 Gloria Barbarani
Gloria Barbarani Cristina Fugazza
Cristina Fugazza John Strouboulis
John Strouboulis Antonella E. Ronchi
Antonella E. Ronchi- 1Dipartimento di Biotecnologie e Bioscienze, Università degli Studi Milano-Bicocca, Milan, Italy
- 2School of Cancer & Pharmaceutical Sciences, Faculty of Life Sciences & Medicine, King’s College London, London, United Kingdom
In the last few years, the advent of new technological approaches has led to a better knowledge of the ontogeny of erythropoiesis during development and of the journey leading from hematopoietic stem cells (HSCs) to mature red blood cells (RBCs). Our view of a well-defined hierarchical model of hematopoiesis with a near-homogeneous HSC population residing at the apex has been progressively challenged in favor of a landscape where HSCs themselves are highly heterogeneous and lineages separate earlier than previously thought. The coordination of these events is orchestrated by transcription factors (TFs) that work in a combinatorial manner to activate and/or repress their target genes. The development of next generation sequencing (NGS) has facilitated the identification of pathological mutations involving TFs underlying hematological defects. The examples of GATA1 and KLF1 presented in this review suggest that in the next few years the number of TF mutations associated with dyserythropoietic disorders will further increase.
Introduction
Erythropoiesis leads to the production of the proper number of RBCs required by the body under homeostatic and stress conditions. In healthy adults, erythropoiesis ensures the release in the blood stream of 2 × 106 RBCs/second, but this number dramatically increases to respond to inadequate tissue oxygenation (Tsiftsoglou et al., 2009; Dzierzak and Philipsen, 2013; Nandakumar et al., 2016).
Insufficient quantitative or qualitative production of fully functional RBCs, whether acquired or inherited, results in a wide spectrum of diseases generally defined as anemias.
The causes of anemias are variable and reflect the complexity of the differentiation and maturation of erythrocytes. In some cases, the number of RBCs is extremely low because of the failure to produce erythroid progenitors, as in Diamond-Blackfan Anemia (DBA) (Da Costa et al., 2018). In other cases, impaired differentiation leads to the accumulation of erythroid precursors in the bone marrow [β-thalassemia (Rivella, 2015), congenital dyserythropoietic anemia, CDA (Iolascon et al., 2011)] or to the unbalanced production of different blood cell types [myelodysplastic syndromes, MDS (Levine et al., 2007; Lefevre et al., 2017)], resulting in insufficient RBC numbers in the bloodstream. In other forms of anemias, RBCs are produced but defects in some crucial gene products [typically specific enzymes (Koralkova et al., 2014; Grace et al., 2018), membrane proteins or cytoskeletal components (Mohandas and Gallagher, 2008; Perrotta et al., 2008), sickle globin chains (Rees et al., 2010), channel proteins (Glogowska and Gallagher, 2015), specific pathways (Bianchi et al., 2009; Schwarz et al., 2009)] result in RBCs with decreased oxygen delivery capacity and/or shortened lifespan. Very often, different diseases share common features: for example imbalanced globin chains in β-thalassemia is accompanied by the accumulation of defective precursors in the bone marrow and by ineffective erythropoiesis (IE), as is also observed in CDA (Libani et al., 2008; Iolascon et al., 2011; Ribeil et al., 2013; Rivella, 2015).
Recently, thanks to the advent of new technologies, including NGS using small pools of cells or single cells (Nestorowa et al., 2016; Paul et al., 2016; Ye et al., 2017; Giladi et al., 2018), the development of improved panels of surface markers (Guo et al., 2013; Notta et al., 2016) and the design of in vivo cell tracing systems (Dykstra and Bystrykh, 2014; Perie et al., 2014; Pei et al., 2017; Rodriguez-Fraticelli et al., 2018; Upadhaya et al., 2018), our understanding of hematopoiesis -and erythropoiesis- has greatly expanded. In parallel, genome wide association approaches (GWAS) (Menzel et al., 2007; Sankaran et al., 2008; Uda et al., 2008; Soranzo et al., 2009; van der Harst et al., 2012), massive genome and exome sequencing (Chami et al., 2016) led to the identification of new variant/modifier alleles influencing erythropoiesis associated with TFs.
In this scenario, TFs not only control lineage commitment transitions but are emerging as key-players underpinning, so far unexplained erythroid diseases. Here, we consider GATA1 and KLF1 as paradigmatic TFs. By focusing on these examples, we aim to provide evidence of their pleiotropic effects rather than to give a complete list of GATA1 or KLF1 mutations identified so far.
Erythropoiesis
Erythropoiesis During Development
The first wave of erythropoiesis originates in the yolk sac, where Primitive Erythroid Cells (EryPs) sustain the oxygenation demand of the growing embryo (Dzierzak and Philipsen, 2013). EryPs are large in size and still nucleated when released in the circulation, where they later enucleate (Isern et al., 2011; Dzierzak and Philipsen, 2013; Palis, 2014). In mouse, at E8.25 a second wave of erythro-myelo-precursors (EMPs) originates in the yolk sac and colonizes the fetal liver, generating the first definitive RBCs (Palis, 2016). Finally, around E10.5, hematopoietic stem cells (HSCs) from aorta-gonad-mesonephros (AGM), placenta and possibly other yet unknown sites, colonize the fetal liver. These cells will sustain definitive hematopoiesis for the remainder of gestation and, around birth, will migrate to the bone marrow, the site of adult hematopoiesis (Dzierzak and Philipsen, 2013).
From HSC to RBC
Until recently, the “classical model” of hematopoiesis was considered a paradigm of a stepwise, hierarchical cellular specification system, whereby HSCs generated multipotent progenitors with progressively restricted lineage potential through a sequence of binary choices. The grand entrance of new single-cell separation technologies, in vivo lineage tracing systems and single-cell analysis, provided novel and surprising insights, prompting the idea that early transcriptional priming develops into the acquisition of specific lineage programs (Cabezas-Wallscheid et al., 2014; Haas et al., 2018). In this context, erythroid cells would originate early in the hematopoietic hierarchy, i.e., from stem/multipotential progenitor stages (Guo et al., 2013; Notta et al., 2016; Tusi et al., 2018), soon after the emergence of the megakaryocytic lineage (Upadhaya et al., 2018).
The first clearly recognizable unipotent erythroid progenitor, identified decades ago in in vitro clonogenic assays, is the BFU-E (burst-forming unit-erythroid), that differentiate into rapidly dividing colony-forming-unit erythroid (CFU-E) (Hattangadi et al., 2011; Koury, 2016; Dulmovits et al., 2017). The entry of CFU-Es into erythroid terminal differentiation marks the transition into final maturation (Hwang et al., 2017; Tusi et al., 2018).
Extracellular and Intracellular Signals
Red blood cell differentiation, their production in homeostatic and stress condition, is governed by an integrated complex interplay of extracellular and cell-cell signals within the microenvironment that activate the appropriate downstream intracellular signals, ultimately converging on key TFs. Although these aspects are beyond the scope of this review, we give a glimpse of the major players in these regulatory networks in Figure 1.
