 Alejandro Gutiérrez
Alejandro Gutiérrez Cristina Contreras
Cristina Contreras Ana Sánchez
Ana Sánchez Dolores Prieto
Dolores Prieto
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 06 February 2019
Sec. Membrane Physiology and Membrane Biophysics
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.00055
This article is part of the Research TopicEvolving Picture of Calcium Handling in Cardiac DiseaseView all 12 articles
Insulin resistance plays a key role in the pathogenesis of type 2 diabetes and is also related to other health problems like obesity, hypertension, and metabolic syndrome. Imbalance between insulin vascular actions via the phosphatidylinositol 3-Kinase (PI3K) and the mitogen activated protein kinase (MAPK) signaling pathways during insulin resistant states results in impaired endothelial PI3K/eNOS- and augmented MAPK/endothelin 1 pathways leading to endothelial dysfunction and abnormal vasoconstriction. The role of PI3K, MAPK, and protein kinase C (PKC) in Ca2+ handling of resistance arteries involved in blood pressure regulation is poorly understood. Therefore, we assessed here whether PI3K, MAPK, and PKC play a role in the Ca2+ signaling pathways linked to adrenergic vasoconstriction in resistance arteries. Simultaneous measurements of intracellular calcium concentration ([Ca2+]i) in vascular smooth muscle (VSM) and tension were performed in endothelium-denuded branches of mesenteric arteries from Wistar rats mounted in a microvascular myographs. Responses to CaCl2 were assessed in arteries activated with phenylephrine (PE) and kept in Ca2+-free solution, in the absence and presence of the selective antagonist of L-type Ca2+ channels nifedipine, cyclopiazonic acid (CPA) to block sarcoplasmic reticulum (SR) intracellular Ca2+ release or specific inhibitors of PI3K, ERK-MAPK, or PKC. Activation of α1-adrenoceptors with PE stimulated both intracellular Ca2+ mobilization and Ca2+ entry along with contraction in resistance arteries. Both [Ca2+]i and contractile responses were inhibited by nifedipine while CPA abolished intracellular Ca2+ mobilization and modestly reduced Ca2+ entry suggesting that α1-adrenergic vasoconstriction is largely dependent Ca2+ influx through L-type Ca2+ channel and to a lesser extent through store-operated Ca2+ channels. Inhibition of ERK-MAPK did not alter intracellular Ca2+ mobilization but largely reduced L-type Ca2+ entry elicited by PE without altering vasoconstriction. The PI3K blocker LY-294002 moderately reduced intracellular Ca2+ release, Ca2+ entry and contraction induced by the α1-adrenoceptor agonist, while PKC inhibition decreased PE-elicited Ca2+ entry and to a lesser extent contraction without affecting intracellular Ca2+ mobilization. Under conditions of ryanodine receptor (RyR) blockade to inhibit Ca2+-induced Ca2+-release (CICR), inhibitors of PI3K, ERK-MAPK, or PKC significantly reduced [Ca2+]i increases but not contraction elicited by high K+ depolarization suggesting an activation of L-type Ca2+ entry in VSM independent of RyR. In summary, our results demonstrate that PI3K, ERK-MAPK, and PKC regulate Ca2+ handling coupled to the α1-adrenoceptor in VSM of resistance arteries and related to both contractile and non-contractile functions. These kinases represent potential pharmacological targets in pathologies associated to vascular dysfunction and abnormal Ca2+ handling such as obesity, hypertension and diabetes mellitus, in which these signaling pathways are profoundly impaired.
Insulin resistance plays a key role in the pathogenesis of type 2 diabetes and is also associated to other metabolic and cardiovascular abnormalities such as obesity, dyslipidemia and hypertension, jointly referred as to metabolic syndrome (Ford, 2005). Imbalance between insulin vascular actions via the phosphatidylinositol 3-Kinase (PI3K) and the mitogen activated protein kinase (MAPK) signaling pathways in insulin resistant states results in impaired endothelial vasodilator PI3K/eNOS/NO and augmented vasoconstrictor MAPK/endothelin 1 (ET1) pathways leading to endothelial dysfunction and exacerbated vasoconstriction (Kim et al., 2006; Prieto et al., 2013). However, altered Ca2+ homeostasis in the arterial wall usually underlies abnormal vasoconstriction and the vascular complications associated to metabolic disease, such as hypertension and coronary artery disease (Okon et al., 2005; Ford et al., 2010; Villalba et al., 2011).
Vascular smooth muscle (VSM) contraction is triggered by the elevation of free intracellular Ca2+ concentration [Ca2+]i due to extracellular Ca2+ influx and/or Ca2+ release from intracellular stores in the sarcoplasmic reticulum (SR), followed by Ca2+-calmodulin-dependent activation of myosin light chain (MLC) kinase (MLCK), MLC phosphorylation and actin/myosin crossbridges formation (Liu and Khalil, 2018). However, increases in force development at a given cytosolic Ca2+ concentration can also occur and hence a dissociation between [Ca2+]i, MLC phosphorylation and vasoconstriction mediated by Ca2+ sensitization mechanisms (Somlyo and Somlyo, 2003; Villalba et al., 2007). Ca2+ release from SR intracellular stores, Ca2+ entry through plasma membrane channels and Ca2+ sensitization mechanisms can differentially contribute to VSM contraction depending on the vasoconstrictor agonist and/or vessel size (Nobe and Paul, 2001; Villalba et al., 2007; Kitazawa and Kitazawa, 2012). Furthermore, relative contribution of protein kinases to Ca2+ handling coupled to receptor-mediated arterial vasoconstriction has also been reported to be size-dependent and thus, involvement of PKC and Rho kinase (RhoK) increase and decrease, respectively, with decreasing arterial size (Kitazawa and Kitazawa, 2012; Martinsen et al., 2012). Although kinases such as PKC and RhoK have traditionally been associated to Ca2+ sensitization mechanisms involved in smooth muscle contraction (Nobe and Paul, 2001; Somlyo and Somlyo, 2003; Villalba et al., 2007), increasing experimental evidence supports a role for protein kinase-mediated regulation of intracellular Ca2+ mobilization and Ca2+ entry mechanisms in VSM and cardiac myocytes (Ghisdal et al., 2003; Villalba et al., 2008; Smani et al., 2010), and differences between large/conductance and small/resistance arteries concerning the role of various kinases in Ca2+ handling have also been demonstrated (Kitazawa and Kitazawa, 2012; Martinsen et al., 2012).
Peripheral small arteries or resistance arteries, whose vasoconstrictor activity is under the sympathetic nervous control, play a key role in blood pressure regulation, hypertension being a common vascular complication in metabolic syndrome and insulin resistant states (Ford, 2005). Both impairment of the signaling pathways including MAPK, PI3K, and PKC in endothelial cells and altered Ca2+ handling in VSM have been reported to underlie abnormal vasoconstriction in metabolic disease (Okon et al., 2005; Kim et al., 2006; Villalba et al., 2011; Prieto et al., 2013). Since the role of MAPK, PI3K, and PKC in Ca2+ handling of resistance arteries is poorly understood, we assessed here whether these kinases are involved in Ca2+ signaling pathways linked to adrenergic vasoconstriction in resistance arteries.
Animal care and experimental protocols conformed to the European Directive for the Protection of Animals Used for Scientific Purposes (European Union Directive 2010/63/EU) and were also supervised by the Animal Care and Use Committee Complutense University of Madrid. Male Wistar rats were housed at the Pharmacy School animal care facility under controlled suitable environmental conditions of temperature (24°C), lighting (12 h light/12 h dark cycle) and humidity (50–60%), and maintained on standard chow and water ad libitum. They were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and euthanized by decapitation and exsanguination at 12-weeks age.
After animals were euthanized, the mesentery was quickly removed and placed on cold physiological saline solution (PSS) of the following composition (mM): NaCl 119, NaHCO3 25, KCl 4.7, KH2PO4 1.17, MgSO4 1.18, CaCl2 1.5, ethylenediaminetetraacetic acid 0.027 and glucose 11, continuously gassed with a mixture of 5% CO2 /95% O2 to maintain pH at 7.4. Mesenteric resistance arteries, third order branches of the superior mesenteric artery, were carefully dissected by removing the surrounding connective and fat tissue. Arterial segments were mounted in parallel in double microvascular myographs (Danish Myo Technology, DMT-Denmark) by inserting two 40 μm tungsten wires and equilibrated for 30 min in PSS at 37°C. The relationship between passive wall tension and internal circumference was determined for each individual artery. The arteries were set to an internal circumference (L1) equal to 90% of that given by a transmural pressure of 100 mmHg for a relaxed vessel in situ, L100 (L1 = 0.9 × L100) at which tension development is maximal (Mulvany and Halpern, 1977). At the beginning of each experiment, arteries were stimulated twice with (KPSS), similar to PSS except that NaCl was substituted for KCl on an equimolar basis, in order to test vessel viability. The endothelium was mechanically removed by inserting a human hair in the vessel lumen and guiding it back and forwards several times. The absence of functional endothelium was confirmed by lack of the relaxation to acetylcholine (10 μM). Arteries were chemically denervated by incubation with guanethidine (10 μM) for 45 min to inhibit adrenergic nerve endings.
Simultaneous measurements of the intracellular calcium concentration ([Ca2+]i) and tension were performed by FURA-2 AM fluorescence in mesenteric resistance arteries as previously reported (Villalba et al., 2007). Arteries were incubated in the dark with 8 μM Fura-2 AM in PSS for 2 h at 37°C. The myograph chamber was mounted on a Zeiss inverted microscope equipped for dual excitation wavelength fluorimetry (Deltascan, Photon Technology). After loading, arteries were illuminated with alternating 340 and 380 nm light using a monochromator-based system (Deltascan, Photon Technology). Fluorescence emission was detected at 510 nm wavelength. The Ratio (R) F340/F380 was taken as a measure of [Ca2+]i. At the end of each experiment fluorescence not related to Ca2+ was measured by bathing the artery in PSS containing 25 mM MnCl2 plus ionomycin (10 μM) to quench Ca2+-insensitive signals and the values obtained were subtracted from those obtained throughout the experiment.
The role of ERK-MAPK, PI3K, and PKC kinases in Ca2+ handling of resistance arteries was assessed in endothelium-denuded arteries kept in Ca2+-free medium. Arteries were exposed for 5 min to Ca2+-free PSS (0 mM Ca2+, 0.1 mM EGTA) to remove all extracellular Ca2+ available for contraction. The myograph solution was then replaced by “nominally Ca2+-free PSS” (0 mM Ca2+, 0 mM EGTA) and concentration-response curves (CRCs) for CaCl2 (10 μM-3 mM) were performed in arteries activated with phenylephrine (PE, 10 μM), in the absence (controls) and presence of the selective blocker of L-type Ca2+ channels nifedipine (0.3 μM), the inhibitor of the Orai1-mediated Ca2+ entry Pyr6 (3 μM) or the specific inhibitors of ERK-MAPK (PD-98059, 3 μM), p38MAPK (SB-203580, 0.3 μM), PI3K (LY-294002, 3 μM) or PKC (GF-109203X, 0.1 μM). The effect of SR Ca2+ store depletion on Ca2+ entry and contraction stimulated by PE was assessed in arteries kept in nominally Ca2+-free medium stimulated with 10 μM PE and then activated with a single Ca2+ concentration (1 mM), before and after SR Ca2+ATPase (SERCA) inhibition with cyclopiazonic acid (CPA, 10 μM), and then treatment with CPA plus nifedipine. The combined effect of SR Ca2+ store depletion with CPA (10 μM) and inhibition of Orai1-mediated Ca2+ entry channels with Pyr6 (3 μM) was also examined on Ca2+ entry and vasoconstriction of mesenteric arteries stimulated with PE (10 μM).
