 Silvia Pregnolato
Silvia Pregnolato Elavazhagan Chakkarapani
Elavazhagan Chakkarapani Anthony R. Isles
Anthony R. Isles Karen Luyt
Karen Luyt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 24 April 2019
Sec. Developmental Physiology
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.00417
This article is part of the Research TopicNeurological Outcomes in Preterm Infants – Current Controversies and Therapies for Brain InjuryView all 32 articles
Preterm birth complications are the leading cause of child death worldwide and a top global health priority. Among the survivors, the risk of life-long disabilities is high, including cerebral palsy and impairment of movement, cognition, and behavior. Understanding the molecular mechanisms of preterm brain injuries is at the core of future healthcare improvements. Glutamate excitotoxicity is a key mechanism in preterm brain injury, whereby the accumulation of extracellular glutamate damages the delicate immature oligodendrocytes and neurons, leading to the typical patterns of injury seen in the periventricular white matter. Glutamate excitotoxicity is thought to be induced by an interaction between environmental triggers of injury in the perinatal period, particularly cerebral hypoxia-ischemia and infection/inflammation, and developmental and genetic vulnerabilities. To avoid extracellular build-up of glutamate, the brain relies on rapid uptake by sodium-dependent glutamate transporters. Astrocytic excitatory amino acid transporter 2 (EAAT2) is responsible for up to 95% of glutamate clearance, and several lines of evidence suggest that it is essential for brain functioning. While in the adult EAAT2 is predominantly expressed by astrocytes, EAAT2 is transiently upregulated in the immature oligodendrocytes and selected neuronal populations during mid-late gestation, at the peak time for preterm brain injury. This developmental upregulation may interact with perinatal hypoxia-ischemia and infection/inflammation and contribute to the selective vulnerability of the immature oligodendrocytes and neurons in the preterm brain. Disruption of EAAT2 may involve not only altered expression but also impaired function with reversal of transport direction. Importantly, elevated EAAT2 levels have been found in the reactive astrocytes and macrophages of human infant post-mortem brains with severe white matter injury (cystic periventricular leukomalacia), potentially suggesting an adaptive mechanism against excitotoxicity. Interestingly, EAAT2 is suppressed in animal models of acute hypoxic-ischemic brain injury at term, pointing to an important and complex role in newborn brain injuries. Enhancement of EAAT2 expression and transport function is gathering attention as a potential therapeutic approach for a variety of adult disorders and awaits exploration in the context of the preterm brain injuries.
Perinatal care has advanced considerably in the last century and has improved survival of many vulnerable newborns, including those born preterm. The World Health Organization estimates that 15 million newborns (1 in 10 live births) are born preterm (<37 weeks of gestation) worldwide each year (World Health Organization, 2012). Despite global improvements, the United Nations Millennium Development Goal to reduce childhood mortality by two-thirds in 2015 was not achieved globally (United Nations, 2015) and 2.7 million children died in the first month of life worldwide in 2015. Of these babies, over 900,000 died due to preterm birth complications – the leading cause of death of newborns and children under 5 years old (Liu et al., 2016). For the newborns who survive, the multi-organ damage can result in life-long disabilities. Globally, preterm birth complications represent the fourth leading cause of years of “healthy” life lost due to disability (i.e., over 102,000 DALYs), above causes such as diarrheal diseases, diabetes, and HIV (World Health Organization, 2016).
Prematurity is a major risk factor for cerebral palsy, “a group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to non-progressive disturbances that occurred in the developing fetal or infant brain” (Bax et al., 2005; Rosenbaum et al., 2007). Cerebral palsy is the most common physical disability in childhood and is a heterogeneous diagnosis, including different clinical types and brain imaging patterns, comorbidities, and multiple causes (Stanley et al., 2000; Locatelli et al., 2010; MacLennan et al., 2015). Preterm birth is clearly an important risk factor and risk is 30 times higher in children born before 33 weeks of gestation than in those born at term (Stanley, 1992; Himpens et al., 2008; Beaino et al., 2010; Mercier et al., 2010; Tronnes et al., 2014; MacLennan et al., 2015; Stavsky et al., 2017). A recent meta-analysis estimated an increase in prevalence from 1.4/1,000 live births in children born at term (>36 weeks of gestation) to 6.8/1,000 live births in moderate to late preterm (32–36 weeks of gestation), rising to 43.2/1,000 live births in very preterm (28–31 weeks of gestation) and 82.3/1,000 live births in extremely preterm infants (<28 weeks of gestation) (Oskoui et al., 2013; Hirvonen et al., 2014). More than a third of the extremely preterm children with cerebral palsy are unable to walk (Moore et al., 2012), and many have multiple disabilities, which may further limit independence and quality of life (Litt et al., 2005; Glass et al., 2008, 2015; Soria-Pastor et al., 2008; Anderson et al., 2011; Moore et al., 2012). A systematic review of international cerebral palsy registers in high-income settings highlighted the extent of these comorbidities: around three quarters of children with cerebral palsy suffer from chronic pain; approximately half have intellectual disabilities (IQ, executive function, language ability); around a quarter have active epilepsy, hip dislocation, bladder control problems, behavioral problems, sleep disorders, and/or speech impairment; 11 and 4% have severe vision and hearing impairment, respectively (Novak et al., 2012). There are less data from low-income settings, but it is likely that comorbidities, as well as mortality, are higher (Khandaker et al., 2015). Preterm birth complications impose a considerable economic burden on the public sector, which was estimated around £2.9 billion in England and Wales in 2006 (Mangham et al., 2009). While administration of magnesium sulfate as a preventative treatment to the mother during preterm labor has been shown to reduce risk of cerebral palsy by a third in very preterm infants (Doyle et al., 2009), no postnatal therapy currently exists for preterm brain injury. This is a global health priority as the increase in both preterm birth and survival rates has not been matched by a decrease in long-term disability (Wilson-Costello et al., 2005).
Preterm birth is associated with smaller brain volumes (Peterson et al., 2003; Inder et al., 2005; Srinivasan et al., 2007) as well as motor, cognitive, and behavioral problems at school age (Peterson et al., 2000, 2003; Abernethy et al., 2004; Nosarti et al., 2005; Gimenez et al., 2006; Anderson and Doyle, 2008; Kesler et al., 2008; Aarnoudse-Moens et al., 2009; Delobel-Ayoub et al., 2009; Soria-Pastor et al., 2009; Anderson et al., 2017). Progress in neuroimaging techniques has been key in linking childhood neurodevelopmental outcomes to perinatal brain injuries and in advancing our knowledge of the underlying neuropathology (Volpe, 2009c; Back, 2017). Both MRI-defined preterm white matter injury (periventricular leukomalacia) and preterm birth are predictive of cerebral palsy (Constantinou et al., 2007; Spittle et al., 2008, 2009, 2018; Duerden et al., 2013). In a large European population study of cerebral palsy, white matter injury was the most common feature found in over 40% of the children (Bax et al., 2006). Originally, cranial ultrasound could only detect the most severe cystic type of white matter injury (cystic periventricular leukomalacia), characterized by focal macroscopic cysts of necrotic tissue in the deep white matter (de Vries et al., 1992) and highly predictive of cerebral palsy (Leviton and Paneth, 1990; De Vries et al., 2004; Serdaroglu et al., 2004; Fetters and Huang, 2007). Necrotic white matter injury can also evolve into microscopic glial scars, which may not be visible with traditional ultrasound. These are a more common type of injury and are sufficient to cause a loss in brain volume (Volpe, 2009c; Volpe et al., 2011). With the development of MRI techniques, a diffuse type of white matter injury has increasingly been recognized in the form of diffuse disturbances of myelination in the central white matter. This has emerged as the predominant type of white matter injury, accounting for over 90% of periventricular leukomalacia cases, as well as the predominant type of preterm brain injury altogether, occurring in 50% preterm newborns (Volpe, 2008). Importantly, while rates of the more severe cystic form have declined to less than 5% with advances in perinatal care, this has not been reflected for the diffuse forms (Maalouf et al., 2001; Counsell et al., 2003; Inder et al., 2003; Miller et al., 2003; Back et al., 2007b; Volpe, 2008). These could be seen as different manifestations of an “encephalopathy of prematurity” (Volpe, 2009c) or even as distinct pathologies (Back and Rosenberg, 2014). In the last two decades, advanced MRI techniques have highlighted that injury is not limited to the white matter but it extends to the deep grey matter, cortex, and cerebellum, all of which contribute to the volume loss (Counsell and Boardman, 2005; Ball et al., 2012). The cerebellum is gathering attention as a key target of injury. This region grows rapidly at the peak time for preterm birth and damage in the form of infarction, atrophy, and poor growth has been reported as common in very preterm infants developing cerebral palsy and long-term motor, cognitive, and behavioral impairment (Mercuri et al., 1997; Abraham et al., 2001; Bodensteiner and Johnsen, 2005; Johnsen et al., 2005; Limperopoulos et al., 2005a,b, 2007; Nosarti et al., 2008; Parker et al., 2008; Lawrence et al., 2014). Indeed, there is a relationship between cerebellar volume loss and white matter injury, pointing to the existence of a common insult, such as hypoxia-ischemia and infection/inflammation, which are known to damage the developing cerebellum (Shah et al., 2006; Volpe, 2009b; Hutton et al., 2014).
Disentangling the spatial and temporal contributions of infection/inflammation and hypoxia-ischemia will be key in understanding brain injuries across the perinatal spectrum. For example, while white matter injury is typical of the preterm newborn, it may be present in a subset of newborns born at term who experienced in utero hypoxic-ischemic insults (e.g., placental insufficiencies) (Mallard et al., 1998; Rees et al., 1998; Zhu et al., 2016). Indeed, newborns born at term with hypoxic-ischemic encephalopathy are also at high risk and up to 40% develop cerebral palsy (Gluckman et al., 2005; Shankaran et al., 2005; Azzopardi et al., 2009; Simbruner et al., 2010; Jacobs et al., 2011). Investigating the molecular basis for divergence between term and preterm injuries is paramount for development of age-appropriate pharmacological therapies.
Brain injury is thought to be more common in preterm than term newborns for several reasons, including developmental and genetic vulnerabilities and differential exposure to adverse perinatal environments. A considerable body of in vitro and in vivo evidence points two potential triggers of injury, hypoxia-ischemia, and infection/inflammation (Volpe, 2008, 2009a; Deng, 2010; Volpe et al., 2011; Back and Rosenberg, 2014; Back, 2017). These insults are thought to interact in the vulnerable immature brain and converge onto three downstream mechanisms of injury: inflammation, glutamate excitotoxicity, and ultimately free radical attack, which directly damages cell components as well as triggering delayed cell death by apoptosis. Severity and temporal profile of hypoxia-ischemia and infection/inflammation, degree of brain maturity, comorbidities, sex, and genetic background may all contribute to individual differences in pathogenesis, clinical presentation, and individual susceptibility to injury. We will review the role of developmental vulnerabilities, infection/inflammation, and hypoxia-ischemia and bring the focus on the common downstream mechanism of glutamate excitotoxicity. We will then review the evidence linking glutamate transport to excitotoxic preterm brain injuries and highlight the current evidence supporting excitatory amino acid transporter 2 (EAAT2) as a potential therapeutic target.
The brain undergoes rapid and critical developmental events during the peak time of premature brain injury (24–32 weeks), including neuronal migration, growth of axons and dendrites, synaptogenesis, development of the vascular system, and myelination. Interference with these natural trajectories determines selective cellular and regional vulnerabilities and may redirect subsequent development. Among their functions, oligodendrocytes are responsible for laying the highly specialized myelin membrane around axons and are therefore key for the development of the white matter. Myelination begins before birth and peaks in the first 2 years of postnatal life, with the intracortical fibers of the cortex being myelinated in the third decade. The process of myelination requires that oligodendrocytes first proliferate and develop into mature oligodendrocytes and then depose myelin around axons (Volpe, 2008). Around the peak time of preterm brain injury (28–32 weeks of gestation), the pre-oligodendrocyte stage still represents the majority of the oligodendrial pool in the very preterm brain (Iida et al., 1995; Back et al., 2001). Pre-oligodendrocytes are more vulnerable than mature oligodendrocytes to hypoxia-ischemia, infection/inflammation, oxidative damage, and ultimately cell death (Back et al., 1998, 2002, 2005, 2007b; Fern and Moller, 2000; Baud et al., 2004; Fragoso et al., 2004; Segovia et al., 2008; Volpe et al., 2011). Indeed, a unique feature of periventricular white matter injury is an arrest in the development of oligodendrocytes at the pre-oligodendrocyte stage, leading to the abnormal myelination patterns typically seen through MRI (Back et al., 2007b; Volpe et al., 2011). More severe necrotic injury extends to all the cell components, leading to cysts and exacerbating myelin injury via focal axonal degeneration (Laptook, 2016; Back, 2017). Concurrent developmental vulnerabilities include the limited ability of the immature brain to synthesize appropriate amounts of growth factors needed for brain development and self-protection, and an immature immune system, potentially promoting an excessive and sustained inflammatory response (Gilles et al., 2018).
Alongside the intrinsic developmental vulnerability of the immature brain, the preterm newborn is exposed to a range of potentially harmful exposures in the perinatal period. Supported by mounting experimental and epidemiological evidence, perinatal infection/inflammation leading to an overly intense inflammatory response, or a “cytokine storm”, has increasingly been recognized as a major risk factor not only for preterm birth but also for preterm white matter injury and long-term neurodisabilities (Yoon et al., 1996, 1997, 2000; Baud et al., 1999; Duggan et al., 2001; Dollner et al., 2002; Heep et al., 2003; Kaukola et al., 2004, 2006; Ellison et al., 2005; Bi et al., 2014). The preterm brain is often exposed to inflammation early during fetal development (e.g., maternal infections and chorioamnionitis) and usually for prolonged periods during postnatal life in the neonatal intensive care environment (e.g., neonatal infections, inflammatory comorbidities such as necrotizing enterocolitis), during critical phases of myelination and brain plasticity (Murphy et al., 1995; Grether and Nelson, 1997; Verma et al., 1997; Alexander et al., 1998; Dammann and Leviton, 1998, 2000, 2004; O’Shea et al., 1998; Leviton et al., 1999; Wu and Colford, 2000; Dammann et al., 2002; Rezaie and Dean, 2002; Stoll et al., 2002; Wu, 2002; Schlapbach et al., 2011; Hagberg et al., 2015; Anblagan et al., 2016). A combination of multiple inflammatory hits, antenatally and postnatally, has been shown to increase risk of brain injury and disability compared to single hits (Korzeniewski et al., 2014; van der Burg et al., 2016; Yanni et al., 2017). Indeed, pharmacological interventions targeting inflammation may have translational potential based on preclinical studies (Hagberg et al., 2015).
The role of hypoxia-ischemia in preterm brain injury is more controversial. In term newborns with hypoxic-ischemic encephalopathy, defined and acute hypoxic-ischemic events before or during birth (e.g., placental abruption, cord occlusion, and uterine rupture) are usually recognized by the clinician and represent the first step of a diagnosis of hypoxic-ischemic encephalopathy, aided by objective clinical and neuroimaging criteria. In the preterm newborn, a sentinel event is rarely recognized, and hypoxia-ischemia is generally assumed to have a more complex temporal profile, with intermittent or chronic nature (Laptook, 2016; Ohshima et al., 2016). However, it remains challenging to determine the individual contribution of hypoxia-ischemia among several coexistent factors, such as infection/inflammation, growth restriction, or hyperoxia (Gopagondanahalli et al., 2016). Physiologically, it is conceivable that the preterm brain is vulnerable to hypoxia-ischemia due to the anatomical and functional immaturity of the periventricular vasculature, which would make the periventricular white matter vulnerable to minor drops in cerebral perfusion (Takashima and Tanaka, 1978; Lou et al., 1979; De Reuck, 1984; Altman et al., 1988; Pryds, 1991; Miyawaki et al., 1998; Inage et al., 2000; Volpe, 2008; Laptook, 2016). The periventricular white matter has lower basal blood flow compared to grey matter regions in both humans (Greisen, 1986; Pryds et al., 1990) and the preterm fetal sheep (Szymonowicz et al., 1988; Gleason et al., 1989; Riddle et al., 2006). Further drops in blood flow are common in sick premature infants with respiratory disease due to lung immaturity (Soul et al., 2007). Mechanical ventilation may contribute to ischemia due to the vasoconstrictive effect of the induced cumulative hypocarbia (Shankaran et al., 2006). Perinatal hypoxic-ischemic episodes are also likely to play a key role, including ongoing placental pathologies, an overlapping risk factor for intrauterine growth restriction, low birthweight, and preterm birth. A meta-analysis recently reported an association between preterm brain injury and perinatal risk factors related to hypoxia-ischemia, including oligohydramnios, acidemia, low Apgar scores, apnea, respiratory distress syndrome, and seizures (Huang et al., 2017). However, the link between regional differences in blood flow and vulnerability to severe white matter injury is not consistent, and even in moderate ischemia, some regions of white matter are spared. This suggests that ischemia is necessary but not sufficient in isolation (Riddle et al., 2006; McClure et al., 2008; Back, 2017). Indeed, it has been suggested that more consistent evidence is needed to ascertain the specific role of hypoxic and ischemic events in preterm brain injury altogether and that future research should take into account contributions and interactions with other biological processes, including infection/inflammation and developmental vulnerability (Gilles et al., 2018). Importantly, the impact of hypoxia-ischemia on the cerebellum is also emerging, as shown by reports of volume loss and death of Purkinje cells and Bergmann glia in term newborns with hypoxic-ischemic encephalopathy and mid-late gestation fetal sheep exposed to asphyxia (Rees et al., 1997; Inage et al., 1998; Castillo-Melendez et al., 2004; Biran et al., 2012; Hutton et al., 2014). In an established mouse model of chronic hypoxia recapitulating perinatal brain injuries, damage to the cerebellum was reported in terms of a significant loss of GABAergic interneurons and a delay in dendritic arborization of Purkinje cells, followed by motor impairment and cerebellar learning deficits (Chahboune et al., 2009; Zonouzi et al., 2015; Sathyanesan et al., 2018).
