 Ambrish Kumar1
Ambrish Kumar1 Jay D. Potts
Jay D. Potts
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 02 July 2019
Sec. Integrative Physiology
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.00821
This article is part of the Research TopicNeuropeptides as Central Mediators of Cardiovascular DiseaseView all 6 articles
α-Calcitonin gene-related peptide (α-CGRP) is a regulatory neuropeptide of 37 amino acids. It is widely distributed in the central and peripheral nervous system, predominantly in cell bodies of the dorsal root ganglion (DRG). It is the most potent vasodilator known to date and has inotropic and chronotropic effects. Using pharmacological and genetic approaches, our laboratory and other research groups established the protective role of α-CGRP in various cardiovascular diseases such as heart failure, experimental hypertension, myocardial infarction, and myocardial ischemia/reperfusion injury (I/R injury). α-CGRP acts as a depressor to attenuate the rise in blood pressure in three different models of experimental hypertension: (1) DOC-salt, (2) subtotal nephrectomy-salt, and (3) L-NAME-induced hypertension during pregnancy. Subcutaneous administration of α-CGRP lowers the blood pressure in hypertensive and normotensive humans and rodents. Recent studies also demonstrated that an α-CGRP analog, acylated α-CGRP, with extended half-life (~7 h) reduces blood pressure in Ang-II-induced hypertensive mouse, and protects against abdominal aortic constriction (AAC)-induced heart failure. Together, these studies suggest that α-CGRP, native or a modified form, may be a potential therapeutic agent to treat patients suffering from cardiac diseases.
α-Calcitonin gene-related peptide (α-CGRP) is a regulatory neuropeptide that belongs to the calcitonin/CGRP peptide family that includes adrenomedullin (ADM), amylin, and calcitonin (Breimer et al., 1988). α-CGRP is generated from the tissue-specific alternative splicing of the primary transcript of the calcitonin gene CALC-I (Amara et al., 1982; Rosenfeld et al., 1983). In thyroid C cells, the CALC-I gene product containing exon 4 forms calcitonin, whereas the CALC-I gene product containing exons 5 and 6 forms α-CGRP in sensory neurons (Figure 1). Peptide α-CGRP is a single polypeptide of 37 amino acids containing a disulfide bridge between positions 2 and 7 and a phenylalanine amide residue at the C-terminal. Another distinct homologue of the CGRP is also known and termed as β-CGRP. However, β-CGRP is encoded from a separate CALC-II gene located on chromosome 11 in humans, and is not produced by alternative splicing of the gene (Steenbergh et al., 1985; Alevizaki et al., 1986). α-CGRP is synthesized in specific regions of the central and peripheral nervous systems, particularly in the sensory neurons of the dorsal root ganglion (DRG). β-CGRP is mainly expressed in the gut, intestine, pituitary gland, and immune cells such as T cells (Petermann et al., 1987; Mulderry et al., 1988; Xing et al., 2000). The α- and β-forms of the CGRP are highly conserved across species and share >90% homology; however, both forms differ by three amino acids in human (Figure 2A; Steenbergh et al., 1986). The biological action of CGRP isoforms is overlapping; moreover, α- and β-CGRPs exerts distinct hemodynamic and gastric effects in humans, respectively (Shekhar et al., 1991; Dubois-Rande et al., 1992). Compared to β-CGRP, α-CGRP has a markedly greater activity in the regulation of cardiovascular functions (Juaneda et al., 2000; Russell et al., 2014).
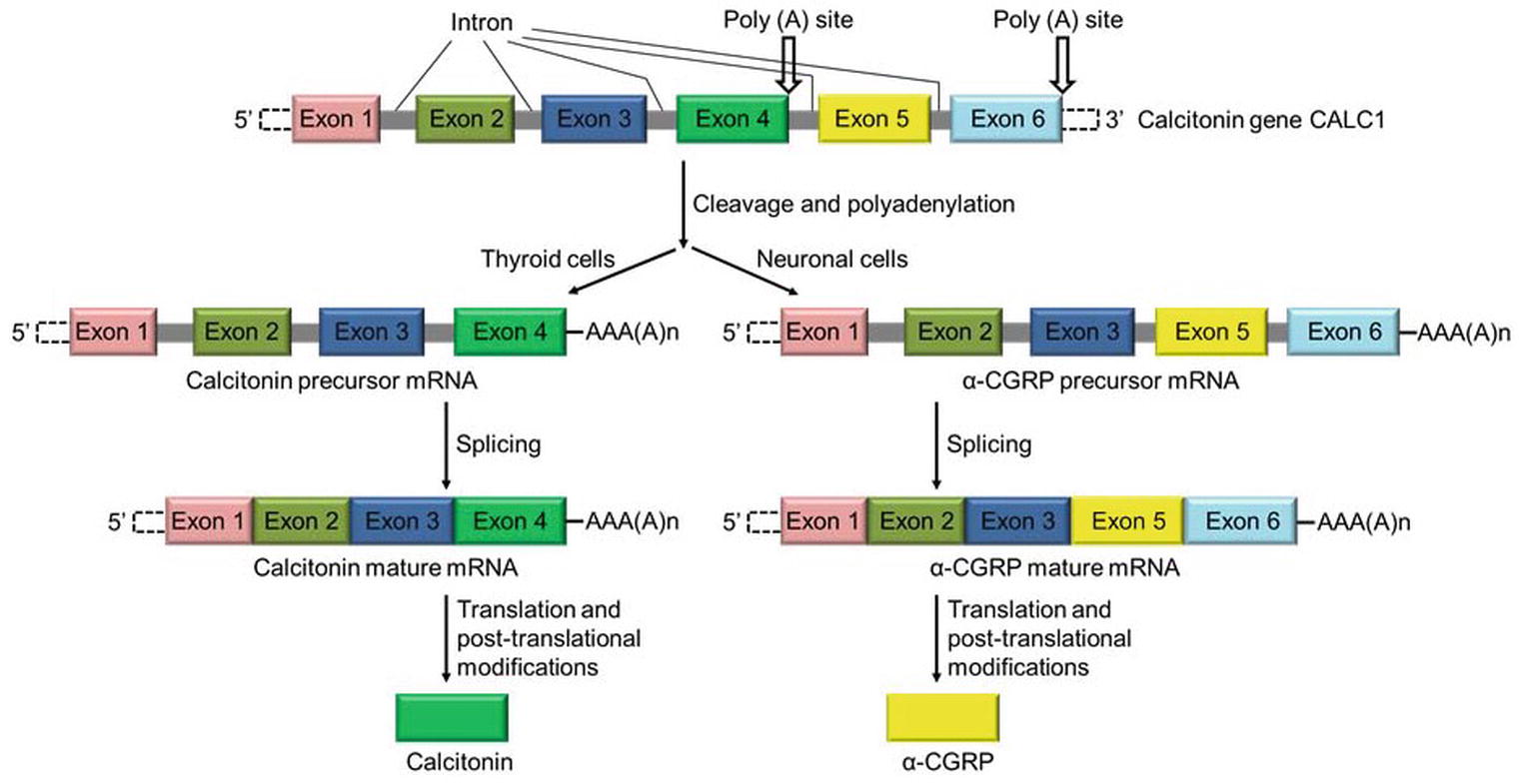
Figure 1. Schematic diagram showing the synthesis of calcitonin and α-CGRP from a common gene CALC-I. Calcitonin gene CALC-I undergoes alternative splicing and forms protein calcitonin in thyroid C cells, and α-CGRP in the sensory neurons.

Figure 2. Amino acid sequence of human α- and β-CGRPs, and rodent α-CGRP. (A) Human α- and β-CGRPs have a disulfide bridge (-S-S-) at cys2 and cys7, and amidated phenylalanine at C-terminus. Both forms of CGRP share >90% homology, and differ by three amino acids at positions 3, 22, and 25 (as shown by boxes). (B) Amino acid sequence of human and rodent (mouse/rat) α-CGRPs. Human and rodent (mouse/rat) α-CGRPs differ at amino acid residues 1, 3, 25, and 35 (as shown by arrows). Both have a disulfide bridge (-S-S-) at cys2 and cys7, and amidated phenylalanine at C-terminus.
Human and rodent (mouse/rat) α-CGRPs differ at four amino acid positions – 1, 3, 25, and 35 (Figure 2B); nevertheless, both share identical biological activities. Structurally, α-CGRP is composed of four domains (Figure 3):
1. Domain 1: N-terminus Domain 1 spans from amino acid residues 1 to 7 and forms a loop/ring-like structure. NMR studies suggest that the cysteine at positions 2 and 7 forms a disulfide bridge (Cys2-Cys7) (Breeze et al., 1991), and it has been shown that loss of the disulfide bond abolishes all the biological activity of the α-CGRP peptide. Domain 1 of α-CGRP binds to the transmembrane domain of the receptor and is essential for receptor activation. The N-terminal truncated peptide, CGRP8–37, acts as a CGRP receptor antagonist that has a high affinity for the CGRP receptor but lacking any biological activity (Chiba et al., 1989).
2. Domain 2: Domain 2 consists of an α-helix and spans the peptide region from amino acids 8-18. Deletion of this region decreases the affinity of the ligand α-CGRP to its receptor by 50- to 100-fold (Rovero et al., 1992).
3. Domain 3: Domain 3 of α-CGRP spans from residues 19-27 and can form either a β- or γ-twist (Conner et al., 2002) and may make additional receptor contacts or may stabilize an appropriate conformation of amino acids 28-37.
4. Domain 4: This C-terminus region of the peptide lies at residues 28-37 and contains two turn regions which form a putative binding epitope. This C-terminal region of the α-CGRP interacts with N-terminal region of the CGRP-receptor (Carpenter et al., 2001).
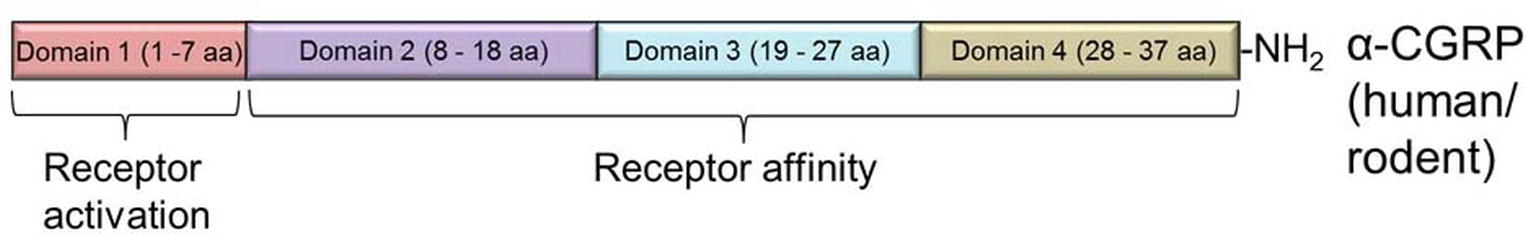
Figure 3. Domain structure of α-CGRP neuropeptide in rodent and human. The peptide is composed of four domains (Domain 1 to Domain 4). Domain 1 is essential for the CGRP receptor activation, while Domains 2–4 are necessary for receptor affinity.
Together, biological assays and deletion studies reveal that the amino acids 8-37 (Domain 2 to Domain 4) are required for the affinity of the peptide to its receptor, and the N-terminal Domain 1 of α-CGRP is essential for the receptor activation.
CGRP mediates its action through a complex heterotrimeric CGRP receptor that is composed of three proteins: (1) calcitonin receptor-like receptor (CLR, also known as CRLR), (2) receptor activity modifying protein 1 (RAMP 1), and (3) receptor component protein (RCP) molecule (Figure 4).