The inhibitory effect of PD-98059 (3 μM), LY-294002 (3 μM), or GF-109203X (0.1 μM) on Ca2+ entry was also assessed in KPSS-depolarized arteries. After a first stimulation with KPSS, arteries were incubated with the inhibitors for at least 30 min before a second stimulation with KPSS was repeated. To evaluate the potential relationship between the PI3K, MAPK, and PKC pathways and the ryanodine receptor (RyR)-mediated Ca2+-induced Ca2+-release (CICR) mechanism in resistance arteries (Sánchez et al., 2018), RyR was blocked by incubation with 10 μM ryanodine for 25 min and then 1.5 mM CaCl2 was added to arteries depolarized with Ca2+-free high K+ solution (KPSS00). The effect of the selective inhibitors of ERK-MAPK, PI3K, or PKC was further assessed in arteries under conditions of RyR blockade.
Ca2+-free PSS and Ca2+-free KPSS solutions were similar to PSS and KPSS, respectively, except that CaCl2 was replaced by 100 μM of EGTA, which was omitted when CaCl2 was administered (“nominally Ca2+-free solution,” 0 mM Ca2+, 0 mM EGTA). Acetylcholine, guanethidine, and phenylephrine were obtained from Sigma-Aldrich (Spain). All of them were dissolved in distilled water. Nifedipine, CPA, Pyr6 and kinase inhibitors (PD-98059, LY-294002, GF-109203X, and SB-203580) were obtained from Tocris Cookson (Bristol, United Kingdom). Stock solutions of Pyr6, PD-98059 and LY-294002 were made in distilled water, and those of CPA Pyr6, ryanodine, SB-203580, and GF-109203X in DMSO and further diluted in water. Nifedipine was initially dissolved in ethanol and further dilutions were made in distilled water.
Results are expressed as either absolute values (units of R F340/F380 or Nm-1 of active tension) or as a percentage of the response to KPSS in each artery, as means ± SEM of 6–10 arteries (one artery from each animal). Arterial sensitivity to agonists was expressed in terms of pEC50, that was the negative value of log EC50, EC50 being the concentration of agonist giving 50% of the maximal response or effect (Emax). Statistically significant differences between means were analyzed by using paired or unpaired Student’s t-test where appropriate, or one-way ANOVA followed by Bonferroni’s post hoc test for comparisons involving more than two groups. Probability levels lower than 5% (P < 0.05) were considered statistically significant. Calculations were made using a standard software package (GraphPad Prism 5.0, GraphPad Software, Inc., San Diego, CA, United States).
In order to assess the involvement of intracellular Ca2+ mobilization and Ca2+ entry mechanisms coupled to the α1-adrenoceptor in resistance arteries, endothelium-denuded mesenteric arteries were kept in a nominally Ca2+-free medium, stimulated with PE and further activated with increasing Ca2+ concentrations (Figure 1A). PE induced an initial rapid increase in VSM [Ca2+]i and a simultaneous phasic contraction showing intracellular Ca2+ mobilization (Figures 1A,C), and a further sustained elevation of [Ca2+]i along with vasoconstriction upon Ca2+ re-addition, indicative of VSM Ca2+ entry (Figures 1A,B). While there were no significant differences in the initial PE-induced [Ca2+]i increases and contraction corresponding to intracellular Ca2+ mobilization (Figure 1C), PE-induced vasoconstriction upon Ca2+ re-addition was larger than the simultaneous sustained [Ca2+]i increases (Figure 1C). Involvement of Ca2+ sensitization in the α1-adrenoceptor-mediated vasoconstriction was further depicted by the steep slope of the [Ca2+]i -tension relationship for PE, showing that large contractions are developed without parallel increases in [Ca2+]i levels (Figure 1D).
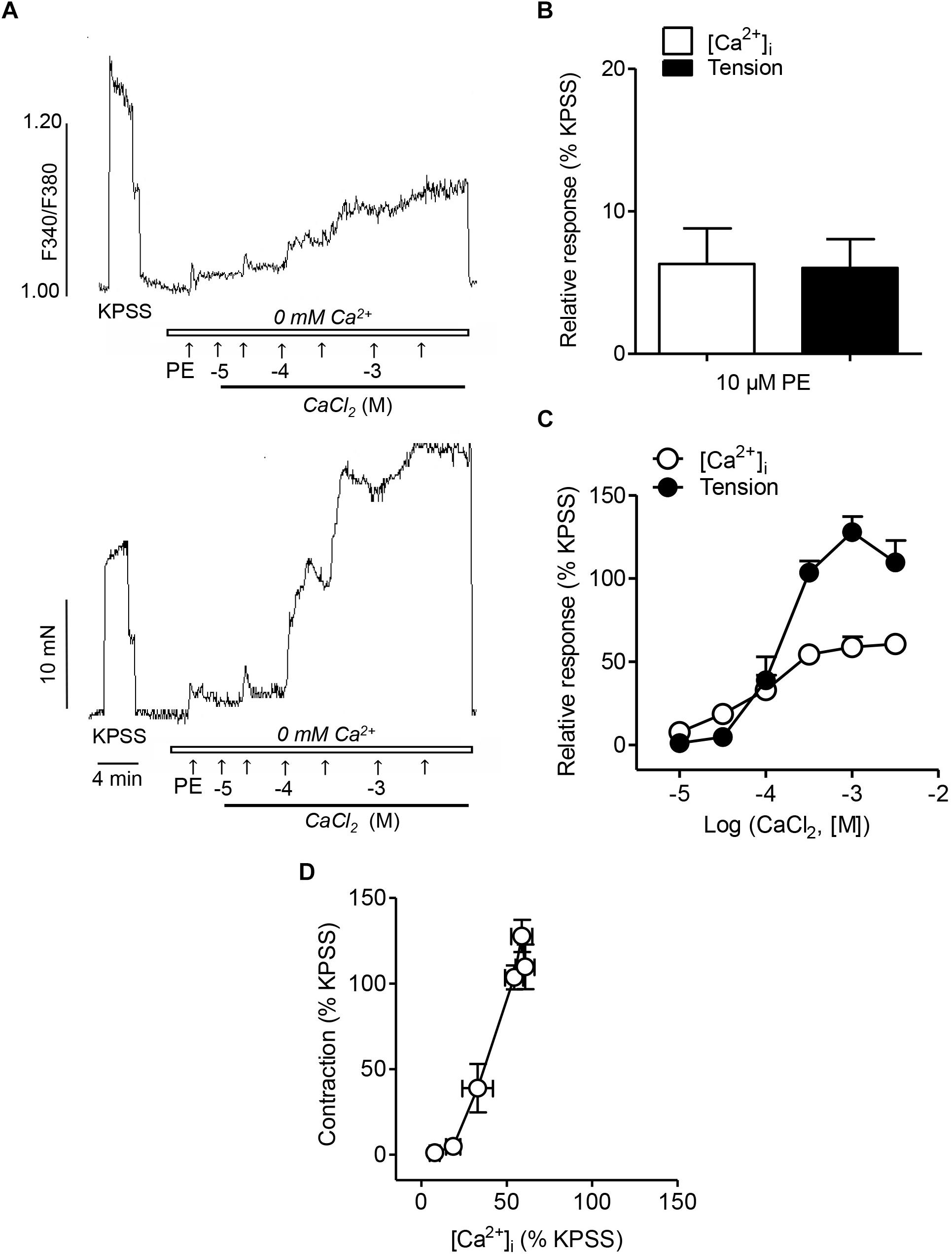
Figure 1. α1-Adrenoceptor activation involves intracelular Ca2+ mobilization, Ca2+ entry and Ca2+ sensitization associated to contraction (A) Representative traces illustrating the changes in both [Ca2+]i mobilization (top) and vasoconstriction (bottom) induced by activation of α1-adrenoceptors by a single dose of Phenylephrine (PE) (10 μM) and further cumulative addition of CaCl2 (closed bar) in endothelium-denuded mesenteric arteries kept in nominally Ca2+-free medium (0 mM Ca2+, open bar). (B,C) Summarized data showing changes in [Ca2+]i and vasoconstrictor responses stimulated by 10 μM PE addition in arteries kept in nominally Ca2+-free medium (B) and further cumulative re-addition of CaCl2 (C). (D) Relationship between [Ca2+]i-contraction in response to cumulative addition of Ca2+ in arteries kept in Ca2+ free medium and stimulated with 10 μM PE. Responses are expressed as absolute values of either [Ca2+]i (ΔF340/F380) or tension (Nm-1) (A) or relative to those elicited by KPSS (B–D). Values are means ± SEM of n = 7 arteries (one from each animal).
Treatment with the blocker of L-type voltage-dependent Ca2+ channels nifedipine largely inhibited the CaCl2 CCR in arteries stimulated with PE (Figure 2A), while the inhibitor of the Orai1-mediated Ca2+ entry Pyr6 only induced a moderate decrease of these responses (Figure 2B). Combined treatment with the SERCA inhibitor CPA to deplete SR Ca2+ stores plus the inhibitor of store-operated Ca2+ channels Pyr6 caused a larger inhibition of PE-induced vasoconstriction (Figure 2B). The effect of SR store depletion by treatment with CPA was further assessed on changes in [Ca2+]i and contraction elicited by PE, in arteries kept in nominally Ca2+-free medium and further stimulated with 1 mM Ca2+ (Figures 2C,D). CPA inhibited PE-induced intracellular Ca2+ mobilization and phasic contraction and reduced the sustained Ca2+ entry and vasoconstriction elicited by the α1-adrenoceptor PE. The latter were abolished by combined treatment with CPA plus the blocker of the L-type voltage-dependent channels nifedipine (Figures 2C,D). These data demonstrate that α1-adrenergic vasoconstriction is largely due to Ca2+ influx through L-type voltage-dependent Ca2+ channels and to lesser extent to Ca2+release from the SR and store-operated Ca2+ entry.
