 Kelly Q. Zhou
Kelly Q. Zhou Colin R. Green
Colin R. Green Laura Bennet
Laura Bennet Alistair J. Gunn
Alistair J. Gunn Joanne O. Davidson
Joanne O. Davidson- 1Department of Physiology, The University of Auckland, Auckland, New Zealand
- 2Department of Ophthalmology, The University of Auckland, Auckland, New Zealand
Perinatal brain injury remains a major cause of death and life-long disability. Perinatal brain injury is typically associated with hypoxia-ischemia and/or infection/inflammation. Both hypoxia-ischemia and infection trigger an inflammatory response in the brain. The inflammatory response can contribute to brain cell loss and chronic neuroinflammation leading to neurological impairments. It is now well-established that brain injury evolves over time, and shows a striking spread from injured to previously uninjured regions of the brain. There is increasing evidence that this spread is related to opening of connexin hemichannels and pannexin channels, both of which are large conductance membrane channels found in almost all cell types in the brain. Blocking connexin hemichannels within the first 3 h after hypoxia-ischemia has been shown to improve outcomes in term equivalent fetal sheep but it is important to also understand the downstream pathways linking membrane channel opening with the development of injury in order to identify new therapeutic targets. Open membrane channels release adenosine triphosphate (ATP), and other neuroactive molecules, into the extracellular space. ATP has an important physiological role, but has also been reported to act as a damage-associated molecular pattern (DAMP) signal mediated through specific purinergic receptors and so act as a primary signal 1 in the innate immune system inflammasome pathway. More crucially, extracellular ATP is a key inflammasome signal 2 activator, with purinergic receptor binding triggering the assembly of the multi-protein inflammasome complex. The inflammasome pathway and complex formation contribute to activation of inflammatory caspases, and the release of inflammatory cytokines, including interleukin (IL)-1β, tumor necrosis factor (TNF)-α, IL-18, and vascular endothelial growth factor (VEGF). We propose that the NOD-like receptor protein-3 (NLRP3) inflammasome, which has been linked to inflammatory responses in models of ischemic stroke and various inflammatory diseases, may be one mechanism by which connexin hemichannel opening especially mediates perinatal brain injury.
Introduction
Perinatal brain injury is associated with death or significant long-term neurodevelopmental impairment, affecting 1.15 million infants in the world in 2010 (Lee et al., 2013). It affects both term and preterm infants (born <37 weeks of completed gestation), with the greatest incidence in the most preterm infants (Larroque et al., 2008). In term born infants, perinatal brain injury is most often linked to hypoxia-ischemia (HI) (Vannucci, 2000; Shankaran et al., 2005), where the incidence of moderate to severe hypoxic-ischemic encephalopathy (HIE) is ~1–3 in 1,000 live births (Edwards et al., 2010). By contrast, in preterm infants, brain injury arises from the complex interaction of HI, infection/inflammation and preterm birth itself (Galinsky et al., 2018b). Strikingly, ~50% of extremely preterm infants (born <26 weeks of gestation) develop moderate to severe disability (Marlow et al., 2005).
Currently, there is only one established neuroprotective treatment for term babies born with HIE, which is therapeutic hypothermia. Clinically, this treatment is only partially effective, with a number needed to treat of ~8, meaning of 8 treated infants with HIE, one additional infant will survive without moderate to severe disability (Gunn et al., 2017; Natarajan et al., 2018). However, there are currently no proven treatments to reduce brain damage for preterm infants with HIE or perinatal infection/inflammation. Therefore, there is significant interest in developing complementary or novel treatments for perinatal brain injury after HI or infection/inflammation. For term infants, it is important to develop interventions with additive benefit to therapeutic hypothermia, as it is now standard therapy. Novel treatments are needed for term and preterm infection/inflammation.
After HI, there is a striking evolution of injury which progresses over time from injured to previously uninjured regions of the brain (Azzopardi et al., 1989; Thornton et al., 1998). The mechanisms behind this spread are not well-understood. Increasing evidence supports the opening of cell membrane channels after HI as one of the mediators of this propagation of injury, particularly connexin hemichannels, which are the unopposed half of a gap junction, forming large conductance channels in the cell membrane (Davidson et al., 2012, 2014; Wang et al., 2014; Li et al., 2015). Pannexin channels may also open under pathological conditions, although they appear to be tightly regulated (Qiu and Dahl, 2009) and their role in the developing brain remains unclear. In this review, we examine the hypothesis that prolonged opening of connexin hemichannels leads to a cascade of injurious mechanism, potentially through inflammation by activation, amplification and perpetuation of the inflammasome pathway.
An inflammatory response is seen in perinatal brain injury after HI as well as infection/inflammation, and if chronic inflammation is established can have deleterious effects (Hagberg et al., 2015; Bennet et al., 2018). Hemichannels release adenosine triphosphate (ATP) (Kang et al., 2008; Chekeni et al., 2010; Orellana et al., 2011; Bennett et al., 2012), which may act as a damage associated molecular pattern (DAMP), but is also a key inflammasome activator signal. The release of ATP through connexin hemichannels can perpetuate inflammation through purinergic signaling (Pedata et al., 2016). Purinergic signaling has been associated with the activation of microglia (Li et al., 2011; Kaiser et al., 2016) and the inflammasome complex—a multi-protein complex involved in initiating an innate immune system inflammatory response (Feng et al., 2015). This review will dissect the evidence for these pathways downstream of connexin hemichannel and pannexin channel opening and how they contribute to inflammation. One other membrane pore forming protein, Gasdermin D, has been implicated in the inflammasome pathway (Groslambert and Py, 2018; Kerur et al., 2018), but that channel is very small in comparison with the pannexin and connexin channels and appears to be primarily associated with apoptosis. As will become evident in this review, both in vitro and animal studies using connexin hemichannel or pannexin channel blockers, would suggest that other channels are likely to play minor roles in relation to ATP release, and inflammasome activation in particular.
Evolution of Injury
Perinatal brain injury after HI is an evolving process that can be characterized into four phases (Bennet et al., 2010; Davidson et al., 2015b). The primary phase of injury occurs during the HI insult itself, when the failure of oxidative metabolism results in anoxic depolarization, edema and necrosis (Wassink et al., 2018). After restoration of blood flow and oxygen supply, there is a period of apparent transient recovery when oxidative metabolism is at least partially restored, known as the latent phase (Davidson et al., 2018a). However, the latent phase is the key time when the deleterious mechanisms leading to the spread of brain injury may be initiated; for example, the opening of connexin hemichannels, which will be discussed further below (Davidson et al., 2012).
Following the latent phase, there is a delayed (“secondary”) deterioration of oxidative metabolism starting ~6–15 h after the insult (Azzopardi et al., 1989; Williams et al., 1991; Gunn et al., 1997). This phase is characterized by delayed cerebral energy failure followed by seizures and secondary cell swelling (Bennet et al., 2006; Davidson et al., 2015c). There is marked neuronal injury after HI at term, with the majority of neuronal loss occurring during the secondary phase, through a continuum of necrosis-apoptosis and autophagy (Northington et al., 2007, 2011).
The tertiary phase is a period of repair and reorganization, persisting for weeks to years after the initial insult (Fleiss and Gressens, 2012). During this period, surviving cells in the brain can rewire, but there may be a low level of ongoing cell death due to the loss of trophic support and problems with connectivity (Ness et al., 2001; Romanko et al., 2004). Long-term impairment in perinatal brain injury may also be associated with epigenetic changes (Fleiss and Gressens, 2012), but also persistent inflammation (Bennet et al., 2018) as reported for other types of brain injury and degenerative diseases (Patterson and Holahan, 2012; Freeman and Ting, 2016).
Inflammation in Perinatal Brain Injury
Perinatal brain injury associated with HI or infection/inflammation can trigger an inflammatory response. The innate immune response is the body's first line of defense against pathogens, reacting rapidly following exposure to invading organisms (Medzhitov, 2007). As part of the innate immune response, pattern recognition receptors (PRRs) expressed on immune cells recognize both the conserved molecular structures found on the pathogen known as pathogen-associated molecular patterns (PAMPs), and the endogenous signals released by damaged tissues known as danger-associated molecular patterns (DAMPs) (Medzhitov, 2007; Takeuchi and Akira, 2010).
The activation of PRRs initiates the inflammatory response leading to release of inflammatory cytokines, such as interleukin (IL)-1β (Turner et al., 2014). This early phase of inflammation targets the invading pathogens and/or clears injured tissue, which is beneficial to the host. However, this inflammatory response also leads to the death of uninjured neural cells in a process known as “bystander cell loss” (Hagberg et al., 2015). It is believed that the initial pro-inflammatory response is followed by anti-inflammatory and reparative processes, and either eventual resolution of inflammation or chronic inflammation (Gilroy and De Maeyer, 2015; Hagberg et al., 2015).
Elevated levels of inflammatory markers after birth are associated with adverse neurodevelopmental outcomes. A prospective cohort study of 73 term infants exposed to perinatal asphyxia showed that those who died or were diagnosed with cerebral palsy at a 1 year follow up, were associated with higher levels of IL-1, IL-6 and tumor necrosis factor (TNF)-α in heel-stick blood samples collected on the first or second day of birth (Foster-Barber et al., 2001). In the same cohort, followed up at 30 months of age, those with higher serum cytokine levels of IL-1β, IL-6, and IL-8 at birth were associated with abnormal cognitive and motor outcomes (Bartha et al., 2004). The extremely low gestational age newborns (born before 28 weeks of gestation) (ELGANs) study is a large multi-center observational study. In this cohort, elevated blood concentrations of inflammatory proteins, measured in the first few weeks of life was associated with mental and motor impairments at 2 years old (O'Shea et al., 2012) and adverse cognitive outcome at 10 years of age (Kuban et al., 2017), as well as increased risk of behavioral problems, including autism (Korzeniewski et al., 2018). Cytokine levels were not measured at later time points to determine if inflammation was sustained. However, a study by Lin and colleagues has suggested that perinatal brain injury is associated with long-lasting alterations to the inflammatory response (Lin et al., 2010). Peripheral blood mononuclear cells from a small cohort of preterm born children with periventricular leukomalacia induced cerebral palsy, were shown to have higher mRNA levels of inflammatory molecules, both before and after lipopolysaccharide (LPS) stimulation (Lin et al., 2010). Non-resolving inflammation could be the result of a prolonged or excessive inflammatory response, which may lead to the disruption of pathways that normally induce inflammatory resolution (Nathan and Ding, 2010; Bennet et al., 2018).
Connexin Hemichannels
Connexin gap junctions connect the intracellular space of two adjacent cells allowing for the exchange of ions and molecules (Kumar and Gilula, 1996; Alexander and Goldberg, 2003; Davidson et al., 2013a). A connexin hemichannel is comprised of six connexin subunits. Connexin hemichannels from adjacent cells dock together to form a gap junction (Unger et al., 1999; Davidson et al., 2013a; Leybaert et al., 2017) (Figure 1). Humans have 21 connexin genes, and 11 of these are expressed in the brain (Theis et al., 2005). Connexin 43 (Cx43) has been of particular interest in the brain as it is abundantly expressed, especially in astrocytes, microglia and microvascular endothelium (Dermietzel et al., 2000; Nagy et al., 2003; Davidson et al., 2013a). Under physiological conditions, prior to forming a gap junction, hemichannels at the cell surface have a low probability of opening, shown by low membrane permeability during resting conditions in cultured cells (Decrock et al., 2009). It is not known whether this is the case in vivo, as cells may be exposed to a wide range of stimuli influencing hemichannel opening (Sáez et al., 2005). When hemichannels do open though, they form a large non-selective membrane channel capable of passing molecules up to about 1 kDa in size (Evans et al., 2006). There is increasing evidence that dysregulated hemichannel opening can be detrimental and they are often referred to as “pathological pores” (Paul et al., 1991; Decrock et al., 2015; Willebrords et al., 2016).
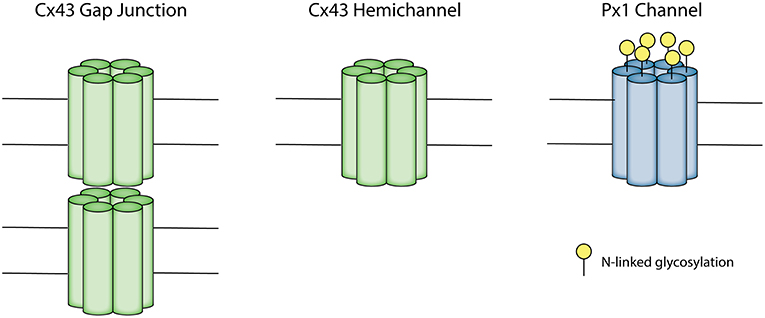
Figure 1. Schematic depicting the Connexin 43 (Cx43) gap junction and hemichannel and pannexin 1 (Px1) channel. A connexon consists of six connexin subunits. A Cx43 gap junction is made up of two opposing connexons, each contributed from two adjacent cells. A Cx43 hemichannel is an unopposed connexon. A Px1 channel is formed by six pannexin subunits. Px1 does not form gap junctions due to N-linked glycosylation on the extracellular loop.
Connexin hemichannels are involved in the spread of brain injury after HI or infection in the adult and immature brain (Davidson et al., 2012, 2013a) (studies are summarized in Table 1). They open in response to stimuli mimicking exposure to ischemia and inflammation in vitro. For example, in cultured cells, oxygen-glucose deprivation (OGD), metabolic inhibition, low extracellular Ca2+, and strong depolarization have all been proven to stimulate hemichannel opening (Contreras et al., 2002; Decrock et al., 2009; Orellana et al., 2010). Conditioned medium from LPS-activated microglia or TNF-α and IL-1β increased hemichannel activity in cultured astrocytes (Retamal et al., 2007). In addition, neuroinflammation induced by S. aureus intracerebral inoculation increased Cx43 hemichannel activity in astrocytes surrounding the abscess (Karpuk et al., 2011). The opening of connexin hemichannels allows for the influx of Na+, Cl−, and Ca2+ and the efflux of K+ ions. This increases cell permeability, leading to the depolarization of the cell, and cell lysis (Paul et al., 1991; Gómez-Hernández et al., 2003). Molecules, such as ATP, glutamate and aspartate can be released; all of these molecules are associated with injury (Ye et al., 2003; Gomes et al., 2005; Zhao et al., 2005; Kang et al., 2008). Additionally, Cx43 hemichannels are likely involved in the propagation of seizures following severe HI in near-term fetal sheep (Davidson et al., 2012).
