
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 21 December 2018
Sec. Membrane Physiology and Membrane Biophysics
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01831
This article is part of the Research TopicEvolving Picture of Calcium Handling in Cardiac DiseaseView all 12 articles
 Shanna Hamilton1
Shanna Hamilton1 Radmila Terentyeva1
Radmila Terentyeva1 Tae Yun Kim1
Tae Yun Kim1 Peter Bronk1
Peter Bronk1 Richard T. Clements2,3
Richard T. Clements2,3 Jin O-Uchi4György Csordás5
Jin O-Uchi4György Csordás5 Bum-Rak Choi1
Bum-Rak Choi1 Dmitry Terentyev1*
Dmitry Terentyev1*In a physiological setting, mitochondria increase oxidative phosphorylation during periods of stress to meet increased metabolic demand. This in part is mediated via enhanced mitochondrial Ca2+ uptake, an important regulator of cellular ATP homeostasis. In a pathophysiological setting pharmacological modulation of mitochondrial Ca2+ uptake or retention has been suggested as a therapeutic strategy to improve metabolic homeostasis or attenuate Ca2+-dependent arrhythmias in cardiac disease states. To explore the consequences of mitochondrial Ca2+ accumulation, we tested the effects of kaempferol, an activator of mitochondrial Ca2+ uniporter (MCU), CGP-37157, an inhibitor of mitochondrial Na+/Ca2+ exchanger, and MCU inhibitor Ru360 in rat ventricular myocytes (VMs) from control rats and rats with hypertrophy induced by thoracic aortic banding (TAB). In periodically paced VMs under β-adrenergic stimulation, treatment with kaempferol (10 μmol/L) or CGP-37157 (1 μmol/L) enhanced mitochondrial Ca2+ accumulation monitored by mitochondrial-targeted Ca2+ biosensor mtRCamp1h. Experiments with mitochondrial membrane potential-sensitive dye TMRM revealed this was accompanied by depolarization of the mitochondrial matrix. Using redox-sensitive OMM-HyPer and ERroGFP_iE biosensors, we found treatment with kaempferol or CGP-37157 increased the levels of reactive oxygen species (ROS) in mitochondria and the sarcoplasmic reticulum (SR), respectively. Confocal Ca2+ imaging showed that accelerated Ca2+ accumulation reduced Ca2+ transient amplitude and promoted generation of spontaneous Ca2+ waves in VMs paced under ISO, suggestive of abnormally high activity of the SR Ca2+ release channel ryanodine receptor (RyR). Western blot analyses showed increased RyR oxidation after treatment with kaempferol or CGP-37157 vs. controls. Furthermore, in freshly isolated TAB VMs, confocal Ca2+ imaging demonstrated that enhancement of mitochondrial Ca2+ accumulation further perturbed global Ca2+ handling, increasing the number of cells exhibiting spontaneous Ca2+ waves, shortening RyR refractoriness and decreasing SR Ca2+ content. In ex vivo optically mapped TAB hearts, kaempferol exacerbated proarrhythmic phenotype. On the contrary, incubation of cells with MCU inhibitor Ru360 (2 μmol/L, 30 min) normalized RyR oxidation state, improved intracellular Ca2+ homeostasis and reduced triggered activity in ex vivo TAB hearts. These findings suggest facilitation of mitochondrial Ca2+ uptake in cardiac disease can exacerbate proarrhythmic disturbances in Ca2+ homeostasis via ROS and enhanced activity of oxidized RyRs, while strategies to reduce mitochondrial Ca2+ accumulation can be protective.
Sudden cardiac death remains the leading global cause of mortality, and over half of patients with heart failure (HF) die suddenly due to the development of ventricular arrhythmia (Benjamin et al., 2018). Arrhythmogenesis in the failing heart is often linked to enhanced Ca2+-dependent triggered activity, in the form of early and delayed afterdepolarizations (Landstrom et al., 2017). These abnormal electrical activities arise in part as a consequence of untimely and dysregulated Ca2+ release from the sarcoplasmic reticulum (SR), through SR Ca2+ release channel, the ryanodine receptor (RyR). Abnormal activity of RyR leads to increased Ca2+ leak and promotes the generation of spontaneous Ca2+ waves (SCWs), that can subsequently propagate to trigger organ-wide arrhythmia (Bers, 2002).
Mitochondria play an essential role in cardiac Ca2+ homeostasis in physiological conditions (Kwong et al., 2015; Luongo et al., 2017). Excitation-contraction coupling consumes large amounts of ATP and mitochondria increase oxidative phosphorylation to meet increased metabolic demand. Influx of Ca2+ into the mitochondria is critical for the availability of ATP as major enzymes in the tricarboxylic acid cycle are activated by Ca2+. Mitochondria are in close spatial proximity to the SR (Dorn and Scorrano, 2010; Eisner et al., 2013; Lu et al., 2013; Seidlmayer et al., 2016; Lopez-Crisosto et al., 2017; Csordás et al., 2018), and it is well established that during higher workload, there is an elevation of cystolic Ca2+ concentration in ventricular myocytes (VMs) that transpires to a small and slow rise in mitochondrial Ca2+ concentration ([Ca2+]m), leading to enhanced energy production (Brandes and Bers, 1997; Luongo et al., 2015). Influx of Ca2+ through the mitochondrial Ca2+ uniporter (MCU) channel complex is driven largely by the negative membrane potential across the inner mitochondrial membrane (Kirichok et al., 2004; Baughman et al., 2011; De Stefani et al., 2011). Conversely, mitochondrial efflux mainly occurs via the mitochondrial Na/Ca2+/Li+-exchanger (NCLX) (Palty et al., 2010; Boyman et al., 2013; Luongo et al., 2017).
Mitochondria are a major source of reactive oxygen species (ROS) in the myocyte, and while an increase in oxidative stress is a prerequisite for many cellular stress responses, excessive ROS production in cardiac disease contributes to ventricular arrhythmogenesis by altering the function of multiple ion channels and transporters (Zima and Blatter, 2006; Niggli et al., 2013; Wagner et al., 2013). RyRs are highly sensitive to ROS, and contain multiple redox-sensitive cysteine residues (Zima and Blatter, 2006). Cysteine thiol oxidation of RyR increases channel activity, and many groups including ours have previously established that increased RyR oxidation in VMs from diseased hearts promotes proarrhythmic spontaneous SR Ca2+ release in the form of propagating Ca2+ waves that underlie increased triggered activity (Terentyev et al., 2008; Belevych et al., 2009; Cooper et al., 2013; Kyrychenko et al., 2013; Bovo et al., 2018). Scavenging of mitochondrial ROS was shown to improve Ca2+ homeostasis and attenuate arrhythmic potential in multiple models of cardiac disease including HF, hypertrophy, diabetic cardiomyopathy or aging (Mochizuki et al., 2007; Terentyev et al., 2008; Belevych et al., 2012; Cooper et al., 2013; Luo et al., 2013; Joseph et al., 2016; Kim et al., 2017).
Given the contribution of mitochondrial dysfunction to multiple cardiac disease states, maintaining mitochondrial Ca2+ homeostasis remains an attractive therapeutic target (Dietl and Maack, 2017). In conditions with defective intracellular Ca2+ and Na2+ homeostasis such as in models of HF, increasing [Ca2+]m above a specific threshold was suggested to improve metabolism and substrate utilization, as well as reduce oxidative stress and ROS overload in the myocyte (Liu and O’Rourke, 2008; Kolhaas et al., 2010; Liu et al., 2014). More recently, Schweitzer et al. (2017) suggested that pharmacological enhancement of [Ca2+]m suppressed arrhythmia in a model of catecholaminergic polymorphic ventricular tachycardia (CPVT), a condition characterized by mutations in the RyR macromolecular complex that renders channels hyperactive. Given mitochondria are in close proximity to SR Ca2+ release sites, it has been proposed that increasing mitochondrial Ca2+ uptake may improve buffering capacity (Seguchi et al., 2005; Drago et al., 2012; Zhao et al., 2013), thereby limiting local Ca2+ release events, Ca2+ sparks, which would result in a decrease in generation and propagation velocity of proarrhythmic SCWs. Conversely, a reduction of mitochondrial Ca2+ uptake may serve as an anti-arrhythmic strategy. In models of HF and ischemia-reperfusion, pathological mitochondrial dysfunction and mitochondria Ca2+ overload contribute to oxidative stress and cell death (Santulli et al., 2015). Pharmacological inhibition (García-Rivas Gde et al., 2006; Xie et al., 2018) or genetic ablation (Kwong et al., 2015; Luongo et al., 2015) of MCU, as well as conditional NCLX overexpression (Luongo et al., 2017) has been shown to protect against ischemia-induced myocyte injury, the development Ca2+-dependent arrhythmia and the progression of HF.
In the present study, we aimed to determine the effects of pharmacological facilitation of [Ca2+]m accumulation and inhibition of mitochondrial Ca2+ uptake on intracellular Ca2+ homeostasis and arrhythmic potential using rat model of cardiac hypertrophy induced by thoracic aortic banding (TAB). To achieve this goal, we utilized whole heart optical mapping, genetically encoded ROS and mitochondrial Ca2+ biosensors, confocal microscopy and biochemistry to dissect the influence of MCU enhancer kaempferol and NCLX inhibitor CGP-37157 on intracellular Ca2+ cycling, in both healthy and hypertrophic VMs. Our results suggest that enhancement of mitochondrial Ca2+ accumulation in either setting elevates mitochondrial ROS emission, increasing oxidation of RyR and aberrant spontaneous Ca2+ release. Attenuating mitochondrial Ca2+ uptake serves as an anti-arrhythmic treatment in hypertrophic hearts, whereby triggered activity was reduced by pharmacological inhibition of MCU with Ru360.
Procedures involving animals were approved by The Rhode Island Hospital Institutional Animal Care and Use Committee and followed the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 2011).
The mitochondrial targeting sequence cytochrome C oxidase subunit IV was fused as the N-terminal of the coding sequence of plasmid RCamp1h (Akerboom et al., 2013) to create a probe to monitor intra-mitochondrial Ca2+. pC1-HyPer-3 was a gift from Vsevolod Belousov (Addgene plasmid # 42131). The mitochondrial localization sequence of mAKAP1 followed by a linker was fused as the N-terminus of the coding sequence of pC1-HyPer-3 (Burns-Hamuro et al., 2003; DiPilato et al., 2004; Bilan et al., 2013). This enables anchoring of the probe to the outer mitochondrial membrane (OMM) to measure H2O2 at the mitochondrial surface, and the subsequent viral construct is thus referred to as OMM-HyPer. ERroGFP_iE_pCDNA3 was a gift from David Ron (Addgene plasmid # 47954). The ERroGFP_iE probe is targeted to the endoplasmic reticulum (ER) by the cleavable signal peptide and C-terminal KDEL ER retrieval signal (Avezov et al., 2013). Adenovirus carrying plasmid constructs were generated utilizing the ViraPower Gateway expression system (Thermo Fisher Scientific, Waltham, MA, United States). Briefly, coding regions were cloned into the pENTRTM 1A entry vector, and recombined into pAd/CMV/V5-DESTTM destination vector by LR recombinase reaction. Once sequence-verified, destination vector plasmids were digested with restriction enzyme PacI and transfected into HEK293A cells using LipofectamineTM 2000 (Thermo Fisher Scientific). Adenoviral stock titer was determined using the Adeno-X qPCR Titration Kit (Takara Bio USA, Inc., Mountain View, CA, United States).
Myocytes were isolated from male 9- to 12-week-old Sprague-Dawley rats (controls) from Harlan Laboratories (Indianapolis, IN, United States). Male Sprague-Dawley rats with TAB surgery were purchased from Charles River Laboratories (Wilmington, MA, United States). Animals were shipped 5–7 days after surgery and acclimatized for 3–4 weeks in the Rhode Island Hospital animal facility. Experiments were performed 4–5 weeks after aortic banding procedure.
Bilateral thoracotomy was performed on euthanized rats and the heart plunged into ice cold Tyrode’s solution. The hearts were mounted on a Langendorff apparatus and retrogradely perfused with Tyrode solution (Terentyev et al., 2009) containing collagenase II (Worthington Biochemical Corp., Lakewood, NJ, United States) at 37°C for 16–17 min. Ventricles were minced and placed in a 37°C water bath shaker in collagenase solution. Isolated VMs were plated onto laminin-coated glass coverslips in 24-well plates.
For experiments with cultured control and TAB rats VMs, myocytes were cultured in serum-free medium 199 (Thermo Fisher Scientific), supplemented with 25 mmol/L NaHCO3, 10 mmol/L HEPES, 5 mmol/L creatine, 5 mmol/L taurine, 10 μ/mL penicillin, 10 μg/mL streptomycin and 10 μg/mL gentamycin (pH 7.3). Unattached cells were removed after 1 h and remaining VMs were cultured for 48 h. Cultured VMs were infected with adenoviruses at multiplicity of infection (MOI) of 10 for all described constructs. Myocytes were cultured at 37°C in 95% air, 5% CO2 for 36–48 h before analysis.
Kaempferol directly activates MCU (Montero et al., 2004; Vay et al., 2007), and was obtained from Millipore Sigma (Burlington, MA, United States), used at 10 μmol/L. CGP-37571 inhibits NCLX (Liu and O’Rourke, 2008; Kolhaas et al., 2010; Liu et al., 2010), and was obtained from Millipore Sigma, used at 1 μmol/L. SB 202190 is an inhibitor of p38 mitogen-activated protein (MAP) kinase, has also been shown to activate MCU (Montero et al., 2004), and was obtained from Millipore Sigma, used at 30 μmol/L. Ru360 specifically inhibits mitochondrial Ca2+ uptake through MCU (Matlib et al., 1998; García-Rivas Gde et al., 2006), and was obtained from Millipore Sigma, used at 2 μmol/L. MitoTEMPO, a specific scavenger of mitochondrial superoxide was obtained from Millipore Sigma, used at 20 μmol/L. Dantrolene, an RyR antagonist (Kobayashi et al., 2009; Maxwell et al., 2012) was obtained from Millipore Sigma and used at 2 μmol/L.
Confocal imaging was performed using a Leica SP5 II confocal microscope equipped with 63 × 1.4 numerical aperture oil objective in linescan and x–y mode. All confocal imaging experiments were performed under β-adrenergic stimulation with 50 nmol/L isoproterenol (ISO, Millipore Sigma). Control VMs were paced via field stimulation at 2 Hz, while TAB VMs were paced at 0.5 Hz using extracellular platinum electrodes. Myocytes were studied in Tyrode’s solution (Terentyev et al., 2009). Confocal imaging data were analyzed using Leica Software, Origin 8.0 (OriginLab, Northampton, MA, United States) and ImageJ (National Institutes of Health, Bethesda, MA, United States).
Intact VMs were loaded with Rhod-2 AM (Thermo Fisher Scientific) at room temperature for 12 min, followed by a 10 min wash. Rhod-2 was excited using 543 nm line of HeNe laser and fluorescence emission was collected at 560–660 nm wavelengths in linescan mode at 200 Hz sampling rate. Calcium transients were recorded at room temperature. To test for the propensity of triggered activity, VMs were paced for 20 s and latency between the last pacing stimulus and the subsequent SCW was calculated. To assess SR Ca2+ load, 10 mmol/L caffeine was applied at the end of experiments. The data is presented as ΔF/F0, where F0 is basal fluorescence and ΔF = F – F0.
Biosensor mtRCamp1h was excited using 543 nm line of HeNe laser and fluorescence emission was collected at 560–660 nm wavelengths. For permeabilized VM experiments, myocytes were saponin-permeabilized (0.001%) and equilibrated with a solution containing thapsigargin (10 μmol/L), cytochalasin D (10 μmol/L), FCCP (20 μmol/L), and ionomycin (5 μmol/L). Tyrode’s solution containing Ca2+ buffer EGTA (2 mmol/L) was applied to obtain minimum mtRCamp1h fluorescence. Maximum fluorescence was achieved by application of Ca2+ (100 μmol/L). The data is presented as as ΔF/F0, where F0 is basal fluorescence and ΔF = F – F0. Biosensors ERroGFP_iE and OMM-HyPer were excited using 488 nm line of Argon laser and fluorescence emission was collected at 500–550 nm wavelengths, measured in x–y mode. Maximum fluorescence (Fmax) was obtained by application 200 μmol/L DTDP and minimum fluorescence was obtained by application ROS scavenger DTT (5 mmol/L). Data is presented as a percentage of ΔF/ΔFmax where ΔF = F – Fmin, and ΔFmax = Fmax–Fmin. Mitochondrial membrane potential was monitored with the voltage-sensitive fluorescent indicator, tetramethylrhodamine, methyl ester (TMRM; Thermo Fisher Scientific). Isolated VMs were loaded with 20 μmol/L for 1 min and washed thoroughly prior to imaging. TMRM was excited using 543 nm line of HeNe laser and fluorescence emission was collected at 560–660 nm wavelengths, measured in x–y mode. Fluorescence of TMRM was normalized to the minimum fluorescence signal obtained by application of mitochondrial uncoupler, carbonyl p-(trifluoromethoxy) phenylhydrazone (FCCP, 50 μmol/L). For experiments with mitoTEMPO, isolated VMs were pretreated with mitoTEMPO (20 μmol/L, 30 min), before loading with TMRM as described.
The emission of ROS was measured in isolated VMs in Tyrode solution using MitoSOX Red mitochondrial superoxide indicator (Thermo Fisher Scientific; 20 μmol/L, 30 min loading). The indicator was excited with 514 nm line of an argon laser and emission was collected at 560–660 nm, measured in x–y mode. Fluorescence of MitoSOX was normalized to the maximum fluorescence signal obtained by application of peroxide H2O2 (10 mmol/L).
Freshly isolated control or TAB rat VMs were treated with ISO (50 nmol/L, 5 min total), kaempferol (10 μmol/L, 5 min total) or CGP (1 μmol/L, 10 min total) and paced for 1 min at 2 Hz at room temperature before immediate lysis in lysis buffer from Cell Signaling (Danvers, MA, United States, Cat#9803S), supplemented with phosphatase (Calbiochem, San Diego, CA, United States, Cat#524625) and protease inhibitor cocktails (Millipore Sigma, Cat#P8340) as described previously (Terentyev et al., 2014). For co-immunoprecipitation, Pierce Co-immunoprecipitation Kit (Thermo Fisher Scientific, Cat#26149) was used. Lysate (500 μl) was pre-cleared with Control Agarose Resin for 30 min at 4°C, centrifuged at 1,000 × g for 1 min. Flow-through was incubated with antibody-coupled resin (anti-RyR2, Thermo Fisher Scientific, Cat#MA3-916 and negative control antibody, normal mouse IgG, Santa Cruz Biotechnology, Cat#sc-2025) for 2 h at 4°C. Columns were washed three times. Protein complexes were eluted with elution buffer provided in the kit. To determine oxidation of RyR, the Oxidized Protein Western Blot Kit (Abcam, Cambridge, MA, United States, Cat#ab178020) was used, whereby carbonyl groups of immunoprecipitated RyR2 were derivatized to 2,4 dinitrophenylhydrazone (DNP) by reaction with 2,4 dinitrophenylhydrazine. For control we used Derivatization Control Solution, provided in the kit. The DNP-RyR2 protein samples were separated on 4–20% Mini-PROTEAN TGX gels (Bio-Rad Laboratories, Hercules, CA, United States, Cat#456-1094) and DNP-associated signal assessed by the kit-provided anti-DNP primary antibody and anti-RyR2 (Thermo Fisher Scientific, Cat#MA3-916), followed by HRP-conjugated secondary antibody and anti-mouse lgG(H+L) HRP secondary antibody (Promega, Madison, WI, United States, Cat#W4021). Abcam antibodies (Cat#ab57602 and Cat#ab101055) were used to assess expression levels of mitofusin 1 and mitofusin 2. Anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies were used for loading control (Abcam, Cat#ab8245). Blots were developed with ECL (Bio-Rad Laboratories) and quantified using ImageJ and Origin 8 software.