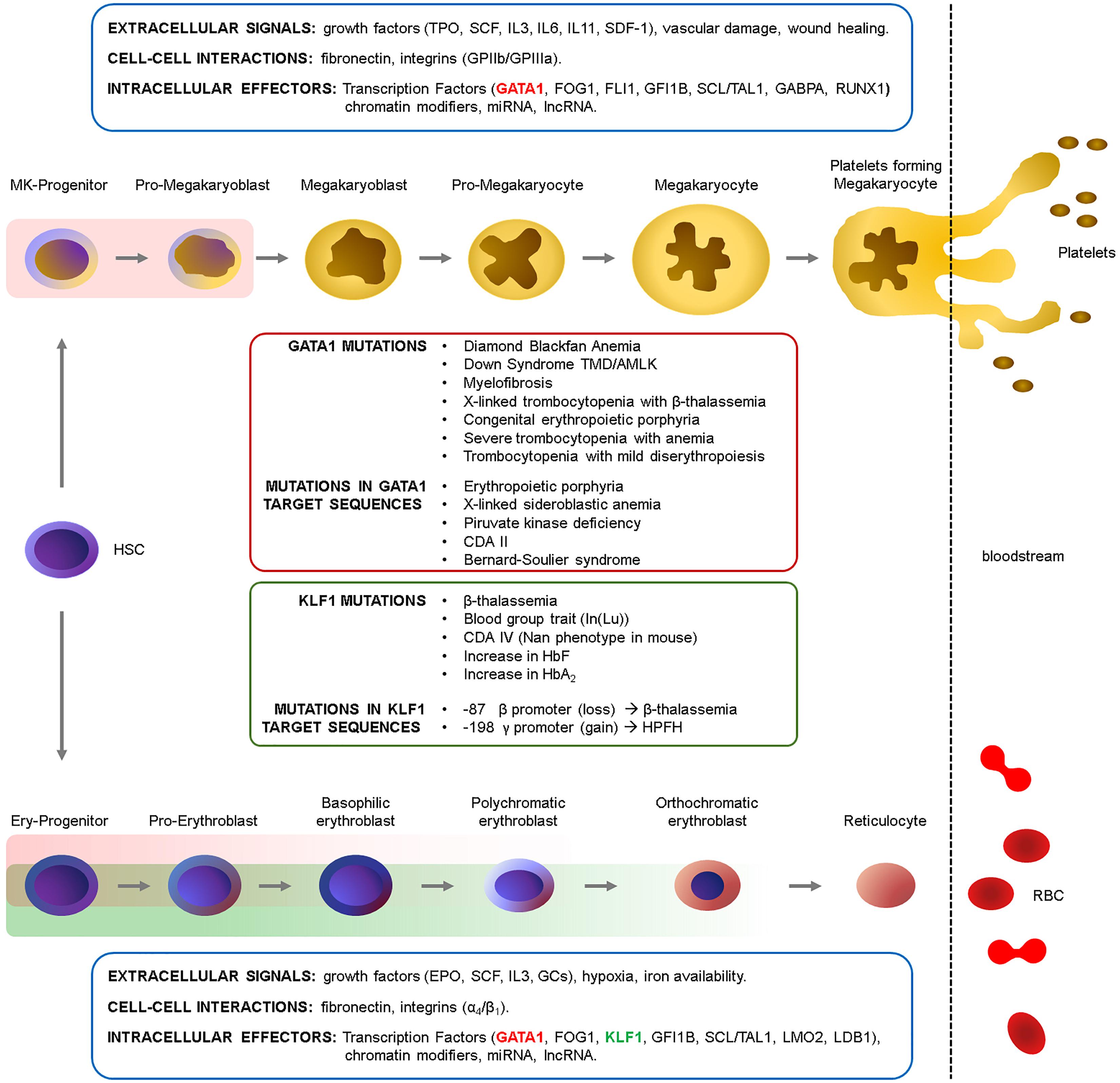
Figure 1. Erythropoiesis and megakaryopoiesis are regulated at multiple levels. A complex network of extracellular signals -activating intracellular signaling pathways-, cell–cell interactions within the niche and intracellular effectors regulate cell differentiation in homeostatic conditions and in response to stress stimuli (Ferreira et al., 2005; Hattangadi et al., 2011; Songdej and Rao, 2017). These signals converge on TFs and chromatin modifiers which ultimately define the transcriptome at each given stage. The main growth factors, integrins and transcription factors involved in these processes are indicated. The GATA1 (red rectangles) and KLF1 (green rectangles) windows of expression are indicated (see also Figure 2B). HSC, Hematopoietic Stem cell; TPO, thrombopoietin; SCF, Stem cell Factor; IL, interleukin; SDF-1, stromal-derived factor-1; GPIIb/IIIa, integrins αIIb/β3 (CD41/CD61); EPO, erythropoietin; GCs, glucocorticoids. α4/β1, integrins α4/β1 (CD49d/CD29).
The Role of Transcription Factors
Transcription factors, together with cofactors and chromatin modifiers, dictate the lineage-specific, stage-specific transcriptional programs by coordinately activating and/or repressing their targets through their binding to DNA (Portela and Esteller, 2010; Dore and Crispino, 2011; Love et al., 2014). The advent of NGS has rapidly expanded our understanding of TFs functions in physiological erythropoiesis, discovering TF mutations as cause of yet unexplained hematological -and dyserythropoietic- defects. Here, we focus on the key examples of GATA1 and KLF1 and their mutations to provide a glimpse of the complexity of their actions (Figure 2).
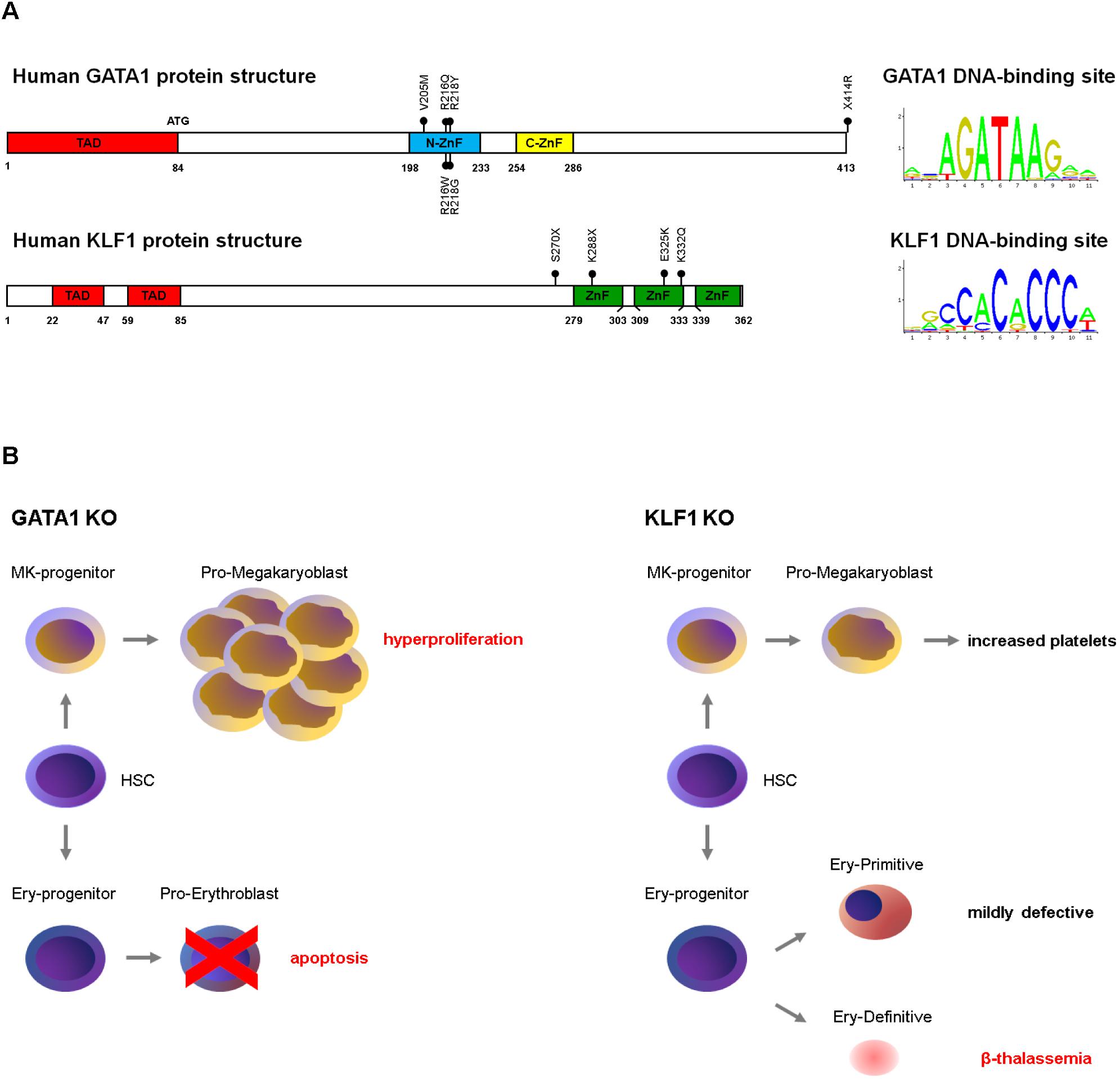
Figure 2. (A) Schematic structure of GATA1 and KLF1 proteins and of their DNA-binding motifs. The position of the mutations discussed in this review are indicated. ZnF, zinc fingers; TAD, transactivation domains. The DNA consensus are from the JASPAR database (http://jaspar2016.genereg.net/). (B) Phenotype of GATA1 (Pevny et al., 1991, 1995; Fujiwara et al., 1996; Shivdasani et al., 1997; Gutierrez et al., 2008) and KLF1 (Nuez et al., 1995; Perkins et al., 1995; Hodge et al., 2006; Nilson et al., 2006; Frontelo et al., 2007; Tallack and Perkins, 2010) gene knockouts in mouse.
The Example of the “Master Regulator” GATA1
The X-linked GATA1 gene encodes a zinc finger TF expressed in the hematopoietic system in erythroid, megakaryocytic and, at lower levels, in eosinophilic, dendritic, and mast cells (Yu et al., 2002a; Ferreira et al., 2005; Gutierrez et al., 2007; Kozma et al., 2010).
GATA1 has three main functional domains: an N-terminal activation domain (N-TAD) and two homologous zinc (Zn) finger domains in the C-terminal half of the protein. The N-terminal Zn finger binds to the GATA1 main cofactor FOG1 (Friend-of-GATA) and modulates the affinity of GATA1 for binding to complex sites in vitro (Trainor et al., 1996; Newton et al., 2001; Yu et al., 2002b). The C-terminal Zn finger (C-ZnF) binds to DNA (WGATAR motif).
GATA1 produces two isoforms: the full length protein (GATA1-FL, 47 kDa) and a shorter variant (GATA1s, 40 kDa), translated from codon 84 within the third exon. GATA1s lacks the N-TAD and results in a protein with a reduced transactivation activity (Calligaris et al., 1995). Gata1 knockout in mice (Pevny et al., 1991) results in embryonic lethality around E10.5–E11.5 due to severe anemia, with GATA1-null cells undergoing massive apoptosis at the proerythroblastic stage (Pevny et al., 1995; Fujiwara et al., 1996). The conditional erythroid knockout in adult mice causes aplastic anemia, revealing its essential role in both steady-state and stress erythropoiesis (Gutierrez et al., 2008).
