 Dominic G. Whittaker
Dominic G. Whittaker Jules C. Hancox
Jules C. Hancox Henggui Zhang
Henggui Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 11 January 2019
Sec. Computational Physiology and Medicine
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01888
This article is part of the Research Topic Cardiac Modeling: Aiming for Optimization of Therapy View all 16 articles
Short QT syndrome variant 1 (SQT1) arises due to gain-of-function mutations to the human Ether-à-go-go-Related Gene (hERG), which encodes the α subunit of channels carrying rapid delayed rectifier potassium current, IKr. In addition to QT interval shortening and ventricular arrhythmias, SQT1 is associated with increased risk of atrial fibrillation (AF), which is often the only clinical presentation. However, the underlying basis of AF and its pharmacological treatment remain incompletely understood in the context of SQT1. In this study, computational modeling was used to investigate mechanisms of human atrial arrhythmogenesis consequent to a SQT1 mutation, as well as pharmacotherapeutic effects of selected class I drugs–disopyramide, quinidine, and propafenone. A Markov chain formulation describing wild type (WT) and N588K-hERG mutant IKr was incorporated into a contemporary human atrial action potential (AP) model, which was integrated into one-dimensional (1D) tissue strands, idealized 2D sheets, and a 3D heterogeneous, anatomical human atria model. Multi-channel pharmacological effects of disopyramide, quinidine, and propafenone, including binding kinetics for IKr/hERG and sodium current, INa, were considered. Heterozygous and homozygous formulations of the N588K-hERG mutation shortened the AP duration (APD) by 53 and 86 ms, respectively, which abbreviated the effective refractory period (ERP) and excitation wavelength in tissue, increasing the lifespan and dominant frequency (DF) of scroll waves in the 3D anatomical human atria. At the concentrations tested in this study, quinidine most effectively prolonged the APD and ERP in the setting of SQT1, followed by disopyramide and propafenone. In 2D simulations, disopyramide and quinidine promoted re-entry termination by increasing the re-entry wavelength, whereas propafenone induced secondary waves which destabilized the re-entrant circuit. In 3D simulations, the DF of re-entry was reduced in a dose-dependent manner for disopyramide and quinidine, and propafenone to a lesser extent. All of the anti-arrhythmic agents promoted pharmacological conversion, most frequently terminating re-entry in the order quinidine > propafenone = disopyramide. Our findings provide further insight into mechanisms of SQT1-related AF and a rational basis for the pursuit of combined IKr and INa block based pharmacological strategies in the treatment of SQT1-linked AF.
The short QT syndrome (SQTS) is characterized primarily by a short QT interval on the ECG, which corresponds to abbreviated ventricular repolarisation. However, symptomatic atrial fibrillation (AF) has also been reported as a common first clinical presentation of the SQTS (Schimpf et al., 2005), suggesting that pathophysiological mechanisms leading to shortening of the QT interval also affect the atria, which can increase susceptibility to AF. The N588K mutation to the human Ether-à-go-go-Related gene (hERG), which encodes the α subunit of channels carrying rapid delayed rectifier potassium current, IKr, underlies a form of SQTS variant 1 (SQT1) (Brugada et al., 2004), and has been associated with a high incidence of AF in affected probands—as high as 50% (Hu et al., 2017). Whilst ventricular arrhythmia substrates in SQT1 have received much attention (Zhang and Hancox, 2004; Patel and Antzelevitch, 2008; Adeniran et al., 2011), there have been comparatively fewer studies investigating mechanisms by which SQT1 mutations promote AF, which can be an important biomarker of the SQTS. Furthermore, effective management of AF remains a challenge, and is incompletely understood in the context of SQT1 (Enriquez et al., 2016; Hancox et al., 2018).
A previous simulation study (Loewe et al., 2014b) used the Courtemanche-Ramirez-Nattel (CRN) mathematical model of the human atrial action potential (AP) (Courtemanche et al., 1998) to demonstrate shortening of the atrial effective refractory period (ERP) and tissue excitation wavelength (WL) consequent to the N588K-hERG mutation. This was shown to facilitate initiation and sustenance of spiral waves in idealized two-dimensional (2D) sheets of human atrial tissue, which is a likely mechanism for increased susceptibility to AF. However, both the intrinsic electrical heterogeneities and complex anatomy of the human atria have also been suggested to influence arrhythmia vulnerability and dynamics in overall response to K+ channel mutations (Colman et al., 2017; Whittaker et al., 2017b). Consequently, the first aim of the present study was to assess the arrhythmogenicity of the N588K-hERG mutation in anatomically-detailed models of the human atria with realistic structure and inclusion of regional differences in electrophysiology.
The class Ia anti-arrhythmic drug quinidine is typically used as the frontline therapy for QT interval normalization in SQT1 (Gaita et al., 2004; Giustetto, 2006; Hu et al., 2017), and disopyramide has been suggested as a possible alternative (Dumaine and Antzelevitch, 2006; Schimpf et al., 2007). Regarding management of atrial arrhythmias, the class Ic anti-arrhythmic drug propafenone has been reported to be effective at preventing recurrent episodes of (paroxysmal) AF mediated by the N588K mutation to hERG (Hong et al., 2005; Bjerregaard et al., 2006), maintaining 2 patients free of arrhythmia recurrence for >2 years (Hong et al., 2005). In an experimental setting in which a hERG activator was used to approximate SQT1, quinidine (a blocker of INa and IKr) was effective at extending atrial AP duration and ERP and preventing AF (Nof et al., 2010), more so than E-4031 (a selective IKr blocker) or lidocaine (an INa blocker) alone, suggesting that K+ and Na+ channel blocking effects combine synergistically for improved management of AF in SQT1. Similarly, in our previous study, a multi-scale computational modeling approach was used to investigate ventricular pharmacological effects of disopyramide and quinidine in SQT1 (Whittaker et al., 2017a), where combined INa and IKr block by both compounds was shown to prolong ERP to a greater extent than INa or IKr block alone. Mechanisms by which Na+ and K+-channel blocking agents may provide beneficial effects in the context of SQT1-mediated human atrial pro-arrhythmia remain unclear. A multi-scale cardiac modeling approach is being used increasingly for optimisation of therapy (Yuan et al., 2015). Consequently, the second aim of the present study was to assess the efficacy of the class I drugs disopyramide, quinidine, and propafenone on rate and rhythm control of human atrial arrhythmias mediated by SQT1, from cell to 3D tissue levels, using drug binding models (including reduced potency of drugs against SQT1 mutant IKr) with multi-channel pharmacology.
Human atrial electrophysiology was simulated using the Colman et al. model (Colman et al., 2013), as updated recently (Ni et al., 2017), and is hereinafter referred to as the CNZ (Colman-Ni-Zhang) model. The equations for IKr in the CNZ model were replaced with previously developed and validated Markov chain formulations of wild type (WT) and N588K mutant IKr/hERG (Whittaker et al., 2017a). The maximal conductance of IKr was set to gKr = 0.0111375, where is the extracellular potassium concentration (Adeniran et al., 2011), resulting in an action potential duration (APD) at 90% repolarisation (APD90) of 248.8 ms at 1 Hz in the baseline human atrial cell model, which is well within the experimentally-measured range of APDs in human atrial myocytes (Bosch et al., 1999; Kim et al., 2002; Dobrev and Ravens, 2003; Katoh et al., 2005; Redpath et al., 2006; Pau et al., 2007). A family of regional human atrial cell models was incorporated into the CNZ model, accounting for distinct differences in electrophysiology of the right and left atrium (RA and LA, respectively), right and left atrial appendages (RAA and LAA, respectively), crista terminalis (CT), pectinate muscles (PM), atrio-ventricular ring (AVR), atrial septum (AS), Bachmann's bundle (BB), and pulmonary veins (PV) (Colman et al., 2013). Changes to maximal ionic conductances relative to the RA model implemented in order to create a family of regional cell models are given in Table S1. The homozygous (N588K) form of SQT1 was modeled as consisting of 100% mutant subunits, whereas the heterozygous (WT-N588K) form was assumed to consist of a 1:1 WT:mutant subunit ratio (Whittaker et al., 2017a).
In our previous study (Whittaker et al., 2017a), the actions of the class Ia anti-arrhythmic drugs disopyramide and quinidine on human ventricles were simulated in the setting of SQT1. State-dependent binding of disopyramide and quinidine to hERG channels was simulated through addition of drug-bound open and inactivated states to Markov chain formulations of IKr, and the guarded receptor model (Starmer et al., 1984) was used to describe use-dependent block of fast sodium current, INa, by both agents. Furthermore, the blocking actions of disopyramide and quinidine on slow delayed rectifier potassium current, IKs, L-type calcium current, ICaL, transient outward potassium current, Ito, inward rectifier potassium current, IK1 (quinidine only), and late sodium current, INaL (quinidine only), were modeled using a simple “pore block” approach. In the present study, the actions of disopyramide and quinidine on human atrial electrophysiology were represented using the same formulations and IC50 (half maximal inhibitory concentration) values presented in Whittaker et al. (2017a). In addition, disopyramide and quinidine have both been reported to block the atrial-specific ultra-rapid delayed rectifier potassium current, IKur. For disopyramide, the IC50 for block of IKur was taken to be 25.0 μM (Aréchiga et al., 2008), and for quinidine the IC50 was taken to be 6.6 μM, as measured in human atrial myocytes (Nenov et al., 1998). As the CNZ model does not include late sodium current, quinidine block of INaL was not included in simulations. Full details of disopyramide and quinidine models can be found in Whittaker et al. (2017a).
Using the same approach as detailed for disopyramide and quinidine in Whittaker et al. (2017a), state-dependent models of drug block by propafenone were developed. Interactions between propafenone and IKr/hERG were developed based on experimental data obtained at 37°C (Paul et al., 2002; McPate et al., 2008), where estimation of parameters for drug-bound states of the Markov chain formulation of IKr was performed using the procedure outlined in Moreno et al. (2016). Binding and unbinding rates to activated and inactivated state channels were allowed to vary freely in order to minimize the difference between simulated and experimental dose-dependent steady state block (WT and N588K), mean fractional block of tail currents during a pulse protocol, and voltage-dependence of tail current block (Paul et al., 2002; McPate et al., 2008). The extended drug-free Markov chain model of IKr/hERG is shown in Figure 1, as well as the close concordance between simulated and experimental data regarding propafenone block of IhERG. Parameters and state affinities are given in Table S2.
