 Tianhong Wang
Tianhong Wang Shiqi Zhang
Shiqi Zhang Tingwei Pei
Tingwei Pei Zhijun Yu
Zhijun Yu Jingze Liu
Jingze Liu- Hebei Key Laboratory of Animal Physiology, Biochemistry and Molecular Biology, College of Life Sciences, Hebei Normal University, Shijiazhuang, China
The tick Haemaphysalis longicornis is widely distributed in eastern Asia, New Zealand and Australia, and is well-known as a vector of multiple zoonotic pathogens. This species exhibits two reproductive strategies, bisexual and obligate parthenogenetic reproduction. Hence, in the current study, the complete mitochondrial genomes of the bisexual and parthenogenetic populations were assembled and analyzed, and the expression of the mitochondrial protein-coding genes was evaluated and compared between the two reproductive populations. The results indicated that the length of the mitochondrial genomes of the two reproductive populations is 14,694 and 14,693 bp in the bisexual and parthenogenetic populations, respectively. The AT content in the mitochondrial genome of the bisexual and obligate parthenogenetic population reached 77.22 and 77.34%, respectively. The phylogenetic tree was constructed combining 13 protein-coding genes, which showed that the genetic distance between the bisexual and parthenogenetic populations was less than that between the subspecies. The expression of the mitochondrial protein-coding genes was quantitatively analyzed at different feeding status for the bisexual and parthenogenetic populations, and the results showed significant differences in the expression patterns of these genes, suggesting that they might trigger specific energy utilization mechanisms due to their different reproductive strategies and environmental pressures.
Introduction
Ticks are obligate blood-sucking ectoparasites with global distribution, and they can feed on a broad range of animals (Kaufman, 2010). They are notorious vectors of zoonotic pathogens. Furthermore, tick-borne diseases (TBDs) are increasingly threatening animal and human health and thus causing great economic damages (Jongejan and Uilenberg, 2004; Kovalev and Mukhacheva, 2013). The global annual financial losses due to ticks and TBDs are estimated in billions of dollars (Burger et al., 2014b). In China, 117 species of ticks in 7 genera have been recorded, and more than 60 tick species have shown vector potential (Yu et al., 2015). The tick Haemaphysalis longicornis is widely distributed in eastern Asia, New Zealand and Australia and is well-known as a vector of multiple zoonotic pathogens (Hoogstraal et al., 1968; Herrin and Oliver, 1974). These pathogens include Anaplasma bovis, Babesia ovate, Borrelia burgdorfer, Rickettsia japonica, Theileria orientalis, and severe fever with thrombocytopenia syndrome virus (SFTSV) (Fujisaki et al., 1994; Jongejan and Uilenberg, 2004; Matsuo et al., 2013; Takahashi et al., 2014; Qin et al., 2018; Zhang et al., 2019). Human infection of tick H. longicornis was reported in at least 23 provinces in China (Chen et al., 2010, 2015; Zhan et al., 2017) with a case fatality rate of 10−30% (Liu et al., 2014). In America, this species has been expanded to 19 counties in 8 states since the first detection in 2013 (Magori, 2018).
In H. longicornis, the life history of a bisexual and parthenogenetic population is different (Hoogstraal et al., 1968). The parthenogenetic population can feed engorged and oviposit without males and thus might have stronger reproductive capacity and spreading ability. However, the distribution of the parthenogenetic population was only reported in Shanghai, Gansu and Sichuan provinces in China (Zhou Z. et al., 2004; Yang et al., 2007; Chen et al., 2010). The parthenogenetic species were historically a critical problem for early taxonomic studies (Monis et al., 2002). The number of eggs laid by the parthenogenetic population was significantly lower than that of the bisexual population (Oliver, 1977), whereas the engorgement body weight of females and egg size of the parthenogenetic population were considerably higher than that of the bisexual population, but the hatching rate of eggs was still lower in the parthenogenetic population (Chen et al., 2014). In recent years, scanning electron microscopy (SEM) has been used to describe the morphological characteristics of the two reproductive populations (Yang et al., 2007; Wang et al., 2013).
Mitochondrial genomes are characterized by simple structure, small molecular weight, rapid evolutionary rate, and matrilineal inheritance, which is particularly crucial in phylogenetic studies (Lin and Danforth, 2004; Gissi et al., 2008; Li and Liang, 2018). To date, more than 20 complete mitochondrial genomes in ixodid ticks have been available for phylogenetic analysis (Burger et al., 2014b). Additionally, structural genomic features, such as secondary structures of tRNA and rRNA, are also applied in comparative and evolutionary genomics (Qin et al., 2015; Kim et al., 2017). Compared with a phylogenetic analysis of a single gene sequence, a combined study of multiple mitochondrial genes can more accurately evaluate the genetic distance among or within species (Papanicolaou et al., 2008; Timmermans et al., 2014). Hence, investigations on the genetic relatedness of mitochondrial genomes will be helpful in making taxonomic determinations. Additionally, the protein-coding genes (PCGs) play a vital role in activity changes of the arthropod mitochondrial complex (Uno et al., 2004; Fontanillas et al., 2010). Evaluation of the differential expression profiling of PCGs will help to elucidate the energy utilization of different reproductive strategies.
In the current study, the complete mitochondrial genomes of the bisexual and parthenogenetic populations of H. longicornis were assembled and analyzed, and the expression of the mitochondrial PCGs was evaluated and compared between the two reproductive populations. The differences in survival climate may have influenced the survival strategies of H. longicornis and may have resulted from mutations in the parthenogenetic population. These results may help to elucidate the possible interconnections among environmental stress, genetic evolution, and parthenogenetic patterns.
Materials and Methods
Sample Collection and DNA Extraction
The bisexual population of H. longicornis was collected from Xiaowutai National Nature Reserve Area of Zhuolu County (40∘ 03′ 03″ N, 115∘ 23′ 15″ E), Zhangjiakou City, Hebei Province, China. The parthenogenetic population was collected in Cangxi County (31∘ 44′ 35″ N, 105∘ 49′ 04″ E), Guangyuan City, Sichuan Province, China. The free-living nymphal collection was conducted in the above two locations using a white cloth flag in April of each year. The collected ticks were placed into perforated, clean centrifugal tubes with ventilated lids on one side through a 4-mm-diameter hole sealed with plastic screen. After the nymphs molted, delimitation between the two populations was performed based on our previous publications (Yang et al., 2007; Chen X. et al., 2012; Chen Z. et al., 2012; Wang et al., 2013). The distinguishing feature between the two populations is whether males appear in the adult population after molting nymphs.
The ticks were fed on the New Zealand white rabbit ear until engorged and were maintained under standard environmental chamber conditions (26 ± 1∘C, 75% ± 5 RH, and 12 h: 12 h L:D). All experimental procedures in this study were approved by the Animal Ethics Committee of the Hebei Normal University (Protocol Number: IACUC-157026).
Three mitochondrial genome samples were sequenced as follows: 10 unfed adults of parthenogenetic female, bisexual female and bisexual male were separately placed in a 1.5-ml centrifuge tube, washed with 75% ethanol for 30 s, and then homogenized under liquid nitrogen. DNA was extracted using the EasyPure® genomic DNA kit (TransGen Biotech Co., Ltd., Beijing, China). DNA concentration was estimated using a TU-1950 spectrophotometer (Xi’an Yima Opto-electrical Technology Co., Ltd., Xi’an, China). The extracted DNA was visualized on 1% agarose gel to ensure strong bands and purified using the EasyPure® quick gel extraction kit (TransGen Biotech Co., Ltd., Beijing, China). All purified DNA was stored at −80∘C until use.
Next Generation Sequencing Library Construction and Sequence Analysis
For the sequencing library construction, the fragmented tick genomic DNA (400−500 bp) was obtained by sharing 2-μg of tick DNA using Covaris M220 Focused-ultrasonicator (Convaris, Inc., Woburn, MA, United States). Subsequent reaction steps using TruSeqTM DNA Sample HT Prep Kit (Illumina Inc., San Diego, CA, United States), which include repaired ends, adenylated 3′ ends, added A-Tailing and ligated adapter, and then the ligation products were purified with agarose electrophoresis and enriched the DNA fragments with a PCR primer cocktail that annealed to the ends of the adapters. Finally, the library was quantified and sequenced on illumina Hiseq X Ten sequencing platform according to the standard operation.
In this study, we annotated our next-generation sequencing (NGS) data with the mitochondrial genome of three different species of Haemaphysalis from the National Center of Biotechnology Information (NCBI) nucleotide database, as the Genebank number: AB075954 (H. flava), JX573135 (H. formosensis) and JX573136 (H. parva). SOAPdenovo v2.041 was used for sequence filtering and assembly (Xie et al., 2014). GapCloser v1.12 software (a SOAP suite tool) was used to perform vulnerability completion and base correction. The splicing of the original mitochondrial genome sequencing data was reflected in the (Supplementary Table S1). The mitochondrial circular map was assembled by Organellar Genome DRAW2 (Lohse et al., 2013). The three complete mitochondrial genome circular maps of H. longicornis were uploaded to the (Supplementary Figures S1−S3). The complete mitochondrial genome sequence was deposited in the NCBI nucleotide database accession numbers: MK450606 (The bisexual population of H. longicornis) and MK439888 (The parthenogenetic population of H. longicornis).
The PCGs, tRNA, rRNA and non-coding regions (NCRs) of the genome were predicted using MITOS3 online analysis software (Bernt et al., 2013). The PCGs were blasted using the GO4 database. MEGA v7.0 for Windows (Kumar et al., 2016) was used to analyze the base content and the similarity. The calculation formula of the base skew is AT skew = (A−T)/(A+T); GC skew = (G−C)/(G+C) (Alexandre et al., 2005). The relative synonymous Codon usage (RSCU) analysis was carried out using Codon W software version 2.7.2.1 for Windows (Cancilla et al., 1995). The tRNAscan-SE5 was used to identify the tRNA structure (Burger et al., 2014a). The genome tandem repeats of NCRs were predicted using Tandem Repeats Finder6 (Benson, 1999).