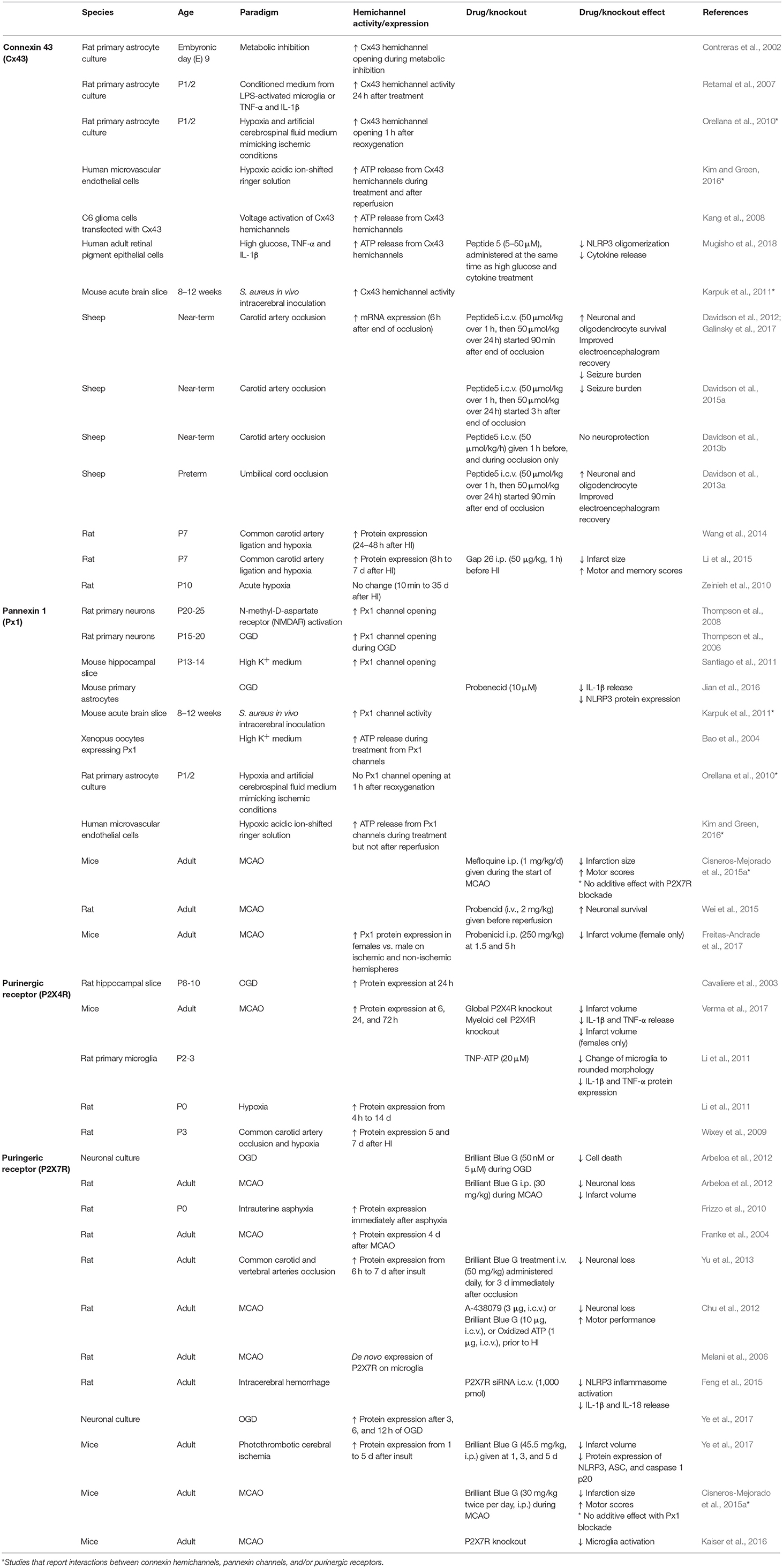
Table 1. Summary of studies of Cx43 hemichannels and Px1 channels and purinergic receptors P2X4 and P2X7.
An upregulation of Cx43 has been shown to occur in adult and perinatal animals after HI and in adult human post-mortem studies of cerebral ischemia (Nakase et al., 2006, 2009; Davidson et al., 2012; Wang et al., 2014; Li et al., 2015). The timing of this Cx43 increase varies between studies, with Cx43 mRNA levels significantly increased at 6 h after carotid artery occlusion in near-term fetal sheep (Davidson et al., 2012). Protein levels progressively increased in the ischemic region from 8 h to 7 days after common carotid artery ligation and hypoxia (commonly referred to as Rice-Vannucci model of HI) in post-natal day (P)7 rats (equivalent to 33–34 weeks of human brain maturation) (Li et al., 2015). In the subventricular zone, however, Wang and colleagues reported Cx43 protein expression was only significantly increased from 24 h after the same insult in P7 rats (Wang et al., 2014). However, there was no change in hippocampal Cx43 protein expression at all time-points between 10 min and 35 days after acute hypoxia in P10 rats (equivalent to human term brain maturation) (Zeinieh et al., 2010). Thus, the changes in Cx43 expression may be dependent on species, brain region, type and/or severity of insult. It should be noted that, the Rice-Vannucci model of HI in rodents produces a unilateral infarction in the brain but only moderate systemic hypoxia (Rice et al., 1981), whereas bilateral carotid artery occlusion in near-term fetal sheep produces a global cerebral insult resulting in a watershed pattern of brain injury, without systemic hypoxia (Williams et al., 1992). The reader should note that protein assays and immunohistochemical labeling do not allow ready discrimination between gap junctions and hemichannels, or channel function.
The blockade of Cx43 hemichannels, however, has been neuroprotective in a variety of different animal models of perinatal HI (Davidson et al., 2012, 2014; Li et al., 2015). Intracerebroventricular (i.c.v.) infusion (50 μmol/kg over 1 h, then 50 μmol/kg over 24 h) of the Cx43 mimetic peptide—Peptide5, started at 90 min after the end of carotid artery occlusion in near-term fetal sheep, was associated with improved neuronal and oligodendrocyte survival and, electroencephalogram (EEG) recovery and reduced seizure burden (Davidson et al., 2012). Similarly, Peptide5 infusion following the same protocol as above in preterm fetal sheep after umbilical cord occlusion improved EEG recovery and reduced neuronal and oligodendrocyte loss (Davidson et al., 2014). Reduced infarct size and improved motor and memory scores were observed in Gap 26 treated P7 rats [a connexin mimetic peptide which blocks hemichannels and gap junctions (Wang et al., 2013)], given i.p., 50 μg/kg, 1 h before HI (Li et al., 2015).
Cx43 hemichannel opening in vivo has been shown to occur after ischemia (in the latent phase), but not during ischemia. Pre-administration of Peptide5 was not neuroprotective when given 1 h before and during ischemia, but was neuroprotective when given after ischemia (Davidson et al., 2012, 2013b). Supporting this, hemichannels in astrocyte cultures subjected to OGD have been shown to open 1 h after OGD (Orellana et al., 2010). In addition, carbenoxolone (100 μmol/L) inhibition of connexin hemichannels did not attenuate ATP release during OGD in hippocampal slices (Frenguelli et al., 2007). Both Cx43 hemichannel and pannexin channel opening was reported to occur during hypoxia in endothelial cells with two thirds of ATP release connexin hemichannel mediated, but upon reperfusion, only Cx43 hemichannel opening occurred (Kim and Green, 2016). These studies indicate that Cx43 hemichannels likely do open after ischemia and may have an ongoing role in the evolution of injury after the insult. This is supported by the demonstration that prolonged infusion of Peptide5 (i.c.v., starting at 90 min) at a dose of 50 μmol/kg over 1 h, then 50 μmol/kg over 24 h of Peptide5 provided greater neuroprotection compared to a 1 h infusion only (50 μmol/kg) (Davidson et al., 2012). Furthermore, delayed administration of another known hemichannel blocker, Gap 26 (i.p., 50 μmol/kg) at 24 h after HI still had neuroprotective effects (Li et al., 2015). Interestingly, when Peptide5 administration (i.c.v., 50 μmol/kg for 1 h followed by 50 μmol/kg for 24 h) was delayed until 3 h after the end of ischemia in near-term fetal sheep, the neuroprotective effects were reduced when compared to earlier administration (Davidson et al., 2012, 2015a). The delayed administration reduced the seizure burden, but there was no clear effect on cell survival or EEG recovery (Davidson et al., 2012, 2015a).
The mechanisms behind how Cx43 hemichannels lead to neuronal and oligodendrocyte loss in HI is not well-understood. During and/or after HI there is impaired intracellular Ca2+ handling. This has been shown to contribute to mitochondrial dysfunction and necrotic and apoptotic cell death (Puka-Sundvall et al., 2000; Mallard et al., 2014). Potentially, the opening of hemichannels could lead to an influx of excessive intracellular Ca2+ accumulation leading directly to the neuronal and/or oligodendrocyte death (Galinsky et al., 2018a). In support, GABAergic striatal neurons expressing intracellular calcium binding proteins are shown to be highly susceptible to HI injury in near-term fetal sheep (Galinsky et al., 2017).
ATP has been widely associated with both inflammasome signal 1 priming which results in transcriptional upregulation of pro-IL-1β and pro-IL-18 and molecules in the inflammasome pathway itself, and inflammasome signal 2 activation that results in assembly of the NLRP3-ASC-pro-caspase1 inflammasome complex within the cell cytoplasm, and activation of caspase 1. Cx43 hemichannel mediated ATP release in particular has been associated with NOD-like receptor protein-3 (NLRP3) inflammasome complex assembly as shown in retinal pigment epithelial cells (Mugisho et al., 2018), and with inflammasome activation in vivo in a model of chronic pain (Tonkin et al., 2018). Neuron and oligodendrocyte damage after perinatal brain injury may also be due to this innate immune system inflammatory response, and is discussed further in section Activation of the Inflammasome below.
Pannexin Channels
Pannexin channels may have similar functions to connexin hemichannels, but their role in perinatal brain injury is not well-understood. Pannexins share homology with the gap junction proteins in invertebrates called innexins (Panchin, 2005), and have a similar topological structure to connexins, despite not sharing sequence homology (Panchin, 2005) (Figure 1). Unlike connexins, pannexins have N-glycosylation on the extracellular loop, which appears to prevent the formation of cell-cell junctions (Boassa et al., 2007) although there is some evidence for pannexin junction formation in C2C12 cells in culture (Ishikawa et al., 2011). There are three subtypes of pannexin, of which pannexin 1 (Px1) and pannexin 2 (Px2) are found to be expressed in the adult and developing brain (Bruzzone et al., 2003; Vogt et al., 2005). Px1 is predominantly expressed in the plasma membrane, whereas Px2 is mainly expressed in intracellular membranes (Boassa et al., 2007).
Opening of pannexin channels can be induced by a range of stimuli in vitro—including OGD, high extracellular K+ concentration, hypoglycemia, cell swelling and the stimulation of NMDA receptors (Thompson et al., 2006, 2008; Kawamura et al., 2010; Santiago et al., 2011). Px1 channels are permeable to ions, and molecules, such as ATP and glucose (Bruzzone et al., 2003; Bao et al., 2004; Riquelme et al., 2013) and the opening of Px1 channels contributes to anoxic depolarization, which can lead to cell death (Thompson et al., 2008; Weilinger et al., 2012). In astrocytes on the border of abscess regions caused by S. aureus intracerebral inoculation in mice, there is increased Cx43 hemichannel and Px1 channel activity (Karpuk et al., 2011). Increased Px1 channel activity in apoptotic lymphocytes in vitro has been associated with providing “find me” signals through ATP release to attract macrophages (Chekeni et al., 2010). In addition, Px1 channel opening has been linked to the activation of the NLRP3 inflammasome and IL-1β release in astrocytes subjected to OGD (Jian et al., 2016).
In adult rodent models of stroke, blocking Px1 has neuroprotective effects (Cisneros-Mejorado et al., 2015a; Wei et al., 2015; Freitas-Andrade et al., 2017). However, to the best of our knowledge, the effect of Px1 blockade after HI has not been examined in the developing brain. Mefloquine (Px1 blocker) injections given during the start of middle cerebral artery occlusion (MCAO) (i.p., 1 mg/kg/day) in adult mice reduced infarction size and improved motor scores (Cisneros-Mejorado et al., 2015a). It should be noted, however, that mefloquine may also inhibit connexin hemichannel opening (Cruikshank et al., 2004). The effectiveness of Px1 blockade is time dependent, as the administration of probenecid, another non-specific Px1 blocker, was most protective against the death of hippocampal CA1 neurons when given before reperfusion (i.v., 2 mg/kg) in adult rats subjected to MCAO (Wei et al., 2015). There was partial protection when administered at 2 h, but no effect when delayed until 6 h after the insult (Wei et al., 2015). Probenecid administration for 7 days starting at 6 h, however, was more protective than a single dose at 6 h. There was also reduced inflammation, shown by reduced astrocytic and microglial immunoreactivity (Wei et al., 2015). The role of Px1 may also be sex dependent, as Px1 protein levels were higher in female than male mice before and after MCAO (Freitas-Andrade et al., 2017). Both Px1 knockout mice and probenecid treated mice (at 1.5 and 5 h, 250 mg/kg, i.p.), had reduced infarct volumes after MCAO compared to control, but the effect was not seen in males (Freitas-Andrade et al., 2017). It is unclear what mechanisms are involved in producing this sex difference, but it may be related to differences in caspase-dependent cell death pathways and estrogen receptor β signaling in females (Freitas-Andrade et al., 2017).
The interaction of connexin hemichannels and pannexin cell channels has been examined in astrocyte cultures exposed to hypoxia and artificial cerebral spinal fluid medium that mimics ischemic conditions in the brain (Orellana et al., 2010). After reoxygenation, there was increased hemichannel activity, indicated by increased uptake of ethidium bromide, which peaked at 1 h after rexoygenation. This hemichannel activity was mediated by Cx43 hemichannels and not pannexin channels, as dye uptake was reduced by Cx43 hemichannel blockade or Cx43 knockout, but not pannexin blockade (Orellana et al., 2010). This is consistent with the endothelial cell hypoxia-reperfusion study referred to above, where pannexin channel opening was reported during hypoxia, but not reperfusion (Kim and Green, 2016). Furthermore, connexin hemichannels, but not pannexin channels, have been implicated in the inflammasome pathway in muscular dystrophy (Cea et al., 2016), and inflammatory cytokine release normally associated with the inflammasome pathway in rodent models of Parkinson's disease (Maatouk et al., 2018) and Alzheimer's disease (Yi et al., 2016). Finally, pannexin channels are self-regulated, and whilst they may release ATP, they are also closed by the presence of ATP in the extracellular milieu (Qiu and Dahl, 2009). Taken overall, it is possible that pannexin channel opening may play a role in inflammasome pathway initiation, but amplification and perpetuation in chronic disease conditions may be primarily connexin hemichannel mediated. This is likely to apply to perinatal brain injury and inflammation too, but remains to be proven.
Astrocytes and Inflammation
Astrocytes play an important physiological role in maintaining the homeostasis of the microenvironment in the brain. Astrocytes are the most abundant cell type and account for half of the cells in the central nervous system (Markiewicz and Lukomska, 2006). They are electrically non-excitable cells, but have a role in propagating Ca2+ waves through gap junctions between astrocytes-astrocytes and astrocytes-neurons (Nedergaard, 1994), and extracellularly between isolated cells (Hassinger et al., 1996). Astrocytes maintain K+ ionic balance (Wallraff et al., 2006), and have an important role in the reuptake and recycling of glutamate (Rothstein et al., 1994). Additionally, astrocytes provide metabolic support for neurons by producing lactate, which is taken up by neurons to produce ATP (Mächler et al., 2016).
Astrocytes can also contribute to the inflammatory response after perinatal brain injury. Reactive astrogliosis can occur where astrocytes undergo hypertrophy, increase expression of glial fibrillary protein and form a glial scar around a focal injury (Romero et al., 2014). However, astrogliosis responses have varied between different experimental paradigms. GFAP expression was increased but numbers of GFAP positive cells did not change in P7 rats at 24 h after HI (Odorcyk et al., 2017). There were increased GFAP positive cells and area fraction in preterm fetal sheep at 3 days after umbilical cord occlusion (Wassink et al., 2017). In contrast, there were no changes in the number and area fraction of GFAP positive cells in near-term fetal sheep at 7 days after carotid artery occlusion (Davidson et al., 2016). Moreover, there was a reduction in area fraction and the size of astrocytes in neonatal pigs at 3 days after hypoxia (Sullivan et al., 2010).
