 Jonas Skogestad
Jonas Skogestad Jan Magnus Aronsen
Jan Magnus Aronsen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 07 November 2018
Sec. Cardiac Electrophysiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01500
This article is part of the Research TopicThe Role of Calcium Handling in Heart Failure and Heart Failure Associated ArrhythmiasView all 14 articles
Routine use of diuretics and neurohumoral activation make hypokalemia (serum K+ < 3. 5 mM) a prevalent electrolyte disorder among heart failure patients, contributing to the increased risk of ventricular arrhythmias and sudden cardiac death in heart failure. Recent experimental studies have suggested that hypokalemia-induced arrhythmias are initiated by the reduced activity of the Na+/K+-ATPase (NKA), subsequently leading to Ca2+ overload, Ca2+/Calmodulin-dependent kinase II (CaMKII) activation, and development of afterdepolarizations. In this article, we review the current mechanistic evidence of hypokalemia-induced triggered arrhythmias and discuss how molecular changes in heart failure might lower the threshold for these arrhythmias. Finally, we discuss how recent insights into hypokalemia-induced arrhythmias could have potential implications for future antiarrhythmic treatment strategies.
Despite continuous improvements in therapies, long-term prognosis in heart failure (HF) remains poor, with overall 5-year mortality reaching 50% (Yancy et al., 2013), and even higher in more advanced stages (NYHA III-IV) (Arnold et al., 2013). Sudden cardiac death (SCD), mostly due to ventricular tachyarrhythmias (VTs), contributes to ~50% of HF deaths (Tomaselli and Zipes, 2004). Hypokalemia is a well-recognized risk factor for VT, and hypokalemia is both common and independently associated with worse clinical outcomes in HF patients (Cleland et al., 1987; Ahmed et al., 2007; Bowling et al., 2010; Kjeldsen, 2010; Aldahl et al., 2017; Nunez et al., 2018), as well as increasing the risk of ventricular arrhythmias and mortality during acute myocardial infarction (Goyal et al., 2012; Colombo et al., 2018; Hoppe et al., 2018).
Here, we review the current evidence for mechanisms of triggered hypokalemia-induced arrhythmias, how cardiac remodeling in HF might lower the threshold for these arrhythmias, and use this to propose future antiarrhythmic drug targets.
Hypokalemia is defined as serum K+ levels (serum-[K+]) < 3.5 mM (Unwin et al., 2011), but several studies report increased risk of SCD and all-cause mortality in HF patients with serum-[K+] <4 mM (Nolan et al., 1998; Macdonald and Struthers, 2004; Bowling et al., 2010; Aldahl et al., 2017). The prevalence of hypokalemia in HF patients varies between 19 and 54% depending on the definition of hypokalemia and patient characteristics (Wester and Dyckner, 1986; Guo et al., 2005; Ahmed et al., 2007; Bowling et al., 2010; Collins et al., 2017). The prevalence was more likely to be higher in patient populations that were studied before the introduction of beta-blockers, ACE-inhibitors, and AT1-antagonists as standard HF therapy, as all of these drugs increase serum K+ levels and thus counteract hypokalemia. In addition, the prevalence of hypokalemia is generally higher in hospitalized patients compared to nonhospitalized patients (Unwin et al., 2011).
The main causes of hypokalemia in HF are use of diuretics and activation of the renin-angiotensin-aldosterone system that causes loss of K+ in the urine (Leier et al., 1994). Increased levels of catecholamines also contribute by shifting K+ into the intracellular compartment (Packer, 1990; Osadchii, 2010; Urso et al., 2015), whereas volume overload in more progressive HF could cause a dilution effect (Leier et al., 1994).
