 Ridwan Babatunde Ibrahim
Ridwan Babatunde Ibrahim Yo-Tsen Liu1,3,4,5†
Yo-Tsen Liu1,3,4,5† Ssu-Yu Yeh
Ssu-Yu Yeh Jin-Wu Tsai
Jin-Wu Tsai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 02 April 2019
Sec. Systems Biology Archive
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.00338
This article is part of the Research Topic Contribution of Translational Animal Models to the Systems Biology of Neurodegenerative Disorders View all 7 articles
Transthyretin amyloidosis (ATTR amyloidosis) is a fatal systemic disease caused by amyloid deposits of misfolded transthyretin, leading to familial amyloid polyneuropathy and/or cardiomyopathy, or a rare oculoleptomeningeal amyloidosis. A good model system that mimic the disease phenotype is crucial for the development of drugs and treatments for this devastating degenerative disorder. The present models using fruit flies, worms, rodents, non-human primates and induced pluripotent stem cells have helped researchers understand important disease-related mechanisms and test potential therapeutic options. However, the challenge of creating an ideal model still looms, for these models did not recapitulates all symptoms, particularly neurological presentation, of ATTR amyloidosis. Recently, knock-in techniques was used to generate two humanized ATTR mouse models, leading to amyloid deposition in the nerves and neuropathic manifestation in these models. This review gives a recent update on the milestone, progress, and challenges in developing different models for ATTR amyloidosis research.
Transthyretin amyloidosis (ATTR) is the most common type of autosomal-dominant hereditary systemic amyloidosis associated with the transthyretin (TTR) protein; with either early- or late-onset presentation in patients (Benson and Kincaid, 2007; Plante-Bordeneuve and Said, 2011; Adams et al., 2012). ATTR can arise either due to senescent event associated with the wild-type TTR gene leading to wild-type amyloidosis (formerly known as senile systemic amyloidosis), or hereditary ATTR due to mutations in the TTR gene, leading to either familial amyloid polyneuropathy (ATTR-FAP), familial amyloid cardiomyopathy (ATTR-FAC), or a rare form of oculoleptomeningeal amyloidosis (OLMA) (Ando et al., 2005; Rapezzi et al., 2010; Leung et al., 2013; Mathieu et al., 2018).
TTR is a secreted homotetrameric protein found largely in the plasma and cerebrospinal fluid (CSF) (Almeida et al., 2004; Buxbaum and Reixach, 2009). It is primarily synthesized in the liver, which provides most plasma pool of the protein (Ingenbleek and Bernstein, 2015), and in the choroid plexus of the brain contributing to its presence in the CSF (Richardson et al., 2015; Johnson et al., 2018). TTR is also synthesized in the retinal pigment epithelium (RPE) of the eye (Pfeffer et al., 2004). Its main function is to transport thyroid hormone and retinol throughout the body (Buxbaum and Reixach, 2009; Prapunpoj and Leelawatwattana, 2009). In ATTR amyloidosis, deposition of unfolded TTR aggregates are found in the heart, peripheral nerves and other organs, leading to multiple dysfunctions of these organs (Buxbaum and Reixach, 2009).
Following its description by Andrade in 1952, the number of identified causative mutations in TTR for transthyretin amyloidosis has increased (Benson, 2017). Currently there are over 100 mutations in the TTR gene known to cause the disease, most being single amino acid substitution (Benson, 2017). The most common ATTR mutation is a substitution of valine residue for methionine at position 30 (p.Val30Met) (Stewart et al., 2018). This mutation alters the native tetrameric structure of circulating TTR, promoting release of amyloidogenic TTR monomers which misfold and deposit as amyloid at target organs (Ando et al., 2005; Coelho et al., 2016). Most people affected with hereditary ATTR amyloidosis are heterozygous, expressing both normal and variant TTR protein (Hamilton and Benson, 2001; Suhr et al., 2003).
The clinical phenotype of variant TTR amyloidosis varies greatly by its mutation, age of onset, disease penetrance and prognosis (Ruberg and Berk, 2012). Clinically, ATTR-FAP patients manifest irreversible progressive sensorimotor and autonomic neuropathic symptoms (Hund, 2012). Symptoms start with discomfort in the feet, spontaneous pain and numbness moving proximally to the thigh and then to the upper limbs (Plante-Bordeneuve and Said, 2011; Hund, 2012). Patient deaths are inevitable around 11 years from disease onset (Koike et al., 2012; Adams, 2013). In ATTR-FAC patients, symptoms like diastolic and systolic dysfunction, arrhythmias and other signs of progressive heart failure are common (Castano et al., 2015). In some cases, patients have both peripheral neuropathy and cardiac involvement (Haagsma et al., 2004). In rare cases of OLMA, seizures, ataxia, vitreous opacity, and glaucoma are common symptoms (McColgan et al., 2015; Mathieu et al., 2018) (Figure 1).