Beating hearts were harvested from anesthetized TAB rats via thoracotomy and were retrogradely perfused through the aorta in a Langendorff perfusion system (Radnoti Glass Technology, Monrovia, CA, United States) with (in mmol/L): 130 NaCl, 24 NaHCO3, 1.0 MgCl2, 5.0 KCl, 1.2 NaH2PO4, 5 dextrose, and 1 CaCl2, at pH 7.4, gassed with 95% O2 and 5% CO2. Constant flow perfusion was set to 10 mL/min with a peristaltic pump. Hearts were placed in a water-heated chamber to maintain temperature at 37 ± 0.2°C, and 5 μmol/L blebbistatin was added to perfusate to reduce movement artifact. Hearts were loaded with Ca2+ indicator Rhod-2 AM (Thermo Fisher Scientific), using 25 μL of stock solution (1 mg/mL of DMSO) delivered through a bubble trap, above the aortic cannula. The ECGs were continuously monitored with a Powerlab system (AD Instrument, Colorado Springs, CO, United States). The optical apparatus has been described previously (Kim et al., 2015). Fluorescence images of Rhod-2 signal were recorded from the anterior surface of the heart using a CMOS camera (100 × 100 pixels, 2000 frames/sec, 1.5 cm × 1.5 cm field of view, Ultima-L, SciMedia, Japan). Drugs (kaempferol and Ru360) were perfused for 20–30 min and ISO (50 nmol/L) was added to investigate the effect of drugs on VT/VF induction in TAB hearts. The fluorescence (F) from Rhod-2 was normalized with ΔF/F. Hearts were stimulated with 150 ms cycle length followed by premature stimulation of 10 beats of S2 until refractoriness or VT induction. Propagation and duration of Ca2+ transients were mapped using (dF/dt)max and 75% recovery, respectively, as previously described (Kim et al., 2015).
Statistical analysis of Ca2+ imaging and biochemical data was performed using Origin 8 (OriginLab). Data are presented as mean ± standard error (SEM) for single cell and ± standard deviation (SD) for intact heart optical mapping. Uppercase n (N) = number of animals, lowercase n = number of VMs. Statistical significance between groups were performed using Student’s t-test (paired and unpaired), Fisher’s exact test and one-way ANOVA with Bonferroni post hoc test where appropriate. For all analyses, a p-value of less than 0.05 was considered significant.
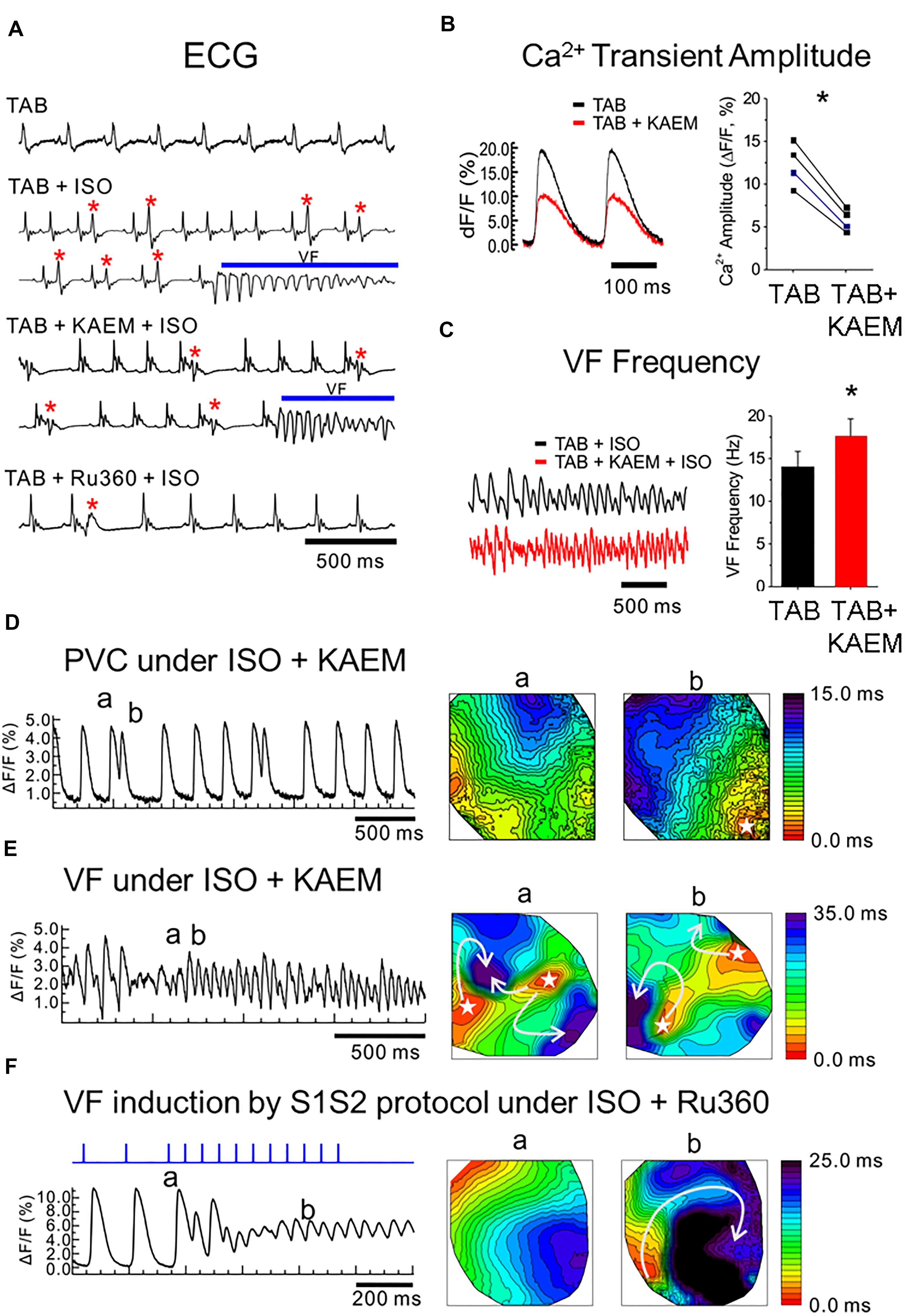
Figure 1. MCU enhancer kaempferol exacerbates ventricular arrhythmias in TAB rat hearts. (A) ECG recordings from ex vivo intact hearts. TAB: ECGs under normal condition. TAB+ISO: ISO (50 nmol/L) perfusion frequently induces PVCs (red stars) and VT/VF in TAB rat hearts. TAB+KAEM+ISO: Pretreatment of kaempferol (KAEM, 1 μmol/L) failed to prevent PVCs and VT/VF induction. TAB+RU360+ISO: Ru360 pretreatment (2 μmol/L) reduced PVC events (red star) and prevented VT/VF induction under ISO in TAB rat hearts. (B) KAEM reduced Ca2+ transient amplitude (12.3 ± 2.6 ΔF/F before KAEM vs. 5.8 ± 1.3 ΔF/F after KAEM, ∗p = 0.0018, paired Student’s t-test, N = 4). (C) KAEM increases VF frequency (14.1 ± 1.8 Hz before KAEM vs. 17.6 ± 2.1 Hz after KAEM). (D,E) Frequent PVCs before VF induction and focal activity in the presence of KAEM. Activation maps are shown in the right panel. (F) Conduction block and reentry formation by S1S2 pacing protocol in the presence of Ru360 (N = 4).
We have previously reported that the rat model hypertrophy induced by TAB is highly arrhythmogenic, with incidences of non-sustained ventricular tachycardia and fibrillation (VT/VF) occurring in 100% of TAB hearts exposed to 50 nmol/L ISO (Kim et al., 2017). We investigated whether the MCU activator, kaempferol (Montero et al., 2004; Vay et al., 2007), increases mitochondrial Ca2+ accumulation to reduce cytosolic Ca2+ and suppress ventricular arrhythmias in this model. Hearts were isolated and perfused retrogradely as described in “Materials and Methods” and echocardiograms (ECGs) were monitored to investigate arrhythmogenesis under kaempferol and Ru360. ISO induced frequent premature ventricular contractions (PVCs) leading to VFs in TAB rat hearts (Figure 1A ECG traces under TAB+ISO). Pretreatment with MCU activator, kaempferol (10 μmol/L), did not prevent PVCs and VF induction. ECG traces in Figure 1A (TAB+KAEM+ISO panel) shows frequent PVCs (red stars) that led to VFs (blue bar) under kaempferol. However, MCU blocker Ru360 (2 μmol/L) suppressed number of PVCs and prevented VFs in TAB rat hearts (Figure 1A, TAB+Ru360+ISO panel).
We investigated the effect of kaempferol on Ca2+ handling and arrhythmogenesis using optical mapping. Kaempferol reduces the Ca2+ transient amplitude by 53% (Figure 1B, ∗p = 0.002) but despite smaller Ca2+ transients, kaempferol induced transient VTs in two of four TAB hearts, even without ISO. Addition of ISO caused frequent PVCs (Figure 1D) and long-lasting VFs (Figure 1E) in three of four hearts. Activation maps of PVCs (Figure 1D) and VFs (Figure 1E) suggest that focal activity play a major role in the initiation and maintenance of VFs. The frequencies of VF were significantly higher with kaempferol (17.6 ± 2.1 Hz, n = 3, in kaempferol vs. 14 ± 1.8 Hz, n = 7, control TAB with ISO, ∗p = 0.042, Figure 1C). In contrast, MCU inhibitor, Ru360 (Matlib et al., 1998; García-Rivas Gde et al., 2006), suppressed spontaneous VT/VFs in the presence of ISO in four of four hearts. S1S2 pacing induced reentry and VFs in two of four hearts in the presence of Ru360 (Figure 1F), suggesting that conduction block, not focal activity, underlies VF induction during S1S2 pacing in Ru360 group. These intact heart optical mapping data suggest that enhancement of mitochondrial Ca2+ accumulation may exacerbate ventricular arrhythmias in TAB rat hearts through increasing focal activity.
To gain mechanistic insights as to how increasing mitochondrial Ca2+ accumulation affects global Ca2+ handling in VMs, we used a genetically encoded fluorescent Ca2+ biosensor, mtRCamp1h. The RCamp1h indicator, with a Kd ∼ 1.3 μM (Akerboom et al., 2013), was fused with an N-terminal cytochrome C oxidase subunit IV tag for targeting to the mitochondrial matrix. We generated adenovirus encoding the sensor, and VMs isolated from control rat hearts were infected with adenovirus at a MOI of 10 and cultured for 48 h prior to experimentation. Rat myocytes are thought to preserve electrical properties and structure including T-tubule organization for the first 48 h of culture (Banyasz et al., 2008).
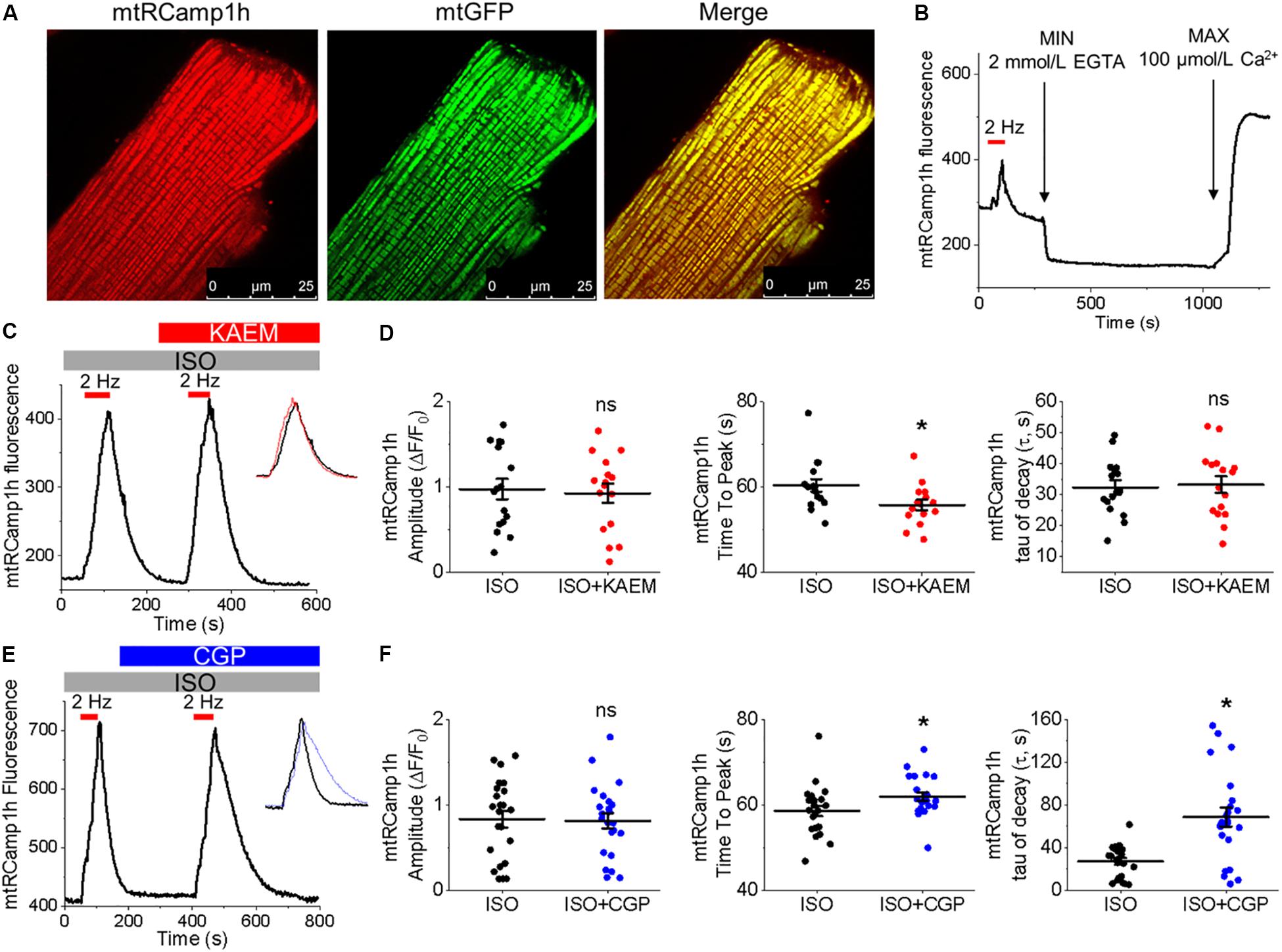
Figure 2. Expression of biosensor mtRCamp1h in cultured control rat VMs reveals mitochondrial Ca2+ accumulation is modified by kaempferol and CGP-37157. (A) Representative images of a cultured VM 48 h after adenoviral infection with mtRCamp1h (left, red, MOI of 10) and mitochondrial-targeted GFP (center, green, MOI of 10). A merged image is indicated on the right. (B) Recording of a permeabilized VM infected with mtRCamp1h. Fmax and Fmin were assessed by addition of EGTA (2 mmol/L) and Ca2+ (100 μmol/L), respectively. (C) Representative trace from cultured VM expressing mtRCamp1h. VMs were treated with ISO (50 nmol/L) and paced at 2 Hz for 60 s intervals (red bars), then treated with kaempferol (KAEM, 10 μmol/L) and paced again peaks are overlaid in the inset for visualization, with ISO in black, ISO+KAEM in red. Graphs in (D) depict mean data ± SEM of mtRCamp1h transient amplitude, time to transient peak and tau of decay (n = 16 per group, N = 5, ∗p < 0.05, paired Student’s t-test). (E) Representative trace from VM expressing mtRCamp1h. VMs were treated with ISO and paced at 2 Hz for 60 s intervals (red bars), then treated with CGP-37157 (CGP, 1 μmol/L) and paced again. Peaks are overlaid in the inset for visualization, with ISO in black, ISO+CGP in blue. Graphs in (F) depict mean data ± SEM from mtRCamp1h transient amplitude, time to transient peak and tau of decay (n = 23, N = 4, ∗p < 0.05, paired Student’s t-test).
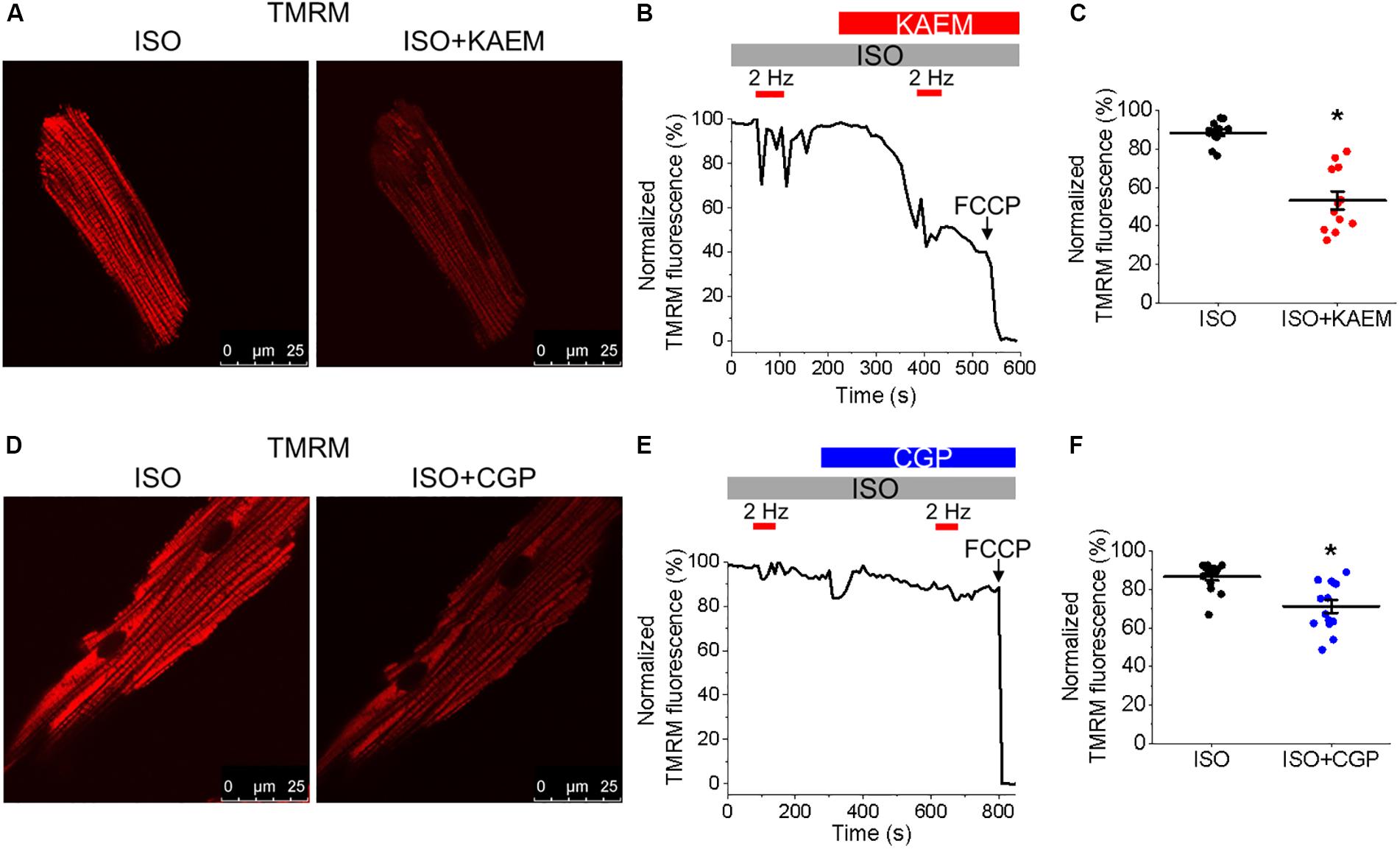
Figure 3. Pharmacological enhancers of mitochondrial Ca2+ accumulation modulate Δψm in cultured control rat VMs. (A) Representative images of a VM treated with ISO (50 nmol/L) before and after application of kaempferol (KAEM, 10 μmol/L). Δψm of VMs was monitored by TMRM fluorescence (20 μmol/L). (B) Representative recording of changes in Δψm in response to application of ISO and pacing at 2 Hz (red bars) for 60 s, followed by application of KAEM. TMRM signal was normalized to minimum fluorescence obtained by application of FCCP (50 μmol/L) and represented as percentage of baseline. Graph in (C) depicts pooled data for minimum fluorescence after pacing and application of ISO or KAEM (n = 12, N = 4, ∗p < 0.05, paired Student’s t-test). (D) Representative images of a VM treated with ISO before and after application of CGP-37157 (CGP, 1 μmol/L). Δψm of VMs was monitored by TMRM fluorescence. (E) Representative recordings of changes in Δψm in response to application of ISO and pacing at 2 Hz for 60 s, followed by application of CGP. Graph in (F) depicts pooled mean data ± SEM for minimum fluorescence after pacing and application of ISO or KAEM (n = 14, N = 3, ∗p < 0.05, paired Student’s t-test).