Several experimental studies have shown that hypoxia-ischemia and infection/inflammation lead to worse brain and behavioral outcomes when they interact, and insults that are individually insufficient to cause injury can lead to injury when combined (Dommergues et al., 2000; Eklind et al., 2001; Lehnardt et al., 2003; Ikeda et al., 2004; Larouche et al., 2005; Favrais et al., 2007; Wang et al., 2007, 2009, 2010; Aden et al., 2010; van Tilborg et al., 2018). This has led to the multiple hit hypothesis of preterm brain injury, whereby a mild first event sensitizes the brain to subsequent insults (Leviton et al., 2013; Van Steenwinckel et al., 2014; Barnett et al., 2018). The current hypothesis is that hypoxia-ischemia triggers an inflammatory response per se. This additional endogenous response combined with the inflammation triggered by infection leads to a pro-inflammatory “cytokine storm,” which is not matched by upregulation of anti-inflammatory cytokines and neurotrophic factors. This in turn sensitizes the brain to hypoxic-ischemic injury by enhancing glutamate excitotoxicity and damaging the blood-brain barrier (Hagberg et al., 2015). Tertiary mechanisms of injury, mediated by epigenetic modifications, may sustain the sensitization in the long term and interfere with remodeling and repair mechanisms (Dammann, 2007; Fleiss and Gressens, 2012).
A substantial body of experimental evidence suggests that glutamate excitotoxicity triggered by hypoxia-ischemia and/or infection/inflammation plays a key role in the pathogenesis of preterm white matter injury (Hagberg et al., 2002; Johnston, 2005; Volpe, 2008; Deng, 2010; Volpe et al., 2011).
Glutamate is the main excitatory neurotransmitter in the mammalian brain (Meldrum, 2000). It is essential for brain function, orchestrating not only fast excitatory neurotransmission but also long-lasting neuronal changes necessary for memory, learning, and cognition. It is also fundamental during brain development, due to its role in regulating formation and elimination of synapses, as well as neuronal migration, proliferation, and viability. Glutamate is abundant inside the brain cells, and most neurons and glial cells have glutamate receptors distributed across most cellular elements, highlighting the importance of glutamatergic systems for normal function (Curtis and Johnston, 1974; Watkins and Evans, 1981; Bliss and Collingridge, 1993; Newcomer et al., 2000; Platt, 2007). Stimulation of a glutamatergic neuron results in Ca2+-dependent release of glutamate in the synapse by vesicular exocytosis. Extracellular glutamate binds to and activates post-synaptic ionotropic (NMDA, AMPA, and kainate receptors) and metabotropic (mGluR) glutamate receptors, stimulating the post-synaptic neurons via Ca2+ or Na+ influx and inducing intracellular signaling cascades that lead to physiological cellular responses, such as regulation of transcription factors and DNA replication (Nicholls and Attwell, 1990; Danbolt, 2001).
Glutamatergic transmission is terminated when glutamate transporters, expressed predominantly by astrocytes, slowly take up glutamate from the synaptic space (30 glutamate molecules per second at Vmax) (Otis and Kavanaugh, 2000; Bergles et al., 2002; Grewer and Rauen, 2005; Takahashi et al., 2015). In the preterm brain, glutamate transporters are also expressed by immature neurons and oligodendrocytes, although their significance is controversial, as reviewed below. In astrocytes, glutamate is converted to glutamine via glutamine synthetase. Glutamine is shuttled back into the pre-synaptic neuron, where it is converted into glutamate via glutaminase (Figure 1). The glutamate-glutamine cycle is not essential for supplying glutamate for neuronal release but is needed for normal glutamatergic transmission (Danbolt, 2001; Takahashi et al., 2015; Danbolt et al., 2016).
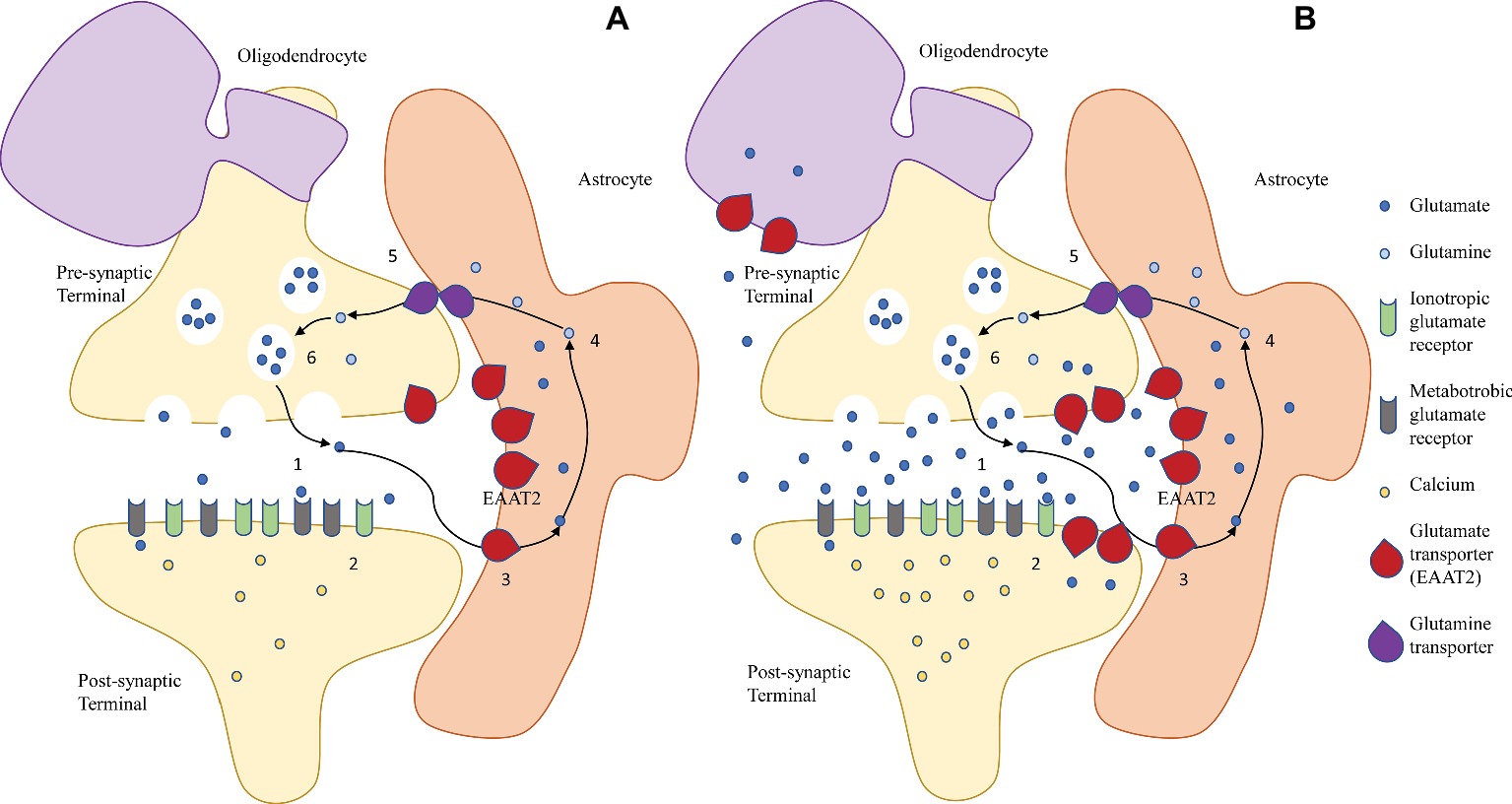
Figure 1. The glutamate/glutamine cycle in (A) physiological conditions and (B) excitotoxic conditions in the immature brain. (A) In the mature healthy brain, glutamate is released by exocytosis from the pre-synaptic neuronal terminal into the synapse (1), and it binds to post-synaptic ionotropic (NMDA, AMPA, and kainate receptors) and metabotropic (mGluR) glutamate receptors, inducing Ca2+-mediated signaling cascades that result in cellular responses (2). Extracellular glutamate is taken up primarily by astroglial EAAT2 (3) and converted to glutamine (4), which is shuttled back to the pre-synaptic terminal via glutamine transporters (5). Here, glutamine is converted back to glutamate (6). (B) During excitotoxicity, a combination of increased neuronal release and decreased astroglial uptake lead to a rise of extracellular glutamate levels, leading to overactivation of the post-synaptic glutamate receptors, Ca2+ overload, and activation of apoptotic pathways. Reversal of transport of astroglial transporters may also contribute to the accumulation of extracellular glutamate. In the immature brain, upregulation of the glutamate transporters in underdeveloped neurons and oligodendrocytes may contribute to their selective vulnerability.
The ubiquity of glutamate is a double-edged sword: when homeostasis is disrupted, glutamate can turn into a potent neurotoxin. If the concentration of glutamate in the extracellular space rises above physiological levels, post-synaptic glutamate receptors are overactivated. This excessive activation, or excitotoxicity, leads to cell death via activation of suicide cell programs (apoptosis) (Danbolt, 2001; Sattler and Tymianski, 2001) (Figure 1). Since it was first proposed in the late 1960s (Olney, 1969), the concept of glutamate excitotoxicity has been implicated in several adult disorders, both acute (e.g., ischemic stroke and traumatic brain injury) and chronic (e.g., amyotrophic lateral sclerosis, Alzheimer’s, Parkinson’s, major depression, and addiction) (Doble, 1999; Takahashi et al., 2015). Consistently, injection of glutamate agonists into the cortex, striatum, and periventricular white matter of newborn rodents, rabbits, and kittens produces patterns of perinatal brain injuries similar to those seen in humans (McDonald et al., 1988; Innocenti and Berbel, 1991a,b; Marret et al., 1995; Gressens et al., 1996; Acarin et al., 1999; Follett et al., 2000). On the other hand, pharmacological inhibition of glutamate receptors before or immediately after an hypoxic-ischemic insult is neuroprotective in both preterm (Follett et al., 2004; Manning et al., 2008) and term (Hagberg et al., 1994; Follett et al., 2000) brain injuries. Indeed, one of the mechanisms through which magnesium sulfate is thought to exert neuroprotection is by preventing excitotoxic damage through NMDA receptor blockade (Lingam and Robertson, 2018).
Evidence of in vivo disturbance of glutamate signaling has been produced for animal models of hypoxic-ischemic brain injury. In a rat model of mild white matter injury near term, a rise in extracellular glutamate is observed in the acute phase after hypoxia-ischemia, with oligodendrocytes and axons representing the major sources of extracellular glutamate and astrocytes failing to take up excess glutamate (Back et al., 2007a). Similarly, repeated umbilical cord occlusion in the near-term fetal sheep causes periventricular white matter injury, the extent of which correlates with extracellular local glutamate levels (Loeliger et al., 2003). Notably, the largest increase in glutamate occurred over the hours after the insult, a delayed increase that suggested impaired glutamate transport. In a piglet model of hypoxic-ischemic encephalopathy at term, glutamate levels in the basal ganglia were shown to change in two phases: an early increase in the first 6 hours was followed by transient and slight recovery by 12 hours, possibly due to the self-protective glutamate transport mechanisms and conversion to glutamine in astrocytes; a further increase occurred after a day, possibly through cells bursting due to reperfusion injury and reversal of glutamate transport in the late stages of disease (Dang et al., 2017). In humans, elevated glutamate levels have been reported in the cerebrospinal fluid and basal ganglia of asphyxiated newborns (Riikonen et al., 1992; Hagberg et al., 1993). Moreover, elevated glutamine levels have been found in MRI-defined punctate necrotic white matter lesions (Wisnowski et al., 2013). Glutamate is taken up into astrocytes for conversion into glutamine and shuttling back to neurons. The finding of elevated glutamine rather than glutamate may be due at least in part to the temporal lag between insult and measurement. An important limitation of in vivo glutamate measurements in preterm newborns is that the peak window of glutamate changes is probably missed, because magnetic resonance measurements are likely to be carried out long after the initial insults in newborns that have already become sick. As such, these findings suggest that disrupted glutamate homeostasis persists in the subacute phase in moderate necrotic white matter injury. Although a relatively small subset of the newborns with punctate lesions also had evidence of cysts, no studies to date have measured glutamatergic metabolism specifically in newborns with severe cystic white matter injury.
Glutamate homeostasis can be disrupted by an acute hypoxic-ischemic event, and the phases of the subsequent excitotoxic injury are well described. During the primary energy failure, oxygen and blood deprivation lead to impairment of ATP production due to failure of oxidative phosphorylation. Astrocytes, with their unique oxidative capacity and ability to upregulate ATP production, are central to maintaining energy metabolism during the first stage of ischemia (Dienel and Hertz, 2005). Impairment of the ATP-dependent Na+/K+ pumps leads to loss of the electrochemical gradient across the cell membrane. If the insult is severe, some cells may die at this early stage via necrosis, due to influx of ions and water, cell swelling, and bursting. Within hours, the necrotic injury due to severe energy failure leads to death of all cellular elements and develops into the white matter cysts (Back, 2017). Depolarization of the cell membrane activates Ca2+ channels in the pre-synaptic terminal, triggering vesicular release of glutamate in the synapse. In astrocytes, hypoxia-ischemia leads to a failure in the astrocytic glutamate uptake system, which also relies on Na+/K+ gradients. The combination of increased synaptic release and reduced astrocytic uptake leads to accumulation of glutamate in the synaptic space and overactivation of post-synaptic ionotropic and metabotropic glutamate receptors (Volpe, 2008). The subsequent intracellular Ca2+ influx triggers activation of phospholipases, endonucleases, proteases, and nitric oxide synthase, with degradation of cellular and extracellular structures, and generation of harmful free radicals and reactive oxygen and nitrogen species. Glutamate leaking outside the synapse activates extrasynaptic NMDA receptors, which, contrarily to the pro-survival action of synaptic NMDA receptors, promotes excitotoxic cell death even further (Parsons and Raymond, 2014). This excitotoxic-oxidative cascade eventually leads to cell damage or death via necrosis, apoptosis, and autophagy in the secondary phase of injury (Olney, 1969; Benveniste et al., 1984; McDonald and Johnston, 1990; Choi, 1992; Thornton et al., 2012; Back, 2017; Descloux et al., 2018) (Figure 1).
In preterm brain injury, comorbidities stimulating inflammation are thought to contribute to disruption of glutamate homeostasis and potentiation of excitotoxicity. TNFα, for example, is one of the most studied cytokines and is emerging as a key link between inflammation and glutamate excitotoxicity (Olmos and Llado, 2014). TNFα has both neuroprotective and neurotoxic effects depending on the different signaling pathways activated by the different receptors. In fact, pharmacological inhibition or genetic deletion after a combined inflammatory and excitotoxic insult is neuroprotective (Aden et al., 2010; Kendall et al., 2011), but knocking out TNFα receptors in the mouse increases susceptibility to hypoxic-ischemic injury (Bruce et al., 1996). TNFα potentiates glutamate excitotoxicity in vitro via complex and interacting mechanisms involving crosstalk between neurons and glial cells and leading to vicious cycles of glutamate and cytokine release. In neurons, TNFα increases the excitatory strength at the synapse by increasing cell surface expression of glutamate receptors and their permeability to Ca2+, while also decreasing expression of inhibitory GABAA receptors (Olmos and Llado, 2014). In microglia, TNFα stimulates autocrine release of TNFα and glutamate by upregulating glutaminase and from hemichannels of gap junctions (Takeuchi et al., 2006). In astrocytes, TNFα stimulates glutamate release via prostaglandin E2 and exacerbates impairment of glutamate transport (Bezzi et al., 1998). Cheung et al. (1998) suggested that glutamate concentration may be key in determining the pathways of cell death, with higher glutamate concentrations preferentially triggering necrosis and lower concentrations leading to apoptosis. Either way, even transient excess of glutamate can start a number of events that ultimately cause death or damage of vulnerable cell populations (Ottersen et al., 1996).
The patterns of excitotoxic injury tend to be different in the preterm and term brain. Experimental evidence suggests that the main cellular target of excitotoxic injury in the preterm brain is pre-oligodendrocytes (Volpe et al., 2011). Glutamate is highly toxic to pre-oligodendrocytes in cell culture and leads to cell death via free radical attack (Oka et al., 1993). The white matter in the rat is much more vulnerable to hypoxia-ischemia at preterm-equivalent age, when pre-oligodendrocytes are predominant, than at term-equivalent age, when mature oligodendrocytes are the major form (Back et al., 2002; Craig et al., 2003; Dean et al., 2011). Indeed, the patterns of hypoxic-ischemic white matter injury seem to be determined primarily by the timing of appearance (Buser et al., 2010) and spatial distribution (Riddle et al., 2006) of pre-oligodendrocytes rather than severity of ischemia itself. Pre-oligodendrocytes are strikingly more vulnerable than immature neurons of the cortex and caudate nucleus in moderate global ischemia in the preterm fetal sheep (Dean et al., 2013; McClendon et al., 2014). Immature neurons are also vulnerable, as NMDA receptors are functionally upregulated, more permeable to calcium and less sensitive to magnesium block (Jantzie et al., 2015).
In physiological conditions, the abundance of glutamate receptors in the white matter is key during early neuronal development, contributing to rapid growth and myelination. However, their abundance also confers increased vulnerability in excitotoxic conditions (Kaindl et al., 2009). Indeed, the selective vulnerability of subplate neurons compared to cortical neurons observed in a preterm model of hypoxia-ischemia has been suggested to originate from an increase of glutamate receptors in these neurons associated with early maturation (McQuillen et al., 2003). Similarly, it has been suggested that selective vulnerability of the deep grey matter and sensorimotor cortex in term hypoxic-ischemic encephalopathy could be related to peaking NMDA receptor expression and proximity to developing glutamatergic circuits (Rocha-Ferreira and Hristova, 2016). As such, developmental expression of key glutamatergic genes in the grey and white matter may contribute to the different patterns of excitotoxic injury (Volpe, 2008).
Overall, the potential sources of extracellular glutamate in the white matter include pre-oligodendrocytes, astrocytes, neurons, ependymal cells, and cells of the choroid plexus (Back and Rosenberg, 2014). While therapies targeting excitotoxicity have so far mostly focused on glutamate receptor blockade, targeting glutamate transport is gathering interest as a potential avenue for neuroprotection by counteracting glutamate accumulation in the first place (Tilleux and Hermans, 2007; Kim et al., 2011; Fontana, 2015; Takahashi et al., 2015).