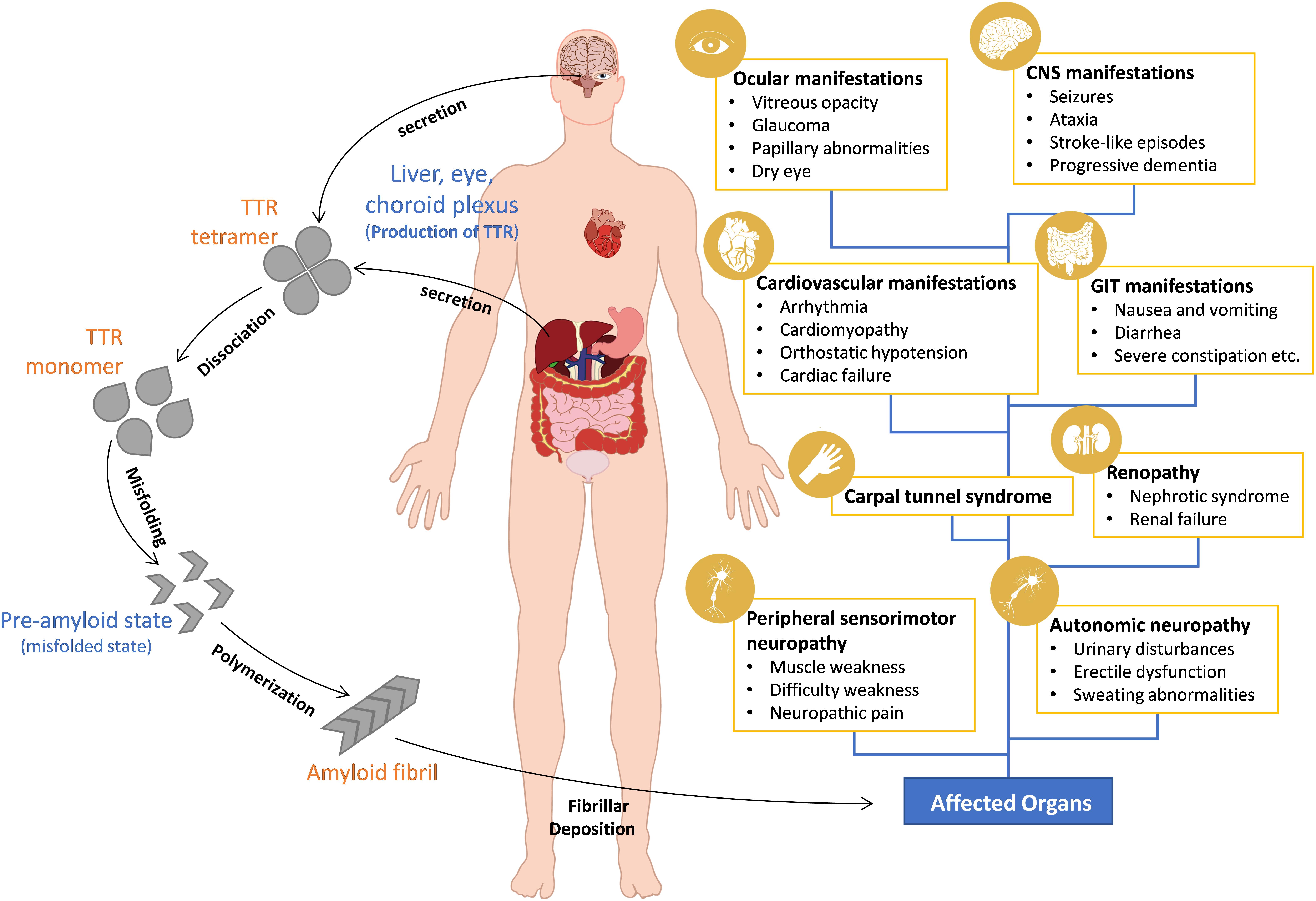
Figure 1. Pathogenesis and clinical manifestations of ATTR amyloidosis. Wild-type and mutant TTR are produced and secreted into the blood or cerebrospinal fluid (CSF) as tetrameric proteins. Mutations in the TTR gene makes the tetrameric TTR protein dissociate into aggregate prone monomers that misfold and aggregate into amyloid fibrils which deposit in the heart, peripheral nerves, and other organs. Clinical manifestations depend mainly on the site of TTR amyloid deposition.
Currently, treatment options for ATTR amyloidosis are limited but liver transplantation (LT) serve as an established option for early-stage disease patients (Gertz, 2017). LT has been shown to improve patient’s survival as it replaces production of mutant TTR with wild-type TTR proteins via the donor organ (Carvalho et al., 2015; Ericzon et al., 2015). However, several limitations to the use of LT have been reported, including toxicities associated with immunosuppression, incidence of hepatic artery thrombosis etc. (Carvalho et al., 2015; Hawkins et al., 2015; Adams et al., 2017) expediting the need for alternative therapies for ATTR amyloidosis. Multiple potential therapies targeting the underlying mechanisms involved in ATTR amyloidosis are emerging (see Ueda and Ando, 2014; Gertz et al., 2015; Nuvolone and Merlini, 2017; Plante-Bordeneuve, 2018 for a review).
Although creating an ideal animal model can be challenging, they play invaluable roles in our understanding of disease pathology and discovery of new drug targets and biomarkers (McGonigle and Ruggeri, 2014; Bolker, 2017). A lot of effort is being put into generating successful models for ATTR amyloidosis that can recapitulate all human characteristics of the disease. However, earlier models could not recapitulate all human pathology of ATTR amyloidosis (see Buxbaum, 2009 for a review). Recently, two models show deposition of TTR deposits in the nerves a major breakthrough in modeling ATTR amyloidosis in mice. This review aims to give an update on the evolution of animal and cell models studied in ATTR amyloidosis, from invertebrate animals, including worms (Caenorhabditis elegans) and flies (Drosophila melanogaster), to mice, non-human primates, and lastly induced pluripotent stem cells (iPSC). We surveyed literatures from PubMed using keywords “Animal models for TTR amyloidosis; Transthyretin FAP animal models; Transthyretin amyloidosis models.” The use of these models in testing potential therapeutic targets have also been highlighted (Figure 2).
