 Astrid Brull
Astrid Brull Blanca Morales Rodriguez
Blanca Morales Rodriguez Gisèle Bonne
Gisèle Bonne Antoine Muchir
Antoine Muchir Anne T. Bertrand
Anne T. Bertrand
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 30 October 2018
Sec. Striated Muscle Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01533
This article is part of the Research TopicNuclear Envelope Function in Striated Muscle Health and DiseaseView all 6 articles
Emery-Dreifuss muscular dystrophy (EDMD) is a genetic condition characterized by early contractures, skeletal muscle weakness, and cardiomyopathy. During the last 20 years, various genetic approaches led to the identification of causal genes of EDMD and related disorders, all encoding nuclear envelope proteins. By their respective localization either at the inner nuclear membrane or the outer nuclear membrane, these proteins interact with each other and establish a connection between the nucleus and the cytoskeleton. Beside this physical link, these proteins are also involved in mechanotransduction, responding to environmental cues, such as increased tension of the cytoskeleton, by the activation or repression of specific sets of genes. This ability of cells to adapt to environmental conditions is altered in EDMD. Increased knowledge on the pathophysiology of EDMD has led to the development of drug or gene therapies that have been tested on mouse models. This review proposed an overview of the functions played by the different proteins involved in EDMD and related disorders and the current therapeutic approaches tested so far.
Since the first description of a new X-linked muscular disorder by A. Emery, F. Dreifuss and G. Hogan about six decades ago (Dreifuss and Hogan, 1961; Emery and Dreifuss, 1966), Emery-Dreifuss muscular dystrophy (EDMD) is now also described as an autosomal dominant and recessive inherited disease (Chakrabarti and Pearce, 1981; Fenichel et al., 1982; Miller et al., 1985; Serratrice and Pouget, 1986; Witt et al., 1988). In the 1990's, positional cloning analyses led to the identification of two major EDMD causative genes: EMD in classical X-linked forms and LMNA in autosomal dominant and recessive forms (Bione et al., 1994; Bonne et al., 1999), elucidating around 60% of EDMD cases. These two genes encode proteins of the nuclear envelope, named emerin and A-type lamins, respectively, raising a new research area around the nuclear membranes. More recently, based on the common localization of these proteins, various genetic strategies, including candidate gene approaches, were undertaken in patients suffering from muscular dystrophies without mutations in EMD or LMNA. This has led to the identification of mutations or variants in genes encoding other nuclear envelope proteins such as FHL1B (Gueneau et al., 2009; Ziat et al., 2016), nesprin 1 and 2 (Zhang et al., 2007), SUN1/2 (Meinke et al., 2014), and LUMA (Liang et al., 2011) associated to a range of muscular dystrophies, some of them sharing clinical features of EDMD. This group of disorders is now collectively named striated muscle laminopathies.
This review focuses on the basal description of: (1) the clinic of striated muscle laminopathies, (2) the functions of the disease-causing proteins, and (3) the different treatment strategies that have been studied so far.
The first EDMD description (Emery and Dreifuss, 1966) concerned a three generation family from Virginia, USA, in which eight males were affected by an unusual type of muscular dystrophy transmitted in an X-linked manner (Dreifuss and Hogan, 1961; Emery and Dreifuss, 1966). It was first considered by McKusick in 1971 as a benign form with contractures of a new X-linked muscular dystrophy (McKusick, 1971) and thereafter included in the Becker's 1972 classification of muscular dystrophies (Becker, 1972).
In its 1986's classification of muscular dystrophies, Becker added the autosomal forms of EDMD to the spectrum of muscular dystrophies known at that time (Becker, 1986). Clinically indistinguishable from the X-linked form, he suggested the Hauptmann-Tannhauser eponym to name these autosomal forms. In view of these various reports, and in agreement with other authors (Serratrice and Pouget, 1986; Witt et al., 1988), Emery suggested to use the term Emery-Dreifuss muscular dystrophy for the disorder characterized by (1) early contractures, (2) humeroperoneal muscle weakness, and (3) cardiac disease characterized by supraventricular arrhythmias, disorders of atrioventricular conduction, and cardiomyopathy (Sanna et al., 2003). Then they were classified as either myopathic or neurogenic in origin, and inherited as an X linked, recessive or autosomal dominant trait (Emery, 1989). This genetic heterogeneity was confirmed thereafter during the 90's and the following decade when the underlying molecular bases of EDMD were identified.
Positional cloning led to the identification of the first molecular defects in X-linked EDMD on chromosome Xq28 in the STA gene (now called EMD) (Bione et al., 1994), followed a few years later by the identification of a mutation on chromosome 1q21 in the LMNA gene (Bonne et al., 1999). These two genes encode proteins of the nuclear envelope (see below), opening a new field of research on striated muscle disorders linked to defects in nuclear envelope proteins. About a decade later, acquired knowledge about the pathophysiology of EDMD has led to the identification of other genes involved in EDMD and EDMD-like disorders. Based on a candidate gene strategy, two different groups identified mutations in the SYNE1 and/or SYNE2 genes (Zhang et al., 2007) and missense variants in the TMEM43 gene (Liang et al., 2011) in the so-called autosomal “EDMD-related myopathy.” Indeed, joint contractures are either lacking or not prominent, and for patients carrying SYNE1 and/or SYNE2 mutations, skeletal muscle involvement was highly variable between patients. Finally, a whole-genome analysis of six EDMD families with X-linked inheritance led to the identification of mutations in FHL1 gene (Gueneau et al., 2009) (Table 1).
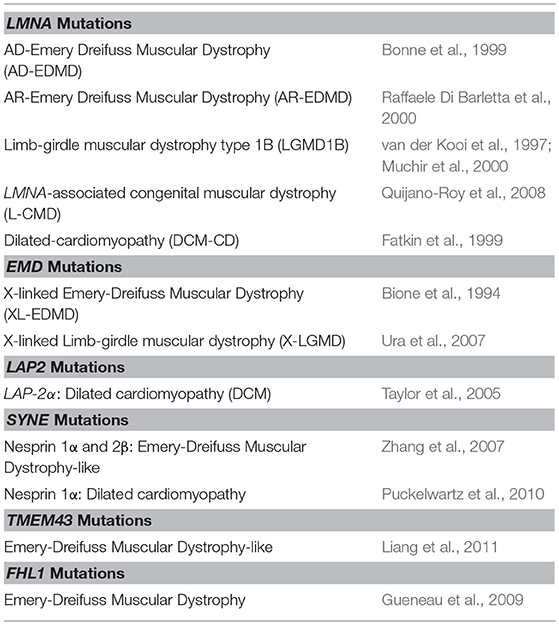
Table 1. Striated and cardiac muscle laminopathies caused by mutations in nuclear envelope proteins.
The nuclear envelope is a lipid bilayer membrane that separates the cytoplasm from the nucleus in eukaryotic cells and that encloses the genetic material (Watson, 1955; Aaronson and Blobel, 1975). The nuclear envelope is composed of the inner nuclear membrane (INM) and the outer nuclear membrane (ONM) (Figure 1). The ONM is continuous with the rough endoplasmic reticulum membrane sharing many of its protein content, with the exception of some integral proteins that are retained at the ONM through specific interactions with INM proteins (like nesprins at the ONM that interact with SUN at the INM, both composing the linker of the nucleoskeleton and the cytoskeleton (LINC) complex) (Crisp et al., 2006). By contrast, the INM contains its own integral proteins, like LEM-domain proteins (LAP2, Emerin, MAN1) or LUMA. INM and ONM interact at the site of nuclear pores and through the LINC complex (Crisp et al., 2006; Starr and Fridolfsson, 2010; Luxton and Starr, 2014). Underneath the INM, the nuclear lamina, a protein meshwork composed of A- and B-type lamins, is connected to the cytoskeleton via the LINC complex. The nuclear lamina is involved in different nuclear functions such as DNA replication and chromatin organization but also has important roles in cytoplasmic organization and cytoskeletal mechanotransduction (Lammerding et al., 2004; Hale et al., 2008; Lombardi et al., 2011).
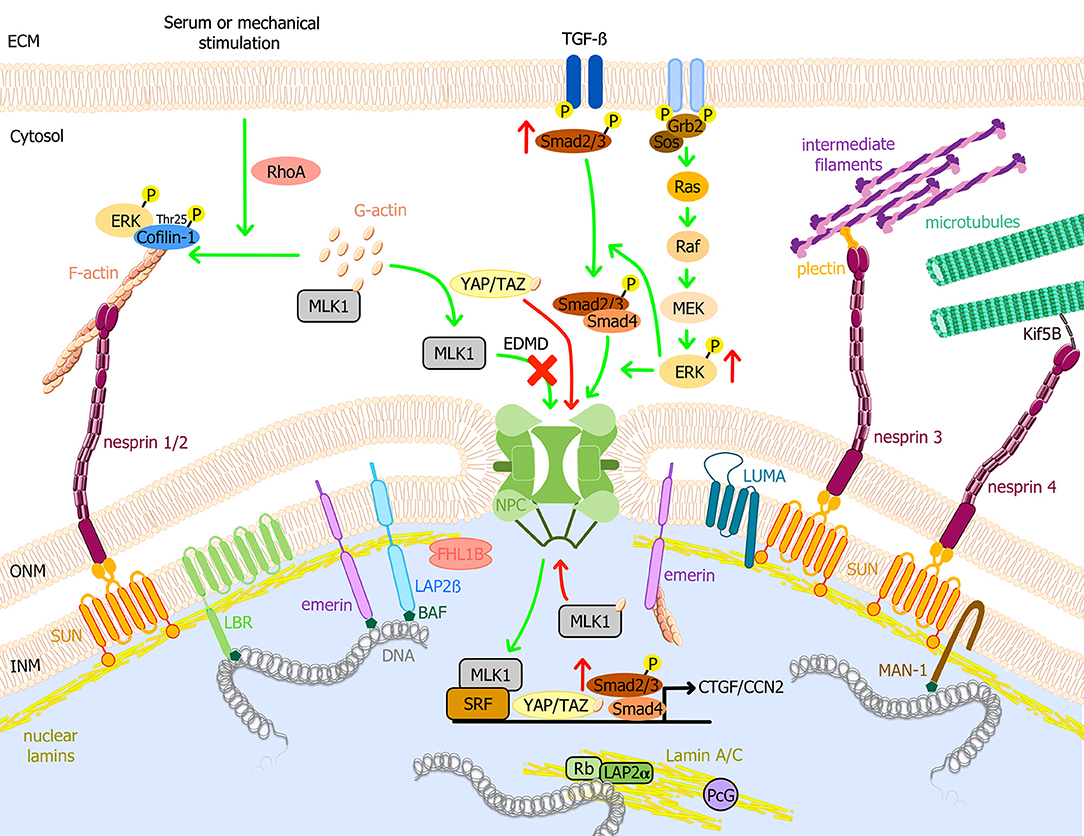
Figure 1. Schematic model of the nuclear envelope proteins and their potential roles in EDMD physiopathology. Nuclear lamins form a meshwork underneath the INM connected with the cytoplasm. It interacts with TM proteins of the nuclear envelope, i.e., emerin, LBR, LAP2, SUN1/2, and MAN1, and with several transcription factors such as Rb. Through the LINC complex, A-type lamins interact with actin microfilaments, microtubules, and cytoplasmic intermediate filaments, connecting the nuclear lamina to the extracellular matrix. MAPK pathways are important transduction cascades initiated by extracellular mitogens, growth factors and cytokines at the cell surface and finalized to the nucleus to control gene expression, regulating cell proliferation and differentiation, survival and apoptosis. LMNA mutations causing EDMD have been related to the activation of ERK, JNK, and p38α cascades, leading to the posterior activation of CTGF/CCN2 via TGF-ß/Smad signaling pathway and cofilin-1, the latter leading to actin depolymerization. After serum or mechanical stimulation, RhoA promotes cytoplasmic actin polymerization, causing the release of MKL1 from cytoplasmic G-actin. MKL1 translocates into the nucleus and together with SRF induces gene expression. In addition, emerin facilitates polymerization of nuclear actin, reducing the nuclear export of MKL1 to the cytoplasm. In EDMD cells, emerin mislocalizes and is unable to modulate nuclear actin polymerization. G-actin binds to MKL1 and it is exported from the nucleus, impairing gene expression. YAP and TAZ are key transcription factors for cell proliferation. YAP/TAZ activation causes their nuclear accumulation, promoting cell proliferation, and inhibiting differentiation. Nuclear localization of YAP/TAZ is increased in patient myoblasts with LMNA mutations. Green arrows indicate reported pathways and red arrows indicate altered EDMD reported pathways. BAF, barrier-to-autointegration factor; ECM, Extracellular matrix; EDMD, Emery-Dreifuss muscular dystrophy; ERK, extracellular signal-regulated kinase; F-actin, actin filament; G-actin, globular actin; Grb2, growth factor receptor-bound protein 2; INM, inner nuclear membrane; Kif5B, kinesin family member 5B; LAP, lamin associated protein; LBR, lamin B receptor; MAPK, mitogen-activated protein kinase; MKL1, megakaryoblastic leukemia 1; NPC, nuclear pore complex; ONM, outer nuclear membrane; Raf, proto-oncogen serine/threonine-protein kinase; Rb, retinoblastoma protein; RhoA, ras homolog family member A; Sos, son of sevenless homolog 1; SRF, serum response factor; TGF-ß, transforming growth factor ß; YAP, yes-associated protein 1; TAZ, transcriptional coactivator with PDZ-binding motif.
Lamins are type V intermediate filament proteins (Fisher et al., 1986; Goldman et al., 1986; McKeon et al., 1986). As all intermediate filament proteins, lamins possess an α-helical coiled-coil central rod domain, composed of heptad repeats of amino acids, bordered by a globular amino-terminal “head” domain and a carboxyl-terminal “tail” domain. This latter also comprises an Ig-like domain and a nuclear localization sequence (NLS) (Parry et al., 1986; Frangioni and Neel, 1993; Dhe-Paganon et al., 2002; Krimm et al., 2002), which make lamins the only intermediate filament protein found in the nucleus. Based on their sequences and structural properties, nuclear lamins can be separated in A-type and B-type lamins (Gerace et al., 1978). In mammals, three genes encode for lamins: LMNA, LMNB1, and LMNB2. LMNA encodes for A-type lamins, with lamins A and C being the main isoforms generated by alternative splicing and expressed in all differentiated cells (Lin and Worman, 1993; Dechat et al., 2010). Lamins AΔ10 and C2 represent minor isoforms specifically expressed in tumoral cells and in male germ line, respectively (Furukawa et al., 1994; Machiels et al., 1996; Alsheimer et al., 2000). Lamins A and C are identical for their first 566 amino acids and vary in their carboxyl terminal tails. The most abundant B-type lamins are lamin B1, encoded by LMNB1, and lamin B2 encoded by LMNB2, both constitutively expressed (Gruenbaum et al., 2005; Burke and Stewart, 2013).
Lamin A and B-type lamins are synthesized as precursors that are post-translationally processed. Firstly a farnesylation of the cysteine of the C-terminal CaaX motif of prelamin A, lamin B1 and B2 occurs, leading to the anchorage of lamins in the nuclear membrane (Farnsworth et al., 1989; Beck et al., 1990). Secondly the aaX amino acids are cleaved by endopeptidases (either Rce1 or Zmpste24) (Nigg et al., 1992; Varela et al., 2005; Young et al., 2005). Thirdly the cysteine residue is methylated by Icmt1 (Maske et al., 2003). Finally, lamin A, but not lamin B1 and B2, is subjected to a last post-transcriptional modification consisting in the cleavage of the last 15 residues containing the farnesylated residue, resulting in mature lamin A, which is release from the INM.