Maintaining the baseline extracellular glutamate concentrations in the nanomolar range is essential to avoid extracellular glutamate build-up. The brain has no known enzymatic mechanism to metabolize glutamate in the extracellular space, and simple diffusion over short distances is thought to bring only a minor contribution. Hence, the brain relies substantially on intracellular glutamate uptake, and astrocytes provide by far the largest contribution to preventing excitotoxicity through expression of glutamate transporters (Danbolt, 2001; Tzingounis and Wadiche, 2007; Vandenberg and Ryan, 2013). Given their crucial role, it is not surprising that expression of astrocytic glutamate transporters is constitutively high (Zhou and Danbolt, 2013). Crosstalk between neurons and glia relies on tightly controlled extracellular glutamate homeostasis, and it is becoming increasingly evident that neuron-glia interactions are central to both the kinetics of glutamatergic synaptic activity in physiological (Fontana, 2015) and excitotoxic conditions (Carmignoto, 2000). Glutamate is released by astrocytes in immature rat optic nerve in ischemia in vitro (Wilke et al., 2004). Moreover, glutamate transport has been observed in immature axons (Arranz et al., 2008), and impairment has been reported in pre-oligodendrocytes during hypoxia-ischemia, providing a potential mechanism of excitotoxic vulnerability (Oka et al., 1993; Domercq et al., 1999; Fern and Moller, 2000; Deng et al., 2003; Desilva et al., 2007, 2009). The importance of glutamate transport to the integrity of oligodendrocytes and white matter is supported by evidence of extensive excitotoxic injury in oligodendrocytes and axons with experimental inhibition of glutamate transport in the optic nerve in vivo (Domercq et al., 2005).
The five members of the excitatory amino acid transporter (EAAT) family carry out most of the glutamate clearance in the central nervous system (Anderson and Swanson, 2000), especially EAAT1 (SLC1A3, rodent orthologue Glast) and EAAT2 (SLC1A2, rodent orthologue Glt-1) (Bristol and Rothstein, 1996). EAAT2 is the major glutamate transporter in the forebrain, except in the cerebellum, circumventricular organs, and retina, where EAAT1 is prevalent. In physiological conditions, both EAAT1 and EAAT2 are expressed predominantly by astrocytes and localized to the cellular membrane in the adult brain (Danbolt, 2001; Roberts et al., 2014; Takahashi et al., 2015). The high concentration (1 mg/g rat brain tissue), ubiquity (1% of total CNS protein in the adult brain), and high degree of conservation across mammalian species are all indications of physiological importance of EAAT2/Glt-1 (Danbolt, 2001; Fontana, 2015; Danbolt et al., 2016). Unsurprisingly, it is expressed at high density near glutamatergic synapses in developing hippocampal astrocytes, with density and vicinity increasing with neuronal activity (Benediktsson et al., 2012). This transmembrane transporter carries out glutamate uptake through a high affinity energy-dependent process driven by Na+ and K+ gradients. Specifically, glutamate and aspartate are co-transported inside the brain cells with 3 Na+ and 1 H+ for the antiport of 1 K+. EAAT2 is also a selective anion channel, transporting Cl− anions during intermediate conformations, uncoupled from the flux of glutamate (Fontana, 2015).
Several lines of evidence support the central role of EAAT2 expression/function in maintaining extracellular glutamate homeostasis. Pharmacological inhibition of glutamate transport, including EAAT2, leads to rapid extracellular glutamate increase in vitro (Jabaudon et al., 1999) and extended post-synaptic activation mediated by NMDA receptors (Lozovaya et al., 1999). Genetic deletion of Glt-1 via constitutive knockout in the mouse leads to lower body weight, seizures, acute cortical injury in the forebrain, and increased mortality from the second/third postnatal week (Tanaka et al., 1997). Brain tissue from this mouse shows much lower (5%) glutamate transport activity than wild-type, suggesting that Glt-1 is responsible for up to 95% of glutamate transport. This is confirmed by the ability of Glt-1 antibodies to remove 90% of the transport activity in forebrain tissue (Haugeto et al., 1996). Other Glt-1 knockouts have confirmed the obvious phenotype, with lower life span, lower body and brain weight, mild loss of CA1 neurons in the hippocampus, and severe focal neuronal loss in layer II of the neocortex and focal gliosis (Kiryk et al., 2008). A conditional knockout mouse with selective deletion of Glt-1 reproduces this phenotype while ruling out developmental adaptations (Zhou et al., 2014). Heterozygote knockouts, on the other hand, show halved concentrations of Glt-1, but no apparent morphological brain changes, despite an increased risk of traumatic spinal cord injury (Kiryk et al., 2008; Lepore et al., 2011). Inhibition with antisense oligonucleotides in vitro and in vivo induces a rise in extracellular glutamate, excitotoxic injury, and progressive paralysis (Rothstein et al., 1996). On the other hand, selective overexpression in astrocytes is neuroprotective during ischemia (Chao et al., 2010).
Studies of EAAT2 expression point to different patterns depending on cell type, region, developmental age, species, and methodology used (DeSilva et al., 2012). In the adult rat, Glt-1 is expressed in the forebrain, especially in the hippocampus, cortex, striatum, and thalamus as well as in fibrous astrocytes in the white matter (Lehre et al., 1995). The transporter is expressed predominantly by astrocytes but also pre-synaptic axon terminals in the rodent hippocampus and somatosensory cortex (Danbolt, 2001; Chen et al., 2004; Furness et al., 2008; Melone et al., 2009; de Vivo et al., 2010; Danbolt et al., 2016). Neuronal EAAT2 represents no more than 10–20% total EAAT2 (Furness et al., 2008; Danbolt et al., 2016), and while being implicated in adult neuropsychiatric disorders (O’Donovan et al., 2017), neuronal knockout barely affects total Glt-1 protein levels and mouse development (Petr et al., 2015). Conversely, astrocytic knockout leads to a reduction of protein levels to a fifth in the forebrain, lower body weight and increased epilepsy and mortality.
The scenario may be at least in part different in the preterm brain, where transient but more prominent neuronal and pre-oligodendrial expression is observed. During development, dynamic and species-specific changes in both cellular and regional expression have been observed, suggesting that glutamate transporters may be both regulated by and involved in brain development (e.g., participation in the development of the topographic organization). As expected, these changes in rodent Glt-1 expression correspond to changes in total glutamate uptake activity (Ullensvang et al., 1997). Briefly, Glt-1 expression is low until after birth, except for a transient peak of expression in developing axons and oligodendrocytes around mid-late gestation. Glt-1 is expressed in vivo in rat pre-oligodendrocytes, whereas it is no longer detectable in mature oligodendrocytes (DeSilva et al., 2009). Transient neuronal expression is also seen around mid-late gestation in the mouse (Sutherland et al., 1996; Yamada et al., 1998), rat (Furuta et al., 1997), and sheep (Northington et al., 1998). In the fetal rat, Glt-1 is expressed in the amygdala and hippocampus, as well as white matter tracts interconnecting neocortex, basal ganglia, and thalamus (Furuta et al., 1997). In the fetal sheep, Glt-1 is found not only in white matter tracts but also in neuronal bodies and extended to the subplate, cranial nerve nuclei, basal ganglia, and cerebellar cortex, highlighting potential species differences in cellular expression during development (Furuta et al., 1997; Northington et al., 1998, 1999). In the newborn rat at P1, Glt-1 levels are the highest in the spinal cord and moderate in the hippocampus and hypothalamus. Expression increases dramatically from the second postnatal week throughout the central nervous system, especially in the cortex, striatum, caudate nucleus, and hippocampus, reaching adult levels by weeks 4–5 (Rothstein et al., 1994; Levy et al., 1995; Shibata et al., 1996; Sutherland et al., 1996; Furuta et al., 1997; Ullensvang et al., 1997). Astrocyte selectivity is established in the postnatal period in rodents and around mid-late gestation in sheep (Furuta et al., 1997; Takasaki et al., 2008). Nonetheless, Glt-1 is still detected in immature axons at P14–17 (Arranz et al., 2008). The significant developmental changes in Glt-1 after birth may explain why the Glt-1 knockout mice seem to develop normally for the first few weeks and develop seizures and brain injury during postnatal week 3, with many dying by week 4 (Tanaka et al., 1997; Takasaki et al., 2008).
A limited number of studies have investigated developmental regulation of EAAT2 in humans. DeSilva et al. (2012) found that, among EAAT1–3, expression of EAAT2 undergoes particularly prominent maturational changes in post-mortem cortex tissue of preterm and term newborns without neurological disease, all the way into childhood. Consistent with animal studies, EAAT2 expression is generally low until birth and is limited to glia limitans, layer I-III fine astrocytes, and some neuron populations. EAAT2 was found not only in axons but also in the cell body and dendrites of certain neuron populations from as early as 23 gestational weeks up until term and, in some cases, until 8 postnatal months. These neuron populations are layer V pyramidal neurons, layer I neurons (putative Cajal-Retzius cells), and subplate neurons (DeSilva et al., 2012). A great proportion of these neuronal populations is glutamatergic, and it has been suggested that this transient neuronal EAAT2 expression is critical for establishing and orchestrating excitatory transmission during maturation and migration of cortical neurons. Similarly, it could also provide the basis for selective vulnerability to premature excitotoxic injury due to expression of glutamate transporters, which may reverse transport and become sources of extracellular glutamate (Takasaki et al., 2008; DeSilva et al., 2012), as discussed below. This is supported by evidence of selective vulnerability of layer V pyramidal neurons and subplate neurons in human and rat preterm white matter injury (McQuillen et al., 2003; Andiman et al., 2010). The same group reported EAAT2 expression in pre-oligodendrocytes in human fetal white matter at 32 weeks of gestation, during the peak time for premature brain injury, but not at 7 months old, consistent with rat studies (Desilva et al., 2007). EAAT2 expression appeared in the astrocytes of the developing cortex at 41 postconceptional weeks, increasing steeply in the first 1.5 years (DeSilva et al., 2012). Taken together, these findings suggest that the expression of EAAT2/Glt-1 undergoes substantial changes during development and that these changes may contribute to the selective vulnerability of cellular (e.g., immature oligodendrocytes and neurons) and regional (e.g., white matter tracts, hippocampus) targets in preterm brain injury.
Following severe energy failure, the dissipation of the transmembrane gradient impairs astrocytic EAAT2, which relies on transmembrane Na+/K+ gradients. This disruption may involve both quantity and quality of transport activity, i.e., it can manifest as decreased expression and/or impairment of glutamate transport function with establishment of reverse transport. Reverse transport has an outward direction and is driven by the transmembrane gradient of excitatory amino acids independently of ATP and Ca2+ (Nicholls and Attwell, 1990; Szatkowski et al., 1990; Levi and Raiteri, 1993). In this scenario, glutamate transporters become themselves a major source of extracellular glutamate, potentially turning into key contributors of excitotoxic injury in any cells expressing them (Domingues et al., 2010) (Figure 1). While its significance to preterm brain injuries remains to be explored, the importance of reverse transport is supported by evidence that ischemic cell death in the rat striatum can be blocked by an inhibitor of reverse Glt-1 transport (Seki et al., 1999). Moreover, knockout mice lacking Glt-1 are more vulnerable to neuronal death after a short, severe episode of ischemia than wild-type mice, suggesting that Glt-1 is essential for neuroprotection when ischemia is acute; on the other hand, wild-type mice expressing Glt-1 are more vulnerable to neuronal death than mice lacking Glt-1 during extended, chronic ischemia, suggesting that Glt-1 (via reverse transport) becomes neurotoxic when ischemia is prolonged (Mitani and Tanaka, 2003).
Consistent with impairment of glutamate transport, a decrease in glutamate uptake is seen in the hippocampus of rat pups exposed to intrauterine hypoxia following caesarean delivery (Frizzo et al., 2010) and in the cortex, basal ganglia and thalamus of newborn piglets exposed to hypoxia (Jantzie et al., 2010). Loss of Glt-1 expression and/or function has been reported in astrocyte cultures during hypoxia (Dallas et al., 2007) as well as in the adult rat cortex and hippocampus after ischemia (Torp et al., 1995; Rao et al., 2001a,b). In a small study of term-equivalent rats, astrocytic Glt-1 was suppressed in the initial 12 hours in the ischemic core of both the hippocampus and the neocortex, recovered after 48 hours only in the hippocampus, followed by astrogliosis at 72 hours (Fukamachi et al., 2001). In a piglet model of hypoxic-ischemic encephalopathy at term, canonical suppression of Glt-1 in astrocytes of the striatum and hippocampus was accompanied by upregulation in neurons of the striatum (Martin et al., 1997b; Danbolt, 2001; Pow et al., 2004; Desilva et al., 2007, 2012). The striatum is known to be selectively vulnerable to excitotoxicity at term, and this may suggest a potential neuronal response to locally increasing extracellular glutamate levels (Martin et al., 1997a). In P6 rats, exposure to hypoxic preconditioning led to upregulation of Glt-1 in the cortex and suppression in the striatum, with no detectable changes in the hippocampus (Cimarosti et al., 2005). Glt-1 was also suppressed in the white matter in a preterm mouse model of chronic hypoxia, although this model was not subjected to ischemia and showed no sign of reactive astrogliosis (Raymond et al., 2011). Moreover, hypoxia has been found to alter the expression of Glt-1 splice variants in mouse brain and neurons of newborn pigs (Munch et al., 2003; Pow et al., 2004).
Exposure of mouse astrocytes, rat microglia, and human blood macrophages to the bacterial endotoxin lipopolysaccharide (LPS) and the pro-inflammatory cytokine TNFα has been found to enhance EAAT2 expression and glutamate uptake function in vitro (Rimaniol et al., 2000; Persson et al., 2005; O’Shea et al., 2006). On the other hand, TNFα suppresses both glutamate uptake and EAAT2 in a dose-dependent manner (via NF-κB) in human fetal astrocytes (Fine et al., 1996; Liao and Chen, 2001; Su et al., 2003). TNFα also selectively suppresses EAAT2 via NF-κB during hypoxia in vitro (Boycott et al., 2008).
An important finding is that EAAT2 is upregulated in the reactive astrocytes and macrophages of post-mortem human brain tissue from preterm babies with white matter injury compared to controls, suggesting a possible response to hypoxia-ischemia and/or inflammation in the preterm brain (Desilva et al., 2008). Pre-oligodendrocytes in both cases and controls expressed EAAT2, with no qualitative differences in expression, although function was not measured. Upregulation of EAAT2 in reactive astrocytes and macrophages in preterm white matter injury may be an adaptive mechanism to counteract excitotoxicity, or it could be a secondary mechanism due to gliosis. Whether in chronic white matter injury, this upregulation contributed to excitotoxicity via transport reversal remains to be established. Further studies are needed to elucidate how perinatal hypoxia-ischemia and infection/inflammation affect EAAT2 homeostasis, separately and in combination. Interestingly, genome-wide gene expression analysis of reactive astrocytes in two adult mouse models of ischemic stroke and LPS-induced neuroinflammation revealed that at least half of the altered gene expression is specific on the insult, with indication that reactive astrocytes may be neuroprotective in ischemia but detrimental in neuroinflammation (Zamanian et al., 2012). Overall, candidacy of EAAT2 is supported by the fact that dysregulation is implicated in several neurological, neurodegenerative, and psychiatric disorders thought to involve glutamate excitotoxicity (i.e., transient cerebral ischemia, ischemic stroke, epilepsy, traumatic brain injury, amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, chronic pain, Huntington’s disease, HIV-associated cognitive disorder, glioma, major depression, schizophrenia, and addiction) (Danbolt, 2001; Beart and O’Shea, 2007; Fontana, 2015; Karki et al., 2015; Takahashi et al., 2015; Verkhratsky et al., 2016; Zhang et al., 2016; Zhou et al., 2016; Goodwani et al., 2017; O’Donovan et al., 2017; Assefa et al., 2018; Fogarty, 2018; Kim et al., 2018; Parkin et al., 2018).
A better understanding of the role of glutamate transport in preterm brain injuries will require further investigations of EAAT1 in the cerebellum. EAAT1 is highly expressed in cerebellar astrocytes, particularly Bergmann’s glia (Lehre et al., 1995; Danbolt, 2001). The processes of these cells ensheath the Purkinje cell synapses, which have been suggested to be selectively vulnerable to excitotoxicity induced by hypoxia-ischemia (Harding et al., 1984; Shibata et al., 1996). Indeed, EAAT1 is developmentally upregulated from 23 weeks gestation, possibly in conjunction with the maturation of the Purkinje cells. Importantly, EAAT1 undergoes rapid changes in hypoxic-ischemic encephalopathy at term, with a decrease in the molecular layer and an increase in the Purkinje and inner granule cell layer at an early stage. This increase becomes marked at a later stage, potentially pointing to an adaptive neuroprotective mechanism against excitotoxicity (Inage et al., 1998).
Mechanisms leading to loss of expression and/or function are likely to be complex. Ying’s (1997) “deleterious network hypothesis” (1997) suggests that glutamate build-up may lead to detrimental vicious cycles. For example, receptor overactivation may lead to increased energy consumption and oxidative damage, which is known to impair glutamate transporters’ activity and expression, potentially leading to reverse transport with further glutamate release. Ion flux may cause cell swelling, leading to impaired energy metabolism (Danbolt, 2001). Inflammation may further potentiate the risks of excitotoxicity via glutamate transport suppression, including selective effects on EAAT2 (Aden et al., 2010; Kapitanovic Vidak et al., 2012). Evidence to date supports the concept of suicide loops in pre-oligodendrocytes, which could provide both the source and the target for excitotoxic injury in the preterm brain. In this context, the combination of developmental upregulation of EAAT2 and establishment of reverse transport in the context of an energy failure could increase vulnerability of pre-oligodendrocytes to excitotoxic death (Back and Rosenberg, 2014). Similarly, transient expression in neuronal populations could feed into suicide loops and explain the loss of layer V pyramidal neurons accompanying necrotic PVL (Andiman et al., 2010). This is a different mechanism to that hypothesized in the mature brain, where the sources of glutamate killing neurons are thought to be other cells, including astrocytes and excitatory terminals (Lipton and Rosenberg, 1994) or, alternatively, retrograde degeneration from axonal injury. Astrocytes may have a delayed response due to their unique ability to use glycogen as a metabolic fuel during the initial stages of energy deprivation. In this scenario, extracellular glutamate concentrations may rise significantly only after depletion of glycogen stores in astrocytes (Grewer et al., 2008), with a subsequent steep rise in extracellular glutamate and excitotoxic cell death (Gouix et al., 2009). In chronic white matter injury, upregulation of astrocytic EAAT2 may be detrimental when accompanied by establishment of reverse transport. Experimental data are needed to evaluate these hypotheses.