It has long been recognized that diuretics, both thiazides and loop-diuretics, increase the risk of hypokalemia and cardiac arrhythmias in patients receiving digitalis (Steiness and Olesen, 1976; Kaplan, 1984). Hypertensive men with baseline ECG abnormalities following an intensive diuretics regime displayed increased mortality compared to the standard regime in the Multiple Risk Factor Intervention Trial (1982). Later trials found no increased mortality with intensive diuretics treatment (1984) or when comparing diuretics to other anti-hypertensive agents (Officers et al., 2002), leading some authors to argue that the anti-hypertensive effect of diuretics compensates for the suggested pro-arrhythmic effect by diuretics alone (Papademetriou, 2006). Nevertheless, one study noted that a minority of patients using thiazides developed marked hypokalemia and cardiac arrhythmias (Siegel et al., 1992), and a case-control study found a dose-response relationship between thiazide dosage and the risk of SCD (Siscovick et al., 1994). Importantly, in patients with left ventricular dysfunction there was 30–40% increased risk of arrhythmic death among patients who used diuretics (Cooper et al., 1999). These results collectively suggest that, even though diuretics are important drugs for blood pressure reduction and prevention of volume overload in HF, being aware of the risk of hypokalemia and cardiac arrhythmias, in particular in the setting of heart disease, is important.
In contrast to thiazides and loop diuretics, mineralocorticoid receptor antagonists limit the renal excretion of K+, increase serum-[K+], and limit the risk for cardiac arrhythmias induced by hypokalemia (Siscovick et al., 1994; Cooper et al., 1999). ACE inhibitors, aldosterone receptor blockers, and beta blockers could potentially prevent hypokalemia by opposing the neurohumoral activation associated with HF that lowers serum-[K+] (Macdonald and Struthers, 2004).
Serum [K+] is altered during and after intensive exercise. During exercise, marked hyperkalemia may develop due to the release of K+ from skeletal muscles (Sejersted and Sjogaard, 2000). Increased levels of catecholamines counteract and decrease recovery time from exercise-induced hyperkalemia (Williams et al., 1985) by stimulating Na+/K+-ATPase (NKA) (Despa et al., 2008). Interestingly, serum-[K+] undershoots during the recovery phase after physical exercise, leading to postexercise hypokalemia (Medbo and Sejersted, 1990; Lindinger, 1995). The combination of hyperkalemia and subsequent hypokalemia with increased catecholamines during physical exercise could potentially contribute to the increased risk of cardiac arrhythmias and SCD observed during exercise in patients with structural or ischemic heart diseases (Siscovick et al., 1984; Thompson et al., 2007). Intriguingly, the risk of arrhythmias is particularly high in the recovery phase after exercise (Young et al., 1984), which coincides with postexercise hypokalemia (Medbo and Sejersted, 1990; Lindinger, 1995). Low serum-[K+] might thus be a cause of arrhythmias in patients even without clinically recognized hypokalemia.
Ventricular tachyarrhythmias are highly prevalent in HF, with 50–80% of patients having nonsustained VT on ambulatory cardiac monitoring (Singh et al., 1997; Teerlink et al., 2000). Re-entry and triggered arrhythmias are the two main types of tachyarrhythmias (Antzelevitch and Burashnikov, 2011). Fibrosis, scarring, and conduction abnormalities promote mechanical and electrophysiological re-entry, whereas reduced repolarization reserve, Ca2+ dysregulation, and altered transmembrane ion currents cause triggered arrhythmias (Tomaselli and Zipes, 2004; Ebinger et al., 2005; Jin et al., 2008).
Triggered arrhythmias are reported to initiate most VTs in nonischemic HF and even half of the VTs in ischemic HF (Pogwizd et al., 1992; Pogwizd and Bers, 2004). Triggered arrhythmias are caused by either early or delayed afterdepolarizations (EADs or DADs), abnormal depolarizations of the membrane potential that could give rise to a spontaneous action potential (AP) between two regular APs (Volders et al., 2000). DADs are caused by spontaneous Ca2+ release in a feed forward reaction that propagates as Ca2+ waves along the sarcoplasmic reticulum (SR) membrane as illustrated in Figure 1. Ca2+ waves can occur due to overload of Ca2+ in the SR and/or reduced threshold for Ca2+ leak through the ryanodine receptors (RyRs) (Venetucci et al., 2008). The spontaneously released Ca2+ activates inward currents named Iti (mainly consisting of INCX), leading to a depolarization of the resting membrane potential (Clusin, 2003; Venetucci et al., 2008). The Ca2+ wave-induced depolarization triggers an extra AP if the resulting inward current depolarizes the membrane sufficiently to trigger opening of voltage-gated Na+ channels.
