
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 03 December 2018
Sec. Vascular Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01731
This article is part of the Research TopicTargeting Angiogenesis to Treat Autoimmune Diseases and Cancer View all 11 articles
 Tarik Smani1,2†
Tarik Smani1,2† Luis J. Gómez3†
Luis J. Gómez3† Sergio Regodon3
Sergio Regodon3 Geoffrey E. Woodard4Geraldine Siegfried5
Geoffrey E. Woodard4Geraldine Siegfried5 Abdel-Majid Khatib5*‡
Abdel-Majid Khatib5*‡ Juan A. Rosado6*‡
Juan A. Rosado6*‡Angiogenesis is the growth of blood vessels mediated by proliferation, migration, and spatial organization of endothelial cells. This mechanism is regulated by a balance between stimulatory and inhibitory factors. Proangiogenic factors include a variety of VEGF family members, while thrombospondin and endostatin, among others, have been reported as suppressors of angiogenesis. Transient receptor potential (TRP) channels belong to a superfamily of cation-permeable channels that play a relevant role in a number of cellular functions mostly derived from their influence in intracellular Ca2+ homeostasis. Endothelial cells express a variety of TRP channels, including members of the TRPC, TRPV, TRPP, TRPA, and TRPM families, which play a relevant role in a number of functions, including endothelium-induced vasodilation, vascular permeability as well as sensing hemodynamic and chemical changes. Furthermore, TRP channels have been reported to play an important role in angiogenesis. This review summarizes the current knowledge and limitations concerning the involvement of particular TRP channels in growth factor-induced angiogenesis.
The endothelium is a monolayer of endothelial cells (ECs) that line the internal surface of the vascular wall. In addition to serve as a barrier between circulation and the vascular smooth muscle cells, the endothelium plays a relevant role sensing hemodynamic and chemical changes in blood, regulating hemostasis and participating in the formation of new blood vessels, a process called angiogenesis. To create new vessels, ECs need to proliferate, to migrate, and to be organized in three dimensions. There are distinct processes of angiogenesis. The most rapid angiogenic mechanism is known as intussusception. Common in vascular remodeling during development, intussusception is the splitting of a preexisting vessel into two new smaller vessels. This occurs by penetration of smooth muscle cells through the endothelial cell layer (Burri et al., 2004). The formation of new vessels in adult during both physiological and pathological angiogenesis was also attributed to circulating bone marrow—derived endothelial precursor cells (EPCs). Although EPCs are mainly found in active sites of angiogenesis following a chemotactic signal (Patenaude et al., 2010), these cells act as collaborator cells in close proximity to the endothelium and are not incorporated into the vessel (Grunewald et al., 2006). Other angiogenic mechanisms occur during sprouting angiogenesis. Indeed, special ECs of a preexisting vessel acquire the capacity to invade the surrounding tissue by forming an angiogenic sprout. The later is composed of leading cells known as tip cells and trailing stalk cells. These cells are required for the orientation and growth toward the source of an angiogenic factor (Gerhardt and Betsholtz, 2005). As soon as two sprouts anastomose, sprouting is accomplished by lumen formation and the initiation of blood circulation (Fantin et al., 2010). The maturation of newly formed sprouts into differentiated blood vessels requires the recruitment of mural cells, the development of the surrounding matrix and specialization of ECs in organ-specific manner. Pericytes participate in the stabilization of the newly formed blood vessels through direct physical contact and paracrine signaling.
Angiogenesis is regulated by a balance between stimulatory and inhibitory factors. When this balance shifts in favor of positive stimuli the “angiogenic switch” occurs (Hickey and Simon, 2006). To date several negative regulators of angiogenesis have been identified, however little is known about their exact role during physiological angiogenesis. Among these regulators, thrombospondin, previously reported to be secreted by epithelial cells, was found to inhibit tumor growth angiogenesis (Henkin and Volpert, 2011). Lately other anti-angiogenesis agents were also identified including endostatin, tumstatin, vasostatin, and lately anti-vascular endothelial growth factor (VEGF) (Norden et al., 2009). In the adult, under physiological conditions blood ECs are quiescent due to the increased levels of anti-angiogenic factors (thrombospondin and endostatin) compared to proangiogenic forces, such as the VEGF-A, placental growth factor (PlGF), platelet-derived growth factor (PDGF), and others. During pathological situations, including carcinogenesis and chronic inflammation, angiogenic factors are upregulated, and become more prominent than anti-angiogenic agents.
The growth factors VEGFs, PDGF-BB, and PlGF are all grouped in the VEGF superfamily (McDonald and Hendrickson, 1993), and contain a cystine knot motif in their amino acid sequence. In mammals five VEGF members have been identified, namely VEGF-A, -B, -C, -D, and PlGF (McDonald and Hendrickson, 1993) (Figure 1). These growth factors mediate their function on vascular and lymphatic ECs through their cognate receptors VEGFR-1, -2, and -3 and the NP co-receptors. VEGF-A is able to activate both VEGFR-1 and VEGFR-2, whereas VEGF-B and PlGF are selective ligands for VEGFR-1 (Takahashi and Shibuya, 2005). VEGF-C and -D are the only known ligands for VEGFR-3 and are also able to activate VEGFR-2 (Tammela et al., 2005). The different expression of these receptors in various tissues seemed to be responsible for the relatively specific function of their ligands. Indeed, VEGFR-1 and VEGFR-2 are mainly found in vascular ECs, VEGFR-3 is largely restricted to lymphatic endothelium. Thus, according to their affinities for VEGFR-1 and -2, VEGF-A, -B, and PlGF exert angiogenic activities, while VEGF-C and -D predominantly act as lymphangiogenic growth factors by activating VEGFR-3. The interaction of VEGF with VEGFR (Jakobsson et al., 2006) leads to receptor dimerization leading to conformational changes and phosphorylation of their tyrosine residues, which is important for downstream signal mediators activation. The activation cascades outcome is the elaboration of various VEGF biological responses such as cell proliferation, survival, migration and ECs arrangement to form vascular tubes. The activation of VEGFR can be repressed by its dephosphorylation mediated by various phosphotyrosine phosphatases (PTPs), including density enhanced phosphatase 1 (DEP1) and vascular endothelial PTP (VEPTP) (Kappert et al., 2005).
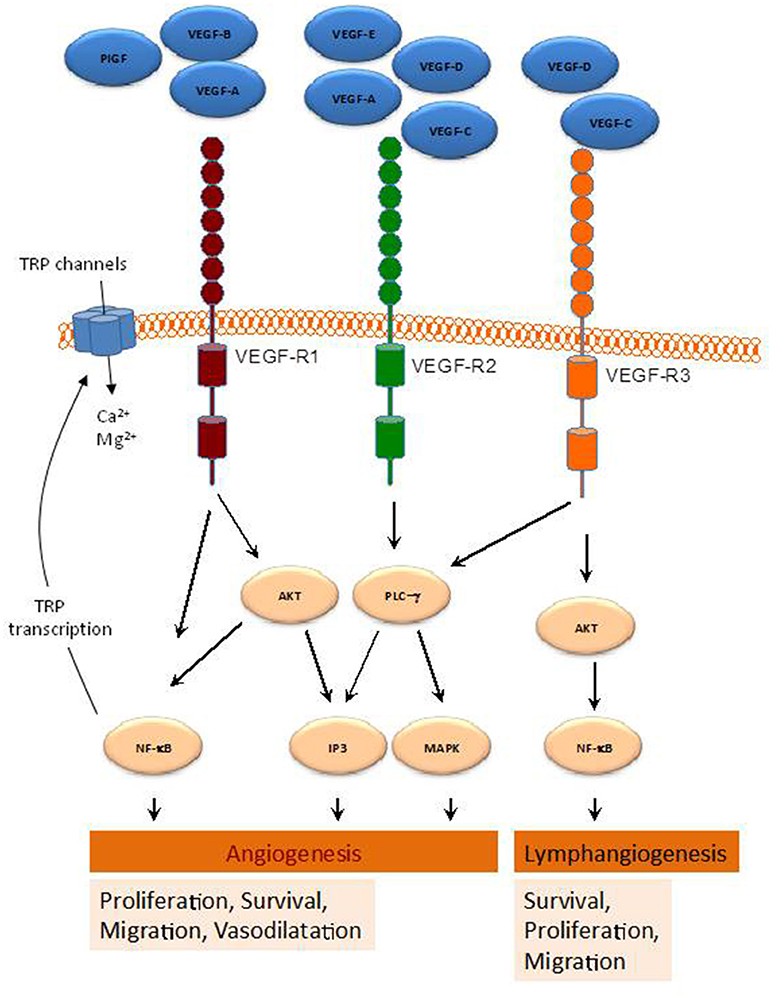
Figure 1. Schematic representation of various pathways activated by the VEGF-family members. By binding to their receptors (VEGFR1-3), indicated VEGF members activate several intracellular pathways involved in a range of cellular functions leading to angiogenesis and lymphangiogenesis.
VEGFR1 (also known as Fms-like tyrosine kinase 1, Flt1,) binds VEGF-A, VEGF-B, and PlGF (Wiesmann et al., 1997). Activation of this receptor was found to induce various kinases including phosphoinositide 3′ kinase (PI3K)/protein kinase B (PKB/AKT), extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK), and the stress kinase p38MAPK (Tchaikovski et al., 2008). VEGFR1 exists as a soluble form (sFlt1) (Kendall and Thomas, 1993), that exhibits higher affinity for VEGFA than VEGFR2. As a result, sFlt1 operates as a negative regulator of angiogenesis by reducing VEGFA/VEGFR2 interaction (Ambati et al., 2006).