Polymorphism Detection and Circular Map Comparison
Based on our previous studies (Chen X. et al., 2012; Chen et al., 2015), we speculated that the differentiation of the bisexual population might generate the parthenogenetic population. In this experiment, we analyzed the mitochondrial genome data of the bisexual population as the treatment group to assess the polymorphism and small insertion-deletion of the parthenogenetic population. Burrows-Wheeler Aligner7 and Genome Analysis Toolkit8 were used to match the genome sequence, and nucleotide polymorphism analysis (Lines et al., 2014; Mccormick et al., 2015). OrthoMCL DB9 was used to draw the three circular maps of the mitochondrial genomes (conditions: E-value: 1−5, E percent identity cutoff: 0, Markov plus index: 1.5) (Fischer et al., 2011).
Phylogenetic Development and Homologous Gene
The mitochondrial genomes of 24 ixodid ticks and Nuttalliella namaqua were obtained from the NCBI10 nucleotide databases. RAxML v8.011 was used to construct a phylogenetic tree with bootstrap replicated evaluation nodes 1000 times (Stamatakis, 2015). Sequence alignments and filtering of the PCGs were carried out using MAFFT v7.012 and Gblocks13. A Venn diagram of homologous genes was drawn using CGV software (Tominski et al., 2009).
Mitochondrial Genome Protein-Coding Genes at Different Feeding Status
The expression profiling of the mitochondrial genome protein-coding genes of H. longicornis at different feeding status was quantitatively analyzed using real-time quantitative PCR (qPCR). The weights of the bisexual and parthenogenetic population samples were similar in this experiment. RNA was extracted using the EasyPure® RNA Kit (TransGen Biotech Co., Ltd., Beijing, China) from H. longicornis at different feeding status, including unfed female (without feeding), partially fed (feed without mating) and engorged (fully engorged and detached).
Then, cDNA was synthesized using 500-ng RNA and 10-μl purity water combined with TranScript® First-Strand cDNA Synthesis SuperMix (TransGen Biotech Co., Ltd., Beijing, China). Samples were incubated at 65∘C for 5 min, chilled on ice for 2 min, and then incubated at 42∘C for 15 min. The cDNA products were stored at −20∘C. The length of the quantitative product was designed between 100 and 200 bp (Supplementary Table S2), and a 20-μl master mix was prepared using SYBRGreen PCR buffer, HotStarTaq DNA polymerase, SYBRGreen I dye, dNTPs (TransGen Biotech Co., Ltd., Beijing, China), PCR stabilizer. PCR was performed using SimpliAmp Thermal Cycler A24811 (Applied Biosystems Shanghai Co., Ltd., Shanghai, China). The qPCR conditions were set as follows: 95∘C for 30 s; 40 cycles of 95∘C for 5 s, 55∘C for 15 s, 72∘C for 10 s. Relative expression of these genes was calculated using the 2–ΔΔCt method. If the relative expression ratio was <1, there was down regulation of the comparison gene in the sample (dotted line group). Conversely, if the expression ratio was >1, there was up regulation in the expression of the sample (dotted line group). All samples were prepared from at least two female individuals, and three parallel experiments were performed to improve the accuracy. The statistical analysis was determined using Duncan’s multiple range test with analysis of variance (ANOVA), and calculations were performed using SPSS v17.0 for Windows (SPSS Inc., Chicago, IL, United States).
Results
Base Features and Gene Composition
The length of the complete genome of the bisexual and parthenogenetic populations H. longicornis is 14,694 and 14,693 bp, respectively. The circular structure of the mitochondrial genomes of the two populations was successfully predicted, and the gene arrangement of the mitochondrial genomes included 13 PCGs, 22 tRNA and 2 rRNA. The contents of the four bases in the base composition of the mitochondrial genome are approximately 38% (A), 39% (T), 13% (C), and 9% (G), respectively. The A+T contents of the mitochondrial genome account for 77.22% in the bisexual population and 77.34% in the parthenogenetic population. The frequency of base used in both reproductive populations was highly similar. The GC-skew and AT-skew of the two reproductive populations were negative; GC-skew in the bisexual population was −0.1446, and AT-skew in the bisexual population was −0.0104. In the parthenogenetic population, GC-skew was −0.1502, and AT-skew was −0.0092 (Table 1).
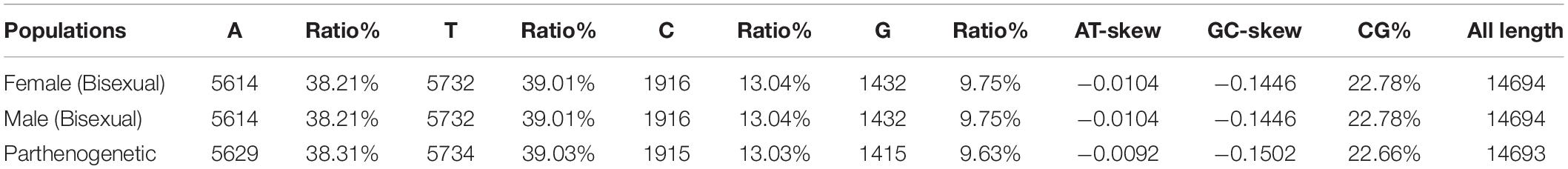
Table 1. Base composition and skewness in the mitochondrial genome of Haemaphysalis longicornis.
Polymorphism and Circular Map of the Mitochondrial Genome
Bisexual female and male ticks share the same mitochondrial genome. Polymorphism analysis of the parthenogenetic population revealed 199 bases differences, but no small indel differences were found (Supplementary Figure S4). Only one site on the nad3 gene displayed a non-synonymous mutation. The circular maps of the mitochondrial genomes were assembled and compared. No significant differences in gene direction and arrangement were observed between the bisexual and parthenogenetic populations (Figure 1).
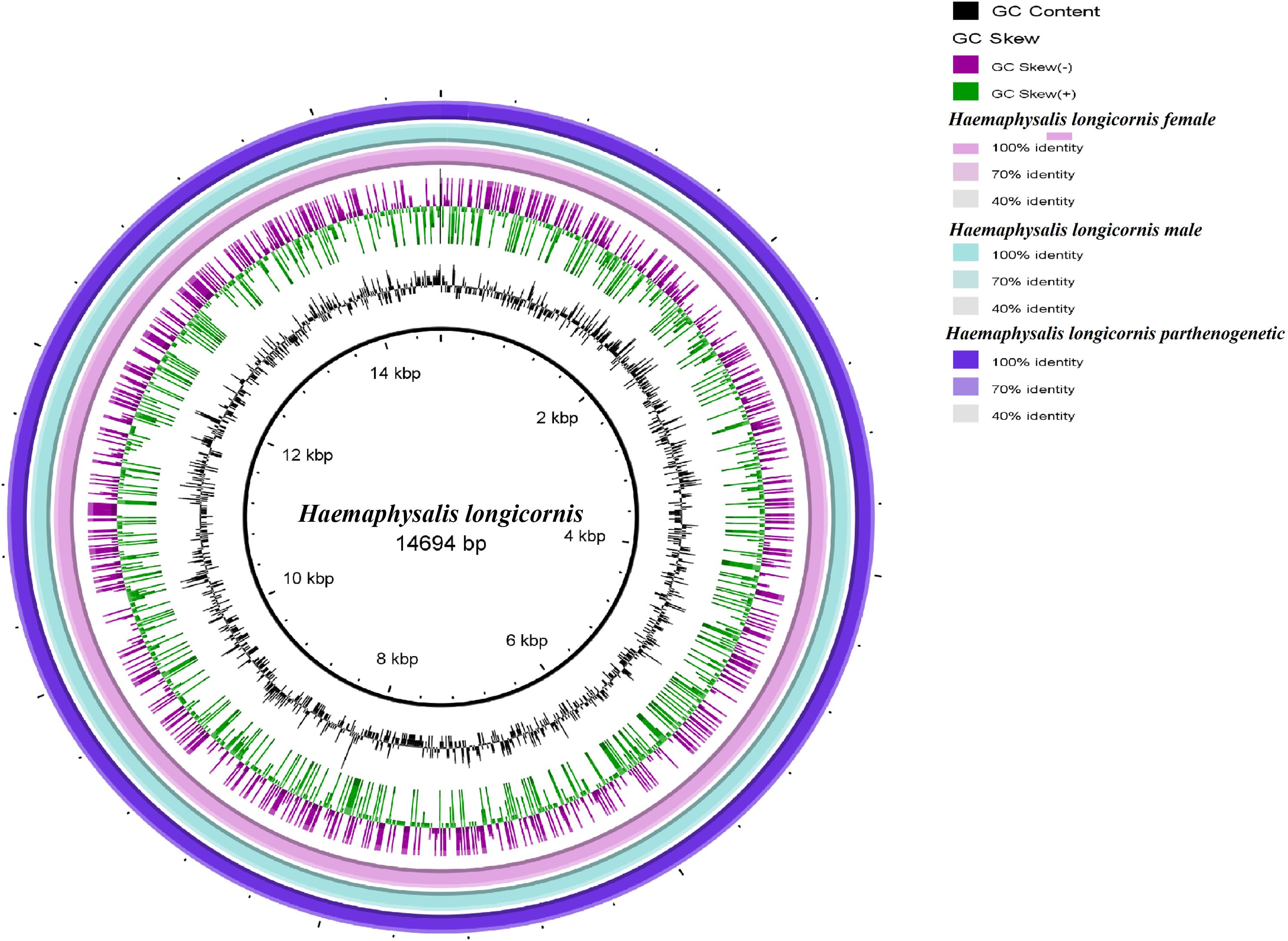
Figure 1. Circular maps of the mitochondrial genome of the bisexual and parthenogenetic Haemaphysalis longicornis. The color change of the ring indicates the level of difference between the bisexual female, bisexual male, and the parthenogenetic population.
Mitochondrial Protein-Coding Gene and Codon Analysis
The length of the PCGs in the bisexual population was 10,434 bp, accounting for 71.01% of the total mitochondrial genome, and was 10,464 bp in the parthenogenetic population, accounting for 71.22%. The heavy strand contains 9 genes (cox1, cox2, cox3, cytb, nad2, nad3, nad6, atp6, and atp8), and the light strand contains 4 genes (nad1, nad4, nad4l, and atp8). Mitochondrial genes of H. longicornis displayed distinct rearrangement characteristics from Haemaphysalis genus (Liu et al., 2018), and the arrangement and distribution of 13 PCGs were completely consistent in the two reproductive populations. Four start codons (ATA, ATT, ATG, and ATC) were detected in H. longicornis. Among these start codons, ATT was adopted by 7 genes (cox1, cox2, nad1, nad2, nad3, nad4l, and nad5), ATA was used by 3 genes (cox3, cytb, and atp8), ATG and ATC were used by 3 genes (nad4, atp6, and nad6). The termination codon TAA was used by 9 PCGs (cox1, cox3, atp6, atp8, nad1, nad2, nad3, nad5, and nad6). The incomplete codon T was used in genes including cox2, nad3, and nad4. Only nad4L used TAG as the termination codon (Supplementary Table S3). The two populations are identical in the usage of the initiation codon and termination codon.