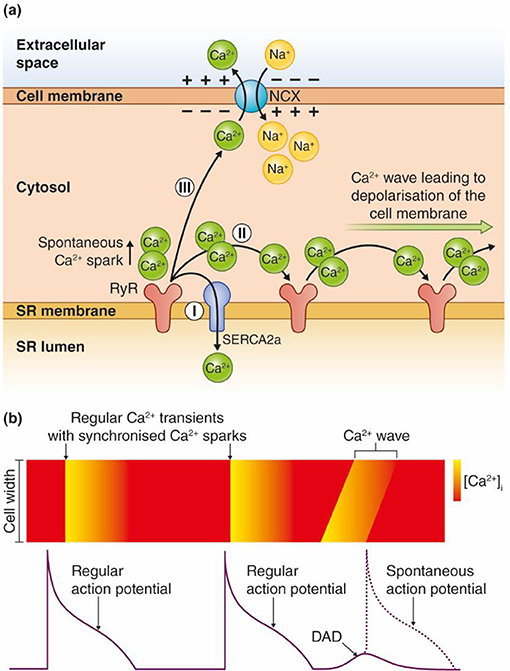
Figure 1. Model for transformation of a Ca2+ wave into an afterdepolarization. (a) Ca2+ ions are released spontaneously from the SR, either due to Ca2+ overload or increased RyR conductance (reduced threshold). The Ca2+ ions released from RyRs have three possible routes: (I) SERCA2 pumps the Ca2+ ions directly back into the SR. The Ca2+ wave is interrupted and no afterdepolarizations occur. (II) The Ca2+ ions diffuse to the neighboring RyRs, which leads to release of new Ca2+ ions. Repetitive events where Ca2+ ions are released and activate the next cluster of RyRs along the SR membrane lead to a Ca2+ wave. (III) Ca2+ can be extruded across the sarcolemma through the Na+/Ca2+ exchanger (NCX). This causes an inward, depolarizing current due to the inward flux of three positively charged Na+ ions per one Ca2+ that is extruded over the cell membrane. (b) Regular APs trigger synchronous Ca2+ release, which leads to cardiomyocyte contraction. Ca2+ waves can lead to DADs between two regular APs, and trigger a spontaneous AP as shown in the figure. Ca2+ waves during an AP can trigger EADs.
EADs typically develop in situations with reduced repolarization reserve, either due to increased inward currents, reduced outward currents, or both (Weiss et al., 2010). EADs occur when inward currents, the L-type Ca2+ current (ICa) or Iti derived from Ca2+ waves during the AP, are larger than the outward currents (mainly K+ currents) during late phases of the AP (Zhao et al., 2012).
Clinically, hypokalemia is associated with triggered arrhythmias such as Torsades De Pointes (TDP), polymorphic VT, ventricular fibrillation (VF), and ventricular ectopy (Nordrehaug et al., 1985). Hypokalemia has been shown to cause regional alterations in conduction velocity (Chah et al., 1982; Smeets et al., 1986; Wolk et al., 1998) and regional action potential duration (APD) heterogeneity (Poelzing and Veeraraghavan, 2007) that establish functional reentry circuits, although a recent study in postinfarction pigs with increased afterload only found slowed conduction velocity with no regional differences in APD (Motloch et al., 2017). Hypokalemia promotes triggered arrhythmias by a reduction in cardiac repolarization reserve and increased intracellular Ca2+ in cardiomyocytes (Weiss et al., 2017). Here, we review evidence for mechanisms coupling hypokalemia to induction of triggered arrhythmias, and argue that this is primarily due to inhibition of NKA (in particular the NKAα2 isoform) leading to development of afterdepolarizations as proposed in Figure 2.