Another piece of complexity in lamins arises from their polymerization. Lamins form coiled-coil homo-dimers that further interact longitudinally through head-to-tail association to form a long polar 10 nm polymer that can further assemble laterally (Aebi et al., 1986; Ben-Harush et al., 2009). Recent studies are however challenging this view and described lamins as being 3.5 nm filaments (Harapin et al., 2015; Turgay et al., 2017). Finally, the different types of lamins assemble in separated networks with B-type lamin network lying close to the nuclear envelope while the A-type lamin network lies closer to the nucleoplasm (Shimi et al., 2015; Xie et al., 2016).
The EMD gene encodes for an ubiquitous type II TM protein called emerin, anchored to the INM through its C-terminal hydrophobic domain (Manilal et al., 1996; Nagano et al., 1996; Tsuchiya et al., 1999). In the nucleoplasm, emerin interacts with numerous proteins, mainly via its LEM domain. It also interacts with lamin A/C, nesprin-1α and LUMA (Ellis, 2006; Liang et al., 2011). Emerin is able to self-assemble in vitro and it was suggested that the LEM domain configuration in the emerin multimers may modulate its interaction with chromatin and nucleoplasmic proteins (Berk et al., 2014; Samson et al., 2017). Depending on tissue, emerin was also described at alternative subcellular locations. This includes endoplasmic or sarcoplasmic reticulum, nucleoplasm, ONM, plasma membrane, and centrosome (Cartegni et al., 1997; Ostlund et al., 1999; Lattanzi et al., 2000; Squarzoni et al., 2000; Manta et al., 2004; Salpingidou et al., 2007). Wheeler et al. proposed that cell type-specific signaling events, such as emerin phosphorylation, might be responsible for targeting emerin to alternative cellular localizations (Wheeler et al., 2010). Emerin functions include regulation of transcription factor localization and function, cell signaling, mechanotransduction, nuclear envelope structure as well as chromatin organization (Koch and Holaska, 2014).
Six different genes named SYNE-1, SYNE-2, SYNE-3, SYNE-4, KASH5, and LRMP encode for multiple spliced isoforms of nesprins (for nuclear envelope spectrin repeat proteins) (Zhou et al., 2018). While nesprin-4, KASH5, and LRMP expression is restricted to epithelial cells, meiotic cells, and a subset of taste receptor cells respectively; nesprin-1, −2, and −3 are ubiquitously expressed with a peculiar abundance of nesprin-1 and−2 in striated muscles. Numerous initiation codons and alternative splicing of SYNE-1 and SYNE-2 genes lead to a wide variety of protein isoforms with great variability in their molecular weight. Some of these smaller isoforms are restricted to specific tissues, like nesprin-1α2 and nesprin-2α1 for striated muscle (Holt et al., 2016).
Giant nesprin-1 and −2 are characterized by a C-terminal Klarsicht/ANC-1/Syne homology (KASH) domain that anchors the proteins to the nuclear envelope, an N-terminal Calponin Homology (CH) domain involved in interactions with actin, and a central domain constituted of multiple spectrin repeats (Rajgor and Shanahan, 2013). Beside interactions with actin, recent data have shown that they are also connected to microtubules through interactions with motor protein kinesin-1 and dynein/dynactin (Wu et al., 2018). Nesprin-3, that lacks the N-terminal actin-binding domain, is involved in the interaction with intermediate filaments through plectin (Ketema and Sonnenberg, 2011). Shorter isoforms may lack some of these domains. While nesprin-1 and−2 giant are set at the ONM where they interact with SUN proteins, the shorter isoforms like nesprin-1α can also be found at the INM and interact directly with lamin A/C and emerin (Mislow et al., 2002).
SUN (Sad1/UNC-84) proteins were named after the identification of their homolog in yeast (Sad1) and C. elegans (UNC-84) (Hagan and Yanagida, 1995; Malone et al., 1999). In mammals, five SUN proteins have been described, SUN1, SUN2, SUN3, SUN4, and SUN5. SUN 1 and SUN 2 are ubiquitously expressed, while other isoforms are only expressed in testes (Sosa et al., 2013).
SUN1 and SUN2 comprise a nucleoplasmic N-terminal region that interacts with lamins (Crisp et al., 2006), a single TM domain that spans the INM, followed by a coiled-coil region that protrude in the perinuclear space, and required for the trimerization of SUN proteins (Sosa et al., 2012; Wang et al., 2012; Zhou et al., 2012; Hennen et al., 2018; Jahed et al., 2018). Finally, the C-terminal SUN domain interacts with the KASH domain of nesprins (Sosa et al., 2012; Wang et al., 2012; Zhou et al., 2012).
The nuclear bridge formed between nesprins and SUN proteins (LINC complex) connect the cytoskeleton (actin, microtubules, and intermediate filaments depending on the nesprin isoform) to the nucleoskeleton (lamin A/C and emerin). It provides structural support to the nucleus and plays an essential role in regulating gene expression and mechanotransduction (Wang et al., 2009; Lombardi et al., 2011).
LUMA (or TMEM43) was first identified in a proteomics screen for nuclear envelope proteins (Dreger et al., 2001). It is characterized by four TM domains involved in its oligomerization and a large fragment protruding in the lumen of the endoplasmic reticulum and the perinuclear space (Bengtsson and Otto, 2008). At the INM, it interacts with lamins A/C and B1 as well as with emerin and SUN2 (Bengtsson and Otto, 2008; Liang et al., 2011). The localization of LUMA at the INM depends on A-type lamins while depletion of LUMA induces emerin mislocalization in HeLa cells but not in mouse neonatal cardiomyocytes (Bengtsson and Otto, 2008; Stroud et al., 2018). In the heart, LUMA is found in the intercalated disks where it has been described as a component of the composite junction (Franke et al., 2014; Siragam et al., 2014). At this specific location, LUMA is supposed to participate in the organization and function of proteins involved in cardiac conduction (Siragam et al., 2014).
The FHL1 gene, localized on the X chromosome, is composed of 8 exons encoding various isoforms of FHL1 (for Four-and-a-Half LIM domain) (Greene et al., 1999). LIM domains (acronym for Lin-11, Isl-1 and Mec3) are highly conserved sequences comprising two zinc fingers in tandem, which are implicated in numerous interactions, essentially with other proteins (Kadrmas and Beckerle, 2004). Each zinc finger is composed of four highly conserved cysteine or histidine linking together one zinc ion (Michelsen et al., 1993; Kosa et al., 1994).
The two first exons are non-coding while exons 6 and 7 are alternatively spliced, which results in three different transcripts: FHL1A, FHL1B and FHL1C; FHL1A being the major isoform in striated muscles. FHL1A is constituted of four and a half LIM domains involved in multiple protein interactions (Lee et al., 1998; Brown et al., 1999a; Greene et al., 1999). FHL1B is composed of only three and a half LIM domain in its N-terminal part, followed by three NLS and one NES, and a binding domain to the RBP-J transcription factor (Brown et al., 1999b). FHL1C is composed of only two and a half LIM domains directly followed by the RBP-J binding domain (Brown et al., 1999b; Ng et al., 2001). Recent data have shown that FHL1B is accumulated at the nuclear lamina where it co-localizes with lamin A/C or emerin (Ziat et al., 2016).
Beside this location at the nuclear envelope, the various FHL1 isoforms have been reported at different locations within the cell: at the focal adhesions where it regulates cell migration via integrins (Robinson et al., 2003); in the sarcomeres where it interacts with MyBP-c as well as with titin and it is involved in sarcomere formation and in the stress-response signaling via activation of the MAPK signaling (McGrath et al., 2006; Sheikh et al., 2008); and in the nucleus where it interacts with various transcriptional factors (like NFAT proteins or RBP-J) involved in cell proliferation and differentiation but also with the pro-apoptotic protein Siva where it is involved in cell survival (Taniguchi et al., 1998; Schulz and Yutzey, 2004; Liang et al., 2008; Cottle et al., 2009).
The use of patient cells and the development of animal models have helped understanding the pathophysiology of EDMD and related disorders, and more specifically, how ubiquitous proteins give rise to striated muscle specific defects. It is now clear that the different proteins involved in EDMD serve as sensors of mechanical tension of the cytoskeleton leading to an adaptive response of the cell via the regulation of the transcription.
EDMD proteins have been implicated in nuclear positioning, nuclear shape and mechanosensing via their interactions with the cytoskeleton (Chang et al., 2015). All cytoskeleton networks (actin, microtubule and intermediate filaments) have been shown to be disorganized in LmnaΔ8−11 mouse embryonic fibroblasts (MEF), in 3T3 Lmna knock-down cells and in various mouse models (Nikolova et al., 2004; Houben et al., 2009; Chatzifrangkeskou et al., 2018). Experiments performed on MEF of LmnaΔ8−11 or Emd-KO mice have shown a disconnection of the nucleus from the rest of cells (Lammerding et al., 2004, 2005; Zwerger et al., 2013) evidenced by the increased centrosome-nucleus distance (Lee et al., 2007; Hale et al., 2008). Shortly after, it was shown that nesprin-2 was a major player in the connection between the nuclear lamina (via interaction with SUN proteins on one side), and the microtubule network (via connection with kinesin light chain-1 on the other side) (Schneider et al., 2011). Cells with disconnection between the cytoplasm and the nucleus have impaired cell polarity, adhesion and migration, which finally impact on myoblasts fusion (Lee et al., 2007; Hale et al., 2008; Chang et al., 2015). In cardiomyocytes, nesprin-2 compensates the loss of nesprin-1, and only nesprin-1/2 double KO mice develop cardiomyopathy (Banerjee et al., 2014).
As mentioned earlier, differentiating myoblasts express specific nesprin isoforms (Randles et al., 2010; Holt et al., 2016). Using various mouse models of mutant nesprin-1 (Table 2), the major roles played by nesprin-1α in nuclear morphology and positioning in skeletal muscles were demonstrated (Zhang et al., 2007, 2010; Puckelwartz et al., 2009; Stroud et al., 2017). Further analyses performed on cultured myoblasts have shown that nesprin-1α isoform is required during muscle differentiation to reorganize the microtubule network from the centrosome in myoblasts to the ONM in myotubes (Gimpel et al., 2017). This reorganization is a prerequisite for the correct nuclear positioning in muscle cells, and is perturbed in C2C12 that are knocked-down for nesprin-1α, in several mouse models, and in patient myoblasts with specific nesprin-1α deletion (Gimpel et al., 2017).
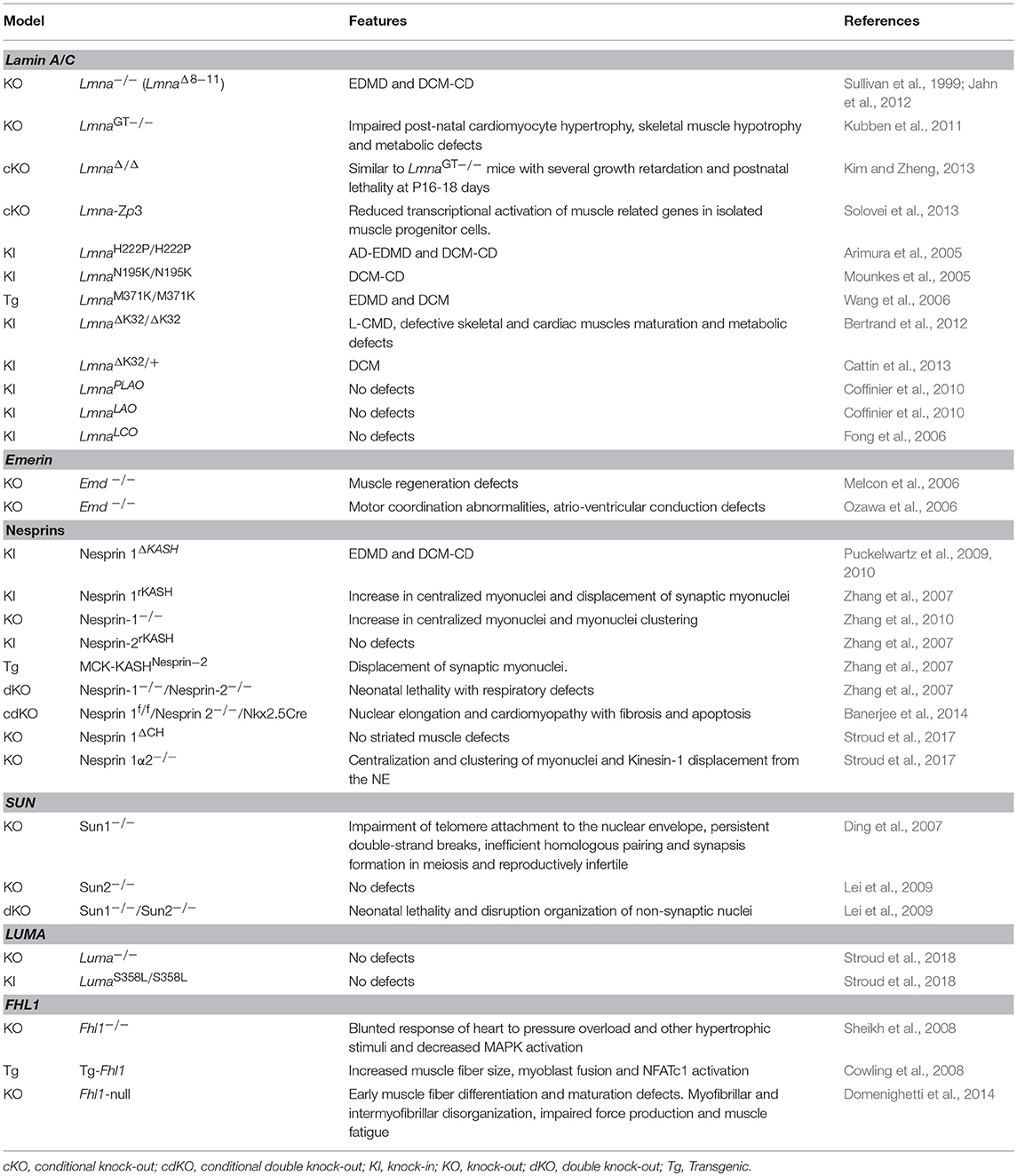
Table 2. List of EDMD and EDMD-like myopathies mouse models.
Alterations of the different cytoskeleton networks have great impact on striated muscle function. This has been more widely explored in the heart, where LmnaΔ8−11/Δ8−11 cardiomyocytes show progressive disorganization and detachment of desmin from the nuclear membrane that parallels the cardiac contractile dysfunction (Nikolova et al., 2004). In LmnaH222P/H222P hearts (Table 2), it was shown that microtubule cytoskeleton alteration and decreased acetylation of α-tubulin lead to connexin-43 displacement from the gap-junction, ultimately leading to electrical conduction disturbances (Le Dour et al., 2017; Macquart et al., 2018).