In summary, it is plausible that both up- and downregulation of EAAT2 contribute to disease, depending on animal model, developmental stage, type and severity of the insult, and comorbidities. Regulation and dysregulation of EAAT2 may occur at the level of transcription (including epigenetic regulation), translation, trafficking, transport, and degradation (Karki et al., 2015; Takahashi et al., 2015). Accordingly, treatments aiming at restoring EAAT2 expression are a current area of research in neuroprotection, alongside enhancement of the transport function (Fontana, 2015). Ceftriaxone, a licensed β-lactam antibiotic safe and tolerable for humans, enhances EAAT2 expression and has been shown to be neuroprotective in animal models of several adult excitotoxic disorders. Although no significant effects have been seen in clinical trials for amyotrophic lateral sclerosis and adult stroke, it is already widely used for the treatment of CNS infections in newborns and would therefore be a feasible drug to explore in the context of preterm neuroprotection. Guanosine enhances EAAT2 transport function and has shown neuroprotective effects in rat models of hypoxic-ischemic encephalopathy (Moretto et al., 2005, 2009) and adult cortical focal ischemia, via multiple mechanisms including prevention of free radical attack and pro-inflammatory response (Hansel et al., 2014, 2015). Several other expression and function enhancers of EAAT2 are currently gathering attention as a potential therapeutic approach for a variety of adult disorders and await exploration in the context of the newborn brain (Fontana, 2015). It is currently unknown whether EAAT2 enhancers would restore glutamate uptake or exacerbate reverse transport in the preterm brain. Combination therapies targeting different mechanisms and therapeutic windows will also need exploring, including more established (i.e., magnesium sulfate) and more exploratory therapies (e.g., anti-inflammatory treatment) (Ofek-Shlomai and Berger, 2014).
Genetic risk stratification and pharmacogenomic approaches focusing on interindividual differences in treatment response are gathering interest and, as our healthcare systems develop, the integration of genomic data in clinical care seems an increasingly achievable goal (Rehm, 2017). Exploratory studies have implicated several functional genetic variants involved in glutamate excitotoxicity and inflammation in neurodevelopmental impairment, including as a sequelae of perinatal brain injuries (O’Callaghan et al., 2009, 2012, 2013; Wu et al., 2011; Kapitanovic Vidak et al., 2012). Among these, common genetic variants altering EAAT2 expression have been reported in association with cerebral palsy and neurodevelopmental delay in very preterm newborns (Rajatileka et al., 2017). Replication in larger samples, genome-wide designs and comparison with term brain injuries are needed to consolidate and expand the finding. Identification of panels of genetic variants that collectively increase risk of injury may be integrated with other types of clinical information and help identify high-risk pregnancies. Moreover, integration of genetic information has the potential to contribute to a more personalized approach to the care of the preterm newborn, with recent studies focusing on the interactions between genetic variants and responsiveness to antenatal magnesium sulfate therapy (Costantine et al., 2012; Clark et al., 2018). EAAT2 variants remain to be evaluated in this context.
Future in vivo studies will need to explore whether dysregulation of the main glutamate transporter, EAAT2, is central to the pathogenesis of preterm brain injuries or if it is a secondary process and whether the different cellular effects represent destructive or compensatory mechanisms. As explained by Danbolt (2001), “as long as one variable is not extreme, it will be the combination of several factors that will determine whether the ship will sink,” and several different primary events/changes may share a final common pathway. Well-designed animal model studies will be needed to provide mechanistic evidence. Human post-mortem studies can provide insights into patterns of dysregulation of expression, function, and localization specific to the different types of perinatal brain injuries, though limited by confounding factors, post-mortem artifacts, reproducibility, and sample size. Promising preliminary findings on the neuroprotective effects of EAAT2 suggest that this is certainly an avenue worth exploring.
KL and SP contributed to the conception and design of the review. SP wrote the first draft of the manuscript. All authors revised, read, and approved the submitted version of the manuscript.
This study was funded by the UK Medical Research Council: S115971–102, funding a 3.5-year PhD studentship and the UK Medical Research Council: MR/L010305/1, funding lab facilities and consumables.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aarnoudse-Moens, C. S., Weisglas-Kuperus, N., van Goudoever, J. B., and Oosterlaan, J. (2009). Meta-analysis of neurobehavioral outcomes in very preterm and/or very low birth weight children. Pediatrics 124, 717–728. doi: 10.1542/peds.2008-2816
Abernethy, L. J., Cooke, R. W. I., and Foulder-Hughes, L. (2004). Caudate and hippocampal volumes, intelligence, and motor impairment in 7-year-old children who were born preterm. Pediatr. Res. 55, 884–893. doi: 10.1203/01.PDR.0000117843.21534.49
Abraham, H., Tornoczky, T., Kosztolanyi, G., and Seress, L. (2001). Cell formation in the cortical layers of the developing human cerebellum. Int. J. Dev. Neurosci. 19, 53–62. doi: 10.1016/S0736-5748(00)00065-4
Acarin, L., Gonzalez, B., Hidalgo, J., Castro, A. J., and Castellano, B. (1999). Primary cortical glial reaction versus secondary thalamic glial response in the excitotoxically injured young brain: astroglial response and metallothionein expression. Neuroscience 92, 827–839. doi: 10.1016/S0306-4522(99)00022-6
Aden, U., Favrais, G., Plaisant, F., Winerdal, M., Felderhoff-Mueser, U., Lampa, J., et al. (2010). Systemic inflammation sensitizes the neonatal brain to excitotoxicity through a pro-/anti-inflammatory imbalance: key role of TNFalpha pathway and protection by etanercept. Brain Behav. Immun. 24, 747–758. doi: 10.1016/j.bbi.2009.10.010
Alexander, J. M., Gilstrap, L. C., Cox, S. M., McIntire, D. M., and Leveno, K. J. (1998). Clinical chorioamnionitis and the prognosis for very low birth weight infants. Obstet. Gynecol. 91, 725–729.
Altman, D. I., Powers, W. J., Perlman, J. M., Herscovitch, P., Volpe, S. L., and Volpe, J. J. (1988). Cerebral blood flow requirement for brain viability in newborn infants is lower than in adults. Ann. Neurol. 24, 218–226. doi: 10.1002/ana.410240208
Anblagan, D., Pataky, R., Evans, M. J., Telford, E. J., Serag, A., Sparrow, S., et al. (2016). Association between preterm brain injury and exposure to chorioamnionitis during fetal life. Sci. Rep. 6:37932. doi: 10.1038/srep37932
Anderson, P. J., De Luca, C. R., Hutchinson, E., Spencer-Smith, M. M., Roberts, G., and Doyle, L. W. (2011). Attention problems in a representative sample of extremely preterm/extremely low birth weight children. Dev. Neuropsychol. 36, 57–73. doi: 10.1080/87565641.2011.540538
Anderson, P. J., and Doyle, L. W. (2008). Cognitive and educational deficits in children born extremely preterm. Semin. Perinatol. 32, 51–58. doi: 10.1053/j.semperi.2007.12.009
Anderson, C. M., and Swanson, R. A. (2000). Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32, 1–14. doi: 10.1002/1098-1136(200010)32:1<1::AID-GLIA10>3.0.CO;2-W
Anderson, P. J., Treyvaud, K., Neil, J. J., Cheong, J. L. Y., Hunt, R. W., Thompson, D. K., et al. (2017). Associations of newborn brain magnetic resonance imaging with long-term neurodevelopmental impairments in very preterm children. J. Pediatr. 187, 58.e1–65.e1. doi: 10.1016/j.jpeds.2017.04.059
Andiman, S. E., Haynes, R. L., Trachtenberg, F. L., Billiards, S. S., Folkerth, R. D., Volpe, J. J., et al. (2010). The cerebral cortex overlying periventricular leukomalacia: analysis of pyramidal neurons. Brain Pathol. 20, 803–814. doi: 10.1111/j.1750-3639.2010.00380.x
Arranz, A. M., Hussein, A., Alix, J. J. P., Pérez-Cerdá, F., Allcock, N., Matute, C., et al. (2008). Functional glutamate transport in rodent optic nerve axons and glia. Glia 56, 1353–1367. doi: 10.1002/glia.20703
Assefa, B. T., Gebre, A. K., and Altaye, B. M. (2018). Reactive astrocytes as drug target in Alzheimer’s disease. Biomed. Res. Int. 2018:10. doi: 10.1155/2018/4160247
Azzopardi, D. V., Strohm, B., Edwards, A. D., Dyet, L., Halliday, H. L., Juszczak, E., et al. (2009). Moderate hypothermia to treat perinatal asphyxial encephalopathy. N. Engl. J. Med. 361, 1349–1358. doi: 10.1056/NEJMoa0900854
Back, S. A. (2017). White matter injury in the preterm infant: pathology and mechanisms. Acta Neuropathol. 134, 331–349. doi: 10.1007/s00401-017-1718-6
Back, S. A., Craig, A., Kayton, R. J., Luo, N. L., Meshul, C. K., Allcock, N., et al. (2007a). Hypoxia – ischemia preferentially triggers glutamate depletion from oligodendroglia and axons in perinatal cerebral white matter. J. Cereb. Blood Flow Metab. 27, 334–347. doi: 10.1038/sj.jcbfm.9600344
Back, S. A., Gan, X., Li, Y., Rosenberg, P. A., and Volpe, J. J. (1998). Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J. Neurosci. 18, 6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998
Back, S. A., Han, B. H., Luo, N. L., Chricton, C. A., Xanthoudakis, S., Tam, J., et al. (2002). Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J. Neurosci. 22, 455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002
Back, S. A., Luo, N. L., Borenstein, N. S., Levine, J. M., Volpe, J. J., and Kinney, H. C. (2001). Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 21, 1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001
Back, S. A., Luo, N. L., Mallinson, R. A., O’Malley, J. P., Wallen, L. D., Frei, B., et al. (2005). Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann. Neurol. 58, 108–120. doi: 10.1002/ana.20530
Back, S. A., Riddle, A., and McClure, M. M. (2007b). Maturation-dependent vulnerability of perinatal white matter in premature birth. Stroke 38, 724–730. doi: 10.1161/01.STR.0000254729.27386.05
Back, S. A., and Rosenberg, P. A. (2014). Pathophysiology of glia in perinatal white matter injury. Glia 62, 1790–1815. doi: 10.1002/glia.22658
Ball, G., Boardman, J. P., Rueckert, D., Aljabar, P., Arichi, T., Merchant, N., et al. (2012). The effect of preterm birth on thalamic and cortical development. Cereb. Cortex 22, 1016–1024. doi: 10.1093/cercor/bhr176
Barnett, M. L., Tusor, N., Ball, G., Chew, A., Falconer, S., Aljabar, P., et al. (2018). Exploring the multiple-hit hypothesis of preterm white matter damage using diffusion MRI. Neuroimage Clin. 17, 596–606. doi: 10.1016/j.nicl.2017.11.017
Baud, O., Emilie, D., Pelletier, E., Lacaze-Masmonteil, T., Zupan, V., Fernandez, H., et al. (1999). Amniotic fluid concentrations of interleukin-1beta, interleukin-6 and TNF-alpha in chorioamnionitis before 32 weeks of gestation: histological associations and neonatal outcome. Br. J. Obstet. Gynaecol. 106, 72–77.
Baud, O., Greene, A. E., Li, J., Wang, H., Volpe, J. J., and Rosenberg, P. A. (2004). Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J. Neurosci. 24, 1531–1540. doi: 10.1523/JNEUROSCI.3989-03.2004
Bax, M., Goldstein, M., Rosenbaum, P., Leviton, A., Paneth, N., Dan, B., et al. (2005). Proposed definition and classification of cerebral palsy, April 2005. Dev. Med. Child Neurol. 47, 571–576. doi: 10.1017/S001216220500112X
Bax, M., Tydeman, C., and Flodmark, O. (2006). Clinical and mri correlates of cerebral palsy: the european cerebral palsy study. JAMA 296, 1602–1608. doi: 10.1001/jama.296.13.1602
Beaino, G., Khoshnood, B., Kaminski, M., Pierrat, V., Marret, S., Matis, J., et al. (2010). Predictors of cerebral palsy in very preterm infants: the EPIPAGE prospective population-based cohort study. Dev. Med. Child Neurol. 52, e119–e125. doi: 10.1111/j.1469-8749.2010.03612.x
Beart, P. M., and O’Shea, R. D. (2007). Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br. J. Pharmacol. 150, 5–17. doi: 10.1038/sj.bjp.0706949
Benediktsson, A. M., Marrs, G. S., Tu, J. C., Worley, P. F., Rothstein, J. D., Bergles, D. E., et al. (2012). Neuronal activity regulates glutamate transporter dynamics in developing astrocytes. Glia 60, 175–188. doi: 10.1002/glia.21249
Benveniste, H., Drejer, J., Schousboe, A., and Diemer, N. H. (1984). Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem. 43, 1369–1374. doi: 10.1111/j.1471-4159.1984.tb05396.x
Bergles, D. E., Tzingounis, A. V., and Jahr, C. E. (2002). Comparison of coupled and uncoupled currents during glutamate uptake by GLT-1 transporters. J. Neurosci. 22, 10153–10162. doi: 10.1523/JNEUROSCI.22-23-10153.2002
Bezzi, P., Carmignoto, G., Pasti, L., Vesce, S., Rossi, D., Rizzini, B. L., et al. (1998). Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 391, 281–285. doi: 10.1038/34651
Bi, D., Chen, M., Zhang, X., Wang, H., Xia, L., Shang, Q., et al. (2014). The association between sex-related interleukin-6 gene polymorphisms and the risk for cerebral palsy. J. Neuroinflammation 11:100. doi: 10.1186/1742-2094-11-100
Biran, V., Verney, C., and Ferriero, D. M. (2012). Perinatal cerebellar injury in human and animal models. Neurol. Res. Int. 2012:858929. doi: 10.1155/2012/858929
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Bodensteiner, J. B., and Johnsen, S. D. (2005). Cerebellar injury in the extremely premature infant: newly recognized but relatively common outcome. J. Child Neurol. 20, 139–142. doi: 10.1177/08830738050200021101
Boycott, H. E., Wilkinson, J. A., Boyle, J. P., Pearson, H. A., and Peers, C. (2008). Differential involvement of TNF alpha in hypoxic suppression of astrocyte glutamate transporters. Glia 56, 998–1004. doi: 10.1002/glia.20673
Bristol, L. A., and Rothstein, J. D. (1996). Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann. Neurol. 39, 676–679. doi: 10.1002/ana.410390519
Bruce, A. J., Boling, W., Kindy, M. S., Peschon, J., Kraemer, P. J., Carpenter, M. K., et al. (1996). Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat. Med. 2, 788–794. doi: 10.1038/nm0796-788
Buser, J. R., Segovia, K. N., Dean, J. M., Nelson, K., Beardsley, D., Gong, X., et al. (2010). Timing of appearance of late oligodendrocyte progenitors coincides with enhanced susceptibility of preterm rabbit cerebral white matter to hypoxia-ischemia. J. Cereb. Blood Flow Metab. 30, 1053–1065. doi: 10.1038/jcbfm.2009.286
Carmignoto, G. (2000). Reciprocal communication systems between astrocytes and neurones. Prog. Neurobiol. 62, 561–581. doi: 10.1016/S0301-0082(00)00029-0
Castillo-Melendez, M., Chow, J. A., and Walker, D. W. (2004). Lipid peroxidation, caspase-3 immunoreactivity, and pyknosis in late-gestation fetal sheep brain after umbilical cord occlusion. Pediatr. Res. 55, 864–871. doi: 10.1203/01.PDR.0000115679.86566.C4
Chahboune, H., Ment, L. R., Stewart, W. B., Rothman, D. L., Vaccarino, F. M., Hyder, F., et al. (2009). Hypoxic injury during neonatal development in murine brain: correlation between in vivo DTI findings and behavioral assessment. Cereb. Cortex 19, 2891–2901. doi: 10.1093/cercor/bhp068
Chao, X. D., Fei, F., and Fei, Z. (2010). The role of excitatory amino acid transporters in cerebral ischemia. Neurochem. Res. 35, 1224–1230. doi: 10.1007/s11064-010-0178-3
Chen, W., Mahadomrongkul, V., Berger, U. V., Bassan, M., DeSilva, T., Tanaka, K., et al. (2004). The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J. Neurosci. 24, 1136–1148. doi: 10.1523/JNEUROSCI.1586-03.2004
Cheung, N. S., Pascoe, C. J., Giardina, S. F., John, C. A., and Beart, P. M. (1998). Micromolar L-glutamate induces extensive apoptosis in an apoptotic-necrotic continuum of insult-dependent, excitotoxic injury in cultured cortical neurones. Neuropharmacology 37, 1419–1429. doi: 10.1016/S0028-3908(98)00123-3
Cimarosti, H., Jones, N. M., O’Shea, R. D., Pow, D. V., Salbego, C., and Beart, P. M. (2005). Hypoxic preconditioning in neonatal rat brain involves regulation of excitatory amino acid transporter 2 and estrogen receptor alpha. Neurosci. Lett. 385, 52–57. doi: 10.1016/j.neulet.2005.05.006
Clark, E. A. S., Weiner, S. J., Rouse, D. J., Mercer, B. M., Reddy, U. M., Iams, J. D., et al. (2018). Genetic variation, magnesium sulfate exposure, and adverse neurodevelopmental outcomes following preterm birth. Am. J. Perinatol. 35, 1012–1022. doi: 10.1055/s-0038-1635109
Constantinou, J. C., Adamson-Macedo, E. N., Mirmiran, M., and Fleisher, B. E. (2007). Movement, imaging and neurobehavioral assessment as predictors of cerebral palsy in preterm infants. J. Perinatol. 27, 225–229. doi: 10.1038/sj.jp.7211664
Costantine, M. M., Clark, E. A., Lai, Y., Rouse, D. J., Spong, C. Y., Mercer, B. M., et al. (2012). Association of polymorphisms in neuroprotection and oxidative stress genes and neurodevelopmental outcomes after preterm birth. Obstet. Gynecol. 120, 542–550. doi: 10.1097/AOG.0b013e318265f232
Counsell, S. J., Allsop, J. M., Harrison, M. C., Larkman, D. J., Kennea, N. L., Kapellou, O., et al. (2003). Diffusion-weighted imaging of the brain in preterm infants with focal and diffuse white matter abnormality. Pediatrics 112, 1–7. doi: 10.1542/peds.112.1.1
Counsell, S. J., and Boardman, J. P. (2005). Differential brain growth in the infant born preterm: current knowledge and future developments from brain imaging. Semin. Fetal Neonatal Med. 10, 403–410. doi: 10.1016/j.siny.2005.05.003
Craig, A., Ling Luo, N., Beardsley, D. J., Wingate-Pearse, N., Walker, D. W., Hohimer, A. R., et al. (2003). Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp. Neurol. 181, 231–240. doi: 10.1016/S0014-4886(03)00032-3
Curtis, D. R., and Johnston, G. A. (1974). Amino acid transmitters in the mammalian central nervous system. Ergeb. Physiol. 69, 97–188.