By contrast, megakaryoblasts lacking GATA1 proliferate abnormally but fail to undergo terminal differentiation (Shivdasani et al., 1997; Vyas et al., 1999). Since these first studies, many other reports revealed the many roles of GATA1 in the erythro/megakaryocytic differentiation (Ferreira et al., 2005). GATA1 mutations identified in patients underscore this pleiotropy: mutations altering the quantity or quality of GATA1 can lead to a variety of phenotypes. Depending on the type of mutation and whether germline or somatic, the severity of the disease and the involvement of the erythroid and/or megakaryocytic compartments greatly varies.
“Quantitative Mutations”: Gene Dosage and Background Effects at Work
Mutations Causing GATA1-FL Loss: Inherited
Diamond-Blackfan anemia (DBA) is an inherited bone marrow failure syndrome characterized by severe anemia due to a great reduction in BFU-Es, without involvement of other hematopoietic lineages. Heterozygous mutations in ribosomal proteins account for about 65% of DBA cases. In 2012 an exome sequencing approach discovered the first GATA1 mutation in a DBA patient (Sankaran et al., 2012). This mutation (c.220G > C transversion) causes the skipping of exon 2, determining GATA1-FL loss, while retaining GATA1s. Unrelated DBA patients were reported to carry the same mutation (Klar et al., 2014), or mutations in the ATG of GATA1-FL (Ludwig et al., 2014; Parrella et al., 2014). Of interest, in a family reported by Hollanda et al. (2006) the inherited loss of GATA1-FL results in macrocytic anemia of various severity in the different patients (with variable involvement of megakaryocytes and neutrophils).
Mutations Causing GATA1-FL Loss: Acquired
Somatic mutations in GATA1, preventing the synthesis of GATA1-FL, predispose newborn Down Syndrome (DS) patients to develop (in 10–20% of cases) transient myeloproliferative disease (TMD) (Wechsler et al., 2002; Xu et al., 2003; Hitzler and Zipursky, 2005). This pre-leukemic condition often spontaneously resolves. However, in about 30% of TMD cases, it develops into acute pediatric megakaryoblastic leukemia (AMKL) (Wechsler et al., 2002; Magalhaes et al., 2006). All the DS-TMD GATA1 mutations identified so far, map in exon 2 and either introduce a STOP codon or alter splicing such that only GATA1s is translated (Mundschau et al., 2003; Rainis et al., 2003). The loss of GATA1-FL in pre-malignant cells characterizes virtually all cases of DS-TMD. The detection of clone-specific GATA1 mutations in DS-TMD and AMKL proves that AMKL derive from the TMD clone (Rainis et al., 2003; Ahmed et al., 2004; Hitzler and Zipursky, 2005). Moreover, GATA1 mutations are extremely rare in AMKL blasts of non-DS patients, clearly indicating a specific cooperation of GATA1 mutations with trisomy 21 (Gruber and Downing, 2015). The restoration of GATA1-FL expression in DS-AMKL-derived cells partially restores erythroid differentiation, further supporting the notion that the loss of GATA1-FL is essential for leukemogenesis (Xu et al., 2003). Importantly, DS-AMKL GATA1 mutations have very little effect on erythropoiesis, suggesting that the co-occurrence trisomy 21 confers the property of specific targeting megakaryoblasts in DS patients.
Various evidences suggest that TMD likely emerges in a yolk sac/fetal liver progenitor in utero (Shimada et al., 2004). In agreement with this hypothesis, in mouse, a knockin allele abolishing GATA1-FL (and leaving GATA1s intact) results in a transient reduction of erythroid cells accompanied by increased megakaryopoiesis that resolves around E14.5 (Li et al., 2005). Despite these observations, the fetal cell type originating TMD and molecular mechanisms by which GATA1 mutations specifically synergizes with trisomy 21 are still unclear (Crispino, 2005).
GATA1 Low Levels and Disease
The notion that low levels of GATA1 lead to the development of myelofibrosis comes from studies in the GATA1-low mouse model, that also develops anemia with age (Vannucchi et al., 2002). In line with this first observation, the majority of patients with primary myelofibrosis (PMF) have GATA1-deficient megakaryocytes (Migliaccio et al., 2005). Of interest, in PMF patients, the reduced level of GATA1 is due to its impaired translation secondary to RPS14 deficiency (Gilles et al., 2017). The connection between GATA1 levels and RP proteins hinges on additional observations: indeed, in cells from DBA patients who are haploinsufficient for RPS19, GATA1 translation is greatly reduced (Ludwig et al., 2014; O’Brien et al., 2017; Khajuria et al., 2018).
Together, these examples again point toward the importance of the correct GATA1 protein dosage and indicates GATA1 post-transcriptional regulation as an important determinant of GATA1 protein level.
“Qualitative Mutations”: the Importance of Protein-Protein Interactions and More
Mutations Abolishing the Interaction With FOG1
In Tsang et al. (1997) identified by yeast two-hybrid a novel zinc finger protein, named FOG1, binding to the N-ZnF of GATA1. GATA1 mutants unable to bind FOG1 (but still retaining DNA binding) do not rescue the severe block in terminal erythroid maturation of GATA1-deficient cells (Tsang et al., 1997). Instead, a compensatory FOG1 mutation restoring the interaction, rescues the GATA1- phenotype, demonstrating that the interaction between the two proteins is essential for erythroid and megakaryocytic differentiation (Crispino et al., 1999; Chang et al., 2002). In Nichols et al. (2000) described a family with dyserythropoietic anemia and thrombocytopenia caused by a GATA1 (V205M) mutation abolishing the GATA1:FOG1 interaction.
Other Allelic Variants, Other Interactions, Other Phenotypes
Remarkably, distinct substitutions at a single residue lead to very different outcomes, underlying the complexity of the GATA1 networks. The R216Q substitution causes X-linked thrombocytopenia with β-Thalassemia (Yu et al., 2002b; Balduini et al., 2004), whereas R216W patients also show features of congenital erythropoietic porphyria (CEP) (Phillips et al., 2007; Di Pierro et al., 2015). The D218Y mutation causes severe thrombocytopenia with anemia (Freson et al., 2002), whereas the D218G substitution causes macrothrombocytopenia with mild dyserythropoiesis and no anemia (Freson et al., 2001; Mehaffey et al., 2001).
Notably, whereas the D218Y diminishes the FOG1:GATA1 interaction, the D218G and R216Q do not, but they rather impair GATA1 ability to recruit the TAL1 cofactor complex (Campbell et al., 2013).
Mutations in the GATA1 DNA Target Sequences as a Cause of Human Erythroid Disorders
Ultimately, TFs elicit their function by binding to DNA motifs on their target genes. Thus, it is expected that mutations creating new -or disrupting- specific binding sites could have phenotypic consequences. Although these mutations remain very elusive, over the years an increasing number of cases has accumulated, implicating these polymorphisms as a source of disease. Such mutations have been associated with congenital erythropoietic porphyria (Solis et al., 2001), X-linked sideroblastic anemia (Campagna et al., 2014; Kaneko et al., 2014), pyruvate kinase deficiency (Manco et al., 2000), CDAII (Russo et al., 2017), Bernard–Soulier syndrome (Ludlow et al., 1996) or linked to erythroid trait variants such as δ-thalassemia (Matsuda et al., 1992) and blood groups (Tournamille et al., 1995; Nakajima et al., 2013; Oda et al., 2015; Moller et al., 2018). Interestingly, a mutation abolishing a GATA1 consensus in the KLF1 promoter (see below), causes a reduction of KLF1, which in turn results in reduced transcription of the KLF1 target genes more sensitive to KLF1 levels, such as BCAM, encoding for the Lutheran (Lu) antigen (Singleton et al., 2008).
E/KLF1: An Unsuspected Key-Player in Various Types of Dyserythropoiesis
KLF1 gene, located on chromosome 19, encodes for a proline-rich protein containing three zinc fingers (Bieker, 1996; Mas et al., 2011; Figure 1B), expressed in the bone marrow and in the erythroid lineage. KLF1 mainly acts by recruiting coactivators and chromatin remodelers, thus contributing to the large epigenetics changes which shape erythroid maturation (Shyu et al., 2014).
As for GATA1, the first evidence for an essential role in erythropoiesis came from the observation that KLF1 knockout mice die in utero around E15 due to fatal anemia (Nuez et al., 1995; Perkins et al., 1995). Given that KLF1 is an important activator of β-globin, lethality was first attributed to β-thalassemia. However, this is not the sole explanation for the defect: the rescue of the α/β imbalance obtained by the transgenic expression of γ-globin is not sufficient to rescue hemolysis, thus pointing to additional roles for KLF1 (Perkins et al., 2000). In 2015, the first case of severe neonatal anemia with kernicterus due to KLF1 compound heterozygosis was described in man (Magor et al., 2015), with an erythroid phenotype largely mirroring that observed in mice: hydrops fetalis, hemolytic anemia, jaundice, hepatosplenomegaly, marked erythroblastosis and high levels of HbF. Another report confirms that in humans, although compatible with life, the loss of KLF1 severely impairs erythropoiesis (Lee et al., 2016).