The correct cellular localization of mtRCamp1h was confirmed with co-expression of mitochondrial matrix-targeted GFP, as shown in Figure 2A. As shown in Figure 2B, basal mtRCamp1h fluorescence appears well within the dynamic range of the probe when adenovirally expressed in VMs. Furthermore, as seen in Figure 2B, we can indeed observe measurable changes in mitochondrial Ca2+ concentration, as indicated by an increase in mtRCamp1h fluorescence, when cultured control VMs are paced at 2 Hz for 1 min under β-adrenergic stimulation with ISO (pacing indicated by red bar). We did not observe significant loading of mitochondria with Ca2+ under baseline conditions with no ISO stimulation (data not shown). To determine Fmin and Fmax for mtRCamp1h, VMs were saponin-permeabilized (0.001%) and equilibrated with a solution containing thapsigargin (10 μmol/L) to deplete SR Ca2+, cytochalasin D (10 μmol/L) to reduce cell contraction, as well as FCCP (20 μmol/L) and ionomycin (5 μmol/L) to dissipate mitochondrial membrane potential (Δψm). Solution containing Ca2+ buffer EGTA at high concentration (2 mmol/L) was applied to obtain minimum mtRCamp1h fluorescence, while maximum fluorescence was achieved by application of Ca2+ (100 μmol/L), as illustrated in Figure 2B.
Measurement of peak mtRCamp1h signal indicated accumulation of [Ca2+]m during workload, but after treatment with MCU enhancer kaempferol (10 μmol/L; Figure 2C) or NCLX blocker CGP-37157 (1 μmol/L; Figure 2E), no significant change in transient amplitude (an increase in [Ca2+]m) was observed (Figures 2D,F, respectively). However, accumulation of mitochondrial Ca2+ during pacing was significantly faster after treatment with kaempferol (Figure 2D, time to peak 59.31 ± 1.01 s. ISO vs. 55.84 ± 1.24 s. ISO and kaempferol, ∗p = 0.03), while the time constant of transient decay, τ, was significantly increased after application of CGP-37157 (Figure 2F, τ = 27.16 ± 3.10 ISO vs. 68.70 ± 8.81 ISO and CGP-37157, ∗p < 0.001). The time to peak of transient after application of CGP-37157 was also increased (58.59 ± 1.23 s. ISO vs. 62.01 ± 0.98 s. ISO and CGP-37157, ∗p = 0.01). This indicates that while pharmacological enhancers of [Ca2+]m do not increase overall mitochondrial Ca2+ loading in cultured control VMs during workload of 1 min pacing, they modify the time course for which those VMs accumulate or retain Ca2+. The finding that the rate of mitochondrial Ca2+ uptake in intact myocytes is only modestly accelerated with kaempferol and even slowed down with CGP-37157 suggest the existence of overriding feedback mechanisms to prevent mitochondrial Ca2+ overload that can cause cell death (Broekemeier et al., 1998; Hüser and Blatter, 1999; Elrod et al., 2010).
It was established that the excessive mitochondrial Ca2+ uptake can be effectively limited by a reduction in Δψm at least in part via activation of mitochondrial Ca2+-dependent K+ channels (O’Rourke et al., 2005; Stowe et al., 2006). While irreversible mPTP opening collapses Δψm, brief openings of the pore may also provide a protective efflux mechanism against mitochondrial Ca2+ overload (Broekemeier et al., 1998; Elrod et al., 2010). We used isolated control rat VMs stained with voltage sensitive dye TMRM (20 μmol/L for 1 min) to determine if enhancement of mitochondrial Ca2+ accumulation modifies Δψm (Figure 3). As in Figure 2, VMs under β-adrenergic stimulation with ISO were paced at 2 Hz for 1 min, as indicated by red bars. Representative images Figure 3A shows VMs before and after the application of kaempferol. A representative trace is shown in Figure 3B, whereby signal was normalized to minimum fluorescence obtained by the application of FCCP (50 μmol/L). Application of kaempferol significantly reduced TMRM fluorescence (Figure 3C, 88.31 ± 1.73% ISO vs. 53.17 ± 4.69% ISO and kaempferol, ∗p < 0.001). Application of CGP-37157 had similar effects (Figures 3D–F, 86.49 ± 1.95% ISO vs. 71.15 ± 3.38% ISO and CGP-37157, ∗p < 0.001, respectively). The decrease in driving force due to the drop in Δψm may explain why kaempferol or CGP-37157 are not able to increase the amplitude of [Ca2+]m effectively limiting mitochondrial Ca2+ uptake, as measured with mtRCamp1h in Figure 2.
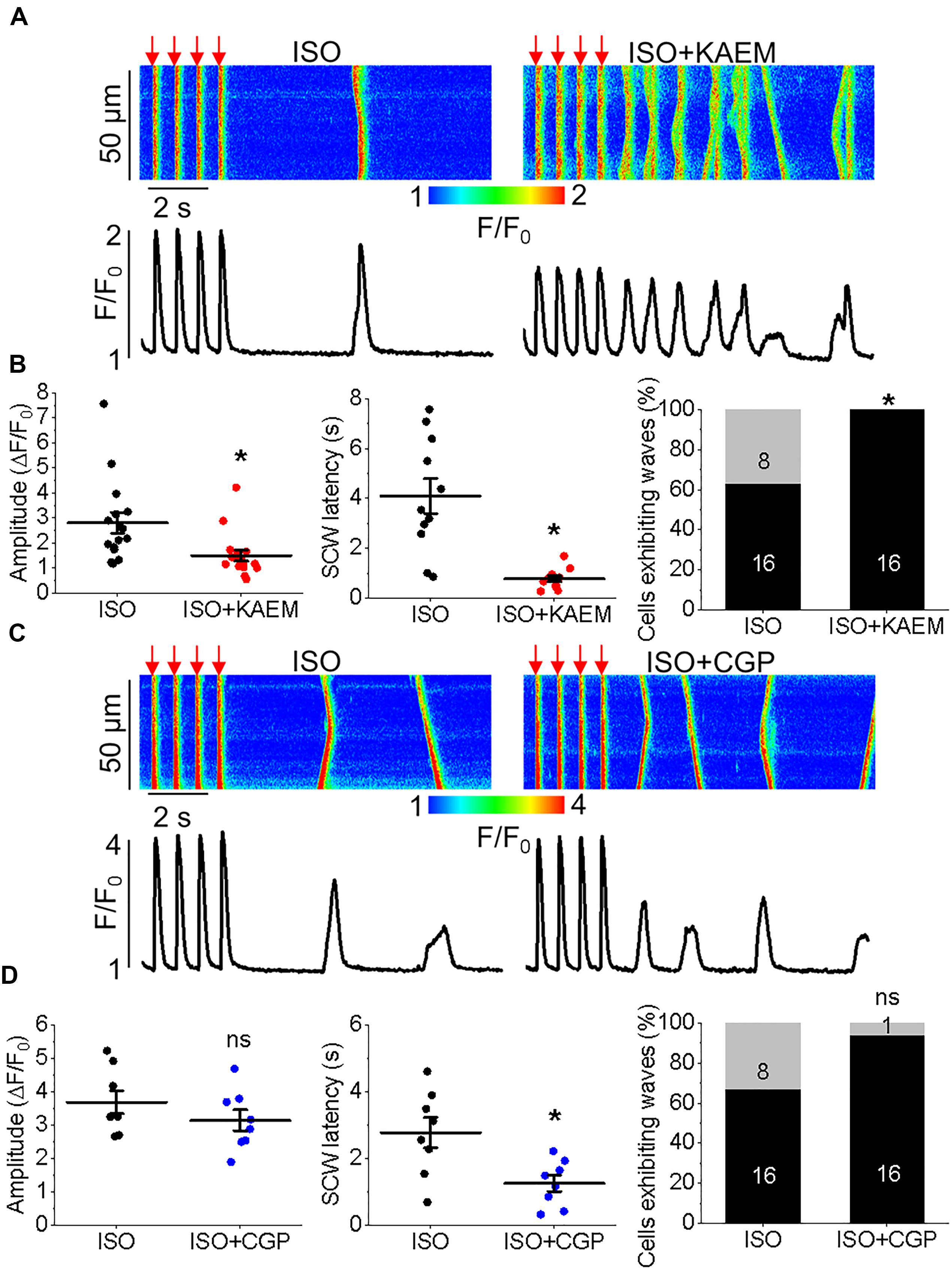
Figure 4. Enhanced mitochondrial Ca2+ accumulation promotes proarrhythmic spontaneous Ca2+ waves in control cultured rat VMs. (A) Representative confocal line scan images of Ca2+ transients and Rhod-2 fluorescence (F/F0) profiles ISO treated (50 nmol/L) rat VM undergoing 2 Hz pace-pause protocol (red arrows) to induce SCW, before and after application of kaempferol (KAEM, 10 μmol/L). Graphs in (B) depict mean data ± SEM from Ca2+ transient amplitude (n = 15, N = 5, ∗p < 0.05, paired Student’s t-test), SCW latency (n = 11, N = 5, paired Student’s t-test) and the percentage of cells exhibiting waves (N = 5, ∗p < 0.05, Fisher’s exact test). (C) Representative confocal line scan images of Ca2+ transients and Rhod-2 fluorescence (F/F0) profiles of ISO treated rat VM undergoing 2 Hz pace-pause protocol before and after application of CGP-37157 (CGP, 1 μmol/L). Graphs in (D) depict mean data ± SEM from Ca2+ transient amplitude (n = 7, N = 4, ns is not significant, paired Student’s t-test), SCW latency (n = 8, N = 4, ∗p < 0.05, paired Student’s t-test) and the percentage of cells exhibiting waves (N = 4, ns is not significant, Fisher’s exact test).
Having demonstrated the effects of kaempferol and CGP-37157 on mitochondrial Ca2+ and Δψm, we next sought to establish the effects of modulating mitochondrial Ca2+ on cytosolic Ca2+ handling in VMs, as illustrated in Figure 4. Cultured control VMs were loaded with the fluorescent Ca2+ indicator Rhod-2 and we recorded cytosolic Ca2+ in the presence of β-adrenergic receptor agonist ISO (50 nmol/L), subjected to a burst-pace pause protocol (2 Hz, 20 s). We assessed Ca2+ transient amplitude and SCW latency as an indication of the propensity for arrhythmogenic Ca2+ release. As shown in Figures 4A,B, VMs treated with ISO and kaempferol have a significantly lower Ca2+ transient amplitude in comparison to ISO alone (2.77 ± 0.44 ΔF/F0 ISO vs. 1.57 ± 0.23 ΔF/F0 ISO and kaempferol, ∗p = 0.006). We observed a fivefold shortening in SCW latency after treatment with kaempferol (∗p < 0.001), as well a significant increase in the percentage of VMs exhibiting SCWs (63% ISO vs. 100% ISO and kaempferol, ∗p < 0.001). A similar pattern was obtained in VMs treated with CGP-37157 (Figures 4C,D), whereby the SCW latency was shortened approximately twofold (∗p = 0.002). Changes in Ca2+ transient amplitude and the percentage of cells exhibiting waves were not significant on treatment with CGP-37157.
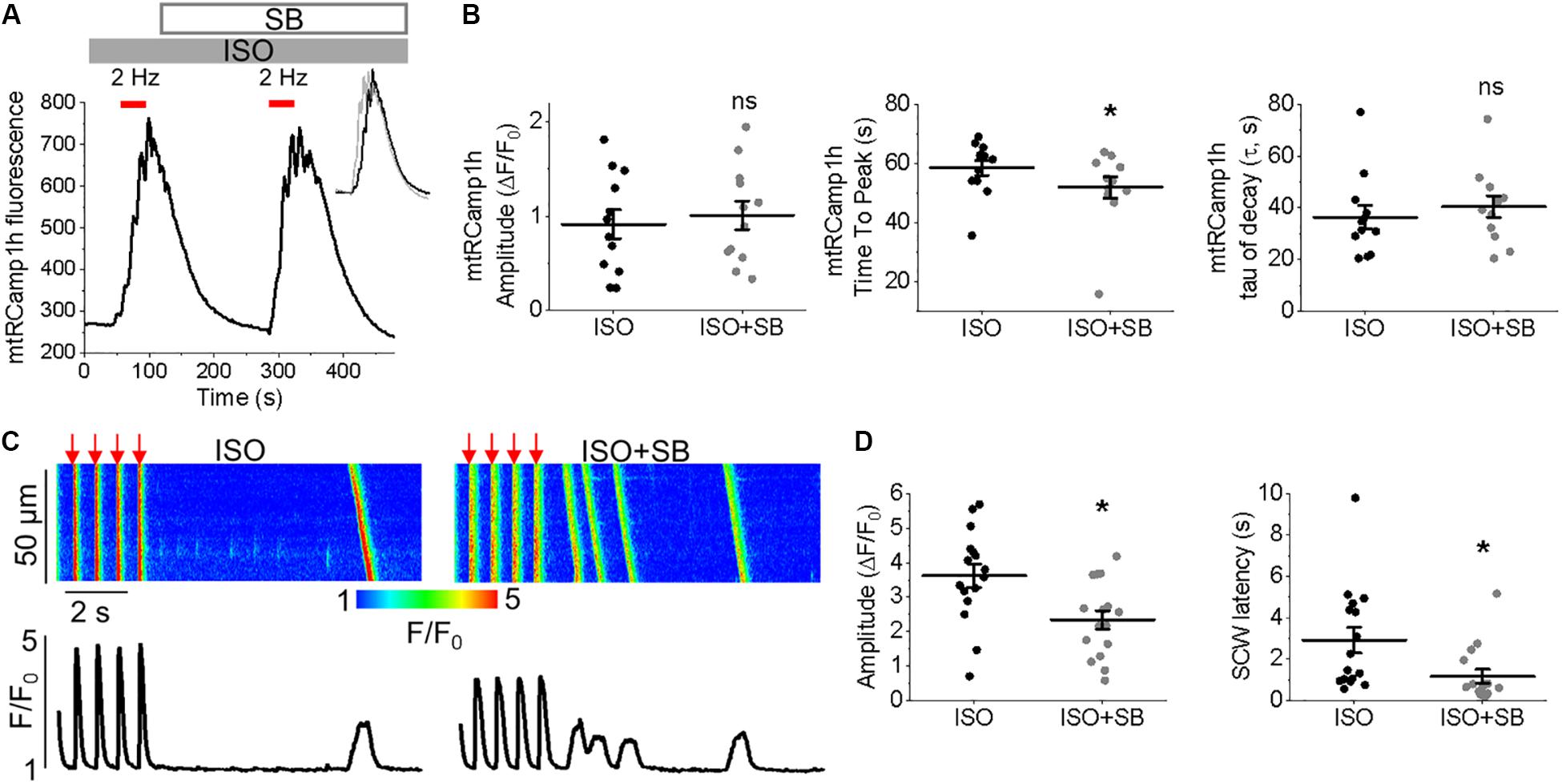
Figure 5. MCU enhancer SB 202190 has similar effects to kaempferol in cultured control rat VMs. (A) Representative trace from cultured VM expressing mtRCamp1h. VMs were treated with ISO (50 nmol/L) and paced at 2 Hz for 60 s intervals (red bars), then treated with SB 202190 (SB) and paced again. Peaks are overlaid in the inset for visualization, with ISO in black, ISO+SB in gray. Graphs in (B) depict mean data ± SEM of mtRCamp1h transient amplitude, time to transient peak and tau of decay (n = 12 per group, N = 3, ∗p < 0.05, paired Student’s t-test). (C) Representative confocal line scan images of Ca2+ transients and Rhod-2 fluorescence (F/F0) profiles ISO treated (50 nmol/L) rat VM undergoing 2 Hz pace-pause protocol (red arrows) to induce SCW, before and after application of SB 202190 (SB, 30 μmol/L). Graphs in (D) depict mean data ± SEM from Ca2+ transient amplitude (n = 16, N = 3, ∗p < 0.05, paired Student’s t-test) and SCW latency (n = 16, N = 3, ∗p < 0.05, paired Student’s t-test).
