic hemodialysis, higher levels of calcium phosphate product were associated with higher CRP levels, and thus with a pro-inflammatory state. In this cohort, intensive lowering of phosphate levels resulted in lower CRP levels, and a significantly improved inflammatory status (Movilli et al., 2005).
Hyperphosphatemia can also directly induce coronary endothelial dysfunction (Di Marco et al., 2013), and also act directly on human vascular smooth muscle cells (VSMC), resulting in VSMC calcification (Jono et al., 2000). Furthermore, hyperphosphatemia can contribute to microvascular dysfunction and HFpEF pathogenesis by reducing prostaglandin synthesis (Ter Maaten et al., 2016). Prostaglandins synthesized in the blood vessel wall act as autocrine or paracrine factors and play a pivotal role in regulation of coronary microvascular function by exerting strong vasodilator effects and by inhibiting platelet aggregation. In clinical practice, supplementation of prostanoids is mostly used in patients with pulmonary hypertension. Prostacyclin analogues are available, such as Selexipag, an oral prostacyclin receptor agonist, which has vasodilator, antiproliferative, and antifibrotic effects. Currently, there is one trial ongoing, which investigates oral Treprostinil, a prostacyclin analogue, in pulmonary hypertension caused by HFpEF (ClinicalTrials.org NCT03037580).
PTH can cause left ventricular interstitial fibrosis and coronary microvascular dysfunction, via its inflammatory effects on monocytes and interstitial fibroblasts (Amann et al., 2003). Interestingly, primary hyperparathyroidism resulted in coronary microvascular dysfunction, which was restored after parathyroidectomy, underlining the effect of PTH on coronary microvascular function (Osto et al., 2012). In hemodialysis patients with secondary hyperparathyroidism, 20 weeks of treatment with cinacalcet ameliorated endothelial dysfunction, diastolic dysfunction, and cardiac hypertrophy by decreasing oxidative stress and increasing nitric oxide production (Figure 2; Choi et al., 2012).
Fibroblast growth factor-23 (FGF-23) is a hormone produced by osteoblasts and osteocytes, which inhibits phosphate reabsorption in the kidneys and suppresses circulating calcitriol, effectively lowering plasma phosphate levels in physiological conditions (Martin et al., 2012). In CKD, FGF-23 is no longer able to reduce phosphate levels due to loss of renal Klotho-FGF receptor 1 complex, resulting in both high phosphate and high FGF-23 levels (Komaba and Fukagawa, 2012). Elevated levels of FGF-23 are associated with an increased cardiovascular risk in patients with CKD (Negri, 2014), and with left ventricular hypertrophy in a cohort of CKD patients (Tanaka et al., 2016). These findings were confirmed in rats, where FGF-23 could directly induce left ventricular hypertrophy while ejection fraction was preserved (Faul et al., 2011). Furthermore, FGF-23 is associated with new-onset HFpEF in a large cohort study of people, who were free of cardiovascular disease at baseline (Almahmoud et al., 2018). Interestingly, in a cohort of HFpEF patients, FGF-23 was not associated with increased mortality, while this was the case for a cohort of HFrEF patients (Koller et al., 2015), suggesting that FGF-23 may be linked to disease onset rather than progression in HFpEF.
Mechanistically, FGF-23 induces chronic inflammation by stimulating cytokine secretion from the liver, but is also locally produced by M1 macrophages, and can thereby further modulate inflammation in the heart (Figure 2; Leifheit-Nestler and Haffner, 2018). FGF-23 inhibits ACE2, resulting in reduced degradation of angiotensin I and II into their vasodilator metabolites angiotensin-(1-9) and angiotensin-(1-7), (Leifheit-Nestler and Haffner, 2018) and consequently increased stimulation of AT1 receptors by angiotensin II. High levels of FGF-23 were further shown to cause endothelial dysfunction, increase superoxide formation, and decrease NO bioavailability in mouse aortas (Silswal et al., 2014). Finally, FGF-23 causes inhibition of 1α-hydroxylase, and can thereby contribute to microvascular damage and cardiac dysfunction due to vitamin D deficiency (Leifheit-Nestler and Haffner, 2018). Hence, elevated FGF-23 levels can contribute to development of HFpEF by attenuating coronary microvascular function and by enhancing angiotensin II induced vascular and myocardial fibrosis. Indeed, preliminary data of Roy et al., suggest that FGF-23 levels correlated with interstitial fibrosis in HFpEF (Roy et al., 2018). Furthermore, FGF-23 counteracted the beneficial effect of paricalcitol on left ventricular hypertrophy, by modulation of the calcineurin/nuclear factor of activated T cell (NFAT) pathway in a rat model of CKD (Czaya et al., 2019). FGF-23 inhibition with KRN23, an anti-FGF antibody, in open label phase 1/2 studies for X-linked hypophosphatemia, showed an increase in serum inorganic phosphate and active vitamin D in all subjects (Imel et al., 2015). Further research into a potential causal role of FGF-23 in HFpEF development is required, prior to embarking on therapeutic interventions.
The kidneys and heart are interdependent organs that are highly connected through multiple systems on both macrovascular and microvascular level. Unfortunately, many studies on the cardiorenal connection have not been conducted in specific HFpEF populations. Pathological processes which are present in CKD, such as vascular changes, deficiencies in kidney produced factors, and impairments in renal filtration can cause and/or contribute to development of HFpEF via several processes, as summarized in Figure 2. Elevated levels of phosphate, PTH, FGF-23, AGEs and uremic toxins, but also anemia and proteinuria can induce a systemic pro-inflammatory state. This state can lead to left ventricular stiffening and coronary microvascular dysfunction by initiating endothelial cell dysfunction, oxidative stress, and vascular smooth muscle cell proliferation. Arterial stiffening, volume expansion, hypertension and RAAS activation, as consequences of CKD, increase left ventricular workload and hypertrophy.
The complexity and multitude of connections between the heart and kidney make it unlikely that there is a single causal contributor for progression from CKD to HFpEF. In addition, although HFpEF is more prevalent in women, and the effect of sex on cardiovascular disease is increasingly recognized (Regitz-Zagrosek, 2006), the specific role of sex in HFpEF pathology still needs to be identified. Multiple large trials have been conducted with treatments for HFpEF, targeting different pathophysiological processes, but unfortunately failed to show clinical benefit. Therefore, current guidelines on treatment of HFpEF focus on lifestyle interventions and the management of comorbidities such as diabetes mellitus, hypertension, obesity and CKD. In addition, it has been proposed that different HFpEF phenotypes exist that should be targeted with different therapeutic strategies. Both male and female CKD patients are interesting and easily identifiable subgroups of HFpEF patients, warranting further investigation both in pathogenesis, as in clinical trials to further investigate cardiorenal connection in HFpEF specifically, and to identify the unique mechanistic pathways involved in various phases of the disease.
This review was designed by JW, MB, and DM. JW and MB have written the largest body of text. OS, JJ, MV, DD, AD, and DM have made a substantial, direct, and intellectual contribution to specific topics. All authors approved the work for publication.
We gratefully acknowledge the support from the Netherlands CardioVascular Research Initiative; an initiative supported by the Dutch Heart Foundation, the Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Science [CVON PHAEDRA (2012-08 to DM), and CVON RECONNECT (2014-11 to DD, DM, JJ, and MV)].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AGE, advanced glycation endproduct; BNP, brain natriuretic peptide; cGMP, cyclic guanosine monophophate; CKD, chronic kidney disease; CRP, C-reactive protein; DM, diabetes mellitus; eGFR, estimated glomerular filtration rate; EPO, erythropoietin; FGF-23, fibroblast growth factor 23; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; LV, left ventricle; NO, nitric oxide; NT-proBNP, N-terminal prohormone of brain natriuretic peptide; PKG, protein kinase G; PTH, parathyroid hormone; RAAS, renin-angiotensin-aldosterone system; ROS, reactive oxygen species; TGF-β, transforming growth factor β.