Astrocytes predominantly express Cx43, with some expression of Cx26, Cx30, Cx40, and Cx45 (Nagy et al., 1997, 2003; Dermietzel et al., 2000). The opening of astrocytic Cx43 hemichannels after HI may compromise the ability of astrocytes to maintain homeostasis and neuronal support. This in turn, can have detrimental effects on neuronal and oligodendrocyte survival, the propagation of seizures and secondary energy failure (Davidson et al., 2013a). Additionally, astrocytes cultures subjected to OGD/reperfusion increased ATP release through Cx43 hemichannels. Further, treating OGD/reperfusion microglial cultures with ATP or medium from astrocytes subjected to OGD/reperfusion induced microglial activation (Yin et al., 2018b). The potential mechanisms of microglial activation through ATP signaling is discussed further below.
Microglia and Inflammation
Microglia are the innate immune cells of the brain; having an important role in immune surveillance under normal conditions (Nimmerjahn et al., 2005). Aside from their immune function, they are important for apoptosis and pruning of excessive neurons and synapses, and the phagocytosis of debris during brain development (Baburamani et al., 2014). After brain injury, however, microglia undergo proliferation and activation during the inflammatory response (Baburamani et al., 2014). Resting microglia commonly have ramified morphology and become amoeboid when activated. However, during development, microglia migrate in the brain in amoeboid form and change into a ramified morphology (Pierre et al., 2017).
Activated microglia have various roles, where the classically activated, or M1-like polarization facilitates the progression of inflammation and the alternatively activated, or M2-like polarization is involved in the resolution of inflammation (Bonestroo et al., 2013; Jaworska et al., 2017). The M1-like microglia express cluster of differentiation (CD)86, CD16, CD32, produce reactive oxygen species and nitric oxide synthase, proteases, a range of interleukins and inflammatory cytokines, such as IL-1β, IL-6, and TNF-α (Czeh et al., 2011; Barakat and Redzic, 2015; Fumagalli et al., 2015). The M2-like polarization express CD206 and arginase 1 and produce anti-inflammatory IL-10 and growth factors, favoring repair (Czeh et al., 2011; Barakat and Redzic, 2015; Fumagalli et al., 2015). However, the view that there is a clear polarization of the microglia phenotypes has been challenged in recent years, and currently it is hypothesized that microglial polarization is a continuum with M1 on one end, and M2 on the other (Cherry et al., 2014; Hellström et al., 2016).
In P7 rats, there was a marked increase in CD45/CD11b positive cells in injured areas at 24 h after transient MCAO. These cells were predominantly microglia and not infiltrated blood monocytes (Denker et al., 2007). Supporting this, microglia/macrophage counts were significantly elevated in the hippocampus from 1 day after common carotid artery ligation and hypoxia in P9 mice (human term brain maturation equivalent) (Ferrazzano et al., 2013). There may be a biphasic response of microglial upregulation, with an initial increase in microglial number in the hippocampus at 2 days, followed by a delayed increased in the striatum and cortex at 9 days, after common carotid artery ligation and hypoxia in P9 mice (Cikla et al., 2016). Similarly, classically activated CD11b/CD86 positive microglia from whole brain homogenates were upregulated at 24 h, followed by a secondary peak at 1 week, which was resolved to control levels by 2 weeks after HI (Rice-Vannucci model) in P10 mice (Winerdal et al., 2012). However, the alternative microglial activation markers were not assessed in that study. Significantly elevated counts of ionized calcium-binding adapter molecule 1 (Iba1) positive microglia in the intragyral white matter of the first and second parasagittal gyrus, and the periventricular white matter were present after 1 week of recovery from global cerebral ischemia in near-term fetal sheep (Davidson et al., 2015c, 2016). Moreover, Iba1 positive microglial number was still elevated in the same white matter regions as above at 21 days after severe HI induced by acute umbilical cord occlusion in preterm fetal sheep (van den Heuij et al., 2019). This longer term recovery highlights the chronic nature of inflammation after perinatal HI insults.
The functions of activated microglia appear to change over time after HI. The activation characterization of microglia and infiltrated macrophages has been assessed in P9 mice after common carotid artery occlusion followed by hypoxia (Hellström et al., 2016). There was an increase in mRNA levels of both pro and anti-inflammatory genes at 24 h after the insult. Furthermore, there was a significant increase in classically activated CD86 positive microglia. In contrast, although absolute numbers of alternatively activated CD206 positive microglia increased, their relative proportion was reduced. Surprisingly, there was a population of microglia which did not express CD86 or CD206, highlighting the complexity of microglia activation polarization (Hellström et al., 2016). The early rise of pro-inflammatory markers is supported by an increase of IL-1β and TNF-α mRNA expression between 1 and 24 h after HI in P7 rats (Rice-Vannucci model) (Bona et al., 1999). In another study, using the same paradigm to induce HI in P7 rats, both pro-inflammatory (IL-1β, TNF-α) and anti-inflammatory (IL-10) mRNA expression increased at 3 h after the insult. At 24 h, TNF-α expression was still elevated, but was lower than at 3 h. In addition, anti-inflammatory cytokine TGF-β expression also increased at 24 h along with an increase of CD206 and Iba1 positive microglia (Bonestroo et al., 2013). However, in P7 rats, the majority of CD86 positive microglia were co-localized with IL-1β and only a small number were co-localized with arginase-1, at 6 days after carotid artery ligation and hypoxia (Jaworska et al., 2017). Although some studies indicate a trend toward resolution of inflammation over time, microglia remain upregulated even 3 weeks after HI in preterm fetal sheep (van den Heuij et al., 2019), and so further experimental studies are needed to determine the function of microglia at these later time points.
The increase in inflammatory cytokine release highlighted in the studies above is consistent with NLRP3 inflammasome activation (Shao et al., 2018). Both astrocytes and microglia are known to play key roles in the inflammasome pathway (for review see Song et al., 2017).
ATP Release From Membrane Channels
Both connexin hemichannels and pannexin channels can contribute to ATP release into the extracellular space (Kang et al., 2008; Chekeni et al., 2010; Orellana et al., 2011; Bennett et al., 2012), although, as discussed above, pannexin channels may also be regulated (closed) by extracellular ATP (Qiu and Dahl, 2009). ATP is a high energy molecule, essential for driving many cellular processes in the body. It also has a crucial neurotransmitter and neuromodulatory role in the brain (Melani et al., 2005; Pedata et al., 2016). ATP can be co-released with other neurotransmitters or function as an extracellular signaling molecule on its own (Burnstock, 2009; Suurväli et al., 2017). However, under pathological conditions, it may have injurious effects acting as a DAMP (Melani et al., 2005; Pedata et al., 2016) but more crucially as an inflammasome signal 2 activator. Extracellular ATP can activate purinergic receptors, which can perpetuate inflammasome activity in a number of brain cell types, including microglia (Bours et al., 2011) (Figure 2).
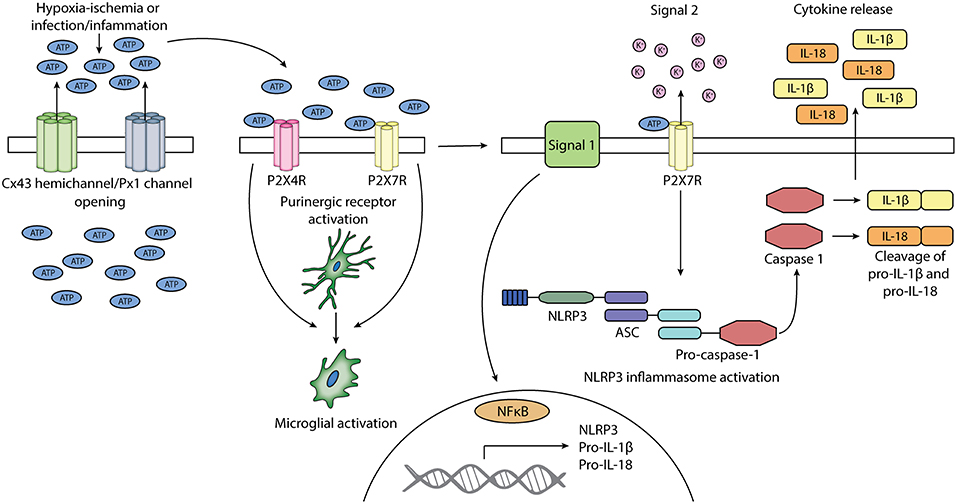
Figure 2. Schematic diagram of the potential involvement of connexin 43 hemichannels and pannexin 1 channels and purinergic receptors P2X4 and P2X7 in the activation of microglia and NLRP3 inflammasome. Cx43 hemichannels and Px1 channels may open after exposure to HI or infection/inflammation. The opening of these channels leads to the release of ATP into the extracellular space. Extracellular ATP can activate purinergic receptors P2X4R and P2X7R. The activation of P2X4R and P2X7R can lead to the activation of microglia. Signal 1 of NLRP3 inflammasome activation is provided by PAMPs or DAMPs, which leads to the upregulation of NLRP3, pro-IL-1β, and pro-IL-18. Signal 2 is mediated by ATP activation of P2X7R and can lead to the assembly of NLRP3, ASC, and pro-caspase 1, forming the NLRP3 inflammasome. Pro-caspase 1 is activated into caspase 1, which cleaves pro-IL-1β and pro-IL-18 into their mature forms. Mature IL-1β and IL-18 is secreted.
The bulk of ATP production occurs in mitochondria through oxidative metabolism, but a small amount is produced through anaerobic glycolysis. The disruption of oxygen and glucose delivery during HI affects oxidative metabolism, attenuating the production of ATP (Bainbridge et al., 2014; Wassink et al., 2014). ATP is critical for the function of Na+/K+-ATPase, which maintain ionic gradients. When ATP is depleted, the failure to maintain cellular ionic homeostasis causes cytotoxic edema, which is indicated by a rise in cortical impedance starting soon after the onset of ischemia in near-term fetal sheep (Williams et al., 1993; Davidson et al., 2018b). We have previously shown that during profound asphyxia in near-term fetal sheep, there was a rapid increase in oxidized cytochrome oxidase at 2–6 min from the start of asphyxia (Drury et al., 2012). The increase in oxidized cytochrome oxidase indicate that there was a decreased availability of reducing equivalents in the mitochondrial electron transport chain, impairing ATP production. Additionally, a phosphorus magnetic resonance spectroscopy study showed that cerebral phosphocreatine and total nucleotide triphosphate decreased, whereas inorganic phosphate increased during HI in neonatal piglets (Bainbridge et al., 2014). During secondary energy failure at 24 and 48 h after HI in neonatal piglets, the cerebral phosphocreatine/inorganic phosphate and nucleotide triphosphate/exchangeable phosphate pool also decreased (Lorek et al., 1994).
Paradoxically, at a time when ATP production is impaired, there is augmented ATP release into the extracellular space after permanent MCAO in the adult rat brain (Melani et al., 2005, 2012). Microdialysis samples collected from the striatum showed that ATP concentrations were an average of 30 nmol/L before ischemia, which increased to an average of 50 nmol/L after permanent MCAO (Melani et al., 2012). When an ecto-ATPase inhibitor—hexapotassium dihydrogen monotitanoundecatungstocobaltate (II) tridecahydrate (PV4, 100 μmol/L) was used, the extracellular ATP concentrations were between 320 and 450 nmol/L (Melani et al., 2012). This is within the range of what was detected in hippocampal slices during reoxygenation after OGD (700 nmol/L), using a biosensor measured in real time (Frenguelli et al., 2007). However, extracellular ATP release has not been examined after HI in the developing brain.
The increase in extracellular ATP can lead to a subsequent increase in adenosine, as ATP is catabolized into adenosine, through the ATP → ADP → AMP → adenosine pathway, mediated by ecto-nucleotidase enzymes (Pedata et al., 2016). Adenosine concentrations increased by 90% during a 1 h period of fetal hypoxia induced by reduction of maternal fraction of inspired oxygen, compared to levels during normoxia in 0.8 gestation fetal sheep (Koos et al., 1997). The rise in adenosine concentrations was attributed to the degradation of AMP, as blocking the degradation of AMP to adenosine with adenosine 5′-(α,β-methylene) diphosphate (AOPCP), an ecto-5′-nucleotidase inhibitor, attenuated the hypoxia-induced rise in adenosine (Koos et al., 1997). In support, in the first hour after permanent MCAO in the adult rat brain, extracellular adenosine was mainly derived from ATP hydrolysis (Melani et al., 2012). This is in contrast to physiological conditions, when adenosine can be directly released from intracellular stores (Melani et al., 2012). Adenosine is very important to the initial metabolic suppression that occurs at the beginning of an HI insult (Hunter et al., 2003). Blocking this response with the adenosine A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) (2.5 mg/mL) in near-term fetal sheep before and during profound asphyxia, was associated with increased neuronal loss compared to the vehicle group (Hunter et al., 2003). Further studies are needed to examine the interaction of ATP and adenosine in vivo, in the developing brain after HI, and to investigate inflammasome complex assembly under those conditions. In parallel, it is necessary to consider the purinergic receptors themselves (see section Purinergic Receptors).
Purinergic Receptors
Purinergic P1 and P2 receptors are activated by adenosine and ATP, respectively. Within the group of P2 receptors, there are P2X receptors, which are ionotropic and P2Y, which are metabotropic (Pedata et al., 2016). In this review, we will focus on the P2X receptors, which form trimeric cationic channels in the cell membrane (Habermacher et al., 2016). Of the seven P2X subtypes (P2X1-7), P2X4 receptor (P2X4R), and P2X7 receptor (P2X7R) have been more widely studied in the brain (Pedata et al., 2016). These channels open in response to ATP binding and are permeable to Na+, K+, and especially Ca2+ (Hattori and Gouaux, 2012). The activation of purinergic receptors can lead to the opening of more connexin hemichannels (and at least potentially pannexin channels), triggering further release of ATP in a process known as “ATP-induced ATP release” in a positive feedback loop (Stout et al., 2002; Baroja-Mazo et al., 2013).
P2X4 Receptor
P2X4R is one of the most sensitive P2X receptors, as it binds ATP in the nanomolar range, whereas P2X7R binds at a micromolar range (Suurväli et al., 2017). It also has the highest permeability to Ca2+, and is the most abundantly expressed P2X subtype in the brain (Egan and Khakh, 2004; Cheng et al., 2014), found on neurons and glial cells, in various brain regions (Bo et al., 2003; Stokes et al., 2017; Suurväli et al., 2017). Under physiological conditions, P2X4R may have an important role in neurotransmission pathways (Suurväli et al., 2017). P2X4 knockout mice show deficits in sensorimotor tasks and social interactions, which was associated with altered subunit expression of glutamate and gamma-aminobutyric acid (GABA) receptors (Wyatt et al., 2013). As P2X4R is expressed abundantly in cells of myeloid origin, such as microglia and monocytes, they may play important roles in pro-inflammatory cytokine release following injury (Cavaliere et al., 2003).
P2X4R has been targeted for neuroprotection after HI. Global P2X4R knockout mice have a reduction in infarct volume and IL-1β and TNF-α release after MCAO compared to wild type mice (Verma et al., 2017). However, when P2X4R was knocked out in myeloid cells only, the neuroprotective effect was only seen in females. The role of P2X4R is complex, as the P2X4R knockout was associated with increased depressive-like behavior compared to wild type mice measured at 30 days after MCAO (Verma et al., 2017). In addition, P2X4R on vascular endothelial cells was involved in neuroprotection via ischemic preconditioning after MCAO in adult mice, as the inhibition of P2X4R abolished this effect (Ozaki et al., 2016).