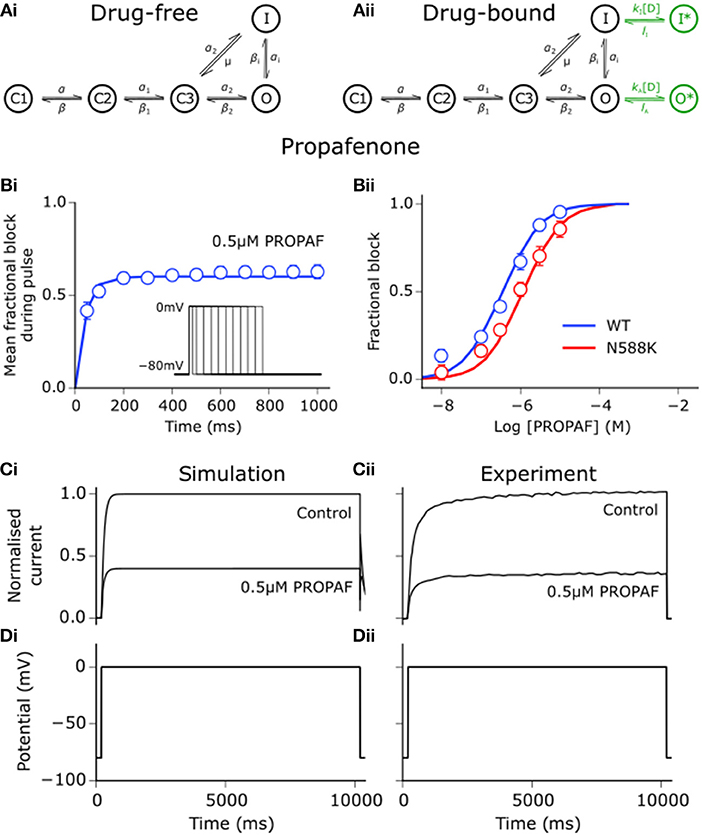
Figure 1. Propafenone interactions with hERG channels. (Ai) Drug-free and (Aii) drug-bound (additional states shown in green) Markov chain models of IKr/hERG. Simulated (solid line) and experimental (points) mean fractional block by 0.5 μM propafenone (PROPAF) of IhERG during pulse protocol (shown inset) (Bi), and dose-response curve under WT (blue) and SQT1 mutant N588K (red) conditions (Bii), where IC50 values are 390 nM and 936 nM (2.4-fold increase), respectively. (Ci) Simulated and (Cii) experimental current traces in response to a 10,000 ms depolarising voltage step to 0 mV from a holding potential of −80 mV (Di,Dii) under control conditions and with application of 0.5 μM PROPAF – these data were used to validate and not to further train the model. Experimental data at 37°C are taken from Paul et al. (2002) and McPate et al. (2008).
Propafenone is a class Ic sodium channel blocking anti-arrhythmic drug (Roden, 2014). Use-dependent block of sodium channels by propafenone was represented using the guarded receptor hypothesis (Starmer et al., 1984), as described for disopyramide and quinidine. Propafenone is predominantly an open state sodium channel blocker with little to no resting state block (Edrich et al., 2005; Burashnikov et al., 2012), and was thus assumed to bind only to activated and inactivated states. Binding and unbinding parameters were constrained based on the dose-dependent, use-dependent, and steady state block of INa by propafenone (Harmer et al., 2008), as shown in Figure 2A. Binding and unbinding parameters and state affinities are given in Table S3.
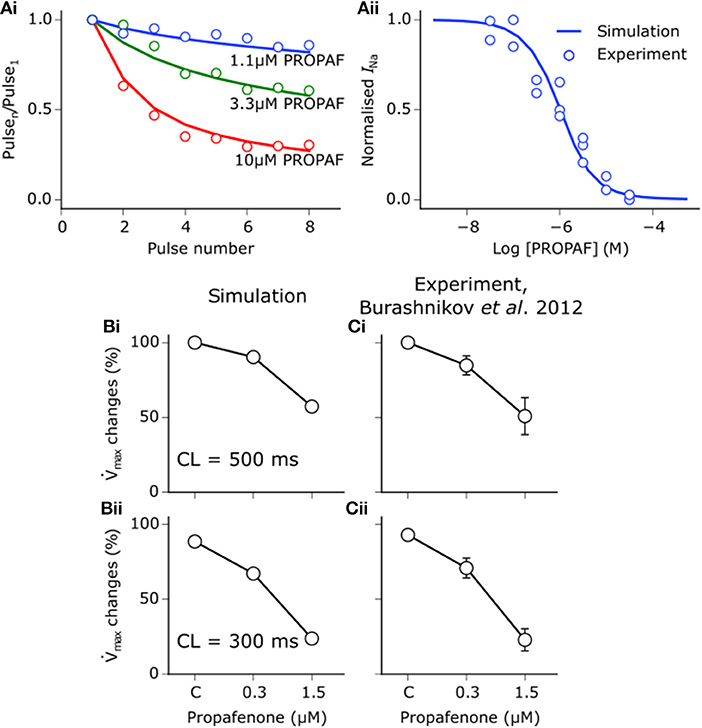
Figure 2. Propafenone interactions with sodium channels. (Ai) Simulated (solid lines) and experimental (points) use-dependent block of sodium channels by propafenone (PROPAF) elicited using a series of 8 pulses from −90 mV to 0 mV at 3 Hz for three different test concentrations. (Aii) Steady state block of sodium channels using pulses from −90 mV to 0 mV at 3 Hz until a steady state was achieved. Experimental data are taken from Harmer et al. (2008), where the steady state block IC50 value is given as 1.4 μM. (B) Simulated and (C) experimentally-measured PROPAF-induced reduction in maximum upstroke velocity () upon application of 0.3 and 1.5 μM PROPAF at a cycle length (CL) of (i) 500 ms and (ii) 300 ms. All reductions are relative to the control (C) value at a CL of 500 ms. Experimental data, recorded from canine atrial myocytes, are taken from Burashnikov et al. (2012).
Whilst propafenone most potently blocks IKr and INa, it is a multi-channel blocker, and thus exerts secondary effects on other ion channels, which were modeled using a simple pore block based on IC50 values from the literature. Propafenone blocks ICaL with an IC50 of 1.5–1.7 μM in mammalian cardiac myocytes (Fei et al., 1993; Hancox and Mitcheson, 1997). In human atrial myocytes, propafenone has been shown to block Ito with an IC50 of 4.8 μM (Gross and Castle, 1998; Seki et al., 1999) and IK1 with an IC50 of 16.8 μM (Amorós et al., 2013). Finally, IKur is blocked with an IC50 of 4.4 μM (Franqueza et al., 1998). The therapeutic range of propafenone has been estimated to be between 2 and 6 μM (Paul et al., 2002). Taking into account plasma protein binding, estimates of the most likely unbound concentrations have been given as 0.15–0.7 μM (Slawsky and Castle, 1994), 0.2–0.6 μM (Duan et al., 1993), and 0.33–1 μM (Seki et al., 1999). In this study, the effects of three equally-spaced concentrations which fell within this range were studied−0.2, 0.5, and 0.8 μM propafenone. It should be noted that these concentrations are not intended to be compared directly with those chosen for disopyramide and quinidine (1, 2, and 5 μM), which were taken from Whittaker et al. (2017a) and are not equally-spaced. A comparison of IC50 values used for disopyramide, quinidine, and propafenone is given in Table S4.
In order to assess whether the combined actions of propafenone in the model induced reasonable rate- and concentration- dependent effects on the AP under control conditions, alterations in the maximum upstroke velocity () upon application of propafenone were compared with experimental data taken from canine atrial cells (Burashnikov et al., 2012). Figure 2B shows changes in upon application of 0.3 and 1.5 μM propafenone at cycle lengths of 500 and 300 ms, where close concordance can be seen between simulation and experiment (Figure 2C). Although experimental data were taken from canine atrial myocytes, the human atrial simulation data nonetheless recapitulated the considerable reduction in at both concentrations and cycle lengths, which is primarily due to the sodium channel blocking actions of propafenone.
Propagation of excitation waves in tissue was described using the monodomain equation,
where V is the transmembrane voltage, D is the global conductivity tensor, Iion is the total ionic current, and Cm is the membrane capacitance. Equation (1) was solved using a finite difference PDE solver based on the explicit forward Euler method and Strang splitting scheme. Effects of the N588K-hERG mutation on ERP, WL, conduction velocity (CV), and spiral wave dynamics were determined using 1D and 2D models of RA tissue, as described previously (Whittaker et al., 2017b, 2018). Spiral waves were initiated in 2D models using an S1-S2 cross-shock protocol, and spiral wave core trajectories over a 5.0 s simulation period were traced by locating phase singularities (Bray and Wikswo, 2002). The behavior of re-entrant excitations in an anatomically-realistic setting was determined using a 3D human atrial structured grid geometry based on the Visible Female dataset (Seemann et al., 2006; Colman et al., 2013; Whittaker et al., 2017b) with rule-based fiber orientations (Krueger et al., 2011). The transverse value of the diffusion coefficient, D⊥, was set to 0.1 mm2 ms−1, where an anisotropy ratio (D||:D⊥) of 3:1 in directions longitudinal and transverse to fibers was applied in the atrial working myocardium, and a ratio of 9:1 along the fast conducting bundles of the BB, CT, and PM (Colman et al., 2013; Whittaker et al., 2017b), which gave global and specific regional activation times in close agreement with experimental measurements (Lemery et al., 2007). The activation (ACT) time was defined as the time required for the membrane potential to reach −20 mV at each point in the geometry (Ni et al., 2017). Activation-recovery interval (ARI) was defined as the time interval between membrane potential depolarisation to −20 mV and repolarisation to −70 mV (adjusted to −65 mV in the PV region due to higher resting membrane potential) (Ni et al., 2017).
Re-entry was initiated using the phase distribution method (Biktashev and Holden, 1998; Whittaker et al., 2017a), by initiating either a clockwise or anti-clockwise scroll wave (as seen from a RA posterior wall view). Where sustained re-entrant activity was initiated, a power spectrum was obtained through fast Fourier transform analysis of pseudo ECG (pECG) time series (recorded from within the RA cavity). The dominant frequency (DF) was computed in Matlab from the largest peak in the power spectrum density. For simulating the effects of disopyramide, quinidine, and propafenone on re-entrant excitation, state variables for each node within the 3D anatomical model were saved after 2.5 s of a 10.0 s episode of sustained re-entrant activity in drug-free conditions. These were then used as initial conditions for new 3D simulations of duration 7.5 s (giving 10.0 s activity overall) in which varying concentrations of anti-arrhythmic drugs (1, 2, or 5 μM disopyramide and quinidine; 0.2, 0.5, or 0.8 μM propafenone) were applied immediately. This gave 18 simulations in total; 3 doses for each of the 3 anti-arrhythmic drugs, with 2 scroll wave initial conditions (both clockwise and anti-clockwise).