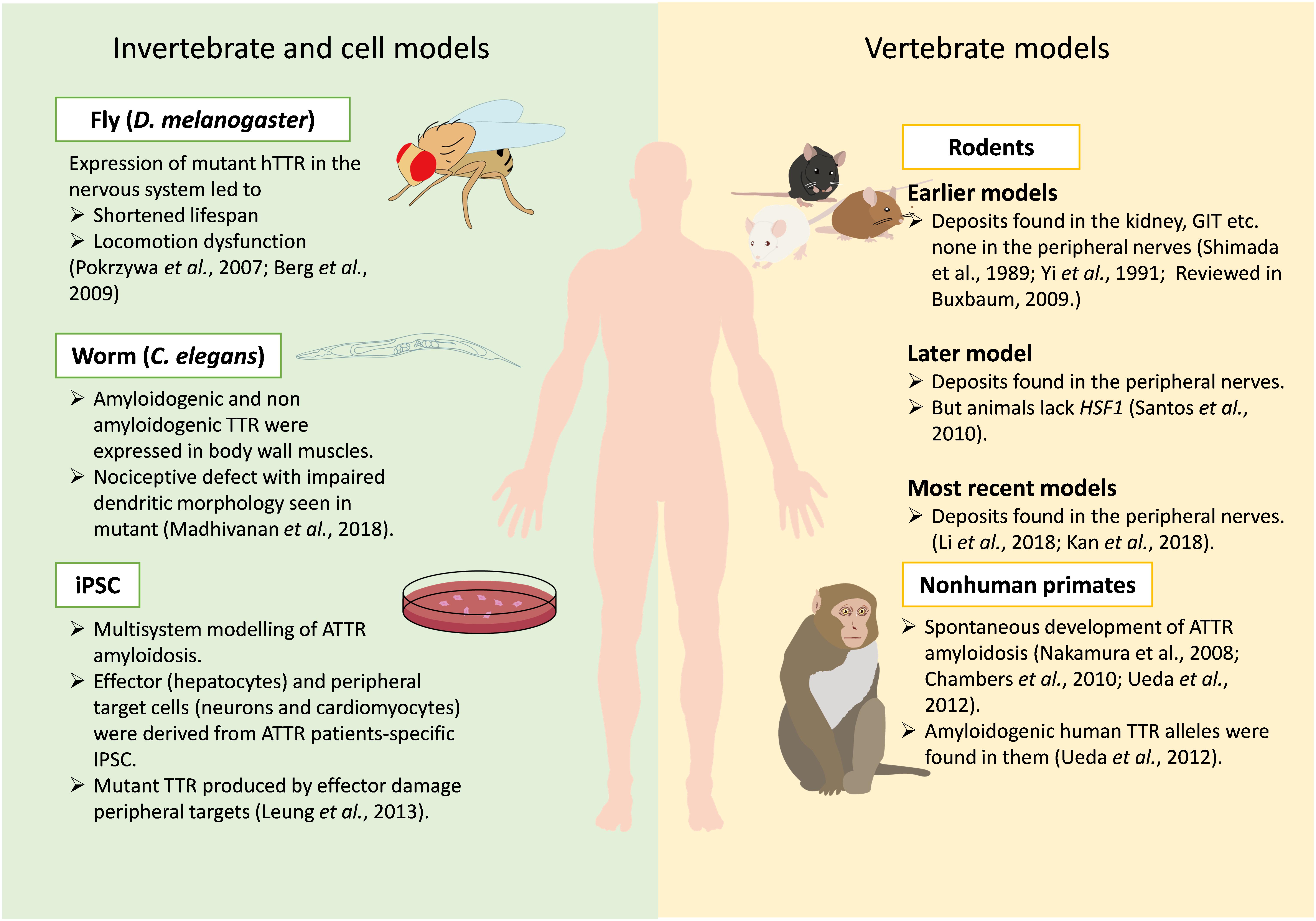
Figure 2. Current models available for studying ATTR amyloidosis. Various models for ATTR amyloidosis are depicted in this figure, including invertebrate, cell and vertebrate models. Key phenotypes and findings from these models are indicated with proper references.
The most widely used invertebrate model organisms for biological research, D. melanogaster and C. elegans, have been used to model ATTR amyloidosis. The existence of conserved molecular pathways between human and invertebrates (Lev et al., 1999; Plowman et al., 1999) coupled with the availability of great genetics, cellular and molecular biology tools makes them good systems for modeling brain diseases and drug discovery research (Alexander et al., 2014; Nussbaum-Krammer and Morimoto, 2014; Ugur et al., 2016).
Since expressing toxic human proteins in invertebrates may mimic some aspects of disease conditions (Muqit and Feany, 2002), the expression of TTR mutants in the fly led to phenotypes partially evocative of the human disease (Figure 2). Expressing ATTR V30M in the nervous system resulted in progressive age-dependent reduced climbing activity and lifespan while TTR WT showed milder phenotype (Berg et al., 2009). Positive amyloid staining by Congo red was also seen in aged ATTR V30M flies (Berg et al., 2009). Similar outcomes were observed with ATTR L55P and an engineered variant TTR-A (mutant with two amino acid replacement TTR V14N/V16E). They showed time-dependent aggregation of misfolded TTR, neurodegeneration, shortened lifespan, and compromised flying ability (Pokrzywa et al., 2007). Although these fly models reveal cell-autonomous toxicity of mutant hTTR instead of cell non-autonomous toxicity as seen in human, they have been used to test peptides which prevent TTR aggregation in vitro (Saelices et al., 2018).
To recapitulate cell non-autonomous toxicity, Madhivanan et al. (2018) created a C. elegans model for ATTR amyloidosis, which expresses human TTR in the body wall muscle. Wild-type TTR and mutants (T119M, V30M, and D18G) were expressed in the body-wall muscle using the unc-54 promoter. Insoluble forms of TTR were detected in C. elegans expressing both ATTR V30M and ATTR D18G mutants (Figure 2). Modeling the differential secretory properties of WT-TTR and other mutants reveal absence of TTR D18G signals from the body cavity, an indication of ineffective secretion due to its highly unstable nature (Hammarstrom et al., 2003; Sekijima et al., 2005; Madhivanan et al., 2018). Expressing ATTR V30M in C. elegans led to nociception impairment analogous to human FAP patients. This also led to significant impairment on dendritic morphology of somatosensory neurons in ATTR V30M animals (Madhivanan et al., 2018). Overall modeling ATTR V30M in C. elegans led to cell non-autonomous neurotoxicity that has not been demonstrated in previous fly models.
Attempts to model ATTR amyloidosis in mice began more than 3 decades ago by random genomic insertion of wild-type or mutant hTTR. However, efforts to produce an ideal mouse model for ATTR amyloidosis have not been as successful as scientists would desire (Buxbaum et al., 2003; Buxbaum, 2009; Kan et al., 2018; Li et al., 2018). To gain insights into the pathogenesis of ATTR amyloidosis, transgenic ATTR V30M mice were created with the mouse metallothionein promoter (Shimada et al., 1989). Despite appreciable serum levels of human TTR in these mice, no amyloid deposits were observed in their peripheral (PNS) and autonomic nervous system (ANS). An improved transgenic line with increased gene copies of TTR V30M was later established (Yi et al., 1991). Slight TTR amyloid deposits were found in the kidneys, gastrointestinal tract (GIT) and cardiovascular organs of these mice at 6 months (Figure 2). Despite extensive amyloid deposition resembling SSA-like amyloid deposits at 24 months, no amyloid deposit was detected in their choroid plexus, PNS and ANS (Yi et al., 1991; Hamilton and Benson, 2001; Buxbaum, 2009). Subsequently, several research groups have generated transgenic animals from wild-type and other mutant TTR gene constructs; none had congophilic deposits in the peripheral nerves (see Buxbaum, 2009 for a review).