Abbate, M., Zoja, C., and Remuzzi, G. (2006). How does proteinuria cause progressive renal damage? J. Am. Soc. Nephrol. 17, 2974–2984. doi: 10.1681/ASN.2006040377
Agrawal, V., Marinescu, V., Agarwal, M., and McCullough, P. A. (2009). Cardiovascular implications of proteinuria: an indicator of chronic kidney disease. Nat. Rev. Cardiol. 6, 301–311. doi: 10.1038/nrcardio.2009.11
Ahmed, A., Rich, M. W., Sanders, P. W., Perry, G. J., Bakris, G. L., Zile, M. R., et al. (2007). Chronic kidney disease associated mortality in diastolic versus systolic heart failure: a propensity matched study. Am. J. Cardiol. 99, 393–398. doi: 10.1016/j.amjcard.2006.08.042
Almahmoud, M. F., Soliman, E. Z., Bertoni, A. G., Kestenbaum, B., Katz, R., Lima, J. A. C., et al. (2018). Fibroblast growth factor-23 and heart failure with reduced versus preserved ejection fraction: MESA. J. Am. Heart Assoc. 7:e008334. doi: 10.1161/JAHA.117.008334
Amann, K., Tornig, J., Kugel, B., Gross, M. L., Tyralla, K., El-Shakmak, A., et al. (2003). Hyperphosphatemia aggravates cardiac fibrosis and microvascular disease in experimental uremia. Kidney Int. 63, 1296–1301. doi: 10.1046/j.1523-1755.2003.00864.x
Anand, I. S., and Gupta, P. (2018). Anemia and iron deficiency in heart failure: current concepts and emerging therapies. Circulation 138, 80–98. doi: 10.1161/CIRCULATIONAHA.118.030099
Anand, I. S., Rector, T. S., Cleland, J. G., Kuskowski, M., Mckelvie, R. S., Persson, H., et al. (2011). Prognostic value of baseline plasma amino-terminal pro-brain natriuretic peptide and its interactions with irbesartan treatment effects in patients with heart failure and preserved ejection fraction: findings from the I-PRESERVE trial. Circ. Heart Fail. 4, 569–577. doi: 10.1161/CIRCHEARTFAILURE.111.962654
Anand, I. S., Claggett, B., Liu, J., Shah, A. M., Rector, T. S., Shah, S. J., et al. (2017). Interaction between spironolactone and natriuretic peptides in patients with heart failure and preserved ejection fraction: from the TOPCAT trial. JACC Heart Fail 5, 241–252. doi: 10.1016/j.jchf.2016.11.015
Antlanger, M., Aschauer, S., Kopecky, C., Hecking, M., Kovarik, J. J., Werzowa, J., et al. (2017). Heart failure with preserved and reduced ejection fraction in hemodialysis patients: prevalence, disease prediction and prognosis. Kidney Blood Press. Res. 42, 165–176. doi: 10.1159/000473868
Bansal, N., Katz, R., Robinson-Cohen, C., Odden, M. C., Dalrymple, L., Shlipak, M. G., et al. (2017). Absolute rates of heart failure, coronary heart disease, and stroke in chronic kidney disease: an analysis of 3 community-based cohort studies. JAMA Cardiol. 2, 314–318. doi: 10.1001/jamacardio.2016.4652
Berg, T. J., Snorgaard, O., Faber, J., Torjesen, P. A., Hildebrandt, P., Mehlsen, J., et al. (1999). Serum levels of advanced glycation end products are associated with left ventricular diastolic function in patients with type 1 diabetes. Diabetes Care 22, 1186–1190. doi: 10.2337/diacare.22.7.1186
Bertero, E., Prates Roma, L., Ameri, P., and Maack, C. (2018). Cardiac effects of SGLT2 inhibitors: the sodium hypothesis. Cardiovasc. Res. 114, 12–18. doi: 10.1093/cvr/cvx149
Bhatia, R. S., Tu, J. V., Lee, D. S., Austin, P. C., Fang, J., Haouzi, A., et al. (2006). Outcome of heart failure with preserved ejection fraction in a population-based study. N. Engl. J. Med. 355, 260–269. doi: 10.1056/NEJMoa051530
Block, G. A., Klassen, P. S., Lazarus, J. M., Ofsthun, N., Lowrie, E. G., and Chertow, G. M. (2004). Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J. Am. Soc. Nephrol. 15, 2208–2218. doi: 10.1097/01.ASN.0000133041.27682.A2
Blood Pressure Lowering Treatment Trialists Collaboration, Ninomiya, T., Perkovic, V., Turnbull, F., Neal, B., Barzi, F., et al. (2013). Blood pressure lowering and major cardiovascular events in people with and without chronic kidney disease: meta-analysis of randomised controlled trials. BMJ 347:f5680. doi: 10.1136/bmj.f5680
Bongartz, L. G., Cramer, M. J., Doevendans, P. A., Joles, J. A., and Braam, B. (2005). The severe cardiorenal syndrome: Guyton revisited. Eur. Heart J. 26, 11–17. doi: 10.1093/eurheartj/ehi020
Borlaug, B. A. (2014). The pathophysiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 11, 507–515. doi: 10.1038/nrcardio.2014.83
Borlaug, B. A., and Kass, D. A. (2011). Ventricular-vascular interaction in heart failure. Cardiol. Clin. 29, 447–459. doi: 10.1016/j.ccl.2011.06.004
Borlaug, B. A., Lewis, G. D., McNulty, S. E., Semigran, M. J., Lewinter, M., Chen, H., et al. (2015). Effects of sildenafil on ventricular and vascular function in heart failure with preserved ejection fraction. Circ. Heart Fail. 8, 533–541. doi: 10.1161/CIRCHEARTFAILURE.114.001915
Borlaug, B. A., Anstrom, K. J., Lewis, G. D., Shah, S. J., Levine, J. A., Koepp, G. A., et al. (2018). Effect of inorganic nitrite vs placebo on exercise capacity among patients with heart failure with preserved ejection fraction: the INDIE-HFpEF randomized clinical trial. JAMA 320, 1764–1773. doi: 10.1001/jama.2018.14852
Bortolotto, L. A., Safar, M. E., Billaud, E., Lacroix, C., Asmar, R., London, G. M., et al. (1999). Plasma homocysteine, aortic stiffness, and renal function in hypertensive patients. Hypertension 34, 837–842. doi: 10.1161/01.HYP.34.4.837
Bozbas, H., Pirat, B., Demirtas, S., Simsek, V., Yildirir, A., Sade, E., et al. (2009). Evaluation of coronary microvascular function in patients with end-stage renal disease, and renal allograft recipients. Atherosclerosis 202, 498–504. doi: 10.1016/j.atherosclerosis.2008.04.043
Brandt, M. C., Mahfoud, F., Reda, S., Schirmer, S. H., Erdmann, E., Bohm, M., et al. (2012). Renal sympathetic denervation reduces left ventricular hypertrophy and improves cardiac function in patients with resistant hypertension. J. Am. Coll. Cardiol. 59, 901–909. doi: 10.1016/j.jacc.2011.11.034
Brenner, B. M., Cooper, M. E., De Zeeuw, D., Keane, W. F., Mitch, W. E., Parving, H. H., et al. (2001). Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 345, 861–869. doi: 10.1056/NEJMoa011161
Briet, M., Boutouyrie, P., Laurent, S., and London, G. M. (2012). Arterial stiffness and pulse pressure in CKD and ESRD. Kidney Int. 82, 388–400. doi: 10.1038/ki.2012.131
Brouwers, F. P., De Boer, R. A., Van Der Harst, P., Voors, A. A., Gansevoort, R. T., Bakker, S. J., et al. (2013). Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur. Heart J. 34, 1424–1431. doi: 10.1093/eurheartj/eht066
Brown, N. J. (2013). Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 9, 459–469. doi: 10.1038/nrneph.2013.110
Brunet, P., Gondouin, B., Duval-Sabatier, A., Dou, L., Cerini, C., Dignat-George, F., et al. (2011). Does uremia cause vascular dysfunction? Kidney Blood Press. Res. 34, 284–290. doi: 10.1159/000327131