Both SQT1 mutant conditions shortened the human atrial APD at a pacing rate of 1 Hz (Figure 3Ai), from 248.8 ms in the WT condition to 195.4 and 163.3 ms in heterozygous (WT-N588K) and homozygous (N588K) mutation conditions, respectively. This was due to loss of inactivation associated with the N588K-hERG mutation, which increased IKr early during the AP and served to accelerate the repolarisation process (Figure 3Aii). Action potential abbreviation reduced considerably the duration of the plateau phase, causing IK1 to contribute to terminal repolarisation earlier during the AP (Figure 3Aiii), and lesser activation and reduced contribution of IKs (Figure 3Aiv). The SQT1 mutation also decreased net ICaL due to the abbreviated plateau phase (Figure 3Av), shortening the APD90 under heterozygous and homozygous conditions (Figure 3Avi). In tissue, accelerated repolarisation under SQT1 conditions shortened the ERP and consequently the excitation WL. At the organ scale (in the 3D anatomical human atria model), both heterozygous and homozygous forms of the N588K-hERG mutation decreased global ARI (Figure 3B), whilst preserving the global dispersion of ARI. Furthermore, whereas ΔARI was decreased between CT/PM regions, it was increased between PV/LA and RA/LA regions under SQT1 conditions (Figure 3C). A summary of the effects of the N588K-hERG mutation on multi-scale human atrial AP biomarkers is given in Table 1.
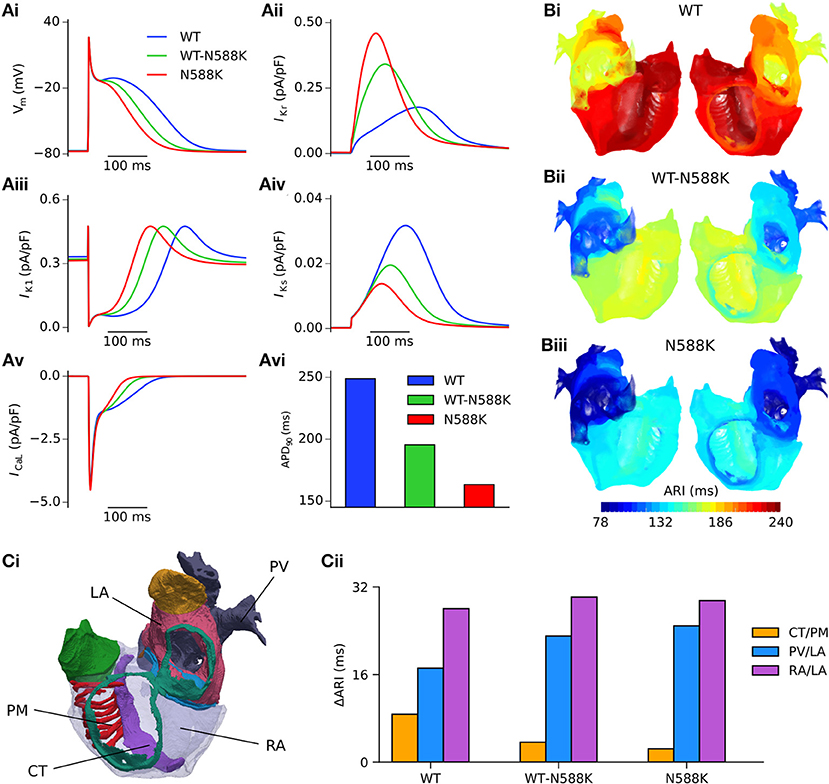
Figure 3. Effects of the N588K-hERG mutation at the single cell and whole human atria level. (Ai) Single cell action potential profiles under WT (blue), WT-N588K (green), and N588K (red) conditions at a pacing frequency of 1 Hz, with corresponding current traces for (Aii) IKr, (Aiii) IK1, (Aiv) IKs, and (Av) ICaL, and (Avi) a summary of the measured APD90. (B) Activation-recovery interval (ARI) maps under (i) WT, (ii) WT-N588K, and (iii) N588K conditions. (Ci) View looking into the cavities of the 3D anatomical human atria model, with the following regions highlighted: crista terminalis (CT), left atrium (LA), pectinate muscles (PM), pulmonary veins (PV), and right atrium (RA). (Cii) Measured dispersion of activation-recovery interval, ΔARI, between different atrial regions under WT and SQT1 mutation conditions.
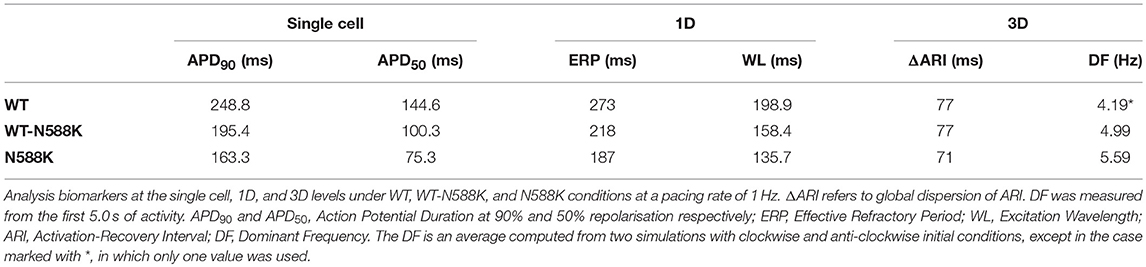
Table 1. A summary of multi-scale simulation results.
A summary of 3D scroll wave simulations in the anatomical human atria model under WT and SQT1 mutation conditions (in a clockwise configuration from a RA posterior wall aspect) is given in Figure 4. In the WT condition, scroll waves followed 2 transient, fragmented circuits around the RA, before self-terminating at ~0.7 s (Video S1), which precluded accurate measurement of the DF. In the WT-N588K condition, re-entrant wave activity sustained for the entire 10.0 s with a DF of 4.99 Hz, and was driven mostly by a macro re-entrant circuit around the right AVR, whilst also showing CT/isthmus driven activity in the RA (Video S2). Under homozygous N588K mutation conditions, scroll wave activity was also driven predominantly by a re-entrant circuit around the AVR, with occasional existence of multiple waves on the RA free wall (Video S3), and a DF of 5.59 Hz. A summary of simulations using anti-clockwise scroll wave initial conditions is shown in Figure S1, and an average of the DF from both simulations (where applicable) measured from the first 5.0 s of activity is given in Table 1.
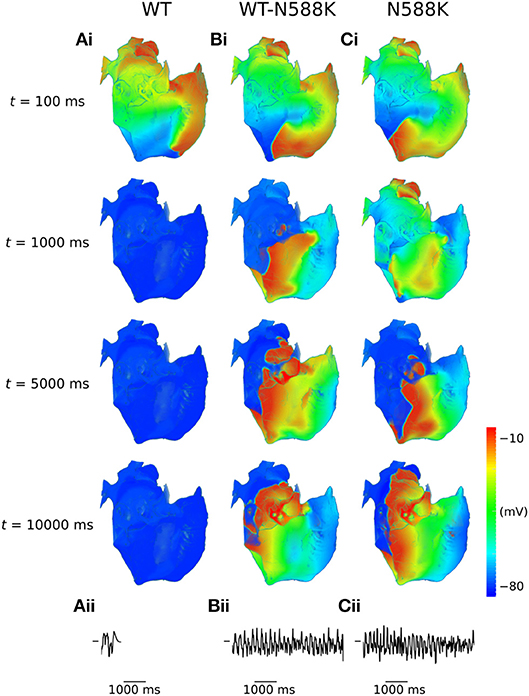
Figure 4. Scroll waves under WT and SQT1 mutation conditions in the 3D anatomical human atria model. (i) Evolution of scroll waves following initiation of re-entry in a clockwise configuration (from a RA posterior wall aspect) at times t = 100 ms, t = 1,000 ms, t = 5,000 ms, and t = 10,000 ms under (A) WT, (B) WT-N588K, and (C) N588K conditions, with (ii) corresponding pseudo ECGs taken from the first 5.0 s of re-entry simulations.
In order to assess the actions of class I anti-arrhythmic drugs on the human atria at the cellular level, the effects of applying various concentrations of disopyramide, quinidine, and propafenone were investigated on the single cell AP under heterozygous WT-N588K (hereinafter referred to simply as SQT1) conditions. Figure 5 shows AP profiles under drug-free SQT1 conditions and upon application of different concentrations of each drug at a pacing frequency of 1 Hz, with corresponding fractional block of IKr and INa. Application of all concentrations of disopyramide and propafenone produced only modest prolongation of the APD, failing to restore it to that of the WT condition. Quinidine, on the other hand, was more effective at prolonging the APD due to extensive IKr block, with the highest concentration (5 μM) prolonging the APD beyond that of the WT level. In contrast, all three agents prolonged the ERP effectively, restoring it to (or exceeding) that of the WT level at the highest respective concentrations tested, due to additional ERP-prolonging effects of INa block.
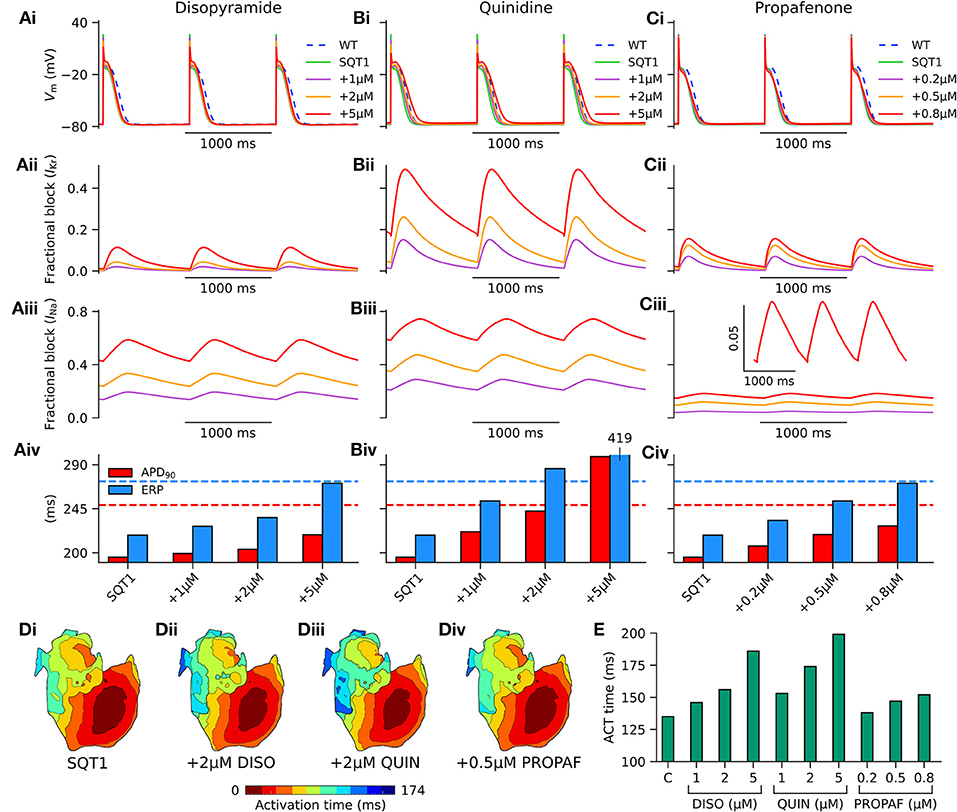
Figure 5. Effects of class I drugs on human atrial electrophysiology in SQT1. Effects of (A) disopyramide (DISO), (B) quinidine (QUIN), and (C) propafenone (PROPAF) on (i) single human atrial cell action potentials (APs) under wild type (WT) conditions (blue, dashed line), drug-free WT-N588K (SQT1) conditions (green, solid line), and SQT1 + varying drug concentrations (purple, orange, and red solid lines). Corresponding fractional block of (ii) IKr and (iii) INa, and (iv) increase in APD90 (red) and effective refractory period (ERP; blue). (D) Activation sequences in the 3D anatomical human atria model under (i) drug-free SQT1 (control – C), (ii) SQT1 + 2 μM DISO, (iii) SQT1 + 2 μM QUIN, and (iv) SQT1 + 0.5 μM PROPAF conditions, with corresponding (E) activation (ACT) time summarized as a barchart. All simulations performed at a basic cycle length of 1,000 ms.