P2X4 Receptor and Activation of Microglia
Microglia and peripheral monocytes have been shown to express P2X4R, and electrical currents can be induced by ATP activation of P2X4R (Wang et al., 2004; Cheng et al., 2014). The expression of P2X4R is upregulated in organotypic hippocampal culture at 24 h after OGD (Cavaliere et al., 2003) In P0 rats, P2X4R protein expression increased from 4 h after hypoxia until 14 days (Li et al., 2011). However, in P3 rats, there was a significant increase in P2X4R protein expression only at 5 days after common carotid artery occlusion and hypoxia (Wixey et al., 2009). In both neonatal rat studies, P2X4R expression was predominantly colocalized with microglia (Wixey et al., 2009; Li et al., 2011). Although the increase in P2X4R at 5 days was not temporally correlated with the early increase in Iba1 protein expression seen at 2 days after HI, it correlated with the delayed increase in Iba1 protein expression from 6 days after HI. It was postulated that these P2X4R positive microglia may be a distinct population of microglia involved in neuroinflammation (Wixey et al., 2009). Furthermore, P2X4R blockade with trinitrophenyl (TNP)-ATP (20 μM) prevented the change of primary microglial cells to a more rounded morphology after hypoxia, and attenuated the increased IL-1β and TNF-α protein expression (Li et al., 2011).
P2X7 Receptor
Although, P2X7R has a lower affinity of 0.1–1 mM for ATP, when compared to P2X4R, the increased extracellular ATP levels after ischemia may be sufficient to activate it (Surprenant and North, 2009; Melani et al., 2012). The deleterious roles of the P2X7R include increasing intracellular Ca2+ concentrations, glutamate release and inflammasome activation (Di Virgilio, 2007; Matute et al., 2007; Rossi and Volterra, 2009; Ye et al., 2017). Additionally, prolonged ATP activation of P2X7R can transform the cationic channel into a large membrane pore, permeable to molecules 900 Da in size (Yan et al., 2010).
Animal studies have shown that P2X7R is upregulated after ischemia in the immature and adult brain. In a fetal rat model of intrauterine asphyxia, P2X7R expression in the hippocampus was upregulated 3-fold immediately after asphyxia, but returned to control levels at 60 min (Frizzo et al., 2010). In adult rats, however, cortical P2X7R expression was increased 5-fold compared to controls at 4 days after permanent MCAO (Franke et al., 2004). Whereas, in adult mice subjected to photothrombotic cerebral ischemia, cortical P2X7R expression was upregulated from 1 day post-ischemia until 5 days (Ye et al., 2017). In cortical neuronal culture, there was a significant upregulation of P2X7R after exposure to OGD (Ye et al., 2017). P2X7R expression was increased in the CA1 region of the hippocampus from 6 h and maintained until 7 days after global cerebral ischemia in adult rats (Yu et al., 2013). The differences in the timing of P2X7R upregulation could be due age, species, brain region and type of insult.
Blocking P2X7R has been shown to be beneficial for ischemic brain injury in vitro and in adult animals. In neuronal culture, Brilliant Blue G—a P2X7R blocker (50 nM or 5 μM), applied during OGD, reduced cell death measured at 24 h after OGD (Arbeloa et al., 2012). This was replicated in vivo; Brilliant Blue G (30 mg/kg, i.p.) administered at 30 min during a 90 min MCAO and continued daily in adult rats reduced infarct volume and neuronal loss when measured at 3 days after the insult (Arbeloa et al., 2012). In support, Brilliant Blue G treatment (50 mg/kg, i.v.) administered daily, for 3 days, starting immediately after occluding bilateral common carotid and vertebral arteries in adult rats, partially attenuated CA1 neuronal loss in the hippocampus at 7 days after injury (Yu et al., 2013). Although Brilliant Blue G has higher affinity for P2X7R, it can also bind to other P2X receptors at higher concentrations (Chu et al., 2012). However, the administration of a more selective P2X7R antagonist—A-438079 (3 μg, i.c.v.), prior to transient common carotid artery occlusion in adult rats, had similar neuroprotective effects to Brilliant Blue G (10 μg, i.c.v.) and Oxidized ATP (1 μg, i.c.v.), in terms of neuronal survival and motor performance (Chu et al., 2012). Additionally, in an adult rat model of intracerebral hemorrhage, silencing the P2X7R gene reduced NLRP3 inflammasome activation and release of IL-1β and IL-18 at 24 h after the insult (Feng et al., 2015). It is difficult to disentangle the timing of when P2X7R is involved in injury, as most of these studies above started blocking P2X7R either during or immediately after ischemia.
Several studies have shown that connexin hemichannels and pannexin channels are involved in purinergic signaling. For example, IL-1β release induced by P2X7R activation was associated with Px1 channels in macrophages (Pelegrin and Surprenant, 2006). When a Px1 blocker—mefloquine (1 mg/kg, i.p.) was co-administered with Brilliant Blue G (30 mg/kg twice per day, i.p.) to block P2X7R, starting at 30 min during 60 min of MCAO, there was no additive benefit when compared to administering either drug alone in adult mice after MCAO (Cisneros-Mejorado et al., 2015a). However, as stated earlier, mefloquine can also target connexin hemichannels (Cruikshank et al., 2004). A potential mechanism of injury may be ATP release, as both the prevention of ATP release, or inhibition of P2X7R may have beneficial effects (Cisneros-Mejorado et al., 2015b). For example, targeting Cx43 using Peptide5 at 0 h (10 mg/kg), 2 h (5 mg/kg), and 4 h (2.5 mg/kg, i.p.), after spinal cord injury in rats was associated with a reduction is tissue damage and improved functional recovery (Mao et al., 2017). Similarly, the administration of Brilliant Blue G to inhibit P2X7R (10 or 50 mg/kg daily for 3 days) starting 10–15 min after spinal cord injury in rats, reduced tissue damage and improved motor performance (Peng et al., 2009).
P2X7 Receptor and Activation of Microglia
In adult rats after MCAO, the expression of P2X7R was colocalized with microglia at 1 and 4 days, neurons at 4 and 7 days, and astrocytes at 4 days after the insult (Franke et al., 2004). De novo expression of P2X7R was observed on activated microglia at 24 h after MCAO in adult rats, with no expression seen in control animals (Melani et al., 2006). It has been debated as to whether the overexpression of P2X7R drives the activation of microglia, or whether the overexpression of P2X7R is a result of microglial activation (Bai and Li, 2013). In support of the role of P2X7R in microglia activation, the overexpression of P2X7R by transfection of microglia in culture resulted in activation and proliferation of microglia (Monif et al., 2009). Additionally, P2X7R knockout mice showed significantly attenuated microglial activation at 72 h following MCAO compared to wild type mice (Kaiser et al., 2016). In the developing brain, both P2X7R and P2X4R were constitutively expressed in microglia in P3 rats, but P2X4R immunofluorescence was more intense than P2X7R. However, only the changes in P2X4R expression was examined after exposure to hypoxia in P0 rats (Li et al., 2011).
Activation of the Inflammasome
The family of PRRs include Toll-like receptors (TRLs), NOD-like receptors (NLRs), and retinoic acid-inducible gene-I-like receptors (RLRs) which recognize PAMPs or DAMPs (Hagberg et al., 2015; Mallard et al., 2018). There is growing interest in the role of NLRs in inflammatory diseases, but relatively little is known about their role in perinatal brain injury. Inflammasomes are a multimeric complex of proteins which oligomerize after sensing PAMP or DAMP signals (Jo et al., 2016). Typically, NLR inflammsomes are formed by the assembly of the NLR family protein, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and pro-caspase 1. The 22 members of the NLR family are distinguished by their N-terminal effector domains, and grouped into NLRA, NLRB, NLRC and NLRP (Ting et al., 2008). One of the better characterized inflammasomes in the NLR family is the NOD-like receptor protein-3 (NLRP3) inflammasome. The activation of the NLRP3 inflammasome leads to the activation of caspase 1, the cleavage and release of mature IL-1β and IL-18 and subsequent release of other cytokines (Jo et al., 2016; Mugisho et al., 2018).
There are two steps to NLRP3 inflammasome activation. Signal 1 is a priming step, induced by PAMPS, such as microbial toxins and surface proteins, TLR ligands and viral RNA (Martinon et al., 2009; Jun et al., 2012; Chakrabarti et al., 2015) or DAMPs, such as amyloid-β, hyaluronan, monosodium urate, calcium pyrophosphate dehydrate and extracellular ATP (Martinon et al., 2009; Gong et al., 2018). Endogenous inflammatory cytokines including TNF-α and IL-1β can also act as priming signals (Franchi et al., 2009; He et al., 2016; Gong et al., 2018). The priming signal leads to the upregulation of NLRP3 and pro-IL-1β and pro-IL-18 expression through the nuclear factor kappa-light-chain-(NF-κB) translocation to the nucleus to trigger their transcription (He et al., 2016). Signal 2 of the NLRP3 inflammasome is the activation signal. This leads to assembly of the inflammasome complex. A clear model for activation remains unclear; much of the literature suggests that many of the same molecules involved in priming may have a secondary role in activation (Shao et al., 2018). That seems unlikely. Thus, is remains a significant question to define which molecular mechanism(s) trigger activation of the NLRP3 inflammasome (He et al., 2016; Jo et al., 2016). More recently, the list of potential activator signals has been narrowed down. Key players are now suggested to include: ATP, calcium signaling, and reactive oxygen species (He et al., 2016; Jo et al., 2016; Groslambert and Py, 2018). Mitochondrial dysfunction, lysosomal rupture and K+ efflux are also reported to be activators (He et al., 2016; Gao et al., 2017), although it is unclear to what extent some of these may be effects of inflammation rather than initiators per se.
Potassium efflux can be mediated through the opening of endogenous ion channels or through bacterial pore forming toxins (Gao et al., 2017). Purinergic receptors have been associated with the efflux of K+ (Yan et al., 2008) as have connexin hemichannels (Schalper et al., 2010; Leybaert et al., 2017). It is unclear how the reduction in intracellular K+ leads to the activation of the inflammasome although studies have shown that it is a necessary step for the kinase Never In Mitosis A-Related Kinase 7 to bind to NLRP3, required for the assembly of the inflammasome (He et al., 2016; Shi et al., 2016). Additionally, K+ efflux can lead to the disruption of mitochondrial function and production of mitochondrial reactive oxygen species (Tang et al., 2017). It is also of note that potassium efflux is induced by ATP suggesting that ATP release may therefore be the primary or upstream activator not only for potassium efflux, but also mitochondrial dysfunction and reactive oxygen species. Our work has shown that reducing ATP release on its own alone (by blocking connexin hemichannels alone) is sufficient to shut down inflammasome complex assembly, while the addition of exogenous ATP sufficient to reestablish assembly (Mugisho et al., 2018), consistent with the evidence that purinergic receptors play a key role, as outlined above and in Table 1.
Inflammasome Activation in Brain Injury
A large body of evidence in the adult brain shows that inflammation is a least in part mediated by the NLRP3 inflammasome in animal models of stroke, traumatic brain injury and subarachnoid hemorrhage with upregulation of mRNA or protein expression of NLRP3, ASC, caspase-1 and associated inflammatory cytokines, such as TNF-α, IL-18 and IL-6 (Gao et al., 2017; Ye et al., 2017; Ismael et al., 2018; Lee et al., 2018; Wang et al., 2018; Xu et al., 2018; Yin et al., 2018a). Additionally, clinical data show that children with severe traumatic brain injury (open/closed head injury not specified) had elevated levels of NLRP3 in cerebrospinal fluid collected between 0 to ~72 h after the injury (Wallisch et al., 2017). NLRP3 and IL-1β mRNA expression was increased in the hippocampus, striatum and the thalamus at 24 h after common carotid artery ligation followed by hypoxia in P9 neonatal mice (Ystgaard et al., 2015). Supporting this, NLRP3 immunofluorescence increased at 24 h after common carotid artery ligation and hypoxia in P7 rats, which was predominantly colocalized with microglia (Chen et al., 2018). Additionally, there was an increase in IL-1β, caspase 1 and NLRP3 protein expression (Chen et al., 2018).
Targeting the NLRP3 inflammasome has neuroprotective effects in ischemic brain injury either through selective inhibition of NLRP3, or through anti-inflammatory drugs which affect the NLRP3 pathway (Ye et al., 2017; Chen et al., 2018; Ismael et al., 2018). The selective inhibition of NLRP3 using MCC950 (50 mg/kg, i.p. in vivo and 1 μM in vitro) reduced neuronal apoptosis when delivered after photothrombotic cerebral ischemia in adult mice, and before OGD in neuronal cultures (Ye et al., 2017). In support, MCC950 (50 mg/kg, i.p.) administration at 1 and 3 h was associated with a reduction in infarction size and cerebral edema, and improved neurological deficit scores, after MCAO in adult mice. These findings were in turn associated with a decrease in protein levels of NLRP3, caspase 1 and ASC at 24 h after the insult (Ismael et al., 2018). Ginkgolide B—a component of Ginkgo biloba extracts (5–10 mg/kg, i.p.), which has anti-inflammatory effects, was administered to P7 rats 30 min prior to common carotid artery ligation and hypoxia and was associated with the attenuation of the increased expression of NLRP3, caspase 1 and IL-1β at 24 h after injury. These changes were accompanied by reduced infarct size and cerebral edema measured at 72 h (Chen et al., 2018). In contrast, NLRP3 knockout was not associated with neuroprotection in P9 mice, as infarction volume was not significantly different compared to wild type mice at 24 h after common carotid artery ligation and hypoxia (Ystgaard et al., 2015) and in some cases other NLRs or other pathways may be implicated.
Inflammasome Activation in Perinatal Infection/Inflammation
The studies discussed in this review predominantly focused on the role of connexin hemichannels and pannexin channels and purinergic receptors in experimental models of HI. Given that HI and infection/inflammation both lead to neuroinflammation, it is possible that both may be perpetuated through inflammasome pathway activation. In this section, we discuss the evidence for inflammasome activation in infection/inflammation in clinical and experimental studies.
Inflammasome activation, at least in adults, is an important inflammatory response to combat infection. For example, adult mice deficient in NLRP3, ASC, or caspase 1 have reduced survival rates compared to wild type mice after Group B Streptococcus infection (Costa et al., 2012). Group B Streptococcus is a Gram-positive bacterium that is common cause of life-threatening sepsis and meningitis in neonates and pregnant women (Henneke and Berner, 2006). Preterm and very-low-birthweight neonates are susceptible to neonatal sepsis likely due to increased risk of exposure to microbes (for example via mechanical ventilation, intravenous catheters and parenteral feeding). Secondly, they may have deficits in their innate and adaptive immune responses leading to insufficient inflammatory mediators (Wynn and Wong, 2010; Strunk et al., 2011). In view of the latter, a study of 21 preterm infants born 24–32 weeks showed that preterm umbilical cord blood monocytes, had a reduction in IL-1β secretion and an induction of NLRP3 expression, compared to term infants and adult peripheral monocytes after ATP/LPS stimulation (Sharma et al., 2015). However, IL-1β secretion from preterm peripheral blood monocytes (24–29 weeks of gestation) collected at an average of 15 post-natal days was comparable to adult monocytes, suggesting that the impairment in IL-1β secretion is restored shortly after birth (Sharma et al., 2015). A study of 72 neonates showed that IL-1β secretion from peripheral blood mononuclear cells did not differ between extremely preterm (born <28 weeks) and very preterm infants (28–32 weeks) on the fifth day of life. However, the peripheral blood mononuclear cells (collected within 24 h of late onset sepsis) from the extremely preterm group secreted higher levels of IL-1β, than the very preterm neonates (Zasada et al., 2018). It is unclear whether this gestational age dependent response is related to the higher counts of circulating mononuclear cells found in the extremely preterm group.