Codons including TTT (Phe), ATT (Ile) and AAA (Lys) showed the highest utilization rate. Codons of GCG (Ala), CCG (Pro), CGG (Arg), CGC (Arg) and ACG (Thr) containing “CG” showed relatively lower utilization rates, that is, less than 10 times (Supplementary Table S4). In tRNA, Phe and Leu2 were used more than 400 times in the bisexual population, and Phe, Ile and Lys were the most frequently used in the parthenogenetic population, being used more than 400 times. The lowest tRNA usage was Arg in the two reproductive populations (Figure 2).
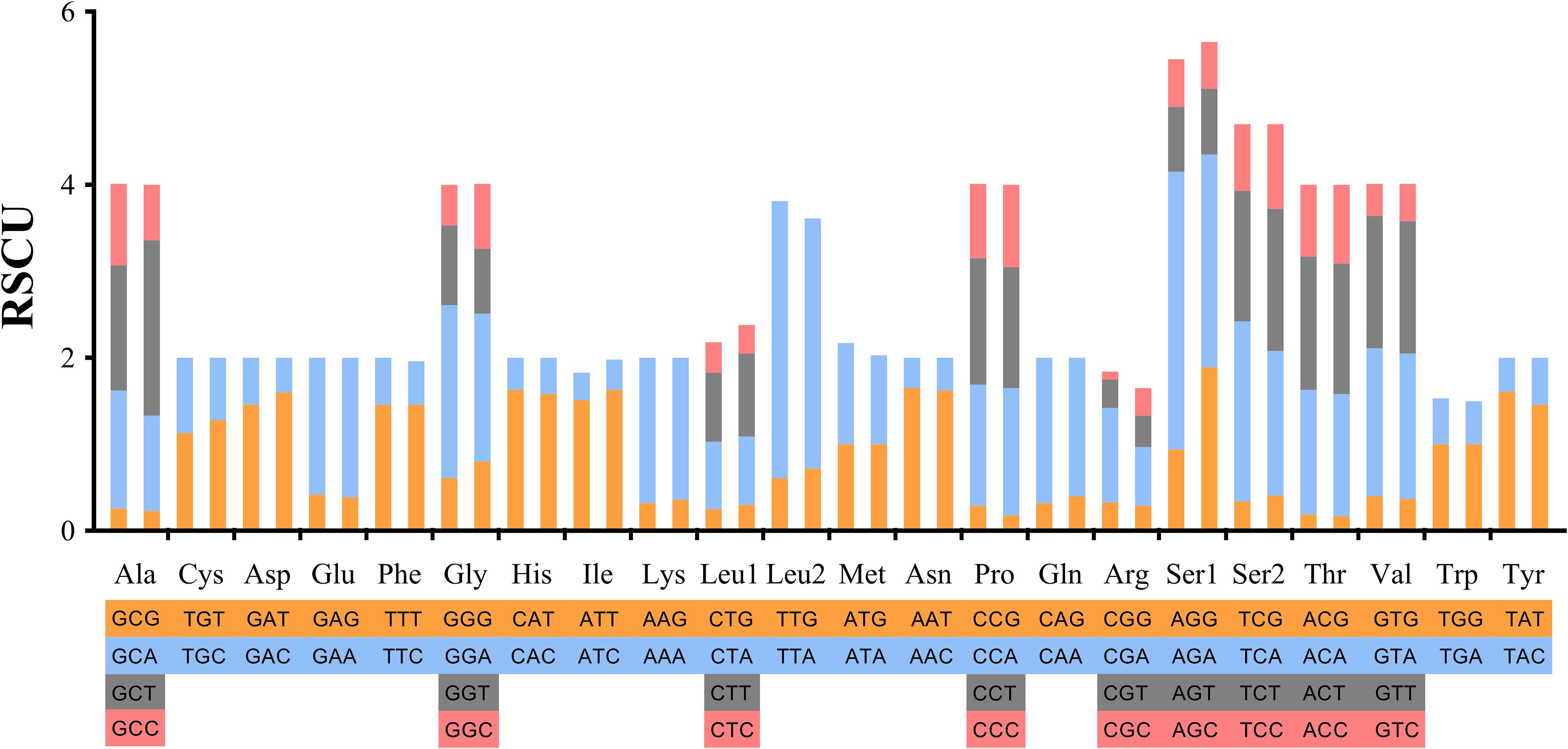
Figure 2. Relative synonymous codon usage in protein-coding genes of Haemaphysalis longicornis bisexual population and parthenogenetic population. The different amino acids are shown in four colors at most.
Transfer RNA and Ribosomal RNA
The mitochondrial genome in H. longicornis contains 22 tRNA and 2 rRNA. The secondary structure of all tRNA was successfully predicted, and the length ranged from 53 to 68 bp, with the longest trnQ, and the shortest trnC (Supplementary Figures S5, S6). The rRNA of H. longicornis displayed a significant AT bias. The total length of the tRNA in the two populations was identical, reaching 1,352 bp. The length of rrnL was 995 bp (AT = 82.21%), and the rrnS length was 778 bp (AT = 79.56%) in the bisexual population. In the parthenogenetic population, the rrnL length was 997 bp (AT = 82.25%), and the rrnL length was 777 bp (AT = 79.51%).
Gene Rearrangement and Position Variance
The mitochondrial genome of H. longicornis was characterized by obvious rearrangement in major genes. Five PCGs, including nad5-nad4-nad4l-nad6-cytb, were rearranged in new regions, which resulted in the displacement of the NCRs. The tRNA genes trnF, trnH, trnT, trnP, trnS2, trnL1, trnC, and trnL2 changed the Drosophila genus arrangement. The displacement of tRNA was also observed in the two reproductive populations (Figure 3).
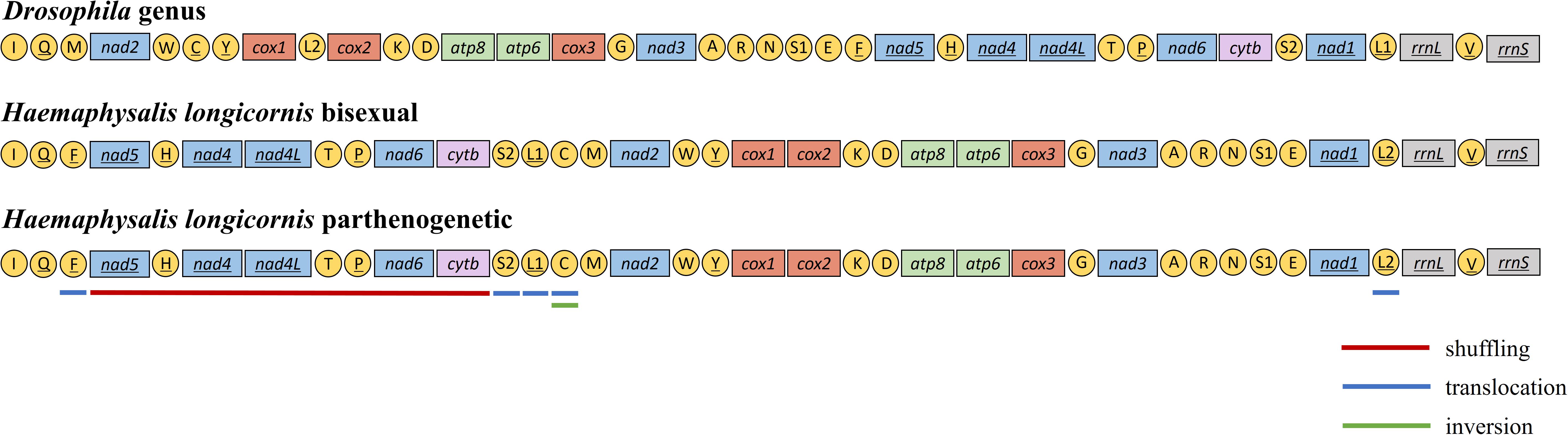
Figure 3. Mitochondrial gene arrangement characteristics of Haemaphysalis longicornis. The different color underlines highlighted the genes and rearrangements.
Non-coding Regions and Gene Overlapping
Most mitochondrial genes of H. longicornis displayed overlapping regions or spacer regions. A total of 22 spacer regions and seven overlapping regions of genes were found in the mitochondrial genome of H. longicornis, with overlapping regions ranging from 1 to 9 bp. The special region was atp6: in the bisexual population, atp6 overlapped by four bases, whereas in the parthenogenetic population, there was an interval region of eight bases. Four regions showed length differences between bisexual and parthenogenetic populations. The mitochondrial genome of H. longicornis contains three NCRs with a total length of more than 100 bp. NCR1 was located between rrnL and trnV, with a length of 159 bp; NCR2 was located between rrnS and trnI, with a length of 240 bp; and NCR3 was located between trnL1 and trnC, with a length of 309 bp (Supplementary Table S3). The content of A+T in the NCRs of H. longicornis was only 60−70%.
Phylogenetic and Homologous Gene Analysis
The phylogeny of the bisexual and the parthenogenetic population of H. longicornis was constructed with 24 tick species with complete mitochondrial genome available from the NCBI database. The results demonstrated that ticks in genera Ixodes, Bothriocroton, Amblyomma and Haemaphysalis were clustered into an independent branch, and ticks in Dermacentor and Rhipicephalus were grouped in Rhipicephalinae. N. namaqua and Ixodidae ticks were in different evolutionary branches. The two reproductive populations of H. longicornis were assigned to the cluster of the Haemaphysalis genus (Figure 4). In addition, homologous gene analysis showed that among 11 orthologous genes in these ticks, only one unique homologous gene was found in 10 ixodid ticks, and two novel homologous genes were found in N. namaqua. There were no unique homologous genes observed in the bisexual and parthenogenetic populations of H. longicornis (Figure 5).