Bucharles, S., Barberato, S. H., Stinghen, A. E., Gruber, B., Meister, H., Mehl, A., et al. (2011). Hypovitaminosis D is associated with systemic inflammation and concentric myocardial geometric pattern in hemodialysis patients with low iPTH levels. Nephron Clin. Pract. 118, c384–c391. doi: 10.1159/000323664
Butler, J., Hamo, C. E., Filippatos, G., Pocock, S. J., Bernstein, R. A., Brueckmann, M., et al. (2017). The potential role and rationale for treatment of heart failure with sodium-glucose co-transporter 2 inhibitors. Eur. J. Heart Fail. 19, 1390–1400. doi: 10.1002/ejhf.933
Capitanio, S., Sambuceti, G., Giusti, M., Morbelli, S., Murialdo, G., Garibotto, G., et al. (2013). 1,25-Dihydroxy vitamin D and coronary microvascular function. Eur. J. Nucl. Med. Mol. Imaging 40, 280–289. doi: 10.1007/s00259-012-2271-0
Charra, B., and Chazot, C. (2003). The neglect of sodium restriction in dialysis patients: a short review. Hemodial. Int. 7, 342–347. doi: 10.1046/j.1492-7535.2003.00060.x
Chitalia, N., Recio-Mayoral, A., Kaski, J. C., and Banerjee, D. (2012). Vitamin D deficiency and endothelial dysfunction in non-dialysis chronic kidney disease patients. Atherosclerosis 220, 265–268. doi: 10.1016/j.atherosclerosis.2011.10.023
Choi, S. R., Lim, J. H., Kim, M. Y., Hong, Y. A., Chung, B. H., Chung, S., et al. (2012). Cinacalcet improves endothelial dysfunction and cardiac hypertrophy in patients on hemodialysis with secondary hyperparathyroidism. Nephron Clin. Pract. 122, 1–8. doi: 10.1159/000347145
Collins, A. J. (2003). Cardiovascular mortality in end-stage renal disease. Am. J. Med. Sci. 325, 163–167. doi: 10.1097/00000441-200304000-00002
Congote, L. F., Sadvakassova, G., Dobocan, M. C., Difalco, M. R., and Li, Q. (2010). Erythropoietin-dependent endothelial proteins: potential use against erythropoietin resistance. Cytokine 51, 113–118. doi: 10.1016/j.cyto.2010.03.020
Cosby, K., Partovi, K. S., Crawford, J. H., Patel, R. P., Reiter, C. D., Martyr, S., et al. (2003). Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 9, 1498–1505. doi: 10.1038/nm954
Cosson, E., Pham, I., Valensi, P., Paries, J., Attali, J. R., and Nitenberg, A. (2006). Impaired coronary endothelium-dependent vasodilation is associated with microalbuminuria in patients with type 2 diabetes and angiographically normal coronary arteries. Diabetes Care 29, 107–112. doi: 10.2337/diacare.29.1.107
Cozzolino, M., Bruschetta, E., Stucchi, A., Ronco, C., and Cusi, D. (2012). Role of vitamin D receptor activators in cardio-renal syndromes. Semin. Nephrol. 32, 63–69. doi: 10.1016/j.semnephrol.2011.11.009
Crawford, J. H., Isbell, T. S., Huang, Z., Shiva, S., Chacko, B. K., Schechter, A. N., et al. (2006). Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood 107, 566–574. doi: 10.1182/blood-2005-07-2668
Crea, F., Bairey Merz, C. N., Beltrame, J. F., Kaski, J. C., Ogawa, H., Ong, P., et al. (2017). The parallel tales of microvascular angina and heart failure with preserved ejection fraction: a paradigm shift. Eur. Heart J. 38, 473–477. doi: 10.1093/eurheartj/ehw461
Czaya, B., Seeherunvong, W., Singh, S., Yanucil, C., Ruiz, P., Quiroz, Y., et al. (2019). Cardioprotective effects of paricalcitol alone and in combination with FGF23 receptor inhibition in chronic renal failure: experimental and clinical studies. Am. J. Hypertens. 32, 34–44. doi: 10.1093/ajh/hpy154
Deckert, T., Feldt-Rasmussen, B., Borch-Johnsen, K., Jensen, T., and Kofoed-Enevoldsen, A. (1989). Albuminuria reflects widespread vascular damage. The Steno hypothesis. Diabetologia 32, 219–226. doi: 10.1007/BF00285287
Dhakal, B. P., Malhotra, R., Murphy, R. M., Pappagianopoulos, P. P., Baggish, A. L., Weiner, R. B., et al. (2015). Mechanisms of exercise intolerance in heart failure with preserved ejection fraction: the role of abnormal peripheral oxygen extraction. Circ. Heart Fail. 8, 286–294. doi: 10.1161/CIRCHEARTFAILURE.114.001825
Di Marco, G. S., Konig, M., Stock, C., Wiesinger, A., Hillebrand, U., Reiermann, S., et al. (2013). High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 83, 213–222. doi: 10.1038/ki.2012.300
Drechsler, C., Pilz, S., Obermayer-Pietsch, B., Verduijn, M., Tomaschitz, A., Krane, V., et al. (2010). Vitamin D deficiency is associated with sudden cardiac death, combined cardiovascular events, and mortality in haemodialysis patients. Eur. Heart J. 31, 2253–2261. doi: 10.1093/eurheartj/ehq246
Faul, C., Amaral, A. P., Oskouei, B., Hu, M. C., Sloan, A., Isakova, T., et al. (2011). FGF23 induces left ventricular hypertrophy. J. Clin. Invest. 121, 4393–4408. doi: 10.1172/JCI46122
Fischer, M., Baessler, A., and Schunkert, H. (2002). Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc. Res. 53, 672–677. doi: 10.1016/S0008-6363(01)00479-5
Foley, R. N., Curtis, B. M., Randell, E. W., and Parfrey, P. S. (2010). Left ventricular hypertrophy in new hemodialysis patients without symptomatic cardiac disease. Clin. J. Am. Soc. Nephrol. 5, 805–813. doi: 10.2215/CJN.07761109
Forrester, S. J., Booz, G. W., Sigmund, C. D., Coffman, T. M., Kawai, T., Rizzo, V., et al. (2018). Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol. Rev. 98, 1627–1738. doi: 10.1152/physrev.00038.2017
Fraenkel, P. G. (2015). Understanding anemia of chronic disease. Hematol. Am. Soc. Hematol. Educ. Program 2015, 14–18. doi: 10.1182/asheducation-2015.1.14
Frank, H., Heusser, K., Hoffken, B., Huber, P., Schmieder, R. E., and Schobel, H. P. (2004). Effect of erythropoietin on cardiovascular prognosis parameters in hemodialysis patients. Kidney Int. 66, 832–840. doi: 10.1111/j.1523-1755.2004.00810.x
Franssen, C., Chen, S., Unger, A., Korkmaz, H. I., De Keulenaer, G. W., Tschope, C., et al. (2016). Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail 4, 312–324. doi: 10.1016/j.jchf.2015.10.007
Fukushima, K., Javadi, M. S., Higuchi, T., Bravo, P. E., Chien, D., Lautamaki, R., et al. (2012). Impaired global myocardial flow dynamics despite normal left ventricular function and regional perfusion in chronic kidney disease: a quantitative analysis of clinical 82Rb PET/CT studies. J. Nucl. Med. 53, 887–893. doi: 10.2967/jnumed.111.099325
Garg, A. X., Kiberd, B. A., Clark, W. F., Haynes, R. B., and Clase, C. M. (2002). Albuminuria and renal insufficiency prevalence guides population screening: results from the NHANES III. Kidney Int. 61, 2165–2175. doi: 10.1046/j.1523-1755.2002.00356.x
Garg, R., Rao, A. D., Baimas-George, M., Hurwitz, S., Foster, C., Shah, R. V., et al. (2015). Mineralocorticoid receptor blockade improves coronary microvascular function in individuals with type 2 diabetes. Diabetes 64, 236–242. doi: 10.2337/db14-0670