Conversely, the excessive activation of the inflammasome and release of pro-inflammatory cytokines in neonatal sepsis, can lead to multi-organ failure and death (Gentile et al., 2015). Caspase-1/11 knockout mice (P5-7) exposed to cecal slurry to induce polymicrobial intra-abdominal neonatal sepsis had greater survival rates compared to wild type mice (Gentile et al., 2015). Additionally, there was reduced IL-1β and IL-18 release at 2, 6, 18, and 24 h after the induction of sepsis. Surprisingly, genetic ablation of ASC or NLRP3 was not associated with a protective effect (Gentile et al., 2015). The knockout of ASC or NLRP3 may be insufficient for the complete elimination of caspase 1 activity as it is not solely activated by the NLRP3 inflammasome (Gentile et al., 2015). Further adverse effects are evidenced from infants who have rare genetic mutations leading to over activation of the NLRP3 inflammasome which manifests as neonatal-onset multi-system inflammatory disease (Aróstegui et al., 2010).
Recent evidence has demonstrated that increased inflammasome activity is associated with chorioamnionitis (Gomez-Lopez et al., 2017). A study of 70 pregnant women who had undergone preterm labor showed that the chorioamniotic membranes from those with acute histological chorioamnionitis compared to those without, had greater mRNA levels of inflammasome components, including NLRP3, and other NLR proteins, caspase 1, IL-1β, IL-18, and increased ASC and caspase 1 complex formation (Gomez-Lopez et al., 2017). Additionally, a study of amniotic fluid from 143 women showed that those who had undergone spontaneous preterm labor, with intra-amniotic infection/inflammation (positive culture for microorganisms in the amniotic fluid, or a white blood cell count of >100 cells/mm3) had significantly higher caspase 1 levels compared to those without intra-amniotic infection/inflammation, delivering at term or preterm (Gotsch et al., 2008). Furthermore, umbilical cord blood monocytes from preterm infants with histological chorioamnionitis showed reduced caspase 1 activity compared to those without histological chorioamnionitis (Sharma et al., 2015).
Although NLRP3 mediated inflammation may be implicated in many diseases, further study of its involvement in perinatal brain injury after HI and infection/inflammation is necessary. Disentangling the temporal effects of inflammation is particularly important, as discussed earlier the role of inflammation can be both beneficial and deleterious.
Conclusion
Connexin and pannexin membrane channels can contribute to the evolution of perinatal brain injury. Connexin hemichannel, and to a lesser extent pannexin channel, blockade, has been associated with neuroprotection in a number of models of brain injury. A potential mechanism of injury perpetuated by connexin hemichannels could be release of what may be a key inflammasome signal 2 activator, ATP. Extracellular ATP activates purinergic receptors, which in turn have been shown to be involved with the activation of microglia and the inflammasome complex. Blockade of purinergic receptors P2X4 and P2X7 has been protective in adult animal models of stroke but further studies are required to investigate the involvement of purinergic receptors in the propagation of perinatal brain injury, and in particular the timing of their contribution to spreading injury. However, although P2X4R and P2X7R have been implicated in microglial activation following HI insults, and could be targets for modulating microglial responses after perinatal brain injury, connexin hemichannel perpetuation of the inflammasome would be upstream of both. Increasing evidence suggests that modulating the cascade upstream of inflammasome activation may attenuate brain injury mediated through inflammation. Inflammasome activation should be investigated further in perinatal brain injury, as it may be an important mediator of deleterious inflammatory responses.
Author Contributions
JD and KZ conceptualized this topical review. KZ, JD, AG, LB, and CG undertook manuscript writing and preparation of figures. All authors reviewed and edited this manuscript.
Funding
The authors' studies discussed in this review was funded by The Health Research Council of New Zealand, grant numbers: 16/003, 12/613, 14/216, 17/601 and the Marsden Fund, grant number 17-UOA232. JD holds a Sir Charles Hercus Fellowship from the Health Research Council of New Zealand (16/003). CG acknowledges support from Wendy and Bruce Hadden. KZ was supported by a University of Auckland Doctoral Scholarship.
Conflict of Interest Statement
CG has intellectual property related to connexin hemichannel and pannexin channel modulation for the treatment of inflammatory disease and regulation of the inflammasome pathway and is a co-founder of OcuNexus Therapeutics, which has a focus on the treatment of chronic disease indications.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alexander, D. B., and Goldberg, G. S. (2003). Transfer of biologically important molecules between cells through gap junction channels. Curr. Med. Chem. 10, 2045–2058. doi: 10.2174/0929867033456927
Arbeloa, J., Pérez-Samartín, A., Gottlieb, M., and Matute, C. (2012). P2X7 receptor blockade prevents ATP excitotoxicity in neurons and reduces brain damage after ischemia. Neurobiol. Dis. 45, 954–961. doi: 10.1016/j.nbd.2011.12.014
Aróstegui, J. I., Lopez Saldana, M. D., Pascal, M., Clemente, D., Aymerich, M., Balaguer, F., et al. (2010). A somatic NLRP3 mutation as a cause of a sporadic case of chronic infantile neurologic, cutaneous, articular syndrome/neonatal-onset multisystem inflammatory disease: novel evidence of the role of low-level mosaicism as the pathophysiologic mechanism underlying mendelian inherited diseases. Arthritis Rheum. 62, 1158–1166. doi: 10.1002/art.27342
Azzopardi, D., Wyatt, J. S., Cady, E. B., Delpy, D. T., Baudin, J., Stewart, A. L., et al. (1989). Prognosis of newborn infants with hypoxic-ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 25, 445–451. doi: 10.1203/00006450-198905000-00004
Baburamani, A. A., Supramaniam, V. G., Hagberg, H., and Mallard, C. (2014). Microglia toxicity in preterm brain injury. Reprod. Toxicol. 48, 106–112. doi: 10.1016/j.reprotox.2014.04.002
Bai, H. Y., and Li, A. P. (2013). P2X(7) receptors in cerebral ischemia. Neurosci. Bull. 29, 390–398. doi: 10.1007/s12264-013-1338-7
Bainbridge, A., Tachtsidis, I., Faulkner, S. D., Price, D., Zhu, T., Baer, E., et al. (2014). Brain mitochondrial oxidative metabolism during and after cerebral hypoxia-ischemia studied by simultaneous phosphorus magnetic-resonance and broadband near-infrared spectroscopy. Neuroimage 102, 173–183. doi: 10.1016/j.neuroimage.2013.08.016
Bao, L., Locovei, S., and Dahl, G. (2004). Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 572, 65–68. doi: 10.1016/j.febslet.2004.07.009
Barakat, R., and Redzic, Z. (2015). Differential cytokine expression by brain microglia/macrophages in primary culture after oxygen glucose deprivation and their protective effects on astrocytes during anoxia. Fluids Barriers CNS 12:6. doi: 10.1186/s12987-015-0002-1
Baroja-Mazo, A., Barberà-Cremades, M., and Pelegrin, P. (2013). The participation of plasma membrane hemichannels to purinergic signaling. Biochim. Biophys. Acta 1828, 79–93. doi: 10.1016/j.bbamem.2012.01.002
Bartha, A. I., Foster-Barber, A., Miller, S. P., Vigneron, D. B., Glidden, D. V., Barkovich, A. J., et al. (2004). Neonatal encephalopathy: association of cytokines with MR spectroscopy and outcome. Pediatr. Res. 56, 960–966. doi: 10.1203/01.PDR.0000144819.45689.BB
Bennet, L., Booth, L., and Gunn, A. J. (2010). Potential biomarkers for hypoxic-ischemic encephalopathy. Semin. Fetal Neonatal Med. 15, 253–260. doi: 10.1016/j.siny.2010.05.007
Bennet, L., Dhillon, S., Lear, C. A., Van Den Heuij, L., King, V., Dean, J. M., et al. (2018). Chronic inflammation and impaired development of the preterm brain. J. Reprod. Immunol. 125, 45–55. doi: 10.1016/j.jri.2017.11.003
Bennet, L., Roelfsema, V., Pathipati, P., Quaedackers, J., and Gunn, A. J. (2006). Relationship between evolving epileptiform activity and delayed loss of mitochondrial activity after asphyxia measured by near-infrared spectroscopy in preterm fetal sheep. J. Physiol. 572, 141–154. doi: 10.1113/jphysiol.2006.105197
Bennett, M. V., Garré, J. M., Orellana, J. A., Bukauskas, F. F., Nedergaard, M., and Saez, J. C. (2012). Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Res. 1487, 3–15. doi: 10.1016/j.brainres.2012.08.042
Bo, X., Kim, M., Nori, S. L., Schoepfer, R., Burnstock, G., and North, R. A. (2003). Tissue distribution of P2X4 receptors studied with an ectodomain antibody. Cell Tissue Res. 313, 159–165. doi: 10.1007/s00441-003-0758-5
Boassa, D., Ambrosi, C., Qiu, F., Dahl, G., Gaietta, G., and Sosinsky, G. (2007). Pannexin1 channels contain a glycosylation site that targets the hexamer to the plasma membrane. J. Biol. Chem. 282, 31733–31743. doi: 10.1074/jbc.M702422200
Bona, E., Andersson, A. L., Blomgren, K., Gilland, E., Puka-Sundvall, M., Gustafson, K., et al. (1999). Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr. Res. 45, 500–509. doi: 10.1203/00006450-199904010-00008
Bonestroo, H. J., Nijboer, C. H., van Velthoven, C. T., Kavelaars, A., Hack, C. E., Van Bel, F., et al. (2013). Cerebral and hepatic inflammatory response after neonatal hypoxia-ischemia in newborn rats. Dev. Neurosci. 35, 197–211. doi: 10.1159/000346685
Bours, M. J., Dagnelie, P. C., Giuliani, A. L., Wesselius, A., and Di Virgilio, F. (2011). P2 receptors and extracellular ATP: a novel homeostatic pathway in inflammation. Front. Biosci. (Schol. Ed.) 3, 1443–1456. doi: 10.2741/s235
Bruzzone, R., Hormuzdi, S. G., Barbe, M. T., Herb, A., and Monyer, H. (2003). Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. U.S.A. 100, 13644–13649. doi: 10.1073/pnas.2233464100
Cavaliere, F., Florenzano, F., Amadio, S., Fusco, F. R., Viscomi, M. T., D'ambrosi, N., et al. (2003). Up-regulation of P2X2, P2X4 receptor and ischemic cell death: prevention by P2 antagonists. Neuroscience 120, 85–98. doi: 10.1016/S0306-4522(03)00228-8
Cea, L. A., Balboa, E., Puebla, C., Vargas, A. A., Cisterna, B. A., Escamilla, R., et al. (2016). Dexamethasone-induced muscular atrophy is mediated by functional expression of connexin-based hemichannels. Biochim. Biophys. Acta 1862, 1891–1899. doi: 10.1016/j.bbadis.2016.07.003
Chakrabarti, A., Banerjee, S., Franchi, L., Loo, Y. M., Gale, M. Jr., Nunez, G., et al. (2015). RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 17, 466–477. doi: 10.1016/j.chom.2015.02.010
Chekeni, F. B., Elliott, M. R., Sandilos, J. K., Walk, S. F., Kinchen, J. M., Lazarowski, E. R., et al. (2010). Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 467, 863–867. doi: 10.1038/nature09413
Chen, A., Xu, Y., and Yuan, J. (2018). Ginkgolide B ameliorates NLRP3 inflammasome activation after hypoxic-ischemic brain injury in the neonatal male rat. Int. J. Dev. Neurosci. 69, 106–111. doi: 10.1016/j.ijdevneu.2018.07.004
Cheng, R. D., Ren, J. J., Zhang, Y. Y., and Ye, X. M. (2014). P2X4 receptors expressed on microglial cells in post-ischemic inflammation of brain ischemic injury. Neurochem. Int. 67, 9–13. doi: 10.1016/j.neuint.2014.01.011
Cherry, J. D., Olschowka, J. A., and O'banion, M. K. (2014). Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation 11:98. doi: 10.1186/1742-2094-11-98
Chu, K., Yin, B., Wang, J., Peng, G., Liang, H., Xu, Z., et al. (2012). Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J. Neuroinflammation 9:69. doi: 10.1186/1742-2094-9-69
Cikla, U., Chanana, V., Kintner, D. B., Covert, L., Dewall, T., Waldman, A., et al. (2016). Suppression of microglia activation after hypoxia-ischemia results in age-dependent improvements in neurologic injury. J. Neuroimmunol. 291, 18–27. doi: 10.1016/j.jneuroim.2015.12.004
Cisneros-Mejorado, A., Gottlieb, M., Cavaliere, F., Magnus, T., Koch-Nolte, F., Scemes, E., et al. (2015a). Blockade of P2X7 receptors or pannexin-1 channels similarly attenuates postischemic damage. J. Cereb. Blood Flow Metab. 35, 843–850. doi: 10.1038/jcbfm.2014.262
Cisneros-Mejorado, A., Pérez-Samartín, A., Gottlieb, M., and Matute, C. (2015b). ATP signaling in brain: release, excitotoxicity and potential therapeutic targets. Cell. Mol. Neurobiol. 35, 1–6. doi: 10.1007/s10571-014-0092-3
Contreras, J. E., Sánchez, H. A., Eugenin, E. A., Speidel, D., Theis, M., Willecke, K., et al. (2002). Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. U.S.A. 99, 495–500. doi: 10.1073/pnas.012589799
Costa, A., Gupta, R., Signorino, G., Malara, A., Cardile, F., Biondo, C., et al. (2012). Activation of the NLRP3 inflammasome by group B streptococci. J. Immunol. 188, 1953–1960. doi: 10.4049/jimmunol.1102543
Cruikshank, S. J., Hopperstad, M., Younger, M., Connors, B. W., Spray, D. C., and Srinivas, M. (2004). Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proc. Natl. Acad. Sci. U.S.A. 101, 12364–12369. doi: 10.1073/pnas.0402044101
Czeh, M., Gressens, P., and Kaindl, A. M. (2011). The yin and yang of microglia. Dev. Neurosci. 33, 199–209. doi: 10.1159/000328989
Davidson, J. O., Dean, J. M., Fraser, M., Wassink, G., Andelius, T. C., Dhillon, S. K., et al. (2018a). Perinatal brain injury: mechanisms and therapeutic approaches. Front. Biosci. (Landmark Ed.) 23, 2204–2226. doi: 10.2741/4700
Davidson, J. O., Draghi, V., Whitham, S., Dhillon, S. K., Wassink, G., Bennet, L., et al. (2018b). How long is sufficient for optimal neuroprotection with cerebral cooling after ischemia in fetal sheep? J. Cereb. Blood Flow Metab. 38, 1047–1059. doi: 10.1177/0271678X17707671