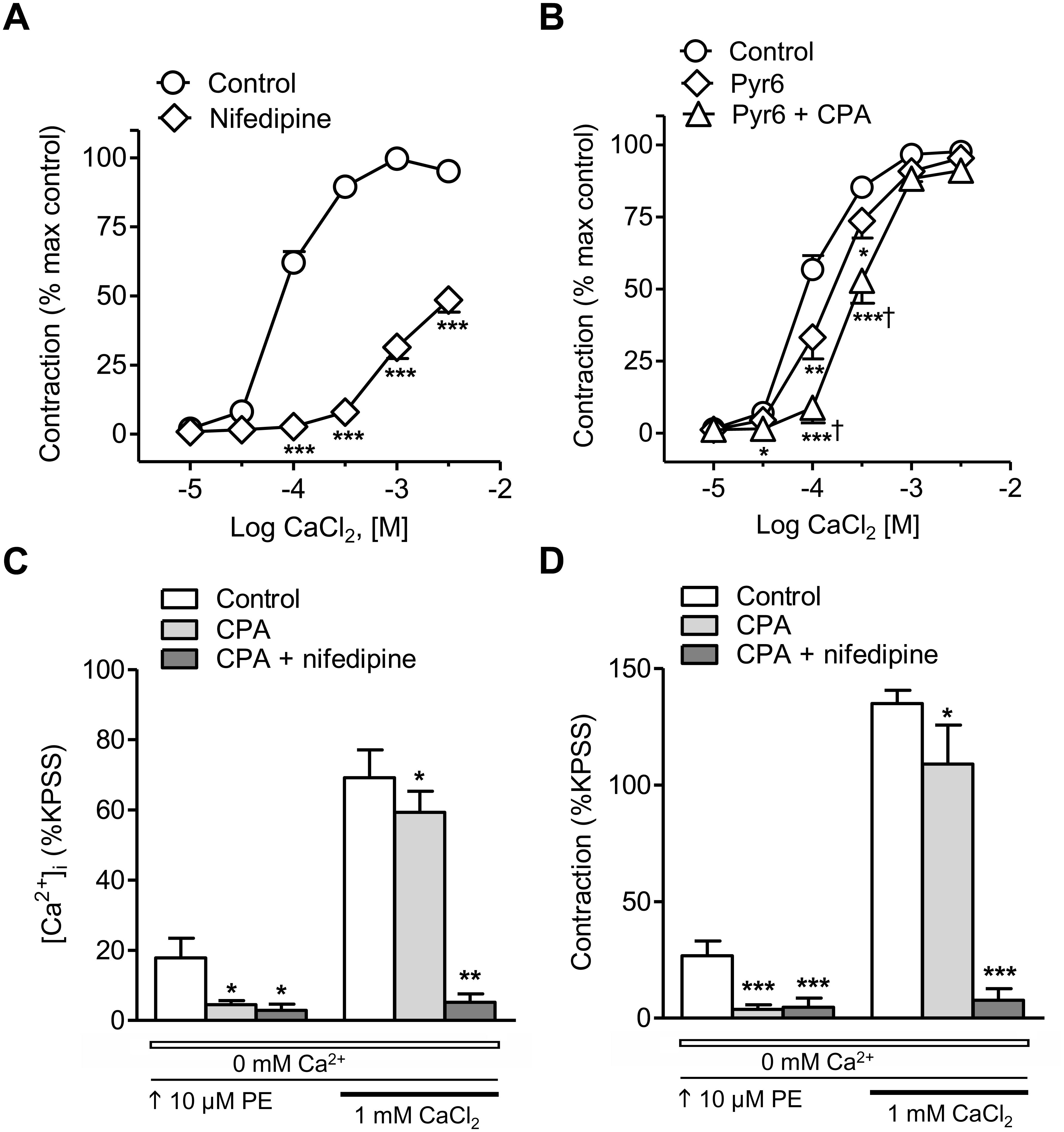
Figure 2. α1-Adrenoceptor-elicited vasoconstriction in resistance arteries is mostly due to Ca2+ entry through voltage-dependent L-type channels. (A,B) Average inhibitory effects of the selective L-type Ca2+ channel blocker nifedipine (0.3 μM) (A) or the inhibitor of the Orai1–mediated Ca2+ entry Pyr6 (3 μM) alone or in combination with the SERCA inhibitor CPA (10 μM) (B) on the CaCl2 concentration-response curves (CRCs) in endothelium-denuded mesenteric resistance arteries kept in nominally Ca2+-free medium and stimulated with 10 μM PE. (C,D) Summarized data showing the effects of SR Ca2+ store depletion by SERCA inhibition with CPA (10 μM) alone or CPA plus nifedipine (0.3 μM) on the changes in [Ca2+]i (C) and contraction (D) in response to 10 μM PE in resistance arteries kept in a nominally Ca2+-free medium (0 mM Ca2+, open bar) after Ca2+ readmission (1 mM CaCl2, closed bar). Results are expressed as a percentage of control maximal responses (A,B) or the KPSS responses (C,D). Values are means ± SEM of n = 6 arteries (one from each animal). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. control. †P < 0.05 vs. Pyr6-treated.
Treatment with the PI3K inhibitor LY-294002 was used to evaluate whether PI3K is involved in Ca2+ entry, Ca2+ mobilization and/or Ca2+ sensitization coupled to the α1-adrenoceptor in resistance arteries. This blocker moderately reduced both Ca2+ entry and vasoconstriction induced by PE (Figures 3A,B and Table 1), without affecting [Ca2+]i-contraction relationships for this agonist which suggests no changes in Ca2+ sensitization (Figure 3C). Interestingly, PI3K inhibition also reduced PE-induced intracellular Ca2+ mobilization and the associated phasic contraction (Figures 3D,E).
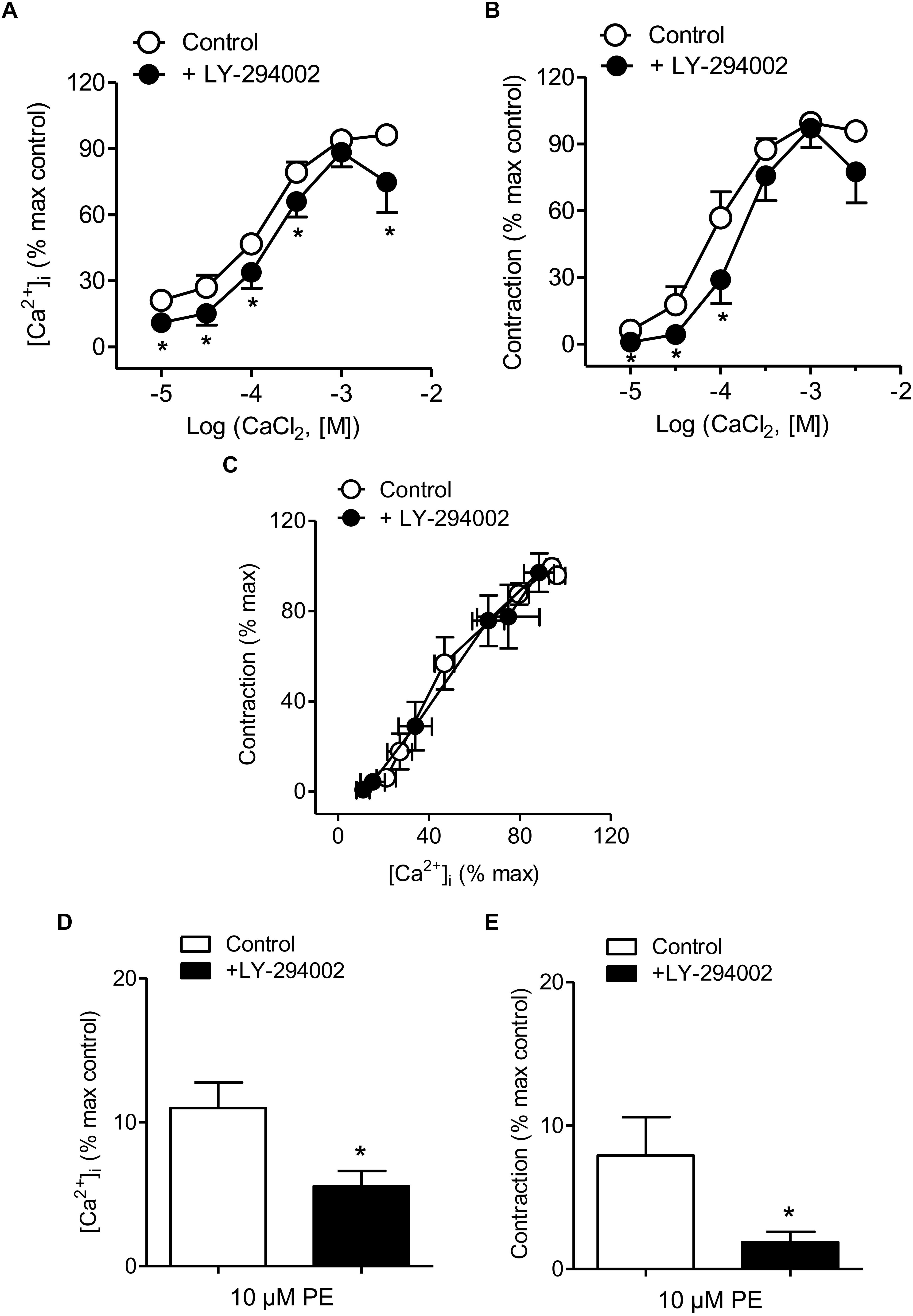
Figure 3. PI3K inhibition reduced Ca2+ entry, vasoconstriction and intracellular Ca2+ mobilization in response to α1-adrenoceptor activation. (A,B) Summarized data showing the effects of the PI3K inhibitor LY-294002 (3 μM) on the increases in [Ca2+]i (A) and contraction (B) elicited by cumulative addition of CaCl2 in endothelium-denuded arteries kept in a nominally Ca2+-free medium and stimulated by 10 μM PE. (C) Effect of LY-294002 on the [Ca2+]i-contraction relationship for 10 μM PE in mesenteric resistance arteries and (D,E) the increases in [Ca2+]i and contraction elicited by 10 μM PE in nominally Ca2+-free PSS. Results are expressed as a percentage of control maximal responses. Values are means ± SEM of n = 8 arteries (one from each animal). ∗P < 0.05 vs. control.
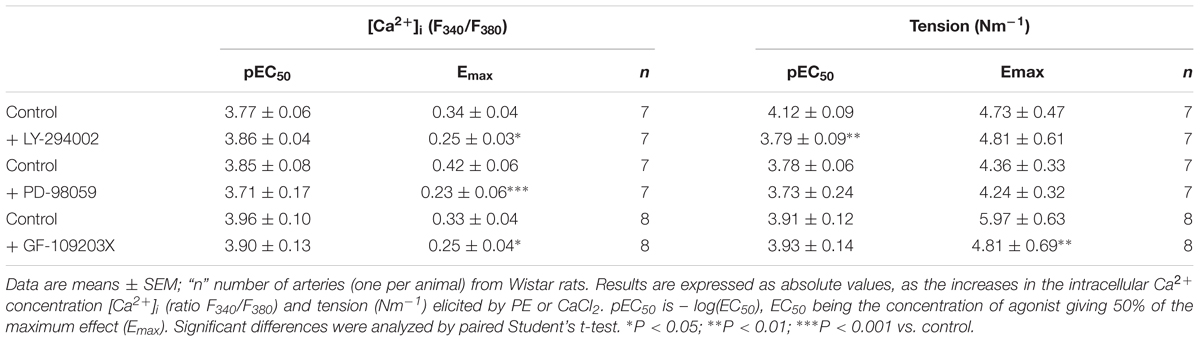
Table 1. Effects of the inhibitors of PI3K LY-294002 (3 μM), ERK-MAPK kinase PD-98059 (3 μM) and PKC GF-109203X (0.1 μM) on the sensitivity and maximal responses of the CaCl2 concentration-response curves in mesenteric arteries stimulated by PE (10 μM) in a Ca2+-free medium.