Ghio, S., Magrini, G., Serio, A., Klersy, C., Fucili, A., Ronaszeki, A., et al. (2006). Effects of nebivolol in elderly heart failure patients with or without systolic left ventricular dysfunction: results of the SENIORS echocardiographic substudy. Eur. Heart J. 27, 562–568. doi: 10.1093/eurheartj/ehi735
GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia) (1997). Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet 349, 1857–1863. doi: 10.1016/S0140-6736(96)11445-8
Gladden, J. D., Linke, W. A., and Redfield, M. M. (2014). Heart failure with preserved ejection fraction. Pflugers Arch. 466, 1037–1053. doi: 10.1007/s00424-014-1480-8
Goicoechea, M., De Vinuesa, S. G., Verdalles, U., Ruiz-Caro, C., Ampuero, J., Rincon, A., et al. (2010). Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin. J. Am. Soc. Nephrol. 5, 1388–1393. doi: 10.2215/CJN.01580210
Goldberg, N., Lundin, A. P., Delano, B., Friedman, E. A., and Stein, R. A. (1992). Changes in left ventricular size, wall thickness, and function in anemic patients treated with recombinant human erythropoietin. Am. Heart J. 124, 424–427. doi: 10.1016/0002-8703(92)90608-X
Gori, M., Lam, C. S., Gupta, D. K., Santos, A. B., Cheng, S., Shah, A. M., et al. (2014a). Sex-specific cardiovascular structure and function in heart failure with preserved ejection fraction. Eur. J. Heart Fail. 16, 535–542. doi: 10.1002/ejhf.67
Gori, M., Senni, M., Gupta, D. K., Charytan, D. M., Kraigher-Krainer, E., Pieske, B., et al. (2014b). Association between renal function and cardiovascular structure and function in heart failure with preserved ejection fraction. Eur. Heart J. 35, 3442–3451. doi: 10.1093/eurheartj/ehu254
Hartog, J. W., De Vries, A. P., Bakker, S. J., Graaff, R., Van Son, W. J., Van Der Heide, J. J., et al. (2006). Risk factors for chronic transplant dysfunction and cardiovascular disease are related to accumulation of advanced glycation end-products in renal transplant recipients. Nephrol. Dial. Transplant. 21, 2263–2269. doi: 10.1093/ndt/gfl132
Hartog, J. W., Voors, A. A., Bakker, S. J., Smit, A. J., and Van Veldhuisen, D. J. (2007). Advanced glycation end-products (AGEs) and heart failure: pathophysiology and clinical implications. Eur. J. Heart Fail. 9, 1146–1155. doi: 10.1016/j.ejheart.2007.09.009
Hernandez, A. F., Hammill, B. G., O’Connor, C. M., Schulman, K. A., Curtis, L. H., and Fonarow, G. C. (2009). Clinical effectiveness of beta-blockers in heart failure: findings from the OPTIMIZE-HF (Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure) Registry. J. Am. Coll. Cardiol. 53, 184–192. doi: 10.1016/j.jacc.2008.09.031
Hickson, L. J., Negrotto, S. M., Onuigbo, M., Scott, C. G., Rule, A. D., Norby, S. M., et al. (2016). Echocardiography criteria for structural heart disease in patients with end-stage renal disease initiating hemodialysis. J. Am. Coll. Cardiol. 67, 1173–1182. doi: 10.1016/j.jacc.2015.12.052
Hillege, H. L., Nitsch, D., Pfeffer, M. A., Swedberg, K., Mcmurray, J. J., Yusuf, S., et al. (2006). Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation 113, 671–678. doi: 10.1161/CIRCULATIONAHA.105.580506
Imel, E. A., Zhang, X., Ruppe, M. D., Weber, T. J., Klausner, M. A., Ito, T., et al. (2015). Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. J. Clin. Endocrinol. Metab. 100, 2565–2573. doi: 10.1210/jc.2015-1551
Jono, S., Mckee, M. D., Murry, C. E., Shioi, A., Nishizawa, Y., Mori, K., et al. (2000). Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 87, E10–E17. doi: 10.1161/01.res.87.7.e10
Jourde-Chiche, N., Dou, L., Cerini, C., Dignat-George, F., and Brunet, P. (2011). Vascular incompetence in dialysis patients – protein-bound uremic toxins and endothelial dysfunction. Semin. Dial. 24, 327–337. doi: 10.1111/j.1525-139X.2011.00925.x
Judd, E., and Calhoun, D. A. (2015). Management of hypertension in CKD: beyond the guidelines. Adv. Chronic Kidney Dis. 22, 116–122. doi: 10.1053/j.ackd.2014.12.001
Kamo, T., Akazawa, H., and Komuro, I. (2015). Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ. Res. 117, 89–98. doi: 10.1161/CIRCRESAHA.117.305349
Kanbay, M., Huddam, B., Azak, A., Solak, Y., Kadioglu, G. K., Kirbas, I., et al. (2011). A randomized study of allopurinol on endothelial function and estimated glomular filtration rate in asymptomatic hyperuricemic subjects with normal renal function. Clin. J. Am. Soc. Nephrol. 6, 1887–1894. doi: 10.2215/CJN.11451210
Kapelios, C. J., Murrow, J. R., Nuhrenberg, T. G., and Montoro Lopez, M. N. (2019). Effect of mineralocorticoid receptor antagonists on cardiac function in patients with heart failure and preserved ejection fraction: a systematic review and meta-analysis of randomized controlled trials. Heart Fail. Rev. 24, 367–377. doi: 10.1007/s10741-018-9758-0
Kasner, M., Aleksandrov, A. S., Westermann, D., Lassner, D., Gross, M., Von Haehling, S., et al. (2013). Functional iron deficiency and diastolic function in heart failure with preserved ejection fraction. Int. J. Cardiol. 168, 4652–4657. doi: 10.1016/j.ijcard.2013.07.185
Katz, D. H., Burns, J. A., Aguilar, F. G., Beussink, L., and Shah, S. J. (2014). Albuminuria is independently associated with cardiac remodeling, abnormal right and left ventricular function, and worse outcomes in heart failure with preserved ejection fraction. JACC Heart Fail 2, 586–596. doi: 10.1016/j.jchf.2014.05.016
Kendrick, J., Chonchol, M., Gnahn, H., and Sander, D. (2010). Higher systolic blood pressure is associated with progression of carotid intima-media thickness in patients with chronic kidney disease. Kidney Int. 77, 794–800. doi: 10.1038/ki.2009.557
Klahr, S., Levey, A. S., Beck, G. J., Caggiula, A. W., Hunsicker, L., Kusek, J. W., et al. (1994). The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of diet in renal disease study group. N. Engl. J. Med. 330, 877–884. doi: 10.1056/NEJM199403313301301
Klein, D. A., Katz, D. H., Beussink-Nelson, L., Sanchez, C. L., Strzelczyk, T. A., and Shah, S. J. (2015). Association of chronic kidney disease with chronotropic incompetence in heart failure with preserved ejection fraction. Am. J. Cardiol. 116, 1093–1100. doi: 10.1016/j.amjcard.2015.06.038
Koller, L., Kleber, M. E., Brandenburg, V. M., Goliasch, G., Richter, B., Sulzgruber, P., et al. (2015). Fibroblast growth factor 23 is an independent and specific predictor of mortality in patients with heart failure and reduced ejection fraction. Circ. Heart Fail. 8, 1059–1067. doi: 10.1161/CIRCHEARTFAILURE.115.002341
Komaba, H., and Fukagawa, M. (2012). The role of FGF23 in CKD – with or without Klotho. Nat. Rev. Nephrol. 8, 484–490.