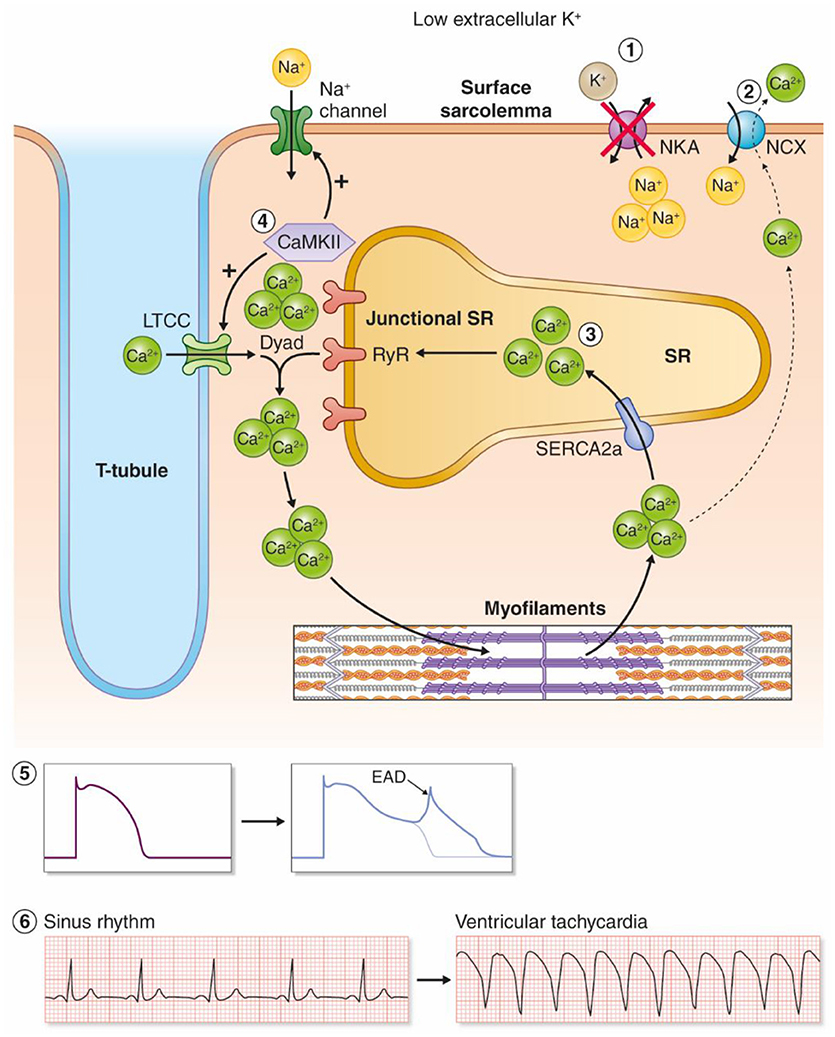
Figure 2. Proposed model for hypokalemia-induced triggered arrhythmias. (1) Low reduces the activity of the NKAα2 isoform. (2) Intracellular Na+ accumulates and leads to reduced inward NCX current, and by this less extrusion of Ca2+. (3) Intracellular and SR Ca2+ increases as a result. (4) Ca2+ overload increases the activity of the Ca2+/calmodulin-dependent kinase (CaMKII), which leads to a vicious cycle by phosphorylation of voltage-gated Na+ channels and L-type Ca2+ channels. (5) Increased influx of Na+ and Ca2+ amplifies Ca2+ overload and triggers EADs. (6) Hypokalemia-induced EADs can trigger ventricular tachyarrhythmias.
The pro-arrhythmic effects of hypokalemia have been linked to reduced outward K+ currents such as IK1, IKr, IKs, and Ito (Yang et al., 1997; Bouchard et al., 2004; Killeen et al., 2007; Osadchii, 2010) and recently to reduced NKA currents (Aronsen et al., 2015; Pezhouman et al., 2015). Reduced outward K+ currents decrease the repolarization reserve, prolong the APD, and increase the risk of afterdepolarizations (Weiss et al., 2017). Low extracellular K+ () leads to hyperpolarization of the resting membrane potential, which paradoxically increases excitability of cardiomyocytes. This effect is ascribed to an increased number of available Na+ channels and the reduced ability of IK1 to generate outward current that protects against membrane depolarization (Bers, 2001).
Pezhouman, Singh, and coworkers observed in an elegant study that reduced NKA activity was necessary and sufficient to develop hypokalemia-induced ventricular arrhythmias. Reduced K+ channel conductance in itself caused only a modest increase in APD and no afterdepolarizations, but potentiated the pro-arrhythmic effect of NKA inhibition in this study (Pezhouman et al., 2015). Comparably, we found that NKA inhibition was necessary and sufficient to increase Ca2+ levels in a factorial analysis where we compared the relative effect of lowering NKA activity vs. all other ion channels and transporters sensitive to . Several lines of evidence suggest that Ca2+ overload caused by NKA inhibition is the main initiating event in hypokalemia-induced ventricular arrhythmias, as discussed in the next sections.