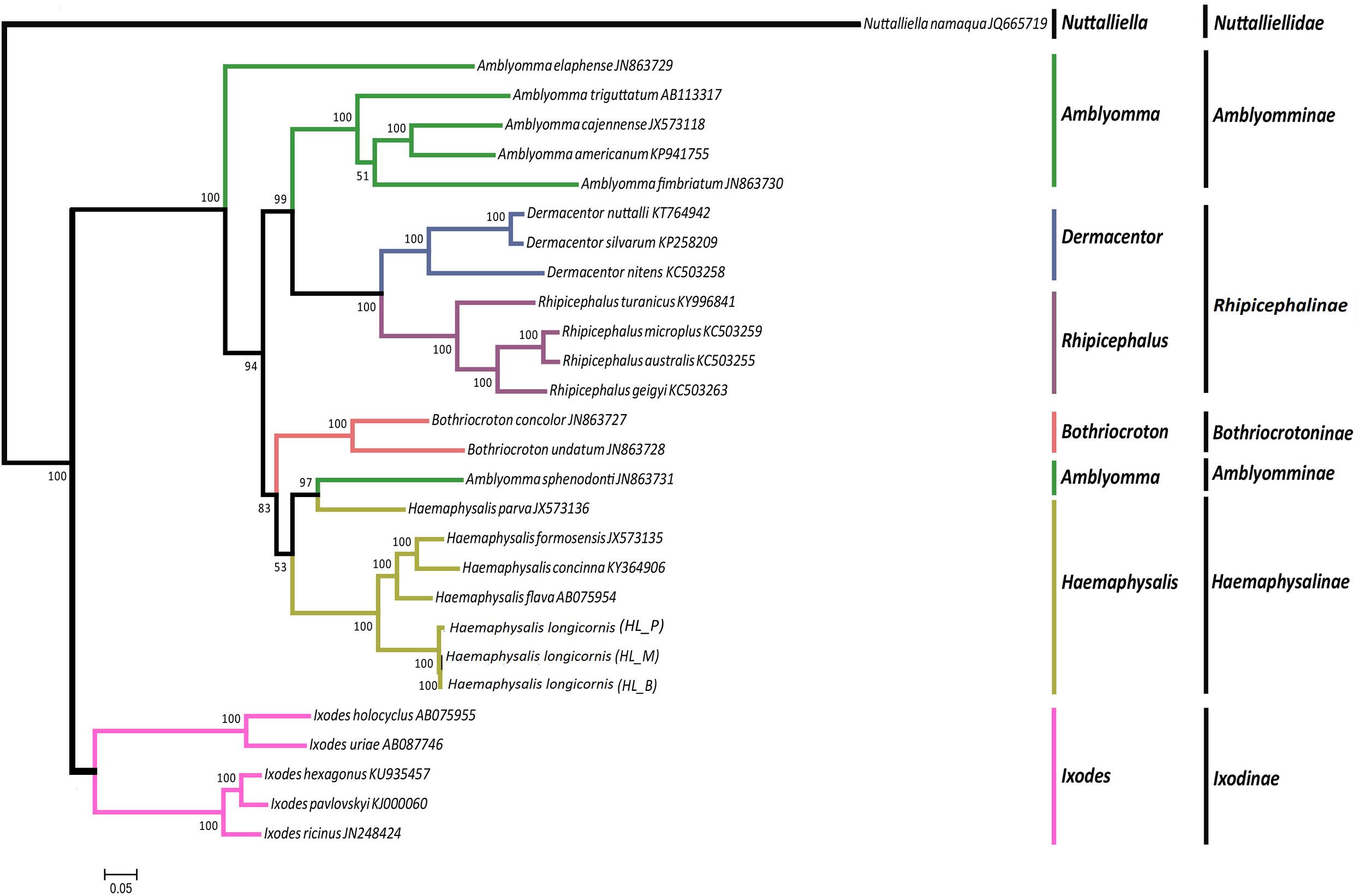
Figure 4. Phylogenetic tree combining 13 protein-coding genes of Haemaphysalis longicornis. Using ML analysis based on 24 Ixodidae mitochondrial genome data, different species were marked. The Nuttalliella namaqua (JQ665719) is the outgroup. In the phylogenetic tree, the scale bar represents the number of expected changes per site. The percentage of the ML bootstrap support was given at each node. The Haemaphysalis longicornis mitochondrial genome of the bisexual and parthenogenetic populations were in the same branch with strong support.
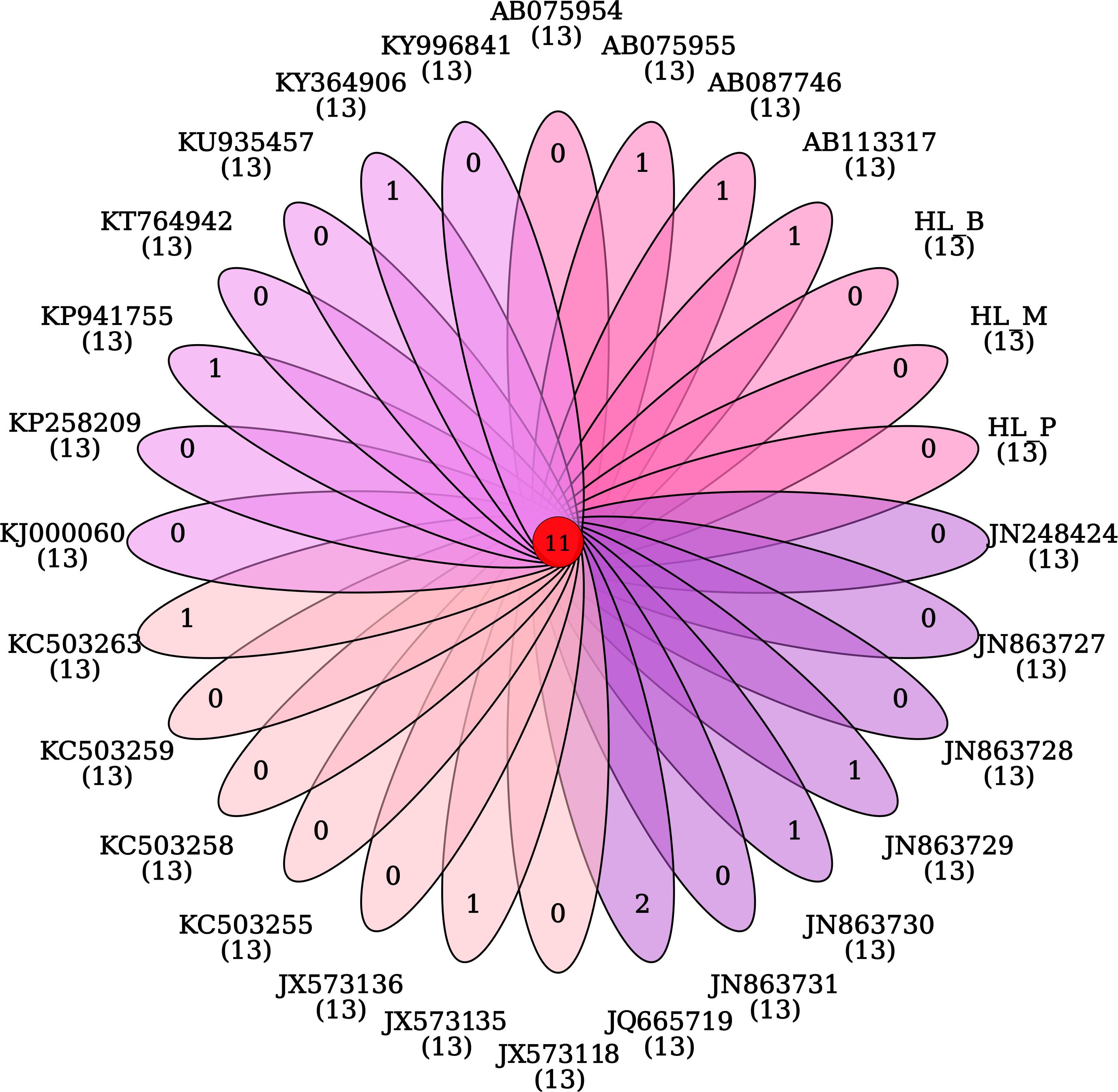
Figure 5. Venn map analysis of the homologous genes in Haemaphysalis longicornis. There are no unique homologous genes in the bisexual and parthenogenetic populations of the Haemaphysalis longicornis mitochondrial genome.
Quantitative Analysis of the Mitochondrial Protein-Coding Genes
The expression profiling of the 13 PCGs was detected in the females of the two populations during unfed, partially fed and engorgement, respectively. In the partially fed stage, the expression levels of most mitochondrial genes were similar to the unfed stage. The expression levels of cox2, nad6, atp6, and atp8 genes were increased in the bisexual population, and cytb and nad4 genes were significantly down regulated (P < 0.01). The expression of the cox1 and atp8 genes of the parthenogenetic population was significantly up regulated (P < 0.01). In contrast, the expression of the nad3 genes was significantly decreased (P < 0.01). The atp8 gene expression was considerably increased in the two populations at the partially fed and engorgement stages (Supplementary Figures S7, S8).
When the gene expression of the parthenogenetic population was used as a reference, all genes in the bisexual population were differentially expressed, among which cox2, cox3, and nad3 were most significantly expressed. The cox3 gene expression of the bisexual population in the unfed stage was 60-fold upregulated, whereas it was 16-fold upregulated to the partially fed and engorgement stages. No changes were observed in the expression level of nad3 in the unfed and engorgement stages in the parthenogenetic population, but it was significantly down regulated in the bisexual population (P < 0.01), which resulted in 20-fold variation between the two populations (Figure 6).