Kottgen, A., Russell, S. D., Loehr, L. R., Crainiceanu, C. M., Rosamond, W. D., Chang, P. P., et al. (2007). Reduced kidney function as a risk factor for incident heart failure: the atherosclerosis risk in communities (ARIC) study. J. Am. Soc. Nephrol. 18, 1307–1315. doi: 10.1681/ASN.2006101159
Lam, C. S., Carson, P. E., Anand, I. S., Rector, T. S., Kuskowski, M., Komajda, M., et al. (2012). Sex differences in clinical characteristics and outcomes in elderly patients with heart failure and preserved ejection fraction: the Irbesartan in Heart Failure with Preserved Ejection Fraction (I-PRESERVE) trial. Circ. Heart Fail. 5, 571–578. doi: 10.1161/CIRCHEARTFAILURE.112.970061
Laurent, S., Cockcroft, J., Van Bortel, L., Boutouyrie, P., Giannattasio, C., Hayoz, D., et al. (2006). Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur. Heart J. 27, 2588–2605. doi: 10.1093/eurheartj/ehl254
Leifheit-Nestler, M., and Haffner, D. (2018). Paracrine effects of FGF23 on the heart. Front. Endocrinol. 9:278. doi: 10.3389/fendo.2018.00278
Leist, M., Ghezzi, P., Grasso, G., Bianchi, R., Villa, P., Fratelli, M., et al. (2004). Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 305, 239–242. doi: 10.1126/science.1098313
Levin, A., and Stevens, P. E. (2014). Summary of KDIGO 2012 CKD Guideline: behind the scenes, need for guidance, and a framework for moving forward. Kidney Int. 85, 49–61. doi: 10.1038/ki.2013.444
Levin, A., Singer, J., Thompson, C. R., Ross, H., and Lewis, M. (1996). Prevalent left ventricular hypertrophy in the predialysis population: identifying opportunities for intervention. Am. J. Kidney Dis. 27, 347–354. doi: 10.1016/S0272-6386(96)90357-1
Liao, L., Jollis, J. G., Anstrom, K. J., Whellan, D. J., Kitzman, D. W., Aurigemma, G. P., et al. (2006). Costs for heart failure with normal vs reduced ejection fraction. Arch. Intern. Med. 166, 112–118. doi: 10.1001/archinte.166.1.112
Lofman, I., Szummer, K., Dahlstrom, U., Jernberg, T., and Lund, L. H. (2017). Associations with and prognostic impact of chronic kidney disease in heart failure with preserved, mid-range, and reduced ejection fraction. Eur. J. Heart Fail. 19, 1606–1614. doi: 10.1002/ejhf.821
London, G. M. (2002). Left ventricular alterations and end-stage renal disease. Nephrol. Dial. Transplant. 17(Suppl. 1), 29–36. doi: 10.1093/ndt/17.suppl_1.29
Martin, A., David, V., and Quarles, L. D. (2012). Regulation and function of the FGF23/klotho endocrine pathways. Physiol. Rev. 92, 131–155. doi: 10.1152/physrev.00002.2011
Masoudi, F. A., Havranek, E. P., Smith, G., Fish, R. H., Steiner, J. F., Ordin, D. L., et al. (2003). Gender, age, and heart failure with preserved left ventricular systolic function. J. Am. Coll. Cardiol. 41, 217–223. doi: 10.1016/S0735-1097(02)02696-7
Matias, P. J., Jorge, C., Ferreira, C., Borges, M., Aires, I., Amaral, T., et al. (2010). Cholecalciferol supplementation in hemodialysis patients: effects on mineral metabolism, inflammation, and cardiac dimension parameters. Clin. J. Am. Soc. Nephrol. 5, 905–911. doi: 10.2215/CJN.06510909
Maurer, M. S., Teruya, S., Chakraborty, B., Helmke, S., and Mancini, D. (2013). Treating anemia in older adults with heart failure with a preserved ejection fraction with epoetin alfa: single-blind randomized clinical trial of safety and efficacy. Circ. Heart Fail. 6, 254–263. doi: 10.1161/CIRCHEARTFAILURE.112.969717
McAlister, F. A., Ezekowitz, J., Tarantini, L., Squire, I., Komajda, M., Bayes-Genis, A., et al. (2012). Renal dysfunction in patients with heart failure with preserved versus reduced ejection fraction: impact of the new Chronic Kidney Disease-Epidemiology Collaboration Group formula. Circ. Heart Fail. 5, 309–314. doi: 10.1161/CIRCHEARTFAILURE.111.966242
McMurray, J. J., Packer, M., Desai, A. S., Gong, J., Lefkowitz, M. P., Rizkala, A. R., et al. (2014). Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 371, 993–1004. doi: 10.1056/NEJMoa1409077
Meems, L. M. G., Brouwers, F. P., Joosten, M. M., Lambers Heerspink, H. J., De Zeeuw, D., Bakker, S. J. L., et al. (2016). Plasma calcidiol, calcitriol, and parathyroid hormone and risk of new onset heart failure in a population-based cohort study. ESC Heart Fail. 3, 189–197. doi: 10.1002/ehf2.12089
Mercadal, L., Metzger, M., Casadevall, N., Haymann, J. P., Karras, A., Boffa, J. J., et al. (2012). Timing and determinants of erythropoietin deficiency in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 7, 35–42. doi: 10.2215/CJN.04690511
Merkus, D., Haitsma, D. B., Sorop, O., Boomsma, F., De Beer, V. J., Lamers, J. M., et al. (2006). Coronary vasoconstrictor influence of angiotensin II is reduced in remodeled myocardium after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 291, H2082–H2089. doi: 10.1152/ajpheart.00861.2005
Middleton, J. P., and Pun, P. H. (2010). Hypertension, chronic kidney disease, and the development of cardiovascular risk: a joint primacy. Kidney Int. 77, 753–755. doi: 10.1038/ki.2010.19
Mitchell, G. F. (2008). Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end-organ damage. J. Appl. Physiol. 105, 1652–1660.
Miura, M., Shiba, N., Nochioka, K., Takada, T., Takahashi, J., Kohno, H., et al. (2012). Urinary albumin excretion in heart failure with preserved ejection fraction: an interim analysis of the CHART 2 study. Eur. J. Heart Fail. 14, 367–376. doi: 10.1093/eurjhf/hfs001
Moradi, H., Sica, D. A., and Kalantar-Zadeh, K. (2013). Cardiovascular burden associated with uremic toxins in patients with chronic kidney disease. Am. J. Nephrol. 38, 136–148. doi: 10.1159/000351758
Movilli, E., Feliciani, A., Camerini, C., Brunori, G., Zubani, R., Scolari, F., et al. (2005). A high calcium-phosphate product is associated with high C-reactive protein concentrations in hemodialysis patients. Nephron Clin. Pract. 101, c161–c167. doi: 10.1159/000087391
Murthy, V. L., Naya, M., Foster, C. R., Hainer, J., Gaber, M., Dorbala, S., et al. (2012). Coronary vascular dysfunction and prognosis in patients with chronic kidney disease. JACC Cardiovasc. Imaging 5, 1025–1034. doi: 10.1016/j.jcmg.2012.06.007
Nangaku, M., and Fujita, T. (2008). Activation of the renin-angiotensin system and chronic hypoxia of the kidney. Hypertens. Res. 31, 175–184. doi: 10.1291/hypres.31.175
Nayor, M., Larson, M. G., Wang, N., Santhanakrishnan, R., Lee, D. S., Tsao, C. W., et al. (2017). The association of chronic kidney disease and microalbuminuria with heart failure with preserved vs. reduced ejection fraction. Eur. J. Heart Fail. 19, 615–623. doi: 10.1002/ejhf.778
Neal, B., Perkovic, V., Mahaffey, K. W., De Zeeuw, D., Fulcher, G., Erondu, N., et al. (2017). Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 377, 644–657. doi: 10.1056/NEJMoa1611925
Negri, A. L. (2014). Fibroblast growth factor 23: associations with cardiovascular disease and mortality in chronic kidney disease. Int. Urol. Nephrol. 46, 9–17. doi: 10.1007/s11255-012-0370-2
Nolte, K., Herrmann-Lingen, C., Platschek, L., Holzendorf, V., Pilz, S., Tomaschitz, A., et al. (2019). Vitamin D deficiency in patients with diastolic dysfunction or heart failure with preserved ejection fraction. ESC Heart Fail. 6, 262–270. doi: 10.1002/ehf2.12413
Noone, D., and Licht, C. (2014). Chronic kidney disease: a new look at pathogenetic mechanisms and treatment options. Pediatr. Nephrol. 29, 779–792. doi: 10.1007/s00467-013-2436-5
Norton, G. R., Candy, G., and Woodiwiss, A. J. (1996). Aminoguanidine prevents the decreased myocardial compliance produced by streptozotocin-induced diabetes mellitus in rats. Circulation 93, 1905–1912. doi: 10.1161/01.CIR.93.10.1905
Nunez, J., Dominguez, E., Ramon, J. M., Nunez, E., Sanchis, J., Santas, E., et al. (2016). Iron deficiency and functional capacity in patients with advanced heart failure with preserved ejection fraction. Int. J. Cardiol. 207, 365–367. doi: 10.1016/j.ijcard.2016.01.187