Conduction slowing due to sodium channel block by the three drugs caused an increase in activation time in the 3D anatomical human atria model (Figure 5D), which was most prominent for quinidine and least prominent for propafenone at the concentrations tested. A summary of the effects of pharmacological modulation across multiple scales is given in Table 2. In addition, in the 1D tissue model, the rate-dependent effects of disopyramide, quinidine, and propafenone on the ERP, CV, and WL are shown in Figure S2. Quinidine was shown to produce the most potent effects on CV and ERP at all pacing rates. Propafenone exerted the weakest effects on ERP at fast pacing rates, but had the greatest propensity to promote beat-to-beat alternans.
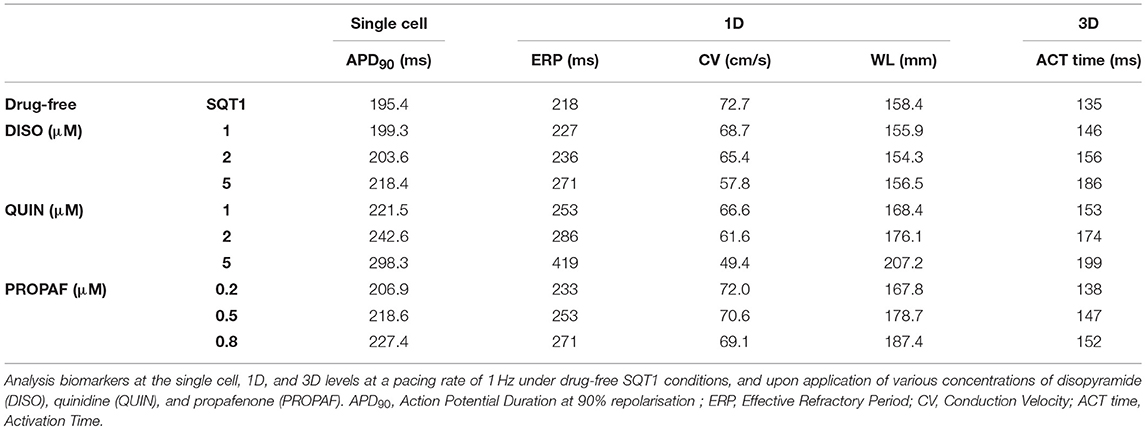
Table 2. A summary of multi-scale pharmacological simulation results.
In 2D simulations, under drug-free WT conditions the initiated spiral wave failed to re-enter the tissue (not shown). Under drug-free SQT1 conditions, the initiated spiral wave sustained for the 5.0 s duration of the simulation, eventually settling into a stationary, epicycloidal trajectory (Figure 6Ai). Application of 5 μM disopyramide was sufficient to terminate re-entry, as the re-entry wavelength was increased to such an extent that the spiral wave meandered out of the tissue (Figure 6Aii). On the other hand, both 1 and 2 μM disopyramide did not terminate re-entrant activity, but did increase the area over which spiral waves meandered (from 0.35 mm2 ms−1 in the WT condition to 0.40 and 0.47 mm2 ms−1, respectively '(Figure 6B). Similarly, 1 μM quinidine increased the area of meander to 0.44 mm2 ms−1 without terminating re-entry, whereas 2 and 5 μM quinidine terminated re-entry without inducing wave break (Figure 6C). Application of 0.2 and 0.5 μM propafenone resulted in termination of re-entry by decreasing both the stationarity and stability of spiral waves (Figure 6D), whereas 0.8 μM propafenone did not terminate re-entry but did destabilize the re-entrant circuit and substantially increase the area of meander (to 0.75 mm2 ms−1).
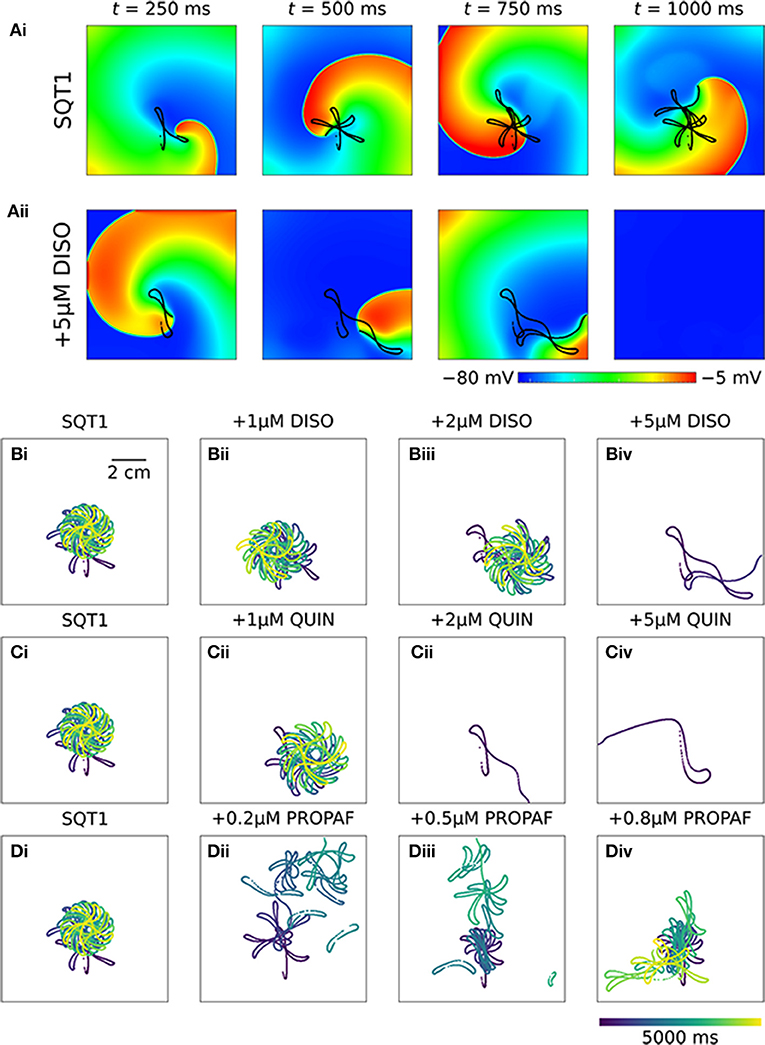
Figure 6. Re-entry dynamics in homogeneous 2D sheets upon application of disopyramide, propafenone, and quinidine in SQT1 mutation conditions. (A) Representative snapshots of re-entrant spiral waves under (i) drug-free SQT1 and (ii) SQT1 + 5 μM disopyramide (DISO) conditions at times, t = 250, 500, 750, and 1,000 ms following re-entry initiation, with spiral wave core trajectories superimposed onto membrane potentials in black. Spiral wave core trajectories over a 5,000 ms period under (Bi, Ci, Di) drug-free SQT1 conditions, and upon application of (Bii) 1 μM, (Biii) 2 μM, and (Biv) 5 μM DISO, (Cii) 1 μM, (Ciii) 2 μM, and (Civ) 5 μM quinidine (QUIN), and (Dii) 0.2 μM, (Diii) 0.5 μM, and (Div) 0.8 μM propafenone (PROPAF).
In 3D simulations using clockwise scroll wave initial conditions, quinidine terminated re-entry at all concentrations, and disopyramide terminated re-entry at concentrations of 1 and 5 μM (arrhythmia termination by 5 μM disopyramide and 2 μM quinidine are shown in Videos S4, S5, respectively). At a concentration of 2 μM disopyramide, the DF was reduced from 4.79 to 3.99 Hz (measured from the final 5.0 s of activity). Propafenone, on the other hand, was comparatively ineffective at reducing the DF at the concentrations tested, but terminated re-entry at a concentration of 0.5 μM (Video S6). Representative examples of arrhythmia termination by disopyramide, propafenone, and quinidine are shown in Figure 7. Disopyramide and quinidine produced a dose-dependent decrease in the DF which was greater than for propafenone (Figure 7C). In an anti-clockwise scroll wave configuration, the efficacy of anti-arrhythmic drugs was less favorable, with pharmacological conversion of re-entrant waves occurring only for 0.5 μM propafenone and 5 μM quinidine. A quantitative summary of all 2D and 3D re-entry simulations is given in Table S5.