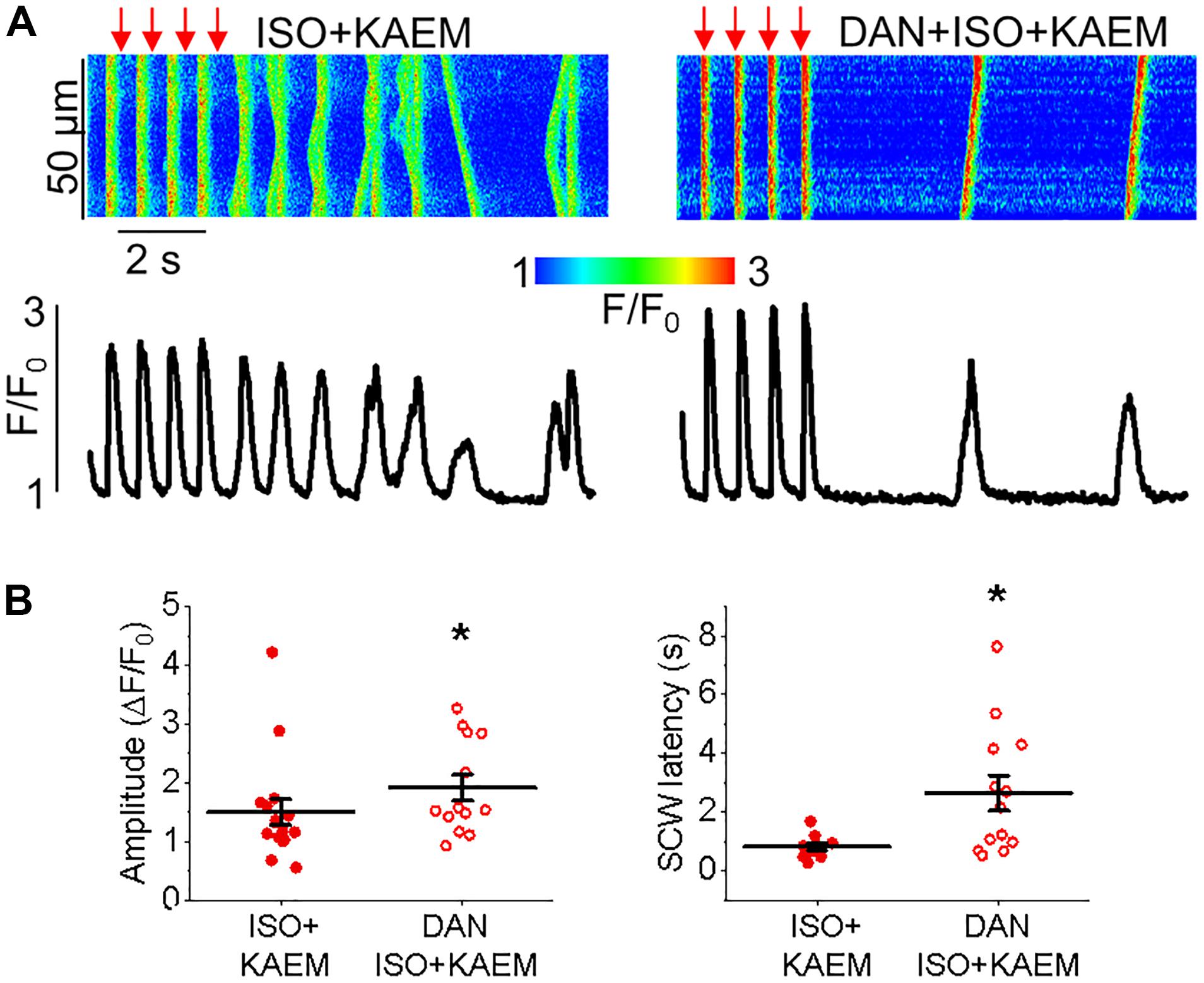
Figure 6. Inhibition of RyR with dantrolene reduces proarrhythmic effects of kaempferol on Ca2+ handling in cultured control rat VMs. (A) Representative confocal line scan images of Ca2+ transients and Rhod-2 fluorescence (F/F0) profiles of TAB rat VMs undergoing 2 Hz pace-pause protocol (red arrows) to induce SCWs. VMs were treated with isoproterenol (ISO, 50 nmol/L) and kaempferol (KAEM, 10 μmol/L), or were pretreated for 5 min with dantrolene (DAN, 2 μmol/L) before ISO and KAEM treatment. Graphs in (B) depict mean data ± SEM from Ca2+ transient amplitude (n = 13–15, N = 3–4, ∗p < 0.05, Student’s t-test) and SCW latency (n = 11–13, N = 3–4, ∗p < 0.05, Student’s t-test).
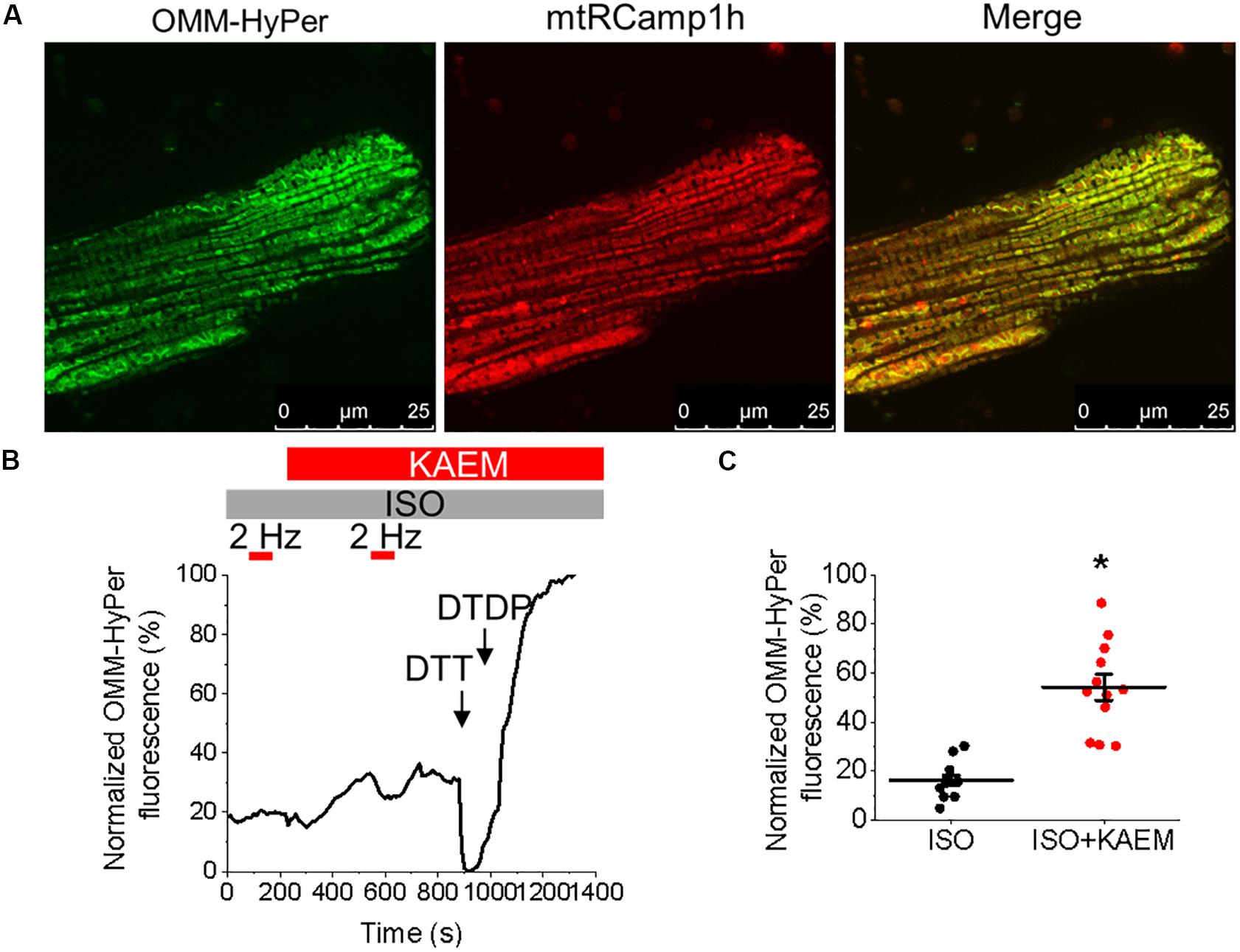
Figure 7. Pharmacological enhancement of mitochondrial Ca2+ accumulation increases mitochondrial ROS in cultured control rat VMs. (A) Representative image of a cultured control VM 48 h after adenoviral infection with biosensor OMM-HyPer (left, green, MOI of 10) and mtRCamp1h (center, red, MOI of 10). A merged image is indicated on the right. (B) Representative trace of OMM-HyPer fluorescence from a VM treated with ISO (50 nmol/L), paced at 2 Hz (red bars) and subsequently treated with kaempferol (KAEM, 10 μmol/L) before further pacing. OMM-HyPer signal was normalized to minimum fluorescence obtained by application of dithiothreitol (DTT, 5 mmol/L) and maximum fluorescence obtained by application of deoxythymidine diphosphate (DTDP, 200 μmol/L). Graph in (C) depicts mean data ± SEM for maximum normalized fluorescence after pacing and application of ISO and KAEM (n = 12, N = 4, ∗p < 0.05, paired Student’s t-test).
To provide additional evidence that facilitation of mitochondrial Ca2+ accumulation exerts strong deleterious effects on intracellular Ca2+ handling we performed experiments using another MCU enhancer SB 202190 (Montero et al., 2004) (Figure 5). As seen from experiments in ISO-treated VMs expressing mtRCamp1h, application of SB 202190 (30 μmol/L) moderately but significantly accelerated rate of mitochondrial Ca2+ uptake while failed to increase overall amplitude of mitochondrial Ca2+ transients (Figures 5A,B), in line with the effects of kaempferol (Figure 2C). Importantly, similar to kaempferol, incubation with SB reduced amplitude of Ca2+ transients and shortened and latency of SCWs in periodically paced ISO-treated cells (Figures 5C,D).
Enhanced propensity to generate SCWs is often attributed to abnormally high activity of SR Ca2+ release channels, RyRs (Bers, 2002). To test whether stabilization of RyRs can attenuate kaempferol–mediated effects on Ca2+ handling, we treated VMs with dantrolene (2 μmol/L, 5 min), a specific inhibitor of RyR. Figure 6 shows that dantrolene restores Ca2+ transient amplitude and significantly reduces kaempferol-mediated shortening of SCW latency in ISO-treated VMs (SCW latency 3.34 ± 0.73 s ISO and kaempferol vs. 2.64 ± 0.60 s ISO, kaempferol and dantrolene, ∗p < 0.001).
Taken together, these data suggest that facilitation of mitochondrial Ca2+ accumulation promotes generation of proarrhythmic SCWs by enhancement of activity of RyRs.
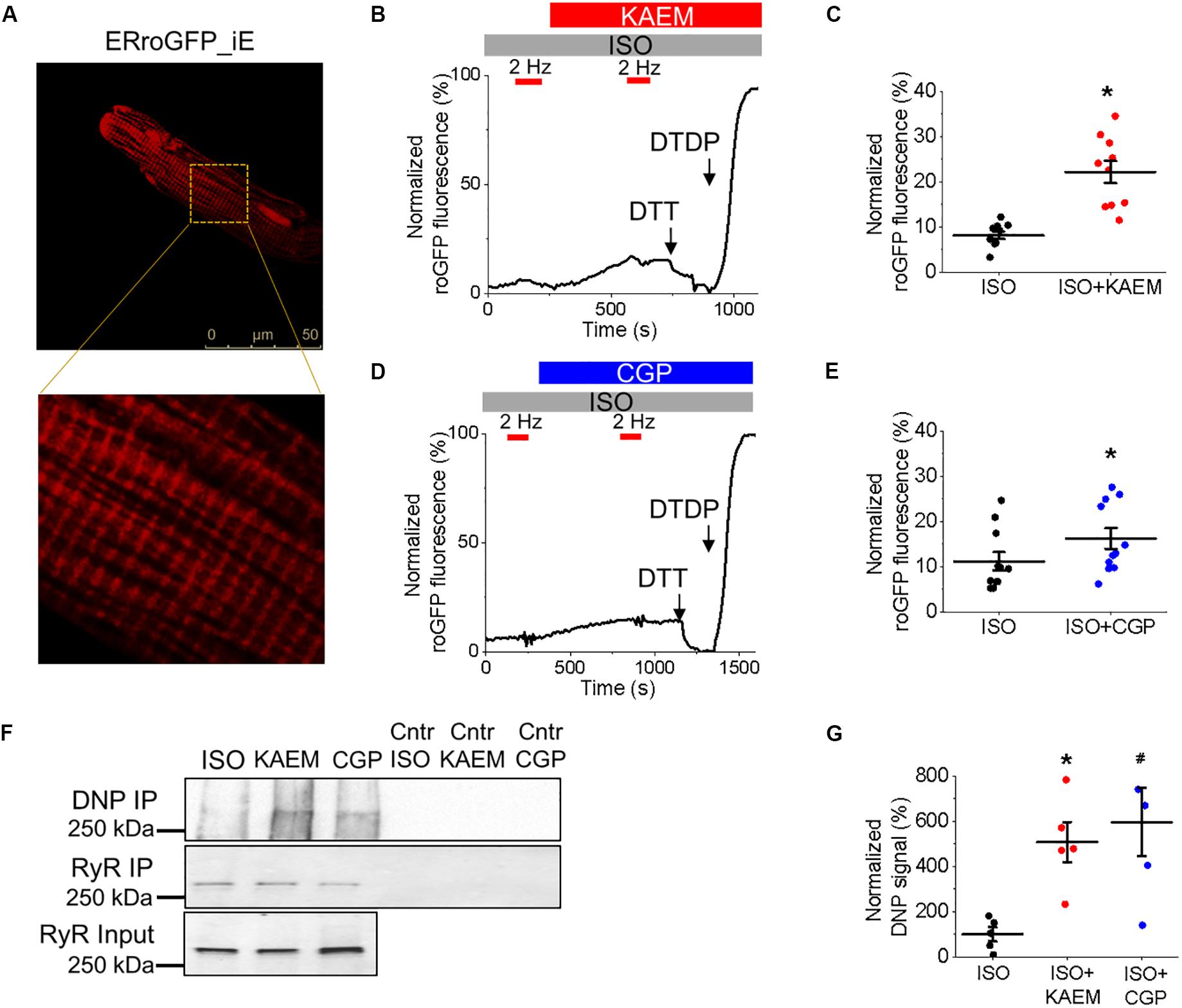
Figure 8. Pharmacological enhancement of mitochondrial Ca2+ accumulation increases ROS in the vicinity of RyR in cultured control rat VMs. (A) Representative image of a cultured control VM 48 h after adenoviral infection with biosensor ERroGFP_iE (MOI of 10). (B) Representative trace of ERroGFP_iE fluorescence from a VM treated with ISO (50 nmol/L), paced at 2 Hz (red bars) and subsequently treated with kaempferol (KAEM, 10 μmol/L) before further pacing. ERroGFP_iE signal was normalized to minimum fluorescence obtained by application of dithiothreitol (DTT, 5 mmol/L) and maximum fluorescence obtained by application of deoxythymidine diphosphate (DTDP, 200 μmol/L). Graph in (C) depicts mean data ± SEM for maximum normalized fluorescence after pacing and application of ISO and KAEM (n = 10, N = 3, ∗p < 0.05, paired Student’s t-test). (D) Representative trace of ERroGFP_iE fluorescence from a VM treated with ISO, paced at 2 Hz and subsequently treated with CGP-37157 (CGP, 1 μmol/L) before further pacing. ERroGFP_iE signal was normalized to minimum fluorescence obtained by application of DTT and maximum fluorescence obtained by application of DTDP. Graph in (E) depicts mean data ± SEM for maximum fluorescence after pacing and application of ISO and CGP (n = 11, N = 3, ∗p < 0.05, paired Student’s t-test). (F) RyR in freshly isolated control VMs was immunoprecipitated (IP) and immunoblotted for oxidation using DNP antibody. Representative images of DNP IP, RyR IP and RyR input signal from VMs treated with ISO, ISO and KAEM, or ISO and CGP, with controls (Cntr). (G) Quantification of normalized DNP signal (%, N = 5, ∗p and #p < 0.05 vs. ISO, one-way ANOVA with Bonferroni post hoc test).
RyR is well established as a ROS sensor, with increased oxidation and thus RyR activity associated with enhanced propensity for spontaneous SR Ca2+ release and proarrhythmic SCWs. Mitochondria is a major intracellular source of ROS and excessive RyR oxidation by mitochondria-derived ROS has been demonstrated in many models of HF and aging (Zima and Blatter, 2006). Facilitation of mitochondrial Ca2+ uptake may accelerate the rate of emission of ROS by stimulating electron transport (Bertero and Maack, 2018). Therefore, to test the possible effects of kaempferol on mitochondrial redox state, control VMs were infected (MOI of 10) with adenovirus construct carrying sequence encoding the novel H2O2 probe OMM-HyPer, and cultured for 48 h prior to imaging. As shown in Figure 7, application of kaempferol significantly increases the signal of mitochondrial-targeted peroxide-sensitive indicator (normalized fluorescence 16.29 ± 2.11% ISO vs. 54.23 ± 5.31% ISO and kaempferol, ∗p < 0.001), confirming that facilitation of mitochondrial Ca2+ accumulation induces mitochondrial ROS release.
Considering the close proximity of mitochondria and SR, we next sought to determine whether facilitation of mitochondrial Ca2+ accumulation leads to an increase in local ROS levels in the vicinity of RyR in cultured control VMs, measured using the ER-tuned redox-sensitive biosensor ERroGFP_iE (Avezov et al., 2013). The GFP sensor contains engineered cysteine residues that enable formation of di-thiol in response to oxidant stress (Cannon and Remington, 2009). After we generated adenovirus encoding the sensor, VMs were infected at a MOI of 10 and cultured for 48 h prior to imaging. As shown in Figure 8A, ERroGFP_iE-infected VMs exhibit a striated pattern indicative of SR targeting of the probe.
Figures 8B,D show representative recordings where signal of ERroGFP_iE was normalized to minimal fluorescence obtained by application of reducing agent dithiothreitol (DTT, 5 mmol/L) and maximal fluorescence obtained by application of oxidizing agent 2,2’-dithiodipyridine (DTDP, 200 μmol/L). At baseline under β-adrenergic stimulation with ISO, little change in oxidation in the form of increased fluorescence and di-thiol formation is observed. However, treatment with kaempferol or CGP-37157 still leads to a measurable and significant increase in ERroGFP_iE signal (8.19 ± 0.83% ISO vs. 22.20 ± 2.49% ISO and kaempferol, ∗p < 0.001 and 11.21 ± 2.03% ISO vs. 16.26 ± 2.32 ISO and CGP-37157, ∗p = 0.006, respectively), indicative of increased oxidation and ROS emission in close proximity of RyR (Figures 8C,E, respectively).
To directly test the hypothesis that pharmacological enhancement of [Ca2+]m accumulation results in increased RyR oxidation, the free thiol content of immunoprecipitated RyRs was measured using DNP-antibody. Figure 8F demonstrates that treatment with kaempferol or CGP-37157 (for 5 and 10 min, respectively) significantly increases oxidation of RyR fivefold and sixfold, respectively (Figure 8G, ∗p = 0.047 and #p = 0.015), shown by increased DNP signal. These data demonstrate that in control VMs, enhanced mitochondrial Ca2+ accumulation leads to increased ROS in the closely situated SR. Subsequent increased RyR oxidation is likely the responsible mechanism for the shortened latency for proarrhythmic RyR-mediated SCWs in VMs with pharmacologically enhanced mitochondrial Ca2+ accumulation.
Kaempferol and CGP-37157 modulate mitochondrial Ca2+ accumulation, membrane potential and ROS emission in cultured VMs from control rat hearts, as well as intracellular Ca2+ cycling and SCW generation. However, in the diseased heart, redox balance is altered and both intracellular and mitochondrial Ca2+ handling can be compromised already (Kim et al., 2017).