Obokata, M., Reddy, Y. N. V., Melenovsky, V., Kane, G. C., Olson, T. P., Jarolim, P., et al. (2018). Myocardial injury and cardiac reserve in patients with heart failure and preserved ejection fraction. J. Am. Coll. Cardiol. 72, 29–40. doi: 10.1016/j.jacc.2018.04.039
Osto, E., Fallo, F., Pelizzo, M. R., Maddalozzo, A., Sorgato, N., Corbetti, F., et al. (2012). Coronary microvascular dysfunction induced by primary hyperparathyroidism is restored after parathyroidectomy. Circulation 126, 1031–1039. doi: 10.1161/CIRCULATIONAHA.111.081307
Owan, T. E., Hodge, D. O., Herges, R. M., Jacobsen, S. J., Roger, V. L., and Redfield, M. M. (2006). Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 355, 251–259. doi: 10.1056/NEJMoa052256
Pandey, A., Kitzman, D. W., Houston, D. K., Chen, H., and Shea, M. K. (2018). Vitamin D status and exercise capacity in older patients with heart failure with preserved ejection fraction. Am. J. Med. 131, 1515.e1511–1515.e1519. doi: 10.1016/j.amjmed.2018.07.009
Paulus, W. J., and Dal Canto, E. (2018). Distinct myocardial targets for diabetes therapy in heart failure with preserved or reduced ejection fraction. JACC Heart Fail 6, 1–7. doi: 10.1016/j.jchf.2017.07.012
Paulus, W. J., and Tschope, C. (2013). A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 62, 263–271. doi: 10.1016/j.jacc.2013.02.092
Pecoits-Filho, R., Bucharles, S., and Barberato, S. H. (2012). Diastolic heart failure in dialysis patients: mechanisms, diagnostic approach, and treatment. Semin. Dial. 25, 35–41. doi: 10.1111/j.1525-139X.2011.01011.x
Pedrinelli, R., Giampietro, O., Carmassi, F., Melillo, E., Dell’Omo, G., Catapano, G., et al. (1994). Microalbuminuria and endothelial dysfunction in essential hypertension. Lancet 344, 14–18. doi: 10.1016/S0140-6736(94)91047-2
Periyasamy, S. M., Chen, J., Cooney, D., Carter, P., Omran, E., Tian, J., et al. (2001). Effects of uremic serum on isolated cardiac myocyte calcium cycling and contractile function. Kidney Int. 60, 2367–2376. doi: 10.1046/j.1523-1755.2001.00053.x
Petrova, R., Yamamoto, Y., Muraki, K., Yonekura, H., Sakurai, S., Watanabe, T., et al. (2002). Advanced glycation endproduct-induced calcium handling impairment in mouse cardiac myocytes. J. Mol. Cell. Cardiol. 34, 1425–1431. doi: 10.1006/jmcc.2002.2084
Pfeffer, M. A., Claggett, B., Assmann, S. F., Boineau, R., Anand, I. S., Clausell, N., et al. (2015). Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial. Circulation 131, 34–42. doi: 10.1161/CIRCULATIONAHA.114.013255
Phan, T. T., Shivu, G. N., Abozguia, K., Davies, C., Nassimizadeh, M., Jimenez, D., et al. (2010). Impaired heart rate recovery and chronotropic incompetence in patients with heart failure with preserved ejection fraction. Circ. Heart Fail. 3, 29–34. doi: 10.1161/CIRCHEARTFAILURE.109.877720
Pieske, B., Maggioni, A. P., Lam, C. S. P., Pieske-Kraigher, E., Filippatos, G., Butler, J., et al. (2017). Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES-PRESERVED) study. Eur. Heart J. 38, 1119–1127. doi: 10.1093/eurheartj/ehw593
Pitt, B., Pfeffer, M. A., Assmann, S. F., Boineau, R., Anand, I. S., Claggett, B., et al. (2014). Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 370, 1383–1392. doi: 10.1056/NEJMoa1313731
Qi, G., Jia, L., Li, Y., Bian, Y., Cheng, J., Li, H., et al. (2011). Angiotensin II infusion-induced inflammation, monocytic fibroblast precursor infiltration, and cardiac fibrosis are pressure dependent. Cardiovasc. Toxicol. 11, 157–167. doi: 10.1007/s12012-011-9109-z
Redfield, M. M., Jacobsen, S. J., Burnett, J. C. Jr., Mahoney, D. W., Bailey, K. R., and Rodeheffer, R. J. (2003). Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA 289, 194–202. doi: 10.1001/jama.289.2.194
Redfield, M. M., Chen, H. H., Borlaug, B. A., Semigran, M. J., Lee, K. L., Lewis, G., et al. (2013). Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 309, 1268–1277. doi: 10.1001/jama.2013.2024
Redfield, M. M., Anstrom, K. J., Levine, J. A., Koepp, G. A., Borlaug, B. A., Chen, H. H., et al. (2015). Isosorbide mononitrate in heart failure with preserved ejection fraction. N. Engl. J. Med. 373, 2314–2324. doi: 10.1056/NEJMoa1510774
Regitz-Zagrosek, V. (2006). Therapeutic implications of the gender-specific aspects of cardiovascular disease. Nat. Rev. Drug Discov. 5, 425–438. doi: 10.1038/nrd2032
Reudelhuber, T. L., Bernstein, K. E., and Delafontaine, P. (2007). Is angiotensin II a direct mediator of left ventricular hypertrophy? Time for another look. Hypertension 49, 1196–1201. doi: 10.1161/HYPERTENSIONAHA.106.075085
Roksnoer, L. C., Van Veghel, R., De Vries, R., Garrelds, I. M., Bhaggoe, U. M., Friesema, E. C., et al. (2015). Optimum AT1 receptor-neprilysin inhibition has superior cardioprotective effects compared with AT1 receptor blockade alone in hypertensive rats. Kidney Int. 88, 109–120. doi: 10.1038/ki.2015.107
Roksnoer, L. C., Van Veghel, R., Van Groningen, M. C., De Vries, R., Garrelds, I. M., Bhaggoe, U. M., et al. (2016). Blood pressure-independent renoprotection in diabetic rats treated with AT1 receptor-neprilysin inhibition compared with AT1 receptor blockade alone. Clin. Sci. 130, 1209–1220. doi: 10.1042/CS20160197
Ronco, C., House, A. A., and Haapio, M. (2008). Cardiorenal and renocardiac syndromes: the need for a comprehensive classification and consensus. Nat. Clin. Pract. Nephrol. 4, 310–311. doi: 10.1038/ncpneph0803
Rosner, M. H., Ronco, C., and Okusa, M. D. (2012). The role of inflammation in the cardio-renal syndrome: a focus on cytokines and inflammatory mediators. Semin. Nephrol. 32, 70–78. doi: 10.1016/j.semnephrol.2011.11.010
Rostand, S. G., and Drueke, T. B. (1999). Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int. 56, 383–392. doi: 10.1046/j.1523-1755.1999.00575.x
Roy, C., Slimani, A., De Meester, C., Amzulescu, M., Ferracin, B., Ginion, A., et al. (2018). 1102 fibroblast growth factor 23 and extracellular volume as markers of myocardial fibrosis and poor outcome in heart failure with preserved ejection fraction. Eur. Heart J. 39(Suppl. 1), 224. doi: 10.1093/eurheartj/ehy565.1102
Ruggenenti, P., Schieppati, A., and Remuzzi, G. (2001). Progression, remission, regression of chronic renal diseases. Lancet 357, 1601–1608. doi: 10.1016/S0140-6736(00)04728-0
Ruggenenti, P., Cravedi, P., and Remuzzi, G. (2012). Mechanisms and treatment of CKD. J. Am. Soc. Nephrol. 23, 1917–1928. doi: 10.1681/ASN.2012040390
Ruiz-Meana, M., Minguet, M., Bou-Teen, D., Miro-Casas, E., Castans, C., Castellano, J., et al. (2019). Ryanodine receptor glycation favors mitochondrial damage in the senescent heart. Circulation 139, 949–964. doi: 10.1161/CIRCULATIONAHA.118.035869
Safar, M. E., Plante, G. E., and Mimran, A. (2015). Arterial stiffness, pulse pressure, and the kidney. Am. J. Hypertens. 28, 561–569. doi: 10.1093/ajh/hpu206
Sallee, M., Dou, L., Cerini, C., Poitevin, S., Brunet, P., and Burtey, S. (2014). The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: a new concept to understand cardiovascular complications of chronic kidney disease. Toxins 6, 934–949. doi: 10.3390/toxins6030934
Salman, I. M. (2015). Cardiovascular autonomic dysfunction in chronic kidney disease: a comprehensive review. Curr. Hypertens. Rep. 17:59. doi: 10.1007/s11906-015-0571-z
Sarkozy, M., Gaspar, R., Zvara, A., Siska, A., Kovari, B., Szucs, G., et al. (2019). Chronic kidney disease induces left ventricular overexpression of the pro-hypertrophic microRNA-212. Sci. Rep. 9:1302. doi: 10.1038/s41598-018-37690-5
Sarnak, M. J., Levey, A. S., Schoolwerth, A. C., Coresh, J., Culleton, B., Hamm, L. L., et al. (2003). Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 108, 2154–2169. doi: 10.1161/01.CIR.0000095676.90936.80
Schroeder, N. J., and Cunningham, J. (2000). What’s new in vitamin D for the nephrologist? Nephrol. Dial. Transplant. 15, 460–466. doi: 10.1093/ndt/15.4.460
Sezer, S., Karakan, S., Atesagaoglu, B., and Acar, F. N. (2014). Allopurinol reduces cardiovascular risks and improves renal function in pre-dialysis chronic kidney disease patients with hyperuricemia. Saudi J. Kidney Dis. Transpl. 25, 316–320.