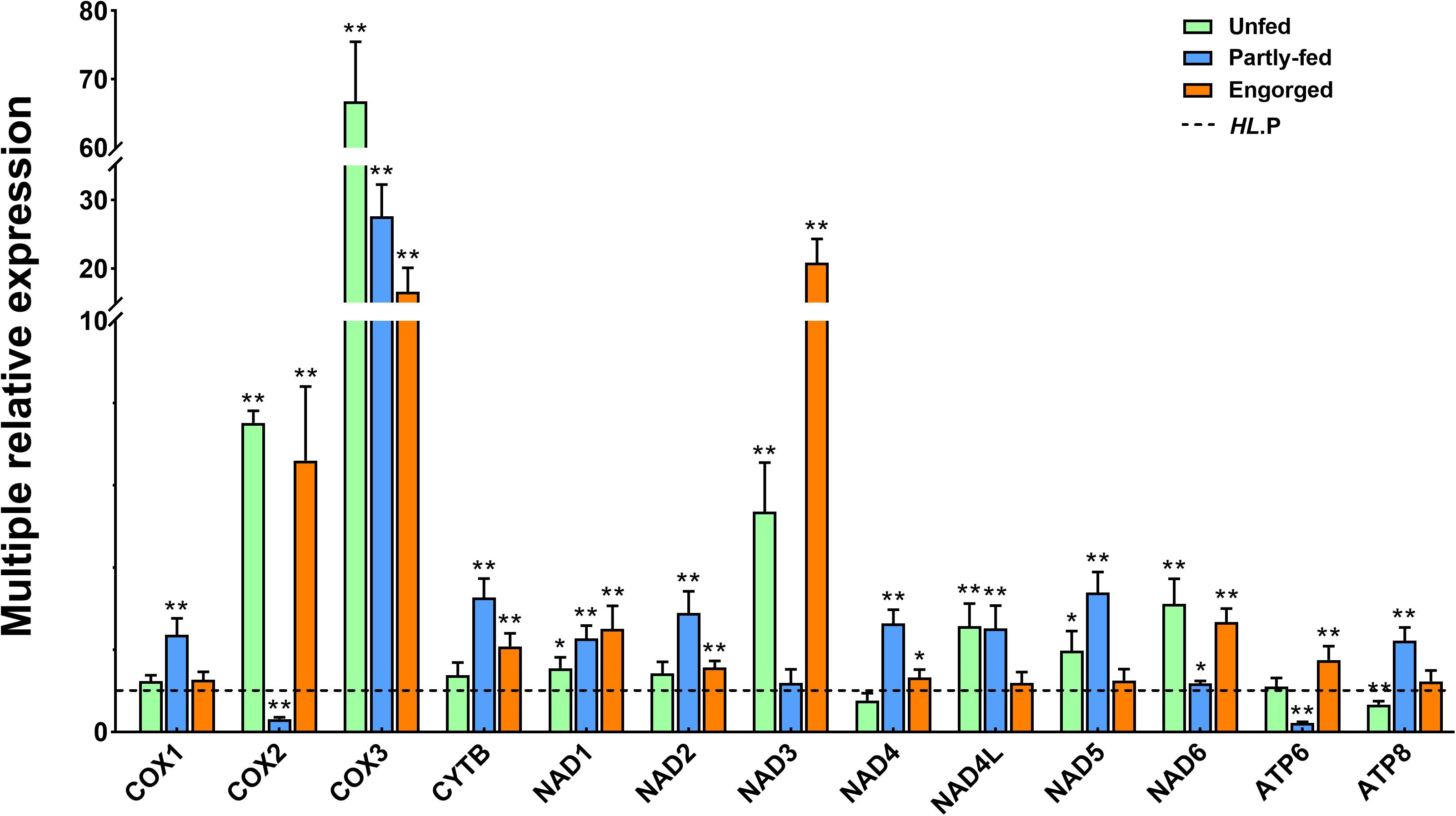
Figure 6. Comparative expression of the different feeding status of Haemaphysalis longicornis. The dotted line represents the expression of the parthenogenetic population. The asterisk indicates a level of significant difference (∗P < 0.05, ∗∗P < 0.01) in gene expression between the different groups.
Discussion
In the current study, the complete mitochondrial genomes of the bisexual and parthenogenetic populations of H. longicornis were assembled. The A+T content in H. longicornis was approximately 77%, and an obvious AT bias was also observed in most tick mitochondrial genomes (Black and Roehrdanz, 1998; Xiong et al., 2013; Williamsnewkirk et al., 2015). The A+T content affected both the codon usage pattern and amino acid composition of proteins (Breinholt and Kawahara, 2013). The GC and AT skew values of the two populations were negative, indicating that the mitochondrial genome has a slight preference in the usage of “T” and “C.” Previous studies have shown that the reversal of GC-skew might be related to the direction of replication but does not affect the direction of genes, whereas AT-skew can vary with changes in the direction of genes, replication and codon location (Wei et al., 2010). In the polymorphism analysis, we found that the mitochondrial sequences were entirely consistent between male and female ticks in the bisexual population, and no special structure was discovered, which maybe attributable to the maternal inheritance of the mitochondrial genome (Burger et al., 2013).
The arrangement of PCGs was similar to other tick species (Burger et al., 2013; Williamsnewkirk et al., 2015; Guo et al., 2016). The starting codon used by H. longicornis contained four common “ATN” types, and no special form of the first codon subtype was found. In H. longicornis, ATT was used as the initial codon for cox1, which is different from “CGA” used in some insect species (Zhu et al., 2013). Three termination codons, including TAA, TAG and “T,” were found in H. longicornis, and the incomplete termination codons “T” were common in mitochondrial genes (Pan et al., 2008; Dai et al., 2016), which might be converted to complete TAA codons by polyadenylation (Ojala et al., 1981).
In the tick H. longicornis, the most frequently used trnS1 and trnS2 displayed A and T bases preference in codon usage. Correspondingly, the TTA codon improved the higher usage of trnL2. A low utilization rate was observed in “CGC” and “CGG,” which resulted in the lower usage of trnR. In addition, trnW showed the lowest utilization rate. Changes in codon usage are generally regarded as important factors affecting protein expression levels (Zhou J. L. et al., 2004; Fang et al., 2015). Although some differences in codon usage were observed between the two reproductive populations, there was no significant difference in genetic structure or polymorphism between the bisexual and parthenogenetic populations.
The secondary structure of tRNA differs only at a few bases between the two reproductive populations. The DHU arms of trnC and trnS1 were absent in the two populations, which were common features in ticks and insects (Jühling et al., 2009; Cameron, 2014; Williamsnewkirk et al., 2015; Yuan et al., 2015; Liu et al., 2018). The base mismatch of mitochondrial tRNA genes is a common phenomenon (Jühling et al., 2012), which mainly appears in four tRNA structures: the amino acid acceptor arm (AA), the TΨC arm (T), the anticodon arm (AC) and the dihydrouridine arm (DHU) (Zhang et al., 2012). A total of 11 base mismatches were observed in the tRNA genes of H. longicornis, among which G-U mismatches occurred 9 times, and the remaining two pairs were U-U mismatches. The mismatch was similar between the bisexual and parthenogenetic populations. The RNA editing process can correct the base mismatch without affecting the tRNA transport function (Bae et al., 2004). However, the base mismatch may jeopardize the survival ability of living organisms (Hendrickson, 2001) and hence result in the evolution of species (Watanabe et al., 1994). The gene length, base skew, and arrangement position of the rRNA in the bisexual and parthenogenetic H. longicornis were similar to those of other insects (Breinholt and Kawahara, 2013).
In most cases, the mitochondrial genomes displayed conservative gene arrangement and stable gene structure. Despite rapid evolution over time, the genomic arrangement of most arthropod mitochondria usually remains unchanged (Boore, 1999), and there is no similarity in mitochondrial gene rearrangement (Cameron et al., 2006). The genetic arrangement in the Drosophila genus was generally considered to be the original form of insects (Boore, 1999). Mitochondrial gene rearrangement includes major gene rearrangement and minor gene rearrangement (Boore and Brown, 2000). Like insects, two patterns of mitochondrial gene rearrangement were found in different genera of ticks. The minor rearrangement type was relatively common and occurred only in exchanges of the tRNA position (Cameron et al., 2007). This kind of rearrangement was common in Ixodes genus, but the rearrangement degree was significantly lower than that observed in other genera of ticks (Liu et al., 2018). The major gene rearrangement referred to the rearrangement or inversion of PCG or rRNA genes and infrequently occurred in insects. Liu et al. reported the major gene rearrangement from some tick genera, such as Haemaphysalis, Amblyomma, and Rhipicephalus (Liu et al., 2018). There were three types of tRNA gene position changes: shuffling (local rearrangements), translocation (cross-gene displacement) and inversion (change the encoding or transcriptional direction) (Dowton and Austin, 1999). Three different displacements were observed in H. longicornis tRNA: shuffling was found in trnH, trnT and trnP, translocation was observed in 5 genes (trnF, trnS2, trnL1, trnC, and trnL2), and trnC experienced gene inversion. The tRNA gene rearrangement was interpreted as a result of tandem duplication and random loss (Jühling et al., 2012; Xia et al., 2016), which could provide important genetic and phylogenetic information (Boore et al., 1998; Cameron et al., 2006).
Mitochondrial genomes generally have no introns. Although there are some intergenic regions, most of the arthropod mitochondrial genes are closely linked (Boore, 1999). The mitochondrial genomes of ticks contain different sizes of overlapping regions, including intergenic regions and NCRs. These regions are generally regarded as being involved in gene expression and regulation. The current study found three NCRs in the mitochondrial genome of H. longicornis. NCR1 was speculated to be a randomly insert fragment, which was also reported previously in Dermacentor and Rhipicephalus and possibly related to the tRNA structure (Cameron and Whiting, 2008; Burger et al., 2014b; Mccooke et al., 2015; Guo et al., 2016). The NCR2 and NCR3 were situated in similar positions with other tick species (Burger et al., 2012; Montagna et al., 2012; Williamsnewkirk et al., 2015). No difference was observed in the length or location of NCRs between the bisexual and parthenogenetic populations of H. longicornis. The NCRs were considered as a gene control region in arthropod, and the number of repeat sequences and conserved structures could directly affect the length and base skew of the NCRs (Simon, 1991; Montagna et al., 2012; Cameron, 2014; Li and Liang, 2018), which may be the reason for the lower A+T content of NCRs (60−70%) in H. longicornis. Recently, a “Tick-Box” structure was found in ticks, and metazoan, which is a 17-bp-long conserved sequence (TTGyrTChwwwTwwGdA) in the mitochondrial genome, was connected to transcription termination and gene alignment (Montagna et al., 2012). This conserved sequence was also found in two reproductive populations of H. longicornis, which were the same as other species of Haemaphysalis (TTGCATCAATTTTTGGA). Also, the conserved “ATGATAA” repeat sequence was found between atp8 and atp6 in H. longicornis, which has been reported in other ticks and insects (Stewart and Beckenbach, 2006; Williamsnewkirk et al., 2015).