Quantitative Mutations of KLF1: Haploinsufficiency/Hypomorphic Alleles
KLF1 is haplosufficient. The loss of one allele is asymptomatic and only genes particularly sensitive to KLF1 gene dosage are affected. This is observed in the Lutheran In(Lu) Blood group, where either frameshift mutations, introducing premature termination, or amino acids substitutions in the zinc binding domain, lead to reduced or ineffective KLF1 production (Singleton et al., 2008; Helias et al., 2013). Interestingly, the search for possible mutations in an erythroid TF -that turned out to be KLF1- as a cause of the In(Lu) phenotype came from transcriptomic analyses showing that In(Lu) cells express reduced levels of many erythroid-specific genes associated with red cell maturation, including BCAM (encoding for the Lu antigen), ALAS2, HBB, SLC4A1, and CD44 (Singleton et al., 2008). More recently, extended serological and FACS analysis of In(Lu) samples also revealed a reduced expression of CD35, ICAM4, and CD147 (Fraser et al., 2018). Interestingly, in one single case the In(Lu) phenotype has been associated with a GATA1 mutation (X414R) (Singleton et al., 2013).
It is now clear that different KLF1 target genes are differentially sensitive not only to KLF1 levels (when one allele carries an inactivating mutation), but also to the type of KLF1 mutation, making it difficult to clearly separate “quantitative” from “qualitative” effects of KLF1 mutations.
Indeed, KLF1 coordinately regulates the expression of a multitude of red cell specific genes including heme biosynthesis genes [ALAS2, HMBS, TFR2 (Singleton et al., 2008)], red cell enzymes [such as pyruvate kinase genes -PKLR (Viprakasit et al., 2014)], globins (see below) or cell cycle proteins (Hodge et al., 2006; Pilon et al., 2008; Tallack et al., 2009; Gnanapragasam et al., 2016). Thus, depending on the type of mutation, a specific subset of targets can be affected, leading to a broad spectrum of phenotypes (Perkins et al., 2016).
The Semi-Dominant Phenotype in Nan (Neonatal Anemia) Mouse and in Human CDAIV
This is particularly evident in the case of the neonatal anemia (Nan) semi-dominant (Nan/+) mouse phenotype (Heruth et al., 2010; Siatecka et al., 2010) and in the phenotype observed in human Congenital dyserythropoietic anemia type IV (CDA IV) (Wickramasinghe et al., 1991; Arnaud et al., 2010; Jaffray et al., 2013; Ravindranath et al., 2018). In the Nan mouse model, the E339D substitution in the second ZnF within the Nan allele, alters Nan-KLF1 binding specificity, resulting in an aberrant transcriptome (Gillinder et al., 2017). The homologous E325K heterozygous mutation in CDA IV patients causes the reduced expression of a subset of KLF1 targets (such as AQP1 and CD44), whereas other targets are normally expressed (such as BCAM) (Singleton et al., 2011). In analogy with the Nan mouse mutation, it is likely that also in man the E325K mutation could alter the mutant-KLF1 DNA-binding specificity, resulting in detrimental gain of function effects. On the basis of the different charge of the variant residues (Aspartic Acid or Lysine) it is possible to speculate that subsets of targets can be differentially affected by the different mutant proteins, likely explaining the distinct human and mice pathologies (Arnaud et al., 2010; Siatecka et al., 2010). On the other hand, traits common to mouse and human phenotypes could likely result from the reduced (50%) WT-KLF1.
The Intricate Link Between KLF1, Globin Expression and the Hemoglobin Switching: Direct and Indirect Effects
KLF1 was originally identified by its ability to bind to the β-globin promoter (Miller and Bieker, 1993) and the connection between KLF1 and β-thalassemia is demonstrated by the paradigmatic -87 mutation in the β-globin promoter CACC box (Feng et al., 1994).
Accordingly, the more evident phenotype of KLF1 knockout mice is a marked β-thalassemia associated with increased HBG1/HBG2, suggesting that KLF1 interferes at different levels with globin genes expression. Indeed, the ablation of KLF1 perturbs the 3-dimensional conformation of the β-globin locus (Noordermeer and de Laat, 2008; Schoenfelder et al., 2010). Moreover, mutations creating de novo KLF1 motifs can also alter the relative expression within the β-locus: this is the case of the -198 mutation in the γ-promoter that introduces a new KLF1 binding site, generating the British type HFPH (Wienert et al., 2017). Besides these direct effects of loss or gain of KLF1 binding, an intricate network of indirect effects downstream to KLF1 haploinsufficiency/mutations must be considered. Borg et al. (2010) reported a Maltese family with HPFH and mild hypochromatic microcytic RBCs, caused by the KLF1 K288X non-sense mutation, ablating the DNA binding domain. Transcription profiling and functional studies in cells from these subjects revealed low levels of BCL11a, the most important known HBG1/HBG2 repressor, suggesting that failure to properly activate BCL11a is the major cause of the observed HPFH (Borg et al., 2011). This was proven true also in the KLF1-deficient mouse model (Zhou et al., 2010). However, the situation is far more complicated: in another family described shortly thereafter, KLF1 haploinsufficiency did not result in HPFH (Satta et al., 2011). Instead, in this family, HPFH was observed only in compound heterozygotes (non-sense S270X and K332Q missense mutations) together with increased red cell protoporphyrin, a trait observed in the Nan mouse phenotype. Large-scale screening of patients with hemoglobinopathies of different ethnic origin supported the association of KLF1 mutations with elevated HbF, thus confirming that KLF1 variants are an important source of HbF variation (Gallienne et al., 2012). Finally, more subtle effects of KLF1 polymorphisms also account for an appreciable proportion of cases with borderline elevated HbA2 (Perseu et al., 2011). Thus, again, the pleiotropic effects of KLF1 are the sum of quantitative and qualitative effects, possibly in combination with other genetic modifiers.
Conclusion and Perspectives
The recent identification of mutations/variants alleles associated with RBC traits involving TFs has greatly increased thanks to new technologies and is expected to further increase in the next few years. This will help not only to explain so far unexplained diseases -and possibly to envisage new therapeutic strategies-, but also to better understand the structure and function of TFs themselves and their involvement in the different gene regulatory networks. This, in turn, will shed light on the contribution of TFs and their target sequences as a source of genetic variability underlying the wide spectrum of the observed erythroid phenotypes.
Author Contributions
AR conceived and wrote the manuscript. GB, CF, and JS contributed with ideas and discussion. CF created figures.
Funding
This work was supported by the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/ under REA grant agreement no. 289611 (HEM_ID Project) to AR and JS and by Fondazione Cariplo grant no. 2012.0517 to AR and JS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Stephan Menzel for critical reading of the manuscript.