The activation of inflammatory and oxidative stress pathways by TTR deposits in patients (Sousa et al., 2000, 2001) led to the hypothesis that disruption of stress response pathway may accelerate TTR deposition. When transgenic mice expressing human TTR V30M were crossed with mice lacking heat shock transcription factor (Hsf1), their offspring showed early and extensive deposition of non-fibrillar TTR in the GIT and subsequent fibrillar non-congophilic deposit in sciatic nerve and dorsal root ganglia for the first time (Santos et al., 2010). Although there are lots of unanswered question about this model due to the absence of Hsf1, making it less optimal for either fine analysis or experimental therapies (Buxbaum, 2009), it has been used in numerous studies to understand pathogenesis of ATTR amyloidogenesis and the preclinical evaluation of pharmacotherapy and gene therapy studies (Cardoso et al., 2010; Ferreira et al., 2014, 2016; Butler et al., 2016).
In hTTR V30M Hsf1+/- mice, TTR-targeting siRNA-mediated silencing of hepatic TTR expression inhibit TTR deposition and facilitate regression of existing TTR deposits in tissues (Butler et al., 2016). In addition, the TTR stabilizer, tafamidis led to regression of TTR deposits across a broad range in affected tissues, together with reducing serum TTR protein concentration in hTTR V30M Hsf1+/- mice. This led to the approval of Patisiran by the US FDA and Tafamidis by the Japanese and European drug agencies for the management of ATTR amyloidosis after clinical trials. Patisiran, an RNAi therapeutic, was documented to successfully improve multiple clinical manifestations of hereditary transthyretin amyloidosis (Adams et al., 2018). Tafamidis on the other hand effectively delay progression of TTR-related polyneuropathy and lower rates of cardiovascular-related hospitalizations in patients (Maurer et al., 2018; Mundayat et al., 2018).
Nevertheless, the search for an ideal mouse model isn’t over until a model which mimic ATTR amyloidosis patients in symptoms and tissue-distribution of congophilic TTR amyloid deposit is achieved. Two groups recently generated mouse models with amyloid deposits in the peripheral nerves without additional genetic changes such as Hsf1 deficiency (Kan et al., 2018; Li et al., 2018). Biophysical analysis alongside biochemical studies in mice reveal that murine TTR inhibit aggregation and deposition of highly unstable human TTR via formation of a highly stable human-mouse heterotetramers (Tagoe et al., 2007; Reixach et al., 2008). To overcome this, Li et al. (2018), produced humanized mouse strains at both the Ttr and Rbp4 loci allowing human TTR (hTTR) to associate with human RBP4 (hRBP4). Two mouse lines; normal (TtrhTTRV 30/hTTRV 30: Rbp4hRBP4/hRBP4, abbreviated as hV/hV:hR/hR) and mutant (TtrhTTRV 30/hTTRM30:Rbp4hRBP4/hRBP4, abbreviated as hV/hM:hR/hR) were generated. Remarkably, even though both hTTR and hRBP4 were found at relatively low levels in the serum of these mouse lines, these mice present with distinct amyloid deposition than previous conventional transgenic models. Amyloid deposits was first noticed in the GIT of hV/hV:hR/hR and hV/hM:hR/hR mice at 12 and 18 months respectively increasingly with age. At 24 months, amyloid deposition was observed in the heart of both lines with more deposit in the mutant mice. Notably at 24 months, Congo red positive amyloid deposits were found in the perineurium of the sciatic nerve of the mutant line (Li et al., 2018). This study further reveal the impact RBP4 might have on amyloid deposition as it is known to prevent TTR amyloid formation at lower pH (White and Kelly, 2001). A limitation to these successful models might be their inability to symbolize human TTR status due to their low serum hTTR level and high proportion of unbound TTR with RBP4 as it is unknown whether amyloid deposition occur in human under such low serum level of hTTR.
Of the earliest signs of ATTR-FAP is gradual degeneration of sensory nerve cells leading to abnormal sensations such as pain (Plante-Bordeneuve and Said, 2011; Ando et al., 2013). Although previous ATTR-FAP mutant mice have helped researchers gain insights into the pathogenesis of the disease (Buxbaum, 2009; Li et al., 2018), none of them replicate ATTR-FAP’s early pain symptoms. A human TTR knock-in mice established by replacing one allele of the mouse Ttr locus with human TTR A97S (hTTRA97S) recapitulate these symptoms (Kan et al., 2018). The TTR-A97S mutation is a hotspot Taiwanese mutation characterized by late onset presentation in patients marked with axonal degeneration (Liu et al., 2008; Yang et al., 2010; Chao et al., 2015). To achieve this, knock-in mice for both wild-type and ATTR A97S were generated by replacing mTtr with hTTR via homologous recombination without altering the promoter and enhancer sequences of the mTtr. The knock-in mice were backcrossed to C57BL/6 females to obtain heterozygotes with genotypes hTTRwt/mTtrwt (abbreviated as hTTRwt) and hTTRA97S/mTtrwt (abbreviated as hTTRA97S) (Kan et al., 2018). Serum and liver hTTR protein levels were detectable in both mice with lower levels in hTTRA97S animals. Congophilic amyloid deposits were detected in the epineurium of the sural nerve, distal convoluted tubule of the kidney and other organs in aging hTTRA97S (>2 years) mice but not in the adult group (8–56 weeks). Aging hTTRA97S mice also exhibit hypersensitivity to mechanical stimuli, less nerve fiber density and less nerve sensation which are early manifestations of ATTR-FAP (Kan et al., 2018). Despite lack of motor deficit in the hTTRA97S mouse, the presence of congophilic deposits plus display of early ATTR-FAP symptoms makes it a good model to test drugs targeting early-stage symptoms of ATTR-FAP.
Spontaneous development of ATTR amyloidosis have been reported in several vervet monkeys (Nakamura et al., 2008; Chambers et al., 2010; Ueda et al., 2012). In the monkeys observed by Ueda et al. (2012) aged vervets were observed to have cardiac arrhythmia, bradycardia and reduced ejection fraction in the heart mimicking human ATTR WT amyloidosis. Sequencing data reveal the presence of the amyloidogenic human TTR Ile 122 allele in these vervets, suggesting its pathogenic role in their development of ATTR amyloidosis. Other human amyloidogenic TTR alleles were also documented in non-human primates: Thr81 in New World monkeys (Callithrix jacchus) and the black-bearded saki (Chiropotes satanas) and Val107 in all New World monkeys (Ueda et al., 2012). These evidences are suggestive of non-human primates as possible valid pathologic models for ATTR amyloidosis.