The effects of the inhibitor of ERK-MAPK PD-98059 on changes in VSM [Ca2+]i and contraction in mesenteric resistance arteries kept in nominally Ca2+-free medium and activated by PE (10 μM) before increasing CaCl2 concentrations were added are shown in Figure 4. Treatment with PD-98059 largely reduced increases in [Ca2+]i elicited by PE upon Ca2+ re-addition (Figure 4A and Table 1) without altering vasoconstriction (Figure 4B and Table 1). PE-induced contractions were not altered either by treatment with the p38MAPK inhibitor SB-203580 (0.3 μM) (Supplementary Figure S1). The relationship [Ca2+]i-contraction for PE was left-shifted upon ERK-MAPK kinase blockade in resistance arteries (Figure 4C), indicating decreased Ca2+ sensitization under conditions of ERK-MAPK blockade and suggesting Ca2+ entry through L-type channels not coupled to vasoconstriction and linked to ERK-MAPK kinase cascade in mesenteric resistance arteries. However, PD-98059 treatment did not affect PE-induced intracellular Ca2+ mobilization and contraction associated to the α1-adrenergic stimulation (Figures 4D,E).
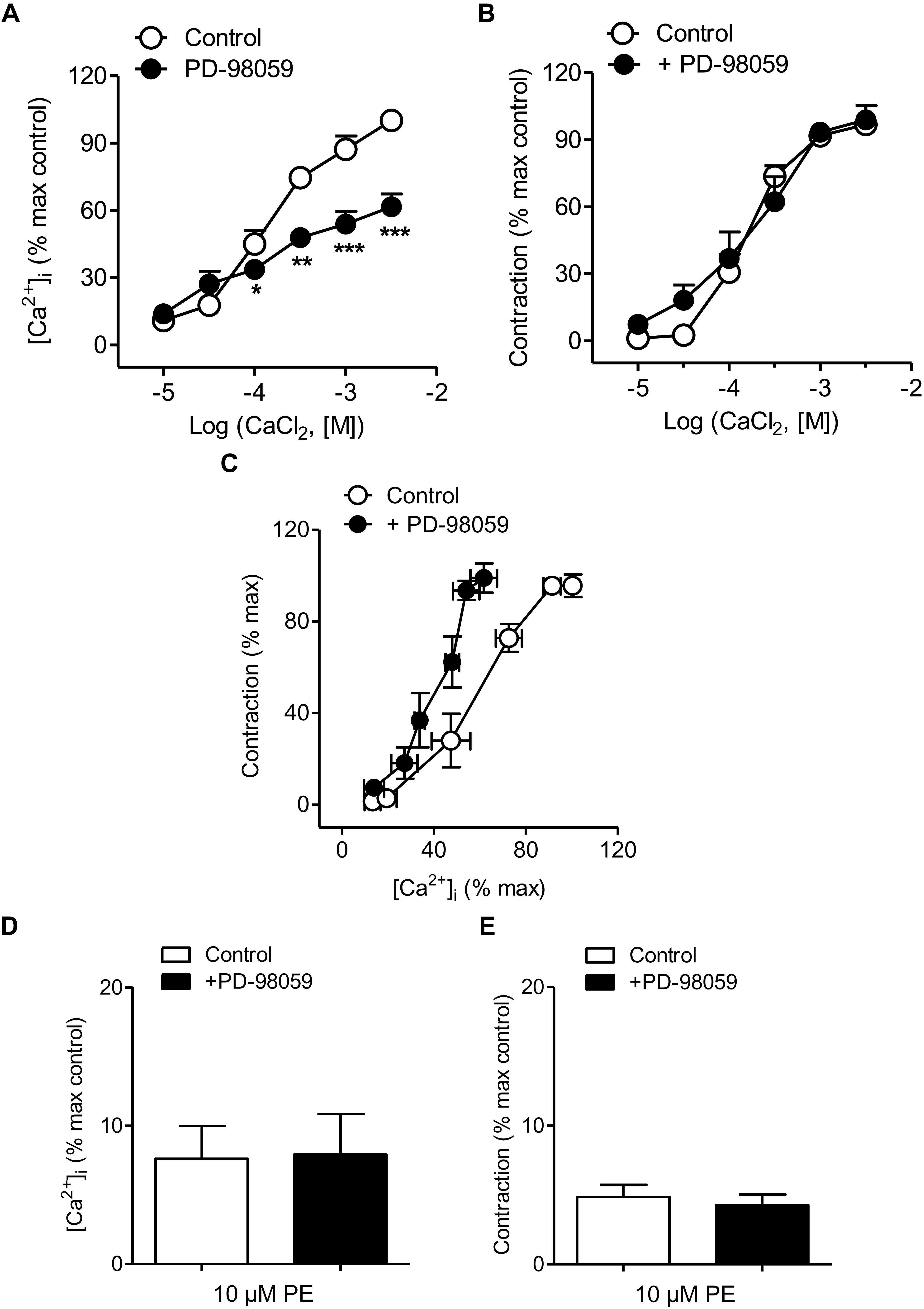
Figure 4. ERK-MAPK inhibition reduced Ca2+ entry but not vasoconstriction and increased Ca2+ sensitization elicited by α1-adrenoceptor activation. (A,B) Average effects of the ERK-MAPK inhibitor PD-98059 (3 μM) on the increases in [Ca2+]i (A) and contraction (B) elicited by cumulative addition of CaCl2 in endothelium-denuded arteries kept in a nominally Ca2+-free medium and stimulated by 10 μM PE. (C) Effect of PD-98059 on the [Ca2+]i-contraction relationship for 10 μM PE in mesenteric resistance arteries and (D,E) on the increases in [Ca2+]i and contraction elicited by 10 μM PE in Ca2+-free PSS. Results are expressed as a percentage of control maximal responses. Values are means ± SEM of n = 7 arteries (one from each animal). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. control.
The PKC inhibitor GF-109203X was used to assess the involvement of PKC in Ca2+ handling coupled to the α1-adrenoceptor in mesenteric resistance arteries. Treatment with GF-109203X reduced the increases in [Ca2+]i and to a minor extent vasoconstriction induced by Ca2+ re-addition in arteries stimulated by PE kept in a nominally Ca2+-free medium (Figures 5A,B and Table 1). Both Ca2+ sensitization (Figure 5C) and PE-induced intracellular Ca2+mobilization and phasic contraction (Figures 5D,E) remained unaffected by PKC inhibition.
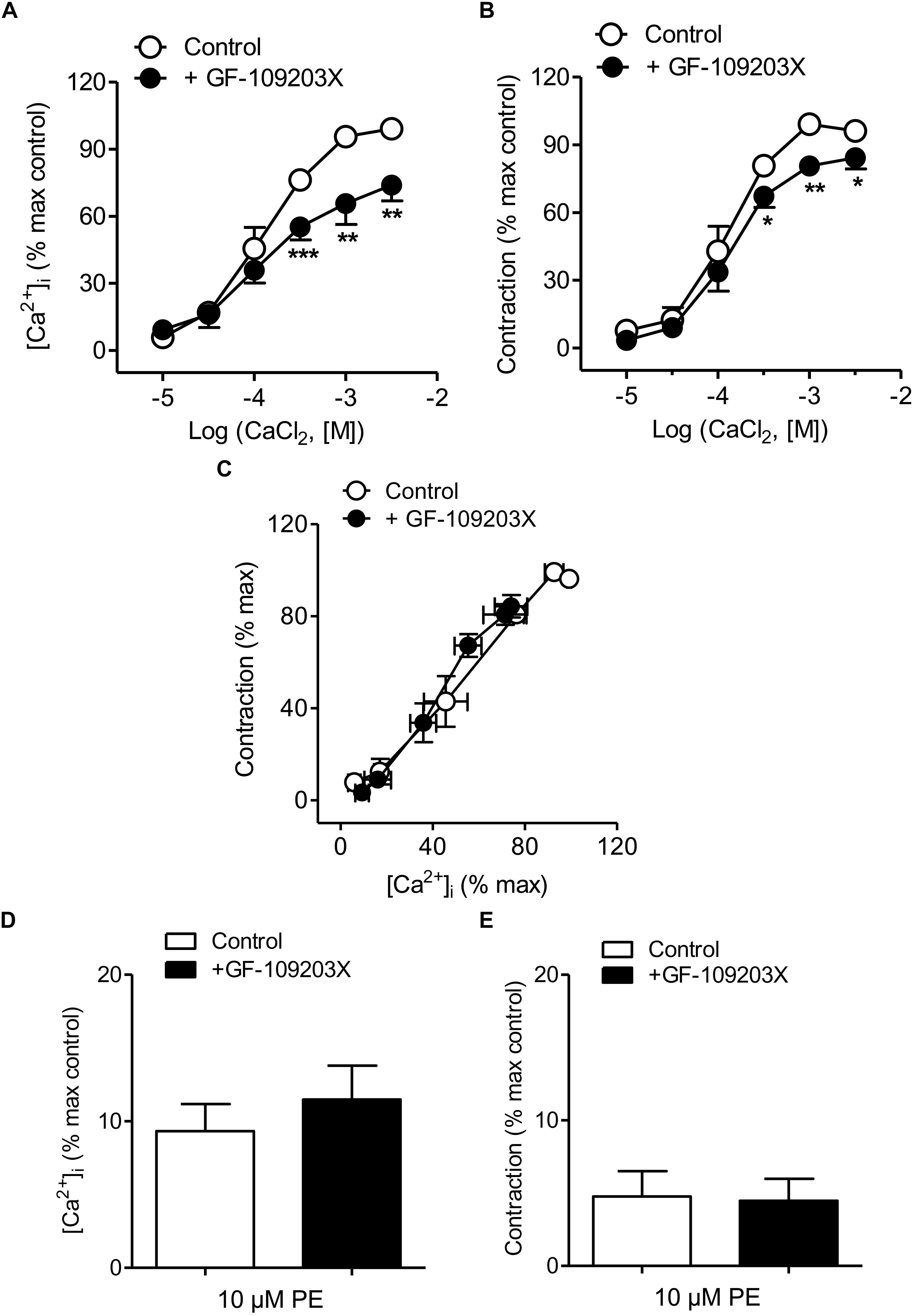
Figure 5. PKC inhibition reduced Ca2+ entry and vasoconstriction without changing Ca2+ sensitization elicited by α1-adrenoceptor activation. (A,B) Average effects of the PKC inhibitor GF-109203X (0.1 μM) on the increases in [Ca2+]i (A) and contraction (B) elicited by cumulative addition of CaCl2 in endothelium-denuded arteries kept in a Ca2+-free medium and stimulated by 10 μM PE. (C) Effect of GF-109203X on the [Ca2+]i-contraction relationships for PE in mesenteric resistance arteries and (D,E) on the increases in [Ca2+]i and contraction elicited by 10 μM PE in in Ca2+-free PSS. Results are expressed as a percentage of control maximal responses. Values are means ± SEM of n = 7 arteries (one from each animal). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. control.