To test whether tethering of mitochondria to the SR is altered in TABs rat VMs, we performed western blot analysis of expression levels of mitofusin 1 and mitofusin 2; proteins that scaffold these two organelles (de Brito and Scorrano, 2008; Chen et al., 2012; Filadi et al., 2015). We did not find changes in expression levels of both these proteins in VMs of TABs vs. those of controls (Figure 9). Next, isolated TAB VMs were infected with adenoviruses to express mitochondrial Ca2+ indicator mtRCamp1h. Figures 10A–D demonstrate that the effects of kaempferol and CGP-37157 on mitochondrial Ca2+ accumulation are qualitatively similar to those in control VMs. As in controls, kaempferol accelerates Ca2+ accumulation (time to peak 72.43 ± 3.75 s ISO vs. 56.14 ± 4.47 s ISO and kaempferol, ∗p < 0.001) but the amplitude does not change significantly, while CGP-37157 impedes mitochondrial Ca2+ transient decay (tau of decay 32.08 ± 7.49 s ISO vs. 72.38 ± 14.99 s ISO and CGP-37157, ∗p = 0.006). In addition, we tested the effects of MCU inhibitor Ru360. Figures 10E,F show representative traces and respective pooled data for mitochondrial Ca2+ transients in ISO-treated control VMs under basal conditions and after 30 min incubation with 2 μmol/L Ru360. Figures 10G,H demonstrates that Ru360 reduces mitochondrial Ca2+ accumulation in TABs (0.29 ± 0.07 ΔF/F0 ISO vs. 0.14 ± 0.03 ΔF/F0 ISO and Ru360, ∗p < 0.001). Notably, the amplitude of pacing-induced mitochondrial Ca2+ transients in ISO-treated TAB VMs is significantly smaller that in controls (amplitude 1.15 ± 0.09 ΔF/F0 ISO Control vs. 0.30 ± 0.07 ΔF/F0 ISO TAB, ∗p < 0.001, Student’s t-test). Given this difference, we next performed experiments using freshly isolated VMs from hypertrophic TAB rat hearts, to test whether kaempferol and CGP-37157 have deleterious effects in a disease setting, or on the contrary could be protective.
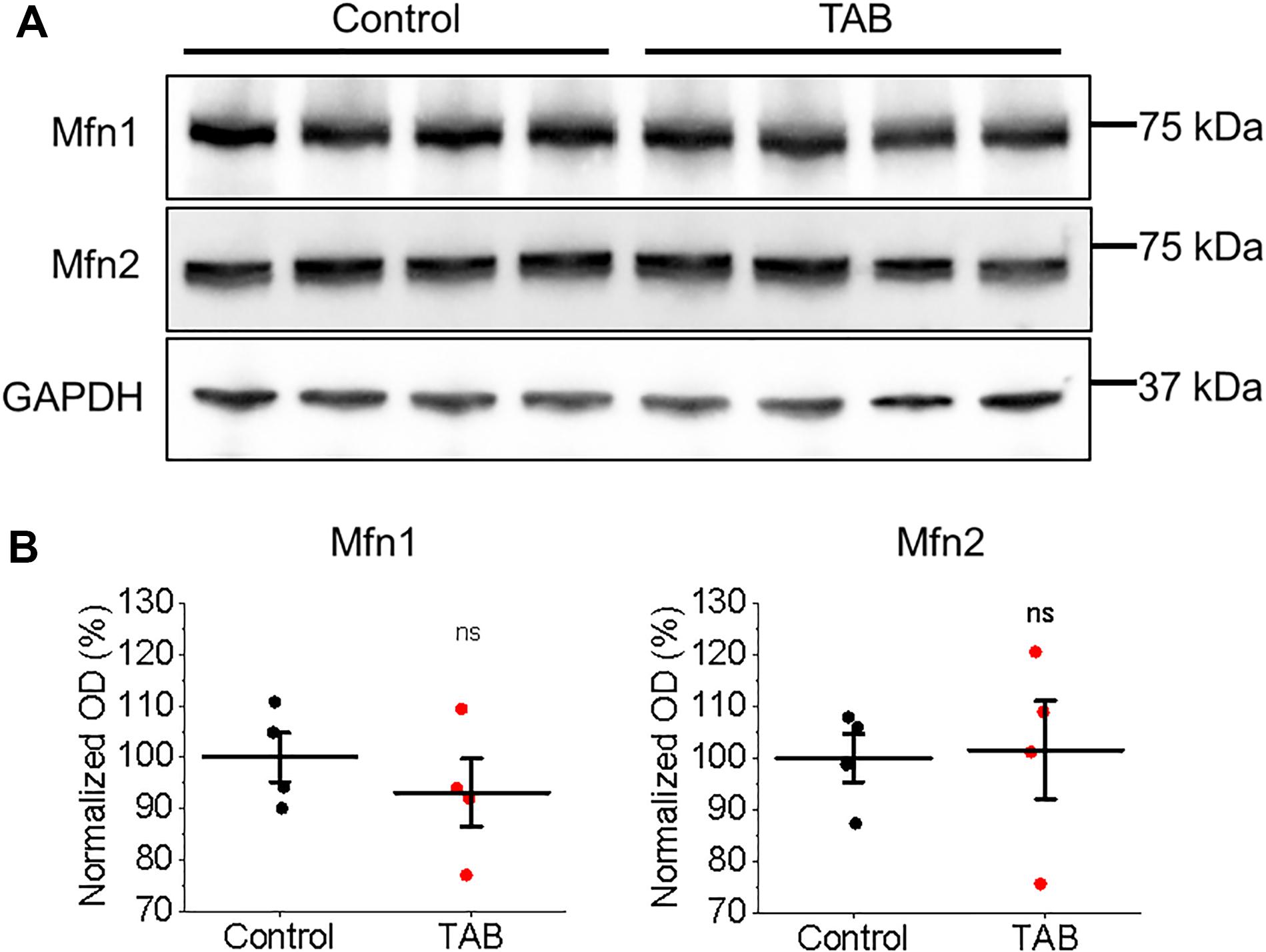
Figure 9. Expression of mitochondrial associated membrane proteins Mfn1 and Mfn2 is not altered in hypertrophic rat VMs. (A) Representative images of control and TAB VMs immunoblotted for expression of mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2). GAPDH was used as a loading control. (B) Quantification of normalized optical densities (N = 4 per group, Student’s t-test).
We studied the effects of enhancing mitochondrial Ca2+ accumulation in TAB VMs on cytosolic Ca2+ cycling using Rhod-2, as illustrated in Figure 11. As with control VMs, the burst pacing-pause protocol was used to assess the propensity of arrhythmogenic Ca2+ release (Figure 11A). After treatment with kaempferol, TAB VMs has a significantly reduced Ca2+ transient amplitude (5.59 ± 0.71 ΔF/F0 ISO vs. 3.08 ± 0.42 ΔF/F0 ISO and kaempferol, ∗p = 0.002), as well as a approximately twofold in decrease in SCW latency (Figure 11C, ∗p = 0.001). The percentage of cells exhibiting SCWs was also significantly increased after kaempferol application (71% ISO vs. 100% ISO and kaempferol, ∗p = 0.01). These changes were accompanied by a significant decrease in SR Ca2+ content assessed by application of 10 mmol/L caffeine (Figure 11B, 5.46 ± 0.77 ΔFcafF/F0 ISO vs. 3.19 ± 0.30 ΔFcafF/F0 ISO and kaempferol, ∗p = 0.01). Decreased Ca2+ transient amplitude and reduced SR Ca2+ load is indicative of increased Ca2+ leak via hyperactive RyRs (Belevych et al., 2007; Terentyev et al., 2008).
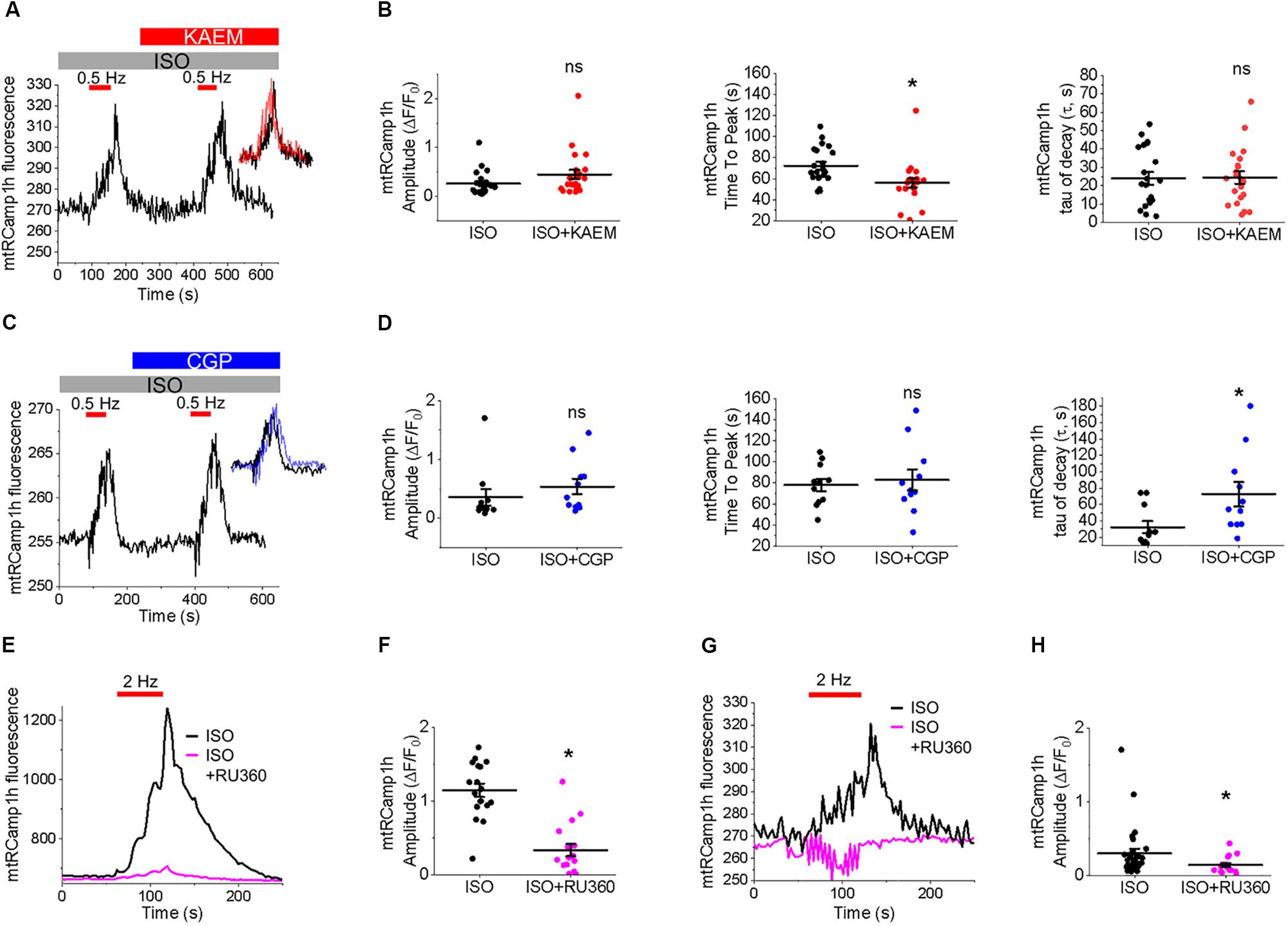
Figure 10. Pharmacological modulators of mitochondrial Ca2+ in TAB VMs exert similar effects to those on controls, despite hypertrophy-related impairment in mitochondrial Ca2+ homeostasis. (A) Representative trace from cultured TAB VM expressing mtRCamp1h. TAB VMs were treated with ISO (50 nmol/L) and paced at 0.5 Hz for 60 s intervals (red bars), before treatment with kaempferol (KAEM, 10 μmol/L) and further pacing. Peaks are overlaid in the inset for visualization, with ISO in black, ISO+KAEM in red. Graphs in (B) depict mean data ± SEM of mtRCamp1h transient amplitude, time to transient peak and tau of decay (n = 22 per group, N = 4, ∗p < 0.05, paired Student’s t-test). (C) Representative trace from cultured TAB VM expressing mtRCamp1h. VMs were treated with ISO and paced at 0.5 Hz for 60 s intervals (red bars), before treatment with CGP-37157 (CGP, 1 μmol/L) and further pacing. Peaks are overlaid in the inset for visualization, with ISO in black, ISO+CGP in blue. Graphs in (D) depict mean data ± SEM from mtRCamp1h transient amplitude, time to transient peak and tau of decay (n = 11, N = 3, ∗p < 0.05, paired Student’s t-test). (E) Representative trace from a cultured control VM expressing mtRCamp1h. Myocytes were treated with ISO (black line) and paced at 2 Hz for 60 s (red bar), or pretreated with Ru360 (2 μmol/L) for 30 min before treatment with ISO and pacing at 2 Hz (pink line). Graph in (F) depicts mean data ± SEM from mtRCamp1h transient amplitude (n = 17–18, N = 3, ∗p < 0.05, Student’s t-test). (G) Representative trace from a cultured TAB VM expressing mtRCamp1h. Myocytes were treated with ISO (black line) and paced at 2 Hz for 60 s (red bar), or pretreated with Ru360 (2 μmol/L) for 30 min before treatment with ISO and pacing at 0.5 Hz (pink line). Graph in (H) depicts mean data ± SEM from mtRCamp1h transient amplitude (n = 17, N = 4, ∗p < 0.05, Student’s t-test).
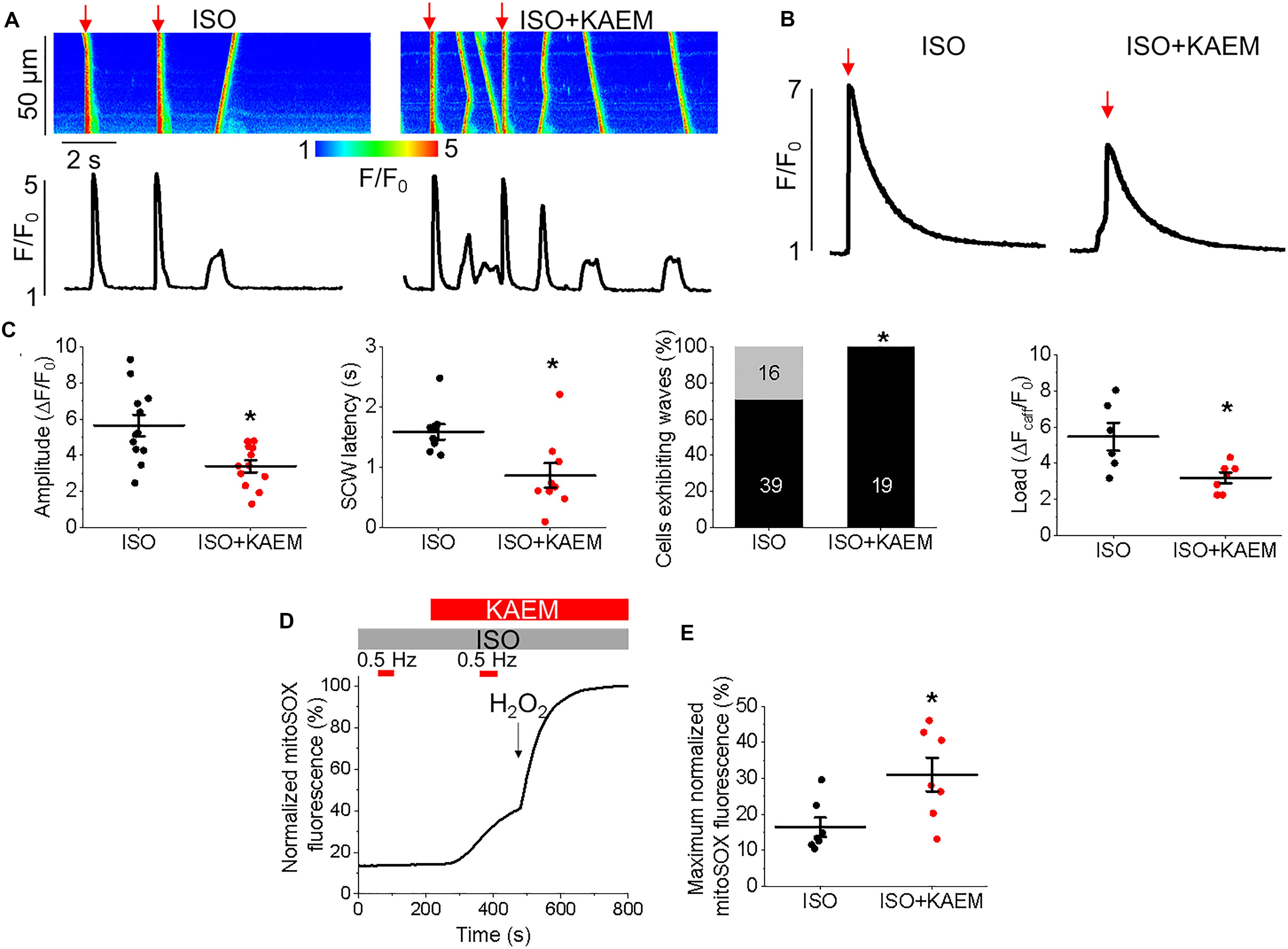
Figure 11. Enhanced mitochondrial Ca2+ accumulation by kaempferol promotes proarrhythmic SCWs in TAB rat VMs. (A) Representative confocal line scan images of Ca2+ transients and Rhod-2 fluorescence (F/F0) profiles of ISO treated (50 nmol/L) TAB rat VMs undergoing 0.5 Hz pace-pause protocol (red arrows) to induce SCWs, before and after application of kaempferol (KAEM, 10 μmol/L). (B) Representative traces of caffeine-induced Ca2+ transients (10 mmol/L). Graphs in C depict mean data ± SEM from Ca2+ transient amplitude (n = 12, N = 4, ∗p < 0.05, paired Student’s t-test), SCW latency (n = 9, N = 4, ∗p < 0.05, paired Student’s t-test), the percentage of cells exhibiting waves (∗p < 0.05, N = 4, Fisher’s exact test) and the caffeine-sensitive Ca2+ store load (n = 6 per group, ∗p < 0.05, Student’s t-test). (D) Representative recording of ROS production measured with MitoSOX in VMs. Signal was normalized to maximum fluorescence obtained on application of 10 mmol/L H2O2. VMs were treated with ISO and kaempferol and paced at 2 Hz (red bars) for 60 s. Graph in (E) depicts mean data ± SEM of normalized MitoSOX fluorescence (n = 7, N = 3, ∗p < 0.05, paired Student’s t-test).