VEGFR2 [KDR (kinase insert domain receptor, human) and Flk1 (fetal liver kinase-1, mouse)]. Actively involved in vascular permeability, this receptor is crucial for ECs function during development. VEGFR2 is expressed most prominently in vascular ECs, with highest expression levels during embryonic vasculogenesis and angiogenesis (Millauer et al., 1993). VEGFR2 expression was also found increased during pathological processes associated with neovascularization such as tumor angiogenesis (Plate et al., 1993). VEGFR2 binds VEGF-A via its extracellular Ig-like domains 2 and 3, but with a lower affinity than VEGFR1 (Fuh et al., 1998). In contrast to VEGFR1, VEGFR2 binds also VEGF-C and VEGF-D (McColl et al., 2003) and represses binding to VEGFR3, which results in the inhibition of the proliferation of lymphatic ECs (Albuquerque et al., 2009). Interaction of VEGF-A and VEGFR2 promotes receptor dimerization (Yang et al., 2010), allowing receptor activation leading to several signaling mediators activation like PLCγ (Cunningham et al., 1997), and the adapter proteins SHB and SCK (Warner et al., 2000). These signals are required for various EC functions including proliferation, cell survival and migration, and vascular permeability.
(also known as Flt4) binds VEGF-C and VEGF-D. Produced as precursor proteins, when proteolytically cleaved show increased affinity for both VEGFR2 and VEGFR3 (Joukov et al., 1997). VEGF-C and VEGFR3 interaction is critical for lymphendothelial function. Expressed in vascular ECs VEGFR3 is up-regulated during active angiogenesis. Binding of VEGF-C or VEGF-D to VEGFR3 leads to various kinases activation in VEGFR3 (Dixelius et al., 2003) and the activation of the PI3K/AKT pathway (Mäkinen et al., 2001), critical in lymphendothelial cell migration and sprouting of lymph EPCs and development of the lymphatic system (Karkkainen et al., 2004). Furthermore, VEGF-C-mediated AKT activation is required for embryonic and adult lymphangiogenesis (Zhou et al., 2010).
Compelling evidence demonstrated that VEGFRs increase intracellular Ca2+ concentration ([Ca2+]i), through the activation of TRP and other Ca2+ channels, which modulates signaling pathways leading to angiogenesis (Simons et al., 2016). For instance, VEGF-A enhances inositol 1,4,5-trisphosphate (IP3) generation, which results in Ca2+ store depletion and the activation of store-operated Ca2+ entry in ECs and EPCs (SOCE) (Faehling et al., 2002; Moccia et al., 2014a). Consistent with this, SOCE inhibition or removal of extracellular Ca2+ has been reported to prevent VEGF-mediated Ca2+ oscillations in endothelial colony forming cells (Dragoni et al., 2011). Moreover, TRPC6 has been found to mediate VEGF-induced Ca2+ influx in microvessel ECs (Pocock et al., 2004), and both TRPC3 and TRPC6 mediate Ca2+ entry by VEGF in human microvascular ECs in vivo (Cheng et al., 2006). Furthermore, Mg2+ influx through TRP family members, such as TRPM6 and TRPM7, has been provided to be relevant for EC proliferation and angiogenesis (Nilius et al., 2003). TRP channels and VEGF signaling exhibit a cross relationship, so that VEGFRs activation has been reported to induce NFκB-mediated activation of transcription of certain TRP genes (Santoni et al., 2011) (Figure 1), while Ca2+ influx via TRP channels has been found to stimulate the transcription of genes encoding different growth factors, including VEGF and PDGF, in ECs (Yao and Garland, 2005). Therefore, TRP channels play a relevant role in VEGF-mediated signaling in ECs, as summarized below.
In 1969, Cosens and Manning reported their findings concerning a blind mutant strain of Drosophila melanogaster whose external appearance and histological sections of retinal structure were indistinguishable from the wild-type strain but exhibited abnormal electroretinogram (Cosens and Manning, 1969). Further studies revealed that while short stimuli induce a similar response in the wild-type and mutant fly, the response in the mutant fly to longer light stimulation was characterized by a marked decay in the receptor potential in the presence of illumination. The trp mutant, called so due to the transient receptor potential in response to light found in the retinular cells of the mutant strain, as compared to the more sustained receptor potential recorded in the wild-type fly, exhibited a defect in the process that links excitation to the membrane conductance (Minke et al., 1975; Minke, 1977). Later on, the light-sensitive conductance in Drosophila photoreceptors was found to be mediated by the Na+ and Ca2+-permeable channel trp and its homolog trpl (Hardie and Minke, 1992; Phillips et al., 1992), and comprises two distinct currents: one is conducted by the highly Ca2+ selective trp channel while the second is conducted by the trpl channel, which is supposed to be responsible for the residual light-sensitive current in the trp mutants (Katz et al., 2017).
The first mammalian homolog of Drosophila trp was identified in mouse in 1995 (Petersen et al., 1995) and two independent groups identified the first human transient receptor potential (TRP) channel, called TRPC1, (Wes et al., 1995; Zhu et al., 1995). Since the discovery of the first TRP channel in mammalian cells 28 TRP genes have been identified, which can be grouped into three subfamilies closely related to Drosophila trp (TRPC, TRPV, and TRPM), two more distantly related subfamilies (TRPP and TRPML), and a less related TRPN group expressed in flies and worms (Montell et al., 2002; Salido et al., 2011).
All TRP channels show a common architecture. They are membrane proteins with six putative transmembrane domains (TM1–TM6) and present a cation-permeable pore region created by a loop between TM5 and TM6 (Figure 2). The N- and C-termini are located intracellularly and show a great variability both in length and amino acid sequence among the different TRP members. The N- and C- terminal sequences include a variety of functional domains (Ramsey et al., 2006), including: (1) a variable number of ankyrin repeats (present in the members of TRPA, TRPC, TRPV, and TRPN subfamilies) that have been found to play a relevant role in channel sensing and gating (Gaudet, 2008); (2) TRPC, TRPM and TRPN exhibit a “TRP domain” sequence adjacent to the TM6, which shows highly conserved sequences called TRP boxes 1 and 2, and has been shown to be required for channel tetramerization and function (Venkatachalam and Montell, 2007). Similarly, the TRPV1, TRPA1, and TRPP channels show a TRP-like domain, which shows a similar α-helical configuration and function to TRP domains (García-Sanz et al., 2004; Zheng et al., 2018); (3) an α-kinase domain present in TRPM6 and TRPM7 that regulates channel function and sensitivity to Mg2+·ATP (Clark et al., 2008; Zhang et al., 2014); (4) an ADPR hydrolase domain (Nudix-like domain or NUDT9 homology domain) in TRPM2, which has been reported to sense ADP-ribose concentration and convey this information to the cell by activation of cation entry (Scharenberg, 2005); (5) a calmodulin- and IP3 receptor (IP3R)-binding site (CIRB, present in TRPC, members), a domain that has been reported to be involved in the modulation of TRPC6 channel function by IP3R and Ca2+/calmodulin (Dionisio et al., 2011) and to modulate plasma membrane location of TRPC3 channels via an IP3R-independent pathway (Wedel et al., 2003); (6) an EF-hand Ca2+-binding domain (present in members of the TRPP, TRPML, and TRPA1) (Zurborg et al., 2007); (7) a large extracellular loop between TM1 and TM2 in TRPP and TRPML, which has recently been reported to play an essential role in channel assembly and function (Salehi-Najafabadi et al., 2017); and, (8) coiled-coil domains located in the C-terminal region (for TRPV, TRPM, TRPA1, and TRPP) or in the N- and C-terminal domains (for TRPC) (García-Sanz et al., 2004; Li et al., 2011a) (Figure 2), which have been found to be involved in subunit-subunit interaction (Launay et al., 2004), as well as in the interaction of TRPs with channel modulators, such as the interaction of TRPC proteins with the endoplasmic reticulum Ca2+ sensor, STIM1 (Lee et al., 2014).