Davidson, J. O., Drury, P. P., Green, C. R., Nicholson, L. F., Bennet, L., and Gunn, A. J. (2014). Connexin hemichannel blockade is neuroprotective after asphyxia in preterm fetal sheep. PLoS ONE 9:e96558. doi: 10.1371/journal.pone.0096558
Davidson, J. O., Green, C. R., Bennet, L., Nicholson, L. F., Danesh-Meyer, H., Carroll, S. J., et al. (2013a). A key role for connexin hemichannels in spreading ischemic brain injury. Curr. Drug Targets 14, 36–46. doi: 10.2174/138945013804806479
Davidson, J. O., Green, C. R., Nicholson, L. F., Bennet, L., and Gunn, A. J. (2013b). Connexin hemichannel blockade is neuroprotective after, but not during, global cerebral ischemia in near-term fetal sheep. Exp. Neurol. 248, 301–308. doi: 10.1016/j.expneurol.2013.06.026
Davidson, J. O., Green, C. R., Nicholson, L. F., O'carroll, S. J., Fraser, M., Bennet, L., et al. (2012). Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann. Neurol. 71, 121–132. doi: 10.1002/ana.22654
Davidson, J. O., Rout, A. L., Wassink, G., Yuill, C. A., Zhang, F. G., Green, C. R., et al. (2015a). Non-additive effects of delayed connexin hemichannel blockade and hypothermia after cerebral ischemia in near-term fetal sheep. J. Cereb. Blood Flow Metab. 35, 2052–2061. doi: 10.1038/jcbfm.2015.171
Davidson, J. O., Wassink, G., Van Den Heuij, L. G., Bennet, L., and Gunn, A. J. (2015b). Therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy–where to from here? Front. Neurol. 6:198. doi: 10.3389/fneur.2015.00198
Davidson, J. O., Wassink, G., Yuill, C. A., Zhang, F. G., Bennet, L., and Gunn, A. J. (2015c). How long is too long for cerebral cooling after ischemia in fetal sheep? J. Cereb. Blood Flow Metab. 35, 751–758. doi: 10.1038/jcbfm.2014.259
Davidson, J. O., Yuill, C. A., Zhang, F. G., Wassink, G., Bennet, L., and Gunn, A. J. (2016). Extending the duration of hypothermia does not further improve white matter protection after ischemia in term-equivalent fetal sheep. Sci. Rep. 6:25178. doi: 10.1038/srep25178
Decrock, E., De Bock, M., Wang, N., Bultynck, G., Giaume, C., Naus, C. C., et al. (2015). Connexin and pannexin signaling pathways, an architectural blueprint for CNS physiology and pathology? Cell. Mol. Life Sci. 72, 2823–2851. doi: 10.1007/s00018-015-1962-7
Decrock, E., De Vuyst, E., Vinken, M., Van Moorhem, M., Vranckx, K., Wang, N., et al. (2009). Connexin 43 hemichannels contribute to the propagation of apoptotic cell death in a rat C6 glioma cell model. Cell Death Differ. 16, 151–163. doi: 10.1038/cdd.2008.138
Denker, S. P., Ji, S., Dingman, A., Lee, S. Y., Derugin, N., Wendland, M. F., et al. (2007). Macrophages are comprised of resident brain microglia not infiltrating peripheral monocytes acutely after neonatal stroke. J. Neurochem. 100, 893–904. doi: 10.1111/j.1471-4159.2006.04162.x
Dermietzel, R., Gao, Y., Scemes, E., Vieira, D., Urban, M., Kremer, M., et al. (2000). Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res. Brain Res. Rev. 32, 45–56. doi: 10.1016/S0165-0173(99)00067-3
Di Virgilio, F. (2007). Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol. Sci. 28, 465–472. doi: 10.1016/j.tips.2007.07.002
Drury, P. P., Bennet, L., Booth, L. C., Davidson, J. O., Wassink, G., and Gunn, A. J. (2012). Maturation of the mitochondrial redox response to profound asphyxia in fetal sheep. PLOS ONE 7:e39273. doi: 10.1371/journal.pone.0039273
Edwards, A. D., Brocklehurst, P., Gunn, A. J., Halliday, H., Juszczak, E., Levene, M., et al. (2010). Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ 340:c363. doi: 10.1136/bmj.c363
Egan, T. M., and Khakh, B. S. (2004). Contribution of calcium ions to P2X channel responses. J. Neurosci. 24, 3413–3420. doi: 10.1523/JNEUROSCI.5429-03.2004
Evans, W. H., De Vuyst, E., and Leybaert, L. (2006). The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem. J. 397, 1–14. doi: 10.1042/BJ20060175
Feng, L., Chen, Y., Ding, R., Fu, Z., Yang, S., Deng, X., et al. (2015). P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J. Neuroinflammation 12:190. doi: 10.1186/s12974-015-0409-2
Ferrazzano, P., Chanana, V., Uluc, K., Fidan, E., Akture, E., Kintner, D. B., et al. (2013). Age-dependent microglial activation in immature brains after hypoxia- ischemia. CNS Neurol. Disord. Drug Targets 12, 338–349. doi: 10.2174/1871527311312030007
Fleiss, B., and Gressens, P. (2012). Tertiary mechanisms of brain damage: a new hope for treatment of cerebral palsy? Lancet Neurol. 11, 556–566. doi: 10.1016/S1474-4422(12)70058-3
Foster-Barber, A., Dickens, B., and Ferriero, D. M. (2001). Human perinatal asphyxia: correlation of neonatal cytokines with MRI and outcome. Dev. Neurosci. 23, 213–218. doi: 10.1159/000046146
Franchi, L., Eigenbrod, T., and Núñez, G. (2009). Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 183, 792–796. doi: 10.4049/jimmunol.0900173
Franke, H., Günther, A., Grosche, J., Schmidt, R., Rossner, S., Reinhardt, R., et al. (2004). P2X7 receptor expression after ischemia in the cerebral cortex of rats. J. Neuropathol. Exp. Neurol. 63, 686–699. doi: 10.1093/jnen/63.7.686
Freeman, L. C., and Ting, J. P. (2016). The pathogenic role of the inflammasome in neurodegenerative diseases. J. Neurochem. 136, 29–38. doi: 10.1111/jnc.13217
Freitas-Andrade, M., Bechberger, J. F., Macvicar, B. A., Viau, V., and Naus, C. C. (2017). Pannexin1 knockout and blockade reduces ischemic stroke injury in female, but not in male mice. Oncotarget 8, 36973–36983. doi: 10.18632/oncotarget.16937
Frenguelli, B. G., Wigmore, G., Llaudet, E., and Dale, N. (2007). Temporal and mechanistic dissociation of ATP and adenosine release during ischaemia in the mammalian hippocampus. J. Neurochem. 101, 1400–1413. doi: 10.1111/j.1471-4159.2006.04425.x
Frizzo, J. K., Cardoso, M. P., De Assis, A. M., Perry, M. L., Volonté, C., and Frizzo, M. E. (2010). Effects of acute perinatal asphyxia in the rat hippocampus. Cell. Mol. Neurobiol. 30, 683–692. doi: 10.1007/s10571-009-9492-1
Fumagalli, S., Perego, C., Pischiutta, F., Zanier, E. R., and De Simoni, M. G. (2015). The ischemic environment drives microglia and macrophage function. Front. Neurol. 6:81. doi: 10.3389/fneur.2015.00081
Galinsky, R., Davidson, J. O., Dean, J. M., Green, C. R., Bennet, L., and Gunn, A. J. (2018a). Glia and hemichannels: key mediators of perinatal encephalopathy. Neural Regen. Res. 13, 181–189. doi: 10.4103/1673-5374.226378
Galinsky, R., Davidson, J. O., Lear, C. A., Bennet, L., Green, C. R., and Gunn, A. J. (2017). Connexin hemichannel blockade improves survival of striatal GABA-ergic neurons after global cerebral ischaemia in term-equivalent fetal sheep. Sci. Rep. 7:6304. doi: 10.1038/s41598-017-06683-1
Galinsky, R., Lear, C. A., Dean, J. M., Wassink, G., Dhillon, S. K., Fraser, M., et al. (2018b). Complex interactions between hypoxia-ischemia and inflammation in preterm brain injury. Dev. Med. Child Neurol. 60, 126–133. doi: 10.1111/dmcn.13629
Gao, L., Dong, Q., Song, Z., Shen, F., Shi, J., and Li, Y. (2017). NLRP3 inflammasome: a promising target in ischemic stroke. Inflamm. Res. 66, 17–24. doi: 10.1007/s00011-016-0981-7
Gentile, L. F., Cuenca, A. L., Cuenca, A. G., Nacionales, D. C., Ungaro, R., Efron, P. A., et al. (2015). Improved emergency myelopoiesis and survival in neonatal sepsis by caspase-1/11 ablation. Immunology 145, 300–311. doi: 10.1111/imm.12450
Gilroy, D., and De Maeyer, R. (2015). New insights into the resolution of inflammation. Semin. Immunol. 27, 161–168. doi: 10.1016/j.smim.2015.05.003
Gomes, P., Srinivas, S. P., Van Driessche, W., Vereecke, J., and Himpens, B. (2005). ATP release through connexin hemichannels in corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 46, 1208–1218. doi: 10.1167/iovs.04-1181
Gómez-Hernández, J. M., De Miguel, M., Larrosa, B., Gonzalez, D., and Barrio, L. C. (2003). Molecular basis of calcium regulation in connexin-32 hemichannels. Proc. Natl. Acad. Sci. U.S.A. 100, 16030–16035. doi: 10.1073/pnas.2530348100
Gomez-Lopez, N., Romero, R., Xu, Y., Garcia-Flores, V., Leng, Y., Panaitescu, B., et al. (2017). Inflammasome assembly in the chorioamniotic membranes during spontaneous labor at term. Am. J. Reprod. Immunol. 77:e12648. doi: 10.1111/aji.12648
Gong, T., Yang, Y., Jin, T., Jiang, W., and Zhou, R. (2018). Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol. 39, 393–406. doi: 10.1016/j.it.2018.01.009
Gotsch, F., Romero, R., Chaiworapongsa, T., Erez, O., Vaisbuch, E., Espinoza, J., et al. (2008). Evidence of the involvement of caspase-1 under physiologic and pathologic cellular stress during human pregnancy: a link between the inflammasome and parturition. J. Matern. Fetal Neonatal Med. 21, 605–616. doi: 10.1080/14767050802212109
Groslambert, M., and Py, B. F. (2018). Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 11, 359–374. doi: 10.2147/JIR.S141220
Gunn, A. J., Gunn, T. R., De Haan, H. H., Williams, C. E., and Gluckman, P. D. (1997). Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J. Clin. Invest. 99, 248–256. doi: 10.1172/JCI119153
Gunn, A. J., Laptook, A. R., Robertson, N. J., Barks, J. D., Thoresen, M., Wassink, G., et al. (2017). Therapeutic hypothermia translates from ancient history in to practice. Pediatr. Res. 81, 202–209. doi: 10.1038/pr.2016.198
Habermacher, C., Dunning, K., Chataigneau, T., and Grutter, T. (2016). Molecular structure and function of P2X receptors. Neuropharmacology 104, 18–30. doi: 10.1016/j.neuropharm.2015.07.032
Hagberg, H., Mallard, C., Ferriero, D. M., Vannucci, S. J., Levison, S. W., Vexler, Z. S., et al. (2015). The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 11, 192–208. doi: 10.1038/nrneurol.2015.13
Hassinger, T. D., Guthrie, P. B., Atkinson, P. B., Bennett, M. V., and Kater, S. B. (1996). An extracellular signaling component in propagation of astrocytic calcium waves. Proc. Natl. Acad. Sci. U.S.A. 93, 13268–13273. doi: 10.1073/pnas.93.23.13268
Hattori, M., and Gouaux, E. (2012). Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 485, 207–212. doi: 10.1038/nature11010
He, Y., Hara, H., and Núñez, G. (2016). Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41, 1012–1021. doi: 10.1016/j.tibs.2016.09.002
Hellström, E. N., Smith, P. L., Fleiss, B., Nair, S., Svedin, P., Wang, W., et al. (2016). Temporal characterization of microglia/macrophage phenotypes in a mouse model of neonatal hypoxic-ischemic brain injury. Front. Cell. Neurosci. 10:286. doi: 10.3389/fncel.2016.00286
Henneke, P., and Berner, R. (2006). Interaction of neonatal phagocytes with group B streptococcus: recognition and response. Infect. Immun. 74, 3085–3095. doi: 10.1128/IAI.01551-05
Hunter, C. J., Bennet, L., Power, G. G., Roelfsema, V., Blood, A. B., Quaedackers, J. S., et al. (2003). Key neuroprotective role for endogenous adenosine A1 receptor activation during asphyxia in the fetal sheep. Stroke 34, 2240–2245. doi: 10.1161/01.STR.0000083623.77327.CE
Ishikawa, M., Iwamoto, T., Nakamura, T., Doyle, A., Fukumoto, S., and Yamada, Y. (2011). Pannexin 3 functions as an ER Ca(2+) channel, hemichannel, and gap junction to promote osteoblast differentiation. J. Cell Biol. 193, 1257–1274. doi: 10.1083/jcb.201101050
Ismael, S., Zhao, L., Nasoohi, S., and Ishrat, T. (2018). Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci. Rep. 8:5971. doi: 10.1038/s41598-018-24350-x
Jaworska, J., Ziemka-Nalecz, M., Sypecka, J., and Zalewska, T. (2017). The potential neuroprotective role of a histone deacetylase inhibitor, sodium butyrate, after neonatal hypoxia-ischemia. J. Neuroinflammation 14:34. doi: 10.1186/s12974-017-0807-8
Jian, Z., Ding, S., Deng, H., Wang, J., Yi, W., Wang, L., et al. (2016). Probenecid protects against oxygen-glucose deprivation injury in primary astrocytes by regulating inflammasome activity. Brain Res. 1643, 123–129. doi: 10.1016/j.brainres.2016.05.002
Jo, E. K., Kim, J. K., Shin, D. M., and Sasakawa, C. (2016). Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol. Immunol. 13, 148–159. doi: 10.1038/cmi.2015.95
Jun, H. K., Lee, S. H., Lee, H. R., and Choi, B. K. (2012). Integrin alpha5beta1 activates the NLRP3 inflammasome by direct interaction with a bacterial surface protein. Immunity 36, 755–768. doi: 10.1016/j.immuni.2012.05.002
Kaiser, M., Penk, A., Franke, H., Krugel, U., Norenberg, W., Huster, D., et al. (2016). Lack of functional P2X7 receptor aggravates brain edema development after middle cerebral artery occlusion. Purinergic Signal. 12, 453–463. doi: 10.1007/s11302-016-9511-x
Kang, J., Kang, N., Lovatt, D., Torres, A., Zhao, Z., Lin, J., et al. (2008). Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 28, 4702–4711. doi: 10.1523/JNEUROSCI.5048-07.2008
Karpuk, N., Burkovetskaya, M., Fritz, T., Angle, A., and Kielian, T. (2011). Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J. Neurosci. 31, 414–425. doi: 10.1523/JNEUROSCI.5247-10.2011
Kawamura, M. Jr., Ruskin, D. N., and Masino, S. A. (2010). Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J. Neurosci. 30, 3886–3895. doi: 10.1523/JNEUROSCI.0055-10.2010
Kerur, N., Fukuda, S., Banerjee, D., Kim, Y., Fu, D., Apicella, I., et al. (2018). cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 24, 50–61. doi: 10.1038/nm.4450
Kim, Y., and Green, C. R. (2016). “Assessing connexin hemichannel function during ischemic injury and reperfusion,” in Gap Junction Channels and Hemichannels, eds D. Bai and J. C. Saez (Boca Raton, FL: CRC Press; Taylor & Francis Group), 169–188.