Shao, C. H., Capek, H. L., Patel, K. P., Wang, M., Tang, K., Desouza, C., et al. (2011). Carbonylation contributes to SERCA2a activity loss and diastolic dysfunction in a rat model of type 1 diabetes. Diabetes 60, 947–959. doi: 10.2337/db10-1145
Sharma, K., and Kass, D. A. (2014). Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ. Res. 115, 79–96. doi: 10.1161/CIRCRESAHA.115.302922
Silberberg, J. S., Barre, P. E., Prichard, S. S., and Sniderman, A. D. (1989). Impact of left ventricular hypertrophy on survival in end-stage renal disease. Kidney Int. 36, 286–290. doi: 10.1038/ki.1989.192
Silswal, N., Touchberry, C. D., Daniel, D. R., Mccarthy, D. L., Zhang, S., Andresen, J., et al. (2014). FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am. J. Physiol. Endocrinol. Metab. 307, E426–E436. doi: 10.1152/ajpendo.00264.2014
Singel, D. J., and Stamler, J. S. (2005). Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu. Rev. Physiol. 67, 99–145. doi: 10.1146/annurev.physiol.67.060603.090918
Smit, A. J., and Lutgers, H. L. (2004). The clinical relevance of advanced glycation endproducts (AGE) and recent developments in pharmaceutics to reduce AGE accumulation. Curr. Med. Chem. 11, 2767–2784. doi: 10.2174/0929867043364342
Smith, D. H., Thorp, M. L., Gurwitz, J. H., Mcmanus, D. D., Goldberg, R. J., Allen, L. A., et al. (2013). Chronic kidney disease and outcomes in heart failure with preserved versus reduced ejection fraction: the Cardiovascular Research Network PRESERVE Study. Circ. Cardiovasc. Qual. Outcomes 6, 333–342. doi: 10.1161/CIRCOUTCOMES.113.000221
Solomon, S. D., Zile, M., Pieske, B., Voors, A., Shah, A., Kraigher-Krainer, E., et al. (2012). The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. Lancet 380, 1387–1395. doi: 10.1016/S0140-6736(12)61227-6
Solomon, S. D., Rizkala, A. R., Gong, J., Wang, W., Anand, I. S., Ge, J., et al. (2017). Angiotensin receptor neprilysin inhibition in heart failure with preserved ejection fraction: rationale and design of the PARAGON-HF trial. JACC Heart Fail 5, 471–482. doi: 10.1016/j.jchf.2017.04.013
Stehouwer, C. D., Henry, R. M., Dekker, J. M., Nijpels, G., Heine, R. J., and Bouter, L. M. (2004). Microalbuminuria is associated with impaired brachial artery, flow-mediated vasodilation in elderly individuals without and with diabetes: further evidence for a link between microalbuminuria and endothelial dysfunction – the Hoorn Study. Kidney Int. Suppl. 66(Suppl. 92), S42–S44. doi: 10.1111/j.1523-1755.2004.09211.x
Steinberg, B. A., Zhao, X., Heidenreich, P. A., Peterson, E. D., Bhatt, D. L., Cannon, C. P., et al. (2012). Trends in patients hospitalized with heart failure and preserved left ventricular ejection fraction: prevalence, therapies, and outcomes. Circulation 126, 65–75. doi: 10.1161/CIRCULATIONAHA.111.080770
Stenvinkel, P., Barany, P., Heimburger, O., Pecoits-Filho, R., and Lindholm, B. (2002). Mortality, malnutrition, and atherosclerosis in ESRD: what is the role of interleukin-6? Kidney Int. Suppl. 61(Suppl. 80), 103–108. doi: 10.1046/j.1523-1755.61.s80.19.x
Stinghen, A. E., Massy, Z. A., Vlassara, H., Striker, G. E., and Boullier, A. (2016). Uremic toxicity of advanced glycation end products in CKD. J. Am. Soc. Nephrol. 27, 354–370. doi: 10.1681/ASN.2014101047
Stirban, A., Negrean, M., Stratmann, B., Gawlowski, T., Horstmann, T., Gotting, C., et al. (2006). Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care 29, 2064–2071. doi: 10.2337/dc06-0531
Streng, K. W., Nauta, J. F., Hillege, H. L., Anker, S. D., Cleland, J. G., Dickstein, K., et al. (2018). Non-cardiac comorbidities in heart failure with reduced, mid-range and preserved ejection fraction. Int. J. Cardiol. 271, 132–139. doi: 10.1016/j.ijcard.2018.04.001
Tadic, M., Pieske-Kraigher, E., Cuspidi, C., Genger, M., Morris, D. A., Zhang, K., et al. (2017). Left ventricular strain and twisting in heart failure with preserved ejection fraction: an updated review. Heart Fail. Rev. 22, 371–379. doi: 10.1007/s10741-017-9618-3
Tanaka, S., Fujita, S., Kizawa, S., Morita, H., and Ishizaka, N. (2016). Association between FGF23, alpha-Klotho, and cardiac abnormalities among patients with various chronic kidney disease stages. PLoS One 11:e0156860. doi: 10.1371/journal.pone.0156860
Ter Maaten, J. M., Damman, K., Verhaar, M. C., Paulus, W. J., Duncker, D. J., Cheng, C., et al. (2016). Connecting heart failure with preserved ejection fraction and renal dysfunction: the role of endothelial dysfunction and inflammation. Eur. J. Heart Fail. 18, 588–598. doi: 10.1002/ejhf.497
Thadhani, R., Appelbaum, E., Pritchett, Y., Chang, Y., Wenger, J., Tamez, H., et al. (2012). Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: the PRIMO randomized controlled trial. JAMA 307, 674–684. doi: 10.1001/jama.2012.120
Thomas, R., Kanso, A., and Sedor, J. R. (2008). Chronic kidney disease and its complications. Prim. Care 35, 329–344, vii. doi: 10.1016/j.pop.2008.01.008
Tonelli, M., Wiebe, N., Culleton, B., House, A., Rabbat, C., Fok, M., et al. (2006). Chronic kidney disease and mortality risk: a systematic review. J. Am. Soc. Nephrol. 17, 2034–2047. doi: 10.1681/ASN.2005101085
Unger, E. D., Dubin, R. F., Deo, R., Daruwalla, V., Friedman, J. L., Medina, C., et al. (2016). Association of chronic kidney disease with abnormal cardiac mechanics and adverse outcomes in patients with heart failure and preserved ejection fraction. Eur. J. Heart Fail. 18, 103–112. doi: 10.1002/ejhf.445
Uthman, L., Baartscheer, A., Bleijlevens, B., Schumacher, C. A., Fiolet, J. W. T., Koeman, A., et al. (2018a). Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia 61, 722–726. doi: 10.1007/s00125-017-4509-7
Uthman, L., Baartscheer, A., Schumacher, C. A., Fiolet, J. W. T., Kuschma, M. C., Hollmann, M. W., et al. (2018b). Direct cardiac actions of sodium glucose cotransporter 2 inhibitors target pathogenic mechanisms underlying heart failure in diabetic patients. Front. Physiol. 9:1575. doi: 10.3389/fphys.2018.01575
van der Putten, K., Braam, B., Jie, K. E., and Gaillard, C. A. (2008). Mechanisms of disease: erythropoietin resistance in patients with both heart and kidney failure. Nat. Clin. Pract. Nephrol. 4, 47–57. doi: 10.1038/ncpneph0655