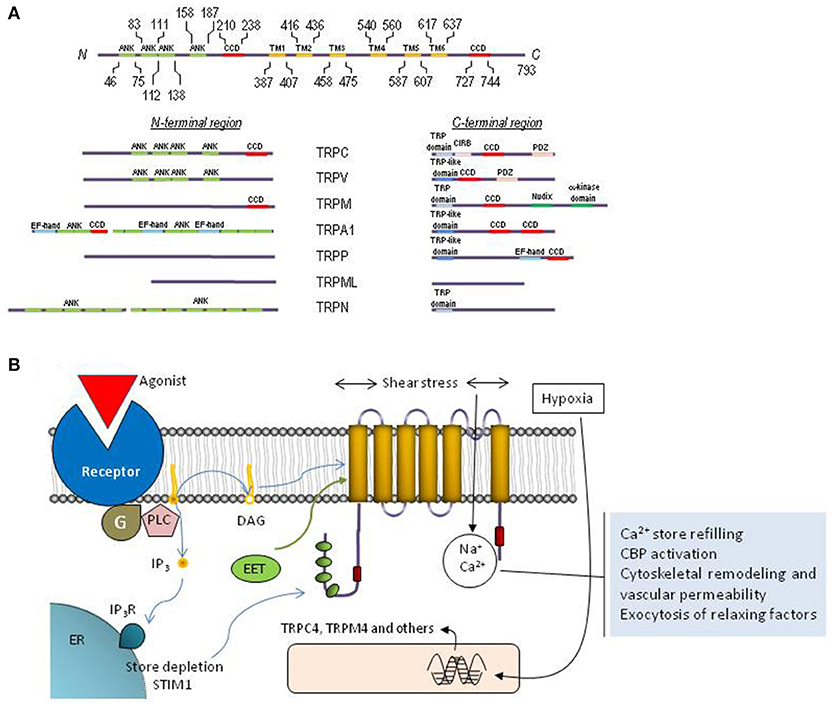
Figure 2. Molecular structure and function of TRP channels. (A) Schematic representation of TRPC1 depicting functionally relevant domains. The cytoplasmic N- and C-termini of each TRP family contain different structural and functional domains as indicated. (B) Cartoon representing the topology of a TRP monomer within the plasma membrane, activation and functions associated to TRP channels in endothelial cells. The protein exhibits six transmembrane domains (TM1-TM6) with the pore located between transmembrane domains 5 and 6, and both N-terminal and C-terminal domains located in the cytosol. Endothelial TRP channels can be activated by receptor occupation, which, in turn, leads to the activation of phospholipase C and the generation of DAG and inositol 1,4,5-trisphosphate (IP3). DAG is an endogenous activator of certain TRP channels and Ca2+ store depletion results in STIM1-dependent activation of TRPC1. Furthermore, TRP channels in endothelial cells can be activated by shear stress and the different EETs or their expression can be upregulated by hypoxia. Calcium influx via TRP channels is involved in Ca2+ store refilling, activation of Ca2+-binding proteins (CBP), cytoskeletal remodeling and the regulation of vascular permeability as well as the exocytosis of smooth muscle cell relaxing factors.
Mammalian TRP channels are permeable to monovalent and divalent cations, with a permeability for Ca2+ over Na+ (ratio PCa/PNa) that ranges from channels that are selective for monovalent cations, such as TRPM4 and TRPM5, to highly Ca2+ selective channels, including TRPV5 and TRPV6, which exhibit a ratio PCa/PNa over 100 (Freichel et al., 2012). It has also been reported that TRP channels are permeable to metal ions, such as manganese, magnesium, zinc, barium, strontium, nickel or cobalt, and, certain TRP members exhibit a greater relative permeability for these ions than for Ca2+ (for an extensive review see Bouron et al., 2015).
TRP channels have been reported to be activated and/or modulated by a number of chemical and physical stimuli, such as extracellular and intracellular ions (including H+, Ca2+ and Mg2+) (Liman, 2007; Zhang et al., 2014) and ligands, both intracellular molecules [such as diacylglycerol (Hofmann et al., 1999), phosphoinositide-4,5-bisphosphate (PIP2) (Nilius et al., 2006; Jardín et al., 2008a)] and exogenous natural and synthetic ligands (for a review see Harteneck et al., 2011; Vetter and Lewis, 2011), temperature and mechanical stretch (Venkatachalam and Montell, 2007). Furthermore, TRPC channels have been reported to be activated by intracellular Ca2+ store depletion via the interaction with STIM1 and Orai1, the key elements for the activation of store-operated Ca2+ entry (SOCE) (Zhang et al., 2005; Feske et al., 2006). Ca2+ entry through SOCE is conducted by two types of channels: the highly Ca2+ selective CRAC (Ca2+ release-activated Ca2+) channel, involving Orai1 subunits, and the less selective store-operated Ca2+ (SOC) channels (Desai et al., 2015). Despite the participation of TRPC channels in SOCE has been a matter of intense debate in the past, there is now a general consensus that TRPC1 is a component of the SOC channels, forming a ternary complex with Orai1 and STIM1, which confers store depletion sensitivity to SOC channels (Huang et al., 2006; Jardin et al., 2008b; Desai et al., 2015; Ambudkar et al., 2017).
ECs have been reported to express at the transcript and/or protein level most of the mammalian TRP isoforms identified, including TRPC1, 3, 4, 5, 6, and 7, TRPV1, 2, and 4, TRPP1 and 2, TRPA1 and TRPM1, 2, 3, 4, 6, 7, and 8, although differences in the expression profile have been reported for different vasculatures and species (Wong and Yao, 2011; Cao et al., 2018).
TRP channels contribute to the Ca2+ influx induced by a plethora of vasoactive agents, including thrombin, ATP, angiotensin II or bradykinin (Bishara and Ding, 2010; Sundivakkam et al., 2013). Ca2+ entry through TRP channels has been found to be involved in the activation of a number of signaling pathways and cellular functions. Among the major functional roles of ECs is the modulation of the vascular tone through the release of a variety of factors that induce relaxation of smooth muscle cells. TRP channels have been reported to play an important role in this process, for instance, irisin, an exercise-induced myokine, has been reported to induce vasodilatation of rat mesenteric arteries through the activation of endothelial TRPV4 channels, which are involved in Ca2+ influx induced by irisin in primary cultured rat mesenteric artery ECs (Ye et al., 2018). Furthermore, TRPV4-deficient mice exhibit attenuated acetylcholine-induced endothelium-dependent vasodilatation associated to a reduced nitric oxide (NO) release (Zhang et al., 2009).
TRP channels have also been found to play a relevant role in vascular permeability, a cellular process that is based on transcellular and paracellular pathways, being the later regulated by the balance between cell-cell adhesive forces and contractile forces generated by the endothelial cytoskeleton (Wong and Yao, 2011). Probably, one of the most widely investigated TRP channels for its implication in endothelial permeability is TRPC6, which has been shown to be involved in lung ischemia-reperfusion-induced edema in mice (Weissmann et al., 2012), as well as in endotoxin-induced lung vascular permeability (Tauseef et al., 2012). TRPC1 and TRPC4 have also been found to be involved in vascular permeability. Expression of TRPC1 induced by TNFα has been reported to enhanced Ca2+ influx and vascular permeability (Paria et al., 2003) and TRPC4-deficient mice where thrombin-evoked Ca2+ signals and endothelial permeability were reduced (Tiruppathi et al., 2002). Other TRP channels, such as TRPV4 or TRPM4, have been reported to play a relevant role in vascular permeability. In isolated rat lung, activation of TRPV4 by 4α-phorbol 12,13-didecanoate (4α-PDD), as well as by 5,6- or 14,15-epoxyeicosatrienoic acids, has been found to increase lung endothelial permeability in a Ca2+ entry-dependent manner, which indicates that TRPV4 is involved in the disruption of the alveolar septal barrier. Consistent with this, the effect of the TRPV4 agonists was impaired in TRPV4-deficient mice (Alvarez et al., 2006). TRPM4 has been reported to be up-regulated in the ECs of blood vessels following spinal cord injury, which has been associated to secondary hemorrhage and progressive hemorrhagic necrosis (Gerzanich et al., 2009). Although the mechanism underlying the role of TRPM4 in vascular permeability remains unclear, there is a body of evidence supporting that TRPM4 expression is involved in post-trauma secondary hemorrhage, i.e., after spinal cord injury in rats, in vivo gene suppression using Trpm4 antisense was found to preserve capillary integrity and impair secondary hemorrhage, and similar results were observed in TRPM4-deficient mice (Gerzanich et al., 2009). Furthermore, 17β-estradiol, which attenuates TRPM4 and sulfonylurea receptor-1, has been reported to suppress disruption of the blood-spinal cord barrier and attenuate secondary hemorrhage after spinal cord injury (Lee et al., 2015).
Finally, TRP channels have also been reported to play a functional role in the ability of the ECs to sense hemodynamic and chemical changes. Flow shear force results in rises in cytosolic Ca2+ concentration ([Ca2+]i), which, in turn, lead to the release of vasodilating factors. A number of TRP channels are sensitive to flow shear stress, such as TRPV4. It has been reported that flow shear stress induces relaxation of the carotid artery, an effect that is mimicked by the TRPV4 activator 4α-PDD and is prevented by the non-selective TRPV4 inhibitor ruthenium red (Köhler et al., 2006). The involvement of TRPV4 in endothelial-dependent vascular dilation was confirmed in TRPV4-deficient mice, which exhibit attenuated response to stimulation with endothelium-derived hyperpolarizing factor (Loot et al., 2008) and, more recently, with studies reporting that TRPV4-TRPC1 heteromeric channels mediate flow shear-induced endothelial Ca2+ influx by a mechanism that might involve an upstream mechanosensitive pathway including phospholipase A2 and cytochrome P450 epoxygenase activity (Loot et al., 2008; Ma et al., 2010). The TRPP1-TRPP2 complex has also been suggested to play a role in flow-induced ECs-mediated vascular dilation, as Ca2+ influx and NO production in response to flow is significantly reduced by TRPP1 or TRPP2 expression silencing (Nauli et al., 2008; AbouAlaiwi et al., 2009); although the mechanism underlying the activation of TRPP2-mediated Ca2+ entry by flow shear forces in ECs remains unclear. A more recent study has identified the formation of a heteromeric channel including the flow-sensitive TRPV4 and both TRPC1 and TRPP2, which mediates the flow-induced Ca2+ influx in native vascular ECs (Du et al., 2014). TRP channels also play a relevant role sensing chemical blood components. For instance, TRPC3, TRPC4, TRPM2, TRPM7, and TRPA1 have been reported to be activated by oxidative stress, leading to Na+ and Ca2+ entry and, thus, mediating the vascular effects associated to reactive oxygen species (ROS) (Wong and Yao, 2011). On the other hand, in addition to sensing ROS, TRPA1 channels have been found to detect molecular oxygen and are essential for hyperoxia- and hypoxia-induced vagal responses (Takahashi et al., 2011). The mechanistic details of the activation of TRPA1 by O2 as well as the transduction pathway remain unclear; however, in cerebral arteries, TRPA1 in the endothelium is mostly located within myoendothelial junction sites, where TRPA1-mediated Ca2+ influx is associated to endothelium-dependent smooth muscle cell vasodilatation through the activation of Ca2+-activated K+ channels (KCa3.1), which, in turns, results in ECs hyperpolarization that is conducted via myoendothelial gap junctions to hyperpolarize the adjacent smooth muscle cell, resulting in myocyte relaxation (Earley, 2012).