Cytoskeleton components and adhesion complexes are known mechanosensitive elements able to activate numerous signaling pathways such as mitogen-activated protein kinase-extracellular signal-regulated kinase (MAPK-ERK) and to induce the nuclear translocation of MKL1 (also known as MAL or MRTF-A) and YAP/TAZ to activate expression of early responsive genes. Interestingly, ERK pathway has been shown to be activated in the heart of LmnaH222P/H222P, LmnaΔ8−11/Δ8−11, and Emd−/y mice at baseline while it was not in Fhl1−/y hearts even after cardiac pressure overload (Muchir et al., 2007a,b; Sheikh et al., 2008; Frock et al., 2012). In muscle cells, while a transient activation of ERK is required few minutes after switching C2C12 myoblasts in differentiation medium, C2C12 where emerin has been knock-down have an increased and prolonged activation of ERK, leading to delayed myotube formation (Huber et al., 2009). Nuclear translocation of MKL1 was inefficient in LmnaΔ8−11/Δ8−11, Emd−/y, and LmnaN195K/N195K MEF as well as in patient skin fibroblasts with LMNA mutation leading to isolated cardiomyopathy (LMNA p.K219T) following serum stimulation, while it was not altered in another patient's skin fibroblasts with LMNA mutations leading to congenital muscular dystrophy (LMNA p.K32del) (Ho et al., 2013a,b). Finally, mRNA of the YAP/TAZ pathway were increased in Emd−/y myoblasts, while at the protein level the nuclear localization of YAP/TAZ was increased in patient myoblasts with LMNA mutations cultured at baseline compared with WT, but not following mechanical stimulation or increase matrix stiffness (Bertrand et al., 2014; Iyer et al., 2017).
In addition, an increasing number of studies are now focusing on the role of the nucleus itself as a mechanosensitive element, mainly through the LINC complex (Kirby and Lammerding, 2018). Analyses made on isolated nuclei have shown that force applied on nesprin-1 is responsible for nuclear stiffening due to a rapid phosphorylation of emerin on Tyr74 and Tyr95, leading to the local recruitment of lamin A/C (Guilluy et al., 2014). Knock-down of lamin A/C, double knock-down of SUN1 and SUN2 or phosphoresistant 74-95FF emerin mutant cells all failed to respond to the stimuli. Absence of mechanotransduction due to mutations or deletion of EDMD proteins is evidenced by the absence of hypertrophy following cardiac pressure overload in LmnaΔ8−11/+, Fhl1−/y or nesprin-1/nespin-2 double knock-out mice (Sheikh et al., 2008; Cupesi et al., 2010; Banerjee et al., 2014). Emerin's function in mechanotransduction might be slightly different as Emd−/y mice subjected to cardiac pressure overload present with normal cardiac hypertrophy, however they still display impaired cardiac function compared to WT (Stubenvoll et al., 2015).
Activation of these pathways may affect downstream processes leading to cardiomyopathy and muscle weakness observed in EDMD. MKL1 activation leads to cardiac fibrosis following myocardial infarction or angiotensin II stimulation in WT heart (Small et al., 2010). Similarly ERK activation is able to induce cardiac fibrosis via activation of CTGF/CCN2 in heart from LmnaH222P/H222P mice (Chatzifrangkeskou et al., 2016). ERK1/2 was also shown to catalyze phosphorylation of cofilin-1 in heart from LmnaH222P/H222P mice leading to actin depolymerisation (Figure 1) (Chatzifrangkeskou et al., 2018). At the opposite, YAP/TAZ-dependent activation of FHOD1 observed in patient myoblasts with mutations in LMNA or SYNE-1 leads to actin polymerization (Bertrand et al., 2014; Schwartz et al., 2017).
As shown by the specific distribution of chromatin and chromosomes in the nucleus, genome organization is not random (Croft et al., 1999) and depends on tissue; e.g., fibroblasts tend to have an uniform distribution of heterochromatin throughout the periphery while epithelial cells present a more patchy distribution (Zuleger et al., 2011). The patterns of chromatin distribution and chromosome positioning are disrupted in cells from patients with nuclear envelope diseases (Sewry et al., 2001; Maraldi et al., 2006), suggesting an important functional role of the spatial genome organization.
Nuclear pore complex proteins, lamins, and several nuclear envelope transmembrane proteins of the INM interact directly with DNA, chromatin and/or chromatin-associated proteins (Mattout-Drubezki and Gruenbaum, 2003). Chromatin contacts with A- or B-type lamins via specific genomic regions called lamina-associated domains (LADs). LADs are gene-poor, AT-rich and heterochromatic regions that dynamically bind to and dissociate from the nuclear lamina at the nuclear periphery (Guelen et al., 2008; Kind et al., 2013; Lund et al., 2015). Other INM proteins such as LEM-domain proteins indirectly interact with chromatin via the barrier-to-autointegration factor protein (BAF) in a highly ordered nucleoprotein complex (Figure 1) (Furukawa, 1999; Zheng et al., 2000; Lee et al., 2001; Mansharamani and Wilson, 2005). Through interactions with BAF, LEM-domain proteins contribute to the tethering of genomic regions to nuclear periphery, connecting interphase chromosomes to the nuclear lamina, thereby intervening in global nuclear organization (Barton et al., 2015).
Transmission of external forces from the extracellular matrix into the nucleus via the LINC complex (Lombardi et al., 2011) causes deformation of the nuclear envelope and lamina, which, in turn, modulates chromatin organization and transcriptional activity (Swift et al., 2013). Paulsen and colleagues showed that the overexpression of Flag-tagged wild-type lamin A/C, the constantly farnesylated mutant p.L647R or lamin A/C p.R388P (causing congenital muscular dystrophy), resulted in different lamin A/C-genome interaction patterns detectable by 3D genome modeling (Chrom3D) from Hi-C and CHIP-seq data, suggesting that laminopathy-causing mutations can also alter interactions with chromatin (Paulsen et al., 2017). Recent studies have evidenced the participation of nuclear envelope proteins regulated in a cell type-specific manner by reversible sequestration of regulatory elements at the nuclear periphery, leading to their repression (Lund et al., 2013; Ronningen et al., 2015; Robson et al., 2016). In link with this observation, Favreau and colleagues showed that the overexpression of lamin A/C p.R453W (the mutation's hot spot for LMNA in EDMD) in C2C12 myoblasts resulted in impaired differentiation to myotubes (Favreau et al., 2004). In addition, PARP1 and CTCF, both altered in LmnaH222P/H222P hearts (Chatzifrangkeskou et al., 2016; Vignier et al., 2018) regulate circadian transcriptional plasticity, mediating chromatin interactions, and promoting the recruitment of circadian genes to the nuclear periphery (Zhao et al., 2015). Whereas lamin B1 is localized in the nuclear periphery and interacts only with heterochromatin LADs, lamin A/C binds to both hetero- and euchromatin (Gesson et al., 2016). Expression of LAP2α, the unique LAP2 isoform that is not anchored at the INM, is essential for the nucleoplamsic localization of A-type lamins (Naetar and Foisner, 2009). Whereas lamin A/C localized at the nuclear periphery binds heterochromatin in a LAP2α-independent manner, the small fraction of A-type lamins localized in the nucleoplasm interacts with euchromatin in a LAP2α-dependent manner (Figure 1) (Dechat et al., 2000; Johnson et al., 2004; Kolb et al., 2011; Gesson et al., 2016). The knock-down of LAP2α resulted in the shift of lamin A/C binding toward more heterochromatic regions. Interestingly, the LMNA p.K32del mutation, responsible for severe muscle weakness and wasting, results in the unique localization of A-type lamins within the nucleoplasm in a LAP2α-independent fashion (Bertrand et al., 2012; Pilat et al., 2013). Likewise, the p.R388P A-type lamins mutant that is also uniquely observed in the nucleoplasm has been shown to have altered interaction with LAD (Paulsen et al., 2017).
In the nucleoplasm, lamin A/C has specific protein partners, including the retinoblastoma protein (Rb), a key regulator of cell proliferation (Figure 1) (Mancini et al., 1994; Novitch et al., 1996; Markiewicz et al., 2002; Pekovic et al., 2007). Studies using LmnaΔ8−11/Δ8−11 mouse MEF showed reduced levels of Rb when compared with controls, concluding that Rb-lamin A/C interactions are required for the stability, localization, and activity of Rb (Johnson et al., 2004) and showing that the localization of lamin A/C within the nucleoplasm is important for cell cycle regulation and cell differentiation (Cohen et al., 2013). Similarly, at the nuclear periphery, emerin binds directly to many transcription regulators, including Lmo7 (Holaska et al., 2006), ß-catenin (Markiewicz et al., 2006) and BAF (Holaska and Wilson, 2007). Lmo7 is a transcriptional factor involved in the activation of myogenic differentiation genes, such as MyoD, Myf5, and Pax3 (Dedeic et al., 2011). Its binding to emerin at the nuclear periphery inhibits the binding and the transcriptional activity of Lmo7. A recent study showed that Lmo7-null mice present with a myopathic phenotype similar to that seen in other EDMD mouse models (Mull et al., 2015). Emerin also participates in the regulation of myogenic differentiation and skeletal muscle regeneration by preventing ß-catenin accumulation to the nucleus and the subsequent upregulation of ß-catenin-target genes (Markiewicz et al., 2006). A massive nuclear accumulation of ß-catenin was observed in fibroblasts from patients with X-EDMD, leading to an autostimulatory growth phenotype (Markiewicz et al., 2006).
EDMD proteins also established interactions with chromatin modifiers. Lamin A/C is required for the correct Polycomb group (PcG) protein nuclear compartmentalization (Cesarini et al., 2015). PcG proteins are epigenetic repressors that control numerous target genes during differentiation (Lanzuolo and Orlando, 2012). Polycomb repressive complex 2 (PCR2), one of the best characterized PcG protein complexes together with PCR1, has been shown to interact with lamin A/C in the nucleoplasm (Figure 1) (Cesarini et al., 2015) and its localization at the nuclear periphery is required for muscle differentiation (Wang et al., 2011). Lamin A/C knock-down leads to the dispersion of PcG proteins in the nucleus, causing an aberrant transcriptional reactivation of PcG targets and consequently leading to premature myogenesis (Cesarini et al., 2015). It has been also reported that FHL1 interacts with RING1, one of the components of PcG protein complex (Qin et al., 2004). FHL1 suppresses the RBP-J-mediated transcription by competing with transactivators for the binding motif RBP-J (Taniguchi et al., 1998) and by recruiting RING1 to RBP-J (Qin et al., 2004). The histone deacetylase HDAC3 binds to LAP2ß and emerin (Somech et al., 2005; Demmerle et al., 2012), the histone acetyltransferase hALP1 binds SUN1 (Chi et al., 2007) and LBR interacts with MeCP2, a DNA methylating enzyme (Guarda et al., 2009). Emerin directly interacts and regulates the activity of HDAC3, regulating indirectly the expression and nuclear localization of myogenic differentiation genes, such as Myf5, MyoD and Pax7. It has been shown that EDMD-causing emerin mutations are not able to bind to HDAC3 (Demmerle et al., 2012, 2013). Treatment of emerin-null cells with theophylline, an HDAC3-specific activator, improves myotube formation, whereas the addition of the HDAC3-specific inhibitor RGFP966 blocks myotube formation in wild-type and emerin-null myogenic progenitors (Collins et al., 2017), suggesting that the altered interaction between emerin and HDAC3 is important in the EDMD disease mechanism.
Lamins are also involved in DNA repair. A-type lamins stabilize 53BP1 protein levels and promote SIRT6-mediated DNA repair, preserving the integrity of the genome (Gonzalez-Suarez et al., 2011; Ghosh et al., 2015). Several LMNA mutations, including some leading to EDMD, have been shown to cause an upregulation of cysteine protease Cathepsin L (CTSL). This upregulation leads to the degradation of 53BP1 and changes in its cellular distribution, resulting in defective double strand breaks (DSB) DNA repair (Gonzalez-Suarez et al., 2011). Moreover, lamin A/C interacts with histones H2AX and its phosphorylated form (γH2AX) (induced by DNA damage), maintaining the positional stability of DNA damage repair foci. By contrast, in LmnaΔ8−11 mouse MEF the mobility of DNA repair foci is increased, evidencing the participation of A-type lamins in the spatial anchor of the genome (Mahen et al., 2013).
Thus, since the first mutation identified in 1994 in EMD, our knowledge of the nuclear envelope proteins functions and related pathomechanisms leading to EDMD and striated muscle laminopathies has tremendously expanded. Each new report confirms and reinforces the combination of the pathomechanistic hypotheses proposed by Hutchison and colleagues (Hutchison et al., 2001), i.e., the structural/mechanical and the gene regulation hypotheses.
No specific treatments exist for EDMD and related striated muscle laminopathies. Reports related to treatments and development of adapted specific therapies focused essentially on EDMD linked to LMNA mutations. Today, therapy is preventive and/or symptomatic. Supportive care, e.g., orthopedic procedures to minimize progression of joint contractures and spinal deformity, has been proposed to patients and in the most severe cases mechanical aids are required (Bonne and Quijano-Roy, 2013). Patients might also present respiratory insufficiency requiring nocturnal or continuous mechanical ventilation, especially in children with the congenital form (L-CMD, Table 1) or in early-onset EDMD patients (Quijano-Roy et al., 2008). All striated muscle laminopathies associated to LMNA mutation share a common denominator, the cardiac involvement, which represents the most life-threating feature of the pathology. Cardiac phenotype is mainly characterized by progressive conduction system defects, manifesting as sick sinus syndrome, atrioventricular block, or bundle branch blocks. Ventricular arrhythmia and systolic dysfunction are also common and all together often lead to impairment of myocardial function, development of heart failure, and cardiac transplant or premature death. Standard cardiac treatments including angiotensin II converting enzyme (ACE) inhibitors or angiotensin receptor blockers are widely used with treatment starting early (Lu et al., 2011). ACE converts angiotensin I into the active vasoconstrictor angiotensin II, indirectly increasing blood pressure and consequently increasing heart rate and vascular tone (Coates, 2003). Despite the use of conventional drugs for cardiovascular diseases (e.g. beta-blockers), heart failure almost always progresses; highlighting the urge to find new treatments for these disorders. Since mutations in LMNA gene were associated with laminopathies in 1999 (Bonne et al., 1999), numerous potential treatment options have emerged and been evaluated in mouse models (Table 2). The treatment strategies have generally been developed to target MAPK signaling pathway, to induce autophagy or to inhibit apoptosis but new therapies are rising (Figure 2). In this section, we review the different treatment strategies that have been studied so far.