References
Ahmed, M., Sternberg, A., Hall, G., Thomas, A., Smith, O., O’Marcaigh, A., et al. (2004). Natural history of GATA1 mutations in Down syndrome. Blood 103, 2480–2489. doi: 10.1182/blood-2003-10-3383
Arnaud, L., Saison, C., Helias, V., Lucien, N., Steschenko, D., Giarratana, M. C., et al. (2010). A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am. J. Hum. Genet. 87, 721–727. doi: 10.1016/j.ajhg.2010.10.010
Balduini, C. L., Pecci, A., Loffredo, G., Izzo, P., Noris, P., Grosso, M., et al. (2004). Effects of the R216Q mutation of GATA-1 on erythropoiesis and megakaryocytopoiesis. Thromb. Haemost. 91, 129–140. doi: 10.1160/TH03-05-0290
Bianchi, P., Fermo, E., Vercellati, C., Boschetti, C., Barcellini, W., Iurlo, A., et al. (2009). Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum. Mutat. 30, 1292–1298. doi: 10.1002/humu.21077
Bieker, J. J. (1996). Isolation, genomic structure, and expression of human erythroid Kruppel-like factor (EKLF). DNA Cell Biol. 15, 347–352. doi: 10.1089/dna.1996.15.347
Borg, J., Papadopoulos, P., Georgitsi, M., Gutierrez, L., Grech, G., Fanis, P., et al. (2010). Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat. Genet. 42, 801–805. doi: 10.1038/ng.630
Borg, J., Patrinos, G. P., Felice, A. E., and Philipsen, S. (2011). Erythroid phenotypes associated with KLF1 mutations. Haematologica 96, 635–638. doi: 10.3324/haematol.2011.043265
Cabezas-Wallscheid, N., Klimmeck, D., Hansson, J., Lipka, D. B., Reyes, A., Wang, Q., et al. (2014). Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522. doi: 10.1016/j.stem.2014.07.005
Calligaris, R., Bottardi, S., Cogoi, S., Apezteguia, I., and Santoro, C. (1995). Alternative translation initiation site usage results in two functionally distinct forms of the GATA-1 transcription factor. Proc. Natl. Acad. Sci. U.S.A. 92, 11598–11602. doi: 10.1073/pnas.92.25.11598
Campagna, D. R., de Bie, C. I., Schmitz-Abe, K., Sweeney, M., Sendamarai, A. K., Schmidt, P. J., et al. (2014). X-linked sideroblastic anemia due to ALAS2 intron 1 enhancer element GATA-binding site mutations. Am. J. Hematol. 89, 315–319. doi: 10.1002/ajh.23616
Campbell, A. E., Wilkinson-White, L., Mackay, J. P., Matthews, J. M., and Blobel, G. A. (2013). Analysis of disease-causing GATA1 mutations in murine gene complementation systems. Blood 121, 5218–5227. doi: 10.1182/blood-2013-03-488080
Chami, N., Chen, M. H., Slater, A. J., Eicher, J. D., Evangelou, E., Tajuddin, S. M., et al. (2016). Exome genotyping identifies pleiotropic variants associated with red blood cell traits. Am. J. Hum. Genet. 99, 8–21. doi: 10.1016/j.ajhg.2016.05.007
Chang, A. N., Cantor, A. B., Fujiwara, Y., Lodish, M. B., Droho, S., Crispino, J. D., et al. (2002). GATA-factor dependence of the multitype zinc-finger protein FOG-1 for its essential role in megakaryopoiesis. Proc. Natl. Acad. Sci. U.S.A. 99, 9237–9242. doi: 10.1073/pnas.142302099
Crispino, J. D. (2005). GATA1 mutations in Down syndrome: implications for biology and diagnosis of children with transient myeloproliferative disorder and acute megakaryoblastic leukemia. Pediatr. Blood Cancer 44, 40–44. doi: 10.1002/pbc.20066
Crispino, J. D., Lodish, M. B., MacKay, J. P., and Orkin, S. H. (1999). Use of altered specificity mutants to probe a specific protein-protein interaction in differentiation: the GATA-1:FOG complex. Mol. Cell 3, 219–228. doi: 10.1016/S1097-2765(00)80312-3
Da Costa, L., Narla, A., and Mohandas, N. (2018). An update on the pathogenesis and diagnosis of Diamond-Blackfan anemia. F1000Res. 7:F1000 Faculty Rev-1350. doi: 10.12688/f1000research.15542.1
Di Pierro, E., Russo, R., Karakas, Z., Brancaleoni, V., Gambale, A., Kurt, I., et al. (2015). Congenital erythropoietic porphyria linked to GATA1-R216W mutation: challenges for diagnosis. Eur. J. Haematol. 94, 491–497. doi: 10.1111/ejh.12452
Dore, L. C., and Crispino, J. D. (2011). Transcription factor networks in erythroid cell and megakaryocyte development. Blood 118, 231–239. doi: 10.1182/blood-2011-04-285981
Dulmovits, B. M., Hom, J., Narla, A., Mohandas, N., and Blanc, L. (2017). Characterization, regulation, and targeting of erythroid progenitors in normal and disordered human erythropoiesis. Curr. Opin. Hematol. 24, 159–166. doi: 10.1097/MOH.0000000000000328
Dykstra, B., and Bystrykh, L. V. (2014). No monkeying around: clonal tracking of stem cells and progenitors in the macaque. Cell Stem Cell 14, 419–420. doi: 10.1016/j.stem.2014.03.006
Dzierzak, E., and Philipsen, S. (2013). Erythropoiesis: development and differentiation. Cold Spring Harb. Perspect. Med. 3:a011601. doi: 10.1101/cshperspect.a011601
Feng, W. C., Southwood, C. M., and Bieker, J. J. (1994). Analyses of beta-thalassemia mutant DNA interactions with erythroid Kruppel-like factor (EKLF), an erythroid cell-specific transcription factor. J. Biol. Chem. 269, 1493–1500.
Ferreira, R., Ohneda, K., Yamamoto, M., and Philipsen, S. (2005). GATA1 function, a paradigm for transcription factors in hematopoiesis. Mol. Cell Biol. 25, 1215–1227. doi: 10.1128/MCB.25.4.1215-1227.2005
Fraser, N. S., Knauth, C. M., Schoeman, E. M., Moussa, A., Perkins, A. C., Walsh, T., et al. (2018). Investigation of the variable In(Lu) phenotype caused by KLF1 variants. Transfusion 58, 2414–2420. doi: 10.1111/trf.14926
Freson, K., Devriendt, K., Matthijs, G., Van Hoof, A., De Vos, R., Thys, C., et al. (2001). Platelet characteristics in patients with X-linked macrothrombocytopenia because of a novel GATA1 mutation. Blood 98, 85–92. doi: 10.1182/blood.V98.1.85
Freson, K., Matthijs, G., Thys, C., Marien, P., Hoylaerts, M. F., Vermylen, J., et al. (2002). Different substitutions at residue D218 of the X-linked transcription factor GATA1 lead to altered clinical severity of macrothrombocytopenia and anemia and are associated with variable skewed X inactivation. Hum. Mol. Genet. 11, 147–152. doi: 10.1093/hmg/11.2.147
Frontelo, P., Manwani, D., Galdass, M., Karsunky, H., Lohmann, F., Gallagher, P. G., et al. (2007). Novel role for EKLF in megakaryocyte lineage commitment. Blood 110, 3871–3880. doi: 10.1182/blood-2007-03-082065
Fujiwara, Y., Browne, C. P., Cunniff, K., Goff, S. C., and Orkin, S. H. (1996). Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc. Natl. Acad. Sci. U.S.A. 93,12355–12358. doi: 10.1073/pnas.93.22.12355
Gallienne, A. E., Dreau, H. M., Schuh, A., Old, J. M., and Henderson, S. (2012). Ten novel mutations in the erythroid transcription factor KLF1 gene associated with increased fetal hemoglobin levels in adults. Haematologica 97, 340–343. doi: 10.3324/haematol.2011.055442
Giladi, A., Paul, F., Herzog, Y., Lubling, Y., Weiner, A., Yofe, I., et al. (2018). Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat. Cell Biol. 20, 836–846. doi: 10.1038/s41556-018-0121-4
Gilles, L., Arslan, A. D., Marinaccio, C., Wen, Q. J., Arya, P., McNulty, M., et al. (2017). Downregulation of GATA1 drives impaired hematopoiesis in primary myelofibrosis. J. Clin. Invest. 127, 1316–1320. doi: 10.1172/JCI82905
Gillinder, K. R., Ilsley, M. D., Nebor, D., Sachidanandam, R., Lajoie, M., Magor, G. W., et al. (2017). Promiscuous DNA-binding of a mutant zinc finger protein corrupts the transcriptome and diminishes cell viability. Nucleic Acids Res. 45, 1130–1143. doi: 10.1093/nar/gkw1014
Glogowska, E., and Gallagher, P. G. (2015). Disorders of erythrocyte volume homeostasis. Int. J. Lab. Hematol. 37(Suppl. 1), 85–91. doi: 10.1111/ijlh.12357
Gnanapragasam, M. N., McGrath, K. E., Catherman, S., Xue, L., Palis, J., and Bieker, J. J. (2016). EKLF/KLF1-regulated cell cycle exit is essential for erythroblast enucleation. Blood 128, 1631–1641. doi: 10.1182/blood-2016-03-706671
Grace, R. F., Bianchi, P., van Beers, E. J., Eber, S. W., Glader, B., Yaish, H. M., et al. (2018). Clinical spectrum of pyruvate kinase deficiency: data from the Pyruvate Kinase Deficiency Natural History Study. Blood 131, 2183–2192. doi: 10.1182/blood-2017-10-810796