TRP channels have also been found to play a relevant role in angiogenesis. Compelling evidence demonstrated that angiogenic growth factors activate TRP channels, causing a subsequent rise in endothelial [Ca2+]i, which modulates the signal transduction pathways leading to angiogenesis (Kwan et al., 2007). It is known that both tumor and physiological angiogenesis are initiated in hypoxic environment principally due to secretion of several growth factors, such as VEGF. These growth factors stimulate proliferation, migration, and tube formation of ECs, resulting in the generation of new capillary (Kohn et al., 1995). Most studies used particularly VEGF to investigate neovascularization in different experimental model. Briefly, tyrosine phosphorylation of VEGFR triggers activation of phospholipase C (PLC), inositol 1,4,5-triphosphate (InsP3) and diacylglycerol (DAG) generation. The consequent Ca2+ entry following the classic Ca2+ release modulates signaling pathways leading to angiogenesis (Simons et al., 2016). Several reports demonstrated that VEGF-induced Ca2+ entry through different isoforms of TRP in several cell types, such as TRPC3 and TRPC6 (Hamdollah Zadeh et al., 2008; Andrikopoulos et al., 2017); TRPM2 through reactive oxygen generation (Mittal et al., 2015); or TRPV1 (Garreis et al., 2016). Certainly, in ECs some TRPs associate to others isoforms forming heteromeric channels (Loot et al., 2008; Nauli et al., 2008; AbouAlaiwi et al., 2009; Ma et al., 2010), however most studies of angiogenesis focused on only one isoform of TRPs as detailed below.
The participation of TRPC3 in angiogenesis has been characterized in Human Umbilical Vein ECs (HUVEC) treated with VEGF. TRPC3 inhibition or its silencing with siRNA attenuated VEGF activation of ERK1/2 phosphorylation, and stimulation of [Ca2+]i transients in HUVEC. Additionally, siRNA of TRPC3 significantly suppressed endothelial tube formation, an indicator of angiogenesis (Andrikopoulos et al., 2017). This study suggests that TRPC3 is activated by the generation of DAG downstream of VEGFR in HUVECs, causing Na+ influx by subsequent activation of the Na+/Ca2+ exchanger in reversal mode, contributing ultimately to angiogenesis (Andrikopoulos et al., 2017). The role of TRPC3 in angiogenesis has also been evaluated in EPCs (Dragoni et al., 2013). As stated above, EPCs are adult stem cells having the ability to differentiate into ECs, and thereby they promote postnatal vasculogenesis and endothelial repair after vascular intima injury (Djohan et al., 2018). Molecular and pharmacological inhibition of TRPC3, using siRNA and Pyr3 respectively, abrogated VEGF-induced Ca2+ response and inhibited proliferation of EPCs (Dragoni et al., 2013). The selectivity of Pyr3 on TRPC3 might be questioned, nevertheless the effect of TRPC3 silencing suggest that this channels might be relevant for vasculogenesis.
Independently of ECs stimulation with VEGF, silencing the expression of TRPC3, TRPC4, or TRPC5 also prevented spontaneous [Ca2+]i oscillations and inhibited tube formation in human umbilical vein-derived EC line EA.hy926 and HUVECs (Antigny et al., 2012). A recent study performed in retina microvascular ECs showed that hypoxia, a potent trigger of angiogenesis, enhanced the expression of TRPC4, whose silencing inhibited VEGF-induced ECs proliferation and migration and in vitro angiogenesis evaluated by tube formation (Song et al., 2015). More recently, silencing of TRPC4 attenuated oxLDL-induced human coronary ECs proliferation; migration and in vitro angiogenesis-tube formation on matrigel, suggesting that suppression of TRPC4 might be an alternative therapeutic strategy for atherosclerotic neovascularization (Qin et al., 2016).
TRPC6 seems also critical for angiogenesis and Ca2+ entry in response to VEGF and 1-oleoyl-2-acetyl-sn-glycerol (OAG, a membrane-permeant DAG analog) in human microvascular ECs and in HUVEC. Experiments using a dominant-negative mutant of TRPC6, made with three mutations in the pore region, reduced ECs proliferation, migration and sprouting in matrigel assay (Hamdollah Zadeh et al., 2008). Similar results were observed in HUVEC, where a dominant-negative form of TRPC6 inhibited VEGF-induced cation current, HUVEC growth and proliferation, as well as VEGF-evoked capillary formation in vitro (Ge et al., 2009). The role of TRPC6 in ECs proliferation and tube formation was also observed when 11,12-EET (11,12-cis-epoxyeicosatrienoic acid) was used to stimulate ECs (Ding et al., 2014).
Other studies have focused on the role of TRPC1 in angiogenesis. Indeed, a proangiogenic role for TRPC1 has been described in vivo in zebrafish, where authors have identified severe angiogenic defects in intersegmental vessel sprouting after knockdown of TRPC1 (Yu et al., 2010). Furthermore, TRPC1 likely controls cell proliferation and tubulogenesis in normal EPCs and in those isolated from peripheral blood of tumor patients (Moccia et al., 2014b). Recently, in vivo matrigel assay confirmed that EPCs isolated from TRPC1 knockout mice has substantially reduced functional activities, including migration and tube formation, indicating that TRPC1 plays an important role in angiogenesis (Du et al., 2018). Nevertheless, other studies suggested that TRPC1 is not relevant for angiogenesis. The use of siRNAs, dominant-negative mutants or neutralizing antibodies, failed to demonstrate that TRPC1 is required for VEGF-induced Ca2+ increase in HUVECs and tube formation (Li et al., 2011b; Antigny et al., 2012). Interestingly, TRPC1 knockout mice developed normal vasculature (Schmidt et al., 2010). Therefore, more investigations are still required to clarify the real role of TRPC1 in the angiogenic processes.
TRPV4 has long been known to regulate angiogenesis and neovascularization by stimulating ECs proliferation and migration as reviewed recently (Moccia, 2018). TRPV4 plays an important role in cytoskeletal reorganization and changes in cell adhesion, which coordinate ECs proliferation and motility via mechanotransduction (Köhler et al., 2006; Reddy et al., 2015; Adapala et al., 2016; Thoppil et al., 2016). TRPV4 is dramatically up-regulated in breast tumor-derived ECs, and is required for arachidonic acid (AA)-evoked Ca2+ entry, which increase the rate of ECs migration and motility as compared to control ECs (Fiorio Pla et al., 2012). Moreover, the absence of TRPV4 in knockout mice was associated with an increase in basal Rho/Rho kinase activity, significant increase in ECs proliferation, migration, and abnormal tube formation in vitro (Thoppil et al., 2016). Interestingly, another study from the same group confirmed that overexpression or pharmacological activation of TRPV4, using GSK1016790, restored the aberrant ECs mechanosensitivity, migration and normalized tube formation in matrigel assay. TRPV4 activation and overexpression likely normalized the abnormal angiogenesis evoked by tumor ECs through the inhibition of the exacerbated Rho activity (Adapala et al., 2016). Therefore, TRPV4 activation seems relevant to normalize tumor angiogenesis via modulation of Rho/Rho kinase pathway.
TRPV1 has been found to be pro-angiogenic. Intraperitoneal injection of mice with a TRPV1 ligand, evodiamine, promoted vascularization in matrigel plugs used in vivo in wild type mice. In contrast, the induced angiogenesis was markedly reduced in TRPV1 knockout mice (Ching et al., 2011). Similarly, using knockout mice TRPV1 appears crucial for 14,15-EET-induced Ca2+ influx, NO production and angiogenesis evaluated by tube formation and in vivo matrigel assays (Su et al., 2014a). In addition, in human microvascular ECs TRPV1 activation is involved in simvastatin-activated Ca2+ influx, which induced the activation of CaMKII signaling and enhanced the formation of TRPV1–eNOS complex, leading to NO production and in vitro angiogenesis-tube formation (Su et al., 2014b).