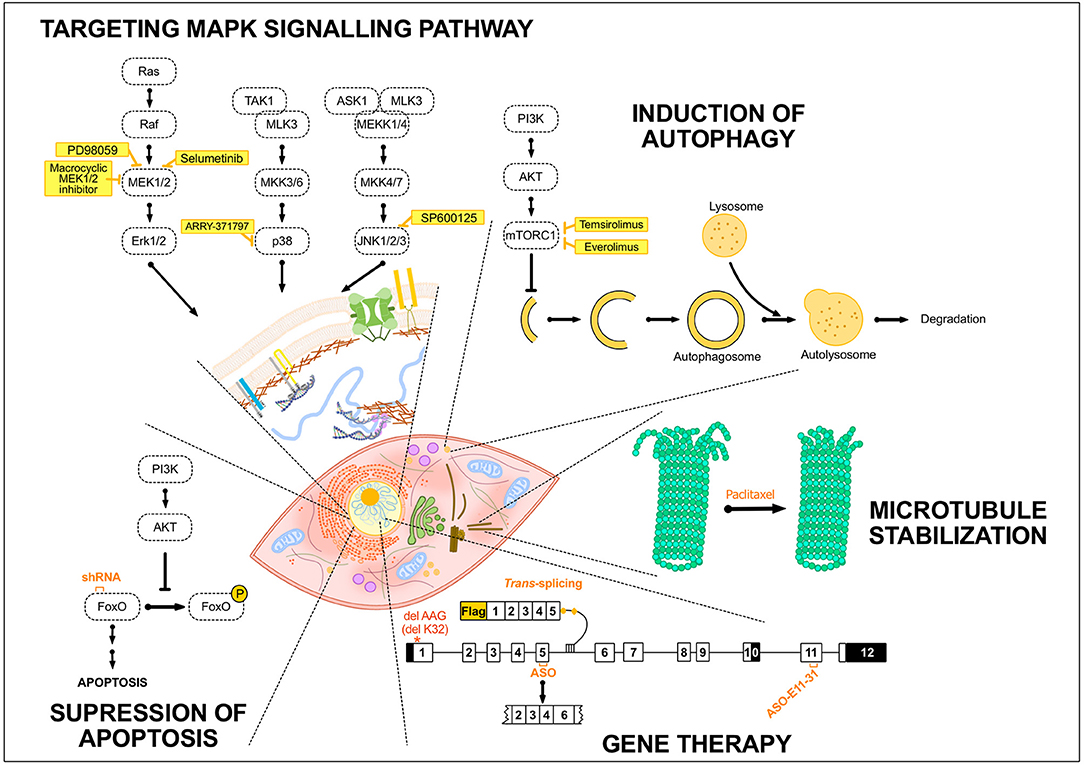
Figure 2. Summary of the potential treatments tested so far in EDMD. Schematic drawing representing several treatment strategies that have been developed for EDMD. The portrayed treatments target diverse mechanisms occurring either in the cytoplasm, e.g., autophagy in the lysosomes, or in the nucleus, including exon-skipping or trans-splicing strategies. The treatment strategies addressed to EDMD have generally been developed to target MAPK signaling pathway or to induce autophagy and these include different inhibitors of components of these pathways such as selumetinib or temsirolimus. To target apoptosis, the utilization of shRNA specific to Fox O1 and 3 has been developed. Others treatments and strategies such as the use of paclitaxel to stabilize microtubules or gene therapy to convert the mutant transcript into a normal transcript or remove an in-frame exon containing a mutation have been studied. AKT, protein kinase B; ASK1, apoptosis signal-regulating kinase 1; ASO, antisense oligonucleotide; ERK, extracellular signal-regulated kinase; FoxO, forkhead box O; MEK1/2, MAPK/ERK kinase 1/2; MEKK1/4, mitogen-activated protein kinase kinase kinase 1/4; MKK3/6, mitogen-activated protein kinase kinase 3/6; MLK3, mitogen-activated protein kinase kinase kinase 11; mTORC1, mammalian target of rapamycin complex 1; PI3K, Phosphoinositide 3-kinase; Raf, proto-oncogen serine/threonine-protein kinase; TAK1, mitogen-activated protein kinase kinase kinase 7.
MAPK signaling pathways are important transduction cascades initiated by extracellular mitogens, growth factors and cytokines at the cell surface and finalized to the nucleus to control gene expression (Davis, 1993). They regulate different cellular processes including cell proliferation and differentiation, survival, and apoptosis. The MAPK pathway is composed of four signaling cascades usually named according to the MAPK central component of each cascade: MAPK/ERK1/2 or classical pathway, JNK, p38 and ERK5 signaling cascades (Keshet and Seger, 2010). As mentioned earlier, Muchir and co-workers observed an activation of ERK, JNK and p38α cascades in hearts from LmnaH222P/H222P mice (Table 2), linking the activation of MAPK pathways due to LMNA mutations to cardiomyopathy (Muchir et al., 2007b, 2012b). Since then, inhibitors of MAPK have become a promising therapeutic option in LMNA cardiomyopathy. Various inhibitors targeting different components of the cascade have been already developed and tested in cancer (Dudley et al., 1995; Zhao and Adjei, 2014; Maik-Rachline and Seger, 2016). Therefore, pharmacological inhibition of ERK1/2 was tested to prevent the cardiac dysfunction in a laminopathic mouse model (Figure 2). The allosteric inhibitor of MEK1/2 (PD098059) was one of the first MEK (the MAPK kinase that phosphorylates ERK1/2) inhibitors to be tested in these mice, delaying the development of left ventricular dilatation and improving function (Muchir et al., 2009). A more potent and selective inhibitor, i.e., Selumetinib (Figure 2) also lead to an improvement in left ventricular dilatation, an increase in cardiac fractional shortening, decreased cardiac fibrosis, as well as, a prolongation in survival compared to placebo-treated mice (Muchir et al., 2012a). More recently, a novel allosteric, macrocyclic MEK1/2 inhibitor was designed and evaluated in LmnaH222P/H222P mice (Wu et al., 2017). This drug also resulted in an improvement of left ventricular systolic function, a decrease in left ventricular fibrosis, as well as, a beneficial effect on skeletal muscle structure and pathology and a prolonged survival.
Given that enhanced activities of JNK and p38α signaling were also observed in hearts from LmnaH222P/H222P mice (Muchir et al., 2007b, 2012b), inhibitors of both JNK and p38α cascades were tested in LMNA cardiomyopathy (Figure 2). LmnaH222P/H222P mice treated with SP600125, an inhibitor of JNK signaling, or with ARRY-371797, a p38α inhibitor, both showed a delay in the development of left ventricular dilatation and a reduction in the decrease of fractional shortening compared to placebo-treated mice (Wu et al., 2010; Muchir et al., 2012b). This led to the first clinical trial in patients with cardiomyopathy caused by LMNA mutations using ARRY-371797 (NCT02057341).
mTOR signaling pathway is involved in many fundamental cellular processes, from protein synthesis to autophagy (Figure 2). mTOR is a serine/threonine kinase that forms the core of two distinct protein complexes, mTORC1 and mTORC2. During cell growth, mTORC1 plays a central role in controlling production of proteins, lipids, and nucleotides as well as downregulating autophagy; mTORC2 also increases cellular proliferation and survival (Saxton and Sabatini, 2017). While mTORC1 is inhibited by rapamycin in an allosteric and specific manner, prolonged rapamycin treatment is needed to abrogate mTORC2 signaling (Sarbassov et al., 2006). Rapamycin and semi-synthetic rapamycin analogs with improved pharmacokinetics properties are widely used in cancer and some of them have reached various stages of clinical development, but only a few, e.g., temsirolimus and everolimus, have been approved by the FDA for their use in cancer treatment (Benjamin et al., 2011; Janku et al., 2018). Choi et al. demonstrated an activation of both AKT (activator of mTORC1) and mTORC1 as well as a defective autophagy in hearts of LmnaH222P/H222P before the onset of clinically detectable cardiomyopathy. The treatment of 14 weeks old LmnaH222P/H222P mice with temsirolimus reactivated autophagy and decreased left ventricular end-systolic dilatation, increased fractional shortening but did not prevent cardiac fibrosis in temsirolimus-treated mice compared to placebo-treated mice (Choi et al., 2012). Additionally, it has been recently published that everolimus is also improving the phenotype in fibroblast cell lines from six laminopathy patients with different LMNA mutations (DuBose et al., 2018). All these studies suggest that rapamycin-related mTORC1 inhibitors such as temsirolimus or everolimus, may be potential treatments for striated muscle laminopathies (Figure 2).
A recent study showed the activation of forkhead box O transcription factors (FoxO TFs) 1 and 3 and a deleterious role for overactivation of FoxO TFs in the hearts of LmnaΔ8−11 mice and cardiac myocytes (Auguste et al., 2018). By contrast, no significant differences in phosphorylated FoxO3a were previously observed in hearts of LmnaH222P/H222P mice compared to control mice (Choi et al., 2012). FoxO TFs are downstream targets of AKT. AKT phosphorylates FoxO TFs inhibiting their function and contributing to cell survival, growth and proliferation (Zhang et al., 2011) (Figure 2). Auguste et al. showed that subcutaneous injection of AAV9 encoding shRNA specific to FoxO1 and 3 in LmnaΔ8−11 newborn mice nonetheless efficiently reduced cardiac FoxO1 and 3 transcripts to wild-type levels but also reduces transcripts levels of FoxO target genes involved in apoptosis. In addition, knockdown of FoxO TFs in the heart resulted on a ~2-fold prolongation of survival in the LmnaΔ8−11 mice compared to untreated LmnaΔ8−11 mice (Auguste et al., 2018). These findings identify another potential target to prevent or ameliorate cardiac phenotype in laminopathies.
It has been largely described in the past that mutant LMNA transcripts have a dominant-negative effect (Azibani et al., 2014). Therefore, the conversion of the mutant transcript into a normal transcript or the removal of an in-frame exon containing a mutation could potentially improve pathogenic phenotypes in laminopathies (Figure 2). Scharner et al. demonstrated that the use of antisense oligonucleotides to skip LMNA exon 5 in human resulted in truncated human Lamin A or C that localized normally, restoring both nuclear shape and endogenous Lamin B1 and emerin localization (Scharner et al., 2015). Additionally, skipping of exon 11 might be another approach to treat other laminopathies caused by missense mutations in exon 11. Lee JM et al. used an exon 11 antisense oligonucleotide (ASO E11-31) to shift the balance between lamin C and prelamin A splicing thus, increasing lamin C production in mouse and human fibroblasts, as well as reducing the expression of progerin in fibroblasts derived from patients with Hutchinson-Gilford progeria syndrome (HGPS) (Lee et al., 2016). Finally, we have recently provided the first evidence of reprogramming LMNA mRNA in vitro by spliceosome-mediated RNA trans-splicing (SMART) as a therapeutic approach for striated muscle laminopathies (Azibani et al., 2018) (Figure 2). This technology allows the incorporation WT exons and the concomitant deletion of the corresponding mutated exons during the step of pre-mRNA maturation via transsplicing. Using this approach in primary myoblasts from LmnaΔK32/ΔK32 mice, we were able to rescue part of the nuclear abnormalities, increasing the proportion of lamin A/C at nuclear periphery, as well as, both emerin and lamin B1 mislocalization. However, we only detected an extremely modest increase in lamin A/C mRNA expression after systemic injection in pups, probably due to the low trans-splicing rate.
Other therapeutic options have been investigated. Arimura et al. showed that the treatment with pyridazinone derivative calcium (Ca2+) sensitizing agent SCH00013 in LmnaH222P/H222P mice resulted in an amelioration of systolic dysfunction, a prolongation in life expectancy, a reduction in cardiac interstitial fibrosis and a modulation of the expression of genes involved in cardiac remodeling (Arimura et al., 2010). The mechanisms for the beneficial effect of SCH00013 have not been elucidated yet and more studies are needed but these findings suggest Ca2+ sensitizer as an option for LMNA cardiomyopathy treatment.
Another approach specifically targets LMNA mutations leading to premature stop codon. Lee et al. showed that treatment of cardiomyocytes derived from human induced pluripotent stem cells carrying different LMNA mutations with PTC124, a pharmaceutical drug that selectively induces translational ribosomal read-through over the premature stop codon, increased the production of full-length lamin A/C proteins although only in one of the patient's cell line (Lee et al., 2017). It has been already tested in a phase 3 trial for Duchenne muscular dystrophy (McDonald et al., 2017), therefore this may be an alternative option for the treatment of nonsense-mutation causing laminopathies.
As mentioned before, we showed an altered distribution of Cx43 in LmnaH222P/H222P mice due to microtubule cytoskeleton alteration and decreased acetylation of α-tubulin, leading to electrical conduction disturbances (Macquart et al., 2018). Paclitaxel treatment, a drug used in therapeutics for several forms of cancer and already tested in patients underlying cardiac dysfunction (Gollerkeri et al., 2001), increased the acetylated form of α-tubulin in LmnaH222P/H222P treated mice, as well as relocalized Cx43 at the intercalated disks (Macquart et al., 2018). These results revealed Paclitaxel as a potential candidate treatment to stabilize microtubule cytoskeleton in EDMD patients.
Finally, another potential treatment in laminopathies arise from the DNA repair defects observed upon loss of A-type lamins (Gonzalez-Suarez et al., 2011). Gonzalez-Suarez and colleagues showed that the inhibition of CTSL by vitamin D treatment resulted in the stabilization of 53BPA and the induction of DNA DSBs repair.
The identification of new proteins causing EDMD and EDMD-like myopathies has provided new insights into the role of nuclear envelope proteins in the cell. The use of animal models has permitted the identification of signaling pathways that are deregulated when these proteins are absent or mutated, helping to understand the mechanisms involved in these pathologies. Therapeutic treatments modulating these pathways and new gene therapy approaches have shown some benefits in mice. However, further studies are needed to better understand how these altered mechanisms lead to these pathologies and how they lead to such different phenotypes.
AsB and BMR original writing and edition of the manuscript. AM edition of the manuscript. GB review and edition of the final manuscript. AnB supervision, writing, and edition of the manuscript.
BMR is employed by Sanofi, a global biopharmaceutical company focused on human health.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), Association Institut de Myologie (AIM) Sorbonne Université- Faculté de Médecine, CURE-CMD foundation and Andrés Marcio Foundation. ERA-CVD Variation as a support to the work.
Aaronson, R. P., and Blobel, G. (1975). Isolation of nuclear pore complexes in association with a lamina. Proc. Natl. Acad. Sci. U.S.A. 72, 1007–1011. doi: 10.1073/pnas.72.3.1007
Aebi, U., Cohn, J., Buhle, L., and Gerace, L. (1986). The nuclear lamina is a meshwork of intermediate-type filaments. Nature 323, 560–564. doi: 10.1038/323560a0
Alsheimer, M., von Glasenapp, E., Schnolzer, M., Heid, H., and Benavente, R. (2000). Meiotic lamin C2: the unique amino-terminal hexapeptide GNAEGR is essential for nuclear envelope association. Proc. Natl. Acad. Sci. U.S.A. 97, 13120–13125. doi: 10.1073/pnas.240466597
Arimura, T., Helbling-Leclerc, A., Massart, C., Varnous, S., Niel, F., Lacene, E., et al. (2005). Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 14, 155–169. doi: 10.1093/hmg/ddi017
Arimura, T., Sato, R., Machida, N., Bando, H., Zhan, D. Y., Morimoto, S., et al. (2010). Improvement of left ventricular dysfunction and of survival prognosis of dilated cardiomyopathy by administration of calcium sensitizer SCH00013 in a mouse model. J. Am. Coll. Cardiol. 55, 1503–1505. doi: 10.1016/j.jacc.2009.10.065
Auguste, G., Gurha, P., Lombardi, R., Coarfa, C., Willerson, J. T., and Marian, A. J. (2018). Suppression of activated FOXO transcription factors in the heart prolongs survival in a mouse model of laminopathies. Circ. Res. 122, 678–692. doi: 10.1161/CIRCRESAHA.117.312052
Azibani, F., Brull, A., Arandel, L., Beuvin, M., Nelson, I., Jollet, A., et al. (2018). Gene Therapy via trans-splicing for LMNA-related congenital muscular dystrophy. Mol. Ther. Nucleic Acids 10, 376–386. doi: 10.1016/j.omtn.2017.12.012
Azibani, F., Muchir, A., Vignier, N., Bonne, G., and Bertrand, A. T. (2014). Striated muscle laminopathies. Semin. Cell Dev. Biol. 29C, 107–115. doi: 10.1016/j.semcdb.2014.01.001
Banerjee, I., Zhang, J., Moore-Morris, T., Pfeiffer, E., Buchholz, K. S., Liu, A., et al. (2014). Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet. 10:e1004114. doi: 10.1371/journal.pgen.1004114
Barton, L. J., Soshnev, A. A., and Geyer, P. K. (2015). Networking in the nucleus: a spotlight on LEM-domain proteins. Curr. Opin. Cell Biol. 34, 1–8. doi: 10.1016/j.ceb.2015.03.005
Beck, L. A., Hosick, T. J., and Sinensky, M. (1990). Isoprenylation is required for the processing of the lamin A precursor. J. Cell Biol. 110, 1489–1499. doi: 10.1083/jcb.110.5.1489
Becker, P. E. (1972). Neues zur genetik und klassifikation der muskeldystrohien. Humangenetik 17, 1–22.