Phylogenetic tree analysis showed that the independent branches of Ixodes, Rhipicephalus, Amblyomma and Dermacentor species were similar to the findings of Burger et al. (2014a). The conjoint analysis using all PCG genes could improve the stability and reliability of phylogenetic reconstruction (Sheffield et al., 2009). The two reproductive populations of H. longicornis were the most closely related and were less than the criteria for subspecies identification, which was consistent with previous results (Chen et al., 2010; Chen X. et al., 2012). Only one non-synonymous site of PCGs was observed in the two populations, whereas nearly 200 polymorphic sites were detected, but no apparent changes were observed in gene structure and arrangement under the evolutionary pressure of different environments.
The mitochondrion is essential for energy metabolism and is also involved in many critical processes, including cell transport, signal transduction, temperature regulation and immune activity (Brand, 2000; Detmer and Chan, 2007). Mitochondrial genes may lead to significant changes in metabolism and adaptability in the process of evolution (Ballard and Pichaud, 2014), and their genetic diversity was mainly formed by random genetic drift and natural selection (Sun et al., 2018). Multiple genes of H. longicornis were significantly upregulated in the partially fed and engorgement stages; among these genes, the most significant changes were detected in atp6 and atp8, which are the core subunits of ATP synthase (Fontanillas et al., 2010). The expression of these genes might provide guarantees for oxidative phosphorylation (OXPHOS) of cells and changes in the metabolism of body temperature. In previous studies, cox1 was considered to be the essential gene for COX activity changes (Uno et al., 2004; Singtripop et al., 2007). In the bisexual H. longicornis, the upregulation of cox1 was not evident compared with the other two subunits (cox2 and cox3). In parthenogenetic H. longicornis, changes in the cox1 gene were noticeable, but down-regulation in cox2 and cox3 was observed. These results suggested that the dynamic changes of the cox subunit were not solely related to the cox1 gene. The cox3 gene in the bisexual population was considerably higher than that in the parthenogenetic population at each feeding status. This differential expression may be attributed to the living environment of the bisexual population, where it is characterized by higher altitude and latitude, and the climatic conditions of temperature and humidity are more severe.
The most specific gene in the NADH complex is nad3 (Sanz et al., 2010). Only one non-synonymous site was observed in this gene, and this mutation was likely to change the gene expression patterns and functions in the two populations. The expression of nad3 was significantly downregulated in the bisexual and parthenogenetic H. longicornis at different status, whereas it was also multiple times higher in the engorgement stage in parthenogenetic H. longicornis compared with that in the bisexual population. The structural changes of PCGs were required events for environmental adaptation, and most genes were maintained or upregulated during blood feeding or at the engorgement stage, which suggested that the metabolic level in the body was increased or the number of mitochondria was increased. These changes will guarantee rapid development and reproduction in H. longicornis.
Conclusion
In the current study, the mitochondrial genomes of the bisexual and parthenogenetic H. longicornis were analyzed, and the gene structure and position arrangement were similar between the two reproductive populations. However, single nucleotide polymorphism analysis showed that approximately 200 bases were different. Phylogenetic analysis suggested that the bisexual and parthenogenetic populations were more closely related than the subspecies. Quantitative results of PCGs showed that the expression patterns of genes in the two reproductive populations were significantly distinctive at different feeding status, which may be similarly associated with environmental differences and reproductive patterns.
Data Availability
The datasets generated for this study can be found in NCBI with the accession numbers: MK450606 (bisexual populations of H. longicornis) and MK439888 (parthenogenetic populations of H. longicornis).
Ethics Statement
All experimental procedures were approved by the Animal Ethics Committee of Hebei Normal University (protocol number: IACUC-157026).
Author Contributions
ZY conceived the experiments. TW performed the experiments. SZ and TP analyzed the data. ZY and TW drafted and edited the manuscript. JL reviewed and corrected the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31672365), the Youth Top Talent Support Program of Hebei Province to ZY, the Natural Science Foundation of Hebei Province (C2019205064), the Financial Assistance for the Introduction of Overseas Researchers (C20190350), the Natural Science Research Programs of the Educational Department of Hebei Province (BJ2016032), the Science Foundation of Hebei Normal University (L2018J04), and the Postgraduate Innovation Foundation of Hebei (CXZZBS2018094).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are very grateful to Dr. Abolfazl Masoudi from our laboratory for reviewing the manuscript and providing valuable comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.00982/full#supplementary-material
FIGURE S1 | Mitochondrial genome circular map of Haemaphysalis longicornis bisexual female.
FIGURE S2 | Mitochondrial genome circular map of Haemaphysalis longicornis bisexual male.
FIGURE S3 | Mitochondrial genome circular map of the Haemaphysalis longicornis parthenogenetic population.
FIGURE S4 | Nucleotide polymorphisms in the parthenogenetic population with the bisexual population considered as the reference.
FIGURE S5 | Secondary structures of tRNA in the bisexual population of Haemaphysalis longicornis.
FIGURE S6 | Secondary structures of tRNAs in the parthenogenetic population of Haemaphysalis longicornis.
FIGURE S7 | Quantitative expression of the different feeding status of the bisexual population. The asterisk indicates a level of significant difference (∗P < 0.05, ∗∗P < 0.01) in gene expression between the different groups.
FIGURE S8 | Quantitative expression of the different feeding status of the parthenogenetic population. The asterisk indicates a level of significant difference (∗P < 0.05, ∗∗P < 0.01) in gene expression between the different groups.
TABLE S1 | Original mitochondrial genome sequencing data of Haemaphysalis longicornis.
TABLE S2 | Quantitative primers in the mitochondrial genome of Haemaphysalis longicornis.
TABLE S3 | Comparison of the mitochondrial gene position and arrangement in the bisexual and parthenogenetic population of Haemaphysalis longicornis.
TABLE S4 | Codon usage of Haemaphysalis longicornis in the mitochondrial protein-coding genes.
Footnotes
- ^ https://sourceforge.net/projects/soapdenovo2/
- ^ http://ogdraw.mpimp-golm.mpg.de/
- ^ http://mitos.bioinf.uni-leipzig.de/index.py
- ^ http://www.geneontology.org
- ^ http://lowelab.ucsc.edu/tRNAscan-SE
- ^ http://tandem.bu.edu/trf/trf.html
- ^ http://bio-bwa.sourceforge.net
- ^ https://software.broadinstitute.org/gatk
- ^ http://orthomcl.org/orthomcl
- ^ https://www.ncbi.nlm.nih.gov/
- ^ https://cme.h-its.org/exelixis/web/software/raxml/index.html
- ^ https://mafft.cbrc.jp/alignment/software/
- ^ http://molevol.cmima.csic.es/castresana/Gblocks.html
References
Alexandre, H., Nelly, L., and Jean, D. J. (2005). Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Syst. Biol. 54, 277–298. doi: 10.1080/10635150590947843
Bae, J. S., Kim, I., Sohn, H. D., and Jin, B. R. (2004). The mitochondrial genome of the firefly, Pyrocoelia rufa: complete DNA sequence, genome organization, and phylogenetic analysis with other insects. Mol. Phylogenet. Evol. 32, 978–985. doi: 10.1016/j.ympev.2004.03.009
Ballard, J. W., and Pichaud, N. J. (2014). Mitochondrial DNA: more than an evolutionary bystander. Funct. Ecol. 28, 218–231. doi: 10.1111/1365-2435.12177
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Black, W. C., and Roehrdanz, R. L. (1998). Mitochondrial gene order is not conserved in arthropods: prostriate and metastriate tick mitochondrial genomes. Mol. Biol. Evol. 15, 1772–1785. doi: 10.1093/oxfordjournals.molbev.a025903
Boore, J. L. (1999). Survey and summary animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. doi: 10.1093/nar/27.8.1767
Boore, J. L., and Brown, W. M. (2000). Mitochondrial genomes of Galathealinum, Helobdella, and Platynereis: sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa. Mol. Biol. Evol. 17, 87–106. doi: 10.1093/oxfordjournals.molbev.a026241
Boore, J. L., Lavrov, D. V., and Brown, W. M. (1998). Gene translocation links insects and crustaceans. Nature 392, 667–668. doi: 10.1038/33577
Brand, M. D. (2000). Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp. Gerontol. 35, 811–820. doi: 10.1016/S0531-5565(00)00135-2
Breinholt, J. W., and Kawahara, A. Y. (2013). Phylotranscriptomics: saturated third codon positions radically influence the estimation of trees based on next-gen data. Genome Biol. Evol. 5, 2082–2092. doi: 10.1093/gbe/evt157
Burger, T. D., Shao, R., and Barker, S. C. (2013). Phylogenetic analysis of the mitochondrial genomes and nuclear rRNA genes of ticks reveals a deep phylogenetic structure within the genus Haemaphysalis and further elucidates the polyphyly of the genus Amblyomma with respect to Amblyomma sphenodonti and Amblyomma elaphense. Ticks Tick Borne Dis. 4, 265–274. doi: 10.1016/j.ttbdis.2013.02.002
Burger, T. D., Shao, R., Beati, L., Miller, H., and Barker, S. C. (2012). Phylogenetic analysis of ticks (Acari: Ixodida) using mitochondrial genomes and nuclear rRNA genes indicates that the genus Amblyomma is polyphyletic. Mol. Phylogenet. Evol. 64, 45–55. doi: 10.1016/j.ympev.2012.03.004
Burger, T. D., Shao, R., and Barker, S. C. (2014a). Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol. Phylogenet. Evol. 76, 241–253. doi: 10.1016/j.ympev.2014.03.017
Burger, T. D., Shao, R., Labruna, M. B., and Barker, S. C. (2014b). Molecular phylogeny of soft ticks (Ixodida: Argasidae) inferred from mitochondrial genome and nuclear rRNA sequences. Ticks Tick Borne Dis. 5, 195–207. doi: 10.1016/j.ttbdis.2013.10.009
Cameron, S. L. (2014). Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117. doi: 10.1146/annurev-ento-011613-162007
Cameron, S. L., Beckenbach, A. T., Dowton, A., and Whiting, M. F. (2006). Evidence from mitochondrial genomics on interordinal relationships. Arthropod Syst. Phylogeny 64, 27–34.