Since [Ca2+]i changes coupled to the α1-adrenoceptor in resistance arteries are largely due to Ca2+ entry through L-type Ca2+ channels, the effects of PD-98059, LY-294002, and GF-109203X were tested on the increase in [Ca2+]i elicited by high K+ depolarization in order to assess whether the modulatory effect of ERK-MAPK, PI3K, and PKC on Ca2+ entry elicited by PE is exerted through L-type Ca2+ channels. Treatment with PD-98059, LY-294002, or GF-109203X reduced Ca2+ entry stimulated by KPSS (Supplementary Figures S2A–C) suggesting that these kinases regulate L-type Ca2+ channel entry in resistance arteries.
RyR-mediated Ca2+-induced Ca2+-release (CICR) upon L-type channel activation has recently been shown in VSM of mesenteric resistance arteries (Sánchez et al., 2018), and therefore we further assessed the potential relationship between PI3K, PKC, and MAPK pathways and RyR-mediated CICR mechanism. Treatment with ryanodine (10 μM) to selectively block the RyR (Meissner, 2017) and the subsequent SR Ca2+ mobilization and amplification of Ca2+ entry through L-type channels, reduced nifedipine-sensitive increases in [Ca2+]i and contraction elicited by Ca2+ readdition in high K+-depolarized endothelium-denuded arteries (Figures 6A,B), thus confirming CICR upon L-type channel activation. On the other hand, in ryanodine-treated arteries to block CICR, selective inhibition of MAPK with PD-98059 (Figures 6C,D), PI3K with LY-294002 (Figure 6E) or PKC with GF-109203X (Figure 6F) resulted in a further significant reduction of the increases in [Ca2+]i but not contraction elicited by Ca2+ re-addition in high K+-depolarized arteries, thus demonstrating a direct stimulatory effect of MAPK, PI3K, and PKC on L-type Ca2+ entry independent of RyR mechanisms.
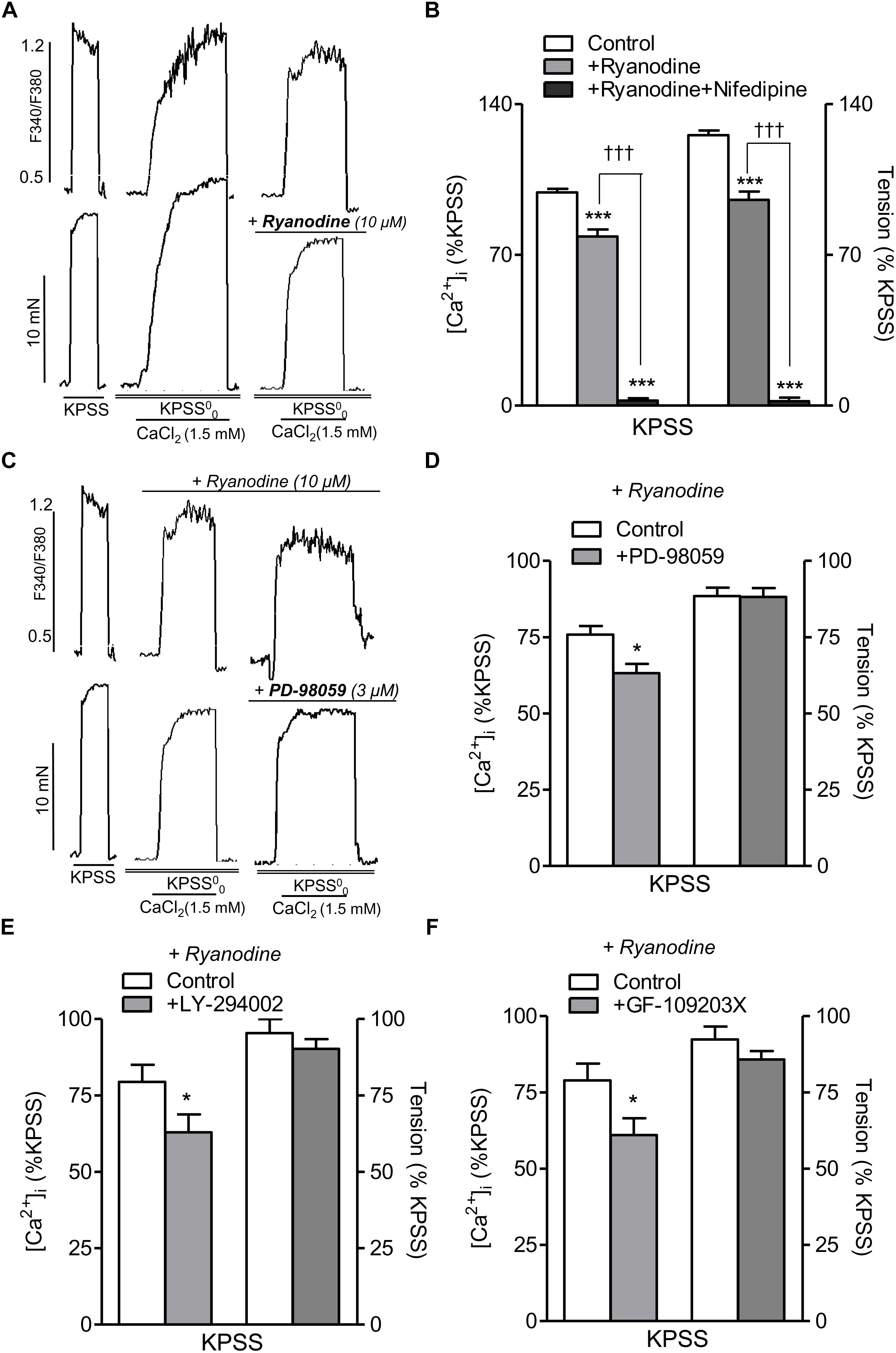
Figure 6. Inhibition of ERK-MAPK, PI3K, or PKC kinase reduces intracelular Ca2+ mobilization elicited by voltage-dependent L-type Ca2+channel activation independently of RyR-mediated Ca2+ release. (A) Representative traces showing the effects of ryanodine (10 μM) treatment on changes in both [Ca2+]i (top) and vasoconstriction (bottom) induced by CaCl2 (1.5 mM) re-addition in endothelium-denuded mesenteric arteries depolarized with high K+ in Ca2+ free medium (KPSS00). (B) Summarized data showing the average effects of ryanodine (10 μM) on changes in [Ca2+]i and contraction elicited by CaCl2 (1.5 mM) re-addition in arteries depolarized with a high K+ solution. (C) Representative traces showing the effects of inhibition of MAPK with PD-98059 (3 μM) on changes in both [Ca2+]i (top) and vasoconstriction (bottom) induced by CaCl2 (1.5 mM) re-addition in endothelium-denuded mesenteric arteries depolarized with high K+ (KPSS00) and treated with ryanodine (30 μM) to block RyR. (D) Summarized data showing the average effects of PD-98059 (3 μM) on changes in [Ca2+]i and vasoconstriction elicited by CaCl2 (1.5 mM) re-addition in arteries depolarized with a high K+ solution under conditions of RyR blockade with ryanodine. Average effects of the PI3K inhibitor LY-294002 (3 μM) (E) or the PKC inhibitor GF-109203X (0.1 μM) (F) on changes in [Ca2+]i and vasoconstriction elicited by CaCl2 (1.5 mM) re-addition in arteries depolarized with a high K+ solution under conditions of RyR blockade. Responses are expressed as absolute values of either [Ca2+]i (ΔF340/F380) or tension (Nm-1) (A,C) or relative to those elicited by KPSS (B,D–F). Values are means ± SEM of n = 5 arteries (one from each animal). Significant differences were analyzed by paired Student’s t-test or one-way ANOVA. ∗P < 0.05 vs. control. ∗∗∗P < 0.001 vs. control. †††P < 0.001 vs. ryanodine-treated.
An increasing body of experimental evidence during the last decade gives support to a key role for protein kinase-mediated regulation of Ca2+ handling in arterial and cardiac myocytes, through phosphorylation of channels involved in either Ca2+ entry through plasma membrane or Ca2+ release from SR intracellular stores. The present study provides new insights into the modulation of Ca2+ handling by PI3K, MAPK, and PKC signaling pathways coupled to the α1-adrenoceptor in resistance arteries and linked to both contractile and non-contractile functions. A graphical summary of our findings is depicted in Figure 7.