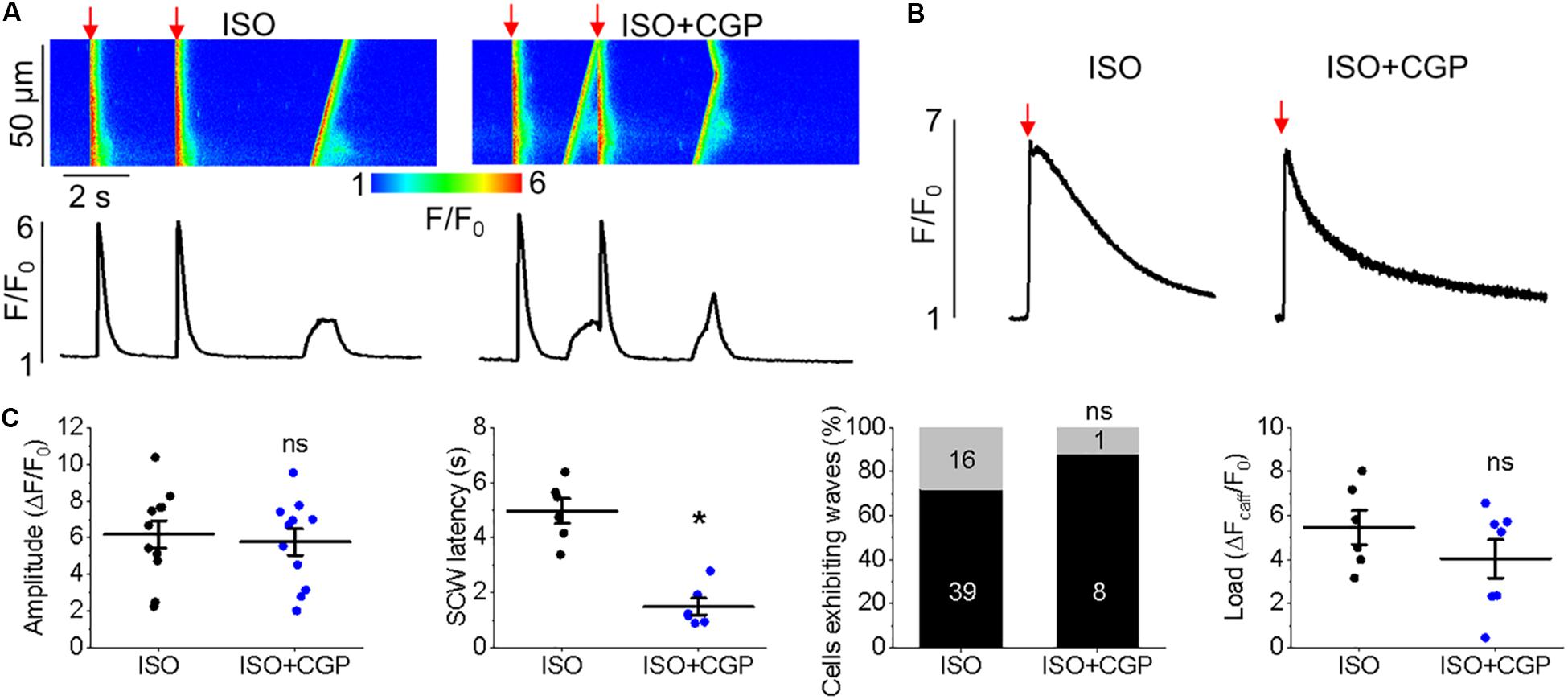
Figure 12. Enhanced mitochondrial Ca2+ accumulation by CGP-37157 promotes proarrhythmic SCWs in TAB rat VMs. (A) Representative confocal line scan images of Ca2+ transients and Rhod-2 fluorescence (F/F0) profiles of ISO treated (50 nmol/L) rat VMs undergoing 0.5 Hz pace-pause protocol before and after application of CGP-37157 (CGP, 1 μmol/L). (B) Representative traces of caffeine-induced Ca2+ transients (10 mmol/L). Graphs in (C) depict mean data ± SEM from Ca2+ transient amplitude (n = 11, N = 5, ns is not significant, paired Student’s t-test), SCW latency (n = 6, N = 5, ∗p < 0.05, paired Student’s t-test), the percentage of cells exhibiting waves (n = 18–55, N = 5, ns is not significant, Fisher’s exact test), and the caffeine-sensitive Ca2+ store load (n = 6, N = 5, ns is not significant, Student’s t-test).
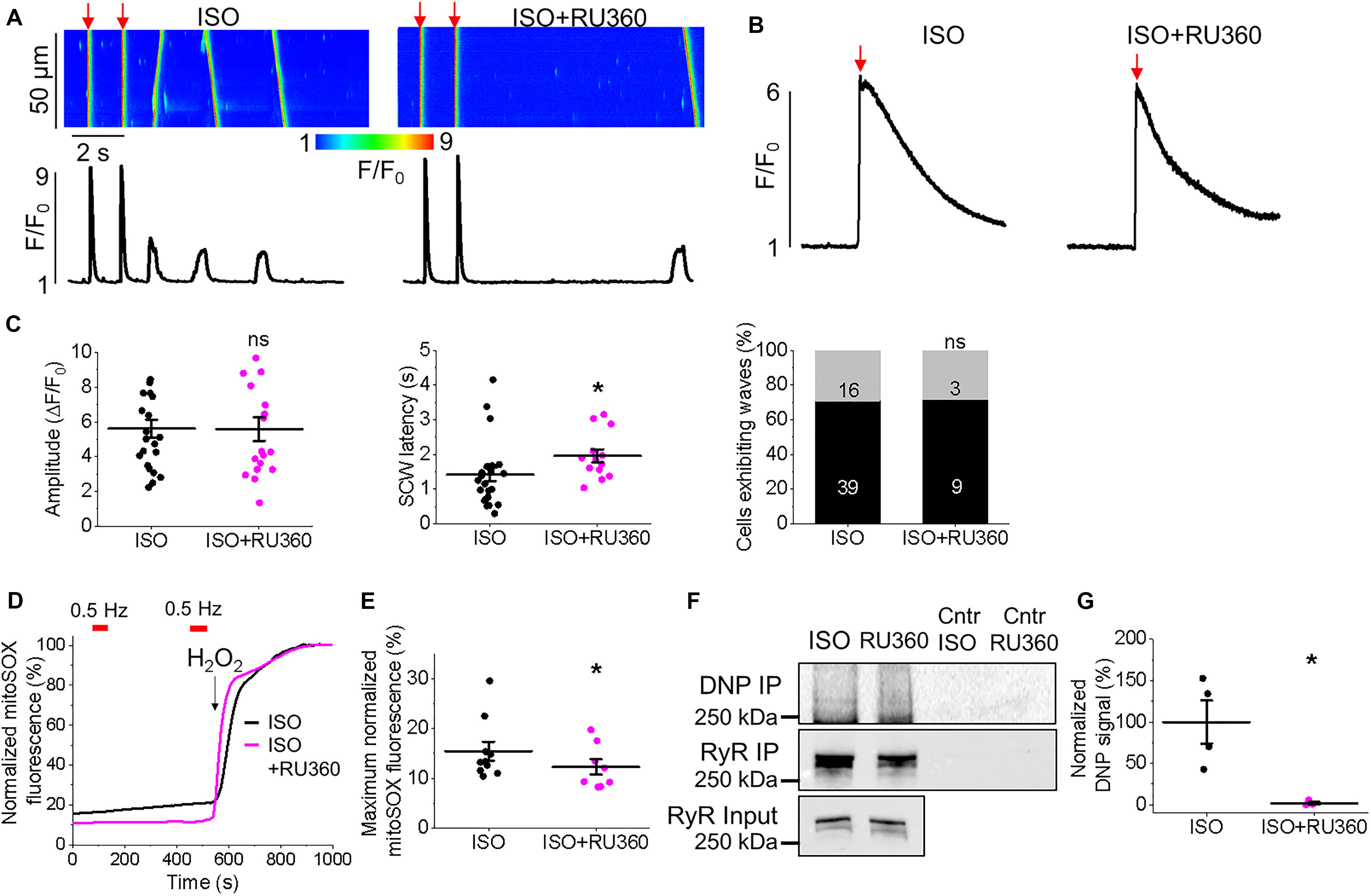
Figure 13. Block of mitochondrial Ca2+ uptake through MCU stabilizes RyRs in TAB rat VMs. (A) Representative confocal line scan images of Ca2+ transients and Rhod-2 fluorescence (F/F0) profiles of ISO-treated (50 nmol/L) TAB VM undergoing 0.5 Hz pace-pause protocol to induce spontaneous Ca2+ waves (SCWs), after 30 min. preincubation with Ru360 (2 μmol/L). (B) Representative traces of caffeine-induced Ca2+ transients (10 mmol/L). Graphs in (C) depict mean data ± SEM from Ca2+ transient amplitude (n = 19–22, N = 4, ns is not significant, Student’s t-test), SCW latency (n = 13–24, N = 4, ns is non-significant, ∗p < 0.05, Student’s t-test), the percentage of cells exhibiting waves (∗p < 0.05, Fisher’s exact test) and the caffeine-sensitive Ca2+ store load (calculated by application of 10 mmol/L caffeine, n = 6, n = 4, ∗p < 0.05, Student’s t-test). N = 4. (D) Representative recording of mitochondrial ROS production measured with MitoSOX in VMs. Signal was normalized to maximum fluorescence obtained on application of H2O2 (10 mmol/L). Myocytes were treated with ISO (black line) or ISO and Ru360 (pink line) and paced at 2 Hz (red bars). Graph in (E) depicts mean data ± SEM of normalized MitoSOX fluorescence (n = 7–10, N = 3, ∗p < 0.05, Student’s t-test). (F) RyR in freshly isolated control and TAB VMs was immunoprecipitated (IP) and immunoblotted for oxidation using DNP antibody. Representative images of DNP IP, RyR IP and RyR input signal from VMs treated with ISO or ISO and Ru360, with controls (Cntr). (G) Quantification of normalized DNP signal (%, N = 4 per group, ∗p < 0.05 vs. ISO, Student’s t-test).
Excessive ROS production is a hallmark of hypertrophy and we have previously established that in TAB VMs, there is increased mitochondrial ROS production in comparison to healthy controls which results in oxidation and thereby abnormally high activity of RyRs (Kim et al., 2017). In the present study, parallel experiments using the mitochondria-specific ROS indicator MitoSOX demonstrated that enhancement of mitochondrial Ca2+ accumulation with kaempferol further increased ROS emission in diseased VMs (Figures 11D,E). Signal was normalized to maximal fluorescence obtained upon application of 10 mmol/L H2O2.
Similar effects on cytosolic Ca2+ transients were obtained when enhancing mitochondrial Ca2+ accumulation in TAB VMs with block of NCLX via CGP-37157 (Figures 12A,B). There was a significant reduction in SCW latency (Figure 12C, 4.79 ± 0.38 s. ISO vs. 1.99 ± 0.45 s. ISO and CGP-37157, ∗p < 0.001), although no significant change in the SR Ca2+ content assessed by caffeine application was observed (Figures 12B,C).
Given that application of MCU-inhibitor Ru360 attenuated triggered activity and arrhythmogenesis in TAB hearts ex vivo (Figure 1), we next assessed intracellular Ca2+ handling of VMs preincubated with 2 μmol/L Ru360 for 30 min. There were no significant differences in the Ca2+ transient amplitude and the percentage of cells exhibiting waves or the caffeine-sensitive SR Ca2+ content (Figures 13A–C). However, there was a significant increase in SCW latency, (Figures 13A,C, 1.42 ± 0.19 s. ISO vs. 1.96 ± 0.19 s. ISO and Ru360, ∗p < 0.001), indicative of stabilization of RyR-mediated Ca2+ release. Myocytes from TAB hearts displayed a decrease in MitoSOX fluorescence after preincubation with Ru360 (Figures 13D,E 15.46 ± 1.91% ISO vs. 14.93 ± 2.15% ISO and Ru360, ∗p < 0.001). There was also a significant reduction in oxidation of immunoprecipitated RyRs after treatment with Ru360 assessed using anti-DNP antibodies (Figures 13F,G). These data suggest that stabilization of SR Ca2+ release stems from attenuation of ROS emission by mitochondria and normalization of RyR redox state.
Regenerative SCWs propagate via the ‘fire-diffuse-fire’ mechanism (Keizer and Smith, 1998; Maxwell and Blatter, 2012), whereby Ca2+ released from one cluster of RyR channels activates Ca2+ release from another. Increasing Ca2+ buffering can intercept Ca2+ diffusing from cluster to cluster and modulate SCW velocity (Ramay et al., 2010; Eisner et al., 2017), as was shown with SR Ca2+-ATPase (SERCa) enhancers (Fernandez-Tenorio and Niggli, 2018). It could be suggested that slower SCW wave propagation after enhancement of mitochondrial Ca2+ accumulation indicates a Ca2+ buffering capacity of mitochondria, serving as a sink for cytosolic Ca2+. However, neither the enhancement (with kaempferol or CGP-37157) nor attenuation (with Ru360) of mitochondrial Ca2+ accumulation altered the velocity of SCWs in either cultured control VMs (Figure 14A) or hypertrophic TAB VMs (Figure 14B). These data suggest that buffering capacity of mitochondria is insufficient to interrupt or slow SCWs, possibly due to concomitant changes in Δψm and local ROS.
The contribution of mitochondrial Ca2+ flux to myocyte excitation-contraction remains the subject of intense research, with both enhancement or reduction of [Ca2+]m posited as therapeutic strategies to improve cardiac function and prevent arrhythmia in cardiac disease (Liu and O’Rourke, 2008; Kolhaas et al., 2010; Liu et al., 2014; Dietl and Maack, 2017; Schweitzer et al., 2017; Xie et al., 2018). Our present study provides evidence that enhanced mitochondrial Ca2+ accumulation dissipates Δψm and drives increased ROS in the mitochondria-SR microdomain. Subsequently, increased modification of RyR by ROS enhances channel activity and increases the propensity for proarrhythmic spontaneous SR Ca2+ release in the form of SCWs. This mechanism further exacerbates proarrhythmic triggered activity in hypertrophic hearts. Conversely, inhibition of MCU is protective against arrhythmogenesis, attenuating oxidative stress and reducing aberrant activity of RyR.
The physical and functional coupling of the SR and mitochondria is critical for matching myocyte workload to mitochondrial ATP generation (Dorn and Maack, 2013; Lopez-Crisosto et al., 2017). The close association facilitates mitochondrial Ca2+ influx upon SR Ca2+ release (Sharma et al., 2000; Szalai et al., 2000; Csordás et al., 2001). To maintain Ca2+ flux balance in the steady state, uptake of mitochondrial Ca2+ is well matched to extrusion, so net [Ca2+]m does not significantly change. We assessed mitochondrial Ca2+ uptake in cultured control and TAB VMs with the genetically encoded Ca2+ probe mtRCamp1h. In comparison to baseline, under β-adrenergic stimulation with ISO we observed an increase in [Ca2+]m during periodic pacing (Figures 2B,E, 10A,C), likely to match the increased demand for ATP.
To modulate mitochondrial Ca2+ influx in cultured control VMs, we applied kaempferol or SB 202190 to directly enhance uptake through MCU, or CGP-37157 to inhibit NCLX thus attenuating Ca2+ efflux. Interestingly, application of these pharmacological agents did not increase [Ca2+]m, but instead altered the dynamics of Ca2+ accumulation or retention (Figures 2D,F). Similar effects were observed in TAB VMs treated with kaempferol and CGP-37157 (Figures 10B,D). The inability to significantly increase total [Ca2+]m under these conditions points to the existence of limiting factors, preventing excessive mitochondrial Ca2+ loading in VMs.
There are three established mechanisms to prevent mitochondrial Ca2+ overload. Activation of mitochondrial Ca2+-activated K+ channels limits depolarization of the mitochondria, reducing the electrochemical inward driving force for Ca2+ (O’Rourke et al., 2005; Clements et al., 2015). Acidification of the mitochondrial matrix by an increased H+ flow through ATP-synthase has been shown to inhibit MCU (Moreau and Parekh, 2009). Additionally, increased matrix Ca2+ also promotes the opening of mPTP, dissipating Δψm and limiting [Ca2+]m accumulation (Broekemeier et al., 1998; Hüser and Blatter, 1999; Elrod et al., 2010). This is evident in Figure 3, whereby application of kaempferol or CGP-37157 to cultured control VMs significantly depolarized Δψm. Interestingly, preincubation of VMs with mitoTEMPO, a specific mitochondria ROS scavenger (20 μmol/L, 30 min), did not prevent dissipation of Δψm by kaempferol (normalized TMRM fluorescence 89.7 ± 2.16% mitoTEMPO and ISO vs. 39.17 ± 3.59% mitoTEMPO, ISO and kaempferol, ∗p < 0.001, paired Student’s t-test, n = 19, N = 3), suggesting that this process is not ROS-dependent.
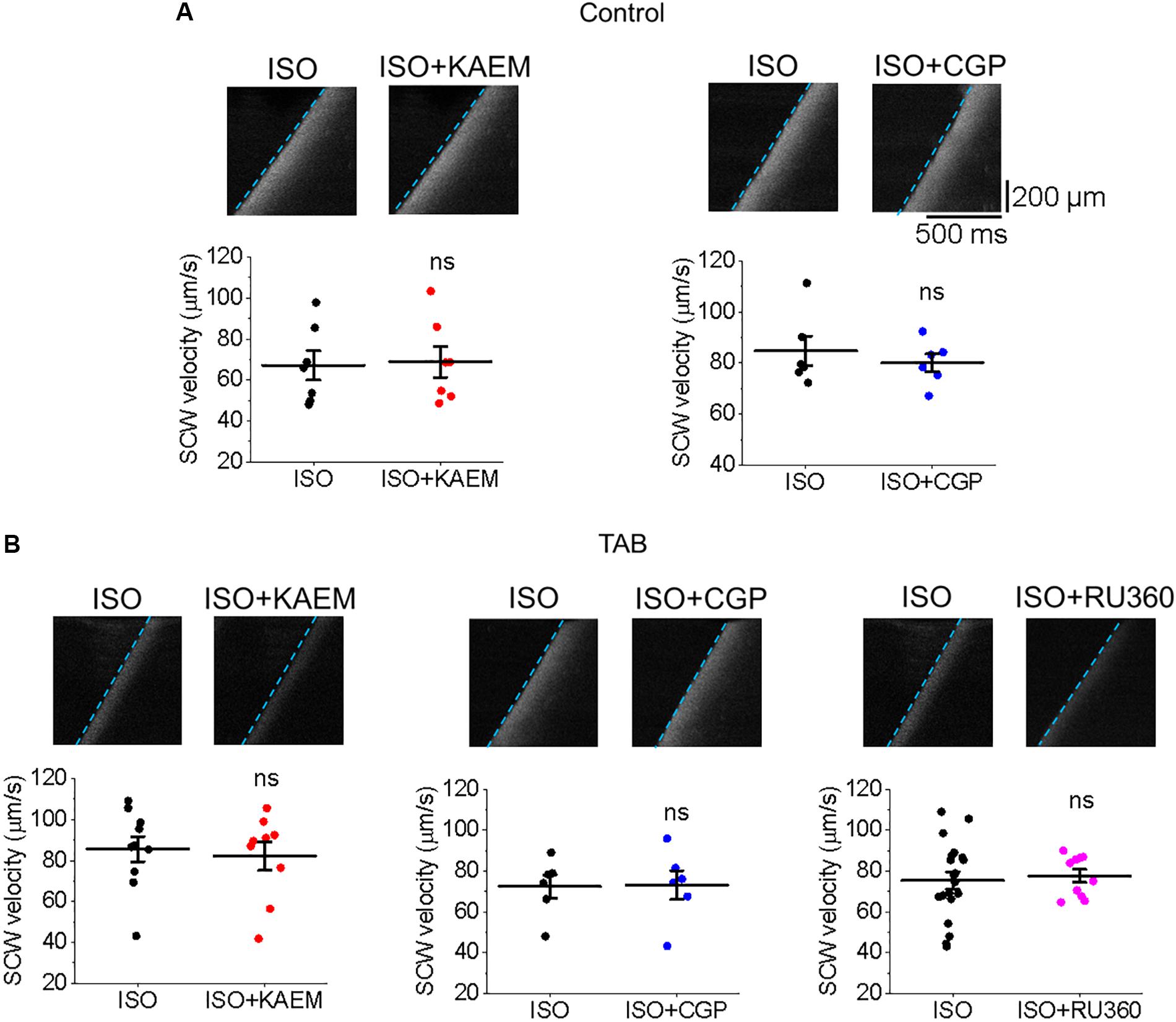
Figure 14. Enhancement or inhibition of mitochondrial Ca2+ accumulation does not modulate SCW propagation. (A) Velocity of SCWs in cultured control VMs. Graphs depict mean ± SEM (n = 6–9, N = 4–5, ns is non-significant, paired Student’s t-test). (B) Velocity of spontaneous Ca2+ waves (SCWs) in TAB VMs. Graphs depict mean ± SEM (n = 10–20, N = 4–5, ns is non-significant, Student’s t-test and paired Student’s t-test where appropriate).