Na+ and Ca2+ homeostasis are coupled through the Na+/Ca2+-exchanger (NCX) that uses the electrochemical gradient of Na+ and Ca2+ to exchange 3 Na+ for 1 Ca2+. This allows NKA to indirectly regulate cardiac Ca2+ fluxes through the regulation of [Na+]i. NKA is an ATP- and voltage-dependent ion transporter that exchanges 3 Na+ ions from the cytosol with 2 K+ ions from the extracellular compartment, leading to a net outward current (Stanley et al., 2015). NKA is the only major Na+ efflux mechanism in cardiomyocytes, and regulates intracellular [Na+] by balancing Na+ efflux against Na+ influx (Aronsen et al., 2013; Despa and Bers, 2013).
NKA activity is regulated by extracellular K+ levels, intracellular Na+ levels, the membrane potential (Glitsch, 2001), posttranslational modifications (Figtree et al., 2009; Poulsen et al., 2010), and the accessory protein phospholemman (PLM), which binds to and inhibits NKA activity (Pavlovic et al., 2007, 2013a). β-adrenergic stimulation leads to phosphorylation of PLM, which relieves PLM inhibition of NKA (Despa et al., 2005; Khafaga et al., 2012). This provides a link between circulating catecholamines and fluxes of Na+ and K+ over the cell membrane. NKA is composed of αβ dimers, where both isoforms exist in three different isoforms (α1-3 and β1-3) (Lingrel and Kuntzweiler, 1994; Sweadner et al., 1994; McDonough et al., 1996; Bers and Despa, 2009). NKAα1 and NKAα2 contribute to 70–95% and 5–30% of the total NKA activity in cardiomyocytes respectively (Lucchesi and Sweadner, 1991; James et al., 1999; Berry et al., 2007; Despa and Bers, 2007; Swift et al., 2007; Despa et al., 2012). Despite being less abundant than NKAα1, several studies strongly suggest that NKAα2, and not NKAα1, is the main NKA isoform that regulates NCX activity and Ca2+ fluxes in cardiomyocytes by limiting the [Na+]i sensed by the NCX (Berry et al., 2007; Despa and Bers, 2007; Swift et al., 2007, 2010; Despa et al., 2012). According to this scheme, reduced activity of NKAα2 increases cellular Ca2+ levels by limiting forward mode NCX activity (that extrudes Ca2+) and by increasing reverse mode NCX activity, which mediates Ca2+ influx during a short time during the early phases of the AP (Bers, 2002; Lines et al., 2006).
Despa et al. demonstrated that a similar degree of NKAα1 and NKAα2 inhibition yielded a comparable rise in intracellular Na+, but only NKAα2 inhibition increased the Ca2+ levels in cardiomyocytes (Despa et al., 2012). This study and others (James et al., 1999; Swift et al., 2007) suggest that the ability of NKAα2 to regulate Ca2+ fluxes and cardiac contractility is most likely due to close localization with NCX, but this remains to be shown. NKAα1 is more abundant at the sarcolemma, whereas NKAα2 preferentially localizes to the transverse T-tubules (Berry et al., 2007; Swift et al., 2007; Yuen et al., 2017). The anchoring protein Ankyrin B coordinates a NKA/NCX microdomain, but both NKAα1 and NKAα2 coprecipitate with Ankyrin-B (Mohler et al., 2005). More studies are needed to determine the precise mechanism through which NKAα2 specifically can control Ca2+ homeostasis and cardiac contractility.
We studied the effect of low (2.7 mM) on intracellular Ca2+ in rat ventricular myocytes, and found that steady-state Ca2+ transients were increased compared to normal K (5.0 mM). The increase in Ca2+ was not present in cells pretreated with a low dose of ouabain to selectively inhibit NKAα2 (Aronsen et al., 2015). This result could be explained by the finding that NKAα2 preferentially regulates intracellular Ca2+, as discussed above, and the different sensitivity to extracellular K+ [K+]e. Changes in [K+]e within the clinical range of hypokalemia are expected to modulate primarily NKAα2 (k0.5 = 2.7 mM) and have relatively small effects on NKAα1 (k0.5 = 1.5 mM) (Han et al., 2009).