Dallas, M., Boycott, H. E., Atkinson, L., Miller, A., Boyle, J. P., Pearson, H. A., et al. (2007). Hypoxia suppresses glutamate transport in astrocytes. J. Neurosci. 27, 3946–3955. doi: 10.1523/JNEUROSCI.5030-06.2007
Dammann, O. (2007). Persistent neuro-inflammation in cerebral palsy: a therapeutic window of opportunity? Acta Paediatr. 96, 6–7. doi: 10.1111/j.1651-2227.2007.00097.x
Dammann, O., Kuban, K. C., and Leviton, A. (2002). Perinatal infection, fetal inflammatory response, white matter damage, and cognitive limitations in children born preterm. Ment. Retard. Dev. Disabil. Res. Rev. 8, 46–50. doi: 10.1002/mrdd.10005
Dammann, O., and Leviton, A. (1998). Infection remote from the brain, neonatal white matter damage, and cerebral palsy in the preterm infant. Semin. Pediatr. Neurol. 5, 190–201. doi: 10.1016/S1071-9091(98)80034-X
Dammann, O., and Leviton, A. (2000). Role of the fetus in perinatal infection and neonatal brain damage. Curr. Opin. Pediatr. 12, 99–104. doi: 10.1097/00008480-200004000-00002
Dammann, O., and Leviton, A. (2004). Inflammatory brain damage in preterm newborns – dry numbers, wet lab, and causal inferences. Early Hum. Dev. 79, 1–15. doi: 10.1016/j.earlhumdev.2004.04.009
Danbolt, N. C. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105. doi: 10.1016/S0301-0082(00)00067-8
Danbolt, N. C., Furness, D. N., and Zhou, Y. (2016). Neuronal vs glial glutamate uptake: resolving the conundrum. Neurochem. Int. 98, 29–45. doi: 10.1016/j.neuint.2016.05.009
Dang, Y. X., Shi, K. N., and Wang, X. M. (2017). Early changes in glutamate metabolism and perfusion in basal ganglia following hypoxia-ischemia in neonatal piglets: a multi-sequence 3.0T MR study. Front. Physiol. 8:237. doi: 10.3389/fphys.2017.00237
De Reuck, J. L. (1984). Cerebral angioarchitecture and perinatal brain lesions in premature and full-term infants. Acta Neurol. Scand. 70, 391–395. doi: 10.1111/j.1600-0404.1984.tb00843.x
de Vivo, L., Melone, M., Rothstein, J. D., and Conti, F. (2010). GLT-1 promoter activity in astrocytes and neurons of mouse hippocampus and somatic sensory cortex. Front. Neuroanat. 3:31. doi: 10.3389/neuro.05.031.2009
de Vries, L. S., Eken, P., and Dubowitz, L. M. S. (1992). The spectrum of leukomalacia using cranial ultrasound. Behav. Brain Res. 49, 1–6. doi: 10.1016/S0166-4328(05)80189-5
De Vries, L. S., Van Haastert, I. L., Rademaker, K. J., Koopman, C., and Groenendaal, F. (2004). Ultrasound abnormalities preceding cerebral palsy in high-risk preterm infants. J. Pediatr. 144, 815–820. doi: 10.1016/j.jpeds.2004.03.034
Dean, J. M., McClendon, E., Hansen, K., Azimi-Zonooz, A., Chen, K., Riddle, A., et al. (2013). Prenatal cerebral ischemia disrupts MRI-defined cortical microstructure through disturbances in neuronal arborization. Sci. Transl. Med. 5:168ra7. doi: 10.1126/scitranslmed.3004669
Dean, J. M., Moravec, M. D., Grafe, M., Abend, N., Ren, J., Gong, X., et al. (2011). Strain-specific differences in perinatal rodent oligodendrocyte lineage progression and its correlation with human. Dev. Neurosci. 33, 251–260. doi: 10.1159/000327242
Delobel-Ayoub, M., Arnaud, C., White-Koning, M., Casper, C., Pierrat, V., Garel, M., et al. (2009). Behavioral problems and cognitive performance at 5 years of age after very preterm birth: the EPIPAGE study. Pediatrics 123, 1485–1492. doi: 10.1542/peds.2008-1216
Deng, W. (2010). Neurobiology of injury to the developing brain. Nat. Rev. Neurol. 6, 328–336. doi: 10.1038/nrneurol.2010.53
Deng, W., Rosenberg, P. A., Volpe, J. J., and Jensen, F. E. (2003). Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc. Natl. Acad. Sci. USA 100, 6801–6806. doi: 10.1073/pnas.1136624100
Descloux, C., Ginet, V., Rummel, C., Truttmann, A. C., and Puyal, J. (2018). Enhanced autophagy contributes to excitotoxic lesions in a rat model of preterm brain injury. Cell Death Dis. 9:853. doi: 10.1038/s41419-018-0916-z
Desilva, T. M., Billiards, S. S., Borenstein, N. S., Trachtenberg, F. L., Volpe, J. J., Kinney, H. C., et al. (2008). Glutamate transporter EAAT2 expression is up-regulated in reactive astrocytes in human periventricular leukomalacia. J. Comp. Neurol. 508, 238–248. doi: 10.1002/cne.21667
DeSilva, T. M., Borenstein, N. S., Volpe, J. J., Kinney, H. C., and Rosenberg, P. A. (2012). Expression of EAAT2 in neurons and protoplasmic astrocytes during human cortical development. J. Comp. Neurol. 520, 3912–3932. doi: 10.1002/cne.23130
DeSilva, T. M., Kabakov, A. Y., Goldhoff, P. E., Volpe, J. J., and Rosenberg, P. A. (2009). Regulation of glutamate transport in developing rat oligodendrocytes. J. Neurosci. 29, 7898–7908. doi: 10.1523/JNEUROSCI.6129-08.2009
Desilva, T. M., Kinney, H. C., Borenstein, N. S., Trachtenberg, F. L., Irwin, N., Volpe, J. J., et al. (2007). The glutamate transporter EAAT2 is transiently expressed in developing human cerebral white matter. J. Comp. Neurol. 501, 879–890. doi: 10.1002/cne.21289
Dienel, G. A., and Hertz, L. (2005). Astrocytic contributions to bioenergetics of cerebral ischemia. Glia 50, 362–388. doi: 10.1002/glia.20157
Doble, A. (1999). The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol. Ther. 81, 163–221. doi: 10.1016/S0163-7258(98)00042-4
Dollner, H., Vatten, L., Halgunset, J., Rahimipoor, S., and Austgulen, R. (2002). Histologic chorioamnionitis and umbilical serum levels of pro-inflammatory cytokines and cytokine inhibitors. BJOG 109, 534–539. doi: 10.1111/j.1471-0528.2002.01028.x
Domercq, M., Etxebarria, E., Perez-Samartin, A., and Matute, C. (2005). Excitotoxic oligodendrocyte death and axonal damage induced by glutamate transporter inhibition. Glia 52, 36–46. doi: 10.1002/glia.20221
Domercq, M., Sánchez-Gómez, M. V., Areso, P., and Matute, C. (1999). Expression of glutamate transporters in rat optic nerve oligodendrocytes. Eur. J. Neurosci. 11, 2226–2236. doi: 10.1046/j.1460-9568.1999.00639.x
Domingues, A. M., Taylor, M., and Fern, R. (2010). Glia as transmitter sources and sensors in health and disease. Neurochem. Int. 57, 359–366. doi: 10.1016/j.neuint.2010.03.024
Dommergues, M. A., Patkai, J., Renauld, J. C., Evrard, P., and Gressens, P. (2000). Proinflammatory cytokines and interleukin-9 exacerbate excitotoxic lesions of the newborn murine neopallium. Ann. Neurol. 47, 54–63. doi: 10.1002/1531-8249(200001)47:1<54::AID-ANA10>3.0.CO;2-Y
Doyle, L. W., Crowther, C. A., Middleton, P., Marret, S., and Rouse, D. (2009). Magnesium sulphate for women at risk of preterm birth for neuroprotection of the fetus. Cochrane Database Syst. Rev. CD004661. doi: 10.1002/14651858.CD004661.pub3
Duerden, E. G., Taylor, M. J., and Miller, S. P. (2013). Brain development in infants born preterm: looking beyond injury. Semin. Pediatr. Neurol. 20, 65–74. doi: 10.1016/j.spen.2013.06.007
Duggan, P. J., Maalouf, E. F., Watts, T. L., Sullivan, M. H., Counsell, S. J., Allsop, J., et al. (2001). Intrauterine T-cell activation and increased proinflammatory cytokine concentrations in preterm infants with cerebral lesions. Lancet 358, 1699–1700. doi: 10.1016/S0140-6736(01)06723-X
Eklind, S., Mallard, C., Leverin, A. L., Gilland, E., Blomgren, K., Mattsby-Baltzer, I., et al. (2001). Bacterial endotoxin sensitizes the immature brain to hypoxic--ischaemic injury. Eur. J. Neurosci. 13, 1101–1106. doi: 10.1046/j.0953-816x.2001.01474.x
Ellison, V. J., Mocatta, T. J., Winterbourn, C. C., Darlow, B. A., Volpe, J. J., and Inder, T. E. (2005). The relationship of CSF and plasma cytokine levels to cerebral white matter injury in the premature newborn. Pediatr. Res. 57, 282–286. doi: 10.1203/01.PDR.0000148286.53572.95
Favrais, G., Schwendimann, L., Gressens, P., and Lelievre, V. (2007). Cyclooxygenase-2 mediates the sensitizing effects of systemic IL-1-beta on excitotoxic brain lesions in newborn mice. Neurobiol. Dis. 25, 496–505. doi: 10.1016/j.nbd.2006.10.012
Fern, R., and Moller, T. (2000). Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J. Neurosci. 20, 34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000
Fetters, L., and Huang, H. H. (2007). Motor development and sleep, play, and feeding positions in very-low-birthweight infants with and without white matter disease. Dev. Med. Child Neurol. 49, 807–813. doi: 10.1111/j.1469-8749.2007.00807.x
Fine, S. M., Angel, R. A., Perry, S. W., Epstein, L. G., Rothstein, J. D., Dewhurst, S., et al. (1996). Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J. Biol. Chem. 271, 15303–15306. doi: 10.1074/jbc.271.26.15303
Fleiss, B., and Gressens, P. (2012). Tertiary mechanisms of brain damage: a new hope for treatment of cerebral palsy? Lancet Neurol. 11, 556–566. doi: 10.1016/S1474-4422(12)70058-3
Fogarty, M. J. (2018). Driven to decay: excitability and synaptic abnormalities in amyotrophic lateral sclerosis. Brain Res. Bull. 140, 318–333. doi: 10.1016/j.brainresbull.2018.05.023
Follett, P. L., Deng, W., Dai, W., Talos, D. M., Massillon, L. J., Rosenberg, P. A., et al. (2004). Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J. Neurosci. 24, 4412–4420. doi: 10.1523/JNEUROSCI.0477-04.2004
Follett, P. L., Rosenberg, P. A., Volpe, J. J., and Jensen, F. E. (2000). NBQX attenuates excitotoxic injury in developing white matter. J. Neurosci. 20, 9235–9241. doi: 10.1523/JNEUROSCI.20-24-09235.2000
Fontana, A. C. (2015). Current approaches to enhance glutamate transporter function and expression. J. Neurochem. 134, 982–1007. doi: 10.1111/jnc.13200
Fragoso, G., Martinez-Bermudez, A. K., Liu, H. N., Khorchid, A., Chemtob, S., Mushynski, W. E., et al. (2004). Developmental differences in HO-induced oligodendrocyte cell death: role of glutathione, mitogen-activated protein kinases and caspase 3. J. Neurochem. 90, 392–404. doi: 10.1111/j.1471-4159.2004.02488.x
Frizzo, J. K., Cardoso, M. P., de Assis, A. M., Perry, M. L., Volonte, C., and Frizzo, M. E. (2010). Effects of acute perinatal asphyxia in the rat hippocampus. Cell. Mol. Neurobiol. 30, 683–692. doi: 10.1007/s10571-009-9492-1
Fukamachi, S., Furuta, A., Ikeda, T., Ikenoue, T., Kaneoka, T., Rothstein, J. D., et al. (2001). Altered expressions of glutamate transporter subtypes in rat model of neonatal cerebral hypoxia-ischemia. Brain Res. Dev. Brain Res. 132, 131–139. doi: 10.1016/S0165-3806(01)00303-0
Furness, D. N., Dehnes, Y., Akhtar, A. Q., Rossi, D. J., Hamann, M., Grutle, N. J., et al. (2008). A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience 157, 80–94. doi: 10.1016/j.neuroscience.2008.08.043
Furuta, A., Rothstein, J. D., and Martin, L. J. (1997). Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J. Neurosci. 17, 8363–8375. doi: 10.1523/JNEUROSCI.17-21-08363.1997
Gilles, F., Gressens, P., Dammann, O., and Leviton, A. (2018). Hypoxia–ischemia is not an antecedent of most preterm brain damage: the illusion of validity. Dev. Med. Child Neurol. 60, 120–125. doi: 10.1111/dmcn.13483
Gimenez, M., Junque, C., Narberhaus, A., Botet, F., Bargallo, N., and Mercader, J. M. (2006). Correlations of thalamic reductions with verbal fluency impairment in those born prematurely. Neuroreport 17, 463–466. doi: 10.1097/01.wnr.0000209008.93846.24
Glass, H. C., Costarino, A. T., Stayer, S. A., Brett, C., Cladis, F., and Davis, P. J. (2015). Outcomes for extremely premature infants. Anesth. Analg. 120, 1337–1351. doi: 10.1213/ANE.0000000000000705
Glass, H. C., Fujimoto, S., Ceppi-Cozzio, C., Bartha, A. I., Vigneron, D. B., Barkovich, A. J., et al. (2008). White-matter injury is associated with impaired gaze in premature infants. Pediatr. Neurol. 38, 10–15. doi: 10.1016/j.pediatrneurol.2007.08.019
Gleason, C. A., Hamm, C., and Jones, M. D. Jr. (1989). Cerebral blood flow, oxygenation, and carbohydrate metabolism in immature fetal sheep in utero. Am. J. Phys. 256, R1264–R1268. doi: 10.1152/ajpregu.1989.256.6.R1264
Gluckman, P. D., Wyatt, J. S., Azzopardi, D., Ballard, R., Edwards, A. D., Ferriero, D. M., et al. (2005). Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet 365, 663–670. doi: 10.1016/S0140-6736(05)70932-6
Goodwani, S., Saternos, H., Alasmari, F., and Sari, Y. (2017). Metabotropic and ionotropic glutamate receptors as potential targets for the treatment of alcohol use disorder. Neurosci. Biobehav. Rev. 77, 14–31. doi: 10.1016/j.neubiorev.2017.02.024
Gopagondanahalli, K. R., Li, J., Fahey, M. C., Hunt, R. W., Jenkin, G., Miller, S. L., et al. (2016). Preterm hypoxic-ischemic encephalopathy. Front. Pediatr. 4:114. doi: 10.3389/fped.2016.00114
Gouix, E., Leveille, F., Nicole, O., Melon, C., Had-Aissouni, L., and Buisson, A. (2009). Reverse glial glutamate uptake triggers neuronal cell death through extrasynaptic NMDA receptor activation. Mol. Cell. Neurosci. 40, 463–473. doi: 10.1016/j.mcn.2009.01.002
Greisen, G. (1986). Cerebral blood flow in preterm infants during the first week of life. Acta Paediatr. Scand. 75, 43–51.
Gressens, P., Marret, S., and Evrard, P. (1996). Developmental spectrum of the excitotoxic cascade induced by ibotenate: a model of hypoxic insults in fetuses and neonates. Neuropathol. Appl. Neurobiol. 22, 498–502. doi: 10.1111/j.1365-2990.1996.tb01123.x
Grether, J. K., and Nelson, K. B. (1997). Maternal infection and cerebral palsy in infants of normal birth weight. JAMA 278, 207–211. doi: 10.1001/jama.1997.03550030047032
Grewer, C., Gameiro, A., Zhang, Z., Tao, Z., Braams, S., and Rauen, T. (2008). Glutamate forward and reverse transport: from molecular mechanism to transporter-mediated release after ischemia. IUBMB Life 60, 609–619. doi: 10.1002/iub.98
Grewer, C., and Rauen, T. (2005). Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J. Membr. Biol. 203, 1–20. doi: 10.1007/s00232-004-0731-6
Hagberg, H., Gilland, E., Diemer, N. H., and Andine, P. (1994). Hypoxia-ischemia in the neonatal rat brain: histopathology after post-treatment with NMDA and non-NMDA receptor antagonists. Biol. Neonate 66, 205–213.