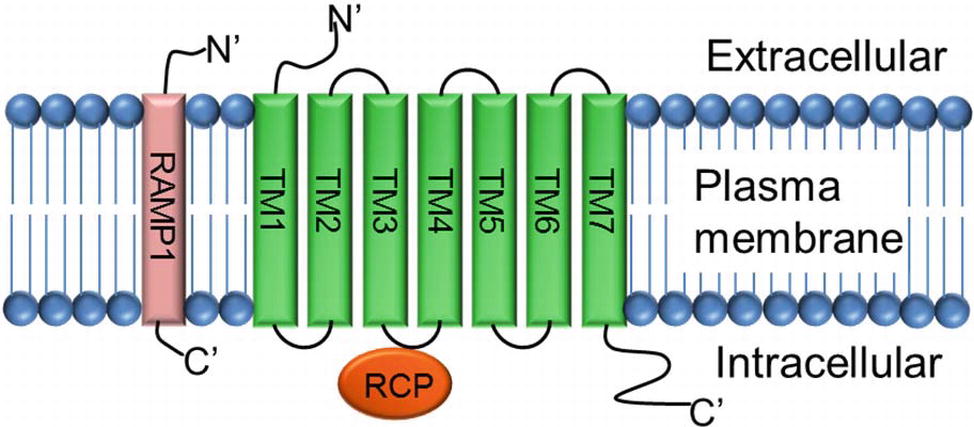
Figure 4. Structure of CGRP receptor. CGRP receptor is a complex heterotrimeric G-protein coupled receptor and requires three protein components: (1) calcitonin receptor-like receptor (CLR; a seven transmembrane protein TM1–TM7), (2) receptor activity modifying protein-1 (RAMP-1; a single transmembrane protein), and (3) receptor component protein (RCP; a small intracellular protein).
The calcitonin receptor-like receptor (CLR) is the ligand-binding portion of the receptor and belongs to the class B “secretin-like” family of the G-protein-coupled receptor (GPCR) (Lin et al., 1991; Barwell et al., 2012). The CLR receptor component possesses seven transmembrane spanning domains (TM1–TM7), a N-terminus extracellular domain, and an intracellular C-terminus (Aiyar et al., 1996). RAMP1 is a single transmembrane-spanning domain protein that has a long extracellular N-terminus of 100 amino acids and a short intracellular C-terminal tail of ~10 amino acids (McLatchie et al., 1998). Heterodimerization of CLR and RAMP1 (CLR/RAMP1) is required for transport of the CLR as a mature glycoprotein from the endoplasmic reticulum/Golgi apparatus to the plasma membrane (Hilairet et al., 2001). Deletion of amino acid residues 91-94, 96-100, and 101-103 totally inhibits the production of cAMP, and substitution of five amino acids at C-terminal of RAMP1, next to transmembrane domain, interrupts dimerization of CLR and RAMP1, and decreases receptor functions (Kuwasako et al., 2003). RCP is a small (~17 kDa) intracellular peripheral membrane protein that is associated with the cell membrane through ionic interactions (Evans et al., 2000). The direct interaction of RCP to the CGRP receptor CLR was verified by immunoprecipitation experiments where RCP was co-immunoprecipitated with the CLR/RAMP complex. Further analyses using a yeast two-hybrid assay and co-immunoprecipitation experiments confirmed that RCP directly interacts with the second intracellular cytoplasmic loop (ICL2) of CLR (Egea and Dickerson, 2012). Studies carried out in NIH3T3 cells expressing a functional CGRP receptor complex CLR-RAMP1 showed that depletion of RCP, using an antisense RNA approach, had no effect on the binding of the α-CGRP ligand to its receptor CLR. However, loss of RCP significantly abolished the α-CGRP-mediated signaling as demonstrated by a significant decrease in the production of cAMP (Evans et al., 2000). Thus, RCP links the receptor CLR to an intracellular G protein-mediated signaling pathway that increases intracellular cAMP levels by activating adenylate cyclase. These studies confirm that CLR, RAMP1, and RCP are required to form a truly functional α-CGRP receptor.
The binding of α-CGRP to its receptor is a two-step process which has been termed the “two-domain model” (Hoare, 2005). This model suggests that the C-terminus of the α-CGRP initially interacts with the N-terminus of the extracellular domain of CLR. Next, the N-terminus of α-CGRP binds to the transmembrane domain and extracellular loop region of the CLR that triggers receptor activation (Kuwasako et al., 2003).
α-CGRP and its receptor are widely distributed in the central and peripheral nervous system and the cardiovascular system (Mulderry et al., 1985). In the peripheral nervous system, the dorsal root ganglia (DRGs) are the prominent sites of α-CGRP synthesis. The DRG contains the cell bodies of sensory nerves that terminate peripherally on blood vessels and centrally in laminae I/II of the spinal cord. A dense perivascular α-CGRP-containing neural network is seen around the blood vessels in all vascular beds (Uddman et al., 1986). It is thought that circulating α-CGRP is largely released from these perivascular nerve terminals. The presence of α-CGRP receptor has been reported in the endothelium, media and intima of vessels, veins, and in multiple tissues and organs.
Several lines of evidence suggest that binding of α-CGRP to its receptor CLR activates a variety of signaling pathways (Figure 5). In most instances, binding of α-CGRP to the CLR/RAMP1 receptor activates adenylate cyclase through the G-protein Gαs that in turn elevates intracellular cAMP level. Increased levels of CGRP-induced cAMP activate protein kinase A (PKA) and increase various downstream signaling molecules, including the potassium-sensitive ATP channels (K-ATP channels), L-type Ca+2 channels, ERK, and cAMP response element-binding protein (CREB). In human embryonic kindney-296 cells, α-CGRP activates ERK by the PI3K-PKA pathways and p38 through PKA signaling. These downstream signaling pathways regulate CGRP actions, including vasodilation, cardiac contraction, and synaptic plasticity (Nelson et al., 1990; Hong et al., 1996; Anderson and Seybold, 2004). In addition, CGRP signaling is also able to act through other G-proteins such as Gαi/o and Gαq/11. It has been reported that activity of CGRP through Gαq/11 stimulates the IP3-PLC pathway leading to an increase in intracellular Ca+2 concentrations, thereby activating protein kinase C (PKC) (Aiyar et al., 1999). Disa et al. (2000) reported that addition of pertussis toxin, which blocks the actions of Gi/Go-type G proteins, blocks CGRP-mediated activation of JNK (c-Jun N-terminal kinase) in the neuroblastoma cell line SK-N-MC cells (Disa et al., 2000). Another study demonstrated that administration of pertussis toxin abolished CGRP-induced stimulation of Ca+2 currents in rat nodose ganglion neurons (Wiley et al., 1992). These data support the involvement of Gαi/o-signaling mediated by CGRP and CGRP receptors. Finally, the CGRP receptor also has an inhibitor role when it couples to Gαi/o, reducing adenylate cyclase (AC) activity and hence reducing intracellular level of cAMP and decreasing protein kinase A (PKA) activity.
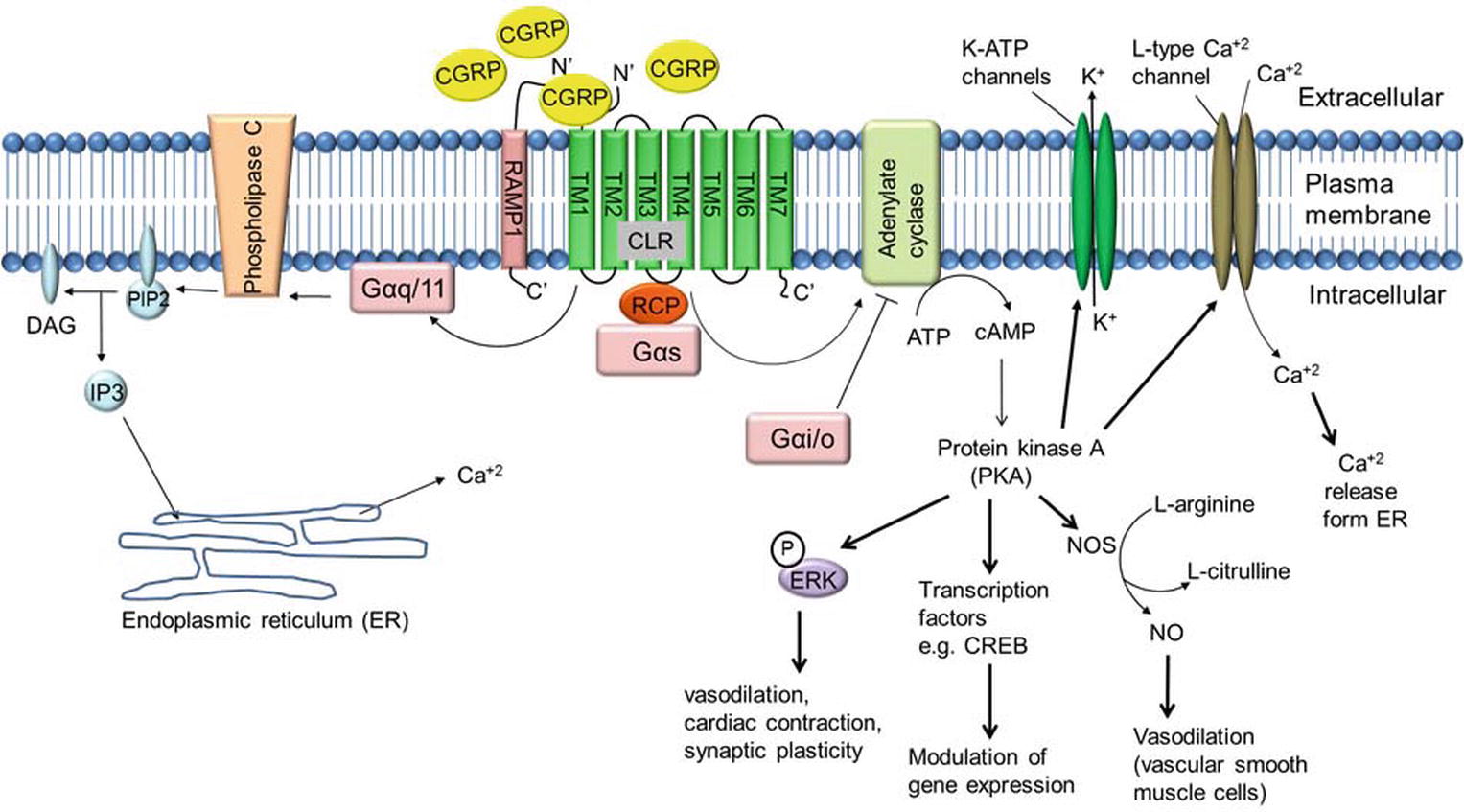
Figure 5. α-CGRP-mediated intracellular signaling. α-CGRP elevates intracellular cAMP level through CLR/RAMP1, G-protein Gαs, and adenylate cyclase (AC) activation. Elevated level of cAMP activates protein kinase A (PKA) and increases various downstream signaling molecules, including the potassium-sensitive ATP channels (K-ATP channels), L-type Ca+2 channel, ERK, and cAMP response element-binding protein (CREB). In addition, α-CGRP signaling is also able to act through other G-proteins such as Gαi/o and Gαq/11. CGRP through Gαq/11 stimulates the IP3-PLC pathway leading to an increase in intracellular Ca+2 concentrations. DAG, diacylglycerol; IP3, inositol triphosphate; PIP2, phosphatidylinositol 4,5-bisphosphate.