Gruber, T. A., and Downing, J. R. (2015). The biology of pediatric acute megakaryoblastic leukemia. Blood 126, 943–949. doi: 10.1182/blood-2015-05-567859
Guo, G., Luc, S., Marco, E., Lin, T. W., Peng, C., Kerenyi, M. A., et al. (2013). Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell Stem Cell 13, 492–505. doi: 10.1016/j.stem.2013.07.017
Gutierrez, L., Nikolic, T., van Dijk, T. B., Hammad, H., Vos, N., Willart, M., et al. (2007). Gata1 regulates dendritic-cell development and survival. Blood 110, 1933–1941. doi: 10.1182/blood-2006-09-048322
Gutierrez, L., Tsukamoto, S., Suzuki, M., Yamamoto-Mukai, H., Yamamoto, M., Philipsen, S., et al. (2008). Ablation of Gata1 in adult mice results in aplastic crisis, revealing its essential role in steady-state and stress erythropoiesis. Blood 111, 4375–4385. doi: 10.1182/blood-2007-09-115121
Haas, S., Trumpp, A., and Milsom, M. D. (2018). Causes and consequences of hematopoietic stem cell heterogeneity. Cell Stem Cell 22, 627–638. doi: 10.1016/j.stem.2018.04.003
Hattangadi, S. M., Wong, P., Zhang, L., Flygare, J., and Lodish, H. F. (2011). From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 118, 6258–6268. doi: 10.1182/blood-2011-07-356006
Helias, V., Saison, C., Peyrard, T., Vera, E., Prehu, C., Cartron, J. P., et al. (2013). Molecular analysis of the rare in(Lu) blood type: toward decoding the phenotypic outcome of haploinsufficiency for the transcription factor KLF1. Hum. Mutat. 34, 221–228. doi: 10.1002/humu.22218
Heruth, D. P., Hawkins, T., Logsdon, D. P., Gibson, M. I., Sokolovsky, I. V., Nsumu, N. N., et al. (2010). Mutation in erythroid specific transcription factor KLF1 causes Hereditary Spherocytosis in the Nan hemolytic anemia mouse model. Genomics 96, 303–307. doi: 10.1016/j.ygeno.2010.07.009
Hitzler, J. K., and Zipursky, A. (2005). Origins of leukaemia in children with Down syndrome. Nat. Rev. Cancer 5, 11–20. doi: 10.1038/nrc1525
Hodge, D., Coghill, E., Keys, J., Maguire, T., Hartmann, B., McDowall, A., et al. (2006). A global role for EKLF in definitive and primitive erythropoiesis. Blood 107, 3359–3370. doi: 10.1182/blood-2005-07-2888
Hollanda, L. M., Lima, C. S., Cunha, A. F., Albuquerque, D. M., Vassallo, J., Ozelo, M. C., et al. (2006). An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat. Genet. 38, 807–812. doi: 10.1038/ng1825
Hwang, Y., Futran, M., Hidalgo, D., Pop, R., Iyer, D. R., Scully, R., et al. (2017). Global increase in replication fork speed during a p57(KIP2)-regulated erythroid cell fate switch. Sci. Adv. 3:e1700298. doi: 10.1126/sciadv.1700298
Iolascon, A., Russo, R., and Delaunay, J. (2011). Congenital dyserythropoietic anemias. Curr. Opin. Hematol. 18, 146–151. doi: 10.1097/MOH.0b013e32834521b0
Isern, J., He, Z., Fraser, S. T., Nowotschin, S., Ferrer-Vaquer, A., Moore, R., et al. (2011). Single-lineage transcriptome analysis reveals key regulatory pathways in primitive erythroid progenitors in the mouse embryo. Blood 117, 4924–4934. doi: 10.1182/blood-2010-10-313676
Jaffray, J. A., Mitchell, W. B., Gnanapragasam, M. N., Seshan, S. V., Guo, X., Westhoff, C. M., et al. (2013). Erythroid transcription factor EKLF/KLF1 mutation causing congenital dyserythropoietic anemia type IV in a patient of Taiwanese origin: review of all reported cases and development of a clinical diagnostic paradigm. Blood Cells Mol. Dis. 51, 71–75. doi: 10.1016/j.bcmd.2013.02.006
Kaneko, K., Furuyama, K., Fujiwara, T., Kobayashi, R., Ishida, H., Harigae, H., et al. (2014). Identification of a novel erythroid-specific enhancer for the ALAS2 gene and its loss-of-function mutation which is associated with congenital sideroblastic anemia. Haematologica 99, 252–261. doi: 10.3324/haematol.2013.085449
Khajuria, R. K., Munschauer, M., Ulirsch, J. C., Fiorini, C., Ludwig, L. S., McFarland, S. K., et al. (2018). Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell 173, 90–103.e19. doi: 10.1016/j.cell.2018.02.036
Klar, J., Khalfallah, A., Arzoo, P. S., Gazda, H. T., and Dahl, N. (2014). Recurrent GATA1 mutations in Diamond-Blackfan anaemia. Br. J. Haematol. 166,949–951. doi: 10.1111/bjh.12919
Koralkova, P., van Solinge, W. W., and van Wijk, R. (2014). Rare hereditary red blood cell enzymopathies associated with hemolytic anemia - pathophysiology, clinical aspects, and laboratory diagnosis. Int. J. Lab. Hematol. 36, 388–397. doi: 10.1111/ijlh.12223
Koury, M. J. (2016). Tracking erythroid progenitor cells in times of need and times of plenty. Exp. Hematol. 44, 653–663. doi: 10.1016/j.exphem.2015.10.007
Kozma, G. T., Martelli, F., Verrucci, M., Gutierrez, L., Migliaccio, G., Sanchez, M., et al. (2010). Dynamic regulation of Gata1 expression during the maturation of conventional dendritic cells. Exp. Hematol. 38, 489–503.e1. doi: 10.1016/j.exphem.2010.03.006
Lee, H. H., Mak, A. S., Kou, K. O., Poon, C. F., Wong, W. S., Chiu, K. H., et al. (2016). An unusual hydrops fetalis associated with compound heterozygosity for kruppel-like factor 1 mutations. Hemoglobin 40, 431–434. doi: 10.1080/03630269.2016.1267017
Lefevre, C., Bondu, S., Le Goff, S., Kosmider, O., and Fontenay, M. (2017). Dyserythropoiesis of myelodysplastic syndromes. Curr. Opin. Hematol. 24, 191–197. doi: 10.1097/MOH.0000000000000325
Levine, R. L., Pardanani, A., Tefferi, A., and Gilliland, D. G. (2007). Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat. Rev. Cancer 7, 673–683. doi: 10.1038/nrc2210
Li, Z., Godinho, F. J., Klusmann, J. H., Garriga-Canut, M., Yu, C., and Orkin, S. H. (2005). Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat. Genet. 37, 613–619. doi: 10.1038/ng1566
Libani, I. V., Guy, E. C., Melchiori, L., Schiro, R., Ramos, P., Breda, L., et al. (2008). Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood 112, 875–885. doi: 10.1182/blood-2007-12-126938
Love, P. E., Warzecha, C., and Li, L. (2014). Ldb1 complexes: the new master regulators of erythroid gene transcription. Trends Genet. 30, 1–9. doi: 10.1016/j.tig.2013.10.001
Ludlow, L. B., Schick, B. P., Budarf, M. L., Driscoll, D. A., Zackai, E. H., Cohen, A., et al. (1996). Identification of a mutation in a GATA binding site of the platelet glycoprotein Ibbeta promoter resulting in the Bernard-Soulier syndrome. J. Biol. Chem. 271, 22076–22080. doi: 10.1074/jbc.271.36.22076
Ludwig, L. S., Gazda, H. T., Eng, J. C., Eichhorn, S. W., Thiru, P., Ghazvinian, R., et al. (2014). Altered translation of GATA1 in diamond-Blackfan anemia. Nat. Med. 20, 748–753. doi: 10.1038/nm.3557
Magalhaes, I. Q., Splendore, A., Emerenciano, M., Figueiredo, A., Ferrari, I., and Pombo-de-Oliveira, M. S. (2006). GATA1 mutations in acute leukemia in children with Down syndrome. Cancer Genet. Cytogenet. 166, 112–116. doi: 10.1016/j.cancergencyto.2005.10.008
Magor, G. W., Tallack, M. R., Gillinder, K. R., Bell, C. C., McCallum, N., Williams, B., et al. (2015). KLF1-null neonates display hydrops fetalis and a deranged erythroid transcriptome. Blood 125, 2405–2417. doi: 10.1182/blood-2014-08-590968
Manco, L., Ribeiro, M. L., Maximo, V., Almeida, H., Costa, A., Freitas, O., et al. (2000). A new PKLR gene mutation in the R-type promoter region affects the gene transcription causing pyruvate kinase deficiency. Br. J. Haematol. 110, 993–997. doi: 10.1046/j.1365-2141.2000.02283.x
Mas, C., Lussier-Price, M., Soni, S., Morse, T., Arseneault, G., Di Lello, P., et al. (2011). Structural and functional characterization of an atypical activation domain in erythroid Kruppel-like factor (EKLF). Proc. Natl. Acad. Sci. U.S.A. 108, 10484–10489. doi: 10.1073/pnas.1017029108
Matsuda, M., Sakamoto, N., and Fukumaki, Y. (1992). Delta-thalassemia caused by disruption of the site for an erythroid-specific transcription factor, GATA-1, in the delta-globin gene promoter. Blood 80, 1347–1351.