Cameron, S. L., Johnson, K. P., and Whiting, M. F. (2007). The mitochondrial genome of the screamer Louse Bothriometopus (Phthiraptera: Ischnocera): effects of extensive gene rearrangements on the evolution of the genome. J. Mol. Evol. 65, 589–604. doi: 10.1007/s00239-007-9042-8
Cameron, S. L., and Whiting, M. F. (2008). The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 408, 112–123. doi: 10.1016/j.gene.2007.10.023
Cancilla, M. R., Hillier, A. J., and Davidson, B. E. (1995). Lactococcus lactis glyceraldehyde 3 phosphate dehydrogenase gene, gap-further evidence for strongly biased codon usage in glycolytic pathway genes. Microbiology 141, 1027–1036. doi: 10.1099/13500872-141-4-1027
Chen, X., Xu, S., Yu, Z., Guo, L., Yang, S., Liu, L., et al. (2014). Multiple lines of evidence on the genetic relatedness of the parthenogenetic and bisexual Haemaphysalis longicornis (Acari: Ixodidae). Infect. Genet. Evol. 21, 308–314. doi: 10.1016/j.meegid.2013.12.002
Chen, X., Yu, Z. Y., Guo, L. D., Li, L. X., Meng, H., Wang, D., et al. (2012). Life cycle of Haemaphysalis doenitzi (Acari: Ixodidae) under laboratory conditions and its phylogeny based on mitochondrial 16S rDNA. Exp. Appl. Acarol. 56, 143–150. doi: 10.1007/s10493-011-9507-8
Chen, Z., Li, Y. Q., Ren, Q. Y., Liu, Z. J., Luo, J., Li, K., et al. (2015). Does Haemaphysalis bispinosa (Acari: Ixodidae) really occur in China? Exp. Appl. Acarol. 65, 249–257. doi: 10.1007/s10493-014-9854-3
Chen, Z., Yang, X. J., Bu, F. J., Yang, X. H., Yang, X. L., and Liu, J. Z. (2010). Ticks (Acari: Ixodoidea: Argasidae, Ixodidae) of China. Exp. Appl. Acarol. 51, 393–404. doi: 10.1007/s10493-010-9335-2
Chen, Z., Yang, X. J., Bu, F. J., Yang, X. L., and Liu, J. Z. (2012). Morphological, biological and molecular characteristics of bisexual and parthenogenetic Haemaphysalis longicornis. Vet. Parasitol. 189, 344–352. doi: 10.1016/j.vetpar.2012.04.021
Dai, L. S., Zhu, B. J., Zhao, Y., Zhang, C. F., and Liu, C. L. (2016). Comparative mitochondrial genome analysis of Eligma narcissus and other Lepidopteran insects reveals conserved mitochondrial genome organization and phylogenetic relationships. Sci. Rep. 6:26387. doi: 10.1038/srep26387
Detmer, S. A., and Chan, D. C. (2007). Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 8, 870–879. doi: 10.1038/nrm2275
Dowton, M., and Austin, A. D. (1999). Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the Hymenoptera. Mol. Biol. Evol. 16, 298–309. doi: 10.1093/oxfordjournals.molbev.a026111
Fang, B., Jiang, W., Zhou, Q., and Wang, S. J. (2015). Codon-optimized NADH oxidase gene expression and gene fusion with glycerol dehydrogenase for bienzyme system with cofactor regeneration. PLoS One 10:e0128412. doi: 10.1371/journal.pone.0128412
Fischer, S., Brunk, B. P., Chen, F., Gao, X., Harb, O. S., and Iodice, J. B. (2011). Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinformatics 6, 6.12.1–6.12.19. doi: 10.1002/0471250953.bi0612s35
Fontanillas, P., Dépraz, A., Giorgi, M. S., and Perrin, N. (2010). Nonshivering thermogenesis capacity associated to mitochondrial DNA haplotypes and gender in the greater white-toothed shrew Crocidura russula. Mol. Ecol. 14, 661–670. doi: 10.1111/j.1365-294X.2004.02414.x
Fujisaki, K., Kawazu, S., and Kamio, T. (1994). The taxonomy of the bovine Theileria spp. Parasitol. Today 10, 31–33. doi: 10.1016/0169-4758(94)90355-7
Gissi, C., Iannelli, F., and Pesole, G. J. (2008). Evolution of the mitochondrial genome of metazoa as exemplified by comparison of congeneric species. Heredity 101, 301–320. doi: 10.1038/hdy.2008.62
Guo, D. H., Zhang, Y., Fu, X., Gao, Y., Liu, Y. T., Qiu, J. H., et al. (2016). Complete mitochondrial genomes of Dermacentor silvarum and comparative analyses with another hard tick Dermacentor nitens. Exp. Parasitol. 169, 22–27. doi: 10.1016/j.exppara.2016.07.004
Hendrickson, T. L. (2001). Recognizing the D-loop of transfer RNAs. Proc. Natl. Acad. Sci. U.S.A. 98, 13473–13475. doi: 10.1073/pnas.251549298
Herrin, C. S., and Oliver, J. H. (1974). Numerical taxonomic studies of parthenogenetic and bisexual populations of Haemaphysalis longicornis and related species (Acari: Ixodidae). J. Parasitol. 60, 1025–1036. doi: 10.2307/3278542
Hoogstraal, H., Roberts, F. H. S., Kohls, G. M., and Tipton, V. J. (1968). Review of Haemaphysalis (Kaiseriana) longicornis Neumann (resurrected) of Australia, New Zealand, New Caledonia, Fiji, Japan, Korea, and Northeastern China and USSR, and its parthenogenetic and bisexual populations (Ixodoidea, Ixodidae). J. Parasitol. 54, 1197–1213. doi: 10.2307/3276992
Jongejan, F., and Uilenberg, G. (2004). The global importance of ticks. Parasitology 129, S3–S14. doi: 10.1017/S0031182004005967
Jühling, F., Mörl, M., Hartmann, R. K., Sprinzl, M., Stadler, P. F., and Pütz, J. J. (2009). tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 37, 159–162. doi: 10.1093/nar/gkn772
Jühling, F., Pütz, J., Bernt, M., Donath, A., Middendorf, M., Florentz, C., et al. (2012). Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 40, 2833–2845. doi: 10.1093/nar/gkr1131
Kaufman, W. R. (2010). Ticks: physiological aspects with implications for pathogen transmission. Ticks Tick Borne Dis. 1, 11–22. doi: 10.1016/j.ttbdis.2009.12.001
Kim, J., Kern, E., Kim, T., Sim, M., Kim, J., Kim, Y., et al. (2017). Phylogenetic analysis of two Plectus mitochondrial genomes (Nematoda: Plectida) supports a sister group relationship between Plectida and Rhabditida within Chromadorea. Mol. Phylogenet. Evol. 107, 90–102. doi: 10.1016/j.ympev.2016.10.010
Kovalev, S. Y., and Mukhacheva, T. A. (2013). Cluster on structure of tick-borne encephalitis virus populations. Infect. Genet. Evol. 14, 22–28. doi: 10.1016/j.meegid.2012.10.011
Kumar, S., Stecher, G., and Tamura, K. J. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Li, K., and Liang, A. P. (2018). Hemiptera mitochondrial control region: new sights into the structural organization, phylogenetic utility, and roles of tandem repetitions of the noncoding segment. Int. J. Mol. Sci. 19:E1292. doi: 10.3390/ijms19051292
Lin, C. P., and Danforth, B. N. (2004). Evolution, how do insect nuclear and mitochondrial gene substitution patterns differ? Insights from bayesian analyses of combined datasets. Mol. Phylogenet. Evol. 30, 686–702. doi: 10.3390/ijms19051292
Lines, M. A., Jobling, R., Brady, L., Marshall, C. R., Scherer, S. W., and Rodriguez, A. R. (2014). Peroxisomal D-bifunctional protein deficiency: three adults diagnosed by whole-exome sequencing. Neurology 82, 963–968. doi: 10.1212/WNL.0000000000000219
Liu, Q., He, B., Huang, S. Y., Wei, F., and Zhu, X. Q. (2014). Severe fever with thrombocytopenia syndrome, an emerging tick-borne zoonosis. Lancet Infect. Dis. 14, 763–772. doi: 10.1016/S1473-3099(14)70718-2
Liu, Z. Q., Liu, Y. F., Kuermanali, N., Wang, D. F., Chen, S. J., Guo, H. L., et al. (2018). Sequencing of complete mitochondrial genomes confirms synonymization of Hyalomma asiaticum asiaticum and kozlovi, and advances phylogenetic hypotheses for the Ixodidae. PLoS One 13:e0197524. doi: 10.1371/journal.pone.0197524
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. J. (2013). OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, 575–581. doi: 10.1093/nar/gkt289
Magori, K. (2018). Preliminary prediction of the potential distribution and consequences of Haemaphysalis longicornis (Ixodida: Ixodidae) in the United States and North America, using a simple rule-based climate envelope model. bioRxiv [Preprint]. doi: 10.1101/389940
Matsuo, T., Okura, N., Kakuda, H., and Yano, Y. (2013). Reproduction in a Metastriata tick, Haemaphysalis longicornis (Acari: Ixodidae). J. Acarol. Soc. Jpn. 22, 1–23. doi: 10.1046/j.1463-6395.2000.00035.x
Mccooke, J. K., Guerrero, F. D., Barrero, R. A., Black, M., Hunter, A., Bell, C., et al. (2015). The mitochondrial genome of a Texas outbreak strain of the cattle tick, Rhipicephalus (Boophilus) microplus, derived from whole genome sequencing Pacific Biosciences and Illumina reads. Gene 571, 135–141. doi: 10.1016/j.gene.2015.06.060
Mccormick, R. F., Truong, S. K., and Mullet, J. E. (2015). RIG: recalibration and interrelation of genomic sequence data with the GATK. G3 5, 655–665. doi: 10.1534/g3.115.017012
Monis, P. T., Andrews, R. H., and Saint, C. P. (2002). Molecular biology techniques in parasite ecology. Int. J. Parasitol. 32, 551–562. doi: 10.1016/s0020-7519(01)00352-6