α-CGRPs containing nerve fibers are components of the primary afferent nervous system, comprising principally of myelinated (Aδ)- and unmyelinated (C)-fiber nerves that respond to chemical, thermal, and mechanical stimuli. Although these nerves have traditionally been thought to “sense” stimuli in the periphery and transmit the information centrally, it is now well established that they have an efferent function as well. It is clear that DRG neuron-derived peptides are released at peripheral sensory nerve terminals in the absence of any stimulation. The continuous expression and release of peptides from DRG neurons may reflect a paracrine function implying that these neurons participate in the continuous regulation of blood flows and other tissue activities.
Various stimuli have been identified that regulate the expression of α-CGRP in different cell types. Multiple chemical and physiological factors such as nerve growth factor (NGF), angiotensin II (Ang-II), brain-derived neurotrophic factor (BDNF), bradykinins and prostaglandins, endothelin, the renin/angiotensin/aldosterone system, and tissue inflammation are known to increase α-CGRP synthesis (Lindsay, 1988; Supowit et al., 1995b, 2001; Freeland et al., 2000; Salio et al., 2007). Freeland et al. (2000) used a primary culture of adult DRG neurons and showed that NGF treatment phosphorylates the CRBP transcription factor that in turn activates the CGRP promoter, and thus increases CGRP expression, via the Ras/Raf/MEK-1/p42/p44 MAPK pathway (Freeland et al., 2000). Supowit et al. (2001) later demonstrated that an intraperitoneal injection of NGF stimulates synthesis and release of CGRP in DRG sensory nerves in spontaneously hypertensive rats (SHRs) (Supowit et al., 2001). Following synthesis, α-CGRP remains stored in the large vesicles within the sensory nerve terminal that, after neuronal depolarization, is released from the neuronal terminal via calcium-dependent exocytosis. The release of α-CGRP from the sensory nerves is increased by capsaicin, low pH, bradykinins and prostaglandins, Ang-II, and norepinephrine. In contrast, steroids and mediators such as opioids inhibit α-CGRP release (Meng et al., 2007). In human plasma, α-CGRP has very short half-life (t1/2 = ~5 min) (MaassenVanDenBrink et al., 2016), and it has been suggested that the endopeptidases endothelin-converting enzyme-1 (ECE-1) and insulin-degrading enzyme (IDE) are responsible for the degradation of α-CGRP (Kim et al., 2012; Hartopo et al., 2013). Using liquid chromatography-tandem mass spectrometry (LC-MS/MS), peptidomics and genetic approaches have demonstrated that IDE primarily cleaves α-CGRP at the Ser17-Arg18 and Asn26-Phe27 sites. LC-MS/MS analysis of mouse spinal cords detected a total of 10 CGRP fragments with the major fragments consisting of two N-terminal fragments, CGRP1–17 and CGRP1–26, and two C-terminal fragments, CGRP18–37 and CGRP26–37. Similar cleaved peptide fragments have been detected by LC-MS/MS when CGRP1–37 was incubated either with the mouse spinal cord lysate or plasma under in vitro conditions. The identification of these four peptide fragments indicates that full-length CGRP1–37 has two major cleavage sites, Ser17-Arg18 and Asn26-Phe27. In order to identify the CGRP-degrading enzyme, a series of biochemical assays were performed and it was ultimately determined that IDE is the only enzyme that cleaves CGRP1–37 at Ser17-Arg18 and Asn26-Phe27. The observed lower level of CGRP1–37 in IDE+/+ mice, in comparison to IDE−/− mice, further established IDE as an endogenous regulator of α-CGRP (Kim et al., 2012).
α-CGRP is the most potent vasodilator known till date and is ~1,000 times more potent than acetylcholine, substance P, and 5-hydroxytriptamine (Brain et al., 1985). An intradermal injection of 10 pmol α-CGRP raised local blood flow, as assessed by laser Doppler flow meter, and induced an erythema in human skin (Brain et al., 1986). It has positive chronotropic, inotropic, and pro-hypertrophic effects in humans (Franco-Cereceda et al., 1987; Gennari et al., 1990; Bell et al., 1995; Al-Rubaiee et al., 2013). The inotropic activity of α-CGRP is mediated by the cAMP/PKA or PKC signaling pathways. It has been shown that α-CGRP dilates multiple vascular beds, including coronary vasculature (Gulbenkian et al., 1993). In addition, α-CGRP administration decreases blood pressure in normotensive and hypertensive animals and humans (DiPette et al., 1989; Dubois-Rande et al., 1992; Supowit et al., 1993). The α-CGRP-mediated reduction in BP is caused by the dilation of peripheral arteries. The biological activity of α-CGRP, i.e., vasodilation/vasorelaxation, can be mediated via NO- and endothelium-independent or NO- and endothelium-dependent pathways (Figure 6). The vasodilation activity of α-CGRP through NO- and endothelium-independent pathway is observed in the majority of tissues (e.g., rat perfused mesentery, porcine coronary artery, and cat cerebral artery) where binding of α-CGRP to its receptor, CLR, increases intracellular cAMP level through G-protein coupled adenylate cyclase (Edvinsson et al., 1985; Han et al., 1990; Yoshimoto et al., 1998). The increased cAMP level activates protein kinase A (PKA), that in turn phosphorylates and opens K+-ATP channels and thus relaxation of vascular smooth muscle cells (Figure 6A) (Nelson et al., 1990). However, there is scant evidence that vasodilation evoked by α-CGRP is mediated, in part, by nitric oxide (NO) release (Figure 6B; Gray and Marshall, 1992a,b). It has been hypothesized that various vascular beds differ in their dilatory response to α-CGRP due to the endothelium releasing α-CGRP (Bergdahl et al., 1999). The vasorelaxation activity of α-CGRP in the rat aorta, human internal mammary artery, and rat pulmonary artery is reported to be mediated by NO- and endothelium-dependent mechanism (Brain et al., 1985; Gray and Marshall, 1992a; Raddino et al., 1997; Wisskirchen et al., 1998). Addition of the inhibitors of NO synthase (NOS) enzyme attenuates the relaxation action of α-CGRP in the rat aorta (Gray and Marshall, 1992b). Gray and Marshall (1992a) further demonstrated that, in the rat aorta, human α-CGRP caused an increase in the level of cAMP and cGMP only in the presence of an intact endothelium. In the absence of an intact endothelium, α-CGRP produced no significant alterations in either cGMP or cAMP, and no subsequent relaxation of the rat aorta (Gray and Marshall, 1992a). At the cellular level, NO is produced from L-arginine via NOS enzymes – eNOS, iNOS, and nNOS. It has also been shown that cAMP can stimulate eNOS activity, and thus increases synthesis and release of NO (Ferro et al., 1999). Another study further demonstrated that the catalytic subunit of PKA can activate and phosphorylate eNOS (Butt et al., 2000). These lines of evidence suggest that NO- and an intact endothelial-dependent pathway are required to elicit α-CGRP-induced relaxation of smooth muscle cells (Figure 6B). In endothelial cells, α-CGRP works through its receptor (CLR) and Gαs protein, via activation of adenylate cyclase (AC), which enhances intracellular levels of cAMP. Increased cAMP levels activate and phosphorylate eNOS via PKA activation, thus increasing synthesis and release of NO. NO freely diffuses into the surrounding smooth muscle cells and activates guanylyl cyclase. Activated guanylyl cyclase increases the intracellular level of cGMP that in turn activates and phosphorylates the K-ATP channel, and thus relaxes smooth muscle cells (Figure 6B). In addition to ATP-sensitive K+ channel, cGMP has also been reported to mediate its vasodilation/vasorelaxation action through cGMP-dependent protein kinase G (PKG), phosphodiesterases (PDEs), and cGMP-gated ion channels (Francis et al., 2010; Takimoto, 2012; Inserte and Garcia-Dorado, 2015). cGMP-dependent protein kinase G (PKG) phosphorylates a variety of proteins including cardiac troponin I, L-type Ca+2 channel, and titin, and accelerates cGMP-mediated relaxation of cardiac myocytes (Takimoto, 2012). PKG promotes, through phosphorylation/activation, opening of large-conductance calcium-activated potassium channels (BK-K+ channels), and thereby loss of intracellular potassium. These events hyperpolarize the plasma membrane and decrease calcium influx through L-type calcium channels (Zhou et al., 1996). PKG also reduces intracellular Ca+2 concentrations by stimulating Ca+2-ATP-dependent calcium efflux via the plasma membrane Ca+2-ATPase pump, thereby causing vascular smooth muscle cells to relax (Francis et al., 2010). Hence, NO/cGMP/PKG signaling pathway lowers intracellular free calcium and decreases the contractile state of smooth muscle cells. The presence of large-conductance calcium-activated potassium channels (BK-K+ channels) in pial arteries suggests that the NO/cGMP-mediated signaling pathway may be involved in CGRP-induced vasodilation (Hong et al., 1996). It has also been shown that NO-induced elevation of cGMP enhances the vasorelaxant responses by impairing cAMP hydrolysis by phosphodiesterase type 3 (PDE3) (Figure 6B; Stangherlin et al., 2011). Eberhardt et al. (2014) reported that NO activating via a neuroendocrine signaling pathway HNO-TRPA1-CGRP could also induce vasodilation. Studies have demonstrated that, in smooth muscle cells, NO reacts with another gasotransmitter, hydrogen sulfide (H2S), and produces a redox molecule, nitroxyl (HNO). HNO activates the transient receptor potential channel (TRPA1) and, thus, increases calcium influx. As a result, CGRP is released, which in turn induces local and systemic vasodilation (Eberhardt et al., 2014).