Mehaffey, M. G., Newton, A. L., Gandhi, M. J., Crossley, M., and Drachman, J. G. (2001). X-linked thrombocytopenia caused by a novel mutation of GATA-1. Blood 98, 2681–2688. doi: 10.1182/blood.V98.9.2681
Menzel, S., Garner, C., Gut, I., Matsuda, F., Yamaguchi, M., Heath, S., et al. (2007). A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat. Genet. 39, 1197–1199. doi: 10.1038/ng2108
Migliaccio, A. R., Rana, R. A., Vannucchi, A. M., and Manzoli, F. A. (2005). Role of GATA-1 in normal and neoplastic hemopoiesis. Ann. N. Y. Acad. Sci. 1044, 142–158. doi: 10.1196/annals.1349.019
Miller, I. J., and Bieker, J. J. (1993). A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol. Cell Biol. 13, 2776–2786. doi: 10.1128/MCB.13.5.2776
Mohandas, N., and Gallagher, P. G. (2008). Red cell membrane: past, present, and future. Blood 112, 3939–3948. doi: 10.1182/blood-2008-07-161166
Moller, M., Lee, Y. Q., Vidovic, K., Kjellstrom, S., Bjorkman, L., Storry, J. R., et al. (2018). Disruption of a GATA1-binding motif upstream of XG/PBDX abolishes Xg(a) expression and resolves the Xg blood group system. Blood 132, 334–338. doi: 10.1182/blood-2018-03-842542
Mundschau, G., Gurbuxani, S., Gamis, A. S., Greene, M. E., Arceci, R. J., and Crispino, J. D. (2003). Mutagenesis of GATA1 is an initiating event in Down syndrome leukemogenesis. Blood 101, 4298–4300. doi: 10.1182/blood-2002-12-3904
Nakajima, T., Sano, R., Takahashi, Y., Kubo, R., Takahashi, K., Kominato, Y., et al. (2013). Mutation of the GATA site in the erythroid cell-specific regulatory element of the ABO gene in a Bm subgroup individual. Transfusion 53(11 Suppl. 2), 2917–2927. doi: 10.1111/trf.12181
Nandakumar, S. K., Ulirsch, J. C., and Sankaran, V. G. (2016). Advances in understanding erythropoiesis: evolving perspectives. Br. J. Haematol. 173,206–218. doi: 10.1111/bjh.13938
Nestorowa, S., Hamey, F. K., Pijuan Sala, B., Diamanti, E., Shepherd, M., Laurenti, E., et al. (2016). A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–e31. doi: 10.1182/blood-2016-05-716480
Newton, A., Mackay, J., and Crossley, M. (2001). The N-terminal zinc finger of the erythroid transcription factor GATA-1 binds GATC motifs in DNA. J. Biol. Chem. 276, 35794–35801. doi: 10.1074/jbc.M106256200
Nichols, K. E., Crispino, J. D., Poncz, M., White, J. G., Orkin, S. H., Maris, J. M., et al. (2000). Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat. Genet. 24, 266–270. doi: 10.1038/73480
Nilson, D. G., Sabatino, D. E., Bodine, D. M., and Gallagher, P. G. (2006). Major erythrocyte membrane protein genes in EKLF-deficient mice. Exp. Hematol. 34, 705–712. doi: 10.1016/j.exphem.2006.02.018
Noordermeer, D., and de Laat, W. (2008). Joining the loops: beta-globin gene regulation. IUBMB Life 60, 824–833. doi: 10.1002/iub.129
Notta, F., Zandi, S., Takayama, N., Dobson, S., Gan, O. I., Wilson, G., et al. (2016). Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 351:aab2116. doi: 10.1126/science.aab2116
Nuez, B., Michalovich, D., Bygrave, A., Ploemacher, R., and Grosveld, F. (1995). Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature 375, 316–318. doi: 10.1038/375316a0
O’Brien, K. A., Farrar, J. E., Vlachos, A., Anderson, S. M., Tsujiura, C. A., Lichtenberg, J., et al. (2017). Molecular convergence in ex vivo models of Diamond-Blackfan anemia. Blood 129, 3111–3120. doi: 10.1182/blood-2017-01-760462
Oda, A., Isa, K., Ogasawara, K., Kameyama, K., Okuda, K., Hirashima, M., et al. (2015). A novel mutation of the GATA site in the erythroid cell-specific regulatory element of the ABO gene in a blood donor with the Am B phenotype. Vox Sang. 108, 425–427. doi: 10.1111/vox.12229
Palis, J. (2014). Primitive and definitive erythropoiesis in mammals. Front. Physiol. 5:3. doi: 10.3389/fphys.2014.00003
Palis, J. (2016). Hematopoietic stem cell-independent hematopoiesis: emergence of erythroid, megakaryocyte, and myeloid potential in the mammalian embryo. FEBS Lett. 590, 3965–3974. doi: 10.1002/1873-3468.12459
Parrella, S., Aspesi, A., Quarello, P., Garelli, E., Pavesi, E., Carando, A., et al. (2014). Loss of GATA-1 full length as a cause of Diamond-Blackfan anemia phenotype. Pediatr. Blood Cancer 61, 1319–1321. doi: 10.1002/pbc.24944
Paul, F., Arkin, Y., Giladi, A., Jaitin, D. A., Kenigsberg, E., Keren-Shaul, H., et al. (2016). Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 164, 325. doi: 10.1016/j.cell.2015.12.046
Pei, W., Feyerabend, T. B., Rossler, J., Wang, X., Postrach, D., Busch, K., et al. (2017). Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 548, 456–460. doi: 10.1038/nature23653
Perie, L., Hodgkin, P. D., Naik, S. H., Schumacher, T. N., de Boer, R. J., and Duffy, K. R. (2014). Determining lineage pathways from cellular barcoding experiments. Cell Rep. 6, 617–624. doi: 10.1016/j.celrep.2014.01.016
Perkins, A., Xu, X., Higgs, D. R., Patrinos, G. P., Arnaud, L., Bieker, J. J., et al. (2016). Kruppeling erythropoiesis: an unexpected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood 127, 1856–1862. doi: 10.1182/blood-2016-01-694331
Perkins, A. C., Peterson, K. R., Stamatoyannopoulos, G., Witkowska, H. E., and Orkin, S. H. (2000). Fetal expression of a human Agamma globin transgene rescues globin chain imbalance but not hemolysis in EKLF null mouse embryos. Blood 95, 1827–1833.
Perkins, A. C., Sharpe, A. H., and Orkin, S. H. (1995). Lethal beta-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature 375, 318–322. doi: 10.1038/375318a0
Perrotta, S., Gallagher, P. G., and Mohandas, N. (2008). Hereditary spherocytosis. Lancet 372, 1411–1426. doi: 10.1016/S0140-6736(08)61588-3
Perseu, L., Satta, S., Moi, P., Demartis, F. R., Manunza, L., Sollaino, M. C., et al. (2011). KLF1 gene mutations cause borderline HbA(2). Blood 118, 4454–4458. doi: 10.1182/blood-2011-04-345736
Pevny, L., Lin, C. S., D’Agati, V., Simon, M. C., Orkin, S. H., and Costantini, F. (1995). Development of hematopoietic cells lacking transcription factor GATA-1. Development 121, 163–172.
Pevny, L., Simon, M. C., Robertson, E., Klein, W. H., Tsai, S. F., D’Agati, V., et al. (1991). Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 349, 257–260. doi: 10.1038/349257a0
Phillips, J. D., Steensma, D. P., Pulsipher, M. A., Spangrude, G. J., and Kushner, J. P. (2007). Congenital erythropoietic porphyria due to a mutation in GATA1: the first trans-acting mutation causative for a human porphyria. Blood 109, 2618–2621. doi: 10.1182/blood-2006-06-022848
Pilon, A. M., Arcasoy, M. O., Dressman, H. K., Vayda, S. E., Maksimova, Y. D., Sangerman, J. I., et al. (2008). Failure of terminal erythroid differentiation in EKLF-deficient mice is associated with cell cycle perturbation and reduced expression of E2F2. Mol. Cell Biol. 28, 7394–7401. doi: 10.1128/MCB.01087-08
Portela, A., and Esteller, M. (2010). Epigenetic modifications and human disease. Nat. Biotechnol. 28, 1057–1068. doi: 10.1038/nbt.1685
Rainis, L., Bercovich, D., Strehl, S., Teigler-Schlegel, A., Stark, B., Trka, J., et al. (2003). Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood 102, 981–986. doi: 10.1182/blood-2002-11-3599
Ravindranath, Y., Johnson, R. M., Goyette, G., Buck, S., Gadgeel, M., and Gallagher, P. G. (2018). KLF1 E325K-associated congenital dyserythropoietic anemia type IV: insights into the variable clinical severity. J. Pediatr. Hematol. Oncol. 40, e405–e409. doi: 10.1097/MPH.0000000000001056
Rees, D. C., Williams, T. N., and Gladwin, M. T. (2010). Sickle-cell disease. Lancet 376, 2018–2031. doi: 10.1016/S0140-6736(10)61029-X
Ribeil, J. A., Arlet, J. B., Dussiot, M., Moura, I. C., Courtois, G., and Hermine, O. (2013). Ineffective erythropoiesis in beta -thalassemia. ScientificWorldJournal. 2013:394295. doi: 10.1155/2013/394295
Rivella, S. (2015). beta-thalassemias: paradigmatic diseases for scientific discoveries and development of innovative therapies. Haematologica 100, 418–430. doi: 10.3324/haematol.2014.114827
Rodriguez-Fraticelli, A. E., Wolock, S. L., Weinreb, C. S., Panero, R., Patel, S. H., Jankovic, M., et al. (2018). Clonal analysis of lineage fate in native haematopoiesis. Nature 553, 212–216. doi: 10.1038/nature25168
Russo, R., Andolfo, I., Gambale, A., De Rosa, G., Manna, F., Arillo, A., et al. (2017). GATA1 erythroid-specific regulation of SEC23B expression and its implication in the pathogenesis of congenital dyserythropoietic anemia type II. Haematologica 102, e371–e374. doi: 10.3324/haematol.2016.162966