Koos, B. J., Kruger, L., and Murray, T. F. (1997). Source of extracellular brain adenosine during hypoxia in fetal sheep. Brain Res. 778, 439–442. doi: 10.1016/S0006-8993(97)01207-9
Korzeniewski, S. J., Allred, E. N., O'shea, T. M., Leviton, A., and Kuban, K. C. K. (2018). Elevated protein concentrations in newborn blood and the risks of autism spectrum disorder, and of social impairment, at age 10 years among infants born before the 28th week of gestation. Transl. Psychiatry 8:115. doi: 10.1038/s41398-018-0156-0
Kuban, K. C., Joseph, R. M., O'shea, T. M., Heeren, T., Fichorova, R. N., Douglass, L., et al. (2017). Circulating inflammatory-associated proteins in the first month of life and cognitive impairment at age 10 years in children born extremely preterm. J. Pediatr. 180, 116–123 e111. doi: 10.1016/j.jpeds.2016.09.054
Kumar, N. M., and Gilula, N. B. (1996). The gap junction communication channel. Cell 84, 381–388. doi: 10.1016/S0092-8674(00)81282-9
Larroque, B., Ancel, P. Y., Marret, S., Marchand, L., Andre, M., Arnaud, C., et al. (2008). Neurodevelopmental disabilities and special care of 5-year-old children born before 33 weeks of gestation (the EPIPAGE study): a longitudinal cohort study. Lancet 371, 813–820. doi: 10.1016/S0140-6736(08)60380-3
Lee, A. C., Kozuki, N., Blencowe, H., Vos, T., Bahalim, A., Darmstadt, G. L., et al. (2013). Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr. Res. 74, 50–72. doi: 10.1038/pr.2013.206
Lee, S. W., Gajavelli, S., Spurlock, M. S., Andreoni, C., De Rivero Vaccari, J. P., Bullock, M. R., et al. (2018). Microglial inflammasome activation in penetrating ballistic-like brain injury. J. Neurotrauma 35, 1681–1693. doi: 10.1089/neu.2017.5530
Leybaert, L., Lampe, P. D., Dhein, S., Kwak, B. R., Ferdinandy, P., Beyer, E. C., et al. (2017). Connexins in cardiovascular and neurovascular health and disease: pharmacological implications. Pharmacol. Rev. 69, 396–478. doi: 10.1124/pr.115.012062
Li, F., Wang, L., Li, J. W., Gong, M., He, L., Feng, R., et al. (2011). Hypoxia induced amoeboid microglial cell activation in postnatal rat brain is mediated by ATP receptor P2X4. BMC Neurosci. 12:111. doi: 10.1186/1471-2202-12-111
Li, X., Zhao, H., Tan, X., Kostrzewa, R. M., Du, G., Chen, Y., et al. (2015). Inhibition of connexin43 improves functional recovery after ischemic brain injury in neonatal rats. Glia 63, 1553–1567. doi: 10.1002/glia.22826
Lin, C. Y., Chang, Y. C., Wang, S. T., Lee, T. Y., Lin, C. F., and Huang, C. C. (2010). Altered inflammatory responses in preterm children with cerebral palsy. Ann. Neurol. 68, 204–212. doi: 10.1002/ana.22049
Lorek, A., Takei, Y., Cady, E. B., Wyatt, J. S., Penrice, J., Edwards, A. D., et al. (1994). Delayed (“secondary”) cerebral energy failure after acute hypoxia-ischemia in the newborn piglet: continuous 48-hour studies by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 36, 699–706. doi: 10.1203/00006450-199412000-00003
Maatouk, L., Yi, C., Carrillo-De Sauvage, M. A., Compagnion, A. C., Hunot, S., Ezan, P., et al. (2018). Glucocorticoid receptor in astrocytes regulates midbrain dopamine neurodegeneration through connexin hemichannel activity. Cell Death Differ. 26, 580–596. doi: 10.1038/s41418-018-0150-3
Mächler, P., Wyss, M. T., Elsayed, M., Stobart, J., Gutierrez, R., Von Faber-Castell, A., et al. (2016). In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 23, 94–102. doi: 10.1016/j.cmet.2015.10.010
Mallard, C., Davidson, J. O., Tan, S., Green, C. R., Bennet, L., Robertson, N. J., et al. (2014). Astrocytes and microglia in acute cerebral injury underlying cerebral palsy associated with preterm birth. Pediatr. Res. 75, 234–240. doi: 10.1038/pr.2013.188
Mallard, C., Tremblay, M. E., and Vexler, Z. S. (2018). Microglia and neonatal brain injury. Neuroscience. doi: 10.1016/j.neuroscience.2018.01.023. [Epub ahead of print].
Mao, Y., Nguyen, T., Tonkin, R. S., Lees, J. G., Warren, C., O'carroll, S. J., et al. (2017). Characterisation of Peptide5 systemic administration for treating traumatic spinal cord injured rats. Exp. Brain Res. 235, 3033–3048. doi: 10.1007/s00221-017-5023-3
Markiewicz, I., and Lukomska, B. (2006). The role of astrocytes in the physiology and pathology of the central nervous system. Acta Neurobiol. Exp. (Warsz). 66, 343–358.
Marlow, N., Wolke, D., Bracewell, M. A., and Samara, M. (2005). Neurologic and developmental disability at six years of age after extremely preterm birth. N. Engl. J. Med. 352, 9–19. doi: 10.1056/NEJMoa041367
Martinon, F., Mayor, A., and Tschopp, J. (2009). The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265. doi: 10.1146/annurev.immunol.021908.132715
Matute, C., Torre, I., Perez-Cerda, F., Pérez-Samartín, A., Alberdi, E., Etxebarria, E., et al. (2007). P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 27, 9525–9533. doi: 10.1523/JNEUROSCI.0579-07.2007
Medzhitov, R. (2007). Recognition of microorganisms and activation of the immune response. Nature 449, 819–826. doi: 10.1038/nature06246
Melani, A., Amadio, S., Gianfriddo, M., Vannucchi, M. G., Volontè, C., Bernardi, G., et al. (2006). P2X7 receptor modulation on microglial cells and reduction of brain infarct caused by middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab. 26, 974–982. doi: 10.1038/sj.jcbfm.9600250
Melani, A., Corti, F., Stephan, H., Müller, C. E., Donati, C., Bruni, P., et al. (2012). Ecto-ATPase inhibition: ATP and adenosine release under physiological and ischemic in vivo conditions in the rat striatum. Exp. Neurol. 233, 193–204. doi: 10.1016/j.expneurol.2011.09.036
Melani, A., Turchi, D., Vannucchi, M. G., Cipriani, S., Gianfriddo, M., and Pedata, F. (2005). ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem. Int. 47, 442–448. doi: 10.1016/j.neuint.2005.05.014
Monif, M., Reid, C. A., Powell, K. L., Smart, M. L., and Williams, D. A. (2009). The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J. Neurosci. 29, 3781–3791. doi: 10.1523/JNEUROSCI.5512-08.2009
Mugisho, O. O., Green, C. R., Kho, D. T., Zhang, J., Graham, E. S., Acosta, M. L., et al. (2018). The inflammasome pathway is amplified and perpetuated in an autocrine manner through connexin43 hemichannel mediated ATP release. Biochim. Biophys. Acta 1862, 385–393. doi: 10.1016/j.bbagen.2017.11.015
Nagy, J. I., Ionescu, A. V., Lynn, B. D., and Rash, J. E. (2003). Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: implications from normal and connexin32 knockout mice. Glia 44, 205–218. doi: 10.1002/glia.10278
Nagy, J. I., Ochalski, P. A., Li, J., and Hertzberg, E. L. (1997). Evidence for the co-localization of another connexin with connexin-43 at astrocytic gap junctions in rat brain. Neuroscience 78, 533–548. doi: 10.1016/S0306-4522(96)00584-2
Nakase, T., Maeda, T., Yoshida, Y., and Nagata, K. (2009). Ischemia alters the expression of connexins in the aged human brain. J. Biomed. Biotechnol. 2009:147946. doi: 10.1155/2009/147946
Nakase, T., Yoshida, Y., and Nagata, K. (2006). Enhanced connexin 43 immunoreactivity in penumbral areas in the human brain following ischemia. Glia 54, 369–375. doi: 10.1002/glia.20399
Natarajan, G., Laptook, A., and Shankaran, S. (2018). Therapeutic hypothermia: how can we optimize this therapy to further improve outcomes? Clin. Perinatol. 45, 241–255. doi: 10.1016/j.clp.2018.01.010
Nathan, C., and Ding, A. (2010). Nonresolving inflammation. Cell 140, 871–882. doi: 10.1016/j.cell.2010.02.029
Nedergaard, M. (1994). Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science 263, 1768–1771. doi: 10.1126/science.8134839
Ness, J. K., Romanko, M. J., Rothstein, R. P., Wood, T. L., and Levison, S. W. (2001). Perinatal hypoxia-ischemia induces apoptotic and excitotoxic death of periventricular white matter oligodendrocyte progenitors. Dev. Neurosci. 23, 203–208. doi: 10.1159/000046144
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Northington, F. J., Chavez-Valdez, R., and Martin, L. J. (2011). Neuronal cell death in neonatal hypoxia-ischemia. Ann. Neurol. 69, 743–758. doi: 10.1002/ana.22419
Northington, F. J., Zelaya, M. E., O'riordan, D. P., Blomgren, K., Flock, D. L., Hagberg, H., et al. (2007). Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience 149, 822–833. doi: 10.1016/j.neuroscience.2007.06.060
Odorcyk, F. K., Nicola, F., Duran-Carabali, L. E., Figueiro, F., Kolling, J., Vizuete, A., et al. (2017). Galantamine administration reduces reactive astrogliosis and upregulates the anti-oxidant enzyme catalase in rats submitted to neonatal hypoxia ischemia. Int. J. Dev. Neurosci. 62, 15–24. doi: 10.1016/j.ijdevneu.2017.07.006
Orellana, J. A., Froger, N., Ezan, P., Jiang, J. X., Bennett, M. V., Naus, C. C., et al. (2011). ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem. 118, 826–840. doi: 10.1111/j.1471-4159.2011.07210.x
Orellana, J. A., Hernandez, D. E., Ezan, P., Velarde, V., Bennett, M. V., Giaume, C., et al. (2010). Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia 58, 329–343. doi: 10.1002/glia.20926
O'Shea, T. M., Allred, E. N., Kuban, K. C., Dammann, O., Paneth, N., Fichorova, R., et al. (2012). Elevated concentrations of inflammation-related proteins in postnatal blood predict severe developmental delay at 2 years of age in extremely preterm infants. J. Pediatr. 160, 395–401 e394. doi: 10.1016/j.jpeds.2011.08.069
Ozaki, T., Muramatsu, R., Sasai, M., Yamamoto, M., Kubota, Y., Fujinaka, T., et al. (2016). The P2X4 receptor is required for neuroprotection via ischemic preconditioning. Sci. Rep. 6:25893. doi: 10.1038/srep25893
Panchin, Y. V. (2005). Evolution of gap junction proteins–the pannexin alternative. J. Exp. Biol. 208, 1415–1419. doi: 10.1242/jeb.01547
Patterson, Z. R., and Holahan, M. R. (2012). Understanding the neuroinflammatory response following concussion to develop treatment strategies. Front. Cell. Neurosci. 6:58. doi: 10.3389/fncel.2012.00058
Paul, D. L., Ebihara, L., Takemoto, L. J., Swenson, K. I., and Goodenough, D. A. (1991). Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J. Cell Biol. 115, 1077–1089. doi: 10.1083/jcb.115.4.1077
Pedata, F., Dettori, I., Coppi, E., Melani, A., Fusco, I., Corradetti, R., et al. (2016). Purinergic signalling in brain ischemia. Neuropharmacology 104, 105–130. doi: 10.1016/j.neuropharm.2015.11.007
Pelegrin, P., and Surprenant, A. (2006). Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 25, 5071–5082. doi: 10.1038/sj.emboj.7601378
Peng, W., Cotrina, M. L., Han, X., Yu, H., Bekar, L., Blum, L., et al. (2009). Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc. Natl. Acad. Sci. U.S.A. 106, 12489–12493. doi: 10.1073/pnas.0902531106
Pierre, W. C., Smith, P. L., Londono, I., Chemtob, S., Mallard, C., and Lodygensky, G. A. (2017). Neonatal microglia: the cornerstone of brain fate. Brain. Behav. Immun. 59, 333–345. doi: 10.1016/j.bbi.2016.08.018
Puka-Sundvall, M., Gajkowska, B., Cholewinski, M., Blomgren, K., Lazarewicz, J. W., and Hagberg, H. (2000). Subcellular distribution of calcium and ultrastructural changes after cerebral hypoxia-ischemia in immature rats. Brain Res. Dev. Brain Res. 125, 31–41. doi: 10.1016/S0165-3806(00)00110-3
Qiu, F., and Dahl, G. (2009). A permeant regulating its permeation pore: inhibition of pannexin 1 channels by ATP. Am. J. Physiol. Cell Physiol. 296, C250–C255. doi: 10.1152/ajpcell.00433.2008
Retamal, M. A., Froger, N., Palacios-Prado, N., Ezan, P., Sáez, P. J., Sáez, J. C., et al. (2007). Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 27, 13781–13792. doi: 10.1523/JNEUROSCI.2042-07.2007
Rice, J. E., Vannucci, R. C., and Brierley, J. B. (1981). The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 9, 131–141. doi: 10.1002/ana.410090206
Riquelme, M. A., Cea, L. A., Vega, J. L., Boric, M. P., Monyer, H., Bennett, M. V., et al. (2013). The ATP required for potentiation of skeletal muscle contraction is released via pannexin hemichannels. Neuropharmacology 75, 594–603. doi: 10.1016/j.neuropharm.2013.03.022
Romanko, M. J., Rothstein, R. P., and Levison, S. W. (2004). Neural stem cells in the subventricular zone are resilient to hypoxia/ischemia whereas progenitors are vulnerable. J. Cereb. Blood Flow Metab. 24, 814–825. doi: 10.1097/01.WCB.0000123906.17746.00
Romero, J., Muñiz, J., Logica Tornatore, T., Holubiec, M., Gonzalez, J., Barreto, G. E., et al. (2014). Dual role of astrocytes in perinatal asphyxia injury and neuroprotection. Neurosci. Lett. 565, 42–46. doi: 10.1016/j.neulet.2013.10.046
Rossi, D., and Volterra, A. (2009). Astrocytic dysfunction: insights on the role in neurodegeneration. Brain Res. Bull. 80, 224–232. doi: 10.1016/j.brainresbull.2009.07.012
Rothstein, J. D., Martin, L., Levey, A. I., Dykes-Hoberg, M., Jin, L., Wu, D., et al. (1994). Localization of neuronal and glial glutamate transporters. Neuron 13, 713–725. doi: 10.1016/0896-6273(94)90038-8
Sáez, J. C., Retamal, M. A., Basilio, D., Bukauskas, F. F., and Bennett, M. V. (2005). Connexin-based gap junction hemichannels: gating mechanisms. Biochim. Biophys. Acta 1711, 215–224. doi: 10.1016/j.bbamem.2005.01.014
Santiago, M. F., Veliskova, J., Patel, N. K., Lutz, S. E., Caille, D., Charollais, A., et al. (2011). Targeting pannexin1 improves seizure outcome. PLoS ONE 6:e25178. doi: 10.1371/journal.pone.0025178