George J. Murphy’s group generated the first known patient-specific induced pluripotent stem cell (iPSC) library for ATTR amyloidosis (Leung and Murphy, 2016; Giadone et al., 2018). This development may help resolve existing variations between patients with identical TTR mutations in readiness for personalized ATTR amyloidosis modeling. IPSC lines from a ATTR L55P patient was generated and subsequently differentiated into hepatocytes (which produce mutant L55P TTR) and peripheral targets (cardiomyocytes and neurons) for ATTR amyloid deposition (Leung et al., 2013). IPSC derived hepatocytes successfully secrete TTRs which were internalized by IPSC neuronal-lineage cells as previously reported (Fleming et al., 2009). Stress-response markers and other known associated genes were upregulated in peripheral targets exposed to ATTR L55P media (Sousa et al., 2005; Leung et al., 2013). This successful patient derived IPSCs could be helpful in understanding complex etiology of the disease.
A good animal model is one which can replicate the human disease by depicting key pathologic and physiologic symptoms. Such model would satisfy the laid criteria for animal validation (Belzung and Lemoine, 2011). Despite recent success in the generation of mouse models with amyloid deposits in the nerves (Kan et al., 2018; Li et al., 2018), discrepancies in key physiologic and behavioral features of ATTR amyloidosis were seen. For example, amyloid deposits were seen earlier in mice carrying the normal human TTR gene (Li et al., 2018), a phenomenon inconsistent with human patient (Ton et al., 2014). Lack of motor deficit was also seen in the model by Kan et al., 2018. Slight modifications of these models may be needed to rectify these discrepancies. Considering the wide genotypic and phenotypic spectrum of this disease, generating mutant-specific ATTR animal models may be required to address specific questions that might allow construction of mutant-specific hypotheses essential to making further breakthroughs.
However, by combining different models, it might be possible to eventually replicate the clinical condition better. The challenge is to identify which aspect of the human disease a particular model addresses. Using these models, we have learnt important pathogenic mechanisms associated with ATTR amyloidosis, including aging-related oxidation, histological inflammation, thermodynamic stability, etc. Based on this knowledge, there have been several mechanism-driven therapeutic targets identified leading to the approval of Patisiran and Tafamidis for the management of this disease. Although this current therapies have achieved significant progress in certain indices for both peripheral neuropathy and cardiopathy, a multi-target drug design may be required to adequately manage the disease.
J-WT finalized the manuscript. All authors prepared the manuscript.
This work was supported by the Ministry of Science and Technology, Taiwan (MOST-105-2633-B-009-003, 106-2321-B-075-001, 106-2628-B-010-002-MY3, 107-2321-B-075-001, and 107-2221-E-010 -014), the Development and Construction Plan of NYMU School of Medicine (107F-M01-0502), and the Brain Research Center, NYMU through the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE), Taiwan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adams, D. (2013). Recent advances in the treatment of familial amyloid polyneuropathy. Ther. Adv. Neurol. Disord. 6, 129–139. doi: 10.1177/1756285612470192
Adams, D., Gonzalez-Duarte, A., O’Riordan, W. D., Yang, C. C., Ueda, M., Kristen, A. V., et al. (2018). Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 11–21. doi: 10.1056/NEJMoa1716153
Adams, D., Lozeron, P., and Lacroix, C. (2012). Amyloid neuropathies. Curr. Opin. Neurol. 25, 564–572. doi: 10.1097/WCO.0b013e328357bdf6
Adams, D., Suhr, O. B., Dyck, P. J., Litchy, W. J., Leahy, R. G., Chen, J., et al. (2017). Trial design and rationale for APOLLO, a Phase 3, placebo-controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol. 17:181. doi: 10.1186/s12883-017-0948-5
Alexander, A. G., Marfil, V., and Li, C. (2014). Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet. 5:279. doi: 10.3389/fgene.2014.00279
Almeida, M. R., Macedo, B., Cardoso, I., Alves, I., Valencia, G., Arsequell, G., et al. (2004). Selective binding to transthyretin and tetramer stabilization in serum from patients with familial amyloidotic polyneuropathy by an iodinated diflunisal derivative. Biochem. J. 381, 351–356. doi: 10.1042/BJ20040011
Ando, Y., Coelho, T., Berk, J. L., Cruz, M. W., Ericzon, B. G., Ikeda, S., et al. (2013). Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J. Rare Dis. 8:31. doi: 10.1186/1750-1172-8-31
Ando, Y., Nakamura, M., and Araki, S. (2005). Transthyretin-related familial amyloidotic polyneuropathy. Arch. Neurol. 62, 1057–1062. doi: 10.1001/archneur.62.7.1057
Belzung, C., and Lemoine, M. (2011). Criteria of validity for animal models of psychiatric disorders: focus on anxiety disorders and depression. Biol. Mood Anxiety Disord. 1:9. doi: 10.1186/2045-5380-1-9
Benson, M. D. (2017). Transthyretin amyloidosis: a little history of hereditary amyloidosis. Amyloid 24, 76–77. doi: 10.1080/13506129.2017.1281116
Benson, M. D., and Kincaid, J. C. (2007). The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 36, 411–423. doi: 10.1002/mus.20821
Berg, I., Thor, S., and Hammarstrom, P. (2009). Modeling familial amyloidotic polyneuropathy (Transthyretin V30M) in Drosophila melanogaster. Neurodegener. Dis. 6, 127–138. doi: 10.1159/000213761
Bolker, J. A. (2017). Animal models in translational research: rosetta stone or stumbling block? BioEssays 39, 1–8. doi: 10.1002/bies.201700089
Butler, J. S., Chan, A., Costelha, S., Fishman, S., Willoughby, J. L., Borland, T. D., et al. (2016). Preclinical evaluation of RNAi as a treatment for transthyretin-mediated amyloidosis. Amyloid 23, 109–118. doi: 10.3109/13506129.2016.1160882
Buxbaum, J., Tagoe, C., Gallo, G., Reixach, N., and French, D. (2003). The pathogenesis of transthyretin tissue deposition: lessons from transgenic mice. Amyloid 10(Suppl. 1), 2–6.