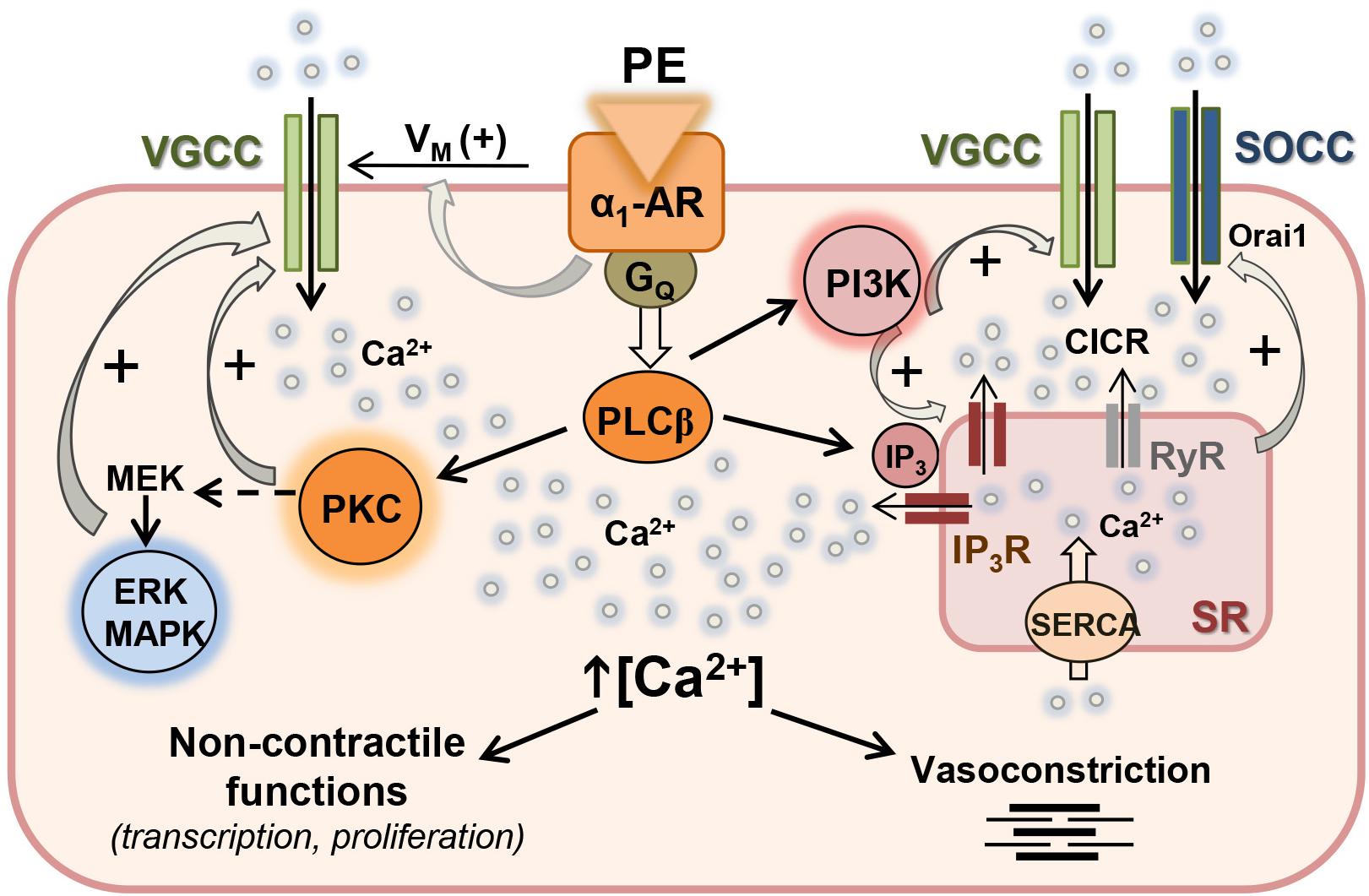
Figure 7. Proposed mechanism for the regulation of Ca2+ handling by PI3K, MAPK, and PKC in resistance arteries VSM. Activation of α1-adrenoceptor (α1-AR) with phenylephrine (PE) is coupled to membrane depolarization (+Vm), stimulation of Ca2+ influx through L-type voltage-gated Ca2+ channels (VGCC) and vasoconstriction. α1-AR is also coupled to PLCβ activation which stimulates Ca2+ release from sarcoplasmic reticulum (RS) Ca2+ store through IP3 receptors (IP3R), and also stimulates PKC which in turn activates the MEK-ERK MAPK intracellular signaling cascade. Both PKC and ERK MAPK can directly act on L-type VGCC and reinforce Ca2+ influx mostly related to non-contractile functions. α1-AR is also coupled to PI3K activation and stimulation of both IP3R-mediated Ca2+ release and Ca2+ entry through VGCC. Ca2+ influx through VGCC amplifies Ca2+ signal through the ryanodine receptor (RyR)-mediated Ca2+-induced Ca2+ release (CICR). Emptying of the SR Ca2+ store leads to store-operated Ca2+ channel (SOCC) entry to refill empty store.
Hypertension and augmented vasoconstriction associated to metabolic disease have traditionally been ascribed to protein kinase-mediated Ca2+ sensitization of the VSM contractile machinery leading to increased vascular tone and systemic vascular resistance (Martínez et al., 2000; Naik et al., 2006; Villalba et al., 2007, 2011; Crestani et al., 2017). However, Ca2+ signaling mechanisms and the relative contribution of Ca2+ sensitization and Ca2+ mobilization mechanisms to arterial contraction are size-dependent. Thus, sensitivity of vasoconstriction and [Ca2+]i changes induced by α1-adrenoceptor activation to pharmacological inhibitors of voltage-dependent L-type Ca2+ entry is higher in resistance arteries compared to large conductance arteries (Prieto et al., 1991; Kitazawa and Kitazawa, 2012; Martinsen et al., 2012). Moreover, contractions of large arteries have been reported to involve kinases such as RhoK and PKC to varying degrees, while vasoconstriction of resistance arteries seem to be mediated exclusively by PKC (Budzyn et al., 2006; Kitazawa and Kitazawa, 2012). The present results confirm early studies demonstrating the involvement of Ca2+ sensitization mechanisms in the noradrenergic vasoconstriction of small mesenteric arteries (Buus et al., 1998; Kitazawa and Kitazawa, 2012), but further demonstrate that PI3K, MAPK, and PKC are involved in the regulation of Ca2+ entry and intracellular Ca2+ mobilization coupled to α1-adrenoceptor activation in resistance arteries.
The PI3K/Akt signaling pathway is stimulated upon activation of the insulin receptor in the vascular endothelium and coupled to eNOS phosphorylation, NO production and vasodilatation. This pathway is impaired and associated to endothelial dysfunction in insulin resistant states (Kim et al., 2006; Contreras et al., 2010). However, PI3Ks play also a main role in the Ca2+ signaling of neurons and cardiac and vascular myocytes (Viard et al., 2004; Ghigo et al., 2017). In VSM, PI3Ks have been involved in the regulation of L-type Ca2+ entry in response to vasoconstrictors and growth factors (Macrez et al., 2001; Le Blanc et al., 2004) but also in the Ca2+-dependent Rho-mediated negative control of MLCP linked to Ca2+ sensitization and contraction (Wang et al., 2006). In the present study, pharmacological inhibition of PI3K moderately reduced both Ca2+ entry and vasoconstriction elicited by PE in resistance arteries, without affecting the relationship Ca2+-tension for the α1-adrenergic agonist indicative of changes in Ca2+ sensitization. These findings are consistent with earlier studies involving PI3K activation in the transduction pathways for the angiotensin II (AII)-induced Ca2+ responses and contraction in VSM (Le Blanc et al., 2004; Rakotoarisoa et al., 2006). In vascular myocytes, PI3Kγ isoform was initially shown to mediate AII-induced activation of L-type voltage-dependent Ca2+ currents, to increase [Ca2+]i and elicit contraction (Quignard et al., 2001; Le Blanc et al., 2004; Rakotoarisoa et al., 2006). This is confirmed in the endothelium-denuded intact resistance arteries of the present study by the inhibitory effect of the PI3K blocker LY294002 on L-type Ca2+ entry elicited by high K+ depolarization. PI3Kγ/Akt phosphorylates Cavβ2 and induces Cavα1C translocation thus increasing L-type Ca2+ currents in arterial myocytes, and pharmacological inhibition PI3Kγ has vasodilator effects and reduces arterial blood pressure (Carnevale and Lembo, 2012). However, our results further demonstrate that PI3K inhibition markedly reduced intracellular Ca2+ release and phasic contraction induced by PE in resistance arteries, which suggests that PI3K might also activate voltage-independent store-operated Ca2+ entry through stimulation of intracellular Ca2+ release. This pathway is linked to the α1-adrenoceptor in resistance arteries, as depicted by the inhibitory effect on Ca2+ entry and contraction induced by SR depletion by blockade of SERCA with CPA, or by treatment with the store-operated Ca2+ entry blocker Pyr6 (Santiago et al., 2015).
α1-Adrenoceptor stimulation causes intracellular Ca2+ mobilization via IP3 receptor-mediated Ca2+ release from SR stores (Liu and Khalil, 2018). Therefore, it seems likely that the reduction of PE-induced intracellular Ca2+ mobilization by the PI3K blocker LY294002 to be due to the inhibition of PI3K-stimulated IP3-dependent intracellular Ca2+ release, as earlier reported for the IGF-1 receptor linked to a G protein-PI3K-phospholipase C signaling pathway in cardiac myocytes (Ibarra et al., 2004). However, we have recently demonstrated RyR-mediated CICR upon activation of L-type channels as a potent amplifying mechanism of Ca2+ entry and contraction in mesenteric resistance arteries (Sánchez et al., 2018). Since RyR1 and RyR2 can be phosphorylated by kinases such as PKA leading to SR Ca2+ leak (Bovo et al., 2017; Meissner, 2017), we further investigated whether PI3K may regulate RyR-mediated CICR stimulated by L-type Ca2+ channel activation. Under conditions of RyR blockade with ryanodine, selective PI3K inhibition with LY294002 significantly reduced increases [Ca2+]i but not contraction elicited by high K+ depolarization. Although RyR phosphorylation cannot be ruled out, the present finding suggest that PI3K activation mainly stimulates Ca2+ entry through direct regulation of L-type Ca2+ channels in resistance arteries, as discussed above and reported for isolated vascular myocytes (Le Blanc et al., 2004).
Enhanced activity of PI3K and up-regulation of PI3Kδ have been found to be associated to augmented L-type Ca2+ entry in arterial myocytes from rat models of type I diabetes (Pinho et al., 2010) and insulin resistance (Sánchez et al., 2018). While in the former PI3K contributed to enhanced vasoconstriction, in the latter PI3K-mediated increased Ca2+ entry compensated for SR Ca2+ store dysfunction. In the heart, PI3Kα/Akt signaling is involved in insulin inotropic actions and activates Ca2+ currents in microdomains containing L-type Ca2+ channels (Lu et al., 2009). This pathway and the corresponding L-type Ca2+ entry is defective in insulin-deficient and resistant states which might contribute to the cardiac contractile dysfunction in diabetic cardiomyopathy (Lu et al., 2007; Ghigo et al., 2017).
On the other hand, ERK1/2 MAPK pathway is involved in insulin mitogenic actions but has also been associated to insulin-mediated enhanced vasoconstriction in insulin resistant states (Kim et al., 2006; Contreras et al., 2010; Prieto et al., 2013). In the present study, the selective inhibitor of ERK-MAPK PD98059 did not alter the initial rapid Ca2+ increase in response to PE corresponding to intracellular Ca2+ mobilization, but caused a profound inhibition of the sustained Ca2+ entry stimulated by PE without altering the associated contraction in resistance arteries. These findings differ from earlier studies involving ERK-MAPK in both Ca2+-dependent vasoconstriction coupled to the AII receptor in human resistance arteries (Touyz et al., 1999), and in Ca2+ sensitization-mediated contraction of coronary artery myocytes linked to the ET1 receptor (Cain et al., 2002). Moreover, the present results differ from reports involving the p38 and JNK limbs of the MAPK pathway in nifedipine-sensitive vasoconstriction linked to α-adrenoceptors in VSM of large arteries (Ok et al., 2011). In contrast, our results suggest a major role of MAPK in the regulation of L-type Ca2+ entry coupled to the α1-adrenergic adrenoceptor in resistance arteries, supported by the marked reduction elicited by PD98059 on the high K+ depolarization-induced Ca2+ entry under conditions of RyR receptor blockade. These findings are in agreement with reports showing that ERK MAPK phosphorylates Cav1.2 channels and enhances L-type currents in cardiac myocytes in response to growth factors involved in cardiac hypertrophy (Takahashi et al., 2004). Differences in the involvement of the ERK-MAPK in Ca2+ handling and contraction in vascular myocytes may be ascribed to the differential activation of this pathway by various receptors. Our results suggest that ERK-MAPK-mediated modulation of Ca2+ handling might be related to VSM non-contractile proliferative pathways coupled to the α1-adrenoceptor in resistance arteries (Kudryavtseva et al., 2013). Further studies are needed to elucidate the role of receptor-coupled MAPK pathways as modulators of both L-type and non- L-type Ca2+ influx involved in cell proliferation.