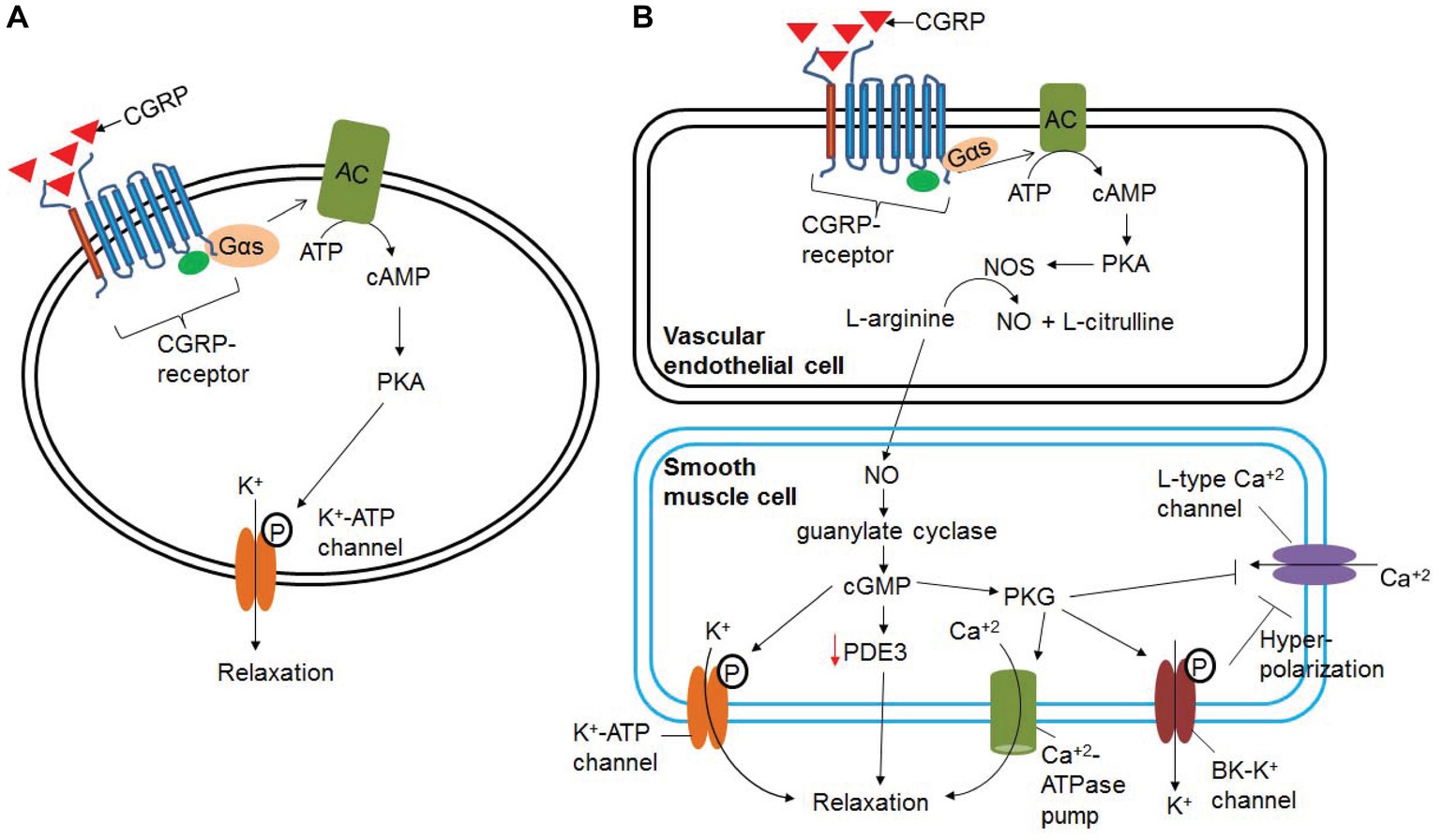
Figure 6. Signaling pathways involved in α-CGRP-induced vasodilation. (A) In NO- and endothelium-independent pathway, binding of α-CGRP to its receptor, CLR, increases intracellular cAMP level through G-protein coupled adenylate cyclase (AC). The increased cAMP level activates protein kinase A (PKA), and that in turn phosphorylates and opens K+-ATP channels and thus relaxation of vascular smooth muscle cells. (B) NO- and endothelium-dependent vasodilatation to α-CGRP. α-CGRP through its receptors activates eNOS enzyme via Gαs-adenylate cyclase-cAMP-PKA pathway, and thus increases the synthesis of NO in the endothelial cells. From the endothelial cells NO freely diffuses to the smooth muscle cells activating guanylate cyclase and increasing intracellular cGMP level. These events activate and phosphorylate K-ATP channel that leads to relaxation of smooth muscle cells. cGMP has also been reported to mediate its vasodilation action through cGMP-dependent protein kinase G (PKG), phosphodiesterases (PDEs), and cGMP-gated ion channels. NO/cGMP/PKG signaling pathway efflux intracellular potassium through BK-K channel, promotes hyperpolarization of the plasma membrane, and thereby decreases influx of calcium through L-type Ca+2 channels. PKG also can efflux intracellular calcium through the plasma membrane Ca+2-ATPase pump. This, in turn, decreases the contractile state of smooth muscle cells.
Several lines of evidence suggest that α-CGRP protects the heart from damage due to hypertension, myocardial infarction, and heart failure (Li et al., 2013), and cardiac and cerebral ischemia (Schebesch et al., 2013). Both In vitro and in vivo studies revealed that α-CGRP protects cardiomyocytes from apoptotic cell death by activation of the PI3K-Akt and Erk 1/2 kinase pathways which in turn activate cell survival pathways (Schaeffer et al., 2003; Sueur et al., 2005). α-CGRP has also been shown to regulate cardiomyocyte survival by inhibiting oxidative stress by the PI3K-AKT and MAPK signaling pathways (Umoh et al., 2014). α-CGRP was demonstrated to protect H9c2 cardiac cells from hypoxia-induced inflammation and apoptosis by modulating NO production (Duan et al., 2016). In these hypoxia-induced H9c2 cells, α-CGRP treatment significantly decreased the levels of pro-inflammatory molecules (IL-6 and TNF-α) as well as apoptotic proteins (caspase-3, and cytochrome-C), and enhanced the expression of basal and phosphorylated eNOS. The role of α-CGRP in various animal models of cardiovascular diseases has been summarized in Supplementary Table S1.
After synthesis, α-CGRP remains in large dense vesicles within the sensory nerves that are located in the perivascular layer of coronary blood vessels and between myofibrillar bundles throughout the myocardium and prominently innervate the heart. This distribution makes them perfectly situated to sense and respond to changes in blood pressure, ischemia, and cytotoxic stress in the heart. Therefore, the efferent function of sensory nerves, exerted via CGRP, could provide a protective function against the myocardial remodeling and dysfunction present in response to hypertension and heart failure.
Studies from our lab have shown the cardioprotective activity of α-CGRP against heart failure (Li et al., 2010, 2013). Elevated plasma levels of α-CGRP and CGRP receptor have been observed in individuals with heart failure, and an acute intravenous infusion of α-CGRP significantly improves cardiac functional parameters in patients with congestive heart failure (Gennari et al., 1990). In humans, circulating levels of α-CGRP are elevated during the initial and middle stages of heart failure but then significantly decline as heart failure progresses (Dubois-Rande et al., 1992). Studies carried out in a non-ischemic rat model of chronic heart failure (CHF) demonstrated that pressure-overload heart failure, induced by ascending aortic banding, increases the expressions of RAMP1 mRNA and protein in both atria and ventricles (Cueille et al., 2002). Furthermore, α-CGRP protects against cardiac hypertrophy in an isoprenaline-induced model of heart failure (Li et al., 2010). Gangula et al. (2000) reported markedly elevated basal systolic blood pressure and the mean arterial pressure (MAP) in α-CGRP KO mice when compared to WT mice (Gangula et al., 2000). Subsequently, Li et al. (2013) performed a series of experiments in α-CGRP KO mice and validated the cardioprotective role of α-CGRP in heart failure (Li et al., 2013). They observed that α-CGRP KO mice that underwent pressure-overload heart failure, induced by transverse aortic constriction (TAC), had a lower survival rate compared to their TAC-WT counterpart. The survival rate of the TAC-WT animals (28 days) was approximately 95%. In comparison, the survival rate of the TAC α-CGRP KO mice between days 0 and 21 was approximately 80% and decreased dramatically from days 21 to 28 to approximately 50%. Echocardiography data demonstrated that TAC α-CGRP KO mice exhibit increased adverse cardiac remodeling and dysfunction compared to the TAC-WT mice counterparts. TAC procedure further lowers fractional shortening and ejection fraction in α-CGRP KO mice compared to TAC-WT mice. The ratios of heart weight to body weight and lung weight to body weight were higher in TAC α-CGRP KO mice compared to the TAC-WT mice. Moreover, TAC α-CGRP KO mice had an increased left ventricular hypertrophy and displayed greater cardiac fibrosis, apoptosis and necrosis, inflammation, and attenuation of angiogenesis compared to TAC-WT hearts. Together, these studies demonstrated that absence of the α-CGRP gene heightened the adverse cardiac remodeling, dysfunction, and mortality in pressure-overload-induced heart failure.
Following acute myocardial infraction, increased immunoreactive α-CGRP levels have been reported in human plasma and nerves (Mair et al., 1990; Roudenok et al., 2001). Myocardial ischemia causes a marked increase in the release of α-CGRP in isolated hearts from guinea pig and rat (Franco-Cereceda, 1988). During cardiac ischemia/reperfusion (I/R) injury, the C-fiber and Aδ-fiber sensory nerves play a possible efferent cardioprotective role that is mediated through the release of α-CGRP (Wang and Wang, 2005). The release of α-CGRP is induced by the activation of the TRPV1 receptor by the stimulation by protons, bradykinin, and prostaglandins (Strecker et al., 2006). A recent study in TRPV1−/− mice showed that transaortic constriction (TAC) surgery enhances the cardiac hypertrophy and cardiac dysfunction in TRPV1-KO mice compared to their wild-type TAC mice. TAC TRPV1−/− mice had developed left ventricular (LV) hypertrophy with increased LV mass, fibrosis, and increased secretion of TNF-α, and interleukin-6. TAC procedure significantly increased α-CGRP release in wild-type mice, but not in TRPV1−/− mice, suggesting that the deteriorated cardiac function after TAC may be due to the decreased CGRP level in absence of TRPV1 (Zhong et al., 2018). Several independent studies demonstrated that α-CGRP significantly reversed myocardial I/R injury and played a role in cardiac preconditioning and remote preconditioning (Wolfrum et al., 2005). α-CGRP was also shown to attenuate I/R injury-induced cardiac mitochondrial dysfunction through inhibition of GSK 3β via PI3k-Akt and ERK 1/2 kinase cascades (Schaeffer et al., 2003; Hausenloy and Yellon, 2004).
Mair et al. (1990) reported that intracoronary infusion of α-CGRP delayed the onset of myocardial ischemia in patients with stable angina pectoris (Mair et al., 1990). In addition, exogenous administration of α-CGRP mimics the cardioprotective effect of preconditioning induced by transient ischemia in a Langendorff-perfused rat heart resulting in an increased release of endogenous α-CGRP. However, administration of BIBN4096BS, a potent α-CGRP receptor antagonist, completely abolished the cardioprotection induced by preconditioning and by exogenous α-CGRP (Wu et al., 2001). Huang et al. (2008) used α-CGRP KO mice to study role of α-CGRP in I/R injury by subjecting isolated-perfused hearts from α-CGRP KO and wild-type mice to an I/R protocol by performing 30 min of ischemia followed by reperfusion (Huang et al., 2008). Following I/R injury, hearts from the α-CGRP KO mice exhibited a significant reduction in cardiac performance compared to their WT counterparts. These α-CGRP KO mice with I/R injury showed an elevated level of creatine kinase (a marker for cell death) and malondialdehyde (a marker for oxidative stress-induced lipid peroxidation) in the heart. Therefore, these studies demonstrate that deletion of α-CGRP makes the heart more vulnerable to I/R injury likely due to increased oxidative stress and inflammation. In summary, these studies suggest an important role for α-CGRP in ischemic preconditioning.
In the hypertensive patients with primary aldosteronism or on a high-salt diet, an increased level of circulating CGRP has been detected (Resnick et al., 1989). Several studies from our laboratory and others have established a direct role for α-CGRP in experimental hypertension (Marquez-Rodas et al., 2006). Our laboratory has demonstrated that α-CGRP acts as a compensatory depressor to attenuate the rise in blood pressure in three different models of experimental hypertension: (1) deoxycorticosterone (DOC)-salt (Supowit et al., 1997), (2) subtotal nephrectomy-salt (Supowit et al., 1998), and (3) L-NAME-induced hypertension during pregnancy (Gangula et al., 1997). A similar role for CGRP has also been shown in a two-kidney one-clip model of hypertension (Supowit et al., 1998), and in chronic hypoxic pulmonary hypertension (Tjen et al., 1992; Bivalacqua et al., 2002). The expression of α-CGRP and α-CGRP receptor is increased in several models of hypertension including an Ang-II model (Li and Wang, 2005).