TRPM2, TRPM4, and TRPM7 have also been found to be involved in angiogenesis (Zhou et al., 2014). Recently, a study demonstrated that VEGF stimulated ECs migration and induced ROS-dependent Ca2+ entry through TRPM2 activation. In addition, they showed that matrigel plugs supplemented with VEGF injected subcutaneously in TRPM2 knockout mice presented significantly reduced vessel formation compared to wild type mice. Using the mouse aortic ring assay, they also observed defective capillary sprouting and reduced capillary lengths isolated from TRPM2 knockout mouse rings as compared with WT mice, indicating that TRPM2 was required for angiogenesis and ischemic neovascularization (Mittal et al., 2015). Moreover, TRPM4 is upregulated in vascular endothelium following hypoxia/ischemia in vitro and in vivo, and in HUVECs following oxygen–glucose deprivation. Pharmacological blocking of TRPM4, or its silencing with siRNA, enhanced tube formation on matrigel and improved capillary integrity in vivo (Loh et al., 2014). Previously, a report demonstrated that silencing of TRPM7, mimics the effect of Mg2+ deficiency in microvascular ECs growth and migration, proposing magnesium and TRPM7 as a modulator of angiogenesis (Baldoli and Maier, 2012).
Little is known regarding the participation of TRPA and TRPP isoforms in the angiogenic process. Few years ago, TRPA1 was suggested as the downstream effector for simvastatin—evoked activation of TRPV1-Ca2+ signaling in ECs, since its inhibition markedly decreased eNOS activation, NO production and in vitro angiogenesis-tube formation (Su et al., 2014b). The role of TRPA1 was further confirmed using matrigel plugs in vivo in TRPA1 knockout mice, whereby simvastatin—induced angiogenesis was partially reduced (Su et al., 2014b).
ECs activity, such as proliferation, migration, and survival is required for angiogenesis under both physiological conditions, (vessel growth and renewal) and pathological conditions, (cardiovascular diseases and tumors initiation and progression). Alteration of these functions resulted from exaggerated or reduced bioavailability of various downstream effectors of VEGF receptors. For example increased Akt and ERK activation following sustained VEGFRs-VEGF interaction induces tumor angiogenesis and growth, whereas, reduced Nitric oxide (NO) production seemed to cause endothelial dysfunction such as deficiency in vascular relaxation. It is now evident that TRP channels are critically involved in physiological and pathological angiogenic process. By controlling Ca2+ homeostasis, different TRP isoforms are activated by pro-angiogenic stimuli that evoke ECs proliferation and migration, as well as the formation of new capillary derived either from ECs or from EPCs. Nevertheless, considerable work is needed to fully understand why many TRPs from different subfamilies are activated by similar pro-angiogenic stimuli such as VEGFs, and whether these TRPs might associate between them to promote their angiogenic effect. To the best of our knowledge, and from the point of view of angiogenesis, the organization and interactions between closely related TRP channels have not been addressed. Several questions still remain unsolved concerning the role or TRP channels in angiogenesis such as are different TRPs located in microdomains with different VEGF-receptors? Are different Ca2+ signals generated by these TRP complexes inducing different cellular functions? Further studies will definitely clarify these and other functional aspects.
In light of the reported findings, the search of selective pharmacological blockers or activator of TRP channels stands out among the strategies for obtaining promising molecular drugs to normalize angiogenesis or for anti-angiogenic therapies to prevent tumor neovascularization.
TS, A-MK, and JR conceived the concept of the review. TS, A-MK, LG, GS, and JR wrote the review. SR, GW, and A-MK designed and formatted the figures. TS, LG, SR, GW, GS, A-MK, and JR read and edited the review manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by MINECO [Grants BFU2016-74932-C2], by the Institute of Carlos III [Grant PI15/00203], by the Andalusia Government [Grant: PI-0313-2016] and Junta de Extremadura-FEDER (IB16046 and GR18061). This study was co-financed by FEDER Funds.
AbouAlaiwi, W. A., Takahashi, M., Mell, B. R., Jones, T. J., Ratnam, S., Kolb, R. J., et al. (2009). Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ. Res. 104, 860–869. doi: 10.1161/CIRCRESAHA.108.192765
Adapala, R. K., Thoppil, R. J., Ghosh, K., Cappelli, H. C., Dudley, A. C., Paruchuri, S., et al. (2016). Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene 35, 314–322. doi: 10.1038/onc.2015.83
Albuquerque, R. J., Hayashi, T., Cho, W. G., Kleinman, M. E., Dridi, S., Takeda, A., et al. (2009). Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat. Med. 15, 1023–1030. doi: 10.1038/nm.2018
Alvarez, D. F., King, J. A., Weber, D., Addison, E., Liedtke, W., and Townsley, M. I. (2006). Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ. Res. 99, 988–995. doi: 10.1161/01.RES.0000247065.11756.19
Ambati, B. K., Nozaki, M., Singh, N., Takeda, A., Jani, P. D., Suthar, T., et al. (2006). Corneal avascularity is due to soluble VEGF receptor-1. Nature 443, 993–997. doi: 10.1038/nature05249
Ambudkar, I. S., de Souza, L. B., and Ong, H. L. (2017). TRPC1, Orai1, and STIM1 in SOCE: friends in tight spaces. Cell Calcium 63, 33–39. doi: 10.1016/j.ceca.2016.12.009
Andrikopoulos, P., Eccles, S. A., and Yaqoob, M. M. (2017). Coupling between the TRPC3 ion channel and the NCX1 transporter contributed to VEGF-induced ERK1/2 activation and angiogenesis in human primary endothelial cells. Cell. Signal. 37, 12–30. doi: 10.1016/j.cellsig.2017.05.013
Antigny, F., Girardin, N., and Frieden, M. (2012). Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J. Biol. Chem. 287, 5917–5927. doi: 10.1074/jbc.M111.295733
Baldoli, E., and Maier, J. A. (2012). Silencing TRPM7 mimics the effects of magnesium deficiency in human microvascular endothelial cells. Angiogenesis 15, 47–57. doi: 10.1007/s10456-011-9242-0
Bishara, N. B., and Ding, H. (2010). Glucose enhances expression of TRPC1 and calcium entry in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 298, H171–H178. doi: 10.1152/ajpheart.00699.2009
Bouron, A., Kiselyov, K., and Oberwinkler, J. (2015). Permeation, regulation and control of expression of TRP channels by trace metal ions. Pflugers Arch. 467, 1143–1164. doi: 10.1007/s00424-014-1590-3
Burri, P. H., Hlushchuk, R., and Djonov, V. (2004). Intussusceptive angiogenesis: its emergence, its characteristics, and its significance. Dev. Dyn. 231, 474–488. doi: 10.1002/dvdy.20184
Cao, S., Anishkin, A., Zinkevich, N. S., Nishijima, Y., Korishettar, A., Wang, Z., et al. (2018). Transient receptor potential vanilloid 4 (TRPV4) activation by arachidonic acid requires protein kinase A-mediated phosphorylation. J. Biol. Chem. 293, 5307–5322. doi: 10.1074/jbc.M117.811075
Cheng, H. W., James, A. F., Foster, R. R., Hancox, J. C., and Bates, D. O. (2006). VEGF activates receptor-operated cation channels in human microvascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 26, 1768–1776. doi: 10.1161/01.ATV.0000231518.86795.0f
Ching, L. C., Kou, Y. R., Shyue, S. K., Su, K. H., Wei, J., Cheng, L. C., et al. (2011). Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc. Res. 91, 492–501. doi: 10.1093/cvr/cvr104
Clark, K., Middelbeek, J., Dorovkov, M. V., Figdor, C. G., Ryazanov, A. G., Lasonder, E., et al. (2008). The alpha-kinases TRPM6 and TRPM7, but not eEF-2 kinase, phosphorylate the assembly domain of myosin IIA, IIB and IIC. FEBS Lett. 582, 2993–2997. doi: 10.1016/j.febslet.2008.07.043
Cosens, D. J., and Manning, A. (1969). Abnormal electroretinogram from a Drosophila mutant. Nature 224, 285–287. doi: 10.1038/224285a0
Cunningham, S. A., Arrate, M. P., Brock, T. A., and Waxham, M. N. (1997). Interactions of FLT-1 and KDR with phospholipase C gamma: identification of the phosphotyrosine binding sites. Biochem. Biophys. Res. Commun. 240, 635–639. doi: 10.1006/bbrc.1997.7719
Desai, P. N., Zhang, X., Wu, S., Janoshazi, A., Bolimuntha, S., Putney, J. W., et al. (2015). Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message. Sci. Signal. 8:ra74. doi: 10.1126/scisignal.aaa8323
Ding, Y., Fromel, T., Popp, R., Falck, J. R., Schunck, W. H., and Fleming, I. (2014). The biological actions of 11,12-epoxyeicosatrienoic acid in endothelial cells are specific to the R/S-enantiomer and require the G(s) protein. J. Pharmacol. Exp. Ther. 350, 14–21. doi: 10.1124/jpet.114.214254
Dionisio, N., Albarran, L., Berna-Erro, A., Hernandez-Cruz, J. M., Salido, G. M., and Rosado, J. A. (2011). Functional role of the calmodulin- and inositol 1,4,5-trisphosphate receptor-binding (CIRB) site of TRPC6 in human platelet activation. Cell. Signal. 23, 1850–1856. doi: 10.1016/j.cellsig.2011.06.022