These results suggest that moderate hypokalemia reduces NKAα2 activity and leads to arrhythmogenic Ca2+ overload by decreased forward mode and/or reduced reverse mode NCX activity (Aronsen et al., 2015). In addition, chronic hypokalemia has been reported to downregulate the expression of cardiac NKAα2, but not the NKAα1 isoform (Azuma et al., 1991). It cannot be excluded that inhibition of NKAα1 also contributes to the arrhythmogenesis in cells exposed , but given the established role of NKAα2 as a regulator of Ca2+ levels in cardiomyocytes, in combination with the present evidence, inhibition of NKAα2 is the most likely mechanism leading to Ca2+ overload in hypokalemia.
In addition to NKA inhibition, which reduces inward NCX current, hypokalemia also alters the NCX activity through hyperpolarization of the resting membrane potential. Hyperpolarization increases the inward NCX current and is associated with reduced intracellular Ca2+ levels (Bouchard et al., 2004), opposing reduced inward NCX current and the increased intracellular Ca2+ following NKA inhibition (Eisner and Lederer, 1979; Aronsen et al., 2013). Both we (Aronsen et al., 2015) and others (Eisner and Lederer, 1979; Bouchard et al., 2004) have reported that myocardial contractility and Ca2+ transients are first reduced, and then subsequently increase to above the basal level after switching from normal to low . In the intact organism, changes in are much slower than in experimental settings, and the Ca2+ levels most likely reach steady state without a biphasic response. Still, it is important to appreciate the biphasic cardiomyocyte response to low , as the steady state in myocardium exposed to low probably reflects the combined effect of membrane hyperpolarization (that reduces Ca2+ levels) and NKA inhibition (that increases Ca2+ levels).
Two studies have reported Ca2+ overload in intact, beating hearts perfused with low K+. One study found that the Ca2+-induced K+ channel was active in hearts exposed to low (corresponding to clinical hypokalemia) and not in hearts exposed to normal (Chan et al., 2015). This was further supported in another study that observed that hearts perfused with normal extracellular Ca2+ levels, in addition to low , developed VT/VF, but not if the hearts were perfused with low extracellular Ca2+. In the latter study, both EADs and DADs appeared in rat hearts exposed to moderate hypokalemia, but only the EADs were followed by episodes of sustained ventricular arrhythmias (Pezhouman et al., 2015). Comparably, only EADs were present in rabbit hearts exposed to moderate hypokalemia (Pezhouman et al., 2015), indicating that that EADs and not DADs are the main trigger of VT/VFs in otherwise normal hearts exposed to hypokalemia.
In addition to the reduction of NKAα2 activity as discussed in the previous sections, other mechanisms contribute to hypokalemia-induced Ca2+ overload in the system-based mechanism presented by Pezhouzman, Singh, and coworkers (Pezhouman et al., 2015). First, hypokalemia reduces outward repolarizing currents, both K+ currents and the NKA current. This increases the APD, allowing more Ca2+ influx through the L-type Ca2+ channels during the plateau phase (Weiss et al., 2017). Second, the initial increase in Ca2+ caused by NKA inhibition and reduced inward NCX currents, combined with APD prolongation, initiates a positive feedback loop (Pezhouman et al., 2015). According to this model, the initial increase in Ca2+ activates CaMKII, which phosphorylates and activates the late Na+ current (INaL) and increases ICa. Activation of INaL and ICa further amplifies the intracellular overload of Na+ and Ca2+, eventually activating CaMKII, in a detrimental, downward spiral, ultimately leading to afterdepolarizations and ventricular arrhythmias. Mathematical modeling supported this model, and both CaMKII inhibitors and inhibitor of INaL prevented development of VT/VF in hearts exposed to hypokalemia. Altogether, the collective data suggest that reduced NKAα2 activity initiates cellular Ca2+ overload in hypokalemia, which further leads to ventricular arrhythmias through a feed forward spiral where the activation of CaMKII amplifies intracellular Na+ and Ca2+ overload, ultimately leading to EADs (Figure 2).