Becker, P. E. (1986). Dominant autosomal muscular dystrophy with early contractures and cardiomyopathy (Hauptmann-Thannhauser). Hum. Genet. 74:184. doi: 10.1007/BF00282089
Bengtsson, L., and Otto, H. (2008). LUMA interacts with emerin and influences its distribution at the inner nuclear membrane. J. Cell Sci. 121(Pt 4), 536–548. doi: 10.1242/jcs.019281
Ben-Harush, K., Wiesel, N., Frenkiel-Krispin, D., Moeller, D., Soreq, E., Aebi, U., et al. (2009). The supramolecular organization of the C. elegans nuclear lamin filament. J. Mol. Biol. 386, 1392–1402. doi: 10.1016/j.jmb.2008.12.024
Benjamin, D., Colombi, M., Moroni, C., and Hall, M. N. (2011). Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 10, 868–880. doi: 10.1038/nrd3531
Berk, J. M., Simon, D. N., Jenkins-Houk, C. R., Westerbeck, J. W., Gronning-Wang, L. M., Carlson, C. R., et al. (2014). The molecular basis of emerin-emerin and emerin-BAF interactions. J. Cell Sci. 127(Pt 18), 3956–3969. doi: 10.1242/jcs.148247
Bertrand, A. T., Renou, L., Papadopoulos, A., Beuvin, M., Lacene, E., Massart, C., et al. (2012). DelK32-lamin A/C has abnormal location and induces incomplete tissue maturation and severe metabolic defects leading to premature death. Hum. Mol. Genet. 21, 1037–1048. doi: 10.1093/hmg/ddr534
Bertrand, A. T., Ziaei, S., Ehret, C., Duchemin, H., Mamchaoui, K., Bigot, A., et al. (2014). Cellular micro-environments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 127(Pt 13), 2873–2884. doi: 10.1242/jcs.144907
Bione, S., Maestrini, E., Rivella, S., Manchini, M., Regis, S., Romei, G., et al. (1994). Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nature Genet. 8, 323–327. doi: 10.1038/ng1294-323
Bonne, G., Di Barletta, M. R., Varnous, S., Becane, H., Hammouda, E. H., Merlini, L., et al. (1999). Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nature Genet. 21, 285–288. doi: 10.1038/6799
Bonne, G., and Quijano-Roy, S. (2013). Emery-Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb. Clin. Neurol. 113, 1367–1376. doi: 10.1016/B978-0-444-59565-2.00007-1
Brown, S., Biben, C., Ooms, L. M., Maimone, M., McGrath, M. J., Gurung, R., et al. (1999a). The cardiac expression of striated muscle LIM protein 1 (SLIM1) is restricted to the outflow tract of the developing heart. J. Mol. Cell Cardiol. 31, 837–843. doi: 10.1006/jmcc.1998.0922
Brown, S., McGrath, M. J., Ooms, L. M., Gurung, R., Maimone, M. M., and Mitchell, C. A. (1999b). Characterization of two isoforms of the skeletal muscle LIM protein 1, SLIM1. Localization of SLIM1 at focal adhesions and the isoform slimmer in the nucleus of myoblasts and cytoplasm of myotubes suggests distinct roles in the cytoskeleton and in nuclear-cytoplasmic communication. J. Biol. Chem. 274, 27083–27091. doi: 10.1074/jbc.274.38.27083
Burke, B., and Stewart, C. L. (2013). The nuclear lamins: flexibility in function. Nat. Rev. Mol. Cell Biol. 14, 13–24. doi: 10.1038/nrm3488
Cartegni, L., di Bareletta, M. R., Barresi, R., Squarzoni, S., Sabatelli, P., Maraldi, N., et al. (1997). Heart-specific localization of emrin: new insights into Emery-Dreifuss muscular dystrophy. Hum. Mol. Genet. 6, 2257–2264. doi: 10.1093/hmg/6.13.2257
Cattin, M. E., Bertrand, A. T., Schlossarek, S., Le Bihan, M. C., Skov Jensen, S., Neuber, C., et al. (2013). Heterozygous LmnadelK32 mice develop dilated cardiomyopathy through a combined pathomechanism of haploinsufficiency and peptide toxicity. Hum. Mol. Genet. 22, 3152–3164. doi: 10.1093/hmg/ddt172
Cesarini, E., Mozzetta, C., Marullo, F., Gregoretti, F., Gargiulo, A., Columbaro, M., et al. (2015). Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J. Cell Biol. 211, 533–551. doi: 10.1083/jcb.201504035
Chakrabarti, A., and Pearce, J. M. S. (1981). Scapuloperoneal syndrome with cardiomyopathy: report of a family with autosomal dominant inheritance and unusual features. J. Neurol. Neurosurg. Psychiat. 44, 1146–1152. doi: 10.1136/jnnp.44.12.1146
Chang, W., Antoku, S., Ostlund, C., Worman, H. J., and Gundersen, G. G. (2015). Linker of nucleoskeleton and cytoskeleton (LINC) complex-mediated actin-dependent nuclear positioning orients centrosomes in migrating myoblasts. Nucleus 6, 77–88. doi: 10.1080/19491034.2015.1004947
Chatzifrangkeskou, M., Le Dour, C., Wu, W., Morrow, J. P., Joseph, L. C., Beuvin, M., et al. (2016). ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum. Mol. Genet. 25, 2220–2233. doi: 10.1093/hmg/ddw090
Chatzifrangkeskou, M., Yadin, D., Marais, T., Chardonnet, S., Cohen-Tannoudji, M., Mougenot, N., et al. (2018). Cofilin-1 phosphorylation catalyzed by ERK1/2 alters cardiac actin dynamics in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 27, 3060–3078. doi: 10.1093/hmg/ddy215
Chi, Y. H., Haller, K., Peloponese, J.M. Jr., and Jeang, K. T. (2007). Histone acetyltransferase hALP and nuclear membrane protein hsSUN1 function in de-condensation of mitotic chromosomes. J. Biol. Chem. 282, 27447–27458. doi: 10.1074/jbc.M703098200
Choi, J. C., Muchir, A., Wu, W., Iwata, S., Homma, S., Morrow, J. P., et al. (2012). Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med 4:144ra102. doi: 10.1126/scitranslmed.3003875
Coates, D. (2003). The angiotensin converting enzyme (ACE). Int J Biochem Cell Biol 35, 769–773. doi: 10.1016/S1357-2725(02)00309-6
Coffinier, C., Jung, H. J., Li, Z., Nobumori, C., Yun, U. J., Farber, E. A., et al. (2010). Direct synthesis of lamin A, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J. Biol. Chem. 285, 20818–20826. doi: 10.1074/jbc.M110.128835
Cohen, T. V., Gnocchi, V. F., Cohen, J. E., Phadke, A., Liu, H., Ellis, J. A., et al. (2013). Defective skeletal muscle growth in lamin A/C-deficient mice is rescued by loss of Lap2alpha. Hum. Mol. Genet. 22, 2852–2869. doi: 10.1093/hmg/ddt135
Collins, C. M., Ellis, J. A., and Holaska, J. M. (2017). MAPK signaling pathways and HDAC3 activity are disrupted during differentiation of emerin-null myogenic progenitor cells. Dis. Model Mech. 10, 385–397. doi: 10.1242/dmm.028787
Cottle, D. L., McGrath, M. J., Wilding, B. R., Cowling, B. S., Kane, J. M., D'Arcy, C. E., et al. (2009). SLIMMER (FHL1B/KyoT3) interacts with the proapoptotic protein Siva-1 (CD27BP) and delays skeletal myoblast apoptosis. J. Biol. Chem. 284, 26964–26977. doi: 10.1074/jbc.M109.036293
Cowling, B. S., McGrath, M. J., Nguyen, M. A., Cottle, D. L., Kee, A. J., Brown, S., et al. (2008). Identification of FHL1 as a regulator of skeletal muscle mass: implications for human myopathy. J. Cell Biol. 183, 1033–1048. doi: 10.1083/jcb.200804077
Crisp, M., Liu, Q., Roux, K., Rattner, J. B., Shanahan, C., Burke, B., et al. (2006). Coupling of the nucleus and cytoplasm: role of the LINC complex. J. Cell Biol. 172, 41–53. doi: 10.1083/jcb.200509124
Croft, J. A., Bridger, J. M., Boyle, S., Perry, P., Teague, P., and Bickmore, W. A. (1999). Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145, 1119–1131. doi: 10.1083/jcb.145.6.1119
Cupesi, M., Yoshioka, J., Gannon, J., Kudinova, A., Stewart, C. L., and Lammerding, J. (2010). Attenuated hypertrophic response to pressure overload in a lamin A/C haploinsufficiency mouse. J. Mol. Cell Cardiol. 48, 1290–1297. doi: 10.1016/j.yjmcc.2009.10.024
Davis, R. J. (1993). The mitogen-activated protein kinase signal transduction pathway. J. Biol. Chem. 268, 14553–14556.
Dechat, T., Adam, S. A., Taimen, P., Shimi, T., and Goldman, R. D. (2010). Nuclear lamins. Cold Spring Harb. Perspect Biol. 2:a000547. doi: 10.1101/cshperspect.a000547
Dechat, T., Korbei, B., Vaughan, O. A., Vlcek, S., Hutchison, C. J., and Foisner, R. (2000). Lamina-associated polypeptide 2alpha binds intranuclear A-type lamins. J. Cell Sci. 113 Pt 19, 3473–3484.
Dedeic, Z., Cetera, M., Cohen, T. V., and Holaska, J. M. (2011). Emerin inhibits Lmo7 binding to the Pax3 and MyoD promoters and expression of myoblast proliferation genes. J. Cell Sci. 124(Pt 10), 1691–1702. doi: 10.1242/jcs.080259
Demmerle, J., Koch, A. J., and Holaska, J. M. (2012). The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J. Biol. Chem. 287, 22080–22088. doi: 10.1074/jbc.M111.325308
Demmerle, J., Koch, A. J., and Holaska, J. M. (2013). Emerin and histone deacetylase 3 (HDAC3) cooperatively regulate expression and nuclear positions of MyoD, Myf5, and Pax7 genes during myogenesis. Chromosome Res. 21, 765–779. doi: 10.1007/s10577-013-9381-9
Dhe-Paganon, S., Werner, E. D., Chi, Y. I., and Shoelson, S. E. (2002). Structure of the globular tail of nuclear lamin. J. Biol. Chem. 277, 17381–17384. doi: 10.1074/jbc.C200038200
Ding, X., Xu, R., Yu, J., Xu, T., Zhuang, Y., and Han, M. (2007). SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev Cell 12, 863–872. doi: 10.1016/j.devcel.2007.03.018
Domenighetti, A. A., Chu, P. H., Wu, T., Sheikh, F., Gokhin, D. S., Guo, L. T., et al. (2014). Loss of FHL1 induces an age-dependent skeletal muscle myopathy associated with myofibrillar and intermyofibrillar disorganization in mice. Hum. Mol. Genet. 23, 209–225. doi: 10.1093/hmg/ddt412
Dreger, M., Bengtsson, L., Schoneberg, T., Otto, H., and Hucho, F. (2001). Nuclear envelope proteomics: Novel integral membrane proteins of the inner nuclear membrane. Proc. Natl. Acad. Sci. U.S.A. 98, 11943–11948. doi: 10.1073/pnas.211201898
Dreifuss, F. E., and Hogan, G. R. (1961). Survival in X-chromosomal muscular dystrophy. Neurology 11, 734–737. doi: 10.1212/WNL.11.8.734
DuBose, A. J., Lichtenstein, S. T., Petrash, N. M., Erdos, M. R., Gordon, L. B., and Collins, F. S. (2018). Everolimus rescues multiple cellular defects in laminopathy-patient fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 115, 4206–4211. doi: 10.1073/pnas.1802811115
Dudley, D. T., Pang, L., Decker, S. J., Bridges, A. J., and Saltiel, A. R. (1995). A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U.S.A. 92, 7686–7689. doi: 10.1073/pnas.92.17.7686
Ellis, J. A. (2006). Emery-Dreifuss muscular dystrophy at the nuclear envelope: 10 years on. Cell Mol. Life Sci. 63, 2702–2709. doi: 10.1007/s00018-006-6247-8
Emery, A. E. (1989). Emery-Dreifuss syndrome. J. Med. Genet. 26, 637–641. doi: 10.1136/jmg.26.10.637
Emery, A. E. H., and Dreifuss, F. E. (1966). Unusual type of benign X-linked muscular dystrophy. J. Neurol. Neurosurg. Psychiat. 29, 338–342. doi: 10.1136/jnnp.29.4.338
Farnsworth, C. C., Wolda, S. L., Gelb, M. H., and Glomset, J. A. (1989). Human lamin B contains a farnesylated cysteine residue. J. Biol. Chem. 264, 20422–20429.
Fatkin, D., MacRae, C., Sasaki, T., Wolff, M. R., Porcu, M., Frenneaux, M., et al. (1999). Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 341, 1715–1724. doi: 10.1056/NEJM199912023412302
Favreau, C., Higuet, D., Courvalin, J. C., and Buendia, B. (2004). Expression of a mutant lamin A that causes Emery-Dreifuss muscular dystrophy inhibits in vitro differentiation of C2C12 myoblasts. Mol. Cell Biol. 24, 1481–1492. doi: 10.1128/MCB.24.4.1481-1492.2004
Fenichel, G. M., Sul, Y. C., Kilroy, A. W., and Blouin, R. (1982). An autosomal-dominant dystrophy with humeropelvic distribution and cardiomyopathy. Neurology 32, 1399–1401. doi: 10.1212/WNL.32.12.1399
Fisher, D. Z., Chaudhary, N., and Blobel, G. (1986). cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. U.S.A. 83, 6450–6454. doi: 10.1073/pnas.83.17.6450
Fong, L. G., Ng, J. K., Lammerding, J., Vickers, T. A., Meta, M., Cote, N., et al. (2006). Prelamin, A., and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Invest. 116, 743–752. doi: 10.1172/JCI27125
Frangioni, J. V., and Neel, B. G. (1993). Use of a general purpose mammalian expression vector for studying intracellular protein targeting: identification of critical residues in the nuclear lamin A/C nuclear localization signal. J. Cell Sci. 105(Pt 2), 481–488.