Buxbaum, J. N. (2009). Animal models of human amyloidoses: are transgenic mice worth the time and trouble? FEBS Lett. 583, 2663–2673. doi: 10.1016/j.febslet.2009.07.031
Buxbaum, J. N., and Reixach, N. (2009). Transthyretin: the servant of many masters. Cell. Mol. Life Sci. 66, 3095–3101. doi: 10.1007/s00018-009-0109-0
Cardoso, I., Martins, D., Ribeiro, T., Merlini, G., and Saraiva, M. J. (2010). Synergy of combined doxycycline/TUDCA treatment in lowering transthyretin deposition and associated biomarkers: studies in FAP mouse models. J. Transl. Med. 8:74. doi: 10.1186/1479-5876-8-74
Carvalho, A., Rocha, A., and Lobato, L. (2015). Liver transplantation in transthyretin amyloidosis: issues and challenges. Liver Transplant. 21, 282–292. doi: 10.1002/lt.24058
Castano, A., Drachman, B. M., Judge, D., and Maurer, M. S. (2015). Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail. Rev. 20, 163–178. doi: 10.1007/s10741-014-9462-7
Chambers, J. K., Kanda, T., Shirai, A., Higuchi, K., Ikeda, S., and Une, Y. (2010). Senile systemic amyloidosis in an aged savannah monkey (Cercopithecus aethiops) with tenosynovial degeneration. J. Vet. Med. Sci. 72, 657–659. doi: 10.1292/jvms.09-0394
Chao, C. C., Huang, C. M., Chiang, H. H., Luo, K. R., Kan, H. W., Yang, N. C., et al. (2015). Sudomotor innervation in transthyretin amyloid neuropathy: pathology and functional correlates. Ann. Neurol. 78, 272–283. doi: 10.1002/ana.24438
Coelho, T., Merlini, G., Bulawa, C. E., Fleming, J. A., Judge, D. P., Kelly, J. W., et al. (2016). Mechanism of Action and clinical application of tafamidis in hereditary transthyretin amyloidosis. Neurol. Ther. 5, 1–25. doi: 10.1007/s40120-016-0040-x
Ericzon, B. G., Wilczek, H. E., Larsson, M., Wijayatunga, P., Stangou, A., Pena, J. R., et al. (2015). Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation 99, 1847–1854. doi: 10.1097/TP.0000000000000574
Ferreira, N., Goncalves, N. P., Saraiva, M. J., and Almeida, M. R. (2016). Curcumin: a multi-target disease-modifying agent for late-stage transthyretin amyloidosis. Sci. Rep. 6:26623. doi: 10.1038/srep26623
Ferreira, N., Pereira-Henriques, A., Attar, A., Klarner, F. G., Schrader, T., Bitan, G., et al. (2014). Molecular tweezers targeting transthyretin amyloidosis. Neurotherapeutics 11, 450–461. doi: 10.1007/s13311-013-0256-8
Fleming, C. E., Mar, F. M., Franquinho, F., Saraiva, M. J., and Sousa, M. M. (2009). Transthyretin internalization by sensory neurons is megalin mediated and necessary for its neuritogenic activity. J. Neurosci. 29, 3220–3232. doi: 10.1523/JNEUROSCI.6012-08.2009
Gertz, M. A. (2017). Hereditary ATTR amyloidosis: burden of illness and diagnostic challenges. Am. J. Manag. Care 23, S107–S112.
Gertz, M. A., Benson, M. D., Dyck, P. J., Grogan, M., Coelho, T., Cruz, M., et al. (2015). Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J. Am. Coll. Cardiol. 66, 2451–2466. doi: 10.1016/j.jacc.2015.09.075
Giadone, R. M., Rosarda, J. D., Akepati, P. R., Thomas, A. C., Boldbaatar, B., James, M. F., et al. (2018). A library of ATTR amyloidosis patient-specific induced pluripotent stem cells for disease modelling and in vitro testing of novel therapeutics. Amyloid 25, 148–155. doi: 10.1080/13506129.2018.1489228
Haagsma, E. B., Hawkins, P. N., Benson, M. D., Lachmann, H. J., Bybee, A., and Hazenberg, B. P. (2004). Familial amyloidotic polyneuropathy with severe renal involvement in association with transthyretin Gly47Glu in Dutch, British and American-Finnish families. Amyloid 11, 44–49. doi: 10.1080/13506120410001682578
Hamilton, J. A., and Benson, M. D. (2001). Transthyretin: a review from a structural perspective. Cell. Mol. Life Sci. 58, 1491–1521. doi: 10.1007/PL00000791
Hammarstrom, P., Sekijima, Y., White, J. T., Wiseman, R. L., Lim, A., Costello, C. E., et al. (2003). D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: a prescription for central nervous system amyloidosis? Biochemistry 42, 6656–6663.