Sankaran, V. G., Ghazvinian, R., Do, R., Thiru, P., Vergilio, J. A., Beggs, A. H., et al. (2012). Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J. Clin. Invest. 122, 2439–2443. doi: 10.1172/JCI63597
Sankaran, V. G., Menne, T. F., Xu, J., Akie, T. E., Lettre, G., Van Handel, B., et al. (2008). Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322, 1839–1842. doi: 10.1126/science.1165409
Satta, S., Perseu, L., Moi, P., Asunis, I., Cabriolu, A., Maccioni, L., et al. (2011). Compound heterozygosity for KLF1 mutations associated with remarkable increase of fetal hemoglobin and red cell protoporphyrin. Haematologica 96, 767–770. doi: 10.3324/haematol.2010.037333
Schoenfelder, S., Sexton, T., Chakalova, L., Cope, N. F., Horton, A., Andrews, S., et al. (2010). Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 42, 53–61. doi: 10.1038/ng.496
Schwarz, K., Iolascon, A., Verissimo, F., Trede, N. S., Horsley, W., Chen, W., et al. (2009). Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat. Genet. 41, 936–940. doi: 10.1038/ng.405
Shimada, A., Xu, G., Toki, T., Kimura, H., Hayashi, Y., and Ito, E. (2004). Fetal origin of the GATA1 mutation in identical twins with transient myeloproliferative disorder and acute megakaryoblastic leukemia accompanying Down syndrome. Blood 103:366. doi: 10.1182/blood-2003-09-3219
Shivdasani, R. A., Fujiwara, Y., McDevitt, M. A., and Orkin, S. H. (1997). A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. EMBO J. 16, 3965–3973. doi: 10.1093/emboj/16.13.3965
Shyu, Y. C., Lee, T. L., Chen, X., Hsu, P. H., Wen, S. C., Liaw, Y. W., et al. (2014). Tight regulation of a timed nuclear import wave of EKLF by PKCtheta and FOE during Pro-E to Baso-E transition. Dev. Cell 28, 409–422. doi: 10.1016/j.devcel.2014.01.007
Siatecka, M., Sahr, K. E., Andersen, S. G., Mezei, M., Bieker, J. J., and Peters, L. L. (2010). Severe anemia in the Nan mutant mouse caused by sequence-selective disruption of erythroid Kruppel-like factor. Proc. Natl. Acad. Sci. U.S.A. 107, 15151–15156. doi: 10.1073/pnas.1004996107
Singleton, B. K., Burton, N. M., Green, C., Brady, R. L., and Anstee, D. J. (2008). Mutations in EKLF/KLF1 form the molecular basis of the rare blood group In(Lu) phenotype. Blood 112, 2081–2088. doi: 10.1182/blood-2008-03-145672
Singleton, B. K., Lau, W., Fairweather, V. S., Burton, N. M., Wilson, M. C., Parsons, S. F., et al. (2011). Mutations in the second zinc finger of human EKLF reduce promoter affinity but give rise to benign and disease phenotypes. Blood 118, 3137–3145. doi: 10.1182/blood-2011-04-349985
Singleton, B. K., Roxby, D. J., Stirling, J. W., Spring, F. A., Wilson, C., Poole, J., et al. (2013). A novel GATA1 mutation (Stop414Arg) in a family with the rare X-linked blood group Lu(a-b-) phenotype and mild macrothrombocytic thrombocytopenia. Br. J. Haematol. 161, 139–142. doi: 10.1111/bjh.12184
Solis, C., Aizencang, G. I., Astrin, K. H., Bishop, D. F., and Desnick, R. J. (2001). Uroporphyrinogen III synthase erythroid promoter mutations in adjacent GATA1 and CP2 elements cause congenital erythropoietic porphyria. J. Clin. Invest. 107, 753–762. doi: 10.1172/JCI10642
Songdej, N., and Rao, A. K. (2017). Hematopoietic transcription factor mutations: important players in inherited platelet defects. Blood 129, 2873–2881. doi: 10.1182/blood-2016-11-709881
Soranzo, N., Spector, T. D., Mangino, M., Kuhnel, B., Rendon, A., Teumer, A., et al. (2009). A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat. Genet. 41, 1182–1190. doi: 10.1038/ng.467
Tallack, M. R., Keys, J. R., Humbert, P. O., and Perkins, A. C. (2009). EKLF/KLF1 controls cell cycle entry via direct regulation of E2f2. J. Biol. Chem. 284, 20966–20974. doi: 10.1074/jbc.M109.006346
Tallack, M. R., and Perkins, A. C. (2010). Megakaryocyte-erythroid lineage promiscuity in EKLF null mouse blood. Haematologica 95, 144–147. doi: 10.3324/haematol.2009.010017
Tournamille, C., Colin, Y., Cartron, J. P., and Le Van Kim, C. (1995). Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy–negative individuals. Nat. Genet. 10, 224–228. doi: 10.1038/ng0695-224
Trainor, C. D., Omichinski, J. G., Vandergon, T. L., Gronenborn, A. M., Clore, G. M., and Felsenfeld, G. (1996). A palindromic regulatory site within vertebrate GATA-1 promoters requires both zinc fingers of the GATA-1 DNA-binding domain for high-affinity interaction. Mol. Cell Biol. 16, 2238–2247. doi: 10.1128/MCB.16.5.2238
Tsang, A. P., Visvader, J. E., Turner, C. A., Fujiwara, Y., Yu, C., Weiss, M. J., et al. (1997). FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell 90, 109–119. doi: 10.1016/S0092-8674(00)80318-9
Tsiftsoglou, A. S., Vizirianakis, I. S., and Strouboulis, J. (2009). Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life 61, 800–830. doi: 10.1002/iub.226
Tusi, B. K., Wolock, S. L., Weinreb, C., Hwang, Y., Hidalgo, D., Zilionis, R., et al. (2018). Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 555, 54–60. doi: 10.1038/nature25741
Uda, M., Galanello, R., Sanna, S., Lettre, G., Sankaran, V. G., Chen, W., et al. (2008). Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc. Natl. Acad. Sci. U.S.A. 105, 1620–1625. doi: 10.1073/pnas.0711566105
Upadhaya, S., Sawai, C. M., Papalexi, E., Rashidfarrokhi, A., Jang, G., Chattopadhyay, P., et al. (2018). Kinetics of adult hematopoietic stem cell differentiation in vivo. J. Exp. Med. 215:2815. doi: 10.1084/jem.20180136
van der Harst, P., Zhang, W., Mateo Leach, I., Rendon, A., Verweij, N., Sehmi, J., et al. (2012). Seventy-five genetic loci influencing the human red blood cell. Nature 492, 369–375. doi: 10.1038/nature11677
Vannucchi, A. M., Bianchi, L., Cellai, C., Paoletti, F., Rana, R. A., Lorenzini, R., et al. (2002). Development of myelofibrosis in mice genetically impaired for GATA-1 expression (GATA-1(low) mice). Blood 100, 1123–1132. doi: 10.1182/blood-2002-06-1913
Viprakasit, V., Ekwattanakit, S., Riolueang, S., Chalaow, N., Fisher, C., Lower, K., et al. (2014). Mutations in Kruppel-like factor 1 cause transfusion-dependent hemolytic anemia and persistence of embryonic globin gene expression. Blood 123, 1586–1595. doi: 10.1182/blood-2013-09-526087
Vyas, P., Ault, K., Jackson, C. W., Orkin, S. H., and Shivdasani, R. A. (1999). Consequences of GATA-1 deficiency in megakaryocytes and platelets. Blood 93, 2867–2875.
Wechsler, J., Greene, M., McDevitt, M. A., Anastasi, J., Karp, J. E., Le Beau, M. M., et al. (2002). Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat. Genet. 32, 148–152. doi: 10.1038/ng955
Wickramasinghe, S. N., Illum, N., and Wimberley, P. D. (1991). Congenital dyserythropoietic anaemia with novel intra-erythroblastic and intra-erythrocytic inclusions. Br. J. Haematol. 79, 322–330. doi: 10.1111/j.1365-2141.1991.tb04541.x
Wienert, B., Martyn, G. E., Kurita, R., Nakamura, Y., Quinlan, K. G. R., and Crossley, M. (2017). KLF1 drives the expression of fetal hemoglobin in British HPFH. Blood 130, 803–807. doi: 10.1182/blood-2017-02-767400
Xu, G., Nagano, M., Kanezaki, R., Toki, T., Hayashi, Y., Taketani, T., et al. (2003). Frequent mutations in the GATA-1 gene in the transient myeloproliferative disorder of Down syndrome. Blood 102, 2960–2968. doi: 10.1182/blood-2003-02-0390
Ye, F., Huang, W., and Guo, G. (2017). Studying hematopoiesis using single-cell technologies. J. Hematol. Oncol. 10:27. doi: 10.1186/s13045-017-0401-7
Yu, C., Cantor, A. B., Yang, H., Browne, C., Wells, R. A., Fujiwara, Y., et al. (2002a). Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J. Exp. Med. 195, 1387–1395.
Yu, C., Niakan, K. K., Matsushita, M., Stamatoyannopoulos, G., Orkin, S. H., and Raskind, W. H. (2002b). X-linked thrombocytopenia with thalassemia from a mutation in the amino finger of GATA-1 affecting DNA binding rather than FOG-1 interaction. Blood 100, 2040–2045.
Keywords: erythropoiesis, dyserythropoiesis, transcription factors, GATA1, KLF1
Citation: Barbarani G, Fugazza C, Strouboulis J and Ronchi AE (2019) The Pleiotropic Effects of GATA1 and KLF1 in Physiological Erythropoiesis and in Dyserythropoietic Disorders. Front. Physiol. 10:91. doi: 10.3389/fphys.2019.00091
Received: 21 November 2018; Accepted: 25 January 2019;
Published: 12 February 2019.
Edited by:
Richard Van Wijk, Utrecht University, NetherlandsReviewed by:
Andrew Charles Perkins, Monash University, AustraliaPatrick G. Gallagher, Yale University, United States
Marije Bartels, University Medical Center Utrecht, Netherlands
Copyright © 2019 Barbarani, Fugazza, Strouboulis and Ronchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonella E. Ronchi, YW50b25lbGxhLnJvbmNoaUB1bmltaWIuaXQ=