Franke, W. W., Dorflinger, Y., Kuhn, C., Zimbelmann, R., Winter-Simanowski, S., Frey, N., et al. (2014). Protein LUMA is a cytoplasmic plaque constituent of various epithelial adherens junctions and composite junctions of myocardial intercalated disks: a unifying finding for cell biology and cardiology. Cell Tissue Res. 357, 159–172. doi: 10.1007/s00441-014-1865-1
Frock, R. L., Chen, S. C., Da, D. F., Frett, E., Lau, C., Brown, C., et al. (2012). Cardiomyocyte-specific expression of lamin a improves cardiac function in Lmna-/- mice. PLoS ONE 7:e42918. doi: 10.1371/annotation/92be6b32-d8e7-44c2-80a9-21097ad27965
Furukawa, K. (1999). LAP2 binding protein 1 (L2BP1/BAF) is a candidate mediator of LAP2-chromatin interaction. J. Cell Sci. 112(Pt 15), 2485–2492.
Furukawa, K., Inagaki, H., and Hotta, Y. (1994). Identification and cloning of an mRNA coding for a germ cell-specificA-type lamin in mice. Exp. Cell Res. 212, 426–430. doi: 10.1006/excr.1994.1164
Gerace, L., Blum, A., and Blobel, G. (1978). Immunocytochemical localization of the major polypeptides of the nuclear pore complex-lamina fraction: Interphase and mitotic distribution. J. Cell. Biol. 79, 546–566. doi: 10.1083/jcb.79.2.546
Gesson, K., Rescheneder, P., Skoruppa, M. P., von Haeseler, A., Dechat, T., and Foisner, R. (2016). A-type lamins bind both hetero- and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 26, 462–473. doi: 10.1101/gr.196220.115
Ghosh, S., Liu, B., Wang, Y., Hao, Q., and Zhou, Z. (2015). Lamin A is an endogenous SIRT6 activator and promotes SIRT6-mediated DNA repair. Cell Rep. 13, 1396–1406. doi: 10.1016/j.celrep.2015.10.006
Gimpel, P., Lee, Y. L., Sobota, R. M., Calvi, A., Koullourou, V., Patel, R., et al. (2017). Nesprin-1alpha-dependent microtubule nucleation from the nuclear envelope via Akap450 is necessary for nuclear positioning in muscle cells. Curr Biol 27, 2999–3009 e2999. doi: 10.1016/j.cub.2017.08.031
Goldman, A. E., Maul, G., Steinert, P. M., Yang, H. Y., and Goldman, R. D. (1986). Keratin-like proteins that coisolate with intermediate filaments of BHK- 21 cells are nuclear lamins. Proc. Natl. Acad. Sci. U.S.A. 83, 3839–3843. doi: 10.1073/pnas.83.11.3839
Gollerkeri, A., Harrold, L., Rose, M., Jain, D., and Burtness, B. A. (2001). Use of paclitaxel in patients with pre-existing cardiomyopathy: a review of our experience. Int J Cancer 93, 139–141. doi: 10.1002/ijc.1295
Gonzalez-Suarez, I., Redwood, A. B., Grotsky, D. A., Neumann, M. A., Cheng, E. H., Stewart, C. L., et al. (2011). A new pathway that regulates 53BP1 stability implicates cathepsin L and vitamin D in DNA repair. EMBO J. 30, 3383–3396. doi: 10.1038/emboj.2011.225
Greene, W. K., Baker, E., Rabbitts, T. H., and Kees, U. R. (1999). Genomic structure, tissue expression and chromosomal location of the LIM-only gene, SLIM1. Gene 232, 203–207. doi: 10.1016/S0378-1119(99)00125-0
Gruenbaum, Y., Margalit, A., Goldman, R. D., Shumaker, D. K., and Wilson, K. L. (2005). The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 6, 21–31. doi: 10.1038/nrm1550
Guarda, A., Bolognese, F., Bonapace, I. M., and Badaracco, G. (2009). Interaction between the inner nuclear membrane lamin B receptor and the heterochromatic methyl binding protein, MeCP2. Exp. Cell Res. 315, 1895–1903. doi: 10.1016/j.yexcr.2009.01.019
Guelen, L., Pagie, L., Brasset, E., Meuleman, W., Faza, M. B., Talhout, W., et al. (2008). Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948–951. doi: 10.1038/nature06947
Gueneau, L., Bertrand, A. T., Jais, J. P., Salih, M. A., Stojkovic, T., Wehnert, M., et al. (2009). Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 85, 338–353. doi: 10.1016/j.ajhg.2009.07.015
Guilluy, C., Osborne, L. D., Van Landeghem, L., Sharek, L., Superfine, R., Garcia-Mata, R., et al. (2014). Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 16, 376–381. doi: 10.1038/ncb2927
Hagan, I., and Yanagida, M. (1995). The product of the spindle formation gene sad1+ associates with the fission yeast spindle pole body and is essential for viability. J. Cell Biol. 129, 1033–1047. doi: 10.1083/jcb.129.4.1033
Hale, C. M., Shrestha, A. L., Khatau, S. B., Stewart-Hutchinson, P. J., Hernandez, L., Stewart, C. L., et al. (2008). Dysfunctional connections between the nucleus and the actin and microtubule networks in laminopathic models. Biophys. J. 95, 5462–5475. doi: 10.1529/biophysj.108.139428
Harapin, J., Bormel, M., Sapra, K. T., Brunner, D., Kaech, A., and Medalia, O. (2015). Structural analysis of multicellular organisms with cryo-electron tomography. Nat. Methods 12, 634–636. doi: 10.1038/nmeth.3401
Hennen, J., Saunders, C. A., Mueller, J. D., and Luxton, G. W. G. (2018). Fluorescence fluctuation spectroscopy reveals differential SUN protein oligomerization in living cells. Mol. Biol. Cell 29, 1003–1011. doi: 10.1091/mbc.E17-04-0233
Ho, C. Y., Jaalouk, D. E., and Lammerding, J. (2013a). Novel insights into the disease etiology of laminopathies. Rare Dis 1:e27002. doi: 10.4161/rdis.27002
Ho, C. Y., Jaalouk, D. E., Vartiainen, M. K., and Lammerding, J. (2013b). Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 497, 507–511. doi: 10.1038/nature12105
Holaska, J. M., Rais-Bahrami, S., and Wilson, K. L. (2006). Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum. Mol. Genet. 15, 3459–3472. doi: 10.1093/hmg/ddl423
Holaska, J. M., and Wilson, K. L. (2007). An emerin “proteome”: purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signaling, mechanosensing, and nuclear architecture. Biochemistry 46, 8897–8908. doi: 10.1021/bi602636m
Holt, I., Duong, N. T., Zhang, Q., Lam le, T., Sewry, C. A., Mamchaoui, K., et al. (2016). Specific localization of nesprin-1-alpha2, the short isoform of nesprin-1 with a KASH domain, in developing, fetal and regenerating muscle, using a new monoclonal antibody. BMC Cell Biol. 17:26. doi: 10.1186/s12860-016-0105-9
Houben, F., Willems, C. H., Declercq, I. L., Hochstenbach, K., Kamps, M. A., Snoeckx, L. H., et al. (2009). Disturbed nuclear orientation and cellular migration in A-type lamin deficient cells. Biochim Biophys Acta 1793, 312–324. doi: 10.1016/j.bbamcr.2008.10.003
Huber, M. D., Guan, T., and Gerace, L. (2009). Overlapping functions of nuclear envelope proteins NET25 (Lem2) and emerin in regulation of extracellular signal-regulated kinase signaling in myoblast differentiation. Mol. Cell Biol. 29, 5718–5728. doi: 10.1128/MCB.00270-09
Hutchison, C. J., Alvarez-Reyes, M., and Vaughan, O. A. (2001). Lamins in disease: why do ubiquitously expressed nuclear envelope proteins give rise to tissue-specific disease phenotypes? J. Cell Sci. 114(Pt 1), 9–19.
Iyer, A., Koch, A. J., and Holaska, J. M. (2017). Expression profiling of differentiating emerin-null myogenic progenitor identifies molecular pathways implicated in their impaired differentiation. Cells 6:38. doi: 10.3390/cells6040038
Jahed, Z., Fadavi, D., Vu, U. T., Asgari, E., Luxton, G. W. G., and Mofrad, M. R. K. (2018). Molecular insights into the mechanisms of SUN1 oligomerization in the nuclear envelope. Biophys J. 114, 1190–1203. doi: 10.1016/j.bpj.2018.01.015
Jahn, D., Schramm, S., Schnolzer, M., Heilmann, C. J., de Koster, C. G., Schutz, W., et al. (2012). A truncated lamin A in the Lmna -/- mouse line: implications for the understanding of laminopathies. Nucleus 3, 463–474. doi: 10.4161/nucl.21676
Janku, F., Yap, T. A., and Meric-Bernstam, F. (2018). Targeting the PI3K pathway in cancer: are we making headway? Nat. Rev. Clin. Oncol. 15, 273–291. doi: 10.1038/nrclinonc.2018.28
Johnson, B. R., Nitta, R. T., Frock, R. L., Mounkes, L., Barbie, D. A., Stewart, C. L., et al. (2004). A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc. Natl. Acad. Sci. U.S.A. 101, 9677–9682. doi: 10.1073/pnas.0403250101
Kadrmas, J. L., and Beckerle, M. C. (2004). The LIM domain: from the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol. 5, 920–931. doi: 10.1038/nrm1499
Keshet, Y., and Seger, R. (2010). The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 661, 3–38. doi: 10.1007/978-1-60761-795-2_1
Ketema, M., and Sonnenberg, A. (2011). Nesprin-3: a versatile connector between the nucleus and the cytoskeleton. Biochem. Soc. Trans. 39, 1719–1724. doi: 10.1042/BST20110669
Kim, Y., and Zheng, Y. (2013). Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem. Biophys. Res. Commun. 440, 8–13. doi: 10.1016/j.bbrc.2013.08.082
Kind, J., Pagie, L., Ortabozkoyun, H., Boyle, S., de Vries, S. S., Janssen, H., et al. (2013). Single-cell dynamics of genome-nuclear lamina interactions. Cell 153, 178–192. doi: 10.1016/j.cell.2013.02.028
Kirby, T. J., and Lammerding, J. (2018). Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 20, 373–381. doi: 10.1038/s41556-018-0038-y
Koch, A. J., and Holaska, J. M. (2014). Emerin in health and disease. Semin. Cell Dev. Biol. 29, 95–106. doi: 10.1016/j.semcdb.2013.12.008
Kolb, T., Maass, K., Hergt, M., Aebi, U., and Herrmann, H. (2011). Lamin, A., and lamin C form homodimers and coexist in higher complex forms both in the nucleoplasmic fraction and in the lamina of cultured human cells. Nucleus 2, 425–433. doi: 10.4161/nucl.2.5.17765
Kosa, J. L., Michelsen, J. W., Louis, H. A., Olsen, J. I., Davis, D. R., Beckerle, M. C., et al. (1994). Common metal ion coordination in LIM domain proteins. Biochemistry 33, 468–477. doi: 10.1021/bi00168a011
Krimm, I., Ostlund, C., Gilquin, B., Couprie, J., Hossenlopp, P., Mornon, J. P., et al. (2002). The Ig-like structure of the C-terminal domain of lamin a/c, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 10, 811–823. doi: 10.1016/S0969-2126(02)00777-3
Kubben, N., Voncken, J. W., Konings, G., van Weeghel, M., van den Hoogenhof, M.M. G., Gijbels, M., et al. (2011). Post-natal myogenic and adipogenic developmental. Defects and metabolic impairment upon loss of A-type lamins. Nucleus 2, 1–13. doi: 10.4161/nucl.2.3.15731
Lammerding, J., Hsiao, J., Schulze, P. C., Kozlov, S., Stewart, C. L., and Lee, R. T. (2005). Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J. Cell Biol. 170, 781–791. doi: 10.1083/jcb.200502148
Lammerding, J., Schulze, P. C., Takahashi, T., Kozlov, S., Sullivan, T., Kamm, R. D., et al. (2004). Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest. 113, 370–378. doi: 10.1172/JCI200419670
Lanzuolo, C., and Orlando, V. (2012). Memories from the polycomb group proteins. Annu Rev Genet 46, 561–589. doi: 10.1146/annurev-genet-110711-155603
Lattanzi, G., Ognibene, A., Sabatelli, P., Capanni, C., Toniolo, D., Columbaro, M., et al. (2000). Emerin expression at the early stages of myogenic differentiation. Differentiation 66, 208–217. doi: 10.1046/j.1432-0436.2000.660407.x
Le Dour, C., Macquart, C., Sera, F., Homma, S., Bonne, G., Morrow, J. P., et al. (2017). Decreased WNT/beta-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/C gene. Hum. Mol. Genet. 26, 333–343. doi: 10.1093/hmg/ddw389
Lee, J. M., Nobumori, C., Tu, Y., Choi, C., Yang, S. H., Jung, H. J., et al. (2016). Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J. Clin. Invest. 126, 1592–1602. doi: 10.1172/JCI85908
Lee, J. S., Hale, C. M., Panorchan, P., Khatau, S. B., George, J. P., Tseng, Y., et al. (2007). Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys J. 93, 2542–2552. doi: 10.1529/biophysj.106.102426
Lee, K. K., Haraguchi, T., Lee, R. S., Koujin, T., Hiraoka, Y., and Wilson, K. L. (2001). Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 114(Pt 24), 4567–4573.
Lee, S. M., Tsui, S. K., Chan, K. K., Garcia-Barcelo, M., Waye, M. M., Fung, K. P., et al. (1998). Chromosomal mapping, tissue distribution and cDNA sequence of four-and-a-half LIM domain protein 1 (FHL1). Gene 216, 163–170. doi: 10.1016/S0378-1119(98)00302-3
Lee, Y. K., Lau, Y. M., Cai, Z. J., Lai, W. H., Wong, L. Y., Tse, H. F., et al. (2017). Modeling treatment response for Lamin A/C related dilated cardiomyopathy in human induced pluripotent stem cells. J. Am. Heart Assoc. 6:e005677. doi: 10.1161/JAHA.117.005677
Lei, K., Zhang, X., Ding, X., Guo, X., Chen, M., Zhu, B., et al. (2009). SUN1 and SUN2 play critical but partially redundant roles in anchoring nuclei in skeletal muscle cells in mice. Proc. Natl. Acad. Sci. U.S.A. 106, 10207–10212. doi: 10.1073/pnas.0812037106
Liang, L., Zhang, H. W., Liang, J., Niu, X. L., Zhang, S. Z., Feng, L., et al. (2008). KyoT3, an isoform of murine FHL1, associates with the transcription factor RBP-J and represses the RBP-J-mediated transactivation. Biochim Biophys Acta 1779, 805–810. doi: 10.1016/j.bbagrm.2008.08.001
Liang, W. C., Mitsuhashi, H., Keduka, E., Nonaka, I., Noguchi, S., Nishino, I., et al. (2011). TMEM43 mutations in emery-dreifuss muscular dystrophy-related myopathy. Ann. Neurol. 69, 1005–1013. doi: 10.1002/ana.22338
Lin, F., and Worman, H. J. (1993). Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 268, 16321–16326.