Hawkins, P. N., Ando, Y., Dispenzeri, A., Gonzalez-Duarte, A., Adams, D., and Suhr, O. B. (2015). Evolving landscape in the management of transthyretin amyloidosis. Ann. Med. 47, 625–638. doi: 10.3109/07853890.2015.1068949
Hund, E. (2012). Familial amyloidotic polyneuropathy: current and emerging treatment options for transthyretin-mediated amyloidosis. Appl. Clin. Genet. 5, 37–41. doi: 10.2147/TACG.S19903
Ingenbleek, Y., and Bernstein, L. H. (2015). Plasma transthyretin as a biomarker of lean body mass and catabolic states. Adv. Nutri. 6, 572–580. doi: 10.3945/an.115.008508
Johnson, B. A., Coutts, M., Vo, H. M., Hao, X., Fatima, N., Rivera, M. J., et al. (2018). Accurate, strong, and stable reporting of choroid plexus epithelial cells in transgenic mice using a human transthyretin BAC. Fluids Barriers CNS 15:22. doi: 10.1186/s12987-018-0107-4
Kan, H. W., Chiang, H., Lin, W. M., Yu, I. S., Lin, S. W., and Hsieh, S. T. (2018). Sensory nerve degeneration in a mouse model mimicking early manifestations of familial amyloid polyneuropathy due to transthyretin Ala97Ser. Neuropathol. Appl. Neurobiol. 44, 673–686. doi: 10.1111/nan.12477
Koike, H., Tanaka, F., Hashimoto, R., Tomita, M., Kawagashira, Y., Iijima, M., et al. (2012). Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late-onset cases from non-endemic areas. J. Neurol. Neurosurg. Psychiatry 83, 152–158. doi: 10.1136/jnnp-2011-301299
Leung, A., and Murphy, G. J. (2016). Multisystemic disease modeling of liver-derived protein folding disorders using induced pluripotent stem cells (iPSCs). Methods Mol. Biol. 1353, 261–270. doi: 10.1007/7651_2014_194
Leung, A., Nah, S. K., Reid, W., Ebata, A., Koch, C. M., Monti, S., et al. (2013). Induced pluripotent stem cell modeling of multisystemic, hereditary transthyretin amyloidosis. Stem Cell Rep. 1, 451–463. doi: 10.1016/j.stemcr.2013.10.003
Lev, S., Hernandez, J., Martinez, R., Chen, A., Plowman, G., and Schlessinger, J. (1999). Identification of a novel family of targets of PYK2 related to drosophila retinal degeneration B (rdgB) protein. Mol. Cell. Biol. 19, 2278–2288. doi: 10.1128/MCB.19.3.2278
Li, X., Lyu, Y., Shen, J., Mu, Y., Qiang, L., Liu, L., et al. (2018). Amyloid deposition in a mouse model humanized at the transthyretin and retinol-binding protein 4 loci. Lab. Invest. 98, 512–524. doi: 10.1038/s41374-017-0019-y
Liu, Y. T., Lee, Y. C., Yang, C. C., Chen, M. L., and Lin, K. P. (2008). Transthyretin Ala97Ser in Chinese-taiwanese patients with familial amyloid polyneuropathy: genetic studies and phenotype expression. J. Neurol. Sci. 267, 91–99. doi: 10.1016/j.jns.2007.10.011
Madhivanan, K., Greiner, E. R., Alves-Ferreira, M., Soriano-Castell, D., Rouzbeh, N., Aguirre, C. A., et al. (2018). Cellular clearance of circulating transthyretin decreases cell-nonautonomous proteotoxicity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 115, E7710–E7719. doi: 10.1073/pnas.1801117115
Mathieu, F., Morgan, E., So, J., Munoz, D. G., Mason, W., and Kongkham, P. (2018). Oculoleptomeningeal amyloidosis secondary to the rare transthyretin c.381TG (p.Ile127Met) mutation. World Neurosurg. 111, 190–193. doi: 10.1016/j.wneu.2017.12.096
Maurer, M. S., Schwartz, J. H., Gundapaneni, B., Elliott, P. M., Merlini, G., Waddington-Cruz, M., et al. (2018). Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 379, 1007–1016. doi: 10.1056/NEJMoa1805689
McColgan, P., Viegas, S., Gandhi, S., Bull, K., Tudor, R., Sheikh, F., et al. (2015). Oculoleptomeningeal amyloidosis associated with transthyretin Leu12Pro in an African patient. J. Neurol. 262, 228–234. doi: 10.1007/s00415-014-7594-2
McGonigle, P., and Ruggeri, B. (2014). Animal models of human disease: challenges in enabling translation. Biochem. Pharmacol. 87, 162–171. doi: 10.1016/j.bcp.2013.08.006
Mundayat, R., Stewart, M., Alvir, J., Short, S., Ong, M. L., Keohane, D., et al. (2018). Positive effectiveness of tafamidis in delaying disease progression in transthyretin familial amyloid polyneuropathy up to 2 years: an analysis from the transthyretin amyloidosis outcomes survey (THAOS). Neurol. Ther. 7, 87–101. doi: 10.1007/s40120-018-0097-9
Muqit, M. M., and Feany, M. B. (2002). Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat. Rev. Neurosci. 3, 237–243. doi: 10.1038/nrn751
Nakamura, S., Okabayashi, S., Ageyama, N., Koie, H., Sankai, T., Ono, F., et al. (2008). Transthyretin amyloidosis and two other aging-related amyloidoses in an aged vervet monkey. Vet. Pathol. 45, 67–72. doi: 10.1354/vp.45-1-67
Nussbaum-Krammer, C. I., and Morimoto, R. I. (2014). Caenorhabditis elegans as a model system for studying non-cell-autonomous mechanisms in protein-misfolding diseases. Dis. Models Mechan. 7, 31–39. doi: 10.1242/dmm.013011
Nuvolone, M., and Merlini, G. (2017). Emerging therapeutic targets currently under investigation for the treatment of systemic amyloidosis. Expert Opin. Ther. Targets 21, 1095–1110. doi: 10.1080/14728222.2017.1398235
Pfeffer, B. A., Becerra, S. P., Borst, D. E., and Wong, P. (2004). Expression of transthyretin and retinol binding protein mRNAs and secretion of transthyretin by cultured monkey retinal pigment epithelium. Mol. Vis. 10, 23–30.