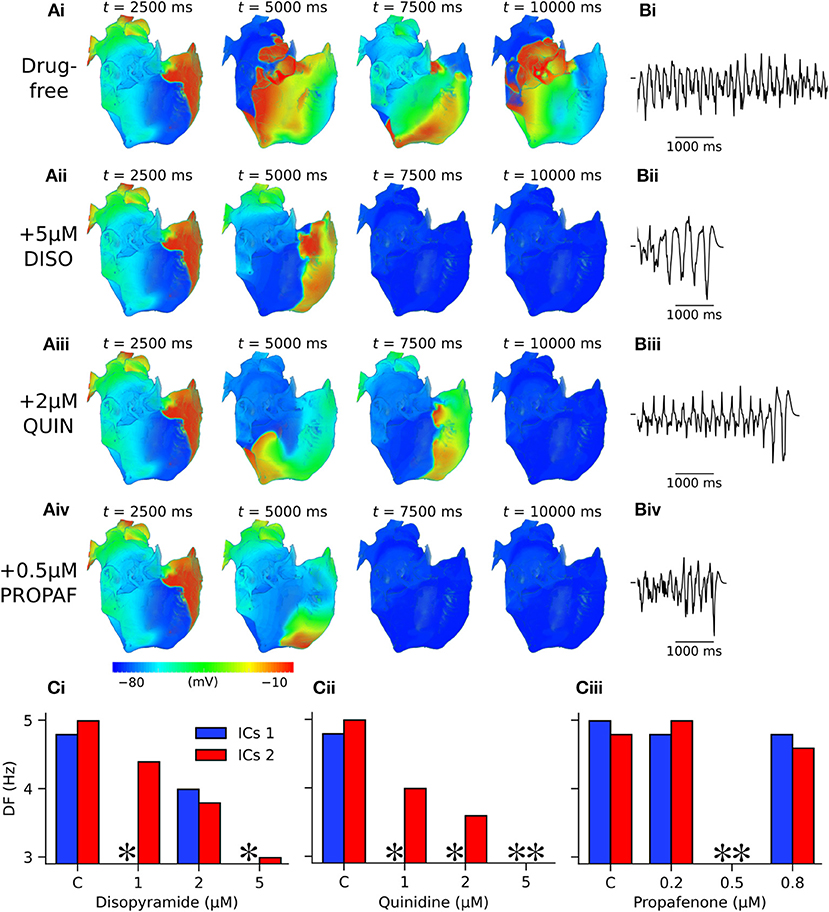
Figure 7. Arrhythmia termination in 3D anatomical human atria by representative concentrations of disopyramide, propafenone, and quinidine under SQT1 mutation conditions. (A) Snapshots of re-entrant scroll waves in a clockwise configuration (viewed from RA posterior wall) at various time points after initiation of a scroll wave in the 3D anatomical human atrial model under (i) drug-free WT-N588K conditions, and upon application of (ii) 5 μM disopyramide (DISO), (iii) 0.5 μM propafenone (PROPAF), and (iv) 2 μM quinidine (QUIN) conditions, with (B) corresponding pseudo ECG recorded during the final 5.0 s. In pharmacological simulation drugs were applied at t = 2,500 ms. (C) Barcharts summarizing DF upon application of various concentrations of (i) DISO, (ii) PROPAF, and (iii) QUIN for clockwise and anti-clockwise initial conditions (ICs 1 and ICs 2, respectively). * denotes simulations in which DF was not calculated due to occurrence of re-entry termination.
In this study, pathophysiological effects of the SQT1-related N588K-hERG mutation on human atrial electrophysiology were assessed, investigating mechanisms of increased susceptibility to the development of atrial arrhythmias in multi-scale cellular and tissue models incorporating electrical and anatomical heterogeneities. Furthermore, the actions of the class I anti-arrhythmic drugs disopyramide, quinidine, and propafenone were assessed in the context of sustained atrial arrhythmic excitations mediated by the SQT1 N588K mutation using drug simulations including binding kinetics, altered potency of IKr/hERG block in SQT1, and multiple ion channel block. Both quinidine and disopyramide have demonstrated efficacy in QT interval prolongation in the setting of SQT1 (Gaita et al., 2004; Schimpf et al., 2007; Mizobuchi et al., 2008; Giustetto et al., 2011; Hu et al., 2017), whereas propafenone has been used in 3 patients for management of episodes of paroxysmal AF mediated by SQT1, which prevented recurrence of arrhythmias but did not prolong the QT interval (Hong et al., 2005; Bjerregaard et al., 2006). This study provides clinically-relevant insights into pharmacology of SQT1 by evaluating and comparing the actions of all three drugs in the context of accelerated atrial repolarisation and arrhythmias mediated by SQT1, offering an important step toward in silico optimisation of pharmacological therapy in this context.
The major findings presented in this study are as follows. (1) Heterozygous and homozygous forms of the SQT1-linked N588K-hERG mutation shortened significantly the APD of human atrial cells, whilst preserving global dispersion of ARI, and increasing ΔARI between certain regions of the human atria. (2) The dominant frequency and lifespan of re-entry in 3D arrhythmia simulations was increased by heterozygous and homozygous forms of the N588K mutation. (3) Disopyramide, quinidine, and propafenone all produced ERP prolongation in the setting of SQT1, the extent of which was greatest for quinidine. (4) In 2D simulations, disopyramide and quinidine terminated re-entry at high concentrations due to increased re-entry wavelength, whereas propafenone terminated re-entry in a non-dose-dependent manner, by inducing secondary waves. (5) In 3D simulations, the DF of re-entry was reduced in a dose-dependent manner for clinically-relevant doses of disopyramide and quinidine, and propafenone to a lesser extent. (6) All three anti-arrhythmic agents demonstrated some efficacy in pharmacological rhythm control, most frequently terminating re-entry in the order quinidine > propafenone = disopyramide. A summary of findings regarding pro-arrhythmic mechanisms of the N588K-hERG mutation in human atria and anti-arrhythmic actions of selected class I drugs is given in Figure 8.
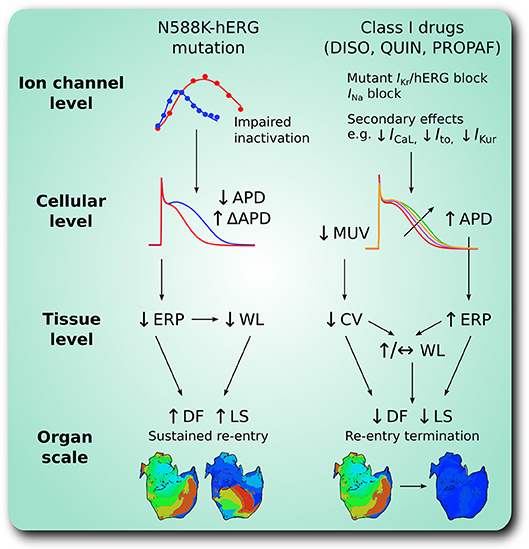
Figure 8. A schematic summary of pro-arrhythmic effects of the N588K-hERG mutation in human atria and anti-arrhythmic effects of class I drugs. At the ion channel level, the N588K mutation impairs hERG channel inactivation, increasing IKr which shortens the action potential duration (APD) at the cellular level, and increases its dispersion, ΔAPD, between certain regions of the human atria. In tissue, shortened APD corresponds to shortened effective refractory period (ERP) and excitation wavelength (WL), promoting sustenance of arrhythmias at the organ scale through higher dominant frequency (DF) and lifespan (LS) of re-entry. Class I drugs disopyramide (DISO), quinidine (QUIN), and propafenone (PROPAF) exert effects at the ion channel level by blocking IKr and INa, in addition to secondary actions. This prolongs APD and ERP, and reduces maximum upstroke velocity (MUV) and conduction velocity (CV) at the cellular and tissue levels, respectively. At the organ scale these pharmacological actions promote arrhythmia rate and rhythm control through reduced DF and LS of re-entry.
The fact that the N588K-hERG mutation is associated with both a short QT interval and reports of AF (Hong et al., 2005) suggests that pathophysiological mechanisms leading to QT interval shortening in SQT1 also affect the atria and may promote AF. In an experimental model of SQT1, application of 20 μM of the IKr agonist PD-118057 shortened the APD in both CT and PM cell types of canine atrial tissue (Nof et al., 2010), as well as increasing APD dispersion between them. In the present study, at the cellular level heterozygous and homozygous forms of the N588K-hERG mutation produced a shortening of the human right atrial APD of 53.4 and 85.5 ms, respectively–values which are consistent with a previous simulation study (Loewe et al., 2014b). At the tissue level, global ΔARI was unaffected by the WT-N588K mutation condition, and was slightly decreased by the homozygous form. The model predicted an increase in ΔARI between PV/LA and RA/LA regions for both heterozygous and homozygous forms of the N588K mutation (Figure 3C), but not between CT/PM regions. This may be due to the fact that the effects of the hERG activator PD-118057 and of the N588K mutation on IKr/hERG are different: the former increases IKr/hERG channel open probability without altering gating kinetics (Zhou et al., 2005), whereas the latter alters kinetics through profound attenuation of inactivation (Cordeiro et al., 2005; McPate et al., 2005).
Interestingly, there is clinical evidence of a LA-RA gradient in the DF in paroxysmal AF (Lazar et al., 2004), which may be underlain by APD differences between the RA and LA in pathological conditions which promote AF (Voigt et al., 2010). Furthermore, regional differences in APD between PV/LA regions have been identified previously as high frequency excitation or microreentrant sources underlying AF (Mandapati et al., 2000; Arora et al., 2003; Kumagai et al., 2004)–this suggests that localized increases in the spatial dispersion of repolarisation could be a mechanism by which atrial arrhythmogenesis is promoted in SQT1, as suggested in the previous experimental study (Nof et al., 2010).
In a previous simulation study (Loewe et al., 2014b), the N588K-hERG mutation in a 1:1 mutant to WT ratio was shown to increase inducibility and lifespan of rotors in idealized 2D sheets of human atrial tissue, but did not permit sustained re-entry. By contrast, in the present study, the heterozygous WT-N588K (SQT1) condition permitted the sustenance of spiral waves in 2D simulations. This may be due to several differences in the modeling, one of which is that the CRN model used in that study (Loewe et al., 2014b) produces an ERP restitution curve which is around 50 ms higher than available experimental data (Wilhelms et al., 2013), making re-entry more difficult to sustain, whereas the CNZ model ERP restitution curve lies mostly within the experimental range (Figure S3). In this study, the presence of realistic structure and heterogeneous electrophysiology, in addition to the cellular electrophysiological substrate induced by the N588K-hERG mutation, also favored the sustenance of re-entrant scroll waves in the 3D anatomical human atria model (using both clockwise and anti-clockwise scroll wave initial conditions), and could both prolong or shorten the lifespan of re-entrant excitations under pharmacological modulation conditions when compared directly to observations in 2D homogeneous sheets. This highlights the value of including both approaches in probing the arrhythmia substrate associated with genetic mutations in human atria, as behavior in heterogeneous 3D anatomical models cannot necessarily be predicted from homogeneous 2D simulations, and vice versa. It should be noted nonetheless that electrical activity observed in 3D re-entry simulations in this study remained relatively organized and generally not driven by multiple wavelets. This lack of complex, chaotic behavior which is observed in persistent AF is consistent with reports of paroxysmal AF in some patients with the N588K-hERG mutation (Hong et al., 2005) – addition of electrical and intracellular gap junction remodeling or fibrosis in patients may be required to promote degeneration of paroxysmal AF into persistent AF.