The level of CGRP in neuronal cells is differentially regulated in several models of hypertension. In the DOC-salt-induced hypertensive rat, elevated levels of immunoreactive CGRP in the spinal cord and α-CGRP mRNA in the DRG were observed (Supowit et al., 1995a). In comparison, decreased levels of immunoreactive CGRP were observed in laminae I and II of the spinal cord, and CGRP mRNA was observed in the DRG in the spontaneously hypertensive rats (SHRs) (Supowit et al., 1993). These results suggest that the lower level of α-CGRP expression in the SHR might contribute to the high blood pressure observed by the relative reduction of vasodilator activity, whereas higher α-CGRP levels in the DOC-salt hypertension animal attenuate the elevated blood pressure by the compensatory vasodilator activity. The latter notion is supported by the fact that in DOC-salt hypertensive rats, the baseline mean arterial pressure was higher than in normotensive rats (175 ± 5 vs. 119 ± 4 mm Hg, p < 0.001). This coupled with the fact that intravenous administration of an α-CGRP receptor antagonist (CGRP8–37) to DOC-salt rats significantly further increased the mean arterial pressure, (Supowit et al., 1997). Similarly, infusion of the antagonist α-CGRP8–37 increased the elevated blood pressure in two other models of experimental hypertension: subtotal nephrectomy (SN)-induced hypertensive rats (Supowit et al., 1998) and N-nitro-L-arginine methyl ester (L-name)-induced hypertension during pregnancy (Gangula et al., 1997). The anti-hypertensive activity of α-CGRP was further confirmed using α-CGRP KO mice (Gangula et al., 2000; Bowers et al., 2005; Supowit et al., 2005). Gangula et al. (2000) reported that basal blood pressure and mean arterial pressure (MAP) were significantly higher in α-CGRP KO mice compared to wild-type mice of a similar strain (Gangula et al., 2000). Another study using a DOC-salt model using α-CGRP KO and WT mice reported that both heart and kidney sections from the α-CGRP KO mice displayed significantly greater cellular damage and had increased levels of pro-inflammatory cytokines and chemokines such as intercellular adhesion molecule-1, vascular adhesion molecule-1, and monocyte chemoattractant protein-1 (Supowit et al., 2005). Compared to the DOC-salt WT mice, oxidative stress measured by urinary microalbumin and isoprostane accumulation were significantly increased in the hypertensive DOC-salt α-CGRP KO mice. These results demonstrated that deletion of α-CGRP makes the heart and kidneys more susceptible to hypertension-induced end organ damage possibly through enhanced oxidative stress and inflammation. These studies further confirm that CGRP found in sensory nerves protects against hypertension-induced heart and kidney damage.
Another study using a conditional deletion of RAMP1 (RAMP1−/−) found that RAMP1-KO mice are hypertensive due to the disruption of α-CGRP signaling (Tsujikawa et al., 2007). Administration of α-CGRP caused a potent relaxation of the arteries in WT mice, but failed to do so in RAMP1-KO mice. Furthermore, Ang-II is a vasoconstrictor and is known to induce hypertension in human (Laragh et al., 1972) and rodents; however, transgenic expression of human RAMP1 blunts the Ang-II-induced adverse effects of hypertension in mice (Sabharwal et al., 2010). It is well known that the renin-angiotensin-aldosterone system (RAAS) is involved in the development of hypertension (Deng and Li, 2005). In α-CGRP KO mice, an increase in the activity of the circulating RAAS is observed, which may be the reason for the higher blood pressure in these mice (Li et al., 2004). It has been shown that addition of a sub-depressor dose of α-CGRP significantly reduced the blood pressure in hypertensive rats, induced by Ang-II or norepinephrine (Fujioka et al., 1991). α-CGRP is also known to inhibit the secretion of aldosterone, induced by Ang-II (Murakami et al., 1991). In spontaneously hypertensive rats (SHRs), aldosterone decreases the vasoconstriction response to electric field stimulation in mesenteric arteries by increasing the vasodilatory response to CGRP (Balfagon et al., 2004). These studies confirm that α-CGRP plays a role in maintaining the blood pressure via interaction with the renin-angiotensin-aldosterone system. A recent study carried out in vascular smooth muscle cells demonstrated that a disintegrin and metalloproteinase 17 (ADAM17) is involved in the protective effect of α-CGRP against Ang-II-induced inflammation via the EGFR-ERK1/2 pathway (Zeng et al., 2018). Moreover, in Ang-II-induced hypertensive mice, increased levels of oxidative stress, apoptosis, and inflammation were observed in the heart and kidney (Aubdool et al., 2017). In these mice, Ang-II upregulates the expression of oxidative stress response proteins, e.g., NADPH detablehydrogenase quinone-1 (NQO1), hypoxia-inducible factor 1α (HIF-α), NADPH oxidase-2 (NOX-2), and heme oxygenase-1 (HO-1), and downregulates the expression of anti-oxidative enzyme glutathione peroxidase-1 (GPX-1) (Aubdool et al., 2017). Ang-II also significantly decreases eNOS expression that in turn causes endothelial dysfunction and vascular hypertrophy (Cai and Harrison, 2000; Aubdool et al., 2017). Finally, Ang-II significantly increases the protein expression of the CGRP receptor RAMP1 in the aorta, mesentery, and heart. These Ang-II adverse effects worsen the cardiac function creating a pathophysiological condition. However, subcutaneous administration of an α-CGRP agonist analog, acylated α-CGRP, significantly attenuated Ang-II-induced adverse effects in heart, kidney, and vessels. The acylated α-CGRP reduced cardiac hypertrophy and remodeling, fibrosis, inflammation and oxidative stress in Ang-II-induced hypertensive mice (Aubdool et al., 2017). These observations from our lab and others provide direct evidence for the involvement of α-CGRP and its receptor in the regulation of blood pressure and their protective functions in hypertension.
The beneficial protective effects of α-CGRP in various cardiovascular diseases have driven research to develop α-CGRP as a therapeutic agent to treat cardiac diseases. Previous studies showed that administration of α-CGRP to normotensive patients decreased blood pressure in a dose-dependent manner (Franco-Cereceda et al., 1987). In addition, intravenous infusion of α-CGRP for 24 h improved myocardial contractility in patients suffering from congestive heart failure (Gennari et al., 1990). In a similar study, infusion of α-CGRP in patients with heart failure decreased systemic arterial pressure in a dose-dependent manner (Dubois-Rande et al., 1992). These human studies confirm the idea that α-CGRP is a promising therapeutic agent to treat patients suffering from cardiovascular diseases. However, the short half-life of α-CGRP (t1/2 = 5 min) in human plasma makes it difficult to use in any long-term treatment regimes. Therefore, novel strategies are needed for α-CGRP delivery and synthesis of innovative α-CGRP agonist analogs. These approaches will be helpful to extend the bioavailability of α-CGRP for longer duration of time in plasma. In recent years, the development of α-CGRP agonist with a longer half-life and bioavailability has been developed (Nilsson et al., 2016; Aubdool et al., 2017; Sheykhzade et al., 2018). Aubdool et al. (2017) tested an acylated form of α-CGRP (developed by Novo Nordisk) in rodent models of hypertension and heart failure. They demonstrated that a daily systemic subcutaneous administration of the α-CGRP analog (50 nmol/kg per mouse) reversed the renal, vascular, and cardiac damage caused by Ang-II-induced hypertension or by abdominal aortic constriction (AAC)-induced heart failure. The acylated αCGRP lowered blood pressure, and reduced cardiac fibrosis, oxidative stress, and cardiac hypertrophy in these mice with hypertension and heart failure (Aubdool et al., 2017). This α analog had an extended half-life in serum (~7 h) and exhibited similar pharmacological properties to native αCGRP in isolated human and rodent vasculature. Similar to native α-CGRP, the acylated αCGRP-induced vasodilation was reversed with similar potencies by the addition of a CGRP receptor antagonist BIBN4096BS (Sheykhzade et al., 2018). Novo Nordisk has also developed another α-CGRP agonist, Serinyl-α-CGRP (2–37)-amide, consisting of an albumin-binding fatty acid moiety in the N-terminus. They demonstrated that serinyl-α-CGRP administration for 2 weeks reduced fasting blood glucose levels and fasting insulin levels in type2 diabetic ob/ob mice. These data suggest that serinyl-α-CGRP may have a therapeutic potential for the treatment of type 2 diabetes (Nilsson et al., 2016). Further research into other modifications is ongoing to develop α-CGRP agonist analogs and warranted given the immense therapeutic potential of αCGRP in a variety of human diseases.
It should also be noted that patients experiencing migraine headaches show an increase in serum level of α-CGRP, and that the increase was observed for both episodic and chronic migraine (Russo, 2015). Clinical trials carried out during the past decade have proved that CGRP receptor antagonists are effective for treating migraine, and antibodies to the receptor and CGRP are currently under investigation. Several antagonists of α-CGRP are currently being developed and tested in order to relieve migraine headache in patients (Maasumi et al., 2018). These antagonists either interact with the α-CGRP receptor or bind to α-CGRP directly to inhibit the interaction between α-CGRP and α-CGRP receptor, thus abolishing the biological activities of α-CGRP. Recently, the Food and Drug Administration (FDA) approved three new monoclonal antibodies that target CGRP or its receptor for migraine prevention. These antibodies are: erenumab, fremanezumab, and galcanezumab. The antibody erenumab (commercial name – Aimovig, developed by Amgen and Novartis) selectively targets the CGRP receptor, and functions as a competitive, reversible inhibitor (Goadsby et al., 2017). It is given as a once monthly treatment to patients to prevent migraine. The antibody fremanezumab (TEV-48125) is a fully humanized IgG2 antibody, and binds to both α and β forms of CGRP ligand and thus blocks peptide binding to its receptor (Bigal et al., 2014; Walter and Bigal, 2015; Silberstein et al., 2017). The antibody is developed by Teva Pharmaceuticals and is marketed under the trade name of Ajovy. Finally, the humanized monoclonal antibody galcanezumab (Emgality) also targets the CGRP peptide and has been developed by Eli Lilly & Co. as a once monthly treatment that helps prevent future migraines (Forderreuther et al., 2018). While these humanized antibodies are showing promising results against migraine, it is important to mention here that while developing new CGRP or its receptor-targeted drugs, it is essential to consider the cardiovascular risks associated with CGRP signaling blockade (MaassenVanDenBrink et al., 2016).
There is overwhelming evidence to support the theory that α-CGRP benefits the heart by decreasing Ang-II activity, increasing cardiac blood flow via its potent vasodilator activity, and protecting cardiomyocytes from ischemia and metabolic stress. Attempts to generate α-CGRP agonists with extended bioavailability and half-life are showing promising results that significantly reverse the adverse effects of hypertension and heart failure in animal models. Thus, α-CGRP, either in its native or a modified form, may have therapeutic benefits to patients suffering from cardiac diseases in near future.
AK wrote the manuscript. JP and DD reviewed, edited, and approved the final version of the manuscript.
This work was supported by the Distinguished Health Sciences Professorship of the University of South Carolina (DD) and by the National Science Foundation EPSCoR Program under NSF Award # OIA-1655740 (JP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.00821/full#supplementary-material
AC, Adenylate cyclase; Ang-II, Angiotensin II; BP, Blood pressure; CGRP, Calcitonin gene-related peptide; CLR, Calcitonin receptor-like receptor; DOC, Deoxycorticosterone; DRG, Dorsal root ganglia; I/R injury, Ischemia/reperfusion injury; KO, Knock-out; L-NAME, N-nitro-L-arginine methyl ester; LV, Left ventricle; NO, Nitric oxide; PKA, Protein kinase A; RAMP, Receptor activity modifying protein; RCP, Receptor component protein; SN-salt, Subtotal nephrectomy-salt; TAC, Transverse aortic constriction; WT, Wild-type.