The RyR-mediated release of Ca2+ from the SR is critical to contractile activation. Termination of SR Ca2+ release allows for Ca2+ released into the cytosol to be resequestered and maintains the refractoriness of Ca2+ signaling during diastole (Terentyev et al., 2002; Szentesi et al., 2004; Sobie et al., 2005). Shortened Ca2+ signaling refractoriness due to hyperactive RyR increases the rate of SCWs in diseased myocytes (Belevych et al., 2012; Brunello et al., 2013), contributing to the pathogenesis of triggered arrhythmias (Pogwizd and Bers, 2004).
We assessed the effects of [Ca2+]m modulation on Ca2+ handling and initiation of triggered activity at the whole organ level, using hearts from rats with TAB-induced hypertrophy, as illustrated in Figure 1. Application of MCU activator kaempferol reduced Ca2+ transient amplitude and exacerbated the proarrhythmic phenotype, with incidences of PVCs and spontaneous VT/VFs with higher VF frequencies. The focal activity and/or transmural outbreak-like activation patterns were frequently seen during PVCs and VFs, suggesting enhanced triggered activity may underlie exacerbated ventricular arrhythmias by kaempferol. In contrast the MCU blocker, Ru360, suppressed spontaneous VT/VFs. The activation maps of pacing induced VFs showed that rapid pacing caused conduction block and reentry formation, suggesting that the protective effect of Ru360 is most likely through suppressing triggered activity. In recordings of intracellular Ca2+ transients, it was evident that kaempferol and NCLX inhibitor CGP-37157 had detrimental effects on both cultured control VMs (Figure 4) and TAB VMs (Figures 11, 12), shortening SCW latency and increasing the propensity for spontaneous Ca2+ release, indicative of increased activity of RyR in both cultured control and hypertrophic TAB VMs. The experiments with RyR inhibitor dantrolene demonstrating attenuation of kaempferol-induced disturbances in Ca2+ cycling (Figure 6) further confirm the central role of dysregulated RyR-mediated Ca2+ release in this process.
Hyperactivity of RyR in cardiac disease is often attributed to posttranslational modifications, including phosphorylation of PKA- and CaMKII-specific sites, and oxidation of many reactive cysteines within the protein (Györke and Carnes, 2008; Niggli et al., 2013; Zima et al., 2014). Mitochondria are a major source of ROS in the myocyte and excessive ROS production is a hallmark of HF, hypertrophy and aging (Zima and Blatter, 2006), in parallel with perturbed Ca2+ homeostasis (Terentyev et al., 2008; Cooper et al., 2013; Kim et al., 2017). Mitochondrial-ROS signaling has also been suggested to directly modulate Ca2+ spark activity (Yan et al., 2008; Zhou et al., 2011). Our experiments using mitochondria-targeted ROS biosensor OMM-HyPer3 show that kaempferol induces surge in mito-ROS production (Figure 7). Furthermore, in the present study, Figure 8 clearly demonstrates increased local ROS in the vicinity of RyR and RyR oxidation in myocytes treated with kaempferol and CGP-37157. Importantly, while oxidative stress is already significant in hypertrophic myocytes, application of kaempferol further increased mitochondrial ROS emission in TAB VMs (Figure 11D), exacerbating Ca2+ mishandling.
Scavenging of mitochondrial ROS has been sufficient to alleviate the arrhythmogenic phenotype in multiple disease states (Mochizuki et al., 2007; Belevych et al., 2012; Cooper et al., 2013; Luo et al., 2013; Joseph et al., 2016) and normalize the redox state of RyR in TAB-induced hypertrophy (Kim et al., 2017). Importantly, inhibition of mitochondrial Ca2+ uptake with Ru360 was sufficient to attenuate Ca2+-dependent arrhythmia in ex vivo TAB hearts (Figure 1) and normalize Ca2+ homeostasis in isolated TAB VMs, stabilizing RyR-mediated Ca2+ release and attenuating proarrhythmic SCWs (Figures 13A–C). Furthermore, Ru360 reduced mitochondrial ROS emission assessed using mitochondria-specific ROS indicator mitoSOX (Figures 13D,E). This indicates that block of mitochondrial Ca2+ influx reduces mitochondrial ROS signaling in TAB VMs resulting in reduction of oxidation levels of RyR (Figures 13F,G). Our data suggest that increased mitochondrial Ca2+ accumulation facilitates increased mitochondrial ROS emission and the oxidation of RyR. This underlies enhanced RyR activity, increased spontaneous Ca2+ release in the form of arrhythmogenic SCWs, and a vicious cycle of Ca2+/ROS-induced myocyte dysfunction.
During pathological mitochondrial Ca2+ overload, opening of mPTP offers an additional Ca2+ efflux pathway (Broekemeier et al., 1998; Hüser and Blatter, 1999; Elrod et al., 2010). Pharmacological inhibition of mPTP with cyclosporine A or genetic ablation of mPTP component cyclophilin D (that reduces opening) has shown to be protective against HF or ischemia-reperfusion injury (Griffiths and Halestrap, 1993; Hausenloy et al., 2010; Yarana et al., 2012; Gordan et al., 2016). However, there is ongoing controversy as to whether inhibition of NCLX can be protective in cardiac disease. In a guinea pig HF model, chronic inhibition of NCLX with CGP-37157 restored diminished [Ca2+]m, thereby improving redox homeostasis and protecting against arrhythmogenesis (Liu et al., 2014). In agreement with these findings, we recorded diminished mitochondrial Ca2+ transients in VMs from diseased hearts in comparison to controls (Figure 2 vs. Figure 10). However, incubation with CGP-37157 did not change significantly mitochondrial Ca2+ transient amplitude despite slowing down transient decay in TABs. It also did not improve but even worsened aberrant intracellular Ca2+ handling (Figure 12). Xie et al. (2018) reported an increased mitochondrial Ca2+ influx during diastolic period in mice with non-ischemic HF and posited that increased mitochondrial Ca2+ efflux drives activation of the sarcolemmal NCX and initiates EADs. In this work, inhibition of both influx and efflux were reported to have anti-arrhythmic effects. In direct contrast, the present study clearly demonstrates that inhibition of NCLX in both control and TAB VMs promotes proarrhythmic spontaneous SR Ca2+ release (Figures 4, 12). Importantly, knockdown of NCLX in a conditional loss-of-function mouse model caused severe myocardial dysfunction, HF and sudden cardiac death (Luongo et al., 2017). This was attributed to substantially increased ROS generation due to mitochondrial Ca2+ overload. Our results are in line with the latter and inhibition of NCLX with CGP-37157 significantly increased ROS emission in cultured control VMs (Figures 7, 8), resulting in defective SR Ca2+ handling in both cultured control (Figure 4) and TAB (Figure 12) VMs.
While mitochondrial Ca2+ flux is closely associated with ATP generation and ROS emission, it has also been suggested that mitochondria may act as buffers that can shape global Ca2+ transients during EC coupling (Maack et al., 2006; Yan et al., 2008; Walsh et al., 2009; Drago et al., 2012; Zhao et al., 2013). Although the low affinity of MCU for Ca2+ (Kd ∼ 10–20 μmol/L Ca2+; Bernardi, 1999) would limit uptake during diastole and normal Ca2+ transients, mitochondrial Ca2+ uptake could occur at high local [Ca2+] near SR Ca2+ release sites (Andrienko et al., 2009). One end of the mitochondria is in close proximity to these sites (∼37–270 nm; Sharma et al., 2000), and is physically tethered to the SR (García-Pérez et al., 2011; Chen et al., 2012) with strategic positioning of MCU near RyR (De La Fuente et al., 2016). This facilitates crosstalk between the organelles and tunneling of Ca2+ between the two has been reported in striated muscle (Shkryl and Shirokova, 2006). In cardiac disease, mitochondria-SR interfaces and therefore Ca2+ transport may be altered due to changes in expression levels of scaffolding proteins including Mfn1 and 2 (Dorn et al., 2015). We tested whether this occurs in our model of hypertrophy and did not find changes in expression levels of these proteins (Figure 9). Mitochondria may act as a buffer, serving as a sink of local Ca2+. Indeed, in a mouse model of CPVT, enhancement of mitochondrial Ca2+ influx reduced frequency of arrhythmogenic Ca2+ waves and incidences of VT/VF (Schweitzer et al., 2017). Also, Zhao et al. (2013) saw a reduction in the frequency of SCWs in myocytes treated with kaempferol after an FCCP-induced reduction in Δψm, while Ru360 increased SCW frequency.
Although SR Ca2+ release events are an important driver of [Ca2+]m, as the total mitochondria Ca2+ flux is small, the overall ability of mitochondria to shape intracellular Ca2+ dynamics remains debated (Dedkova and Blatter, 2008, 2013; O’Rourke and Blatter, 2009; Hohendanner et al., 2013; Williams et al., 2013; Eisner et al., 2017). Troponin I and SERCa are significant Ca2+ buffers in the myocyte, and enhancement or increased expression of SERCa has been shown to significantly improve Ca2+ buffering capacity and attenuate arrhythmogenic spontaneous Ca2+ release (Lyon et al., 2011; Briston et al., 2014). In recent work of Fernandez-Tenorio and Niggli (2018), specific enhancers of SERCa activity reduced the frequency and velocity of SCWs in mouse VMs. In the present study, enhancement of mitochondrial Ca2+ influx significantly increases the propensity for SCW in both cultured control and TAB VMs (Figures 4, 5, 11, 12), while neither enhancement or inhibition of [Ca2+]m modulated the velocity and propagation of SCWs (Figure 14). Importantly, we do not find any differences in the rate of propagation of SCWs between control and TAB VMs where impairment of mitochondria to sequester Ca2+ leads to profound approximately fourfold decrease in the amplitude of pacing-induced mitochondrial Ca2+ transient (Figures 2, 10). These data are in line with previous studies that suggest while Ca2+ modulates mitochondrial function, mitochondria do not serve as a significant buffer of intracellular cytosolic Ca2+ (Bers et al., 1993; Negretti et al., 1993; Lu et al., 2013; Williams et al., 2013). The effects of increased ROS emission on intracellular Ca2+ homeostasis are likely overwhelming and cannot be compensated for by an increase in local Ca2+ buffering capacity by mitochondria.
In conclusion, our data suggest that pharmacological enhancement of mitochondrial Ca2+ accumulation produces deleterious effects on Ca2+ homeostasis under β-adrenergic stimulation. It promotes excessive ROS that enhances RyR activity thereby proarrhythmic spontaneous Ca2+ release. In cardiac hypertrophy, where ROS defenses are weakened, it exacerbates the proarrhythmic alterations in Ca2+ handling. Inhibition of mitochondria Ca2+ uptake is protective because it reduces emission of ROS by mitochondria.
SH, B-RC, JO-U, GC, and DT participated in the study design. SH wrote first draft of manuscript. SH, RT, TYK, PB, RC, JO-U performed the experiments. SH, RT, TYK, B-RC, DT conducted data interpretation and analyses. SH, RT, TYK, PB, RC, JO-U, GC, B-RC and DT reviewed the manuscript submitted for publication. All authors revised and approved the final version of the manuscript.
This work was supported by American Heart Association Grant #18POST33960456 to SH, National Heart, Lung, and Blood Institute at the National Institutes of Health (NIH) RO1HL135236 to RC, NIH 7R01HL136757 to JO-U, NIH 1R01HL142864 to GC, NIH R01HL096669 to B-RC, American Heart Association Grant in Aid 15GRNT25650002 and NIH R01HL121796 to DT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Akerboom, J., Carreras Calderón, N., Tian, L., Wabnig, S., Prigge, M., Tolö, J., et al. (2013). Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front. Mol. Neurosci. 4:2. doi: 10.3389/fnmol.2013.00002
Andrienko, T. N., Picht, E., and Bers, D. M. (2009). Mitochondrial free calcium regulation during sarcoplasmic reticulum calcium release in rat cardiac myocytes. J. Mol. Cell Cardiol. 46, 1027–1036. doi: 10.1016/j.yjmcc.2009.03.015
Avezov, E., Cross, B. C., Kaminski Schierle, G. S., Winters, M., Harding, H. P., Melo, E. P., et al. (2013). Lifetime imaging of a fluorescent protein sensor reveals surprising stability of ER thiol redox. J. Cell Biol. 201, 337–349. doi: 10.1083/jcb.201211155
Banyasz, T., Lozinskiy, I., Payne, C. E., Edelmann, S., Norton, B., Chen, B., et al. (2008). Transformation of adult rat cardiac myocytes in primary culture. Exp. Physiol. 93, 370–382. doi: 10.1113/expphysiol.2007.040659
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Belevych, A., Kubalova, Z., Terentyev, D., Hamlin, R. L., Carnes, C. A., and Györke, S. (2007). Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys. J. 93, 4083–4092. doi: 10.1529/biophysj.107.114546
Belevych, A. E., Terentyev, D., Terentyeva, R., Ho, H. T., Gyorke, I., Bonilla, I. M., et al. (2012). Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ. Res. 110, 569–577. doi: 10.1161/CIRCRESAHA.111.260455
Belevych, A. E., Terentyev, D., Viatchenko-Karpinski, S., Terentyeva, R., Sridhar, A., Nishijima, Y., et al. (2009). Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc. Res. 84, 387–395. doi: 10.1093/cvr/cvp246
Benjamin, E. J., Virani, S. S., Callaway, C. W., Chamberlain, A. M., Chang, A. R., Cheng, S., et al. (2018). Heart disease and stroke statistics-2018 update: a report from the american heart association. Circulation 137, e67–e492. doi: 10.1161/CIR.0000000000000558
Bernardi, P. (1999). Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 79, 1127–1155. doi: 10.1152/physrev.1999.79.4.1127
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 10, 198–205. doi: 10.1038/415198a
Bers, D. M., Bassani, J. W., and Bassani, R. A. (1993). Competition and redistribution among calcium transport systems in rabbit cardiac myocytes. Cardiovasc. Res. 27, 1772–1777. doi: 10.1093/cvr/27.10.1772
Bertero, E., and Maack, C. (2018). Calcium signaling and reactive oxygen species in mitochondria. Circ. Res. 122, 1460–1478. doi: 10.1161/CIRCRESAHA.118.310082
Bilan, D. S., Pase, L., Joosen, L., Gorokhovatsky, A. Y., Ermakova, Y. G., Gadella, T. W., et al. (2013). HyPer-3: a genetically encoded H(2)O(2) probe with improved performance for ratiometric and fluorescence lifetime imaging. ACS Chem. Biol. 8, 535–542. doi: 10.1021/cb300625g
Bovo, E., Mazurek, S. R., and Zima, A. V. (2018). Oxidation of ryanodine receptor following ischemia/reperfusion increases propensity of Ca2+ waves during β-adrenergic receptor stimulation. Am. J. Physiol. Heart Circ. Physiol. 315, H1032–H1040. doi: 10.1152/ajpheart.00334.2018
Boyman, L., Williams, G. S., Khananshvili, D., Sekler, I., and Lederer, W. J. (2013). NCLX: the mitochondrial sodium calcium exchanger. J. Mol. Cell. Cardiol. 59, 205–213. doi: 10.1016/j.yjmcc.2013.03.012
Brandes, R., and Bers, D. M. (1997). Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ. Res. 80, 82–87. doi: 10.1161/01.RES.80.1.82
Briston, S. J., Dibb, K. M., Solaro, R. J., Eisner, D. A., and Trafford, A. W. (2014). Balanced changes in Ca buffering by SERCA and troponin contribute to Ca handling during β-adrenergic stimulation in cardiac myocytes. Cardiovasc. Res. 104, 347–354. doi: 10.1093/cvr/cvu201
Broekemeier, K. M., Klocek, C. K., and Pfeiffer, D. R. (1998). Proton selective substate of the mitochondrial permeability transition pore: regulation by the redox state of the electron transport chain. Biochemistry 22, 13059–13065. doi: 10.1021/bi980820c
Brunello, L., Slabaugh, J. L., Radwanski, P. B., Ho, H. T., Belevych, A. E., Lou, Q., et al. (2013). Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2+ release in a genetic model of arrhythmia. Proc. Natl. Acad. Sci. U.S.A. 110, 10312–10317. doi: 10.1073/pnas.1300052110
Burns-Hamuro, L. L., Ma, Y., Kammerer, S., Reineke, U., Self, C., Cook, C., et al. (2003). Designing isoform-specific peptide disruptors of protein kinase a localization. Proc. Natl. Acad. Sci. U.S.A. 100, 4072–4077. doi: 10.1073/pnas.2628038100
Cannon, M. B., and Remington, S. J. (2009). Redox-sensitive green fluorescent protein: probes for dynamic intracellular redox responses. A review. Methods Mol. Biol. 476, 50–64. doi: 10.1007/978-1-59745-129-1_4
Chen, Y., Csordás, G., Jowdy, C., Schneider, T. G., Csordás, N., Wang, W., et al. (2012). Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 111, 863–875. doi: 10.1161/CIRCRESAHA.112.266585
Clements, R. T., Terentyev, D., and Sellke, F. W. (2015). Ca(2+)-activated K(+) channels as therapeutic targets for myocardial and vascular protection. Circ. J. 79, 455–462. doi: 10.1253/circj.CJ-15-0015
Cooper, L. L., Li, W., Lu, Y., Centracchio, J., Terentyeva, R., Koren, G., et al. (2013). Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J. Physiol. 591(Pt 23), 5895–5911. doi: 10.1113/jphysiol.2013.260521
Csordás, G., Thomas, A. P., and Hajnóczky, G. (2001). Calcium signal transmission between ryanodine receptors and mitochondria in cardiac muscle. Trends Cardiovasc. Med. 11, 269–275. doi: 10.1016/S1050-1738(01)00123-2
Csordás, G., Weaver, D., and Hajnóczky, G. (2018). Endoplasmic reticulum-mitochondrial contactology: structure and signaling functions. Trends Cell Biol. 28, 523–540. doi: 10.1016/j.tcb.2018.02.009
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
De La Fuente, S., Fernandez-Sanz, C., Vail, C., Agra, E. J., Holmstrom, K., Sun, J., et al. (2016). Strategic positioning and biased activity of the mitochondrial calcium uniporter in cardiac muscle. J. Biol. Chem. 28, 23343–23362. doi: 10.1074/jbc.M116.755496
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi: 10.1038/nature10230