Hagberg, H., Mallard, C., Ferriero, D. M., Vannucci, S. J., Levison, S. W., Vexler, Z. S., et al. (2015). The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 11, 192–208. doi: 10.1038/nrneurol.2015.13
Hagberg, H., Peebles, D., and Mallard, C. (2002). Models of white matter injury: comparison of infectious, hypoxic-ischemic, and excitotoxic insults. Ment. Retard. Dev. Disabil. Res. Rev. 8, 30–38. doi: 10.1002/mrdd.10007
Hagberg, H., Thornberg, E., Blennow, M., Kjellmer, I., Lagercrantz, H., Thiringer, K., et al. (1993). Excitatory amino acids in the cerebrospinal fluid of asphyxiated infants: relationship to hypoxic-ischemic encephalopathy. Acta Paediatr. 82, 925–929. doi: 10.1111/j.1651-2227.1993.tb12601.x
Hansel, G., Ramos, D. B., Delgado, C. A., Souza, D. G., Almeida, R. F., Portela, L. V., et al. (2014). The potential therapeutic effect of guanosine after cortical focal ischemia in rats. PLoS One 9:e90693. doi: 10.1371/journal.pone.0090693
Hansel, G., Tonon, A. C., Guella, F. L., Pettenuzzo, L. F., Duarte, T., Duarte, M. M., et al. (2015). Guanosine protects against cortical focal ischemia. Involvement of inflammatory response. Mol. Neurobiol. 52, 1791–1803. doi: 10.1007/s12035-014-8978-0
Harding, R., Rawson, J. A., Griffiths, P. A., and Thorburn, G. D. (1984). The influence of acute hypoxia and sleep states on the electrical activity of the cerebellum in the sheep fetus. Electroencephalogr. Clin. Neurophysiol. 57, 166–173. doi: 10.1016/0013-4694(84)90175-5
Haugeto, O., Ullensvang, K., Levy, L. M., Chaudhry, F. A., Honore, T., Nielsen, M., et al. (1996). Brain glutamate transporter proteins form homomultimers. J. Biol. Chem. 271, 27715–27722. doi: 10.1074/jbc.271.44.27715
Heep, A., Behrendt, D., Nitsch, P., Fimmers, R., Bartmann, P., and Dembinski, J. (2003). Increased serum levels of interleukin 6 are associated with severe intraventricular haemorrhage in extremely premature infants. Arch. Dis. Child. Fetal Neonatal Ed. 88, F501–F504. doi: 10.1136/fn.88.6.F501
Himpens, E., Van den Broeck, C., Oostra, A., Calders, P., and Vanhaesebrouck, P. (2008). Prevalence, type, distribution, and severity of cerebral palsy in relation to gestational age: a meta-analytic review. Dev. Med. Child Neurol. 50, 334–340. doi: 10.1111/j.1469-8749.2008.02047.x
Hirvonen, M., Ojala, R., Korhonen, P., Haataja, P., Eriksson, K., Gissler, M., et al. (2014). Cerebral palsy among children born moderately and late preterm. Pediatrics 134, e1584–e1593. doi: 10.1542/peds.2014-0945
Huang, J., Zhang, L., Kang, B., Zhu, T., Li, Y., Zhao, F., et al. (2017). Association between perinatal hypoxic-ischemia and periventricular leukomalacia in preterm infants: a systematic review and meta-analysis. PLoS One 12:e0184993. doi: 10.1371/journal.pone.0189461
Hutton, L. C., Yan, E., Yawno, T., Castillo-Melendez, M., Hirst, J. J., and Walker, D. W. (2014). Injury of the developing cerebellum: a brief review of the effects of endotoxin and asphyxial challenges in the late gestation sheep fetus. Cerebellum 13, 777–786. doi: 10.1007/s12311-014-0602-3
Iida, K., Takashima, S., and Ueda, K. (1995). Immunohistochemical study of myelination and oligodendrocyte in infants with periventricular leukomalacia. Pediatr. Neurol. 13, 296–304. doi: 10.1016/0887-8994(95)00192-1
Ikeda, T., Mishima, K., Aoo, N., Egashira, N., Iwasaki, K., Fujiwara, M., et al. (2004). Combination treatment of neonatal rats with hypoxia-ischemia and endotoxin induces long-lasting memory and learning impairment that is associated with extended cerebral damage. Am. J. Obstet. Gynecol. 191, 2132–2141. doi: 10.1016/j.ajog.2004.04.039
Inage, Y. W., Itoh, M., and Takashima, S. (2000). Correlation between cerebrovascular maturity and periventricular leukomalacia. Pediatr. Neurol. 22, 204–208. doi: 10.1016/S0887-8994(99)00153-8
Inage, Y. W., Itoh, M., Wada, K., and Takashima, S. (1998). Expression of two glutamate transporters, GLAST and EAAT4, in the human cerebellum: their correlation in development and neonatal hypoxic-ischemic damage. J. Neuropathol. Exp. Neurol. 57, 554–562. doi: 10.1097/00005072-199806000-00003
Inder, T. E., Anderson, N. J., Spencer, C., Wells, S., and Volpe, J. J. (2003). White matter injury in the premature infant: a comparison between serial cranial sonographic and MR findings at term. AJNR Am. J. Neuroradiol. 24, 805–809.
Inder, T. E., Warfield, S. K., Wang, H., Huppi, P. S., and Volpe, J. J. (2005). Abnormal cerebral structure is present at term in premature infants. Pediatrics 115, 286–294. doi: 10.1542/peds.2004-0326
Innocenti, G. M., and Berbel, P. (1991a). Analysis of an experimental cortical network: I. Architectonics of visual areas 17 and 18 after neonatal injections of ibotenic acid; similarities with human microgyria. J. Neural Transplant. Plast. 2, 1–28.
Innocenti, G. M., and Berbel, P. (1991b). Analysis of an experimental cortical network: II. Connections of visual areas 17 and 18 after neonatal injections of ibotenic acid. J. Neural Transplant. Plast. 2, 29–54.
Jabaudon, D., Shimamoto, K., Yasuda-Kamatani, Y., Scanziani, M., Gahwiler, B. H., and Gerber, U. (1999). Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc. Natl. Acad. Sci. USA 96, 8733–8738.
Jacobs, S. E., Morley, C. J., Inder, T. E., Stewart, M. J., Smith, K. R., McNamara, P. J., et al. (2011). Whole-body hypothermia for term and near-term newborns with hypoxic-ischemic encephalopathy: a randomized controlled trial. Arch. Pediatr. Adolesc. Med. 165, 692–700. doi: 10.1001/archpediatrics.2011.43
Jantzie, L. L., Cheung, P. Y., Johnson, S. T., Bigam, D. L., and Todd, K. G. (2010). Cerebral amino acid profiles after hypoxia-reoxygenation and N-acetylcysteine treatment in the newborn piglet. Neonatology 97, 195–203. doi: 10.1159/000252972
Jantzie, L. L., Talos, D. M., Jackson, M. C., Park, H. K., Graham, D. A., Lechpammer, M., et al. (2015). Developmental expression of N-methyl-D-aspartate (NMDA) receptor subunits in human white and gray matter: potential mechanism of increased vulnerability in the immature brain. Cereb. Cortex 25, 482–495. doi: 10.1093/cercor/bht246
Johnsen, S. D., Bodensteiner, J. B., and Lotze, T. E. (2005). Frequency and nature of cerebellar injury in the extremely premature survivor with cerebral palsy. J. Child Neurol. 20, 60–64. doi: 10.1177/08830738050200011001
Johnston, M. V. (2005). Excitotoxicity in perinatal brain injury. Brain Pathol. 15, 234–240. doi: 10.1111/j.1750-3639.2005.tb00526.x
Kaindl, A. M., Favrais, G., and Gressens, P. (2009). Molecular mechanisms involved in injury to the preterm brain. J. Child Neurol. 24, 1112–1118. doi: 10.1177/0883073809337920
Kapitanovic Vidak, H., Catela Ivkovic, T., Jokic, M., Spaventi, R., and Kapitanovic, S. (2012). The association between proinflammatory cytokine polymorphisms and cerebral palsy in very preterm infants. Cytokine 58, 57–64. doi: 10.1016/j.cyto.2011.12.018
Karki, P., Smith, K., Johnson, J., Aschner, M., and Lee, E. Y. (2015). Genetic dys-regulation of astrocytic glutamate transporter EAAT2 and its implications in neurological disorders and manganese toxicity. Neurochem. Res. 40, 380–388. doi: 10.1007/s11064-014-1391-2
Kaukola, T., Herva, R., Perhomaa, M., Paakko, E., Kingsmore, S., Vainionpaa, L., et al. (2006). Population cohort associating chorioamnionitis, cord inflammatory cytokines and neurologic outcome in very preterm, extremely low birth weight infants. Pediatr. Res. 59, 478–483. doi: 10.1203/01.pdr.0000182596.66175.ee
Kaukola, T., Satyaraj, E., Patel, D. D., Tchernev, V. T., Grimwade, B. G., Kingsmore, S. F., et al. (2004). Cerebral palsy is characterized by protein mediators in cord serum. Ann. Neurol. 55, 186–194. doi: 10.1002/ana.10809
Kendall, G. S., Hristova, M., Horn, S., Dafou, D., Acosta-Saltos, A., Almolda, B., et al. (2011). TNF gene cluster deletion abolishes lipopolysaccharide-mediated sensitization of the neonatal brain to hypoxic ischemic insult. Lab. Investig. 91, 328–341. doi: 10.1038/labinvest.2010.192
Kesler, S. R., Reiss, A. L., Vohr, B., Watson, C., Schneider, K. C., Katz, K. H., et al. (2008). Brain volume reductions within multiple cognitive systems in male preterm children at age twelve. J. Pediatr. 152, 513–520, 520.e1. doi: 10.1016/j.jpeds.2007.08.009
Khandaker, G., Smithers-Sheedy, H., Islam, J., Alam, M., Jung, J., Novak, I., et al. (2015). Bangladesh cerebral palsy register (BCPR): a pilot study to develop a national cerebral palsy (CP) register with surveillance of children for CP. BMC Neurol. 15:173. doi: 10.1186/s12883-015-0427-9
Kim, R., Healey, K. L., Sepulveda-Orengo, M. T., and Reissner, K. J. (2018). Astroglial correlates of neuropsychiatric disease: from astrocytopathy to astrogliosis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 87, 126–146. doi: 10.1016/j.pnpbp.2017.10.002
Kim, K., Lee, S.-G., Kegelman, T. P., Su, Z.-Z., Das, S. K., Dash, R., et al. (2011). Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J. Cell. Physiol. 226, 2484–2493. doi: 10.1002/jcp.22609
Kiryk, A., Aida, T., Tanaka, K., Banerjee, P., Wilczynski, G. M., Meyza, K., et al. (2008). Behavioral characterization of GLT1 (+/−) mice as a model of mild glutamatergic hyperfunction. Neurotox. Res. 13, 19–30. doi: 10.1007/BF03033364
Korzeniewski, S. J., Romero, R., Cortez, J., Pappas, A., Schwartz, A. G., Kim, C. J., et al. (2014). A “multi-hit” model of neonatal white matter injury: cumulative contributions of chronic placental inflammation, acute fetal inflammation and postnatal inflammatory events. J. Perinat. Med. 42, 731–743. doi: 10.1515/jpm-2014-0250
Laptook, A. R. (2016). Birth asphyxia and hypoxic-ischemic brain injury in the preterm infant. Clin. Perinatol. 43, 529–545. doi: 10.1016/j.clp.2016.04.010
Larouche, A., Roy, M., Kadhim, H., Tsanaclis, A. M., Fortin, D., and Sebire, G. (2005). Neuronal injuries induced by perinatal hypoxic-ischemic insults are potentiated by prenatal exposure to lipopolysaccharide: animal model for perinatally acquired encephalopathy. Dev. Neurosci. 27, 134–142. doi: 10.1159/000085985
Lawrence, E. J., Froudist-Walsh, S., Neilan, R., Nam, K. W., Giampietro, V., McGuire, P., et al. (2014). Motor fMRI and cortical grey matter volume in adults born very preterm. Dev. Cogn. Neurosci. 10, 1–9. doi: 10.1016/j.dcn.2014.06.002
Lehnardt, S., Massillon, L., Follett, P., Jensen, F. E., Ratan, R., Rosenberg, P. A., et al. (2003). Activation of innate immunity in the CNS triggers neurodegeneration through a toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 100, 8514–8519. doi: 10.1073/pnas.1432609100
Lehre, K. P., Levy, L. M., Ottersen, O. P., Storm-Mathisen, J., and Danbolt, N. C. (1995). Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J. Neurosci. 15, 1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995
Lepore, A. C., O’Donnell, J., Kim, A. S., Yang, E. J., Tuteja, A., Haidet-Phillips, A., et al. (2011). Reduction in expression of the astrocyte glutamate transporter, GLT1, worsens functional and histological outcomes following traumatic spinal cord injury. Glia 59, 1996–2005. doi: 10.1002/glia.21241
Levi, G., and Raiteri, M. (1993). Carrier-mediated release of neurotransmitters. Trends Neurosci. 16, 415–419.
Leviton, A., Fichorova, R. N., O’Shea, T. M., Kuban, K., Paneth, N., Dammann, O., et al. (2013). Two-hit model of brain damage in the very preterm newborn: small for gestational age and postnatal systemic inflammation. Pediatr. Res. 73, 362–370. doi: 10.1038/pr.2012.188
Leviton, A., and Paneth, N. (1990). White matter damage in preterm newborns – an epidemiologic perspective. Early Hum. Dev. 24, 1–22. doi: 10.1016/0378-3782(90)90002-Z
Leviton, A., Paneth, N., Reuss, M. L., Susser, M., Allred, E. N., Dammann, O., et al. (1999). Maternal infection, fetal inflammatory response, and brain damage in very low birth weight infants. Developmental epidemiology network investigators. Pediatr. Res. 46, 566–575. doi: 10.1203/00006450-199911000-00013
Levy, L. M., Lehre, K. P., Walaas, S. I., Storm-Mathisen, J., and Danbolt, N. C. (1995). Down-regulation of glial glutamate transporters after glutamatergic denervation in the rat brain. Eur. J. Neurosci. 7, 2036–2041. doi: 10.1111/j.1460-9568.1995.tb00626.x
Liao, S. L., and Chen, C. J. (2001). Differential effects of cytokines and redox potential on glutamate uptake in rat cortical glial cultures. Neurosci. Lett. 299, 113–116. doi: 10.1016/S0304-3940(01)01499-9
Limperopoulos, C., Bassan, H., Gauvreau, K., Robertson, R. L. Jr., Sullivan, N. R., Benson, C. B., et al. (2007). Does cerebellar injury in premature infants contribute to the high prevalence of long-term cognitive, learning, and behavioral disability in survivors? Pediatrics 120, 584–593. doi: 10.1542/peds.2007-1041
Limperopoulos, C., Soul, J. S., Gauvreau, K., Huppi, P. S., Warfield, S. K., Bassan, H., et al. (2005a). Late gestation cerebellar growth is rapid and impeded by premature birth. Pediatrics 115, 688–695. doi: 10.1542/peds.2004-1169
Limperopoulos, C., Soul, J. S., Haidar, H., Huppi, P. S., Bassan, H., Warfield, S. K., et al. (2005b). Impaired trophic interactions between the cerebellum and the cerebrum among preterm infants. Pediatrics 116, 844–850. doi: 10.1542/peds.2004-2282
Lingam, I., and Robertson, N. J. (2018). Magnesium as a neuroprotective agent: a review of its use in the fetus, term infant with neonatal encephalopathy, and the adult stroke patient. Dev. Neurosci. 40, 1–12. doi: 10.1159/000484891
Lipton, S. A., and Rosenberg, P. A. (1994). Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 330, 613–622. doi: 10.1056/NEJM199403033300907
Litt, J., Taylor, H. G., Klein, N., and Hack, M. (2005). Learning disabilities in children with very low birthweight: prevalence, neuropsychological correlates, and educational interventions. J. Learn. Disabil. 38, 130–141. doi: 10.1177/00222194050380020301
Liu, L., Oza, S., Hogan, D., Chu, Y., Perin, J., Zhu, J., et al. (2016). Global, regional, and national causes of under-5 mortality in 2000-15: an updated systematic analysis with implications for the sustainable development goals. Lancet 388, 3027–3035. doi: 10.1016/S0140-6736(16)31593-8
Locatelli, A., Incerti, M., Paterlini, G., Doria, V., Consonni, S., Provero, C., et al. (2010). Antepartum and intrapartum risk factors for neonatal encephalopathy at term. Am. J. Perinatol. 27, 649–654. doi: 10.1055/s-0030-1249761
Loeliger, M., Watson, C. S., Reynolds, J. D., Penning, D. H., Harding, R., Bocking, A. D., et al. (2003). Extracellular glutamate levels and neuropathology in cerebral white matter following repeated umbilical cord occlusion in the near term fetal sheep. Neuroscience 116, 705–714. doi: 10.1016/S0306-4522(02)00756-X
Lou, H. C., Lassen, N. A., and Friis-Hansen, B. (1979). Impaired autoregulation of cerebral blood flow in the distressed newborn infant. J. Pediatr. 94, 118–121. doi: 10.1016/S0022-3476(79)80373-X
Lozovaya, N. A., Kopanitsa, M. V., Boychuk, Y. A., and Krishtal, O. A. (1999). Enhancement of glutamate release uncovers spillover-mediated transmission by N-methyl-D-aspartate receptors in the rat hippocampus. Neuroscience 91, 1321–1330. doi: 10.1016/S0306-4522(98)00638-1
Maalouf, E. F., Duggan, P. J., Counsell, S. J., Rutherford, M. A., Cowan, F., Azzopardi, D., et al. (2001). Comparison of findings on cranial ultrasound and magnetic resonance imaging in preterm infants. Pediatrics 107, 719–727. doi: 10.1542/peds.107.4.719
MacLennan, A. H., Thompson, S. C., and Gecz, J. (2015). Cerebral palsy: causes, pathways, and the role of genetic variants. Am. J. Obstet. Gynecol. 213, 779–788. doi: 10.1016/j.ajog.2015.05.034
Mallard, E. C., Rees, S., Stringer, M., Cock, M. L., and Harding, R. (1998). Effects of chronic placental insufficiency on brain development in fetal sheep. Pediatr. Res. 43, 262–270. doi: 10.1203/00006450-199802000-00018
Mangham, L. J., Petrou, S., Doyle, L. W., Draper, E. S., and Marlow, N. (2009). The cost of preterm birth throughout childhood in England and Wales. Pediatrics 123, e312–e327. doi: 10.1542/peds.2008-1827
Manning, S. M., Talos, D. M., Zhou, C., Selip, D. B., Park, H.-K., Park, C.-J., et al. (2008). NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J. Neurosci. 28, 6670–6678. doi: 10.1523/JNEUROSCI.1702-08.2008
Marret, S., Mukendi, R., Gadisseux, J. F., Gressens, P., and Evrard, P. (1995). Effect of ibotenate on brain development: an excitotoxic mouse model of microgyria and posthypoxic-like lesions. J. Neuropathol. Exp. Neurol. 54, 358–370. doi: 10.1097/00005072-199505000-00009
Martin, L. J., Brambrink, A., Koehler, R. C., and Traystman, R. J. (1997a). Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. J. Comp. Neurol. 377, 262–285.