Plante-Bordeneuve, V. (2018). Transthyretin familial amyloid polyneuropathy: an update. J. Neurol. 265, 976–983. doi: 10.1007/s00415-017-8708-4
Plante-Bordeneuve, V., and Said, G. (2011). Familial amyloid polyneuropathy. Lancet Neurol. 10, 1086–1097. doi: 10.1016/S1474-4422(11)70246-0
Plowman, G. D., Sudarsanam, S., Bingham, J., Whyte, D., and Hunter, T. (1999). The protein kinases of Caenorhabditis elegans: a model for signal transduction in multicellular organisms. Proc. Natl. Acad. Sci. U.S.A. 96, 13603–13610. doi: 10.1073/pnas.96.24.13603
Pokrzywa, M., Dacklin, I., Hultmark, D., and Lundgren, E. (2007). Misfolded transthyretin causes behavioral changes in a Drosophila model for transthyretin-associated amyloidosis. Eur. J. Neurosci. 26, 913–924. doi: 10.1111/j.1460-9568.2007.05728.x
Prapunpoj, P., and Leelawatwattana, L. (2009). Evolutionary changes to transthyretin: structure-function relationships. FEBS J. 276, 5330–5341. doi: 10.1111/j.1742-4658.2009.07243.x
Rapezzi, C., Quarta, C. C., Riva, L., Longhi, S., Gallelli, I., Lorenzini, M., et al. (2010). Transthyretin-related amyloidoses and the heart: a clinical overview. Nat. Rev. Cardiol. 7, 398–408. doi: 10.1038/nrcardio.2010.67
Reixach, N., Foss, T. R., Santelli, E., Pascual, J., Kelly, J. W., and Buxbaum, J. N. (2008). Human-murine transthyretin heterotetramers are kinetically stable and non-amyloidogenic. A lesson in the generation of transgenic models of diseases involving oligomeric proteins. J. Biol. Chem. 283, 2098–2107. doi: 10.1074/jbc.M708028200
Richardson, S. J., Wijayagunaratne, R. C., D’Souza, D. G., Darras, V. M., and Van Herck, S. L. (2015). Transport of thyroid hormones via the choroid plexus into the brain: the roles of transthyretin and thyroid hormone transmembrane transporters. Front. Neurosci. 9:66. doi: 10.3389/fnins.2015.00066
Ruberg, F. L., and Berk, J. L. (2012). Transthyretin (TTR) cardiac amyloidosis. Circulation 126, 1286–1300. doi: 10.1161/CIRCULATIONAHA.111.078915
Saelices, L., Pokrzywa, M., Pawelek, K., and Eisenberg, D. S. (2018). Assessment of the effects of transthyretin peptide inhibitors in Drosophila models of neuropathic ATTR. Neurobiol. Dis. 120, 118–125. doi: 10.1016/j.nbd.2018.09.007
Santos, S. D., Fernandes, R., and Saraiva, M. J. (2010). The heat shock response modulates transthyretin deposition in the peripheral and autonomic nervous systems. Neurobiol. Aging 31, 280–289. doi: 10.1016/j.neurobiolaging.2008.04.001
Sekijima, Y., Wiseman, R. L., Matteson, J., Hammarstrom, P., Miller, S. R., Sawkar, A. R., et al. (2005). The biological and chemical basis for tissue-selective amyloid disease. Cell 121, 73–85. doi: 10.1016/j.cell.2005.01.018
Shimada, K., Maeda, S., Murakami, T., Nishiguchi, S., Tashiro, F., Yi, S., et al. (1989). Transgenic mouse model of familial amyloidotic polyneuropathy. Mol. Biol. Med. 6, 333–343.
Sousa, M. M., do Amaral, J. B., Guimarães, A., and Saraiva, M. J. (2005). Up-regulation of the extracellular matrix remodeling genes, biglycan, neutrophil gelatinase-associated lipocalin, and matrix metalloproteinase-9 in familial amyloid polyneuropathy. FASEB J. 19, 124–126. doi: 10.1096/fj.04-2022fje
Sousa, M. M., Du Yan, S., Fernandes, R., Guimaraes, A., Stern, D., and Saraiva, M. J. (2001). Familial amyloid polyneuropathy: receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. J. Neurosci. 21, 7576–7586. doi: 10.1523/JNEUROSCI.21-19-07576.2001
Sousa, M. M., Yan, S. D., Stern, D., and Saraiva, M. J. (2000). Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab. Invest. 80, 1101–1110. doi: 10.1038/labinvest.3780116
Stewart, M., Shaffer, S., Murphy, B., Loftus, J., Alvir, J., Cicchetti, M., et al. (2018). Characterizing the high disease burden of transthyretin amyloidosis for patients and caregivers. Neurol. Ther. 7, 349–364. doi: 10.1007/s40120-018-0106-z
Suhr, O. B., Svendsen, I. H., Andersson, R., Danielsson, A., Holmgren, G., and Ranlov, P. J. (2003). Hereditary transthyretin amyloidosis from a Scandinavian perspective. J. Intern. Med. 254, 225–235. doi: 10.1046/j.1365-2796.2003.01173.x
Tagoe, C. E., Reixach, N., Friske, L., Mustra, D., French, D., Gallo, G., et al. (2007). In vivo stabilization of mutant human transthyretin in transgenic mice. Amyloid 14, 227–236. doi: 10.1080/13506120701464396
Ton, V. K., Mukherjee, M., and Judge, D. P. (2014). Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin. Med. Insights Cardiol. 8, 39–44. doi: 10.4137/CMC.S15719
Ueda, M., Ageyama, N., Nakamura, S., Nakamura, M., Chambers, J. K., Misumi, Y., et al. (2012). Aged vervet monkeys developing transthyretin amyloidosis with the human disease-causing Ile122 allele: a valid pathological model of the human disease. Lab. Invest. 92, 474–484. doi: 10.1038/labinvest.2011.195
Ueda, M., and Ando, Y. (2014). Recent advances in transthyretin amyloidosis therapy. Transl. Neurodegener. 3:19. doi: 10.1186/2047-9158-3-19
Ugur, B., Chen, K., and Bellen, H. J. (2016). Drosophila tools and assays for the study of human diseases. Dis. Models Mech. 9, 235–244. doi: 10.1242/dmm.023762
White, J. T., and Kelly, J. W. (2001). Support for the multigenic hypothesis of amyloidosis: the binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc. Natl. Acad. Sci. U.S.A. 98, 13019–13024. doi: 10.1073/pnas.241406698
Yang, N. C., Lee, M. J., Chao, C. C., Chuang, Y. T., Lin, W. M., Chang, M. F., et al. (2010). Clinical presentations and skin denervation in amyloid neuropathy due to transthyretin Ala97Ser. Neurology 75, 532–538. doi: 10.1212/WNL.0b013e3181ec7fda
Keywords: transthyretin, familial amyloid polyneuropathy, C. elegans, D. melanogaster, transgenic mouse, iPSC, neurodegenerative disorder, animal model
Citation: Ibrahim RB, Liu Y-T, Yeh S-Y and Tsai J-W (2019) Contributions of Animal Models to the Mechanisms and Therapies of Transthyretin Amyloidosis. Front. Physiol. 10:338. doi: 10.3389/fphys.2019.00338
Received: 10 December 2018; Accepted: 13 March 2019;
Published: 02 April 2019.
Edited by:
Wael M. Y. Mohamed, International Islamic University Malaysia, MalaysiaReviewed by:
Yahya M. Naguib, Menofia University, EgyptCopyright © 2019 Ibrahim, Liu, Yeh and Tsai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin-Wu Tsai, dHNhaWp3QHltLmVkdS50dw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.