Schalper, K. A., Sanchez, H. A., Lee, S. C., Altenberg, G. A., Nathanson, M. H., and Saez, J. C. (2010). Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol. 299, C1504–C1515. doi: 10.1152/ajpcell.00015.2010
Shankaran, S., Laptook, A. R., Ehrenkranz, R. A., Tyson, J. E., Mcdonald, S. A., Donovan, E. F., et al. (2005). Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N. Engl. J. Med. 353, 1574–1584. doi: 10.1056/NEJMcps050929
Shao, B. Z., Cao, Q., and Liu, C. (2018). Targeting NLRP3 inflammasome in the treatment of CNS diseases. Front. Mol. Neurosci. 11:320. doi: 10.3389/fnmol.2018.00320
Sharma, A. A., Jen, R., Kan, B., Sharma, A., Marchant, E., Tang, A., et al. (2015). Impaired NLRP3 inflammasome activity during fetal development regulates IL-1beta production in human monocytes. Eur. J. Immunol. 45, 238–249. doi: 10.1002/eji.201444707
Shi, H., Wang, Y., Li, X., Zhan, X., Tang, M., Fina, M., et al. (2016). NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 17, 250–258. doi: 10.1038/ni.3333
Song, L., Pei, L., Yao, S., Wu, Y., and Shang, Y. (2017). NLRP3 inflammasome in neurological diseases, from functions to therapies. Front. Cell. Neurosci. 11:63. doi: 10.3389/fncel.2017.00063
Stokes, L., Layhadi, J. A., Bibic, L., Dhuna, K., and Fountain, S. J. (2017). P2X4 receptor function in the nervous system and current breakthroughs in pharmacology. Front. Pharmacol 8:291. doi: 10.3389/fphar.2017.00291
Stout, C. E., Costantin, J. L., Naus, C. C., and Charles, A. C. (2002). Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 277, 10482–10488. doi: 10.1074/jbc.M109902200
Strunk, T., Currie, A., Richmond, P., Simmer, K., and Burgner, D. (2011). Innate immunity in human newborn infants: prematurity means more than immaturity. J. Matern. Fetal Neonatal Med. 24, 25–31. doi: 10.3109/14767058.2010.482605
Sullivan, S. M., Björkman, S. T., Miller, S. M., Colditz, P. B., and Pow, D. V. (2010). Morphological changes in white matter astrocytes in response to hypoxia/ischemia in the neonatal pig. Brain Res. 1319, 164–174. doi: 10.1016/j.brainres.2010.01.010
Surprenant, A., and North, R. A. (2009). Signaling at purinergic P2X receptors. Annu. Rev. Physiol. 71, 333–359. doi: 10.1146/annurev.physiol.70.113006.100630
Suurväli, J., Boudinot, P., Kanellopoulos, J., and Ruutel Boudinot, S. (2017). P2X4: A fast and sensitive purinergic receptor. Biomed. J. 40, 245–256. doi: 10.1016/j.bj.2017.06.010
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. doi: 10.1016/j.cell.2010.01.022
Tang, T., Lang, X., Xu, C., Wang, X., Gong, T., Yang, Y., et al. (2017). CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 8:202. doi: 10.1038/s41467-017-00227-x
Theis, M., Sohl, G., Eiberger, J., and Willecke, K. (2005). Emerging complexities in identity and function of glial connexins. Trends Neurosci. 28, 188–195. doi: 10.1016/j.tins.2005.02.006
Thompson, R. J., Jackson, M. F., Olah, M. E., Rungta, R. L., Hines, D. J., Beazely, M. A., et al. (2008). Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science 322, 1555–1559. doi: 10.1126/science.1165209
Thompson, R. J., Zhou, N., and Macvicar, B. A. (2006). Ischemia opens neuronal gap junction hemichannels. Science 312, 924–927. doi: 10.1126/science.1126241
Thornton, J. S., Ordidge, R. J., Penrice, J., Cady, E. B., Amess, P. N., Punwani, S., et al. (1998). Temporal and anatomical variations of brain water apparent diffusion coefficient in perinatal cerebral hypoxic-ischemic injury: relationships to cerebral energy metabolism. Magn. Reson. Med. 39, 920–927. doi: 10.1002/mrm.1910390609
Ting, J. P., Lovering, R. C., Alnemri, E. S., Bertin, J., Boss, J. M., Davis, B. K., et al. (2008). The NLR gene family: a standard nomenclature. Immunity 28, 285–287. doi: 10.1016/j.immuni.2008.02.005
Tonkin, R. S., Bowles, C., Perera, C. J., Keating, B. A., Makker, P. G. S., Duffy, S. S., et al. (2018). Attenuation of mechanical pain hypersensitivity by treatment with Peptide5, a connexin-43 mimetic peptide, involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp. Neurol. 300, 1–12. doi: 10.1016/j.expneurol.2017.10.016
Turner, M. D., Nedjai, B., Hurst, T., and Pennington, D. J. (2014). Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 1843, 2563–2582. doi: 10.1016/j.bbamcr.2014.05.014
Unger, V. M., Kumar, N. M., Gilula, N. B., and Yeager, M. (1999). Three-dimensional structure of a recombinant gap junction membrane channel. Science 283, 1176–1180. doi: 10.1126/science.283.5405.1176
van den Heuij, L. G., Fraser, M., Miller, S. L., Jenkin, G., Wallace, E. M., Davidson, J. O., et al. (2019). Delayed intranasal infusion of human amnion epithelial cells improves white matter maturation after asphyxia in preterm fetal sheep. J. Cereb. Blood Flow Metab. 39, 223–239. doi: 10.1177/0271678X17729954
Vannucci, R. C. (2000). Hypoxic-ischemic encephalopathy. Am. J. Perinatol. 17, 113–120. doi: 10.1055/s-2000-9293
Verma, R., Cronin, C. G., Hudobenko, J., Venna, V. R., Mccullough, L. D., and Liang, B. T. (2017). Deletion of the P2X4 receptor is neuroprotective acutely, but induces a depressive phenotype during recovery from ischemic stroke. Brain. Behav. Immun. 66, 302–312. doi: 10.1016/j.bbi.2017.07.155
Vogt, A., Hormuzdi, S. G., and Monyer, H. (2005). Pannexin1 and Pannexin2 expression in the developing and mature rat brain. Brain Res. Mol. Brain Res. 141, 113–120. doi: 10.1016/j.molbrainres.2005.08.002
Wallisch, J. S., Simon, D. W., Bayir, H., Bell, M. J., Kochanek, P. M., and Clark, R. S. B. (2017). Cerebrospinal fluid NLRP3 is increased after severe traumatic brain injury in infants and children. Neurocrit. Care 27, 44–50. doi: 10.1007/s12028-017-0378-7
Wallraff, A., Kohling, R., Heinemann, U., Theis, M., Willecke, K., and Steinhauser, C. (2006). The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 26, 5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006
Wang, D., Xu, X., Wu, Y. G., Lyu, L., Zhou, Z. W., and Zhang, J. N. (2018). Dexmedetomidine attenuates traumatic brain injury: action pathway and mechanisms. Neural Regen. Res. 13, 819–826. doi: 10.4103/1673-5374.232529
Wang, J., Ma, A., Xi, J., Wang, Y., and Zhao, B. (2014). Connexin 43 and its hemichannels mediate hypoxia-ischemia-induced cell death in neonatal rats. Child Neurol. Open 1:2329048X14544955. doi: 10.1177/2329048X14544955
Wang, L., Jacobsen, S. E., Bengtsson, A., and Erlinge, D. (2004). P2 receptor mRNA expression profiles in human lymphocytes, monocytes and CD34+ stem and progenitor cells. BMC Immunol. 5:16. doi: 10.1186/1471-2172-5-16
Wang, N., De Bock, M., Decrock, E., Bol, M., Gadicherla, A., Bultynck, G., et al. (2013). Connexin targeting peptides as inhibitors of voltage- and intracellular Ca2+-triggered Cx43 hemichannel opening. Neuropharmacology 75, 506–516. doi: 10.1016/j.neuropharm.2013.08.021
Wassink, G., Davidson, J. O., Dhillon, S. K., Fraser, M., Galinsky, R., Bennet, L., et al. (2017). Partial white and grey matter protection with prolonged infusion of recombinant human erythropoietin after asphyxia in preterm fetal sheep. J. Cereb. Blood Flow Metab. 37, 1080–1094. doi: 10.1177/0271678X16650455
Wassink, G., Davidson, J. O., Lear, C. A., Juul, S. E., Northington, F., Bennet, L., et al. (2018). A working model for hypothermic neuroprotection. J. Physiol. 596, 5641–5654 doi: 10.1113/JP274928
Wassink, G., Gunn, E. R., Drury, P. P., Bennet, L., and Gunn, A. J. (2014). The mechanisms and treatment of asphyxial encephalopathy. Front. Neurosci. 8:40. doi: 10.3389/fnins.2014.00040
Wei, R., Wang, J., Xu, Y., Yin, B., He, F., Du, Y., et al. (2015). Probenecid protects against cerebral ischemia/reperfusion injury by inhibiting lysosomal and inflammatory damage in rats. Neuroscience 301, 168–177. doi: 10.1016/j.neuroscience.2015.05.070
Weilinger, N. L., Tang, P. L., and Thompson, R. J. (2012). Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J. Neurosci. 32, 12579–12588. doi: 10.1523/JNEUROSCI.1267-12.2012
Willebrords, J., Crespo Yanguas, S., Maes, M., Decrock, E., Wang, N., Leybaert, L., et al. (2016). Connexins and their channels in inflammation. Crit. Rev. Biochem. Mol. Biol. 51, 413–439. doi: 10.1080/10409238.2016.1204980
Williams, C. E., Gunn, A., and Gluckman, P. D. (1991). Time course of intracellular edema and epileptiform activity following prenatal cerebral ischemia in sheep. Stroke 22, 516–521. doi: 10.1161/01.STR.22.4.516
Williams, C. E., Gunn, A. J., Mallard, C., and Gluckman, P. D. (1992). Outcome after ischemia in the developing sheep brain: an electroencephalographic and histological study. Ann. Neurol. 31, 14–21. doi: 10.1002/ana.410310104
Williams, C. E., Mallard, C., Tan, W., and Gluckman, P. D. (1993). Pathophysiology of perinatal asphyxia. Clin. Perinatol. 20, 305–325. doi: 10.1016/S0095-5108(18)30395-6
Winerdal, M., Winerdal, M. E., Kinn, J., Urmaliya, V., Winqvist, O., and Aden, U. (2012). Long lasting local and systemic inflammation after cerebral hypoxic ischemia in newborn mice. PLoS ONE 7:e36422. doi: 10.1371/journal.pone.0036422
Wixey, J. A., Reinebrant, H. E., Carty, M. L., and Buller, K. M. (2009). Delayed P2X4R expression after hypoxia-ischemia is associated with microglia in the immature rat brain. J. Neuroimmunol. 212, 35–43. doi: 10.1016/j.jneuroim.2009.04.016
Wyatt, L. R., Godar, S. C., Khoja, S., Jakowec, M. W., Alkana, R. L., Bortolato, M., et al. (2013). Sociocommunicative and sensorimotor impairments in male P2X4-deficient mice. Neuropsychopharmacology 38, 1993–2002. doi: 10.1038/npp.2013.98
Wynn, J. L., and Wong, H. R. (2010). Pathophysiology and treatment of septic shock in neonates. Clin. Perinatol. 37, 439–479. doi: 10.1016/j.clp.2010.04.002
Xu, X., Yin, D., Ren, H., Gao, W., Li, F., Sun, D., et al. (2018). Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol. Dis. 117, 15–27. doi: 10.1016/j.nbd.2018.05.016
Yan, Z., Khadra, A., Li, S., Tomic, M., Sherman, A., and Stojilkovic, S. S. (2010). Experimental characterization and mathematical modeling of P2X7 receptor channel gating. J. Neurosci. 30, 14213–14224. doi: 10.1523/JNEUROSCI.2390-10.2010
Yan, Z., Li, S., Liang, Z., Tomic, M., and Stojilkovic, S. S. (2008). The P2X7 receptor channel pore dilates under physiological ion conditions. J. Gen. Physiol. 132, 563–573. doi: 10.1085/jgp.200810059
Ye, X., Shen, T., Hu, J., Zhang, L., Zhang, Y., Bao, L., et al. (2017). Purinergic 2X7 receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp. Neurol. 292, 46–55. doi: 10.1016/j.expneurol.2017.03.002
Ye, Z. C., Wyeth, M. S., Baltan-Tekkok, S., and Ransom, B. R. (2003). Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J. Neurosci. 23, 3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003
Yi, C., Mei, X., Ezan, P., Mato, S., Matias, I., Giaume, C., et al. (2016). Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer's disease. Cell Death Differ. 23, 1691–1701. doi: 10.1038/cdd.2016.63
Yin, D., Zhou, S., Xu, X., Gao, W., Li, F., Ma, Y., et al. (2018a). Dexmedetomidine attenuated early brain injury in rats with subarachnoid haemorrhage by suppressing the inflammatory response: the TLR4/NF-kappaB pathway and the NLRP3 inflammasome may be involved in the mechanism. Brain Res. 1698, 1–10. doi: 10.1016/j.brainres.2018.05.040
Yin, X., Feng, L., Ma, D., Yin, P., Wang, X., Hou, S., et al. (2018b). Roles of astrocytic connexin-43, hemichannels, and gap junctions in oxygen-glucose deprivation/reperfusion injury induced neuroinflammation and the possible regulatory mechanisms of salvianolic acid B and carbenoxolone. J. Neuroinflammation 15:97. doi: 10.1186/s12974-018-1127-3
Ystgaard, M. B., Sejersted, Y., Loberg, E. M., Lien, E., Yndestad, A., and Saugstad, O. D. (2015). Early upregulation of NLRP3 in the brain of neonatal mice exposed to hypoxia-ischemia: no early neuroprotective effects of NLRP3 deficiency. Neonatology 108, 211–219. doi: 10.1159/000437247
Yu, Q., Guo, Z., Liu, X., Ouyang, Q., He, C., Burnstock, G., et al. (2013). Block of P2X7 receptors could partly reverse the delayed neuronal death in area CA1 of the hippocampus after transient global cerebral ischemia. Purinergic Signal. 9, 663–675. doi: 10.1007/s11302-013-9379-y
Zasada, M., Lenart, M., Rutkowska-Zapala, M., Stec, M., Mol, N., Czyz, O., et al. (2018). Analysis of selected aspects of inflammasome function in the monocytes from neonates born extremely and very prematurely. Immunobiology 223, 18–24. doi: 10.1016/j.imbio.2017.10.019
Zeinieh, M. P., Talhouk, R. S., El-Sabban, M. E., and Mikati, M. A. (2010). Differential expression of hippocampal connexins after acute hypoxia in the developing brain. Brain Dev. 32, 810–817. doi: 10.1016/j.braindev.2009.11.003
Keywords: connexin, pannexin, hemichannel, ischemia, inflammation, inflammasome, ATP
Citation: Zhou KQ, Green CR, Bennet L, Gunn AJ and Davidson JO (2019) The Role of Connexin and Pannexin Channels in Perinatal Brain Injury and Inflammation. Front. Physiol. 10:141. doi: 10.3389/fphys.2019.00141
Received: 12 November 2018; Accepted: 07 February 2019;
Published: 27 February 2019.
Edited by:
Mary Tolcos, RMIT University, AustraliaReviewed by:
Julie Wixey, University of Queensland, AustraliaMeredith Anne Kelleher, Oregon Health & Science University, United States
Copyright © 2019 Zhou, Green, Bennet, Gunn and Davidson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanne O. Davidson, am9hbm5lLmRhdmlkc29uQGF1Y2tsYW5kLmFjLm56