Martin, L. J., Brambrink, A. M., Lehmann, C., Portera-Cailliau, C., Koehler, R., Rothstein, J., et al. (1997b). Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann. Neurol. 42, 335–348.
McClendon, E., Chen, K., Gong, X., Sharifnia, E., Hagen, M., Cai, V., et al. (2014). Prenatal cerebral ischemia triggers dysmaturation of caudate projection neurons. Ann. Neurol. 75, 508–524. doi: 10.1002/ana.24100
McClure, M., Riddle, A., Manese, M., Luo, N. L., Rorvik, D. A., Kelly, K. A., et al. (2008). Cerebral blood flow heterogeneity in preterm sheep: lack of physiological support for vascular boundary zones in fetal cerebral white matter. J. Cereb. Blood Flow Metab. 28, 995–1008. doi: 10.1038/sj.jcbfm.9600597
McDonald, J. W., and Johnston, M. V. (1990). Pharmacology of N-methyl-D-aspartate-induced brain injury in an in vivo perinatal rat model. Synapse 6, 179–188. doi: 10.1002/syn.890060210
McDonald, J. W., Silverstein, F. S., and Johnston, M. V. (1988). Neurotoxicity of N-methyl-D-aspartate is markedly enhanced in developing rat central nervous system. Brain Res. 459, 200–203. doi: 10.1016/0006-8993(88)90306-X
McQuillen, P. S., Sheldon, R. A., Shatz, C. J., and Ferriero, D. M. (2003). Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J. Neurosci. 23, 3308–3315. doi: 10.1523/JNEUROSCI.23-08-03308.2003
Meldrum, B. S. (2000). Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J. Nutr. 130, 1007s–1015s.
Melone, M., Bellesi, M., and Conti, F. (2009). Synaptic localization of GLT-1a in the rat somatic sensory cortex. Glia 57, 108–117. doi: 10.1002/glia.20744
Mercier, C. E., Dunn, M. S., Ferrelli, K. R., Howard, D. B., and Soll, R. F. (2010). Neurodevelopmental outcome of extremely low birth weight infants from the Vermont Oxford network: 1998-2003. Neonatology 97, 329–338. doi: 10.1159/000260136
Mercuri, E., He, J., Curati, W. L., Dubowitz, L. M., Cowan, F. M., and Bydder, G. M. (1997). Cerebellar infarction and atrophy in infants and children with a history of premature birth. Pediatr. Radiol. 27, 139–143. doi: 10.1007/s002470050085
Miller, S. P., Cozzio, C. C., Goldstein, R. B., Ferriero, D. M., Partridge, J. C., Vigneron, D. B., et al. (2003). Comparing the diagnosis of white matter injury in premature newborns with serial MR imaging and transfontanel ultrasonography findings. AJNR Am. J. Neuroradiol. 24, 1661–1669.
Mitani, A., and Tanaka, K. (2003). Functional changes of glial glutamate transporter GLT-1 during ischemia: an in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. J. Neurosci. 23, 7176–7182. doi: 10.1523/JNEUROSCI.23-18-07176.2003
Miyawaki, T., Matsui, K., and Takashima, S. (1998). Developmental characteristics of vessel density in the human fetal and infant brains. Early Hum. Dev. 53, 65–72.
Moore, T., Hennessy, E. M., Myles, J., Johnson, S. J., Draper, E. S., Costeloe, K. L., et al. (2012). Neurological and developmental outcome in extremely preterm children born in England in 1995 and 2006: the EPICure studies. BMJ 345, 274–275. doi: 10.1136/bmj.e7961
Moretto, M. B., Arteni, N. S., Lavinsky, D., Netto, C. A., Rocha, J. B., Souza, D. O., et al. (2005). Hypoxic-ischemic insult decreases glutamate uptake by hippocampal slices from neonatal rats: prevention by guanosine. Exp. Neurol. 195, 400–406. doi: 10.1016/j.expneurol.2005.06.005
Moretto, M. B., Boff, B., Lavinsky, D., Netto, C. A., Rocha, J. B., Souza, D. O., et al. (2009). Importance of schedule of administration in the therapeutic efficacy of guanosine: early intervention after injury enhances glutamate uptake in model of hypoxia-ischemia. J. Mol. Neurosci. 38, 216–219. doi: 10.1007/s12031-008-9154-7
Munch, C., Zhu, B. G., Leven, A., Stamm, S., Einkorn, H., Schwalenstocker, B., et al. (2003). Differential regulation of 5′ splice variants of the glutamate transporter EAAT2 in an in vivo model of chemical hypoxia induced by 3-nitropropionic acid. J. Neurosci. Res. 71, 819–825. doi: 10.1002/jnr.10536
Murphy, D. J., Sellers, S., MacKenzie, I. Z., Yudkin, P. L., and Johnson, A. M. (1995). Case-control study of antenatal and intrapartum risk factors for cerebral palsy in very preterm singleton babies. Lancet 346, 1449–1454. doi: 10.1016/S0140-6736(95)92471-X
Newcomer, J. W., Farber, N. B., and Olney, J. W. (2000). NMDA receptor function, memory, and brain aging. Dialogues Clin. Neurosci. 2, 219–232.
Nicholls, D., and Attwell, D. (1990). The release and uptake of excitatory amino acids. Trends Pharmacol. Sci. 11, 462–468. doi: 10.1016/0165-6147(90)90129-V
Northington, F. J., Traystman, R. J., Koehler, R. C., and Martin, L. J. (1999). GLT1, glial glutamate transporter, is transiently expressed in neurons and develops astrocyte specificity only after midgestation in the ovine fetal brain. J. Neurobiol. 39, 515–526. doi: 10.1002/(SICI)1097-4695(19990615)39:4<515::AID-NEU5>3.0.CO;2-U
Northington, F. J., Traystman, R. J., Koehler, R. C., Rothstein, J. D., and Martin, L. J. (1998). Regional and cellular expression of glial (GLT1) and neuronal (EAAC1) glutamate transporter proteins in ovine fetal brain. Neuroscience 85, 1183–1194. doi: 10.1016/S0306-4522(97)00673-8
Nosarti, C., Allin, M. P., Frangou, S., Rifkin, L., and Murray, R. M. (2005). Hyperactivity in adolescents born very preterm is associated with decreased caudate volume. Biol. Psychiatry 57, 661–666. doi: 10.1016/j.biopsych.2004.12.003
Nosarti, C., Giouroukou, E., Healy, E., Rifkin, L., Walshe, M., Reichenberg, A., et al. (2008). Grey and white matter distribution in very preterm adolescents mediates neurodevelopmental outcome. Brain 131, 205–217. doi: 10.1093/brain/awm282
Novak, I., Hines, M., Goldsmith, S., and Barclay, R. (2012). Clinical prognostic messages from a systematic review on cerebral palsy. Pediatrics 130, e1285–e1312. doi: 10.1542/peds.2012-0924
O’Callaghan, M. E., Maclennan, A. H., Gibson, C. S., McMichael, G. L., Haan, E. A., Broadbent, J. L., et al. (2013). Genetic and clinical contributions to cerebral palsy: a multi-variable analysis. J. Paediatr. Child Health 49, 575–581. doi: 10.1111/jpc.12279
O’Callaghan, M. E., Maclennan, A. H., Gibson, C. S., McMichael, G. L., Haan, E. A., Broadbent, J. L., et al. (2012). Fetal and maternal candidate single nucleotide polymorphism associations with cerebral palsy: a case-control study. Pediatrics 129, e414–e423. doi: 10.1542/peds.2011-0739
O’Callaghan, M. E., MacLennan, A. H., Haan, E. A., and Dekker, G. (2009). The genomic basis of cerebral palsy: a HuGE systematic literature review. Hum. Genet. 126, 149–172. doi: 10.1007/s00439-009-0638-5
O’Donovan, S. M., Sullivan, C. R., and McCullumsmith, R. E. (2017). The role of glutamate transporters in the pathophysiology of neuropsychiatric disorders. NPJ Schizophr. 3:32. doi: 10.1038/s41537-017-0037-1
Ofek-Shlomai, N., and Berger, I. (2014). Inflammatory injury to the neonatal brain – what can we do? Front. Pediatr. 2:30. doi: 10.3389/fped.2014.00030
Ohshima, M., Coq, J. O., Otani, K., Hattori, Y., Ogawa, Y., Sato, Y., et al. (2016). Mild intrauterine hypoperfusion reproduces neurodevelopmental disorders observed in prematurity. Sci. Rep. 6:39377. doi: 10.1038/srep39377
Oka, A., Belliveau, M. J., Rosenberg, P. A., and Volpe, J. J. (1993). Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J. Neurosci. 13, 1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993
Olmos, G., and Llado, J. (2014). Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014:861231. doi: 10.1155/2014/861231
Olney, J. W. (1969). Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 164, 719–721. doi: 10.1126/science.164.3880.719
O’Shea, T. M., Klinepeter, K. L., and Dillard, R. G. (1998). Prenatal events and the risk of cerebral palsy in very low birth weight infants. Am. J. Epidemiol. 147, 362–369. doi: 10.1093/oxfordjournals.aje.a009458
O’Shea, R. D., Lau, C. L., Farso, M. C., Diwakarla, S., Zagami, C. J., Svendsen, B. B., et al. (2006). Effects of lipopolysaccharide on glial phenotype and activity of glutamate transporters: evidence for delayed up-regulation and redistribution of GLT-1. Neurochem. Int. 48, 604–610. doi: 10.1016/j.neuint.2005.12.028
Oskoui, M., Coutinho, F., Dykeman, J., Jetté, N., and Pringsheim, T. (2013). An update on the prevalence of cerebral palsy: a systematic review and meta-analysis. Dev. Med. Child Neurol. 55, 509–519. doi: 10.1111/dmcn.12080
Otis, T. S., and Kavanaugh, M. P. (2000). Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J. Neurosci. 20, 2749–2757.
Ottersen, O. P., Laake, J. H., Reichelt, W., Haug, F. M., and Torp, R. (1996). Ischemic disruption of glutamate homeostasis in brain: quantitative immunocytochemical analyses. J. Chem. Neuroanat. 12, 1–14. doi: 10.1016/S0891-0618(96)00178-0
Parker, J., Mitchell, A., Kalpakidou, A., Walshe, M., Jung, H. Y., Nosarti, C., et al. (2008). Cerebellar growth and behavioural & neuropsychological outcome in preterm adolescents. Brain 131, 1344–1351. doi: 10.1093/brain/awn062
Parkin, G. M., Udawela, M., Gibbons, A., and Dean, B. (2018). Glutamate transporters, EAAT1 and EAAT2, are potentially important in the pathophysiology and treatment of schizophrenia and affective disorders. World J. Psychiatry 8, 51–63. doi: 10.5498/wjp.v8.i2.51
Parsons, M. P., and Raymond, L. A. (2014). Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82, 279–293. doi: 10.1016/j.neuron.2014.03.030
Persson, M., Brantefjord, M., Hansson, E., and Ronnback, L. (2005). Lipopolysaccharide increases microglial GLT-1 expression and glutamate uptake capacity in vitro by a mechanism dependent on TNF-alpha. Glia 51, 111–120. doi: 10.1002/glia.20191
Peterson, B. S., Anderson, A. W., Ehrenkranz, R., Staib, L. H., Tageldin, M., Colson, E., et al. (2003). Regional brain volumes and their later neurodevelopmental correlates in term and preterm infants. Pediatrics 111, 939–948. doi: 10.1542/peds.111.5.939
Peterson, B. S., Vohr, B., Staib, L. H., Cannistraci, C. J., Dolberg, A., Schneider, K. C., et al. (2000). Regional brain volume abnormalities and long-term cognitive outcome in preterm infants. JAMA 284, 1939–1947.
Petr, G. T., Sun, Y., Frederick, N. M., Zhou, Y., Dhamne, S. C., Hameed, M. Q., et al. (2015). Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J. Neurosci. 35, 5187–5201. doi: 10.1523/JNEUROSCI.4255-14.2015
Platt, S. R. (2007). The role of glutamate in central nervous system health and disease – a review. Vet. J. 173, 278–286. doi: 10.1016/j.tvjl.2005.11.007
Pow, D. V., Naidoo, T., Lingwood, B. E., Healy, G. N., Williams, S. M., Sullivan, R. K., et al. (2004). Loss of glial glutamate transporters and induction of neuronal expression of GLT-1B in the hypoxic neonatal pig brain. Brain Res. Dev. Brain Res. 153, 1–11. doi: 10.1016/j.devbrainres.2004.06.019
Pryds, O. (1991). Control of cerebral circulation in the high-risk neonate. Ann. Neurol. 30, 321–329. doi: 10.1002/ana.410300302
Pryds, O., Andersen, G. E., and Friis-Hansen, B. (1990). Cerebral blood flow reactivity in spontaneously breathing, preterm infants shortly after birth. Acta Paediatr. Scand. 79, 391–396.
Rajatileka, S., Odd, D., Robinson, M. T., Spittle, A. C., Dwomoh, L., Williams, M., et al. (2017). Variants of the EAAT2 glutamate transporter gene promoter are associated with cerebral palsy in preterm infants. Mol. Neurobiol. 55, 2013–2024. doi: 10.1007/s12035-017-0462-1
Rao, V. L., Bowen, K. K., and Dempsey, R. J. (2001a). Transient focal cerebral ischemia down-regulates glutamate transporters GLT-1 and EAAC1 expression in rat brain. Neurochem. Res. 26, 497–502. doi: 10.1023/A:1010956711295
Rao, V. L., Dogan, A., Bowen, K. K., Todd, K. G., and Dempsey, R. J. (2001b). Antisense knockdown of the glial glutamate transporter GLT-1 exacerbates hippocampal neuronal damage following traumatic injury to rat brain. Eur. J. Neurosci. 13, 119–128. doi: 10.1111/j.1460-9568.2001.01367.x
Raymond, M., Li, P., Mangin, J. M., Huntsman, M., and Gallo, V. (2011). Chronic perinatal hypoxia reduces glutamate-aspartate transporter function in astrocytes through the Janus kinase/signal transducer and activator of transcription pathway. J. Neurosci. 31, 17864–17871. doi: 10.1523/JNEUROSCI.3179-11.2011
Rees, S., Mallard, C., Breen, S., Stringer, M., Cock, M., and Harding, R. (1998). Fetal brain injury following prolonged hypoxemia and placental insufficiency: a review. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 119, 653–660. doi: 10.1016/S1095-6433(98)01001-0
Rees, S., Stringer, M., Just, Y., Hooper, S. B., and Harding, R. (1997). The vulnerability of the fetal sheep brain to hypoxemia at mid-gestation. Brain Res. Dev. Brain Res. 103, 103–118. doi: 10.1016/S0165-3806(97)81787-7
Rehm, H. L. (2017). Evolving health care through personal genomics. Nat. Rev. Genet. 18, 259–267. doi: 10.1038/nrg.2016.162
Rezaie, P., and Dean, A. (2002). Periventricular leukomalacia, inflammation and white matter lesions within the developing nervous system. Neuropathology 22, 106–132. doi: 10.1046/j.1440-1789.2002.00438.x
Riddle, A., Luo, N. L., Manese, M., Beardsley, D. J., Green, L., Rorvik, D. A., et al. (2006). Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J. Neurosci. 26, 3045–3055. doi: 10.1523/JNEUROSCI.5200-05.2006
Riikonen, R. S., Kero, P. O., and Simell, O. G. (1992). Excitatory amino acids in cerebrospinal fluid in neonatal asphyxia. Pediatr. Neurol. 8, 37–40. doi: 10.1016/0887-8994(92)90050-9
Rimaniol, A. C., Haik, S., Martin, M., Le Grand, R., Boussin, F. D., Dereuddre-Bosquet, N., et al. (2000). Na+−dependent high-affinity glutamate transport in macrophages. J. Immunol. 164, 5430–5438. doi: 10.4049/jimmunol.164.10.5430
Roberts, R. C., Roche, J. K., and McCullumsmith, R. E. (2014). Localization of excitatory amino acid transporters EAAT1 and EAAT2 in human postmortem cortex: a light and electron microscopic study. Neuroscience 277, 522–540. doi: 10.1016/j.neuroscience.2014.07.019
Rocha-Ferreira, E., and Hristova, M. (2016). Plasticity in the neonatal brain following hypoxic-ischaemic injury. Neural Plast. 2016:4901014. doi: 10.1155/2016/4901014
Rosenbaum, P., Paneth, N., Leviton, A., Goldstein, M., Bax, M., Damiano, D., et al. (2007). A report: the definition and classification of cerebral palsy April 2006. Dev. Med. Child Neurol. Suppl. 109, 8–14. doi: 10.1111/j.1469-8749.2007.tb12610.x
Rothstein, J. D., Dykes-Hoberg, M., Pardo, C. A., Bristol, L. A., Jin, L., Kuncl, R. W., et al. (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16, 675–686. doi: 10.1016/S0896-6273(00)80086-0
Rothstein, J. D., Martin, L., Levey, A. I., Dykes-Hoberg, M., Jin, L., Wu, D., et al. (1994). Localization of neuronal and glial glutamate transporters. Neuron 13, 713–725. doi: 10.1016/0896-6273(94)90038-8
Sathyanesan, A., Kundu, S., Abbah, J., and Gallo, V. (2018). Neonatal brain injury causes cerebellar learning deficits and Purkinje cell dysfunction. Nat. Commun. 9:3235. doi: 10.1038/s41467-018-05656-w
Sattler, R., and Tymianski, M. (2001). Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol. Neurobiol. 24, 107–129. doi: 10.1385/MN:24:1-3:107
Schlapbach, L. J., Aebischer, M., Adams, M., Natalucci, G., Bonhoeffer, J., Latzin, P., et al. (2011). Impact of sepsis on neurodevelopmental outcome in a Swiss National Cohort of extremely premature infants. Pediatrics 128, e348–e357. doi: 10.1542/peds.2010-3338
Segovia, K. N., McClure, M., Moravec, M., Luo, N. L., Wan, Y., Gong, X., et al. (2008). Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann. Neurol. 63, 520–530. doi: 10.1002/ana.21359
Seki, Y., Feustel, P. J., Keller, R. W. Jr., Tranmer, B. I., and Kimelberg, H. K. (1999). Inhibition of ischemia-induced glutamate release in rat striatum by dihydrokinate and an anion channel blocker. Stroke 30, 433–440. doi: 10.1161/01.STR.30.2.433
Serdaroglu, G., Tekgul, H., Kitis, O., Serdaroglu, E., and Gökben, S. (2004). Correlative value of magnetic resonance imaging for neurodevelopmental outcome in periventricular leukomalacia. Dev. Med. Child Neurol. 46, 733–739. doi: 10.1111/j.1469-8749.2004.tb00992.x
Shah, D. K., Anderson, P. J., Carlin, J. B., Pavlovic, M., Howard, K., Thompson, D. K., et al. (2006). Reduction in cerebellar volumes in preterm infants: relationship to white matter injury and neurodevelopment at two years of age. Pediatr. Res. 60, 97–102. doi: 10.1203/01.pdr.0000220324.27597.f0
Shankaran, S., Langer, J. C., Kazzi, S. N., Laptook, A. R., and Walsh, M. (2006). Cumulative index of exposure to hypocarbia and hyperoxia as risk factors for periventricular leukomalacia in low birth weight infants. Pediatrics 118, 1654–1659. doi: 10.1542/peds.2005-2463
Shankaran, S., Laptook, A. R., Ehrenkranz, R. A., Tyson, J. E., McDonald, S. A., Donovan, E. F., et al. (2005). Whole-body hypothermia for neonates with hypoxic–ischemic encephalopathy. N. Engl. J. Med. 353, 1574–1584. doi: 10.1056/NEJMcps050929
Shibata, T., Watanabe, M., Tanaka, K., Wada, K., and Inoue, Y. (1996). Dynamic changes in expression of glutamate transporter mRNAs in developing brain. Neuroreport 7, 705–709. doi: 10.1097/00001756-199602290-00006
Simbruner, G., Mittal, R. A., Rohlmann, F., and Muche, R. (2010). Systemic hypothermia after neonatal encephalopathy: outcomes of neo.nEURO.Network RCT. Pediatrics 126, e771–e778. doi: 10.1542/peds.2009-2441
Soria-Pastor, S., Gimenez, M., Narberhaus, A., Falcon, C., Botet, F., Bargallo, N., et al. (2008). Patterns of cerebral white matter damage and cognitive impairment in adolescents born very preterm. Int. J. Dev. Neurosci. 26, 647–654. doi: 10.1016/j.ijdevneu.2008.08.001
Soria-Pastor, S., Padilla, N., Zubiaurre-Elorza, L., Ibarretxe-Bilbao, N., Botet, F., Costas-Moragas, C., et al. (2009). Decreased regional brain volume and cognitive impairment in preterm children at low risk. Pediatrics 124, e1161–e1170. doi: 10.1542/peds.2009-0244
Soul, J. S., Hammer, P. E., Tsuji, M., Saul, J. P., Bassan, H., Limperopoulos, C., et al. (2007). Fluctuating pressure-passivity is common in the cerebral circulation of sick premature infants. Pediatr. Res. 61, 467–473. doi: 10.1203/pdr.0b013e31803237f6
Spittle, A. J., Boyd, R. N., Inder, T. E., and Doyle, L. W. (2009). Predicting motor development in very preterm infants at 12 months’ corrected age: the role of qualitative magnetic resonance imaging and general movements assessments. Pediatrics 123, 512–517. doi: 10.1542/peds.2008-0590
Spittle, A. J., Brown, N. C., Doyle, L. W., Boyd, R. N., Hunt, R. W., Bear, M., et al. (2008). Quality of general movements is related to white matter pathology in very preterm infants. Pediatrics 121, e1184–e1189. doi: 10.1542/peds.2007-1924
Spittle, A. J., Morgan, C., Olsen, J. E., Novak, I., and Cheong, J. L. Y. (2018). Early diagnosis and treatment of cerebral palsy in children with a history of preterm birth. Clin. Perinatol. 45, 409–420. doi: 10.1016/j.clp.2018.05.011
Srinivasan, L., Dutta, R., Counsell, S. J., Allsop, J. M., Boardman, J. P., Rutherford, M. A., et al. (2007). Quantification of deep gray matter in preterm infants at term-equivalent age using manual volumetry of 3-tesla magnetic resonance images. Pediatrics 119, 759–765. doi: 10.1542/peds.2006-2508
Stanley, F. J. (1992). Survival and cerebral palsy in low birthweight infants: implications for perinatal care. Paediatr. Perinat. Epidemiol. 6, 298–310. doi: 10.1111/j.1365-3016.1992.tb00769.x
Stanley, F., Blair, E., and Alberman, E. (2000). Cerebral palsies: Epidemiology and causal pathways. (London: MacKeith Press).