van Heerebeek, L., Hamdani, N., Handoko, M. L., Falcao-Pires, I., Musters, R. J., Kupreishvili, K., et al. (2008). Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 117, 43–51. doi: 10.1161/CIRCULATIONAHA.107.728550
van Heerebeek, L., Hamdani, N., Falcao-Pires, I., Leite-Moreira, A. F., Begieneman, M. P., Bronzwaer, J. G., et al. (2012). Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 126, 830–839. doi: 10.1161/CIRCULATIONAHA.111.076075
Van Tassell, B. W., Arena, R., Biondi-Zoccai, G., Canada, J. M., Oddi, C., Abouzaki, N. A., et al. (2014). Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am. J. Cardiol. 113, 321–327. doi: 10.1016/j.amjcard.2013.08.047
Van Tassell, B. W., Trankle, C. R., Canada, J. M., Carbone, S., Buckley, L., Kadariya, D., et al. (2018). IL-1 blockade in patients with heart failure with preserved ejection fraction. Circ. Heart Fail. 11:e005036. doi: 10.1161/CIRCHEARTFAILURE.118.005036
van Veldhuisen, D. J., Cohen-Solal, A., Bohm, M., Anker, S. D., Babalis, D., Roughton, M., et al. (2009). Beta-blockade with nebivolol in elderly heart failure patients with impaired and preserved left ventricular ejection fraction: data from SENIORS (Study of Effects of Nebivolol Intervention on Outcomes and Rehospitalization in Seniors With Heart Failure). J. Am. Coll. Cardiol. 53, 2150–2158. doi: 10.1016/j.jacc.2009.02.046
Vanholder, R., Van Laecke, S., and Glorieux, G. (2008). What is new in uremic toxicity? Pediatr. Nephrol. 23, 1211–1221. doi: 10.1007/s00467-008-0762-9
Wang, S., Qin, L., Wu, T., Deng, B., Sun, Y., Hu, D., et al. (2014). Elevated cardiac markers in chronic kidney disease as a consequence of hyperphosphatemia-induced cardiac myocyte injury. Med. Sci. Monit. 20, 2043–2053. doi: 10.12659/MSM.890909
Westenbrink, B. D., Visser, F. W., Voors, A. A., Smilde, T. D., Lipsic, E., Navis, G., et al. (2007). Anaemia in chronic heart failure is not only related to impaired renal perfusion and blunted erythropoietin production, but to fluid retention as well. Eur. Heart J. 28, 166–171. doi: 10.1093/eurheartj/ehl419
Westermann, D., Lindner, D., Kasner, M., Zietsch, C., Savvatis, K., Escher, F., et al. (2011). Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 4, 44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451
Wolf, M., Shah, A., Gutierrez, O., Ankers, E., Monroy, M., Tamez, H., et al. (2007). Vitamin D levels and early mortality among incident hemodialysis patients. Kidney Int. 72, 1004–1013. doi: 10.1038/sj.ki.5002451
Wong, W. T., Tian, X. Y., and Huang, Y. (2013). Endothelial dysfunction in diabetes and hypertension: cross talk in RAS, BMP4, and ROS-dependent COX-2-derived prostanoids. J. Cardiovasc. Pharmacol. 61, 204–214. doi: 10.1097/FJC.0b013e31827fe46e
Yan, P., Li, H., Hao, C., Shi, H., Gu, Y., Huang, G., et al. (2011). 2D-speckle tracking echocardiography contributes to early identification of impaired left ventricular myocardial function in patients with chronic kidney disease. Nephron Clin. Pract. 118, c232–c240. doi: 10.1159/000321383
Yancy, C. W., Lopatin, M., Stevenson, L. W., De Marco, T., Fonarow, G. C., Committee, A. S. A., et al. (2006). Clinical presentation, management, and in-hospital outcomes of patients admitted with acute decompensated heart failure with preserved systolic function: a report from the Acute Decompensated Heart Failure National Registry (ADHERE) database. J. Am. Coll. Cardiol. 47, 76–84. doi: 10.1016/j.jacc.2005.09.022
Ye, M., Tian, N., Liu, Y., Li, W., Lin, H., Fan, R., et al. (2016). High serum phosphorus level is associated with left ventricular diastolic dysfunction in peritoneal dialysis patients. PLoS One 11:e0163659. doi: 10.1371/journal.pone.0163659
Yilmaz, R., Akoglu, H., Altun, B., Yildirim, T., Arici, M., and Erdem, Y. (2012). Dietary salt intake is related to inflammation and albuminuria in primary hypertensive patients. Eur. J. Clin. Nutr. 66, 1214–1218. doi: 10.1038/ejcn.2012.110
Zhang, L., Liebelt, J. J., Madan, N., Shan, J., and Taub, C. C. (2017). Comparison of predictors of heart failure with preserved versus reduced ejection fraction in a multiracial cohort of preclinical left ventricular diastolic dysfunction. Am. J. Cardiol. 119, 1815–1820. doi: 10.1016/j.amjcard.2017.03.005
Zhang, Q., Chen, Y., Liu, Q., and Shan, Q. (2016). Effects of renin-angiotensin-aldosterone system inhibitors on mortality, hospitalization, and diastolic function in patients with HFpEF. A meta-analysis of 13 randomized controlled trials. Herz 41, 76–86. doi: 10.1007/s00059-015-4346-8
Zieman, S. J., and Kass, D. A. (2004). Advanced glycation endproduct crosslinking in the cardiovascular system: potential therapeutic target for cardiovascular disease. Drugs 64, 459–470. doi: 10.2165/00003495-200464050-00001
Zieman, S. J., Melenovsky, V., Clattenburg, L., Corretti, M. C., Capriotti, A., Gerstenblith, G., et al. (2007). Advanced glycation endproduct crosslink breaker (alagebrium) improves endothelial function in patients with isolated systolic hypertension. J. Hypertens. 25, 577–583. doi: 10.1097/HJH.0b013e328013e7dd
Zinman, B., Wanner, C., Lachin, J. M., Fitchett, D., Bluhmki, E., Hantel, S., et al. (2015). Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 373, 2117–2128. doi: 10.1056/NEJMoa1504720
Keywords: chronic kidney disease, heart failure, HFpEF, vasculature, microcirculation, therapy
Citation: van de Wouw J, Broekhuizen M, Sorop O, Joles JA, Verhaar MC, Duncker DJ, Danser AHJ and Merkus D (2019) Chronic Kidney Disease as a Risk Factor for Heart Failure With Preserved Ejection Fraction: A Focus on Microcirculatory Factors and Therapeutic Targets. Front. Physiol. 10:1108. doi: 10.3389/fphys.2019.01108
Edited by:
Rui Plácido, University of Lisbon, PortugalReviewed by:
Francisco Altamirano, UT Southwestern Medical Center, United StatesCopyright © 2019 van de Wouw, Broekhuizen, Sorop, Joles, Verhaar, Duncker, Danser and Merkus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daphne Merkus, ZC5tZXJrdXNAZXJhc211c21jLm5s
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.