Montagna, M., Sassera, D., Griggio, F., Epis, S., Bandi, C., and Gissi, C. J. (2012). Tick-Box for 3′-End formation of mitochondrial transcripts in Ixodida, basal Chelicerates and Drosophila. PLoS One 7:e47538. doi: 10.1371/journal.pone.0047538
Ojala, D., Montoya, J., and Attardi, G. J. (1981). tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474. doi: 10.1038/290470a0
Oliver, H. J. (1977). Cytogenetics of mites and ticks. Annu. Rev. Entomol. 22, 407–429. doi: 10.1146/annurev.en.22.010177.002203
Pan, M. H., Yu, Q. Y., Xia, Y. L., Dai, F. Y., Liu, Y. Q., and Lu, C. (2008). Characterization of mitochondrial genome of Chinese wild mulberry silkworm, Bombyx mandarina (Lepidoptera: Bombycidae). Sci. China Life Sci. 51, 693–701. doi: 10.1007/s11427-008-0097-6
Papanicolaou, A., Gebauerjung, S., Blaxter, M. L., Mcmillan, W. O., and Jiggins, C. D. (2008). ButterflyBase: a platform for lepidopteran genomics. Nucleic Acids Res. 36, D582–D587. doi: 10.1093/nar/gkm853
Qin, J., Zhang, Y., Zhou, X., Kong, X., Wei, S., Ward, R. D., et al. (2015). Mitochondrial phylogenomics and genetic relationships of closely related pine moth (Lasiocampidae: Dendrolimus) species in China, using whole mitochondrial genomes. BMC Genomics 16:428. doi: 10.1186/s12864-015-1566-5
Qin, X. R., Han, F. J., Luo, L. M., Zhao, F. M., Han, H. J., Zhang, Z. T., et al. (2018). Anaplasma species detected in Haemaphysalis longicornis tick from china. Ticks Tick Borne Dis. 9, 840–843. doi: 10.1016/j.ttbdis.2018.03.014
Sanz, A., Soikkeli, M., Portero-Otín, M., Wilson, A., Kemppainen, E., and Mcilroy, G. (2010). Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc. Natl. Acad. Sci. U.S.A. 107, 9105–9110. doi: 10.1073/pnas.0911539107
Sheffield, N. C., Song, H., Cameron, S. L., and Whiting, M. F. (2009). Nonstationary evolution and compositional heterogeneity in beetle mitochondrial phylogenomics. Syst. Biol. 58, 381–394. doi: 10.1093/sysbio/syp037
Simon, C. (1991). “Molecular systematics at the species boundary: exploiting conserved and variable regions of the mitochondrial genome of animals via direct sequencing from amplified DNA,” in Molecular Techniques in Taxonomy, eds G. M. Hewitt, A. W. B. Johnston, and J. P. W. Young (Berlin: Springer-Verlag), 33–71. doi: 10.1007/978-3-642-83962-7_4
Singtripop, T., Saeangsakda, M., Tatun, N., Kaneko, Y., and Sakurai, S. (2007). Correlation of oxygen consumption, cytochrome c oxidase, and cytochrome c oxidase subunit I gene expression in the termination of larval diapause in the bamboo borer, Omphisa fuscidentalis. J. Insect Physiol. 53, 933–939. doi: 10.1016/j.jinsphys.2007.03.005
Stamatakis, A. (2015). Using RAxML to infer phylogenies. Curr. Protoc. Bioinformatics 51, 6.14.1–6.14.14. doi: 10.1002/0471250953.bi0614s51
Stewart, J. B., and Beckenbach, A. T. (2006). Insect mitochondrial genomics 2: the complete mitochondrial genome sequence of a giant stonefly, Pteronarcys princeps, asymmetric directional mutation bias, and conserved plecopteran A+T-region elements. Genome 52, 815–824. doi: 10.1139/G08-098
Sun, J. T., Jin, P. Y., Hoffmann, A. A., Duan, X. Z., Dai, J., Hu, G., et al. (2018). Evolutionary divergence of mitochondrial genomes in two Tetranychus species distributed across different climates. Insect Mol. Biol. 27, 698–709. doi: 10.1111/imb.12501
Takahashi, T., Maeda, K., Suzuki, T., and Ishido, A. (2014). The first identification and retrospective study of severe fever with thrombocytopenia syndrome in Japan. J. Infect. Dis. 209, 816–827. doi: 10.1093/infdis/jit603
Timmermans, M. J., Lees, D. C., and Simonsen, T. J. (2014). Towards a mitogenomic phylogeny of Lepidoptera. Mol. Phylogenet. Evol. 79, 169–178. doi: 10.1016/j.ympev.2014.05.031
Tominski, C., Abello, J., and Schumann, H. J. (2009). Technical section: CGV-An interactive graph visualization system. Comput. Graph. 33, 660–678. doi: 10.1016/j.cag.2009.06.002
Uno, T., Nakasuji, A., Shimoda, M., and Aizono, Y. J. (2004). Expression of cytochrome c oxidase subunit 1 gene in the brain at an early stage in the termination of pupal diapause in the sweet potato hornworm, Agrius convolvuli. J. Insect Physiol. 50, 35–42. doi: 10.1016/j.jinsphys.2003.09.011
Wang, D., Hu, Y. H., and Liu, J. Z. (2013). Ultrastructure and development of the Haller’s organ of parthenogenetic Haemaphysalis longicornis (Acari: Ixodidae). Acta Entomol. Sin. 56, 306–311.
Watanabe, Y., Kawai, G., Yokogawa, T., Hayashi, N., Kumazawa, Y., Ueda, T., et al. (1994). Higher-order structure of bovine mitochondrial tRNASerUGA: chemical modification and computer modeling. Nucleic Acids Res. 22, 5378–5384. doi: 10.1093/nar/22.24.5378
Wei, S. J., Shi, M., Chen, X. X., Sharkey, M. J., Achterberg, C. V., Ye, G. Y., et al. (2010). New views on strand asymmetry in insect mitochondrial genomes. PLoS One 5:e12708. doi: 10.1371/journal.pone.0012708
Williamsnewkirk, A. J., Burroughs, M., Changayil, S. S., and Dasch, G. A. (2015). The mitochondrial genome of the lone star tick (Amblyomma americanum). Ticks Tick Borne Dis. 6, 793–801. doi: 10.1016/j.ttbdis.2015.07.006
Xia, Y., Zheng, Y., Murphy, R. W., and Zeng, X. (2016). Intraspecific rearrangement of mitochondrial genome suggests the prevalence of the tandem duplication-random loss (TDLR) mechanism in Quasipaa boulengeri. BMC Genomics 17:965. doi: 10.1186/s12864-016-3309-7
Xie, Y., Wu, G., Tang, J., Luo, R., Patterson, J., Liu, S., et al. (2014). SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 1660–1666. doi: 10.1093/bioinformatics/btu077
Xiong, H., Barker, S. C., Burger, T. D., Raoult, D., and Shao, R. F. (2013). Heteroplasmy in the mitochondrial genomes of human lice and ticks revealed by high throughput sequencing. PLoS One 8:e73329. doi: 10.1371/journal.pone.0073329
Yang, C. M., Yang, G. Y., Xie, Y. X., Wu, K. B., and Yong, J. X. (2007). Morphological observation of parthenogenetic Haemaphysalis longicornis with scanning electron microscope. Acta Parasitol. Med. Entomol. Sin. 14, 104–109.
Yu, Z. J., Wang, H., Wang, T. H., Sun, W. Y., Yang, X. L., and Liu, J. Z. (2015). Tick-borne pathogens and the vector potential of ticks in China. Parasit. Vectors 8:24. doi: 10.1186/s13071-014-0628-x
Yuan, M. L., Zhang, Q. L., Guo, Z. L., Wang, J., and Shen, Y. Y. (2015). Comparative mitogenomic analysis of the superfamily Pentatomoidea (Insecta: Hemiptera: Heteroptera) and phylogenetic implications. BMC Genomics 16:460. doi: 10.1186/s12864-015-1679-x
Zhan, J., Wang, Q., Cheng, J., Hu, B., Li, J., Zhan, F., et al. (2017). Current status of severe fever with thrombocytopenia syndrome in china. Virol. Sin. 32, 51–62. doi: 10.1007/s12250-016-3931-1
Zhou, J. L., Zhou, Y. Z., Gong, H. Y., Chen, L. Z., and Cao, J. (2004). Discovery of parthenogenesis population of Haemaphysalis longicornis in China and its biological characteristic. Chin J. Vector Biol. Control 15, 173–174.
Zhang, M., Nie, X., Cao, T., Wang, J., Li, T., Zhang, X., et al. (2012). The complete mitochondrial genome of the butterfly Apatura metis (Lepidoptera: Nymphalidae). Mol. Biol. Rep. 39, 6529–6536. doi: 10.1007/s11033-012-1481-7
Zhang, R. L., Huang, Z. D., Yu, G. F., and Zhang, Z. (2019). Characterization of microbiota diversity of field-collected Haemaphysalis longicornis (Acari: Ixodidae) with regard to sex and blood meals. J. Basic Microbiol. 59, 215–223. doi: 10.1002/jobm.201800372
Zhou, Z., Schnake, P., Xiao, L., and Lal, A. A. (2004). Enhanced expression of a recombinant malaria candidate vaccine in Escherichia coli by codon optimization. Protein Expr. Purif. 34, 87–94. doi: 10.1016/j.pep.2003.11.006
Keywords: Haemaphysalis longicornis, mitochondrial genome, bisexual and parthenogenetic, mitochondrial protein-coding genes, expression profile
Citation: Wang T, Zhang S, Pei T, Yu Z and Liu J (2019) The Complete Mitochondrial Genome and Expression Profile of Mitochondrial Protein-Coding Genes in the Bisexual and Parthenogenetic Haemaphysalis longicornis. Front. Physiol. 10:982. doi: 10.3389/fphys.2019.00982
Received: 03 April 2019; Accepted: 15 July 2019;
Published: 30 July 2019.
Edited by:
Abid Ali, Abdul Wali Khan University Mardan, PakistanReviewed by:
Shahid Karim, The University of Southern Mississippi, United StatesYuxin Yin, University of California, Los Angeles, United States
Maria Dolores Esteve-Gasent, Texas A&M University, United States
Copyright © 2019 Wang, Zhang, Pei, Yu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhijun Yu, eXV6aGlqdW5AaGVidHUuZWR1LmNu; Jingze Liu, bGl1amluZ3plQGhlYnR1LmVkdS5jbg==