In cardiac myocytes, ERK MAPK phosphorylation of Cav1.2 channels and increased L-type currents are activated upstream by PKC linked to activation of G protein-coupled receptors (Smani et al., 2010). The role of PKC in arterial Ca2+ handling and vasoconstriction is well-documented, although this kinase has mostly been involved in Ca2+ sensitization mechanisms coupled to the α1-adrenoceptor in resistance arteries (Buus et al., 1998; Villalba et al., 2007; Kitazawa and Kitazawa, 2012). PKC phosphorylates the phosphoprotein CPI-17, a potent inhibitor of MLCP, rendering it inactive and therefore promoting increased vascular tone (Somlyo and Somlyo, 2003). However, in our study, the PKC inhibitor GF-109203X did not alter the initial Ca2+ rise but markedly reduced the sustained [Ca2+]i increase in response to PE, and to a lesser extent the simultaneous vasoconstriction of mesenteric resistance arteries, which suggests a major role for PKC in the regulation of Ca2+ entry rather than in the enhancement of myofilament Ca2+ sensitivity of arterial myocytes, as also supported by the unchanged Ca2+-tension relationship for the α1-adrenergic agonist in presence of the PKC inhibitor. Involvement of PKC in Ca2+ entry in resistance arteries partially agrees with that recently reported by Kitazawa and Kitazawa (2012). However, these authors showed that GF-109203X induced a small reduction of PE-induced Ca2+ entry, while it abolished vasoconstriction and reduced phosphorylation of MLC, CPI-17 and MYPT1, supporting a major involvement of PKC in Ca2+ sensitization. The discrepancies between our data showing a minor inhibitory effect of GF-109203X on Ca2+ sensitization coupled to PE vasoconstriction and those in the study by Kitazawa and Kitazawa (2012) might be ascribed to the fact that different PKC isoforms mediate Ca2+ sensitization and Ca2+ entry pathways in arterial smooth muscle (Liu and Khalil, 2018). Thus, the conventional Ca2+-dependent PKC isoforms (α, -β1, -β2, and -γ) are activated by cytosolic Ca2+ and diacylglycerol, and in turn phosphorylate a wide array of substrates implicated in the regulation of Ca2+ fluxes; specifically, PKCα has been involved in the regulation of L-type Ca2+ entry in both vascular and cardiac myocytes (Yang et al., 2009; Gulia et al., 2013). In contrast, the novel PKC isoforms appear to mediate vasoconstriction coupled to Ca2+ sensitizing pathways (Liu and Khalil, 2018). GF-109203X is selective for the conventional PKCα and β1 isoforms but it may also inhibit the novel δ and ε PKC isoforms at the higher micromolar range used in the study of Kitazawa and Kitazawa (2012) and Liu and Khalil (2018).
In the present study, involvement of PKC in the sustained Ca2+ entry induced by PE and sensitive to L-type Ca2+ channel blockade is further supported by the marked inhibitory effect found for GF-109203X on the L-type channel-mediated Ca2+ entry elicited by high K+ depolarization, thus suggesting that PKC modulates voltage-dependent L-type Ca2+ entry coupled to the α1-adrenergic vasoconstriction in resistance arteries, and supporting that PKC-mediated modulation of L-type channels in arterial myocytes (Gulia et al., 2013) contributes not only to myogenic tone but also to agonist-induced vasoconstriction (Cobine et al., 2007; Potts et al., 2012). Activity of both conventional and novel PKC isoforms is chronically enhanced by hyperglycemia, lipotoxicity and oxidative stress, which has been associated to the cardiovascular complications in the insulin resistant states (Geraldes and King, 2010; Turban and Hajduch, 2011). Abnormal PKC activity contributes to the augmented arterial L-type Ca2+ entry and enhanced vasoconstriction in diabetic arteries (Ungvari et al., 1999) and arteries from genetically obese rats (Sánchez et al., 2018), and in the latter compensates for SR Ca2+ store dysfunction.
The present findings demonstrate that PI3K, MAPK, and PKC are involved in the regulation of Ca2+ entry and intracellular Ca2+ mobilization coupled to α1-adrenoceptor activation in resistance arteries and mostly associated to non-contractile functions of VSM. Under conditions of vascular disease, vascular myocytes change from a contractile to a synthetic phenotype that is able to proliferate and migrate. The present results showing a protein kinase-mediated regulation of Ca2+ handling linked to the α1-adrenoceptor in resistance arteries suggest that changes in PI3K, MAPK, and PKC signaling pathways involving enhanced Ca2+ mobilization not coupled to contraction, might participate in the changes toward a VSM proliferative phenotype and be involved in vascular remodeling in hypertension and other insulin resistant states. Further studies are needed to elucidate this issue.
DP conceived, designed, and discussed the experiments. AG and AS performed the experiments and analyzed the data. AG, AS, and DP wrote the manuscript. AG, CC, and AS contributed to discussion, and edited the manuscript.
This work was supported by grants SAF2012-31631 and SAF2016-77526R from MINECO (Spain) cofounded by the FEDER Program of EU.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Manuel Perales and Francisco Puente are thanked for expert technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.00055/full#supplementary-material
FIGURE S1 | p38MAPK inhibition does not alter PE-induced vasososntriction. Average effects of the p38MAPK inhibitor SB-203580 (0.3 μM) on the contractions elicited by cumulative addition of CaCl2 in endothelium-denuded arteries kept in a nominally Ca2+-free medium and stimulated by 10 μM PE. Results are expressed as a percentage of control maximal responses. Values means ± SEM of n = 6 arteries (two from each animal).
FIGURE S2 | ERK-MAPK, PI3K, or PKC kinase inhibitors reduce Ca2+ entry through voltage-dependent L-type channels. Average inhibitory effects ERK-MAPK inhibitor PD-98059 (3 μM) (A), the PI3K inhibitor LY-294002 (3 μM) (B) or the PKC inhibitor GF-109203X (0.1 μM) (C), on the increases in [Ca2+]i elicited by depolarization high K+ (KPSS). Results are expressed as absolute values of [Ca2+]i (ΔF340/F380). Values are means ± SEM of five arteries (one from each animal). Significant differences were analyzed by paired Student’s t-test; ∗P < 0.05 vs. control.
Bovo, E., Huke, S., Blatter, L. A., and Zima, A. V. (2017). The effect of PKA-mediated phosphorylation of ryanodine receptor on SR Ca2+ leak in ventricular myocytes. J. Mol. Cell. Cardiol. 104, 9–16. doi: 10.1016/j.yjmcc.2017.01.015
Budzyn, K., Paull, M., Marley, P. D., and Sobey, C. G. (2006). Segmental differences in the roles of rho-kinase and protein kinase C in mediating vasoconstriction. J. Pharmacol. Exp. Ther. 317, 791–796. doi: 10.1124/jpet.105.100040
Buus, C. L., Aalkjaer, C., Nilsson, H., Juul, B., Moller, J. V., and Mulvany, M. J. (1998). Mechanisms of Ca2+ sensitization of force production by noradrenaline in rat mesenteric small arteries. J. Physiol. 510, 577–590. doi: 10.1111/j.1469-7793.1998.577bk.x
Cain, A. E., Tanner, D. M., and Khalil, R. A. (2002). Endothelin-1–induced enhancement of coronary smooth muscle contraction via MAPK-dependent and MAPK-independent [Ca(2+)](i) sensitization pathways. Hypertension 39, 543–549. doi: 10.1161/hy0202.103129
Carnevale, D., and Lembo, G. (2012). PI3Kγ in hypertension: a novel therapeutic target controlling vascular myogenic tone and target organ damage. Cardiovasc. Res. 95, 403–408. doi: 10.1093/cvr/cvs166
Cobine, C. A., Callaghan, B. P., and Keef, K. D. (2007). Role of L-type calcium channels and PKC in active tone development in rabbit coronary artery. Am. J. Physiol. Heart Circ. Physiol. 292, H3079–H3088. doi: 10.1152/ajpheart.01261.2006
Contreras, C., Sánchez, A., Martínez, P., Raposo, R., Climent, B., García-Sacristán, A., et al. (2010). Insulin resistance in penile arteries from a rat model of metabolic syndrome. Br. J. Pharmacol. 161, 350–364. doi: 10.1111/j.1476-5381.2010.00825.x
Crestani, S., Webb, R. C., and da Silva-Santos, J. E. (2017). High-salt intake augments the activity of the RhoA/rock pathway and reduces intracellular calcium in arteries from rats. Am. J. Hypertens 30, 389–399. doi: 10.1093/ajh/hpw201
Ford, E. S. (2005). Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care 28, 1769–1778. doi: 10.2337/diacare.28.7.1769
Ford, E. S., Zhao, G., and Li, C. (2010). Pre-diabetes and the risk for cardiovascular disease: a systematic review of the evidence. J. Am. Cell. Cardiol. 55, 1310–1317. doi: 10.1016/j.jacc.2009.10.060
Geraldes, P., and King, G. L. (2010). Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 106, 1319–1331. doi: 10.1161/CIRCRESAHA.110.217117
Ghigo, A., Laffargue, M., Li, M., and Hirsch, E. (2017). PI3K and calcium signaling in cardiovascular disease. Circ. Res. 121, 282–292. doi: 10.1161/CIRCRESAHA.117.310183
Ghisdal, P., Vandenberg, G., and Morel, N. (2003). Rho-dependent kinase is involved in agonist-activated calcium entry in rat arteries. J. Physiol. 551, 855–867. doi: 10.1113/jphysiol.2003.047050
Gulia, J., Navedo, M. F., Gui, P., Chao, J. T., Mercado, J. L., Santana, L. F., et al. (2013). Regulation of L-type calcium channel sparklet activity by c-Src and PKC-α. Am. J. Physiol. Cell. Physiol. 305, C568–C577. doi: 10.1152/ajpcell.00381.2011
Ibarra, C., Estrada, M., Carrasco, L., Chiong, M., Liberona, J. L., Cardenas, C., et al. (2004). Insulin-like growth factor-1 induces an inositol 1,4,5-trisphosphate-dependent increase in nuclear and cytosolic calcium in cultured rat cardiac myocytes. J. Biol. Chem. 27, 7554–7565. doi: 10.1074/jbc.M311604200