Dedkova, E. N., and Blatter, L. A. (2008). Mitochondrial Ca2+ and the heart. Cell Calcium 44, 77–91. doi: 10.1016/j.ceca.2007.11.002
Dedkova, E. N., and Blatter, L. A. (2013). Calcium signaling in cardiac mitochondria. J. Mol. Cell. Cardiol. 58, 125–133. doi: 10.1016/j.yjmcc.2012.12.021
Dietl, A., and Maack, C. (2017). Targeting mitochondrial calcium handling and reactive oxygen species in heart failure. Curr. Heart Fail. Rep. 14, 338–349. doi: 10.1007/s11897-017-0347-7
DiPilato, L. M., Cheng, X., and Zhang, J. (2004). Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci. U.S.A. 23, 16513–16518. doi: 10.1073/pnas.0405973101
Dorn, G. W., and Maack, C. (2013). SR and mitochondria: calcium cross-talk between kissing cousins. J. Mol. Cell. Cardiol. 55, 42–49. doi: 10.1016/j.yjmcc.2012.07.015
Dorn, G. W., and Scorrano, L. (2010). Two close, too close: sarcoplasmic reticulum-mitochondrial crosstalk and cardiomyocyte fate. Circ. Res. 107, 689–699. doi: 10.1161/CIRCRESAHA.110.225714
Dorn, G. W., Song, M., and Walsh, K. (2015). Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J. Mol. Cell. Cardiol. 78, 123–128. doi: 10.1016/j.yjmcc.2014.09.015
Drago, I., De Stefani, D., Rizzuto, R., and Pozzan, T. (2012). Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 109, 12986–12991. doi: 10.1073/pnas.1210718109
Eisner, D. A., Caldwell, J. L., Kistamás, K., and Trafford, A. W. (2017). Calcium and excitation- contraction coupling in the heart. Circ. Res. 121, 181–195. doi: 10.1161/CIRCRESAHA.117.310230
Eisner, V., Csordás, G., and Hajnóczky, G. (2013). Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca2+ and reactive oxygen species signaling. J. Cell Sci. 126(Pt 14), 2965–2978. doi: 10.1242/jcs.093609
Elrod, J. W., Wong, R., Mishra, S., Vagnozzi, R. J., Sakthievel, B., Goonasekera, S. A., et al. (2010). Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120, 3680–3687. doi: 10.1172/JCI43171
Fernandez-Tenorio, M., and Niggli, E. (2018). Stabilization of Ca2+ signaling in cardiac muscle by stimulation of SERCA. J. Mol. Cell Cardiol. 119, 87–95. doi: 10.1016/j.yjmcc.2018.04.015
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2015). Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. U.S.A. 112, E2174–E2181. doi: 10.1073/pnas.1504880112
García-Pérez, C., Schneider, T. G., Hajnóczky, G., and Csordás, G. (2011). Alignment of sarcoplasmic reticulum-mitochondrial junctions with mitochondrial contact points. Am. J. Physiol. Heart Circ. Physiol. 301, H1907–H1915. doi: 10.1152/ajpheart.00397.2011
García-Rivas Gde, J., Carvajal, K., Correa, F., and Zazueta, C. (2006). Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 149, 829–837. doi: 10.1038/sj.bjp.0706932
Gordan, R., Fefelova, N., Gwathmey, J. K., and Xie, L. H. (2016). Involvement of mitochondrial permeability transition pore (mPTP) in cardiac arrhythmias: evidence from cyclophilin D knockout mice. Cell Calcium 60, 363–372. doi: 10.1016/j.ceca.2016.09.001
Griffiths, E. J., and Halestrap, A. P. (1993). Protection by cyclosporin a of ischemia/reperfusion- induced damage in isolated rat hearts. J. Mol. Cell Cardiol. 25, 1461–1469. doi: 10.1006/jmcc.1993.1162
Györke, S., and Carnes, C. (2008). Dysregulated sarcoplasmic reticulum calcium release: potential pharmacological target in cardiac disease. Pharmacol. Ther. 119, 340–354. doi: 10.1016/j.pharmthera.2008.06.002
Hausenloy, D. J., Lim, S. Y., Ong, S. G., Davidson, S. M., and Yellon, D. M. (2010). Mitochondrial cyclophilin-D as a critical mediator of ischaemic preconditioning. Cardiovasc. Res. 88, 67–74. doi: 10.1093/cvr/cvq113
Hohendanner, F., Ljubojević, S., MacQuaide, N., Sacherer, M., Sedej, S., Biesmans, L., et al. (2013). Intracellular dyssynchrony of diastolic cytosolic [Ca2+] decay in ventricular cardiomyocytes in cardiac remodeling and human heart failure. Circ. Res. 113, 527–538. doi: 10.1161/CIRCRESAHA.113.300895
Hüser, J., and Blatter, L. A. (1999). Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J. 343(Pt 2), 311–317. doi: 10.1042/bj3430311
Joseph, L. C., Subramanyam, P., Radlicz, C., Trent, C. M., Iyer, V., Colecraft, H. M., et al. (2016). Mitochondrial oxidative stress during cardiac lipid overload causes intracellular calcium leak and arrhythmia. Heart Rhythm. 13, 1699–1706. doi: 10.1016/j.hrthm.2016.05.002
Keizer, J., and Smith, G. D. (1998). Spark-to-wave transition: saltatory transmission of calcium waves in cardiac myocytes. Biophys. Chem. 5, 87–100. doi: 10.1016/S0301-4622(98)00125-2
Kim, T. Y., Kunitomo, Y., Pfeiffer, Z., Patel, D., Hwang, J., Harrison, K., et al. (2015). Complex excitation dynamics underlie polymorphic ventricular tachycardia in a transgenic rabbit model of long QT syndrome type 1. Heart Rhythm. 12, 220–228. doi: 10.1016/j.hrthm.2014.10.003
Kim, T. Y., Terentyeva, R., Roder, K. H., Li, W., Liu, M., Greener, I., et al. (2017). SK channel enhancers attenuate Ca2+-dependent arrhythmia in hypertrophic hearts by regulating mito-ROS-dependent oxidation and activity of RyR. Cardiovasc. Res. 113, 343–353. doi: 10.1093/cvr/cvx005
Kirichok, Y., Krapivinsky, G., and Clapham, D. E. (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 22, 360–364. doi: 10.1038/nature02246
Kobayashi, S., Yano, M., Suetomi, T., Ono, M., Tateishi, H., Mochizuki, M., et al. (2009). Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. J. Am. Coll. Cardiol. 53, 1993–2005. doi: 10.1016/j.jacc.2009.01.065
Kolhaas, M., Liu, T., Knopp, A., Zeller, T., Ong, M. F., Böhm, M., et al. (2010). Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121, 1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911
Kwong, J. Q., Lu, X., Correll, R. N., Schwanekamp, J. A., Vagnozzi, R. J., Sargent, M. A., et al. (2015). The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 12, 15–22. doi: 10.1016/j.celrep.2015.06.002
Kyrychenko, S., Poláková, E., Kang, C., Pocsai, K., Ullrich, N. D., Niggli, E., et al. (2013). Hierarchical accumulation of RyR post-translational modifications drives disease progression in dystrophic cardiomyopathy. Cardiovasc. Res. 97, 666–675. doi: 10.1093/cvr/cvs425
Landstrom, A. P., Dobrev, D., and Wehrens, X. H. T. (2017). Calcium signaling and cardiac arrhythmias. Circ. Res. 120, 1969–1993. doi: 10.1161/CIRCRESAHA.117.310083
Liu, T., Brown, D. A., and O’Rourke, B. (2010). Role of mitochondrial dysfunction in cardiac glycoside toxicity. J. Mol. Cell Cardiol. 49, 728–736. doi: 10.1016/j.yjmcc.2010.06.012
Liu, T., and O’Rourke, B. (2008). Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ. Res. 103, 279–288. doi: 10.1161/CIRCRESAHA.108.175919
Liu, T., Takimoto, E., Dimaano, V. L., DeMazumder, D., Kettlewell, S., Smith, G., et al. (2014). Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 115, 44–54. doi: 10.1161/CIRCRESAHA.115.303062
Lopez-Crisosto, C., Pennanen, C., Vasquez-Trincado, C., Morales, P. E., Bravo-Sagua, R., Quest, A. F. G., et al. (2017). Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 14, 342–360. doi: 10.1038/nrcardio.2017.23
Lu, X., Ginsburg, K. S., Kettlewell, S., Bossuyt, J., Smith, G. L., and Bers, D. M. (2013). Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release. Circ. Res. 112, 424–431. doi: 10.1161/CIRCRESAHA.111.300501
Luo, M., Guan, X., Luczak, E. D., Lang, D., Kutschke, W., Gao, Z., et al. (2013). Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J. Clin. Invest. 123, 1262–1274. doi: 10.1172/JCI65268
Luongo, T. S., Lambert, J. P., Gross, P., Nwokedi, M., Lombardi, A. A., Shanmughapriya, S., et al. (2017). The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545, 93–97. doi: 10.1038/nature22082
Luongo, T. S., Lambert, J. P., Yuan, A., Zhang, X., Gross, P., Song, J., et al. (2015). The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 12, 23–34. doi: 10.1016/j.celrep.2015.06.017
Lyon, A. R., Bannister, M. L., Collins, T., Pearce, E., Sepehripour, A. H., Dubb, S. S., et al. (2011). SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ. Arrhythm. Electrophysiol. 4, 362–372. doi: 10.1161/CIRCEP.110.961615
Maack, C., Cortassa, S., Aon, M. A., Ganesan, A. N., Liu, T., and O’Rourke, B. (2006). Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 21, 172–182. doi: 10.1161/01.RES.0000232546.92777.05
Matlib, M. A., Zhou, Z., Knight, S., Ahmed, S., Choi, K. M., Krause-Bauer, J., et al. (1998). Ru360 specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in cardiac myocytes. J. Biol. Chem. 24, 10223–10231. doi: 10.1074/jbc.273.17.10223
Maxwell, J. T., and Blatter, L. A. (2012). Facilitation of cytosolic calcium wave propagation by local calcium uptake into the sarcoplasmic reticulum in cardiac myocytes. J. Physiol. 590, 6037–6045. doi: 10.1113/jphysiol.2012.239434
Maxwell, J. T., Domeier, T. L., and Blatter, L. A. (2012). Dantrolene prevents arrhythmogenic Ca2+ release in heart failure. Am. J. Physiol. Heart Circ. Physiol. 302, H953–H963. doi: 10.1152/ajpheart.00936.2011
Mochizuki, M., Yano, M., Oda, T., Tateishi, H., Kobayashi, S., Yamamoto, T., et al. (2007). Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J. Am. Coll. Cardiol. 24, 1722–1732. doi: 10.1016/j.jacc.2007.01.064
Montero, M., Lobatón, C. D., Hernández-Sanmiguel, E., Santodomingo, J., Vay, L., Moreno, A., et al. (2004). Direct activation of the mitochondrial calcium uniporter by natural plant flavonoids. Biochem. J. 384(Pt 1), 19–24. doi: 10.1042/BJ20040990
Moreau, B., and Parekh, A. B. (2009). Ca2+ -dependent inactivation of the mitochondrial Ca2+ uniporter involves proton flux through the ATP synthase. Curr. Biol. 18, 855–859. doi: 10.1016/j.cub.2008.05.026
Negretti, N., O’Neill, S. C., and Eisner, D. A. (1993). The relative contributions of different intracellular and sarcolemmal systems to relaxation in rat ventricular myocytes. Cardiovasc. Res. 27, 1826–1830. doi: 10.1093/cvr/27.10.1826
Niggli, E., Ullrich, N. D., Gutierrez, D., Kyrychenko, S., Poláková, E., and Shirokova, N. (2013). Posttranslational modifications of cardiac ryanodine receptors: Ca(2+) signaling and EC- coupling. Biochim. Biophys. Acta 1833, 866–875. doi: 10.1016/j.bbamcr.2012.08.016
O’Rourke, B., and Blatter, L. A. (2009). Mitochondrial Ca2+ uptake: tortoise or hare? J. Mol. Cell. Cardiol. 46, 767–774. doi: 10.1016/j.yjmcc.2008.12.011
O’Rourke, B., Cortassa, S., and Aon, M. (2005). Mitochondrial ion channels: gatekeepers of life and death. Physiology 20, 303–315. doi: 10.1152/physiol.00020.2005
Palty, R., Silverman, W. F., Hershfinkel, M., Caporale, T., Sensi, S. L., Parnis, J., et al. (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. U.S.A. 107, 436–441. doi: 10.1073/pnas.0908099107
Pogwizd, S. M., and Bers, D. M. (2004). Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc. Med. 14, 61–66. doi: 10.1016/j.tcm.2003.12.002
Ramay, H. R., Jafri, M. S., Lederer, W. J., and Sobie, E. A. (2010). Predicting local SR Ca(2+) dynamics during Ca(2+) wave propagation in ventricular myocytes. Biophys. J. 98, 2515–2523. doi: 10.1016/j.bpj.2010.02.038
Santulli, G., Xie, W., Reiken, S. R., and Marks, A. R. (2015). Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. U.SA. 112, 11389–11394. doi: 10.1073/pnas.1513047112
Schweitzer, M. K., Wilting, F., Sedej, S., Dreizehnter, L., Dupper, N. J., Tian, Q., et al. (2017). Suppression of arrhythmia by enhancing mitochondrial Ca2+ uptake in catecholaminergic ventricular tachycardia models. JACC Basic Transl. Sci. 2, 737–747. doi: 10.1016/j.jacbts.2017.06.008
Seguchi, H., Ritter, M., Shizukuishi, M., Ishida, H., Chokoh, G., Nakazawa, H., et al. (2005). Propagation of Ca2+ release in cardiac myocytes: role of mitochondria. Cell Calcium 38, 1–9. doi: 10.1016/j.ceca.2005.03.004
Seidlmayer, L. K., Kuhn, J., Berbner, A., Arias-Loza, P. A., Williams, T., Kaspar, M., et al. (2016). Inositol 1,4,5-trisphosphate-mediated sarcoplasmic reticulum-mitochondrial crosstalk influences adenosine triphosphate production via mitochondrial Ca2+ uptake through the mitochondrial ryanodine receptor in cardiac myocytes. Cardiovasc. Res. 112, 491–501. doi: 10.1093/cvr/cvw185
Sharma, V. K., Ramesh, V., Franzini-Armstrong, C., and Sheu, S. S. (2000). Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J. Bioenerg. Biomembr. 32, 97–104. doi: 10.1023/A:1005520714221
Shkryl, V. M., and Shirokova, N. (2006). Transfer and tunneling of Ca2+ from sarcoplasmic reticulum to mitochondria in skeletal muscle. J. Biol. Chem. 20, 1547–1554. doi: 10.1074/jbc.M505024200
Sobie, E. A., Song, L. S., and Lederer, W. J. (2005). Local recovery of Ca2+ release in rat ventricular myocytes. J. Physiol. 565(Pt 2), 441–447. doi: 10.1113/jphysiol.2005.086496
Stowe, D. F., Aldakkak, M., Camara, A. K., Riess, M. L., Heinen, A., Varadarajan, S. G., et al. (2006). Cardiac mitochondrial preconditioning by Big Ca2+-sensitive K+ channel opening requires superoxide radical generation. Am. J. Physiol. Heart Circ. Physiol. 290, H434–H440. doi: 10.1152/ajpheart.00763.2005
Szalai, G., Csordás, G., Hantash, B. M., Thomas, A. P., and Hajnóczky, G. (2000). Calcium signal transmission between ryanodine receptors and mitochondria. J. Biol. Chem. 19, 15305–15313. doi: 10.1074/jbc.275.20.15305
Szentesi, P., Pignier, C., Egger, M., Kranias, E. G., and Niggli, E. (2004). Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+-induced Ca2+ release refractoriness in heart muscle. Circ. Res. 15, 807–813. doi: 10.1161/01.RES.0000146029.80463.7d
Terentyev, D., Belevych, A. E., Terentyeva, R., Martin, M. M., Malana, G. E., Kuhn, D. E., et al. (2009). miR-1 overexpression enhances Ca(2+) release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII- dependent hyperphosphorylation of RyR2. Circ. Res. 104, 514–521. doi: 10.1161/CIRCRESAHA.108.181651
Terentyev, D., Györke, I., Belevych, A. E., Terentyeva, R., Sridhar, A., Nishijima, Y., et al. (2008). Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 103, 1466–1472. doi: 10.1161/CIRCRESAHA.108.184457
Terentyev, D., Rochira, J. A., Terentyeva, R., Roder, K., Koren, G., and Li, W. (2014). Sarcoplasmic reticulum Ca2+ release is both necessary and sufficient for SK channel activation in ventricular myocytes. Am. J. Physiol. Heart. Circ. Physiol. 306, H738-H746. doi: 10.1152/ajpheart.00621.2013
Terentyev, D., Viatchenko-Karpinski, S., Valdivia, H. H., Escobar, A. L., and Györke, S. (2002). Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ. Res. 6, 414–420. doi: 10.1161/01.RES.0000032490.04207.BD
Vay, L., Hernández-SanMiguel, E., Santo-Domingo, J., Lobatón, C. D., Moreno, A., Montero, M., et al. (2007). Modulation of Ca2+ release and Ca2+ oscillations in HeLa cells and fibroblasts by mitochondrial Ca2+ uniporter stimulation. J. Physiol. 580, 39–49. doi: 10.1113/jphysiol.2006.126391
Wagner, S., Rokita, A. G., Anderson, M. E., and Maier, L. S. (2013). Redox regulation of sodium and calcium handling. Antioxid. Redox Signal. 18, 1063–1077. doi: 10.1089/ars.2012.4818
Walsh, C., Barrow, S., Voronina, S., Chvanov, M., Petersen, O. H., and Tepikin, A. (2009). Modulation of calcium signalling by mitochondria. Biochim. Biophys. Acta 1787, 1374–1382. doi: 10.1016/j.bbabio.2009.01.007
Williams, G. S., Boyman, L., Chikando, A. C., Khairallah, R. J., and Lederer, W. J. (2013). Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. U.S.A. 110, 10479–10486. doi: 10.1073/pnas.1300410110
Xie, A., Song, Z., Liu, H., Zhou, A., Shi, G., Wang, Q., et al. (2018). Mitochondrial Ca2+ influx contributes to arrhythmic risk in nonischemic cardiomyopathy. J. Am. Heart Assoc. 7:e007805. doi: 10.1161/JAHA.117.007805
Yan, Y., Liu, J., Wei, C., Li, K., Xie, W., Wang, Y., et al. (2008). Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc. Res. 15, 432–441. doi: 10.1093/cvr/cvm047
Yarana, C., Sripetchwandee, J., Sanit, J., Chattipakorn, S., and Chattipakorn, N. (2012). Calcium- induced cardiac mitochondrial dysfunction is predominantly mediated by cyclosporine A- dependent mitochondrial permeability transition pore. Arch. Med. Res. 43, 333–338. doi: 10.1016/j.arcmed.2012.06.010
Zhao, Z., Gordan, R., Wen, H., Fefelova, N., Zang, W. J., and Xie, L. H. (2013). Modulation of intracellular calcium waves and triggered activities by mitochondrial ca flux in mouse cardiomyocytes. PLoS One 8:e80574. doi: 10.1371/journal.pone.0080574
Zhou, L., Aon, M. A., Liu, T., and O’Rourke, B. (2011). Dynamic modulation of Ca2+ sparks by mitochondrial oscillations in isolated guinea pig cardiomyocytes under oxidative stress. J. Mol. Cell. Cardiol. 51, 632–639. doi: 10.1016/j.yjmcc.2011.05.007
Zima, A. V., and Blatter, L. A. (2006). Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 71, 310–321. doi: 10.1016/j.cardiores.2006.02.019
Keywords: mitochondria, reactive oxygen species, ryanodine receptor, hypertrophy, ventricular arrhythmia, Ca2+-induced Ca2+ release
Citation: Hamilton S, Terentyeva R, Kim TY, Bronk P, Clements RT, O-Uchi J, Csordás G, Choi B-R and Terentyev D (2018) Pharmacological Modulation of Mitochondrial Ca2+ Content Regulates Sarcoplasmic Reticulum Ca2+ Release via Oxidation of the Ryanodine Receptor by Mitochondria-Derived Reactive Oxygen Species. Front. Physiol. 9:1831. doi: 10.3389/fphys.2018.01831
Received: 21 September 2018; Accepted: 06 December 2018;
Published: 21 December 2018.
Edited by:
Angélica Rueda, Centro de Investigación y de Estudios Avanzados (CINVESTAV), MexicoReviewed by:
Diego De Stefani, Università degli Studi di Padova, ItalyCopyright © 2018 Hamilton, Terentyeva, Kim, Bronk, Clements, O-Uchi, Csordás, Choi and Terentyev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dmitry Terentyev, ZG1pdHJ5X3RlcmVudHlldkBicm93bi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.