Despite the prevalence of AF and decades of research, anti-arrhythmic therapies for AF continue to have limited efficacy and safety (Workman et al., 2011). In this study, the class I anti-arrhythmic drugs disopyramide, quinidine, and propafenone were shown to be only moderately effective in the management of atrial arrhythmias mediated by the SQT1-linked N588K-hERG mutation, although it should be noted that the 7.5 s duration of 3D drug simulations is much shorter than clinical time scales. Pharmacological rhythm control occurred under application of both disopyramide and quinidine, but in an unpredictable, non-dose-dependent way, as observed in our previous in silico study (Whittaker et al., 2017a). One mechanism of re-entry termination involving reduction of the excitable gap under application of 5 μM quinidine is shown in Figure S4.
Both disopyramide and quinidine reduced consistently the DF of re-entry in a dose-dependent manner, although as in (Whittaker et al., 2017a) quinidine was more effective than disopyramide at rate control in the setting of SQT1, likely due to its more potent block of IKr (McPate et al., 2008) and INa (Koumi et al., 1992). Propafenone was comparatively ineffective at controlling the rate of atrial arrhythmias under the WT-N588K mutation condition, although did demonstrate some efficacy in re-entry termination. Figure S5 shows a mechanism of pharmacological rhythm control by 0.5 μM propafenone, in which secondary waves induced by propafenone extinguished all re-entrant pathways. This is consistent with the use of propafenone to manage paroxysmal AF in SQT1 patients (Hong et al., 2005; Bjerregaard et al., 2006), which maintained 3 patients free of arrhythmia recurrence during follow up. However, as propafenone has been reported to have no prolonging effect on the QT interval in the setting of SQT1 (Hong et al., 2005)— an effect which was demonstrated with variable success using two leading human ventricle models (ten Tusscher and Panfilov, 2006; O'Hara et al., 2011) in Figure S6—it is likely that quinidine and disopyramide will remain more suitable pharmacotherapies.
In this study, cellular and tissue level simulations (in a 1D model) alone did not serve as good predictors of the overall effects of anti-arrhythmic drugs in the electrically-heterogeneous, 3D anatomical whole atria model, consistent with previous modeling studies (Varela et al., 2016; Whittaker et al., 2017b). For example, disopyramide was shown not to strongly affect the excitation WL in RA tissue (Table 2; Figure S2), and yet it decreased reliably the DF of re-entrant excitations under heterozygous N588K mutation conditions in 3D simulations. This is likely due to the fact that scroll waves were largely anatomically driven by a typical macro re-entrant atrial flutter/tachycardia circuit (Lee et al., 2012) which was larger than the functional WL; effects of disopyramide on CV were thus more important. In contrast, propafenone produced only modest changes to the CV and was thus less effective at rate control in the model, but did terminate re-entry under conditions in which the re-entrant circuit was abolished by interaction with secondary waves (Figure S5).
Further support for quinidine as a useful pharmacotherapy for AF in the setting of SQT1 comes from the experimental use of the IKr agonist PD-118057 model (Nof et al., 2010). That study suggested that the combined blocking actions on IKr and INa are what made quinidine effective at preventing sustained AF in canine right atrial tissue preparations, as neither IKr nor INa block alone (by E-4031 or lidocaine, respectively) was effective at terminating AF. Similarly, in our previous simulation study of ventricular pharmacotherapy for SQT1 we showed that combined block of INa and IKr by disopyramide and quinidine prolonged the ERP to a greater extent than block of INa or IKr alone (Whittaker et al., 2017a). In this study, we sought to determine whether the greater ERP prolongation associated with the synergistic combination of INa and IKr block by class I drugs would also translate to greater efficacy in terminating re-entry in a 2D sheet of SQT1 mutant human atrial tissue. Figure S7 shows that the actions of 5 μM of a hypothetical drug with disopyramide kinetics for INa and IKr block were able to terminate re-entry, whereas INa or IKr block alone did not, as combined block produced the greatest prolongation of the ERP. The spiral wave trajectories highlight the fact that INa block in particular destabilized the re-entrant circuit, which, when combined with IKr block, caused the spiral wave to meander out of the tissue boundaries (Video S7). The findings from in silico and in vitro experiments thus suggest that the combination of prolonged APD and refractoriness arising from K+ and Na+ channel inhibition is likely to be valuable in the setting of SQT1. Though selective blockers of IKs and IK1 are not yet clinically-available, this combination may also warrant investigation as a pharmacotherapeutic strategy for other forms of K+-linked SQTS (SQT2 and SQT3).
There are a number of limitations of the simulations presented in this study. The 3D anatomical human atria model which was employed for simulations in this study incorporated rule-based fiber orientations (Krueger et al., 2011), which may not capture sufficiently the complex cardiac microstructure of the human atria (Stephenson et al., 2017) which could contribute to the arrhythmia substrate. Whilst several hERG mutations have now been identified in SQT1 (Hancox et al., 2018), the present study focused only on the N588K-hERG mutation (which produces a more marked effect on hERG kinetics than some other mutations, Hancox et al., 2018), so the results here may not apply to all forms of SQT1. However, recently the S631A-hERG mutation (previously engineered for hERG structure-function studies), which produces a similar extent of attenuation of inactivation to N588K-hERG (McPate et al., 2008) has been reported in an SQT1 family (Akdis et al., 2018), and thus the findings of this study are likely also to be relevant to that form of SQT1. An additional potential limitation arises from the fact that mechanical contraction was not considered, which has been reported previously to be impaired in the setting of SQT1 in single human atrial cell simulations (Whittaker et al., 2015) as well as in organ-scale ventricle simulations (Adeniran et al., 2013), and based on clinical measurements (Frea et al., 2015).
The models of disopyramide and quinidine used in this study were adopted from (Whittaker et al., 2017a), in which the effects of anti-arrhythmic drugs were assessed in the setting of SQT1 in the human ventricles. However, both agents also exhibit modest anti-cholinergic effects in the atria (Nakajima et al., 1989) which were not accounted for, due to the absence of a formulation for acetylcholine-activated potassium current, IK, ACh, in the CNZ model. Similarly, the β-adrenergic receptor blocking effects of propafenone were not considered (McLeod et al., 1984). Inclusion of these factors could give some insight into the response of the SQT1 phenotype to anti-arrhythmic drugs in the presence of autonomic modulation, and could give more favorable results in terms of arrhythmia prevention, especially for propafenone, which may be particularly effective against AF triggered by increased vagal tone. In addition, dynamic effects of agents due to the influence of circadian variations in drug concentration and heart rate on bioavailability were not investigated, which can give further insights into differing clinical efficacies of drugs (Loewe et al., 2014a). In relation to this, a further, more general limitation is that translation of drug concentrations from experiments and computer simulations to meaningful clinical concentrations is potentially problematic. Finally, whilst the 1:1 mutant to WT ratio, which was intended to represent the heterozygous state of the proband, may represent an oversimplification of the real hERG channel population in SQT1, the approach adopted in this and our previous studies (Adeniran et al., 2011, 2017; Whittaker et al., 2017a) reproduced quantitatively QT interval shortening and T wave morphology in SQTS conditions which was concordant with clinical observations.
The simulations performed in this study further substantiate a causative link between the SQTS-related N588K mutation and APD/ERP shortening in human atria, as well as increased spatial dispersion of repolarisation, which promotes development of AF. In 3D human atria simulations which included heterogeneous anatomy and electrophysiology, the N588K mutation was shown to increase the dominant frequency and lifespan of re-entrant excitation beyond that observed in WT conditions. Pharmacological simulations demonstrated that disopyramide and quinidine were more effective at rate control than propafenone in the setting of SQT1, and quinidine was most effective at rhythm control. Combined block of INa and IKr by a hypothetical drug was shown to be more beneficial in terms of re-entry termination in a 2D sheet of human atrial tissue than IKr or INa block alone in this context, suggesting useful targets for future pharmacotherapies. The multi-scale integrative cardiac modeling approach adopted in this study serves as a useful paradigm for optimisation of pharmacological therapy, allowing investigation of how genetic defects at the ion channel level influence organ scale propagation, arrhythmogenesis, and response to pharmacotherapies.
DW, JH, and HZ conceived the experiments and wrote the manuscript. DW developed and validated computer models and performed numerical experiments and analysis.