Lombardi, M. L., Jaalouk, D. E., Shanahan, C. M., Burke, B., Roux, K. J., and Lammerding, J. (2011). The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J. Biol. Chem. 286, 26743–26753. doi: 10.1074/jbc.M111.233700
Lu, J. T., Muchir, A., Nagy, P. L., and Worman, H. J. (2011). LMNA cardiomyopathy: cell biology and genetics meet clinical medicine. Dis. Model Mech. 4, 562–568. doi: 10.1242/dmm.006346
Lund, E., Oldenburg, A. R., Delbarre, E., Freberg, C. T., Duband-Goulet, I., Eskeland, R., et al. (2013). Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. 23, 1580–1589. doi: 10.1101/gr.159400.113
Lund, E. G., Duband-Goulet, I., Oldenburg, A., Buendia, B., and Collas, P. (2015). Distinct features of lamin A-interacting chromatin domains mapped by ChIP-sequencing from sonicated or micrococcal nuclease-digested chromatin. Nucleus 6, 30–39. doi: 10.4161/19491034.2014.990855
Luxton, G. W., and Starr, D. A. (2014). KASHing up with the nucleus: novel functional roles of KASH proteins at the cytoplasmic surface of the nucleus. Curr. Opin. Cell Biol. 28, 69–75. doi: 10.1016/j.ceb.2014.03.002
Machiels, B. M., Zorenc, A. H., Endert, J. M., Kuijpers, H. J., van Eys, G. J., Ramaekers, F. C., et al. (1996). An alternative splicing product of the lamin A/C gene lacks exon 10. J. Biol. Chem. 271, 9249–9253. doi: 10.1074/jbc.271.16.9249
Macquart, C., Juttner, R., Le Dour, C., Chatzifrangkeskou, M., Schmitt, A., Gotthardt, M., et al. (2018). Microtubule cytoskeleton regulates connexin 43 localization and cardiac conduction in cardiomyopathy caused by mutation in A-type lamins gene. Hum. Mol. Genet. doi: 10.1093/hmg/ddy227. [Epub ahead of print].
Mahen, R., Hattori, H., Lee, M., Sharma, P., Jeyasekharan, A. D., and Venkitaraman, A. R. (2013). A-type lamins maintain the positional stability of DNA damage repair foci in mammalian nuclei. PLoS ONE 8:e61893. doi: 10.1371/journal.pone.0061893
Maik-Rachline, G., and Seger, R. (2016). The ERK cascade inhibitors: towards overcoming resistance. Drug. Resist. Updat. 25, 1–12. doi: 10.1016/j.drup.2015.12.001
Malone, C. J., Fixsen, W. D., Horvitz, H. R., and Han, M. (1999). UNC-84 localizes to the nuclear envelope and is required for nuclear migration and anchoring during C. elegans development. Development 126, 3171–3181.
Mancini, M. A., Shan, B., Nickerson, J. A., Penman, S., and Lee, W. H. (1994). The retinoblastoma gene product is a cell cycle-dependent, nuclear matrix-associated protein. Proc. Natl. Acad. Sci. U.S.A. 91, 418–422. doi: 10.1073/pnas.91.1.418
Manilal, S., Nguyen, T. M., Sewry, C. A., and Morris, G. E. (1996). The Emery-Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum. Mol. Genet. 5, 801–808. doi: 10.1093/hmg/5.6.801
Mansharamani, M., and Wilson, K. L. (2005). Direct binding of nuclear membrane protein MAN1 to emerin in vitro and two modes of binding to barrier-to-autointegration factor. J. Biol. Chem. 280, 13863–13870. doi: 10.1074/jbc.M413020200
Manta, P., Terzis, G., Papadimitriou, C., Kontou, C., and Vassilopoulos, D. (2004). Emerin expression in tubular aggregates. Acta Neuropathol 107, 546–552. doi: 10.1007/s00401-004-0851-1
Maraldi, N. M., Lattanzi, G., Capanni, C., Columbaro, M., Mattioli, E., Sabatelli, P., et al. (2006). Laminopathies: a chromatin affair. Adv. Enzyme Regul. 46, 33–49. doi: 10.1016/j.advenzreg.2006.01.001
Markiewicz, E., Dechat, T., Foisner, R., Quinlan, R. A., and Hutchison, C. J. (2002). Lamin A/C binding protein LAP2alpha is required for nuclear anchorage of retinoblastoma protein. Mol. Biol. Cell 13, 4401–4413. doi: 10.1091/mbc.e02-07-0450
Markiewicz, E., Tilgner, K., Barker, N., van de Wetering, M., Clevers, H., Dorobek, M., et al. (2006). The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. Embo J. 25, 3275–3285. doi: 10.1038/sj.emboj.7601230
Maske, C. P., Hollinshead, M. S., Higbee, N. C., Bergo, M. O., Young, S. G., and Vaux, D. J. (2003). A carboxyl-terminal interaction of lamin B1 is dependent on the CAAX endoprotease Rce1 and carboxymethylation. J. Cell Biol. 162, 1223–1232. doi: 10.1083/jcb.200303113
Mattout-Drubezki, A., and Gruenbaum, Y. (2003). Dynamic interactions of nuclear lamina proteins with chromatin and transcriptional machinery. Cell Mol. Life Sci. 60, 2053–2063. doi: 10.1007/s00018-003-3038-3
McDonald, C. M., Campbell, C., Torricelli, R. E., Finkel, R. S., Flanigan, K. M., Goemans, N., et al. (2017). Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 1489–1498. doi: 10.1016/S0140-6736(17)31611-2
McGrath, M. J., Cottle, D. L., Nguyen, M. A., Dyson, J. M., Coghill, I. D., Robinson, P. A., et al. (2006). Four and a half LIM protein 1 binds myosin-binding protein C and regulates myosin filament formation and sarcomere assembly. J. Biol. Chem. 281, 7666–7683. doi: 10.1074/jbc.M512552200
McKeon, F. D., Kirschner, M. W., and Caput, D. (1986). Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 319, 463–468. doi: 10.1038/319463a0
McKusick, V. A. (1971). X-linked muscular dystrophy, benign form with contractures. Birth Defects Orig. Artic. Ser. 1971, 113–115.
Meinke, P., Mattioli, E., Haque, F., Antoku, S., Columbaro, M., Straatman, K. R., et al. (2014). Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLoS Genet 10:e1004605. doi: 10.1371/journal.pgen.1004605
Melcon, G., Kozlov, S., Cutler, D. A., Sullivan, T., Hernandez, L., Zhao, P., et al. (2006). Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum. Mol. Genet. 15, 637–651. doi: 10.1093/hmg/ddi479
Michelsen, J. W., Schmeichel, K. L., Beckerle, M. C., and Winge, D. R. (1993). The LIM motif defines a specific zinc-binding protein domain. Proc. Natl. Acad. Sci. U.S.A. 90, 4404–4408. doi: 10.1073/pnas.90.10.4404
Miller, R. G., Layzer, R. B., Mellenthin, M. A., Golabi, M., Francoz, R. A., and Mall, J. C. (1985). Emery-Dreifuss muscular dystrophy with autosomal dominant transmission. Neurology 35, 1230–1233. doi: 10.1212/WNL.35.8.1230
Mislow, J. M., Holaska, J. M., Kim, M. S., Lee, K. K., Segura-Totten, M., Wilson, K. L., et al. (2002). Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett. 525, 135–140. doi: 10.1016/S0014-5793(02)03105-8
Mounkes, L. C., Kozlov, S. V., Rottman, J. N., and Stewart, C.L. (2005). Expression of a LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum. Mol. Genet. 14, 2167–2180. doi: 10.1093/hmg/ddi221
Muchir, A., Bonne, G., van der Kooi, A. J., van Meegen, M., Baas, F., Bolhuis, P. A., et al. (2000). Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet. 9, 1453–1459. doi: 10.1093/hmg/9.9.1453
Muchir, A., Pavlidis, P., Bonne, G., Hayashi, Y. K., and Worman, H.J. (2007a). Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum. Mol. Genet. 16, 1884–1895. doi: 10.1093/hmg/ddm137
Muchir, A., Pavlidis, P., Decostre, V., Herron, A. J., Arimura, T., Bonne, G., et al. (2007b). Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Invest. 117, 1282–1293. doi: 10.1172/JCI29042
Muchir, A., Reilly, S. A., Wu, W., Iwata, S., Homma, S., Bonne, G., et al. (2012a). Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc Res. 93, 311–319. doi: 10.1093/cvr/cvr301
Muchir, A., Shan, J., Bonne, G., Lehnart, S. E., and Worman, H. J. (2009). Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum. Mol. Genet. 18, 241–247. doi: 10.1093/hmg/ddn343
Muchir, A., Wu, W., Choi, J. C., Iwata, S., Morrow, J., Homma, S., et al. (2012b). Abnormal p38alpha mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 21, 4325–4333. doi: 10.1093/hmg/dds265
Mull, A., Kim, G., and Holaska, J. M. (2015). LMO7-null mice exhibit phenotypes consistent with emery-dreifuss muscular dystrophy. Muscle Nerve 51, 222–228. doi: 10.1002/mus.24286
Naetar, N., and Foisner, R. (2009). Lamin complexes in the nuclear interior control progenitor cell proliferation and tissue homeostasis. Cell Cycle 8, 1488–1493. doi: 10.4161/cc.8.10.8499
Nagano, A., Koga, R., Ogawa, M., Kurano, Y., Kawada, J., Okada, R., et al. (1996). Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nature Genet. 12, 254–259. doi: 10.1038/ng0396-254
Ng, E. K., Lee, S. M., Li, H. Y., Ngai, S. M., Tsui, S. K., Waye, M. M., et al. (2001). Characterization of tissue-specific LIM domain protein (FHL1C) which is an alternatively spliced isoform of a human LIM-only protein (FHL1). J Cell Biochem 82, 1–10. doi: 10.1002/jcb.1110
Nigg, E. A., Kitten, G. T., and Vorburger, K. (1992). Targeting lamin proteins to the nuclear envelope: the role of CaaX box modifications. Biochem. Soc. Trans. 20, 500–504. doi: 10.1042/bst0200500
Nikolova, V., Leimena, C., McMahon, A. C., Tan, J. C., Chandar, S., Jogia, D., et al. (2004). Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Invest. 113, 357–369. doi: 10.1172/JCI200419448
Novitch, B. G., Mulligan, G. J., Jacks, T., and Lassar, A. B. (1996). Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J. Cell Biol. 135, 441–456. doi: 10.1083/jcb.135.2.441
Ostlund, C., Ellenberg, J., Hallberg, E., Lippincott-Schwartz, J., and Worman, H. J. (1999). Intracellular trafficking of emerin, the Emery-Dreifuss muscular dystrophy protein. J. Cell Sci. 112(Pt 11), 1709–1719.
Ozawa, R., Hayashi, Y. K., Ogawa, M., Kurokawa, R., Matsumoto, H., Noguchi, S., et al. (2006). Emerin-lacking mice show minimal motor and cardiac dysfunctions with nuclear-associated vacuoles. Am. J. Pathol. 168, 907-917. doi: 10.2353/ajpath.2006.050564
Parry, D. A., Conway, J. F., and Steinert, P. M. (1986). Structural studies on lamin. Similarities and differences between lamin and intermediate-filament proteins. Biochem. J. 238, 305–308. doi: 10.1042/bj2380305
Paulsen, J., Sekelja, M., Oldenburg, A. R., Barateau, A., Briand, N., Delbarre, E., et al. (2017). Chrom3D: three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biology 18:21. doi: 10.1186/s13059-016-1146-2
Pekovic, V., Harborth, J., Broers, J. L., Ramaekers, F. C., van Engelen, B., Lammens, M., et al. (2007). Nucleoplasmic LAP2alpha-lamin A complexes are required to maintain a proliferative state in human fibroblasts. J. Cell Biol. 176, 163–172. doi: 10.1083/jcb.200606139
Pilat, U., Dechat, T., Bertrand, A. T., Woisetschlager, N., Gotic, I., Spilka, R., et al. (2013). The muscle dystrophy-causing DeltaK32 lamin A/C mutant does not impair the functions of the nucleoplasmic lamin-A/C-LAP2alpha complex in mice. J. Cell Sci. 126(Pt 8), 1753–1762. doi: 10.1242/jcs.115246
Puckelwartz, M. J., Kessler, E., Zhang, Y., Hodzic, D., Randles, K. N., Morris, G., et al. (2009). Disruption of nesprin-1 produces an Emery Dreifuss muscular dystrophy-like phenotype in mice. Hum. Mol. Genet. 18, 607–620. doi: 10.1093/hmg/ddn386
Puckelwartz, M. J., Kessler, E. J., Kim, G., Dewitt, M. M., Zhang, Y., Earley, J. U., et al. (2010). Nesprin-1 mutations in human and murine cardiomyopathy. J. Mol. Cell Cardiol. 48, 600–608. doi: 10.1016/j.yjmcc.2009.11.006
Qin, H., Wang, J., Liang, Y., Taniguchi, Y., Tanigaki, K., and Han, H. (2004). RING1 inhibits transactivation of RBP-J by Notch through interaction with LIM protein KyoT2. Nucleic Acids Res. 32, 1492–1501. doi: 10.1093/nar/gkh295
Quijano-Roy, S., Mbieleu, B., Bonnemann, C. G., Jeannet, P. Y., Colomer, J., Clarke, N. F., et al. (2008). De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 64, 177–186. doi: 10.1002/ana.21417
Raffaele Di Barletta, M., Ricci, E., Galluzzi, G., Tonali, P., Mora, M., Morandi, L., et al. (2000). Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 66, 1407–1412. doi: 10.1086/302869
Rajgor, D., and Shanahan, C. M. (2013). Nesprins: from the nuclear envelope and beyond. Expert Rev Mol. Med. 15:e5. doi: 10.1017/erm.2013.6
Randles, K. N., Lam le, T., Sewry, C. A., Puckelwartz, M., Furling, D., Wehnert, M., et al. (2010). Nesprins, but not sun proteins, switch isoforms at the nuclear envelope during muscle development. Dev Dyn 239, 998–1009. doi: 10.1002/dvdy.22229
Robinson, P. A., Brown, S., McGrath, M. J., Coghill, I. D., Gurung, R., and Mitchell, C. A. (2003). Skeletal muscle LIM protein 1 regulates integrin-mediated myoblast adhesion, spreading, and migration. Am. J. Physiol. Cell Physiol. 284, C681–695. doi: 10.1152/ajpcell.00370.2002
Robson, M. I., de Las Heras, J. I., Czapiewski, R., Le Thanh, P., Booth, D. G., Kelly, D. A., et al. (2016). Tissue-specific gene repositioning by muscle nuclear membrane proteins enhances repression of critical developmental genes during myogenesis. Mol. Cell 62, 834–847. doi: 10.1016/j.molcel.2016.04.035
Ronningen, T., Shah, A., Oldenburg, A. R., Vekterud, K., Delbarre, E., Moskaug, J. O., et al. (2015). Prepatterning of differentiation-driven nuclear lamin A/C-associated chromatin domains by GlcNAcylated histone H2B. Genome Res. 25, 1825–1835. doi: 10.1101/gr.193748.115
Salpingidou, G., Smertenko, A., Hausmanowa-Petrucewicz, I., Hussey, P. J., and Hutchison, C.J. (2007). A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. J. Cell Biol. 178, 897–904. doi: 10.1083/jcb.200702026
Samson, C., Celli, F., Hendriks, K., Zinke, M., Essawy, N., Herrada, I., et al. (2017). Emerin self-assembly mechanism: role of the LEM domain. FEBS J. 284, 338–352. doi: 10.1111/febs.13983
Sanna, T., Dello Russo, A., Toniolo, D., Vytopil, M., Pelargonio, G., De Martino, G., et al. (2003). Cardiac features of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutations. Eur. Heart J. 24, 2227–2236. doi: 10.1016/j.ehj.2003.09.020
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J. H., Hsu, P. P., Bagley, A. F., et al. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168. doi: 10.1016/j.molcel.2006.03.029
Saxton, R. A., and Sabatini, D. M. (2017). mTOR Signaling in growth, metabolism, and disease. Cell 169, 361–371. doi: 10.1016/j.cell.2017.03.035
Scharner, J., Figeac, N., Ellis, J. A., and Zammit, P. S. (2015). Ameliorating pathogenesis by removing an exon containing a missense mutation: a potential exon-skipping therapy for laminopathies. Gene Ther. 22, 503–515. doi: 10.1038/gt.2015.8
Schneider, M., Lu, W., Neumann, S., Brachner, A., Gotzmann, J., Noegel, A. A., et al. (2011). Molecular mechanisms of centrosome and cytoskeleton anchorage at the nuclear envelope. Cell Mol. Life Sci. 68, 1593–1610. doi: 10.1007/s00018-010-0535-z
Schulz, R. A., and Yutzey, K. E. (2004). Calcineurin signaling and NFAT activation in cardiovascular and skeletal muscle development. Dev. Biol. 266, 1–16. doi: 10.1016/j.ydbio.2003.10.008
Schwartz, C., Fischer, M., Mamchaoui, K., Bigot, A., Lok, T., Verdier, C., et al. (2017). Lamins and nesprin-1 mediate inside-out mechanical coupling in muscle cell precursors through FHOD1. Sci Rep 7:1253. doi: 10.1038/s41598-017-01324-z
Serratrice, G., and Pouget, J. (1986). Emery-Dreifuss myopathy or amyotrophy with early contractures of muscles and secondary anomalies of cardiac conduction with variable heredity. Rev Neurol. 142, 766–770.