Stavsky, M., Mor, O., Mastrolia, S. A., Greenbaum, S., Than, N. G., and Erez, O. (2017). Cerebral palsy – trends in epidemiology and recent development in prenatal mechanisms of disease, treatment, and prevention. Front. Pediatr. 5:21. doi: 10.3389/fped.2017.00021
Stoll, B. J., Hansen, N., Fanaroff, A. A., Wright, L. L., Carlo, W. A., Ehrenkranz, R. A., et al. (2002). Late-onset sepsis in very low birth weight neonates: the experience of the NICHD neonatal research network. Pediatrics 110, 285–291. doi: 10.1542/peds.110.2.285
Su, Z. Z., Leszczyniecka, M., Kang, D. C., Sarkar, D., Chao, W., Volsky, D. J., et al. (2003). Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2). Proc. Natl. Acad. Sci. USA 100, 1955–1960. doi: 10.1073/pnas.0136555100
Sutherland, M. L., Delaney, T. A., and Noebels, J. L. (1996). Glutamate transporter mRNA expression in proliferative zones of the developing and adult murine CNS. J. Neurosci. 16, 2191–2207. doi: 10.1523/JNEUROSCI.16-07-02191.1996
Szatkowski, M., Barbour, B., and Attwell, D. (1990). Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 348, 443–446. doi: 10.1038/348443a0
Szymonowicz, W., Walker, A. M., Cussen, L., Cannata, J., and Yu, V. Y. (1988). Developmental changes in regional cerebral blood flow in fetal and newborn lambs. Am. J. Phys. 254, H52–H58. doi: 10.1152/ajpheart.1988.254.1.H52
Takahashi, K., Foster, J. B., and Lin, C.-L. G. (2015). Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell. Mol. Life Sci. 72, 3489–3506. doi: 10.1007/s00018-015-1937-8
Takasaki, C., Okada, R., Mitani, A., Fukaya, M., Yamasaki, M., Fujihara, Y., et al. (2008). Glutamate transporters regulate lesion-induced plasticity in the developing somatosensory cortex. J. Neurosci. 28, 4995–5006. doi: 10.1523/JNEUROSCI.0861-08.2008
Takashima, S., and Tanaka, K. (1978). Development of cerebrovascular architecture and its relationship to periventricular leukomalacia. Arch. Neurol. 35, 11–16. doi: 10.1001/archneur.1978.00500250015003
Takeuchi, H., Jin, S., Wang, J., Zhang, G., Kawanokuchi, J., Kuno, R., et al. (2006). Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 281, 21362–21368. doi: 10.1074/jbc.M600504200
Tanaka, K., Watase, K., Manabe, T., Yamada, K., Watanabe, M., Takahashi, K., et al. (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702. doi: 10.1126/science.276.5319.1699
Thornton, C., Rousset, C. I., Kichev, A., Miyakuni, Y., Vontell, R., Baburamani, A. A., et al. (2012). Molecular mechanisms of neonatal brain injury. Neurol. Res. Int. 2012:506320. doi: 10.1155/2012/506320
Tilleux, S., and Hermans, E. (2007). Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J. Neurosci. Res. 85, 2059–2070. doi: 10.1002/jnr.21325
Torp, R., Lekieffre, D., Levy, L. M., Haug, F. M., Danbolt, N. C., Meldrum, B. S., et al. (1995). Reduced postischemic expression of a glial glutamate transporter, GLT1, in the rat hippocampus. Exp. Brain Res. 103, 51–58. doi: 10.1007/BF00241964
Tronnes, H., Wilcox, A. J., Lie, R. T., Markestad, T., and Moster, D. (2014). Risk of cerebral palsy in relation to pregnancy disorders and preterm birth: a national cohort study. Dev. Med. Child Neurol. 56, 779–785. doi: 10.1111/dmcn.12430
Tzingounis, A. V., and Wadiche, J. I. (2007). Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat. Rev. Neurosci. 8, 935–947. doi: 10.1038/nrn2274
Ullensvang, K., Lehre, K. P., Storm-Mathisen, J., and Danbolt, N. C. (1997). Differential developmental expression of the two rat brain glutamate transporter proteins GLAST and GLT. Eur. J. Neurosci. 9, 1646–1655. doi: 10.1111/j.1460-9568.1997.tb01522.x
van der Burg, J. W., Sen, S., Chomitz, V. R., Seidell, J. C., Leviton, A., and Dammann, O. (2016). The role of systemic inflammation linking maternal BMI to neurodevelopment in children. Pediatr. Res. 79, 3–12. doi: 10.1038/pr.2015.179
Van Steenwinckel, J., Schang, A. L., Sigaut, S., Chhor, V., Degos, V., Hagberg, H., et al. (2014). Brain damage of the preterm infant: new insights into the role of inflammation. Biochem. Soc. Trans. 42, 557–563. doi: 10.1042/BST20130284
van Tilborg, E., Achterberg, E. J. M., van Kammen, C. M., van der Toorn, A., Groenendaal, F., Dijkhuizen, R. M., et al. (2018). Combined fetal inflammation and postnatal hypoxia causes myelin deficits and autism-like behavior in a rat model of diffuse white matter injury. Glia 66, 78–93. doi: 10.1002/glia.23216
Vandenberg, R. J., and Ryan, R. M. (2013). Mechanisms of glutamate transport. Physiol. Rev. 93, 1621–1657. doi: 10.1152/physrev.00007.2013
Verkhratsky, A., Steardo, L., Parpura, V., and Montana, V. (2016). Translational potential of astrocytes in brain disorders. Prog. Neurobiol. 144, 188–205. doi: 10.1016/j.pneurobio.2015.09.003
Verma, U., Tejani, N., Klein, S., Reale, M. R., Beneck, D., Figueroa, R., et al. (1997). Obstetric antecedents of intraventricular hemorrhage and periventricular leukomalacia in the low-birth-weight neonate. Am. J. Obstet. Gynecol. 176, 275–281. doi: 10.1016/S0002-9378(97)70485-X
Volpe, J. J. (2009a). Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 8, 110–124. doi: 10.1016/S1474-4422(08)70294-1
Volpe, J. J. (2009b). Cerebellum of the premature infant: rapidly developing, vulnerable, clinically important. J. Child Neurol. 24, 1085–1104. doi: 10.1177/0883073809338067
Volpe, J. J. (2009c). The encephalopathy of prematurity--brain injury and impaired brain development inextricably intertwined. Semin. Pediatr. Neurol. 16, 167–178. doi: 10.1016/j.spen.2009.09.005
Volpe, J. J., Kinney, H. C., Jensen, F. E., and Rosenberg, P. A. (2011). The developing oligodendrocyte: key cellular target in brain injury in the premature infant. Int. J. Dev. Neurosci. 29, 423–440. doi: 10.1016/j.ijdevneu.2011.02.012
Wang, L. W., Chang, Y. C., Lin, C. Y., Hong, J. S., and Huang, C. C. (2010). Low-dose lipopolysaccharide selectively sensitizes hypoxic ischemia-induced white matter injury in the immature brain. Pediatr. Res. 68, 41–47. doi: 10.1203/PDR.0b013e3181df5f6b
Wang, X., Hagberg, H., Nie, C., Zhu, C., Ikeda, T., and Mallard, C. (2007). Dual role of intrauterine immune challenge on neonatal and adult brain vulnerability to hypoxia-ischemia. J. Neuropathol. Exp. Neurol. 66, 552–561. doi: 10.1097/01.jnen.0000263870.91811.6f
Wang, X., Stridh, L., Li, W., Dean, J., Elmgren, A., Gan, L., et al. (2009). Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J. Immunol. 183, 7471–7477. doi: 10.4049/jimmunol.0900762
Watkins, J. C., and Evans, R. H. (1981). Excitatory amino acid transmitters. Annu. Rev. Pharmacol. Toxicol. 21, 165–204. doi: 10.1146/annurev.pa.21.040181.001121
Wilke, S., Thomas, R., Allcock, N., and Fern, R. (2004). Mechanism of acute ischemic injury of oligodendroglia in early myelinating white matter: the importance of astrocyte injury and glutamate release. J. Neuropathol. Exp. Neurol. 63, 872–881.
Wilson-Costello, D., Friedman, H., Minich, N., Fanaroff, A. A., and Hack, M. (2005). Improved survival rates with increased neurodevelopmental disability for extremely low birth weight infants in the 1990s. Pediatrics 115, 997–1003. doi: 10.1542/peds.2004-0221
Wisnowski, J. L., Bluml, S., Paquette, L., Zelinski, E., Nelson, M. D. Jr., Painter, M. J., et al. (2013). Altered glutamatergic metabolism associated with punctate white matter lesions in preterm infants. PLoS One 8:e56880. doi: 10.1371/journal.pone.0056880
World Health Organization (2012) in Born too soon: The global action report on preterm birth. eds. C. P. Howson, M. V. Kinney, and J. E. Lawn (Geneva: World Health Organization).
World Health Organization (2016). Global Health estimates 2015: Disease burden by cause, age, sex, by country and by region, 2000–2015. (Geneva: World Health Organization).
Wu, Y. W. (2002). Systematic review of chorioamnionitis and cerebral palsy. Ment. Retard. Dev. Disabil. Res. Rev. 8, 25–29. doi: 10.1002/mrdd.10003
Wu, Y. W., and Colford, J. M. Jr. (2000). Chorioamnionitis as a risk factor for cerebral palsy: a meta-analysis. JAMA 284, 1417–1424.
Wu, D., Zou, Y. F., Xu, X. Y., Feng, X. L., Yang, L., Zhang, G. C., et al. (2011). The association of genetic polymorphisms with cerebral palsy: a meta-analysis. Dev. Med. Child Neurol. 53, 217–225. doi: 10.1111/j.1469-8749.2010.03884.x
Yamada, K., Watanabe, M., Shibata, T., Nagashima, M., Tanaka, K., and Inoue, Y. (1998). Glutamate transporter GLT-1 is transiently localized on growing axons of the mouse spinal cord before establishing astrocytic expression. J. Neurosci. 18, 5706–5713. doi: 10.1523/JNEUROSCI.18-15-05706.1998
Yanni, D., Korzeniewski, S. J., Allred, E. N., Fichorova, R. N., O’Shea, T. M., Kuban, K., et al. (2017). Both antenatal and postnatal inflammation contribute information about the risk of brain damage in extremely preterm newborns. Pediatr. Res. 82, 691–696. doi: 10.1038/pr.2017.128
Ying, W. (1997). Deleterious network: a testable pathogenetic concept of Alzheimer’s disease. Gerontology 43, 242–253.
Yoon, B. H., Jun, J. K., Romero, R., Park, K. H., Gomez, R., Choi, J. H., et al. (1997). Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1beta, and tumor necrosis factor-alpha), neonatal brain white matter lesions, and cerebral palsy. Am. J. Obstet. Gynecol. 177, 19–26. doi: 10.1016/S0002-9378(97)70432-0
Yoon, B. H., Romero, R., Park, J. S., Kim, C. J., Kim, S. H., Choi, J. H., et al. (2000). Fetal exposure to an intra-amniotic inflammation and the development of cerebral palsy at the age of three years. Am. J. Obstet. Gynecol. 182, 675–681. doi: 10.1067/mob.2000.104207
Yoon, B. H., Romero, R., Yang, S. H., Jun, J. K., Kim, I. O., Choi, J. H., et al. (1996). Interleukin-6 concentrations in umbilical cord plasma are elevated in neonates with white matter lesions associated with periventricular leukomalacia. Am. J. Obstet. Gynecol. 174, 1433–1440. doi: 10.1016/S0002-9378(96)70585-9
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
Zhang, Y., Tan, F., Xu, P., and Qu, S. (2016). Recent advance in the relationship between excitatory amino acid transporters and Parkinson’s disease. Neural Plast. 2016:8. doi: 10.1155/2016/8941327
Zhou, Y., and Danbolt, N. C. (2013). GABA and glutamate transporters in brain. Front. Endocrinol. 4:165. doi: 10.3389/fendo.2013.00165
Zhou, Y., Waanders, L. F., Holmseth, S., Guo, C., Berger, U. V., Li, Y., et al. (2014). Proteome analysis and conditional deletion of the EAAT2 glutamate transporter provide evidence against a role of EAAT2 in pancreatic insulin secretion in mice. J. Biol. Chem. 289, 1329–1344. doi: 10.1074/jbc.M113.529065
Zhou, X.-w., Wang, X., Yang, Y., Luo, J.-w., Dong, H., Liu, Y.-h., et al. (2016). Biomarkers related with seizure risk in glioma patients: a systematic review. Clin. Neurol. Neurosurg. 151, 113–119. doi: 10.1016/j.clineuro.2016.10.001
Zhu, M. Y., Milligan, N., Keating, S., Windrim, R., Keunen, J., Thakur, V., et al. (2016). The hemodynamics of late-onset intrauterine growth restriction by MRI. Am. J. Obstet. Gynecol. 214, 367.e1–367.e17. doi: 10.1016/j.ajog.2015.10.004
Keywords: preterm infant, brain injury, glutamate, excitotoxicity, inflammation, EAAT2, SLC1A2, GLT-1
Citation: Pregnolato S, Chakkarapani E, Isles AR and Luyt K (2019) Glutamate Transport and Preterm Brain Injury. Front. Physiol. 10:417. doi: 10.3389/fphys.2019.00417
Edited by:
Carina Mallard, University of Gothenburg, SwedenReviewed by:
Pierre Gressens, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2019 Pregnolato, Chakkarapani, Isles and Luyt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Pregnolato, c3AxNjAyN0BicmlzdG9sLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.