DW was supported by the British Heart Foundation (FS/14/5/30533–HZ and JH) and is now supported by a Wellcome Trust ISSF fellowship (204825/Z/16/Z). This work was also supported by grants from EPSRC (UK) (EP/J00958X/1; EP/I029826/1), MC-IRSES CORDIS3D (317766), NSFC (61179009), Shenzhen Science and Technology Innovation Committee (JCYJ20151029173639477; JSGG20160229125049615. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
JH gratefully acknowledges receipt of a University of Bristol research fellowship.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2018.01888/full#supplementary-material
Adeniran, I., Hancox, J. C., and Zhang, H. (2013). In silico investigation of the short QT syndrome, using human ventricle models incorporating electromechanical coupling. Front. Physiol. 4:166. doi: 10.3389/fphys.2013.00166
Adeniran, I., McPate, M. J., Witchel, H. J., Hancox, J. C., and Zhang, H. (2011). Increased vulnerability of human ventricle to re-entrant excitation in hERG-linked variant 1 short QT syndrome. PLoS Comput. Biol. 7:e1002313. doi: 10.1371/journal.pcbi.1002313
Adeniran, I., Whittaker, D. G., Harchi, A. E., Hancox, J. C., and Zhang, H. (2017). In silico investigation of a KCNQ1 mutation associated with short QT syndrome. Sci. Rep. 7:8469. doi: 10.1038/s41598-017-08367-2
Akdis, D., Saguner, A. M., Medeiros-Domingo, A., Schaller, A., Balmer, C., Steffel, J., et al. (2018). Multiple clinical profiles of families with the short QT syndrome. EP Eur. 20, f113–f121. doi: 10.1093/europace/eux186
Amorós, I., Dolz-Gaitón, P., Gómez, R., Matamoros, M., Barana, A., de la Fuente, M. G., et al. (2013). Propafenone blocks human cardiac Kir2.x channels by decreasing the negative electrostatic charge in the cytoplasmic pore. Biochem. Pharmacol. 86, 267–278. doi: 10.1016/j.bcp.2013.04.023
Aréchiga, I. A., Barrio-Echavarria, G. F., Rodríguez-Menchaca, A. A., Moreno-Galindo, E. G., Decher, N., Tristani-Firouzi, M., et al. (2008). Kv1.5 open channel block by the antiarrhythmic drug disopyramide: molecular determinants of block. J. Pharmacol. Sci. 108, 49–55. doi: 10.1254/jphs.08084FP
Arora, R., Verheule, S., Scott, L., Navarrete, A., Katari, V., Wilson, E., et al. (2003). Arrhythmogenic substrate of the pulmonary veins assessed by high-resolution optical mapping. Circulation 107, 1816–1821. doi: 10.1161/01.CIR.0000058461.86339.7E
Biktashev, V. N., and Holden, A. V. (1998). Reentrant waves and their elimination in a model of mammalian ventricular tissue. Chaos. Interdiscip. J. Nonlinear Sci. 8, 48–56. doi: 10.1063/1.166307
Bjerregaard, P., Jahangir, A., and Gussak, I. (2006). Targeted therapy for short QT syndrome. Expert Opin. Ther. Targets 10, 393–400. doi: 10.1517/14728222.10.3.393
Bosch, R. F., Zeng, X., Grammer, J. B., Popovic, K., Mewis, C., and Kühlkamp, V. (1999). Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc. Res. 44, 121–131. doi: 10.1016/S0008-6363(99)00178-9
Bray, M.-A., and Wikswo, J. P. (2002). Use of topological charge to determine filament location and dynamics in a numerical model of scroll wave activity. IEEE Trans. Biomed. Eng. 49, 1086–1093. doi: 10.1109/TBME.2002.803516
Brugada, R., Hong, K., Dumaine, R., Cordeiro, J., Gaita, F., Borggrefe, M., et al. (2004). Sudden death associated with Short-QT syndrome linked to mutations in HERG. Circulation 109, 30–35. doi: 10.1161/01.CIR.0000109482.92774.3A
Burashnikov, A., Belardinelli, L., and Antzelevitch, C. (2012). Atrial-selective sodium channel block strategy to suppress atrial fibrillation: ranolazine versus propafenone. J. Pharmacol. Exp. Ther. 340, 161–168. doi: 10.1124/jpet.111.186395
Colman, M. A., Aslanidi, O. V., Kharche, S., Boyett, M. R., Garratt, C., Hancox, J. C., et al. (2013). Pro-arrhythmogenic effects of atrial fibrillation-induced electrical remodelling: insights from the three-dimensional virtual human atria. J. Physiol. 591, 4249–4272. doi: 10.1113/jphysiol.2013.254987
Colman, M. A., Ni, H., Liang, B., Schmitt, N., and Zhang, H. (2017). In silico assessment of genetic variation in KCNA5 reveals multiple mechanisms of human atrial arrhythmogenesis. PLOS. Comput. Biol. 13:e1005587. doi: 10.1371/journal.pcbi.1005587
Cordeiro, J. M., Brugada, R., Wu, Y. S., Hong, K., and Dumaine, R. (2005). Modulation of IKr inactivation by mutation N588K in KCNH2: A link to arrhythmogenesis in short QT syndrome. Cardiovasc. Res. 67, 498–509. doi: 10.1016/j.cardiores.2005.02.018
Courtemanche, M., Ramirez, R. J., and Nattel, S. (1998). Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am. J. Physiol. Heart Circ. Physiol. 275, H301–H321.
Dobrev, D., and Ravens, U. (2003). Remodeling of cardiomyocyte ion channels in human atrial fibrillation. Basic Res. Cardiol. 98, 137–148. doi: 10.1007/s00395-003-0409-8
Duan, D., Fermini, B., and Nattel, S. (1993). Potassium channel blocking properties of propafenone in rabbit atrial myocytes. J. Pharmacol. Exp. Ther. 264, 1113–1123.
Dumaine, R., and Antzelevitch, C. (2006). Disopyramide: although potentially life-threatening in the setting of long QT, could it be life-saving in short QT syndrome? J. Mol. Cell. Cardiol. 41, 421–423. doi: 10.1016/j.yjmcc.2006.06.070
Edrich, T., Wang, S.-Y., and Wang, G. K. (2005). State-dependent block of human cardiac hNav1.5 sodium channels by propafenone. J. Membr. Biol. 207, 35–43. doi: 10.1007/s00232-005-0801-4
Enriquez, A., Antzelevitch, C., Bismah, V., and Baranchuk, A. (2016). Atrial fibrillation in inherited cardiac channelopathies: from mechanisms to management. Heart Rhythm. 13, 1878–1884. doi: 10.1016/j.hrthm.2016.06.008
Fei, L., Gill, J. S., McKenna, W. J., and Camm, A. J. (1993). Effects of propafenone on calcium currents in single ventricular myocytes of guinea-pig. Br. J. Pharmacol. 109, 178–182. doi: 10.1111/j.1476-5381.1993.tb13550.x
Franqueza, L., Valenzuela, C., Delpón, E., Longobardo, M., Caballero, R., and Tamargo, J. (1998). Effects of propafenone and 5-hydroxy-propafenone on hKv1.5 channels. Br. J. Pharmacol. 125, 969–978. doi: 10.1038/sj.bjp.0702129
Frea, S., Giustetto, C., Capriolo, M., Scrocco, C., Fornengo, C., Benedetto, S., et al. (2015). New echocardiographic insights in short QT syndrome: more than a channelopathy? Heart Rhythm. 12, 2096–2105. doi: 10.1016/j.hrthm.2015.05.024
Gaita, F., Giustetto, C., Bianchi, F., Schimpf, R., Haissaguerre, M., Cal,ò, L., et al. (2004). Short QT syndrome: pharmacological treatment. J. Am. Coll. Cardiol. 43, 1494–1499. doi: 10.1016/j.jacc.2004.02.034
Giustetto, C. (2006). “Quinidine to treat short QT syndrome: a real alternative to ICD?,” in Cardiac Arrhythmias 2005, ed. A. R. MD (Milan: Springer), 333–335.
Giustetto, C., Schimpf, R., Mazzanti, A., Scrocco, C., Maury, P., Anttonen, O., et al. (2011). Long-term follow-Up of patients with short QT syndrome. J. Am. Coll. Cardiol. 58, 587–595. doi: 10.1016/j.jacc.2011.03.038
Gross, G. J., and Castle, N. A. (1998). Propafenone inhibition of human atrial myocyte repolarizing currents. J. Mol. Cell. Cardiol. 30, 783–793. doi: 10.1006/jmcc.1998.0643
Hancox, J. C., and Mitcheson, J. S. (1997). Inhibition of L-type calcium current by propafenone in single myocytes isolated from the rabbit atrioventricular node. Br. J. Pharmacol. 121, 7–14. doi: 10.1038/sj.bjp.0701086
Hancox, J. C., Whittaker, D. G., Du, C., Stuart, A. G., and Zhang, H. (2018). Emerging therapeutic targets in the short QT syndrome. Expert Opin. Ther. Targets 22:439–51. doi: 10.1080/14728222.2018.1470621
Harmer, A. R., Abi-Gerges, N., Easter, A., Woods, A., Lawrence, C. L., Small, B. G., et al. (2008). Optimisation and validation of a medium-throughput electrophysiology-based hNav1.5 assay using IonWorksTM. J. Pharmacol. Toxicol. Methods 57, 30–41. doi: 10.1016/j.vascn.2007.09.002
Hong, K., Bjerregaard, P., Gussak, I., and Brugada, R. (2005). Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J. Cardiovasc. Electrophysiol. 16, 394–396. doi: 10.1046/j.1540-8167.2005.40621.x
Hu, D., Li, Y., Zhang, J., Pfeiffer, R., Gollob, M. H., Healey, J., et al. (2017). The phenotypic spectrum of a mutation hotspot responsible for the short QT syndrome. JACC. Clin. Electrophysiol. 3, 727–43. doi: 10.1016/j.jacep.2016.11.013
Katoh, H., Shinozaki, T., Baba, S., Satoh, S., Kagaya, Y., Watanabe, J., et al. (2005). Monophasic action potential duration at the crista terminalis in patients with sinus node disease. Circ. J. 69, 1361–1367. doi: 10.1253/circj.69.1361
Kim, B.-S., Kim, Y.-H., Hwang, G.-S., Pak, H.-N., Lee, S. C., Shim, W. J., et al. (2002). Action potential duration restitution kinetics in human atrial fibrillation. J. Am. Coll. Cardiol. 39, 1329–1336. doi: 10.1016/S0735-1097(02)01760-6
Koumi, S., Sato, R., Katori, R., Hisatome, I., Nagasawa, K., and Hayakawa, H. (1992). Sodium channel states control binding and unbinding behaviour of antiarrhythmic drugs in cardiac myocytes from the guinea pig. Cardiovasc. Res. 26, 1199–1205. doi: 10.1093/cvr/26.12.1199
Krueger, M. W., Schmidt, V., Tobón, C., Weber, F. M., Lorenz, C., Keller, D. U. J., et al. (2011). “Modeling atrial fiber orientation in patient-specific geometries: a semi-automatic rule-based approach,” in Functional Imaging and Modeling of the Heart Lecture Notes in Computer Science. (Springer, Berlin, Heidelberg), 223–232.
Kumagai, K., Ogawa, M., Noguchi, H., Yasuda, T., Nakashima, H., and Saku, K. (2004). Electrophysiologic properties of pulmonary veins assessed using a multielectrode basket catheter. J. Am. Coll. Cardiol. 43, 2281–2289. doi: 10.1016/j.jacc.2004.01.051
Lazar, S., Dixit, S., Marchlinski, F. E., Callans, D. J., and Gerstenfeld, E. P. (2004). Presence of left-to-right atrial frequency gradient in paroxysmal but not persistent atrial fibrillation in humans. Circulation 110, 3181–3186. doi: 10.1161/01.CIR.0000147279.91094.5E
Lee, G., Sanders, P., and Kalman, J. M. (2012). Catheter ablation of atrial arrhythmias: state of the art. Lancet 380, 1509–1519. doi: 10.1016/S0140-6736(12)61463-9
Lemery, R., Birnie, D., Tang, A. S., Green, M., Gollob, M., Hendry, M., et al. (2007). Normal atrial activation and voltage during sinus rhythm in the human heart: an endocardial and epicardial mapping study in patients with a history of atrial fibrillation. J. Cardiovasc. Electrophysiol. 18, 402–408. doi: 10.1111/j.1540-8167.2007.00762.x
Loewe, A., Lutz, Y., Wilhelms, M., Sinnecker, D., Barthel, P., Scholz, E. P., et al. (2014a). In-silico assessment of the dynamic effects of amiodarone and dronedarone on human atrial patho-electrophysiology. Europace 16, iv30–iv38. doi: 10.1093/europace/euu230
Loewe, A., Wilhelms, M., Fischer, F., Scholz, E. P., Dössel, O., and Seemann, G. (2014b). Arrhythmic potency of human ether-à-go-go-related gene mutations L532P and N588K in a computational model of human atrial myocytes. Europace 16, 435–443. doi: 10.1093/europace/eut375
Mandapati, R., Skanes, A., Chen, J., Berenfeld, O., and Jalife, J. (2000). Stable microreentrant sources as a mechanism of atrial fibrillation in the isolated sheep heart. Circulation 101, 194–199. doi: 10.1161/01.CIR.101.2.194
McLeod, A. A., Stiles, G. L., and Shand, D. G. (1984). Demonstration of beta adrenoceptor blockade by propafenone hydrochloride: clinical pharmacologic, radioligand binding and adenylate cyclase activation studies. J. Pharmacol. Exp. Ther. 228, 461–466.