Aiyar, N., Disa, J., Stadel, J. M., and Lysko, P. G. (1999). Calcitonin gene-related peptide receptor independently stimulates 3′,5′-cyclic adenosine monophosphate and Ca2+ signaling pathways. Mol. Cell. Biochem. 197, 179–185. doi: 10.1023/A:1006962221332
Aiyar, N., Rand, K., Elshourbagy, N. A., Zeng, Z., Adamou, J. E., Bergsma, D. J., et al. (1996). A cDNA encoding the calcitonin gene-related peptide type 1 receptor. J. Biol. Chem. 271, 11325–11329. doi: 10.1074/jbc.271.19.11325
Alevizaki, M., Shiraishi, A., Rassool, F. V., Ferrier, G. J., MacIntyre, I., and Legon, S. (1986). The calcitonin-like sequence of the beta CGRP gene. FEBS Lett. 206, 47–52. doi: 10.1016/0014-5793(86)81338-2
Al-Rubaiee, M., Gangula, P. R., Millis, R. M., Walker, R. K., Umoh, N. A., Cousins, V. M., et al. (2013). Inotropic and lusitropic effects of calcitonin gene-related peptide in the heart. Am. J. Physiol. Heart Circ. Physiol. 304, H1525–H1537. doi: 10.1152/ajpheart.00874.2012
Amara, S. G., Jonas, V., Rosenfeld, M. G., Ong, E. S., and Evans, R. M. (1982). Alternative RNA processing in calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature 298, 240–244. doi: 10.1038/298240a0
Anderson, L. E., and Seybold, V. S. (2004). Calcitonin gene-related peptide regulates gene transcription in primary afferent neurons. J. Neurochem. 91, 1417–1429. doi: 10.1111/j.1471-4159.2004.02833.x
Aubdool, A. A., Thakore, P., Argunhan, F., Smillie, S. J., Schnelle, M., Srivastava, S., et al. (2017). A novel alpha-calcitonin gene-related peptide analogue protects against end-organ damage in experimental hypertension, cardiac hypertrophy, and heart failure. Circulation 136, 367–383. doi: 10.1161/CIRCULATIONAHA.117.028388
Balfagon, G., Marquez-Rodas, I., Alvarez, Y., Alonso, M. J., Cachofeiro, V., Salaices, M., et al. (2004). Aldosterone modulates neural vasomotor response in hypertension: role of calcitonin gene-related peptide. Regul. Pept. 120, 253–260. doi: 10.1016/j.regpep.2004.03.016
Barwell, J., Gingell, J. J., Watkins, H. A., Archbold, J. K., Poyner, D. R., and Hay, D. L. (2012). Calcitonin and calcitonin receptor-like receptors: common themes with family B GPCRs? Br. J. Pharmacol. 166, 51–65. doi: 10.1111/j.1476-5381.2011.01525.x
Bell, D., Schluter, K. D., Zhou, X. J., McDermott, B. J., and Piper, H. M. (1995). Hypertrophic effects of calcitonin gene-related peptide (CGRP) and amylin on adult mammalian ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 27, 2433–2443. doi: 10.1006/jmcc.1995.0231
Bergdahl, A., Valdemarsson, S., Nilsson, T., Sun, X. Y., Hedner, T., and Edvinsson, L. (1999). Dilatory responses to acetylcholine, calcitonin gene-related peptide and substance P in the congestive heart failure rat. Acta Physiol. Scand. 165, 15–23. doi: 10.1046/j.1365-201x.1999.00456.x
Bigal, M. E., Escandon, R., Bronson, M., Walter, S., Sudworth, M., Huggins, J. P., et al. (2014). Safety and tolerability of LBR-101, a humanized monoclonal antibody that blocks the binding of CGRP to its receptor: results of the phase 1 program. Cephalalgia 34, 483–492. doi: 10.1177/0333102413517775
Bivalacqua, T. J., Hyman, A. L., Kadowitz, P. J., Paolocci, N., Kass, D. A., and Champion, H. C. (2002). Role of calcitonin gene-related peptide (CGRP) in chronic hypoxia-induced pulmonary hypertension in the mouse. Influence of gene transfer in vivo. Regul. Pept. 108, 129–133. doi: 10.1016/S0167-0115(02)00100-3
Bowers, M. C., Katki, K. A., Rao, A., Koehler, M., Patel, P., Spiekerman, A., et al. (2005). Role of calcitonin gene-related peptide in hypertension-induced renal damage. Hypertension 46, 51–57. doi: 10.1161/01.HYP.0000168926.44648.ed
Brain, S. D., Tippins, J. R., Morris, H. R., MacIntyre, I., and Williams, T. J. (1986). Potent vasodilator activity of calcitonin gene-related peptide in human skin. J. Invest. Dermatol. 87, 533–536. doi: 10.1111/1523-1747.ep12455620
Brain, S. D., Williams, T. J., Tippins, J. R., Morris, H. R., and MacIntyre, I. (1985). Calcitonin gene-related peptide is a potent vasodilator. Nature 313, 54–56. doi: 10.1038/313054a0
Breeze, A. L., Harvey, T. S., Bazzo, R., and Campbell, I. D. (1991). Solution structure of human calcitonin gene-related peptide by 1H NMR and distance geometry with restrained molecular dynamics. Biochemistry 30, 575–582. doi: 10.1021/bi00216a036
Breimer, L. H., MacIntyre, I., and Zaidi, M. (1988). Peptides from the calcitonin genes: molecular genetics, structure and function. Biochem. J. 255, 377–390. doi: 10.1042/bj2550377
Butt, E., Bernhardt, M., Smolenski, A., Kotsonis, P., Frohlich, L. G., Sickmann, A., et al. (2000). Endothelial nitric-oxide synthase (type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J. Biol. Chem. 275, 5179–5187. doi: 10.1074/jbc.275.7.5179
Cai, H., and Harrison, D. G. (2000). Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ. Res. 87, 840–844. doi: 10.1161/01.RES.87.10.840
Carpenter, K. A., Schmidt, R., von Mentzer, B., Haglund, U., Roberts, E., and Walpole, C. (2001). Turn structures in CGRP C-terminal analogues promote stable arrangements of key residue side chains. Biochemistry 40, 8317–8325. doi: 10.1021/bi0102860
Chiba, T., Yamaguchi, A., Yamatani, T., Nakamura, A., Morishita, T., Inui, T., et al. (1989). Calcitonin gene-related peptide receptor antagonist human CGRP-(8-37). Am. J. Phys. 256, E331–E335. doi: 10.1152/ajpendo.1989.256.2.E331
Conner, A. C., Hay, D. L., Howitt, S. G., Kilk, K., Langel, U., Wheatley, M., et al. (2002). Interaction of calcitonin-gene-related peptide with its receptors. Biochem. Soc. Trans. 30, 451–455. doi: 10.1042/bst0300451
Cueille, C., Pidoux, E., de Vernejoul, M. C., Ventura-Clapier, R., and Garel, J. M. (2002). Increased myocardial expression of RAMP1 and RAMP3 in rats with chronic heart failure. Biochem. Biophys. Res. Commun. 294, 340–346. doi: 10.1016/S0006-291X(02)00487-4
Deng, P. Y., and Li, Y. J. (2005). Calcitonin gene-related peptide and hypertension. Peptides 26, 1676–1685. doi: 10.1016/j.peptides.2005.02.002
DiPette, D. J., Schwarzenberger, K., Kerr, N., and Holland, O. B. (1989). Dose-dependent systemic and regional hemodynamic effects of calcitonin gene-related peptide. Am J Med Sci 297, 65–70. doi: 10.1097/00000441-198902000-00001
Disa, J., Parameswaran, N., Nambi, P., and Aiyar, N. (2000). Involvement of cAMP-dependent protein kinase and pertussis toxin-sensitive G-proteins in CGRP mediated JNK activation in human neuroblastoma cell line. Neuropeptides 34, 229–233. doi: 10.1054/npep.2000.0810
Duan, L., Lei, H., Zhang, Y., Wan, B., Chang, J., Feng, Q., et al. (2016). Calcitonin gene-related peptide improves hypoxia-induced inflammation and apoptosis via nitric oxide in H9c2 cardiomyoblast cells. Cardiology 133, 44–53. doi: 10.1159/000439123
Dubois-Rande, J. L., Merlet, P., Benvenuti, C., Sediame, S., Macquin-Mavier, I., Chabrier, E., et al. (1992). Effects of calcitonin gene-related peptide on cardiac contractility, coronary hemodynamics and myocardial energetics in idiopathic dilated cardiomyopathy. Am. J. Cardiol. 70, 906–912. doi: 10.1016/0002-9149(92)90736-I
Eberhardt, M., Dux, M., Namer, B., Miljkovic, J., Cordasic, N., Will, C., et al. (2014). H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat. Commun. 5:4381. doi: 10.1038/ncomms5381
Edvinsson, L., Fredholm, B. B., Hamel, E., Jansen, I., and Verrecchia, C. (1985). Perivascular peptides relax cerebral arteries concomitant with stimulation of cyclic adenosine monophosphate accumulation or release of an endothelium-derived relaxing factor in the cat. Neurosci. Lett. 58, 213–217. doi: 10.1016/0304-3940(85)90166-1
Egea, S. C., and Dickerson, I. M. (2012). Direct interactions between calcitonin-like receptor (CLR) and CGRP-receptor component protein (RCP) regulate CGRP receptor signaling. Endocrinology 153, 1850–1860. doi: 10.1210/en.2011-1459
Evans, B. N., Rosenblatt, M. I., Mnayer, L. O., Oliver, K. R., and Dickerson, I. M. (2000). CGRP-RCP, a novel protein required for signal transduction at calcitonin gene-related peptide and adrenomedullin receptors. J. Biol. Chem. 275, 31438–31443. doi: 10.1074/jbc.M005604200
Ferro, A., Queen, L. R., Priest, R. M., Xu, B., Ritter, J. M., Poston, L., et al. (1999). Activation of nitric oxide synthase by beta 2-adrenoceptors in human umbilical vein endothelium in vitro. Br. J. Pharmacol. 126, 1872–1880. doi: 10.1038/sj.bjp.0702512
Forderreuther, S., Zhang, Q., Stauffer, V. L., Aurora, S. K., and Lainez, M. J. A. (2018). Preventive effects of galcanezumab in adult patients with episodic or chronic migraine are persistent: data from the phase 3, randomized, double-blind, placebo-controlled EVOLVE-1, EVOLVE-2, and REGAIN studies. J. Headache Pain 19:121. doi: 10.1186/s10194-018-0951-2
Francis, S. H., Busch, J. L., Corbin, J. D., and Sibley, D. (2010). cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 62, 525–563. doi: 10.1124/pr.110.002907
Franco-Cereceda, A. (1988). Calcitonin gene-related peptide and tachykinins in relation to local sensory control of cardiac contractility and coronary vascular tone. Acta Physiol. Scand. Suppl. 569, 1–63.