Dixelius, J., Makinen, T., Wirzenius, M., Karkkainen, M. J., Wernstedt, C., Alitalo, K., et al. (2003). Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. J. Biol. Chem. 278, 40973–40979. doi: 10.1074/jbc.M304499200
Djohan, A. H., Sia, C. H., Lee, P. S., and Poh, K. K. (2018). Endothelial progenitor cells in heart failure: an authentic expectation for potential future use and a lack of universal definition. J. Cardiovasc. Transl. Res. 11, 393–402. doi: 10.1007/s12265-018-9810-4
Dragoni, S., Laforenza, U., Bonetti, E., Lodola, F., Bottino, C., Berra-Romani, R., et al. (2011). Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 29, 1898–1907. doi: 10.1002/stem.734
Dragoni, S., Laforenza, U., Bonetti, E., Lodola, F., Bottino, C., Guerra, G., et al. (2013). Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 22, 2561–2580. doi: 10.1089/scd.2013.0032
Du, J., Ma, X., Shen, B., Huang, Y., Birnbaumer, L., and Yao, X. (2014). TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 28, 4677–4685. doi: 10.1096/fj.14-251652
Du, L. L., Shen, Z., Li, Z., Ye, X., Wu, M., Hong, L., et al. (2018). TRPC1 deficiency impairs the endothelial progenitor cell function via inhibition of calmodulin/eNOS pathway. J. Cardiovasc. Transl. Res. 11, 339–345. doi: 10.1007/s12265-018-9798-9
Earley, S. (2012). TRPA1 channels in the vasculature. Br. J. Pharmacol. 167, 13–22. doi: 10.1111/j.1476-5381.2012.02018.x
Faehling, M., Kroll, J., Fohr, K. J., Fellbrich, G., Mayr, U., Trischler, G., et al. (2002). Essential role of calcium in vascular endothelial growth factor A-induced signaling: mechanism of the antiangiogenic effect of carboxyamidotriazole. FASEB J. 16, 1805–1807. doi: 10.1096/fj.01-0938fje
Fantin, A., Vieira, J. M., Gestri, G., Denti, L., Schwarz, Q., Prykhozhij, S., et al. (2010). Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 116, 829–840. doi: 10.1182/blood-2009-12-257832
Feske, S., Gwack, Y., Prakriya, M., Srikanth, S., Puppel, S. H., Tanasa, B., et al. (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185. doi: 10.1038/nature04702
Fiorio Pla, A., Ong, H. L., Cheng, K. T., Brossa, A., Bussolati, B., Lockwich, T., et al. (2012). TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 31, 200–212. doi: 10.1038/onc.2011.231
Freichel, M., Almering, J., and Tsvilovskyy, V. (2012). The role of TRP proteins in mast cells. Front. Immunol. 3:150. doi: 10.3389/fimmu.2012.00150
Fuh, G., Li, B., Crowley, C., Cunningham, B., and Wells, J. A. (1998). Requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J. Biol. Chem. 273, 11197–11204. doi: 10.1074/jbc.273.18.11197
García-Sanz, N., Fernandez-Carvajal, A., Morenilla-Palao, C., Planells-Cases, R., Fajardo-Sanchez, E., Fernandez-Ballester, G., et al. (2004). Identification of a tetramerization domain in the C terminus of the vanilloid receptor. J. Neurosci. 24, 5307–5314. doi: 10.1523/JNEUROSCI.0202-04.2004
Garreis, F., Schroder, A., Reinach, P. S., Zoll, S., Khajavi, N., Dhandapani, P., et al. (2016). Upregulation of transient receptor potential vanilloid type-1 channel activity and Ca2+ influx dysfunction in human pterygial cells. Invest. Ophthalmol. Vis. Sci. 57, 2564–2577. doi: 10.1167/iovs.16-19170
Gaudet, R. (2008). A primer on ankyrin repeat function in TRP channels and beyond. Mol. Biosyst. 4, 372–379. doi: 10.1039/b801481g
Ge, R., Tai, Y., Sun, Y., Zhou, K., Yang, S., Cheng, T., et al. (2009). Critical role of TRPC6 channels in VEGF-mediated angiogenesis. Cancer Lett. 283, 43–51. doi: 10.1016/j.canlet.2009.03.023
Gerhardt, H., and Betsholtz, C. (2005). How do endothelial cells orientate? EXS 94, 3–15. doi: 10.1007/3-7643-7311-3_1
Gerzanich, V., Woo, S. K., Vennekens, R., Tsymbalyuk, O., Ivanova, S., Ivanov, A., et al. (2009). De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat. Med. 15, 185–191. doi: 10.1038/nm.1899
Grunewald, M., Avraham, I., Dor, Y., Bachar-Lustig, E., Itin, A., Jung, S., et al. (2006). VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell 124, 175–189. doi: 10.1016/j.cell.2005.10.036
Hamdollah Zadeh, M. A., Glass, C. A., Magnussen, A., Hancox, J. C., and Bates, D. O. (2008). VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 15, 605–614. doi: 10.1080/10739680802220323
Hardie, R. C., and Minke, B. (1992). The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron 8, 643–651. doi: 10.1016/0896-6273(92)90086-S
Harteneck, C., Klose, C., and Krautwurst, D. (2011). Synthetic modulators of TRP channel activity. Adv. Exp. Med. Biol. 704, 87–106. doi: 10.1007/978-94-007-0265-3_4
Henkin, J., and Volpert, O. V. (2011). Therapies using anti-angiogenic peptide mimetics of thrombospondin-1. Expert Opin. Ther. Targets 15, 1369–1386. doi: 10.1517/14728222.2011.640319
Hickey, M. M., and Simon, M. C. (2006). Regulation of angiogenesis by hypoxia and hypoxia-inducible factors. Curr. Top. Dev. Biol. 76, 217–257. doi: 10.1016/S0070-2153(06)76007-0
Hofmann, T., Obukhov, A. G., Schaefer, M., Harteneck, C., Gudermann, T., and Schultz, G. (1999). Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397, 259–263. doi: 10.1038/16711
Huang, G. N., Zeng, W., Kim, J. Y., Yuan, J. P., Han, L., Muallem, S., et al. (2006). STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 8, 1003–1010. doi: 10.1038/ncb1454
Jakobsson, L., Kreuger, J., Holmborn, K., Lundin, L., Eriksson, I., Kjellen, L., et al. (2006). Heparan sulfate in trans potentiates VEGFR-mediated angiogenesis. Dev. Cell. 10, 625–634. doi: 10.1016/j.devcel.2006.03.009
Jardin, I., Lopez, J. J., Salido, G. M., and Rosado, J. A. (2008b). Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J. Biol. Chem. 283, 25296–25304. doi: 10.1074/jbc.M802904200
Jardín, I., Redondo, P. C., Salido, G. M., and Rosado, J. A. (2008a). Phosphatidylinositol 4,5-bisphosphate enhances store-operated calcium entry through hTRPC6 channel in human platelets. Biochim. Biophys. Acta 1783, 84–97. doi: 10.1016/j.bbamcr.2007.07.007
Joukov, V., Sorsa, T., Kumar, V., Jeltsch, M., Claesson-Welsh, L., Cao, Y., et al. (1997). Proteolytic processing regulates receptor specificity and activity of VEGF-C. EMBO J. 16, 3898–3911. doi: 10.1093/emboj/16.13.3898
Kappert, K., Peters, K. G., Bohmer, F. D., and Ostman, A. (2005). Tyrosine phosphatases in vessel wall signaling. Cardiovasc. Res. 65, 587–598. doi: 10.1016/j.cardiores.2004.08.016
Karkkainen, M. J., Haiko, P., Sainio, K., Partanen, J., Taipale, J., Petrova, T. V., et al. (2004). Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 5, 74–80. doi: 10.1038/ni1013
Katz, B., Payne, R., and Minke, B. (2017). “TRP channels in vision,” in Neurobiology of TRP Channels, 2nd Edn., ed T. L. R. Emir (Boca Raton, FL: CRC Press/Taylor & Francis) 27–63.
Kendall, R. L., and Thomas, K. A. (1993). Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. U.S.A. 90, 10705–10709. doi: 10.1073/pnas.90.22.10705
Köhler, R., Heyken, W. T., Heinau, P., Schubert, R., Si, H., Kacik, M., et al. (2006). Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler. Thromb. Vasc. Biol. 26, 1495–1502. doi: 10.1161/01.ATV.0000225698.36212.6a
Kohn, E. C., Alessandro, R., Spoonster, J., Wersto, R. P., and Liotta, L. A. (1995). Angiogenesis: role of calcium-mediated signal transduction. Proc. Natl. Acad. Sci. U.S.A. 92, 1307–1311. doi: 10.1073/pnas.92.5.1307
Kwan, H. Y., Huang, Y., and Yao, X. (2007). TRP channels in endothelial function and dysfunction. Biochim. Biophys. Acta 1772, 907–914. doi: 10.1016/j.bbadis.2007.02.013
Launay, P., Cheng, H., Srivatsan, S., Penner, R., Fleig, A., and Kinet, J. P. (2004). TRPM4 regulates calcium oscillations after T cell activation. Science 306, 1374–1377. doi: 10.1126/science.1098845
Lee, J. Y., Choi, H. Y., Na, W. H., Ju, B. G., and Yune, T. Y. (2015). 17beta-estradiol inhibits MMP-9 and SUR1/TrpM4 expression and activation and thereby attenuates BSCB disruption/hemorrhage after spinal cord injury in male rats. Endocrinology 156, 1838–1850. doi: 10.1210/en.2014-1832
Lee, K. P., Choi, S., Hong, J. H., Ahuja, M., Graham, S., Ma, R., et al. (2014). Molecular determinants mediating gating of Transient Receptor Potential Canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J. Biol. Chem. 289, 6372–6382. doi: 10.1074/jbc.M113.546556
Li, J., Cubbon, R. M., Wilson, L. A., Amer, M. S., McKeown, L., Hou, B., et al. (2011b). Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 108, 1190–1198. doi: 10.1161/CIRCRESAHA.111.243352
Li, M., Yu, Y., and Yang, J. (2011a). Structural biology of TRP channels. Adv. Exp. Med. Biol. 704, 1–23. doi: 10.1007/978-94-007-0265-3_1
Liman, E. R. (2007). “The Ca(2+)-activated TRP channels: TRPM4 and TRPM5,” in TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades, eds W. B. Liedtke and S. Heller (Boca Raton: CRC Press/Taylor & Francis).