McPate, M. J., Duncan, R. S., Hancox, J. C., and Witchel, H. J. (2008). Pharmacology of the short QT syndrome N588K-hERG K+ channel mutation: differential impact on selected class I and class III antiarrhythmic drugs. Br. J. Pharmacol. 155, 957–966. doi: 10.1038/bjp.2008.325
McPate, M. J., Duncan, R. S., Milnes, J. T., Witchel, H. J., and Hancox, J. C. (2005). The N588K-HERG K+ channel mutation in the ‘short QT syndrome': mechanism of gain-in-function determined at 37 °C. Biochem. Biophys. Res. Commun. 334, 441–449. doi: 10.1016/j.bbrc.2005.06.112
Mizobuchi, M., Enjoji, Y., Yamamoto, R., Ono, T., Funatsu, A., Kambayashi, D., et al. (2008). Nifekalant and disopyramide in a patient with short QT syndrome: evaluation of pharmacological effects and electrophysiological properties. Pacing Clin. Electrophysiol. 31, 1229–1232. doi: 10.1111/j.1540-8159.2008.01169.x
Moreno, J. D., Lewis, T. J., and Clancy, C. E. (2016). Parameterization for in-silico modeling of ion channel interactions with drugs. PLoS ONE 11:e0150761. doi: 10.1371/journal.pone.0150761
Nakajima, T., Kurachi, Y., Ito, H., Takikawa, R., and Sugimoto, T. (1989). Anti-cholinergic effects of quinidine, disopyramide, and procainamide in isolated atrial myocytes: mediation by different molecular mechanisms. Circ. Res. 64, 297–303. doi: 10.1161/01.RES.64.2.297
Nenov, N. I., Crumb, W. J., Pigott, J. D., Harrison, L. H., and Clarkson, C. W. (1998). Quinidine interactions with human atrial potassium channels. Circ. Res. 83, 1224–1231. doi: 10.1161/01.RES.83.12.1224.
Ni, H., Whittaker, D. G., Wang, W., Giles, W. R., Narayan, S. M., and Zhang, H. (2017). Synergistic anti-arrhythmic effects in human atria with combined use of sodium blockers and acacetin. Front. Physiol. 8:946. doi: 10.3389/fphys.2017.00946
Nof, E., Burashnikov, A., and Antzelevitch, C. (2010). Cellular basis for atrial fibrillation in an experimental model of short QT1: Implications for a pharmacological approach to therapy. Heart Rhythm 7, 251–257. doi: 10.1016/j.hrthm.2009.10.017
O'Hara, T., Virág, L., Varr,ó, A., and Rudy, Y. (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput. Biol. 7:e1002061. doi: 10.1371/journal.pcbi.1002061
Patel, C., and Antzelevitch, C. (2008). Cellular basis for arrhythmogenesis in an experimental model of the SQT1 form of the short QT syndrome. Heart Rhythm 5, 585–590. doi: 10.1016/j.hrthm.2008.01.022
Pau, D., Workman, A. J., Kane, K. A., and Rankin, A. C. (2007). Electrophysiological and arrhythmogenic effects of 5-hydroxytryptamine on human atrial cells are reduced in atrial fibrillation. J. Mol. Cell. Cardiol. 42, 54–62. doi: 10.1016/j.yjmcc.2006.08.007
Paul, A. A., Witchel, H. J., and Hancox, J. C. (2002). Inhibition of the current of heterologously expressed HERG potassium channels by flecainide and comparison with quinidine, propafenone and lignocaine. Br. J. Pharmacol. 136, 717–729. doi: 10.1038/sj.bjp.0704784
Redpath, C. J., Rankin, A. C., Kane, K. A., and Workman, A. J. (2006). Anti-adrenergic effects of endothelin on human atrial action potentials are potentially anti-arrhythmic. J. Mol. Cell. Cardiol. 40, 717–724. doi: 10.1016/j.yjmcc.2006.01.012
Roden, D. M. (2014). Pharmacology and toxicology of Nav1.5-class 1 anti-arrhythmic drugs. Card. Electrophysiol. Clin. 6, 695–704. doi: 10.1016/j.ccep.2014.07.003
Schimpf, R., Veltmann, C., Giustetto, C., Gaita, F., Borggrefe, M., and Wolpert, C. (2007). In vivo effects of mutant HERG K+ channel inhibition by disopyramide in patients with a short QT-1 syndrome: a pilot study. J. Cardiovasc. Electrophysiol. 18, 1157–1160. doi: 10.1111/j.1540-8167.2007.00925.x
Schimpf, R., Wolpert, C., Gaita, F., Giustetto, C., and Borggrefe, M. (2005). Short QT syndrome. Cardiovasc. Res. 67, 357–366. doi: 10.1016/j.cardiores.2005.03.026
Seemann, G., Höper, C., Sachse, F. B., Dössel, O., Holden, A. V., and Zhang, H. (2006). Heterogeneous three-dimensional anatomical and electrophysiological model of human atria. Philos. Trans. R. Soc. Lond. Math. Phys. Eng. Sci. 364, 1465–1481. doi: 10.1098/rsta.2006.1781
Seki, A., Hagiwara, N., and Kasanuki, H. (1999). Effects of propafenone on K currents in human atrial myocytes. Br. J. Pharmacol. 126, 1153–1162. doi: 10.1038/sj.bjp.0702428
Slawsky, M. T., and Castle, N. A. (1994). K+ channel blocking actions of flecainide compared with those of propafenone and quinidine in adult rat ventricular myocytes. J. Pharmacol. Exp. Ther. 269, 66–74.
Starmer, C. F., Grant, A. O., and Strauss, H. C. (1984). Mechanisms of use-dependent block of sodium channels in excitable membranes by local anesthetics. Biophys. J. 46, 15–27. doi: 10.1016/S0006-3495(84)83994-6
Stephenson, R. S., Atkinson, A., Kottas, P., Perde, F., Jafarzadeh, F., Bateman, M., et al. (2017). High resolution 3-Dimensional imaging of the human cardiac conduction system from microanatomy to mathematical modeling. Sci. Rep. 7:7188. doi: 10.1038/s41598-017-07694-8
ten Tusscher, K. H., and Panfilov, A. V. (2006). Alternans and spiral breakup in a human ventricular tissue model. Am. J. Physiol. - Heart Circ. Physiol. 291, H1088–H1100. doi: 10.1152/ajpheart.00109.2006
Varela, M., Colman, M. A., Hancox, J. C., and Aslanidi, O. V. (2016). Atrial heterogeneity generates re-entrant substrate during atrial fibrillation and anti-arrhythmic drug action: mechanistic insights from canine atrial models. PLOS. Comput. Biol. 12:e1005245. doi: 10.1371/journal.pcbi.1005245.
Voigt, N., Trausch, A., Knaut, M., Matschke, K., Varró, A., Wagoner, D. R. V., et al. (2010). Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillationclinical perspective. Circ. Arrhythm. Electrophysiol. 3, 472–480. doi: 10.1161/CIRCEP.110.954636
Whittaker, D. G., Colman, M. A., Ni, H., Hancox, J. C., and Zhang, H. (2015). In silico investigation of short QT syndrome-linked potassium channel mutations on electro-mechanical function of human atrial cells. Comput. Cardiol. Conference 2015, 853–856. doi: 10.1109/CIC.2015.7411045.
Whittaker, D. G., Colman, M. A., Ni, H., Hancox, J. C., and Zhang, H. (2018). Human atrial arrhythmogenesis and sinus bradycardia in KCNQ1-linked short QT syndrome: insights from computational modelling. Front. Physiol. 9:1402. doi: 10.3389/fphys.2018.01402
Whittaker, D. G., Ni, H., Benson, A. P., Hancox, J. C., and Zhang, H. (2017a). Computational analysis of the mode of action of disopyramide and quinidine on hERG-linked short QT syndrome in human ventricles. Front. Physiol. 8:759. doi: 10.3389/fphys.2017.00759
Whittaker, D. G., Ni, H., Harchi, A. E., Hancox, J. C., and Zhang, H. (2017b). Atrial arrhythmogenicity of KCNJ2 mutations in short QT syndrome: insights from virtual human atria. PLOS. Comput. Biol. 13:e1005593. doi: 10.1371/journal.pcbi.1005593
Wilhelms, M., Hettmann, H., Maleckar, M. M., Koivumäki, J. T., Dössel, O., and Seemann, G. (2013). Benchmarking electrophysiological models of human atrial myocytes. Front. Physiol. 3:487. doi: 10.3389/fphys.2012.00487
Workman, A. J., Smith, G. L., and Rankin, A. C. (2011). Mechanisms of termination and prevention of atrial fibrillation by drug therapy. Pharmacol. Ther. 131, 221–241. doi: 10.1016/j.pharmthera.2011.02.002
Yuan, Y., Bai, X., Luo, C., Wang, K., and Zhang, H. (2015). The virtual heart as a platform for screening drug cardiotoxicity. Br. J. Pharmacol. 172, 5531–5547. doi: 10.1111/bph.12996
Zhang, H., and Hancox, J. C. (2004). In silico study of action potential and QT interval shortening due to loss of inactivation of the cardiac rapid delayed rectifier potassium current. Biochem. Biophys. Res. Commun. 322, 693–699. doi: 10.1016/j.bbrc.2004.07.176
Keywords: arrhythmia, short QT syndrome, atrial fibrillation, hERG, class I anti-arrhythmics, human atria, potassium channels
Citation: Whittaker DG, Hancox JC and Zhang H (2019) In silico Assessment of Pharmacotherapy for Human Atrial Patho-Electrophysiology Associated With hERG-Linked Short QT Syndrome. Front. Physiol. 9:1888. doi: 10.3389/fphys.2018.01888
Received: 01 August 2018; Accepted: 12 December 2018;
Published: 11 January 2019.
Edited by:
Olaf Doessel, Karlsruhe Institute of Technology (KIT), GermanyReviewed by:
Richard Gray, United States Food and Drug Administration, United StatesCopyright © 2019 Whittaker, Hancox and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jules C. Hancox, anVsZXMuaGFuY294QGJyaXN0b2wuYWMudWs=
Henggui Zhang, aGVuZ2d1aS56aGFuZ0BtYW5jaGVzdGVyLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.