Franco-Cereceda, A., Gennari, C., Nami, R., Agnusdei, D., Pernow, J., Lundberg, J. M., et al. (1987). Cardiovascular effects of calcitonin gene-related peptides I and II in man. Circ. Res. 60, 393–397. doi: 10.1161/01.RES.60.3.393
Freeland, K., Liu, Y. Z., and Latchman, D. S. (2000). Distinct signalling pathways mediate the cAMP response element (CRE)-dependent activation of the calcitonin gene-related peptide gene promoter by cAMP and nerve growth factor. Biochem. J. 345, 233–238. doi: 10.1042/bj3450233
Fujioka, S., Sasakawa, O., Kishimoto, H., Tsumura, K., and Morii, H. (1991). The antihypertensive effect of calcitonin gene-related peptide in rats with norepinephrine- and angiotensin II-induced hypertension. J. Hypertens. 9, 175–179. doi: 10.1097/00004872-199102000-00013
Gangula, P. R., Supowit, S. C., Wimalawansa, S. J., Zhao, H., Hallman, D. M., DiPette, D. J., et al. (1997). Calcitonin gene-related peptide is a depressor in NG-nitro-L-arginine methyl ester-induced hypertension during pregnancy. Hypertension 29, 248–253. doi: 10.1161/01.HYP.29.1.248
Gangula, P. R., Zhao, H., Supowit, S. C., Wimalawansa, S. J., Dipette, D. J., Westlund, K. N., et al. (2000). Increased blood pressure in alpha-calcitonin gene-related peptide/calcitonin gene knockout mice. Hypertension 35, 470–475. doi: 10.1161/01.HYP.35.1.470
Gennari, C., Nami, R., Agnusdei, D., and Fischer, J. A. (1990). Improved cardiac performance with human calcitonin gene related peptide in patients with congestive heart failure. Cardiovasc. Res. 24, 239–241. doi: 10.1093/cvr/24.3.239
Goadsby, P. J., Reuter, U., Hallstrom, Y., Broessner, G., Bonner, J. H., Zhang, F., et al. (2017). A controlled trial of erenumab for episodic migraine. N. Engl. J. Med. 377, 2123–2132. doi: 10.1056/NEJMoa1705848
Gray, D. W., and Marshall, I. (1992a). Human alpha-calcitonin gene-related peptide stimulates adenylate cyclase and guanylate cyclase and relaxes rat thoracic aorta by releasing nitric oxide. Br. J. Pharmacol. 107, 691–696. doi: 10.1111/j.1476-5381.1992.tb14508.x
Gray, D. W., and Marshall, I. (1992b). Nitric oxide synthesis inhibitors attenuate calcitonin gene-related peptide endothelium-dependent vasorelaxation in rat aorta. Eur. J. Pharmacol. 212, 37–42. doi: 10.1016/0014-2999(92)90069-G
Gulbenkian, S., Saetrum Opgaard, O., Ekman, R., Costa Andrade, N., Wharton, J., Polak, J. M., et al. (1993). Peptidergic innervation of human epicardial coronary arteries. Circ. Res. 73, 579–588. doi: 10.1161/01.RES.73.3.579
Han, S. P., Naes, L., and Westfall, T. C. (1990). Calcitonin gene-related peptide is the endogenous mediator of nonadrenergic-noncholinergic vasodilation in rat mesentery. J. Pharmacol. Exp. Ther. 255, 423–428.
Hartopo, A. B., Emoto, N., Vignon-Zellweger, N., Suzuki, Y., Yagi, K., Nakayama, K., et al. (2013). Endothelin-converting enzyme-1 gene ablation attenuates pulmonary fibrosis via CGRP-cAMP/EPAC1 pathway. Am. J. Respir. Cell Mol. Biol. 48, 465–476. doi: 10.1165/rcmb.2012-0354OC
Hausenloy, D. J., and Yellon, D. M. (2004). New directions for protecting the heart against ischaemia-reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)-pathway. Cardiovasc. Res. 61, 448–460. doi: 10.1016/j.cardiores.2003.09.024
Hilairet, S., Foord, S. M., Marshall, F. H., and Bouvier, M. (2001). Protein-protein interaction and not glycosylation determines the binding selectivity of heterodimers between the calcitonin receptor-like receptor and the receptor activity-modifying proteins. J. Biol. Chem. 276, 29575–29581. doi: 10.1074/jbc.M102722200
Hoare, S. R. (2005). Mechanisms of peptide and nonpeptide ligand binding to class B G-protein-coupled receptors. Drug Discov. Today 10, 417–427. doi: 10.1016/S1359-6446(05)03370-2
Hong, K. W., Yoo, S. E., Yu, S. S., Lee, J. Y., and Rhim, B. Y. (1996). Pharmacological coupling and functional role for CGRP receptors in the vasodilation of rat pial arterioles. Am. J. Phys. 270, H317–H323. doi: 10.1152/ajpheart.1996.270.1.H317
Huang, R., Karve, A., Shah, I., Bowers, M. C., DiPette, D. J., Supowit, S. C., et al. (2008). Deletion of the mouse alpha-calcitonin gene-related peptide gene increases the vulnerability of the heart to ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 294, H1291–H1297. doi: 10.1152/ajpheart.00749.2007
Inserte, J., and Garcia-Dorado, D. (2015). The cGMP/PKG pathway as a common mediator of cardioprotection: translatability and mechanism. Br. J. Pharmacol. 172, 1996–2009. doi: 10.1111/bph.12959
Juaneda, C., Dumont, Y., and Quirion, R. (2000). The molecular pharmacology of CGRP and related peptide receptor subtypes. Trends Pharmacol. Sci. 21, 432–438. doi: 10.1016/S0165-6147(00)01555-8
Kim, Y. G., Lone, A. M., Nolte, W. M., and Saghatelian, A. (2012). Peptidomics approach to elucidate the proteolytic regulation of bioactive peptides. Proc. Natl. Acad. Sci. USA 109, 8523–8527. doi: 10.1073/pnas.1203195109
Kuwasako, K., Kitamura, K., Nagoshi, Y., Cao, Y. N., and Eto, T. (2003). Identification of the human receptor activity-modifying protein 1 domains responsible for agonist binding specificity. J. Biol. Chem. 278, 22623–22630. doi: 10.1074/jbc.M302571200
Laragh, J. H., Baer, L., Brunner, H. R., Buhler, F. R., Sealey, J. E., and Vaughan, E. D. Jr. (1972). Renin, angiotensin and aldosterone system in pathogenesis and management of hypertensive vascular disease. Am. J. Med. 52, 633–652. doi: 10.1016/0002-9343(72)90054-X
Li, J., Levick, S. P., DiPette, D. J., Janicki, J. S., and Supowit, S. C. (2013). Alpha-calcitonin gene-related peptide is protective against pressure overload-induced heart failure. Regul. Pept. 185, 20–28. doi: 10.1016/j.regpep.2013.06.008
Li, J. Z., Peng, J., Xiao, L., Zhang, Y. S., Liao, M. C., Li, X. H., et al. (2010). Reversal of isoprenaline-induced cardiac remodeling by rutaecarpine via stimulation of calcitonin gene-related peptide production. Can. J. Physiol. Pharmacol. 88, 949–959. doi: 10.1139/y10-067
Li, J., and Wang, D. H. (2005). Development of angiotensin II-induced hypertension: role of CGRP and its receptor. J. Hypertens. 23, 113–118. doi: 10.1097/00004872-200501000-00020
Li, J., Zhao, H., Supowit, S. C., DiPette, D. J., and Wang, D. H. (2004). Activation of the renin-angiotensin system in alpha-calcitonin gene-related peptide/calcitonin gene knockout mice. J. Hypertens. 22, 1345–1349. doi: 10.1097/01.hjh.0000125409.50839.f1
Lin, H. Y., Harris, T. L., Flannery, M. S., Aruffo, A., Kaji, E. H., Gorn, A., et al. (1991). Expression cloning of an adenylate cyclase-coupled calcitonin receptor. Science 254, 1022–1024. doi: 10.1126/science.1658940
Lindsay, R. M. (1988). Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J. Neurosci. 8, 2394–2405. doi: 10.1523/JNEUROSCI.08-07-02394.1988
MaassenVanDenBrink, A., Meijer, J., Villalon, C. M., and Ferrari, M. D. (2016). Wiping out CGRP: potential cardiovascular risks. Trends Pharmacol. Sci. 37, 779–788. doi: 10.1016/j.tips.2016.06.002
Maasumi, K., Michael, R. L., and Rapoport, A. M. (2018). CGRP and migraine: the role of blocking calcitonin gene-related peptide ligand and receptor in the management of migraine. Drugs 78, 913–928. doi: 10.1007/s40265-018-0923-5
Mair, J., Lechleitner, P., Langle, T., Wiedermann, C., Dienstl, F., and Saria, A. (1990). Plasma CGRP in acute myocardial infarction. Lancet 335:168. doi: 10.1016/0140-6736(90)90040-C
Marquez-Rodas, I., Longo, F., Rothlin, R. P., and Balfagon, G. (2006). Pathophysiology and therapeutic possibilities of calcitonin gene-related peptide in hypertension. J. Physiol. Biochem. 62, 45–56. doi: 10.1007/BF03165805
McLatchie, L. M., Fraser, N. J., Main, M. J., Wise, A., Brown, J., Thompson, N., et al. (1998). RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 393, 333–339. doi: 10.1038/30666
Meng, J., Wang, J., Lawrence, G., and Dolly, J. O. (2007). Synaptobrevin I mediates exocytosis of CGRP from sensory neurons and inhibition by botulinum toxins reflects their anti-nociceptive potential. J. Cell Sci. 120, 2864–2874. doi: 10.1242/jcs.012211
Mulderry, P. K., Ghatei, M. A., Rodrigo, J., Allen, J. M., Rosenfeld, M. G., Polak, J. M., et al. (1985). Calcitonin gene-related peptide in cardiovascular tissues of the rat. Neuroscience 14, 947–954. doi: 10.1016/0306-4522(85)90156-3
Mulderry, P. K., Ghatei, M. A., Spokes, R. A., Jones, P. M., Pierson, A. M., Hamid, Q. A., et al. (1988). Differential expression of alpha-CGRP and beta-CGRP by primary sensory neurons and enteric autonomic neurons of the rat. Neuroscience 25, 195–205. doi: 10.1016/0306-4522(88)90018-8
Murakami, M., Suzuki, H., Nakamoto, H., Kageyama, Y., Naitoh, M., Sakamaki, Y., et al. (1991). Calcitonin gene-related peptide modulates adrenal hormones in conscious dogs. Acta Endocrinol. 124, 346–352. doi: 10.1530/acta.0.1240346
Nelson, M. T., Huang, Y., Brayden, J. E., Hescheler, J., and Standen, N. B. (1990). Arterial dilations in response to calcitonin gene-related peptide involve activation of K+ channels. Nature 344, 770–773. doi: 10.1038/344770a0
Nilsson, C., Hansen, T. K., Rosenquist, C., Hartmann, B., Kodra, J. T., Lau, J. F., et al. (2016). Long acting analogue of the calcitonin gene-related peptide induces positive metabolic effects and secretion of the glucagon-like peptide-1. Eur. J. Pharmacol. 773, 24–31. doi: 10.1016/j.ejphar.2016.01.003
Petermann, J. B., Born, W., Chang, J. Y., and Fischer, J. A. (1987). Identification in the human central nervous system, pituitary, and thyroid of a novel calcitonin gene-related peptide, and partial amino acid sequence in the spinal cord. J. Biol. Chem. 262, 542–545.
Raddino, R., Pela, G., Manca, C., Barbagallo, M., D'Aloia, A., Passeri, M., et al. (1997). Mechanism of action of human calcitonin gene-related peptide in rabbit heart and in human mammary arteries. J. Cardiovasc. Pharmacol. 29, 463–470. doi: 10.1097/00005344-199704000-00006
Resnick, L. M., Preibisz, J. J., and Laragh, J. H. (1989). “Calcitonin gene-related peptide-like immunoreactivity in hypertension: relation to blood pressure, sodium and calcium metabolism”, in Salt and Hypertension. eds. R. Rettig, D. Ganten, and P. Luft. (NewYork: Springer-Verla), 190–199.