Loh, K. P., Ng, G., Yu, C. Y., Fhu, C. K., Yu, D., Vennekens, R., et al. (2014). TRPM4 inhibition promotes angiogenesis after ischemic stroke. Pflugers Arch. 466, 563–576. doi: 10.1007/s00424-013-1347-4
Loot, A. E., Popp, R., Fisslthaler, B., Vriens, J., Nilius, B., and Fleming, I. (2008). Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc. Res. 80, 445–452. doi: 10.1093/cvr/cvn207
Ma, X., Qiu, S., Luo, J., Ma, Y., Ngai, C. Y., Shen, B., et al. (2010). Functional role of vanilloid transient receptor potential 4-canonical transient receptor potential 1 complex in flow-induced Ca2+ influx. Arterioscler. Thromb. Vasc. Biol. 30, 851–858. doi: 10.1161/ATVBAHA.109.196584
Mäkinen, T., Veikkola, T., Mustjoki, S., Karpanen, T., Catimel, B., Nice, E. C., et al. (2001). Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 20, 4762–4773. doi: 10.1093/emboj/20.17.4762
McColl, B. K., Baldwin, M. E., Roufail, S., Freeman, C., Moritz, R. L., Simpson, R. J., et al. (2003). Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J. Exp. Med. 198, 863–868. doi: 10.1084/jem.20030361
McDonald, N. Q., and Hendrickson, W. A. (1993). A structural superfamily of growth factors containing a cystine knot motif. Cell 73, 421–424. doi: 10.1016/0092-8674(93)90127-C
Millauer, B., Wizigmann-Voos, S., Schnurch, H., Martinez, R., Moller, N. P., Risau, W., et al. (1993). High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 72, 835–846. doi: 10.1016/0092-8674(93)90573-9
Minke, B. (1977). Drosophila mutant with a transducer defect. Biophys. Struct. Mech. 3, 59–64. doi: 10.1007/BF00536455
Minke, B., Wu, C., and Pak, W. L. (1975). Induction of photoreceptor voltage noise in the dark in Drosophila mutant. Nature 258, 84–87. doi: 10.1038/258084a0
Mittal, M., Urao, N., Hecquet, C. M., Zhang, M., Sudhahar, V., Gao, X. P., et al. (2015). Novel role of reactive oxygen species-activated Trp melastatin channel-2 in mediating angiogenesis and postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 35, 877–887. doi: 10.1161/ATVBAHA.114.304802
Moccia, F. (2018). Endothelial Ca(2+) signaling and the resistance to anticancer treatments: partners in crime. Int. J. Mol. Sci. 19:E217. doi: 10.3390/ijms19010217
Moccia, F., Dragoni, S., Poletto, V., Rosti, V., Tanzi, F., Ganini, C., et al. (2014b). Orai1 and transient receptor potential channels as novel molecular targets to impair tumor neovascularization in renal cell carcinoma and other malignancies. Anticancer. Agents Med. Chem. 14, 296–312. doi: 10.2174/18715206113139990315
Moccia, F., Tanzi, F., and Munaron, L. (2014a). Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 14, 457–480. doi: 10.2174/1566524013666131118113410
Montell, C., Birnbaumer, L., Flockerzi, V., Bindels, R. J., Bruford, E. A., Caterina, M. J., et al. (2002). A unified nomenclature for the superfamily of TRP cation channels. Mol. Cell 9, 229–231. doi: 10.1016/S1097-2765(02)00448-3
Nauli, S. M., Kawanabe, Y., Kaminski, J. J., Pearce, W. J., Ingber, D. E., and Zhou, J. (2008). Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 117, 1161–1171. doi: 10.1161/CIRCULATIONAHA.107.710111
Nilius, B., Droogmans, G., and Wondergem, R. (2003). Transient receptor potential channels in endothelium: solving the calcium entry puzzle? Endothelium 10, 5–15. doi: 10.1080/10623320303356
Nilius, B., Mahieu, F., Prenen, J., Janssens, A., Owsianik, G., Vennekens, R., et al. (2006). The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. EMBO J. 25, 467–478. doi: 10.1038/sj.emboj.7600963
Norden, A. D., Drappatz, J., Muzikansky, A., David, K., Gerard, M., McNamara, M. B., et al. (2009). An exploratory survival analysis of anti-angiogenic therapy for recurrent malignant glioma. J. Neurooncol. 92, 149–155. doi: 10.1007/s11060-008-9745-8
Paria, B. C., Malik, A. B., Kwiatek, A. M., Rahman, A., May, M. J., Ghosh, S., et al. (2003). Tumor necrosis factor-alpha induces nuclear factor-kappaB-dependent TRPC1 expression in endothelial cells. J. Biol. Chem. 278, 37195–37203. doi: 10.1074/jbc.M304287200
Patenaude, A., Parker, J., and Karsan, A. (2010). Involvement of endothelial progenitor cells in tumor vascularization. Microvasc. Res. 79, 217–223. doi: 10.1016/j.mvr.2010.01.007
Petersen, C. C., Berridge, M. J., Borgese, M. F., and Bennett, D. L. (1995). Putative capacitative calcium entry channels: expression of Drosophila trp and evidence for the existence of vertebrate homologues. Biochem. J. 311 (Pt 1), 41–44. doi: 10.1042/bj3110041
Phillips, A. M., Bull, A., and Kelly, L. E. (1992). Identification of a Drosophila gene encoding a calmodulin-binding protein with homology to the trp phototransduction gene. Neuron 8, 631–642. doi: 10.1016/0896-6273(92)90085-R
Plate, K. H., Breier, G., Millauer, B., Ullrich, A., and Risau, W. (1993). Up-regulation of vascular endothelial growth factor and its cognate receptors in a rat glioma model of tumor angiogenesis. Cancer Res. 53, 5822–5827.