Kim, J. A., Montagnani, M., Koh, K. K., and Kuon, M. J. (2006). Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 113, 1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213
Kitazawa, T., and Kitazawa, K. (2012). Size-dependent heterogeneity of contractile Ca2+ sensitization in rat arterial smooth muscle. J. Physiol. 590, 5401–5423. doi: 10.1113/jphysiol.2012.241315
Kudryavtseva, O., Aalkjaer, C., and Matchkov, V. V. (2013). Vascular smooth muscle cell phenotype is defined by Ca2+-dependent transcription factors. FEBS J. 280, 5488–5499. doi: 10.1111/febs.12414
Le Blanc, C., Mironneau, C., Barbot, C., Henaff, M., Bondeva, T., Wetzker, R., et al. (2004). Regulation of vascular L-type Ca2+ channels by phosphatidylinositol 3,4,5-trisphosphate. Circ. Res. 95, 300–307. doi: 10.1161/01.RES.0000138017.76125.8b
Liu, Z., and Khalil, R. A. (2018). Evolving mechanisms of vascular smooth muscle contraction highlight key targets in vascular disease. Biochem. Pharmacol. 153, 91–122. doi: 10.1016/j.bcp.2018.02.012
Lu, Z., Jiang, Y. P., Wang, W., Xu, X. H., Mathias, R. T., Entcheva, E., et al. (2009). Loss of cardiac phosphoinositide 3-kinase p110 alpha results in contractile dysfunction. Circulation 120, 318–325. doi: 10.1161/CIRCULATIONAHA.109.873380
Lu, Z., Jiang, Y. P., Xu, X. H., Ballou, L. M., Cohen, I. S., and Lin, R. Z. (2007). Decreased L-type Ca2+ current in cardiac myocytes of type 1 diabetic akita mice due to reduced phosphatidylinositol 3-kinase signaling. Diabetes 56, 2780–2789. doi: 10.2337/db06-1629
Macrez, N., Mironneau, C., Carricaburu, V., Quignard, J. F., Babich, A., Czupalla, C., et al. (2001). Phosphoinositide 3-kinase isoforms selectively couple receptors to vascular L-type Ca(2+) channels. Circ. Res. 89, 692–699. doi: 10.1161/hh2001.097864
Martínez, M. C., Randriamboavonjy, V., Ohlmann, P., Komas, N., Duarte, J., Schneider, F., et al. (2000). Involvement of protein kinase C, tyrosine kinases, and Rho kinase in Ca2+ handling of human small arteries. Am. J. Physiol. Heart Circ. Physiol. 279, H1228–H1238. doi: 10.1152/ajpheart.2000.279.3.H1228
Martinsen, A., Yerna, X., Rath, G., Gomez, E. L., Dessy, C., and Morel, N. (2012). Different effect of Rho kinase inhibition on calcium signaling in rat isolated large and small arteries. J. Vasc. Res. 49, 522–533. doi: 10.1159/000341230
Meissner, G. (2017). The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 149, 1065–1089. doi: 10.1085/jgp.201711878
Mulvany, M. J., and Halpern, W. (1977). Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 41, 19–26. doi: 10.1161/01.RES.41.1.19
Naik, J. S., Xiang, L., and Hester, R. L. (2006). Enhanced role for RhoA-associated kinase in adrenergic-mediated vasoconstriction in gracilis arteries from obese Zucker rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R154–R161. doi: 10.1152/ajpregu.00245.2005
Nobe, K., and Paul, R. J. (2001). Distinct pathways of Ca(2+) sensitization in porcine coronary artery: effects of Rho-related kinase and protein kinase C inhibition on force and intracellular Ca(2+). Circ. Res. 88, 1283–1290. doi: 10.1161/hh1201.092035
Ok, S. H., Jeong, Y. S., Kim, J. G., Lee, S. M., Sung, H. J., Kim, H. J., et al. (2011). c-Jun NH2-terminal kinase contributes to dexmedetomidine-induced contraction in isolated rat aortic smooth muscle. Yonsei. Med. J. 52, 420–428. doi: 10.3349/ymj.2011.52.3.420
Okon, E. B., Chung, A. W., Rauniyar, P., Padilla, E., Tejerina, T., McManus, B. M., et al. (2005). Compromised arterial function in human type 2 diabetic patient. Diabetes 54, 2415–2423. doi: 10.2337/diabetes.54.8.2415
Pinho, J. F., Medeiros, M. A., Capettini, L. S., Rezende, B. A., Campos, P. P., Andrade, S. P., et al. (2010). Phosphatidylinositol 3-kinase-δ up-regulates L-type Ca2+ currents and increases vascular contractility in a mouse model of type 1 diabetes. Br. J. Pharmacol. 161, 1458–1471. doi: 10.1111/j.1476-5381.2010.00955.x
Potts, L. B., Ren, Y., Lu, G., Kuo, E., Ngo, E., Kuo, L., et al. (2012). Constriction of retinal arterioles to endothelin-1: requisite role of rho kinase independent of protein kinase C and L-type calcium channels. Invest. Ophthalmol. Visc. Sci. 53, 2904–2912. doi: 10.1167/iovs.12-9542
Prieto, D., Contreras, C., and Sánchez, A. (2013). Endothelial dysfunction, obesity and insulin resistance. Curr. Vasc. Pharmacol. 11, 412–426.
Prieto, D., Mulvany, M. J., and Nyborg, N. C. (1991). (+)-S-12967 and (-)-S-12968: 1,4-dihydropyridine stereoisomers with calcium channel agonistic and antagonistic properties in rat resistance arteries. Br. J. Pharmacol. 103, 1703–1708. doi: 10.1111/j.1476-5381.1991.tb09850.x
Quignard, J. F., Mironneau, J., Carricaburu, V., Fournier, B., Babich, A., Nurnberg, B., et al. (2001). Phosphoinositide 3-kinase gamma mediates angiotensin II-induced stimulation of L-type calcium channels in vascular myocytes. J. Biol. Chem. 276, 32545–32551. doi: 10.1074/jbc.M102582200
Rakotoarisoa, L., Carricaburu, V., Leblanc, C., Mironneau, C., Mironneau, J., and Macrez, N. (2006). Angiotensin II-induced delayed stimulation of phospholipase C gamma1 requires activation of both phosphatidylinositol 3-kinase gamma and tyrosine kinase in vascular myocytes. J. Cell. Mol. Med. 10, 734–748.
Sánchez, A., Contreras, C., Climent, B., Gutiérrez, A., Muñoz, M., García-Sacristán, A., et al. (2018). Impaired Ca2+ handling in resistance arteries from genetically obese zucker rats: role of the PI3K, ERK1/2 and PKC signaling pathways. Biochem. Pharmacol. 152, 114–128. doi: 10.1016/j.bcp.2018.03.020
Santiago, E., Climent, B., Muñoz, M., García-Sacristán, A., Rivera, L., and Prieto, D. (2015). Hydrogen peroxide activates store-operated Ca(2+) entry in coronary arteries. Br. J. Pharmacol. 172, 5318–5332. doi: 10.1111/bph.13322
Smani, T., Calderón-Sanchez, E., Gómez-Hurtado, N., Fernández-Velasco, M., Cachofeiro, V., Lahera, V., et al. (2010). Mechanisms underlying the activation of L-type calcium channels by urocortin in rat ventricular myocytes. Cardiovasc Res. 87, 459–466. doi: 10.1093/cvr/cvq063
Somlyo, A. P., and Somlyo, A. V. (2003). Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358. doi: 10.1152/physrev.00023.2003
Takahashi, E., Fukuda, K., Miyoshi, S., Murata, M., Kato, T., Ita, M., et al. (2004). Leukemia inhibitory factor activates cardiac L-Type Ca2+ Channels via Phosphorylation of Serine 1829 in the rabbit cav1.2 Subunit. Circ. Res. 94, 1242–1248. doi: 10.1161/01.RES.0000126405.38858.BC
Touyz, R. M., El Mabrouk, M., He, G., Wu, X. H., and Schiffrin, E. L. (1999). Mitogen-activated protein/extracellular signal-regulated kinase inhibition attenuates angiotensin II-mediated signaling and contraction in spontaneously hypertensive rat vascular smooth muscle cells. Circ. Res. 84, 505–515. doi: 10.1161/01.RES.84.5.505
Turban, S., and Hajduch, E. (2011). Protein kinase C isoforms: mediators of reactive lipid metabolites in the development of insulin resistance. FEBS Lett. 585, 269–274. doi: 10.1016/j.febslet.2010.12.022
Ungvari, Z., Pacher, P., Kecskemeti, V., Papp, G., Szollár, L., and Koller, A. (1999). Increased myogenic tone in skeletal muscle arterioles of diabetic rats. Possible role of increased activity of smooth muscle Ca2+ channels and protein kinase, C. Cardiovasc. Res. 43, 1018–1028. doi: 10.1016/S0008-6363(99)00106-6
Viard, P., Butcher, A. J., Halet, G., Davies, A., Nürnberg, B., Heblich, F., et al. (2004). PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat. Neurosci. 7, 939–946. doi: 10.1038/nn1300
Villalba, N., Contreras, C., Hernández, M., García-Sacristán, A., and Prieto, D. (2011). Impaired Ca2+ handling in penile arteries from prediabetic zucker rats: involvement of rho kinase. Am. J. Physiol. Heart Circ. Physiol. 300, H2044–H2053. doi: 10.1152/ajpheart.01204.2010
Villalba, N., Stankevicius, E., Garcia-Sacristán, A., Simonsen, U., and Prieto, D. (2007). Contribution of both Ca2+ entry and Ca2+ sensitization to the alpha1-adrenergic vasoconstriction of rat penile small arteries. Am. J. Physiol. Heart Circ. Physiol. 292, H1157–H1169. doi: 10.1152/ajpheart.01034.2006
Villalba, N., Stankevicius, E., Simonsen, U., and Prieto, D. (2008). Rho kinase is involved in Ca2+ entry of rat penile small arteries. Am. J. Physiol. Heart Circ. Physiol. 294, H1923–H1932. doi: 10.1152/ajpheart.01221.2007
Wang, Y., Yoshioka, K., Azam, M. A., Takuwa, N., Sakurada, S., Kayaba, Y., et al. (2006). Class II phosphoinositide 3-kinase alpha-isoform regulates Rho, myosin phosphatase and contraction in vascular smooth muscle. Biochem. J. 394, 581–592. doi: 10.1042/BJ20051471
Keywords: ERK-MAPK, PI3K, PKC, L-type Ca2+ channel, RyR, intracellular Ca2+ mobilization, α1-adrenergic vasoconstriction, resistance arteries
Citation: Gutiérrez A, Contreras C, Sánchez A and Prieto D (2019) Role of Phosphatidylinositol 3-Kinase (PI3K), Mitogen-Activated Protein Kinase (MAPK), and Protein Kinase C (PKC) in Calcium Signaling Pathways Linked to the α1-Adrenoceptor in Resistance Arteries. Front. Physiol. 10:55. doi: 10.3389/fphys.2019.00055
Received: 17 September 2018; Accepted: 17 January 2019;
Published: 06 February 2019.
Edited by:
Maria Fernandez-Velasco, Hospital Universitario La Paz, SpainReviewed by:
Peter S. Reinach, Wenzhou Medical University, ChinaCopyright © 2019 Gutiérrez, Contreras, Sánchez and Prieto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dolores Prieto, ZHByaWV0b0B1Y20uZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.