Sewry, C. A., Brown, S. C., Mercuri, E., Bonne, G., Feng, L., Camici, G., et al. (2001). Skeletal muscle pathology in autosomal dominant Emery-Dreifuss muscular dystrophy with lamin A/C mutations. Neuropathol. Appl. Neurobiol. 27, 281–290. doi: 10.1046/j.0305-1846.2001.00323.x
Sheikh, F., Raskin, A., Chu, P. H., Lange, S., Domenighetti, A. A., Zheng, M., et al. (2008). An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J. Clin. Invest. 118, 3870–3880. doi: 10.1172/JCI34472
Shimi, T., Kittisopikul, M., Tran, J., Goldman, A. E., Adam, S. A., Zheng, Y., et al. (2015). Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 26, 4075–4086. doi: 10.1091/mbc.e15-07-0461
Siragam, V., Cui, X., Masse, S., Ackerley, C., Aafaqi, S., Strandberg, L., et al. (2014). TMEM43 mutation p.S358L alters intercalated disc protein expression and reduces conduction velocity in arrhythmogenic right ventricular cardiomyopathy. PLoS ONE 9:e109128. doi: 10.1371/journal.pone.0109128
Small, E. M., Thatcher, J. E., Sutherland, L. B., Kinoshita, H., Gerard, R. D., Richardson, J. A., et al. (2010). Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res 107, 294–304. doi: 10.1161/CIRCRESAHA.110.223172
Solovei, I., Wang, A. S., Thanisch, K., Schmidt, C. S., Krebs, S., Zwerger, M., et al. (2013). LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152, 584–598. doi: 10.1016/j.cell.2013.01.009
Somech, R., Shaklai, S., Geller, O., Amariglio, N., Simon, A. J., Rechavi, G., et al. (2005). The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. J. Cell Sci. 118(Pt 17), 4017–4025. doi: 10.1242/jcs.02521
Sosa, B. A., Kutay, U., and Schwartz, T. U. (2013). Structural insights into LINC complexes. Curr. Opin. Struct. Biol. 23, 285–291. doi: 10.1016/j.sbi.2013.03.005
Sosa, B. A., Rothballer, A., Kutay, U., and Schwartz, T. U. (2012). LINC complexes form by binding of three KASH peptides to domain interfaces of trimeric SUN proteins. Cell 149, 1035–1047. doi: 10.1016/j.cell.2012.03.046
Squarzoni, S., Sabatelli, P., Capanni, C., Petrini, S., Ognibene, A., Toniolo, D., et al. (2000). Emerin presence in platelets. Acta Neuropathol. 100, 291–298. doi: 10.1007/s004019900169
Starr, D. A., and Fridolfsson, H. N. (2010). Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annu. Rev. Cell Dev. Biol. 26, 421–444. doi: 10.1146/annurev-cellbio-100109-104037
Stroud, M. J., Fang, X., Zhang, J., Guimaraes-Camboa, N., Veevers, J., Dalton, N. D., et al. (2018). Luma is not essential for murine cardiac development and function. Cardiovasc Res. 114, 378–388. doi: 10.1093/cvr/cvx205
Stroud, M. J., Feng, W., Zhang, J., Veevers, J., Fang, X., Gerace, L., et al. (2017). Nesprin 1alpha2 is essential for mouse postnatal viability and nuclear positioning in skeletal muscle. J. Cell Biol. 216, 1915–1924. doi: 10.1083/jcb.201612128
Stubenvoll, A., Rice, M., Wietelmann, A., Wheeler, M., and Braun, T. (2015). Attenuation of Wnt/beta-catenin activity reverses enhanced generation of cardiomyocytes and cardiac defects caused by the loss of emerin. Hum. Mol. Genet. 24, 802–813. doi: 10.1093/hmg/ddu498
Sullivan, T., Escalante-Alcalde, D., Bhatt, H., Anver, M., Bhat, N., Nagashima, K., et al. (1999). Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147, 913–920. doi: 10.1083/jcb.147.5.913
Swift, J., Ivanovska, I. L., Buxboim, A., Harada, T., Dingal, P. C., Pinter, J., et al. (2013). Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341:1240104. doi: 10.1126/science.1240104
Taniguchi, Y., Furukawa, T., Tun, T., Han, H., and Honjo, T. (1998). LIM protein KyoT2 negatively regulates transcription by association with the RBP-J DNA-binding protein. Mol. Cell Biol. 18, 644–654. doi: 10.1128/MCB.18.1.644
Taylor, M. R., Slavov, D., Gajewski, A., Vlcek, S., Ku, L., Fain, P. R., et al. (2005). Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum. Mutat. 26, 566–574. doi: 10.1002/humu.20250
Tsuchiya, Y., Hase, A., Ogawa, M., Yorifuji, H., and Arahata, K. (1999). Distinct regions specify the nuclear membrane targeting of emerin, the responsible protein for Emery-Dreifuss muscular dystrophy. Eur J Biochem 259, 859–865. doi: 10.1046/j.1432-1327.1999.00112.x
Turgay, Y., Eibauer, M., Goldman, A. E., Shimi, T., Khayat, M., Ben-Harush, K., et al. (2017). The molecular architecture of lamins in somatic cells. Nature 543, 261–264. doi: 10.1038/nature21382
Ura, S., Hayashi, Y. K., Goto, K., Astejada, M. N., Murakami, T., Nagato, M., et al. (2007). Limb-girdle muscular dystrophy due to emerin gene mutations. Arch. Neurol. 64, 1038–1041. doi: 10.1001/archneur.64.7.1038
van der Kooi, A. J., van Meegen, M., Ledderhof, T. M., McNally, E. M., de Visser, M., and Bolhuis, P. A. (1997). Genetic localization of a newly recognized autosomal dominant limb-girdle muscular dystrophy with cardiac involvement (LGMD1B) to chromosome 1q11–21. Am. J. Hum. Genet. 60, 891–895.
Varela, I., Cadinanos, J., Pendas, A. M., Gutierrez-Fernandez, A., Folgueras, A. R., Sanchez, L. M., et al. (2005). Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 437, 564–568. doi: 10.1038/nature04019
Vignier, N., Chatzifrangkeskou, M., Rodriguez, B. M., Mericskay, M., Mougenot, N., Bonne, G., et al. (2018). Rescue of biosynthesis of nicotinamide adenine dinucleotide (NAD+) protects the heart in cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. doi: 10.1093/hmg/ddy278. [Epub ahead of print].
Wang, J., Kumar, R. M., Biggs, V. J., Lee, H., Chen, Y., Kagey, M. H., et al. (2011). The Msx1 homeoprotein recruits polycomb to the nuclear periphery during development. Dev. Cell 21, 575–588. doi: 10.1016/j.devcel.2011.07.003
Wang, N., Tytell, J. D., and Ingber, D. E. (2009). Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 10, 75–82. doi: 10.1038/nrm2594
Wang, W., Shi, Z., Jiao, S., Chen, C., Wang, H., Liu, G., et al. (2012). Structural insights into SUN-KASH complexes across the nuclear envelope. Cell Res. 22, 1440–1452. doi: 10.1038/cr.2012.126
Wang, Y., Herron, A. J., and Worman, H. J. (2006). Pathology and nuclear abnormalities in hearts of transgenic mice expressing M371K lamin A encoded by an LMNA mutation causing Emery-Dreifuss muscular dystrophy. Hum. Mol. Genet. 15, 2479–2489. doi: 10.1093/hmg/ddl170
Watson, M. L. (1955). The nuclear envelope; its structure and relation to cytoplasmic membranes. J. Biophys. Biochem. Cytol. 1, 257–270. doi: 10.1083/jcb.1.3.257
Wheeler, M. A., Warley, A., Roberts, R. G., Ehler, E., and Ellis, J. A. (2010). Identification of an emerin-beta-catenin complex in the heart important for intercalated disc architecture and beta-catenin localisation. Cell Mol. Life Sci. 67, 781–796. doi: 10.1007/s00018-009-0219-8
Witt, T. N., Garner, C. G., Pongratz, D., and Baur, X. (1988). Autosomal dominant Emery-Dreifuss syndrome: evidence of a neurogenic variant of the disease. Europ. Arch. Psychiat. Neurol. Sci. 237, 230–236. doi: 10.1007/BF00449912
Wu, W., Chordia, M. D., Hart, B. P., Kumarasinghe, E. S., Ji, M. K., Bhargava, A., et al. (2017). Macrocyclic MEK1/2 inhibitor with efficacy in a mouse model of cardiomyopathy caused by lamin A/C gene mutation. Bioorg. Med. Chem. 25, 1004–1013. doi: 10.1016/j.bmc.2016.12.014
Wu, W., Shan, J., Bonne, G., Worman, H. J., and Muchir, A. (2010). Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim. Biophys. Acta 1802, 632–638. doi: 10.1016/j.bbadis.2010.04.001
Wu, Y. K., Umeshima, H., Kurisu, J., and Kengaku, M. (2018). Nesprins and opposing microtubule motors generate a point force that drives directional nuclear motion in migrating neurons. Development 145:dev158782. doi: 10.1242/dev.158782
Xie, W., Chojnowski, A., Boudier, T., Lim, J. S., Ahmed, S., Ser, Z., et al. (2016). A-type lamins form distinct filamentous networks with differential nuclear pore complex associations. Curr. Biol. 26, 2651–2658. doi: 10.1016/j.cub.2016.07.049
Young, S. G., Fong, L. G., and Michaelis, S. (2005). Prelamin, A., Zmpste24, misshapen cell nuclei, and progeria–new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid. Res. 46, 2531–2558. doi: 10.1194/jlr.R500011-JLR200
Zhang, J., Felder, A., Liu, Y., Guo, L. T., Lange, S., Dalton, N. D., et al. (2010). Nesprin 1 is critical for nuclear positioning and anchorage. Hum. Mol. Genet. 19, 329–341. doi: 10.1093/hmg/ddp499
Zhang, Q., Bethmann, C., Worth, N. F., Davies, J. D., Wasner, C., Feuer, A., et al. (2007). Nesprin-1 and−2 are involved in the pathogenesis of Emery-Dreifuss Muscular Dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 16, 2816–2833. doi: 10.1093/hmg/ddm238
Zhang, X., Tang, N., Hadden, T. J., and Rishi, A. K. (2011). Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 1813, 1978–1986. doi: 10.1016/j.bbamcr.2011.03.010
Zhao, H., Sifakis, E. G., Sumida, N., Millan-Arino, L., Scholz, B. A., Svensson, J. P., et al. (2015). PARP1- and CTCF-mediated interactions between active and repressed chromatin at the lamina promote oscillating transcription. Mol Cell 59, 984–997. doi: 10.1016/j.molcel.2015.07.019
Zhao, Y., and Adjei, A. A. (2014). The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 11, 385–400. doi: 10.1038/nrclinonc.2014.83
Zheng, R., Ghirlando, R., Lee, M. S., Mizuuchi, K., Krause, M., and Craigie, R. (2000). Barrier-to-autointegration factor (BAF) bridges DNA in a discrete, higher-order nucleoprotein complex. Proc. Natl. Acad. Sci. U.S.A. 97, 8997–9002. doi: 10.1073/pnas.150240197
Zhou, C., Rao, L., Shanahan, C. M., and Zhang, Q. (2018). Nesprin-1/2: roles in nuclear envelope organisation, myogenesis and muscle disease. Biochem. Soc. Trans. 46, 311–320. doi: 10.1042/BST20170149
Zhou, Z., Du, X., Cai, Z., Song, X., Zhang, H., Mizuno, T., et al. (2012). Structure of Sad1-UNC84 homology (SUN) domain defines features of molecular bridge in nuclear envelope. J. Biol. Chem. 287, 5317–5326. doi: 10.1074/jbc.M111.304543
Ziat, E., Mamchaoui, K., Beuvin, M., Nelson, I., Azibani, F., Spuler, S., et al. (2016). FHL1B interacts with lamin A/C and emerin at the nuclear lamina and is misregulated in Emery-Dreifuss Muscular Dystrophy. J. Neuromuscul. Dis. 3, 497–510. doi: 10.3233/JND-160169
Zuleger, N., Robson, M. I., and Schirmer, E. C. (2011). The nuclear envelope as a chromatin organizer. Nucleus-Austin 2, 339–349. doi: 10.4161/nucl.2.5.17846
Keywords: Emery-Dreifuss muscular dystrophy (EDMD), A-type lamins, nuclear envelope (NE), muscular dystrophy (MD), cardiomyopathy, emerin, nesprin protein, luma
Citation: Brull A, Morales Rodriguez B, Bonne G, Muchir A and Bertrand AT (2018) The Pathogenesis and Therapies of Striated Muscle Laminopathies. Front. Physiol. 9:1533. doi: 10.3389/fphys.2018.01533
Received: 17 August 2018; Accepted: 11 October 2018;
Published: 30 October 2018.
Edited by:
Julien Ochala, King's College London, United KingdomReviewed by:
Anna Kostareva, Karolinska Institutet (KI), SwedenCopyright © 2018 Brull, Morales Rodriguez, Bonne, Muchir and Bertrand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne T. Bertrand, YS5iZXJ0cmFuZEBpbnN0aXR1dC1teW9sb2dpZS5vcmc=
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.