Pocock, T. M., Foster, R. R., and Bates, D. O. (2004). Evidence of a role for TRPC channels in VEGF-mediated increased vascular permeability in vivo. Am. J. Physiol. Heart Circ. Physiol. 286, H1015–1026. doi: 10.1152/ajpheart.00826.2003
Qin, W., Xie, W., Xia, N., He, Q., and Sun, T. (2016). Silencing of transient receptor potential channel 4 alleviates oxLDL-induced angiogenesis in human coronary artery endothelial cells by inhibition of VEGF and NF-kappaB. Med. Sci. Monit. 22, 930–936. doi: 10.12659/MSM.897634
Ramsey, I. S., Delling, M., and Clapham, D. E. (2006). An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647. doi: 10.1146/annurev.physiol.68.040204.100431
Reddy, K., Khaliq, A., and Henning, R. J. (2015). Recent advances in the diagnosis and treatment of acute myocardial infarction. World J. Cardiol. 7, 243–276. doi: 10.4330/wjc.v7.i5.243
Salehi-Najafabadi, Z., Li, B., Valentino, V., Ng, C., Martin, H., Yu, Y., et al. (2017). Extracellular loops are essential for the assembly and function of polycystin receptor-ion channel complexes. J. Biol. Chem. 292, 4210–4221. doi: 10.1074/jbc.M116.767897
Salido, G. M., Jardin, I., and Rosado, J. A. (2011). The TRPC ion channels: association with Orai1 and STIM1 proteins and participation in capacitative and non-capacitative calcium entry. Adv. Exp. Med. Biol. 704, 413–433. doi: 10.1007/978-94-007-0265-3_23
Santoni, G., Morelli, M. B., Santoni, M., and Nabissi, M. (2011). New deals on the transcriptional and post-transcriptional regulation of TRP channel target genes during the angiogenesis of glioma. J. Exp. Integr. Med. 1, 221–234. doi: 10.5455/jeim.290711.ir.006
Scharenberg, A. M. (2005). TRPM2 and TRPM7: channel/enzyme fusions to generate novel intracellular sensors. Pflugers Arch. 451, 220–227. doi: 10.1007/s00424-005-1444-0
Schmidt, K., Dubrovska, G., Nielsen, G., Fesus, G., Uhrenholt, T. R., Hansen, P. B., et al. (2010). Amplification of EDHF-type vasodilatations in TRPC1-deficient mice. Br. J. Pharmacol. 161, 1722–1733. doi: 10.1111/j.1476-5381.2010.00985.x
Simons, M., Gordon, E., and Claesson-Welsh, L. (2016). Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 17, 611–625. doi: 10.1038/nrm.2016.87
Song, H. B., Jun, H. O., Kim, J. H., and Fruttiger, M. (2015). Suppression of transient receptor potential canonical channel 4 inhibits vascular endothelial growth factor-induced retinal neovascularization. Cell Calcium 57, 101–108. doi: 10.1016/j.ceca.2015.01.002
Su, K. H., Lee, K. I., Shyue, S. K., Chen, H. Y., Wei, J., and Lee, T. S. (2014a). Implication of transient receptor potential vanilloid type 1 in 14,15-epoxyeicosatrienoic acid-induced angiogenesis. Int. J. Biol. Sci. 10, 990–996. doi: 10.7150/ijbs.9832
Su, K. H., Lin, S. J., Wei, J., Lee, K. I., Zhao, J. F., Shyue, S. K., et al. (2014b). The essential role of transient receptor potential vanilloid 1 in simvastatin-induced activation of endothelial nitric oxide synthase and angiogenesis. Acta Physiol. (Oxf). 212, 191–204. doi: 10.1111/apha.12378
Sundivakkam, P. C., Natarajan, V., Malik, A. B., and Tiruppathi, C. (2013). Store-operated Ca2+ entry (SOCE) induced by protease-activated receptor-1 mediates STIM1 protein phosphorylation to inhibit SOCE in endothelial cells through AMP-activated protein kinase and p38beta mitogen-activated protein kinase. J. Biol. Chem. 288, 17030–17041. doi: 10.1074/jbc.M112.411272
Takahashi, H., and Shibuya, M. (2005). The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci.109, 227–241. doi: 10.1042/CS20040370
Takahashi, N., Kuwaki, T., Kiyonaka, S., Numata, T., Kozai, D., Mizuno, Y., et al. (2011). TRPA1 underlies a sensing mechanism for O2. Nat. Chem. Biol. 7, 701–711. doi: 10.1038/nchembio.640
Tammela, T., Enholm, B., Alitalo, K., and Paavonen, K. (2005). The biology of vascular endothelial growth factors. Cardiovasc. Res. 65, 550–563. doi: 10.1016/j.cardiores.2004.12.002
Tauseef, M., Knezevic, N., Chava, K. R., Smith, M., Sukriti, S., Gianaris, N., et al. (2012). TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 209, 1953–1968. doi: 10.1084/jem.20111355
Tchaikovski, V., Fellbrich, G., and Waltenberger, J. (2008). The molecular basis of VEGFR-1 signal transduction pathways in primary human monocytes. Arterioscler. Thromb. Vasc. Biol. 28, 322–328. doi: 10.1161/ATVBAHA.107.158022
Thoppil, R. J., Cappelli, H. C., Adapala, R. K., Kanugula, A. K., Paruchuri, S., and Thodeti, C. K. (2016). TRPV4 channels regulate tumor angiogenesis via modulation of Rho/Rho kinase pathway. Oncotarget 7, 25849–25861. doi: 10.18632/oncotarget.8405
Tiruppathi, C., Freichel, M., Vogel, S. M., Paria, B. C., Mehta, D., Flockerzi, V., et al. (2002). Impairment of store-operated Ca2+ entry in TRPC4(-/-) mice interferes with increase in lung microvascular permeability. Circ. Res. 91, 70–76. doi: 10.1161/01.RES.0000023391.40106.A8
Venkatachalam, K., and Montell, C. (2007). TRP channels. Annu. Rev. Biochem. 76, 387–417. doi: 10.1146/annurev.biochem.75.103004.142819
Vetter, I., and Lewis, R. J. (2011). Natural product ligands of TRP channels. Adv. Exp. Med. Biol. 704, 41–85. doi: 10.1007/978-94-007-0265-3_3
Warner, A. J., Lopez-Dee, J., Knight, E. L., Feramisco, J. R., and Prigent, S. A. (2000). The Shc-related adaptor protein, Sck, forms a complex with the vascular-endothelial-growth-factor receptor KDR in transfected cells. Biochem. J. 347, 501–509. doi: 10.1042/bj3470501
Wedel, B. J., Vazquez, G., McKay, R. R., St, J. B. G., and Putney, J. W. Jr. (2003). A calmodulin/inositol 1,4,5-trisphosphate (IP3) receptor-binding region targets TRPC3 to the plasma membrane in a calmodulin/IP3 receptor-independent process. J. Biol. Chem. 278, 25758–25765. doi: 10.1074/jbc.M303890200
Weissmann, N., Sydykov, A., Kalwa, H., Storch, U., Fuchs, B., Mederos y Schnitzler, M., et al. (2012). Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat. Commun. 3:649. doi: 10.1038/ncomms1660
Wes, P. D., Chevesich, J., Jeromin, A., Rosenberg, C., Stetten, G., and Montell, C. (1995). TRPC1, a human homolog of a Drosophila store-operated channel. Proc. Natl. Acad. Sci. U.S.A. 92, 9652–9656. doi: 10.1073/pnas.92.21.9652
Wiesmann, C., Fuh, G., Christinger, H. W., Eigenbrot, C., Wells, J. A., and de Vos, A. M. (1997). Crystal structure at 1.7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 91, 695–704. doi: 10.1016/S0092-8674(00)80456-0
Wong, C. O., and Yao, X. (2011). TRP channels in vascular endothelial cells. Adv. Exp. Med. Biol. 704, 759–780. doi: 10.1007/978-94-007-0265-3_40
Yang, Y., Xie, P., Opatowsky, Y., and Schlessinger, J. (2010). Direct contacts between extracellular membrane-proximal domains are required for VEGF receptor activation and cell signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 1906–1911. doi: 10.1073/pnas.0914052107
Yao, X., and Garland, C. J. (2005). Recent developments in vascular endothelial cell transient receptor potential channels. Circ. Res. 97, 853–863. doi: 10.1161/01.RES.0000187473.85419.3e
Ye, L., Xu, M., Hu, M., Zhang, H., Tan, X., Li, Q., et al. (2018). TRPV4 is involved in irisin-induced endothelium-dependent vasodilation. Biochem. Biophys. Res. Commun. 495, 41–45. doi: 10.1016/j.bbrc.2017.10.160
Yu, P. C., Gu, S. Y., Bu, J. W., and Du, J. L. (2010). TRPC1 is essential for in vivo angiogenesis in zebrafish. Circ. Res. 106, 1221–1232. doi: 10.1161/CIRCRESAHA.109.207670
Zhang, D. X., Mendoza, S. A., Bubolz, A. H., Mizuno, A., Ge, Z. D., Li, R., et al. (2009). Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 53, 532–538. doi: 10.1161/HYPERTENSIONAHA.108.127100
Zhang, S. L., Yu, Y., Roos, J., Kozak, J. A., Deerinck, T. J., Ellisman, M. H., et al. (2005). STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905. doi: 10.1038/nature04147
Zhang, Z., Yu, H., Huang, J., Faouzi, M., Schmitz, C., Penner, R., et al. (2014). The TRPM6 kinase domain determines the Mg.ATP sensitivity of TRPM7/M6 heteromeric ion channels. J. Biol. Chem. 289, 5217–5227. doi: 10.1074/jbc.M113.512285
Zheng, W., Cai, R., Hofmann, L., Nesin, V., Hu, Q., Long, W., et al. (2018). Direct binding between pre-S1 and TRP-like domains in TRPP channels mediates gating and functional regulation by PIP2. Cell Rep. 22, 1560–1573. doi: 10.1016/j.celrep.2018.01.042
Zhou, F., Chang, Z., Zhang, L., Hong, Y. K., Shen, B., Wang, B., et al. (2010). Akt/Protein kinase B is required for lymphatic network formation, remodeling, and valve development. Am. J. Pathol. 177, 2124–2133. doi: 10.2353/ajpath.2010.091301
Zhou, W., Guo, S., Xiong, Z., and Liu, M. (2014). Oncogenic role and therapeutic target of transient receptor potential melastatin 7 channel in malignancy. Expert Opin. Ther. Targets 18, 1177–1196. doi: 10.1517/14728222.2014.940894
Zhu, X., Chu, P. B., Peyton, M., and Birnbaumer, L. (1995). Molecular cloning of a widely expressed human homologue for the Drosophila trp gene. FEBS Lett. 373, 193–198. doi: 10.1016/0014-5793(95)01038-G
Keywords: angiogenesis, endothelial cells, VEGF, TRP channels, TRPC, TRPV, TRPM
Citation: Smani T, Gómez LJ, Regodon S, Woodard GE, Siegfried G, Khatib A-M and Rosado JA (2018) TRP Channels in Angiogenesis and Other Endothelial Functions. Front. Physiol. 9:1731. doi: 10.3389/fphys.2018.01731
Received: 23 June 2018; Accepted: 16 November 2018;
Published: 03 December 2018.
Edited by:
Vijaya Iragavarapu-Charyulu, Florida Atlantic University, United StatesReviewed by:
Alexander Dietrich, Ludwig Maximilian University of Munich, GermanyCopyright © 2018 Smani, Gómez, Regodon, Woodard, Siegfried, Khatib and Rosado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdel-Majid Khatib, bWFqaWQua2hhdGliQGluc2VybS5mcg==
Juan A. Rosado, amFyb3NhZG9AdW5leC5lcw==
†These authors have contributed equally to this work
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.