Rosenfeld, M. G., Mermod, J. J., Amara, S. G., Swanson, L. W., Sawchenko, P. E., Rivier, J., et al. (1983). Production of a novel neuropeptide encoded by the calcitonin gene via tissue-specific RNA processing. Nature 304, 129–135. doi: 10.1038/304129a0
Roudenok, V., Gutjar, L., Antipova, V., and Rogov, Y. (2001). Expression of vasoactive intestinal polypeptide and calcitonin gene-related peptide in human stellate ganglia after acute myocardial infarction. Ann. Anat. 183, 341–344. doi: 10.1016/S0940-9602(01)80177-1
Rovero, P., Giuliani, S., and Maggi, C. A. (1992). CGRP antagonist activity of short C-terminal fragments of human alpha CGRP, CGRP(23-37) and CGRP(19-37). Peptides 13, 1025–1027. doi: 10.1016/0196-9781(92)90067-D
Russell, F. A., King, R., Smillie, S. J., Kodji, X., and Brain, S. D. (2014). Calcitonin gene-related peptide: physiology and pathophysiology. Physiol. Rev. 94, 1099–1142. doi: 10.1152/physrev.00034.2013
Russo, A. F. (2015). Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu. Rev. Pharmacol. Toxicol. 55, 533–552. doi: 10.1146/annurev-pharmtox-010814-124701
Sabharwal, R., Zhang, Z., Lu, Y., Abboud, F. M., Russo, A. F., and Chapleau, M. W. (2010). Receptor activity-modifying protein 1 increases baroreflex sensitivity and attenuates angiotensin-induced hypertension. Hypertension 55, 627–635. doi: 10.1161/HYPERTENSIONAHA.109.148171
Salio, C., Averill, S., Priestley, J. V., and Merighi, A. (2007). Costorage of BDNF and neuropeptides within individual dense-core vesicles in central and peripheral neurons. Dev. Neurobiol. 67, 326–338. doi: 10.1002/dneu.20358
Schaeffer, C., Vandroux, D., Thomassin, L., Athias, P., Rochette, L., and Connat, J. L. (2003). Calcitonin gene-related peptide partly protects cultured smooth muscle cells from apoptosis induced by an oxidative stress via activation of ERK1/2 MAPK. Biochim. Biophys. Acta 1643, 65–73. doi: 10.1016/j.bbamcr.2003.09.005
Schebesch, K. M., Herbst, A., Bele, S., Schodel, P., Brawanski, A., Stoerr, E. M., et al. (2013). Calcitonin-gene related peptide and cerebral vasospasm. J. Clin. Neurosci. 20, 584–586. doi: 10.1016/j.jocn.2012.07.006
Shekhar, Y. C., Anand, I. S., Sarma, R., Ferrari, R., Wahi, P. L., and Poole-Wilson, P. A. (1991). Effects of prolonged infusion of human alpha calcitonin gene-related peptide on hemodynamics, renal blood flow and hormone levels in congestive heart failure. Am. J. Cardiol. 67, 732–736. doi: 10.1016/0002-9149(91)90531-O
Sheykhzade, M., Abdolalizadeh, B., Koole, C., Pickering, D. S., Dreisig, K., Johansson, S. E., et al. (2018). Vascular and molecular pharmacology of the metabolically stable CGRP analogue, SAX. Eur. J. Pharmacol. 829, 85–92. doi: 10.1016/j.ejphar.2018.04.007
Silberstein, S. D., Dodick, D. W., Bigal, M. E., Yeung, P. P., Goadsby, P. J., Blankenbiller, T., et al. (2017). Fremanezumab for the preventive treatment of chronic migraine. N. Engl. J. Med. 377, 2113–2122. doi: 10.1056/NEJMoa1709038
Stangherlin, A., Gesellchen, F., Zoccarato, A., Terrin, A., Fields, L. A., Berrera, M., et al. (2011). cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ. Res. 108, 929–939. doi: 10.1161/CIRCRESAHA.110.230698
Steenbergh, P. H., Hoppener, J. W., Zandberg, J., Lips, C. J., and Jansz, H. S. (1985). A second human calcitonin/CGRP gene. FEBS Lett. 183, 403–407.
Steenbergh, P. H., Hoppener, J. W., Zandberg, J., Visser, A., Lips, C. J., and Jansz, H. S. (1986). Structure and expression of the human calcitonin/CGRP genes. FEBS Lett. 209, 97–103. doi: 10.1016/0014-5793(86)81091-2
Strecker, T., Reeh, P. W., Weyand, M., and Messlinger, K. (2006). Release of calcitonin gene-related peptide from the isolated mouse heart: methodological validation of a new model. Neuropeptides 40, 107–113. doi: 10.1016/j.npep.2005.11.005
Sueur, S., Pesant, M., Rochette, L., and Connat, J. L. (2005). Antiapoptotic effect of calcitonin gene-related peptide on oxidative stress-induced injury in H9c2 cardiomyocytes via the RAMP1/CRLR complex. J. Mol. Cell. Cardiol. 39, 955–963. doi: 10.1016/j.yjmcc.2005.09.008
Supowit, S. C., Gururaj, A., Ramana, C. V., Westlund, K. N., and DiPette, D. J. (1995a). Enhanced neuronal expression of calcitonin gene-related peptide in mineralocorticoid-salt hypertension. Hypertension 25, 1333–1338.
Supowit, S. C., Ramana, C. V., Westlund, K. N., and DiPette, D. J. (1993). Calcitonin gene-related peptide gene expression in the spontaneously hypertensive rat. Hypertension 21, 1010–1014. doi: 10.1161/01.HYP.21.6.1010
Supowit, S. C., Rao, A., Bowers, M. C., Zhao, H., Fink, G., Steficek, B., et al. (2005). Calcitonin gene-related peptide protects against hypertension-induced heart and kidney damage. Hypertension 45, 109–114. doi: 10.1161/01.HYP.0000151130.34874.fa
Supowit, S. C., Zhao, H., and DiPette, D. J. (2001). Nerve growth factor enhances calcitonin gene-related peptide expression in the spontaneously hypertensive rat. Hypertension 37, 728–732. doi: 10.1161/01.HYP.37.2.728
Supowit, S. C., Zhao, H., Hallman, D. M., and DiPette, D. J. (1997). Calcitonin gene-related peptide is a depressor of deoxycorticosterone-salt hypertension in the rat. Hypertension 29, 945–950. doi: 10.1161/01.HYP.29.4.945
Supowit, S. C., Zhao, H., Hallman, D. M., and DiPette, D. J. (1998). Calcitonin gene-related peptide is a depressor in subtotal nephrectomy hypertension. Hypertension 31, 391–396. doi: 10.1161/01.HYP.31.1.391
Supowit, S. C., Zhao, H., Wang, D. H., and DiPette, D. J. (1995b). Regulation of neuronal calcitonin gene-related peptide expression. Role of increased blood pressure. Hypertension 26, 1177–1180.
Takimoto, E. (2012). Cyclic GMP-dependent signaling in cardiac myocytes. Circ. J. 76, 1819–1825. doi: 10.1253/circj.CJ-12-0664
Tjen, A. L. S., Ekman, R., Lippton, H., Cary, J., and Keith, I. (1992). CGRP and somatostatin modulate chronic hypoxic pulmonary hypertension. Am. J. Phys. 263, H681–H690. doi: 10.1152/ajpheart.1992.263.3.H681
Tsujikawa, K., Yayama, K., Hayashi, T., Matsushita, H., Yamaguchi, T., Shigeno, T., et al. (2007). Hypertension and dysregulated proinflammatory cytokine production in receptor activity-modifying protein 1-deficient mice. Proc. Natl. Acad. Sci. USA 104, 16702–16707. doi: 10.1073/pnas.0705974104
Uddman, R., Edvinsson, L., Ekblad, E., Hakanson, R., and Sundler, F. (1986). Calcitonin gene-related peptide (CGRP): perivascular distribution and vasodilatory effects. Regul. Pept. 15, 1–23. doi: 10.1016/0167-0115(86)90071-6
Umoh, N. A., Walker, R. K., Millis, R. M., Al-Rubaiee, M., Gangula, P. R., and Haddad, G. E. (2014). Calcitonin gene-related peptide regulates cardiomyocyte survival through regulation of oxidative stress by PI3K/Akt and MAPK signaling pathways. Ann. Clin. Exp. Hypertens. 2:1007.
Walter, S., and Bigal, M. E. (2015). TEV-48125: a review of a monoclonal CGRP antibody in development for the preventive treatment of migraine. Curr. Pain Headache Rep. 19:6. doi: 10.1007/s11916-015-0476-1
Wang, L., and Wang, D. H. (2005). TRPV1 gene knockout impairs postischemic recovery in isolated perfused heart in mice. Circulation 112, 3617–3623. doi: 10.1161/CIRCULATIONAHA.105.556274
Wiley, J. W., Gross, R. A., and MacDonald, R. L. (1992). The peptide CGRP increases a high-threshold Ca2+ current in rat nodose neurones via a pertussis toxin-sensitive pathway. J. Physiol. 455, 367–381. doi: 10.1113/jphysiol.1992.sp019306
Wisskirchen, F. M., Burt, R. P., and Marshall, I. (1998). Pharmacological characterization of CGRP receptors mediating relaxation of the rat pulmonary artery and inhibition of twitch responses of the rat vas deferens. Br. J. Pharmacol. 123, 1673–1683. doi: 10.1038/sj.bjp.0701783
Wolfrum, S., Nienstedt, J., Heidbreder, M., Schneider, K., Dominiak, P., and Dendorfer, A. (2005). Calcitonin gene related peptide mediates cardioprotection by remote preconditioning. Regul. Pept. 127, 217–224. doi: 10.1016/j.regpep.2004.12.008
Wu, D. M., van Zwieten, P. A., and Doods, H. N. (2001). Effects of calcitonin gene-related peptide and BIBN4096BS on myocardial ischemia in anesthetized rats. Acta Pharmacol. Sin. 22, 588–594.
Xing, L., Guo, J., and Wang, X. (2000). Induction and expression of beta-calcitonin gene-related peptide in rat T lymphocytes and its significance. J. Immunol. 165, 4359–4366. doi: 10.4049/jimmunol.165.8.4359
Yoshimoto, R., Mitsui-Saito, M., Ozaki, H., and Karaki, H. (1998). Effects of adrenomedullin and calcitonin gene-related peptide on contractions of the rat aorta and porcine coronary artery. Br. J. Pharmacol. 123, 1645–1654. doi: 10.1038/sj.bjp.0701805
Zeng, S. Y., Yang, L., Hong, C. L., Lu, H. Q., Yan, Q. J., Chen, Y., et al. (2018). Evidence that ADAM17 mediates the protective action of CGRP against angiotensin II-induced inflammation in vascular smooth muscle cells. Mediat. Inflamm. 2018:2109352. doi: 10.1155/2018/2109352
Zhong, B., Rubinstein, J., Ma, S., and Wang, D. H. (2018). Genetic ablation of TRPV1 exacerbates pressure overload-induced cardiac hypertrophy. Biomed. Pharmacother. 99, 261–270. doi: 10.1016/j.biopha.2018.01.065
Zhou, X. B., Ruth, P., Schlossmann, J., Hofmann, F., and Korth, M. (1996). Protein phosphatase 2A is essential for the activation of Ca2+−activated K+ currents by cGMP-dependent protein kinase in tracheal smooth muscle and Chinese hamster ovary cells. J. Biol. Chem. 271, 19760–19767. doi: 10.1074/jbc.271.33.19760
Keywords: heart failure, calcitonin gene-related peptide (CGRP), neuropeptide, cardiovascular diseases, hypertension, myocardial infarction
Citation: Kumar A, Potts JD and DiPette DJ (2019) Protective Role of α-Calcitonin Gene-Related Peptide in Cardiovascular Diseases. Front. Physiol. 10:821. doi: 10.3389/fphys.2019.00821
Edited by:
Scott Levick, University of Sydney, AustraliaReviewed by:
Claudia Penna, University of Turin, ItalyCopyright © 2019 Kumar, Potts and DiPette. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Donald J. DiPette, ZG9uYWxkLmRpcGV0dGVAdXNjbWVkLnNjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.