Some evidence suggests that reduced NKA activity and subsequent development of Ca2+ overload and CaMKII activity could be involved in the arrhythmogenesis in other cardiac diseases besides hypokalemia. Transgenic mice with unphosphorylatable PLM have increased incidence of pacing-induced arrhythmias (Pavlovic et al., 2013b), corresponding to the finding that PLM KO have increased amount of triggered arrhythmias after beta-adrenergic stimulation, as PLM phosphorylation protects against Na+ and Ca2+ overload (Despa et al., 2008). Reduced NKA activity, increased intracellular Na+, and reduced inward NCX current are the main mechanisms of digitalis-induced arrhythmias (Wasserstrom and Aistrup, 2005). It has also been reported that digitalis increases CaMKII activity with pro-arrhythmogenic downstream effects, which is a further indication that the pathophysiological mechanisms of digitalis- and hypokalemia-induced arrhythmias are similar (Gonano et al., 2011). The same model could also explain the triggered arrhythmias observed in the Ankyrin B syndrome (LQTS4), a rare genetic syndrome characterized by conductance abnormalities and high risk of ventricular tachyarrhythmias (Cunha and Mohler, 2009). NKA expression is mildly reduced in Ankyrin B+/− mice (Mohler et al., 2003, 2005), and the NKA current is lower than in healthy controls (Camors et al., 2012). Further, forward mode NCX is reduced (Camors et al., 2012), leading to increased CaMKII activity (Popescu et al., 2016) and higher frequency of Ca2+ waves (Camors et al., 2012) in Ankyrin B+/− mice, suggesting that the mechanisms discussed for hypokalemia also partly could explain induction of ventricular arrhythmias in the Ankyrin B syndrome.
Molecular remodeling (e.g., transcriptional alterations, posttranscriptional regulation, and posttranslation regulation of proteins) and structural remodeling (cardiac hypertrophy and fibrosis) are hallmarks of HF (Fedak et al., 2005; Kehat and Molkentin, 2010). The remodeling is associated with increased risk of arrhythmias, and might potentiate the pro-arrhythmic effects of hypokalemia. However, there is a paucity of mechanistic data on hypokalemia in HF. Given the high prevalence of hypokalemia in HF patients, more studies are needed to clarify how the remodeling-associated HF influences the risk of hypokalemia-induced arrhythmias. Here, we discuss key alterations in HF that we consider relevant for the expected effect of hypokalemia in failing hearts.
Intracellular Na+ is increased in both human and animal models of hypertrophy and HF (Pieske et al., 2002; Pogwizd et al., 2003), and one study also found increased Na+ in the subsarcolemmal space in cells from dogs with cardiac hypertrophy (Verdonck et al., 2003). In theory, the increased Na+ levels could be due to increased Na+ influx or less Na+ extrusion. Although NKA expression and/or activity generally is shown to be reduced in HF, for example by reducing the phosphorylation of PLM (Shamraj et al., 1993; Semb et al., 1998; Bossuyt et al., 2005; Boguslavskyi et al., 2014), one study found no alterations in NKA activity (Despa et al., 2002). Several studies have also reported upregulated INaL in HF cardiomyocytes (Undrovinas et al., 1999; Moreno and Clancy, 2012), indicating increased Na+ influx in HF cardiomyocytes. In a study designed to determine the mechanism for increased Na+ concentration in HF, Despa and coworkers found that increased [Na+]i in failing rabbit cardiomyocytes (that more closely resembles human cardiomyocytes than rodents) primarily is due to higher TTX-sensitive Na+ influx and not due to reduced NKA activity (Despa et al., 2002). Altogether, it seems likely that increased Na+ influx and possibly reduced Na+ efflux could contribute to the increased intracellular Na+ during HF.
Increased intracellular Na+ in HF leads to increased intracellular Ca2+, by favoring less Ca2+ extrusion through forward mode NCX and/or more Ca2+ influx through reverse mode NCX. NCX is often upregulated in human (Studer et al., 1994; Reinecke et al., 1996) and experimental (Pogwizd et al., 1999; Sipido et al., 2000) HF, whereas Sarco(Endo)plasmic Reticulum Calcium ATPase 2 (SERCA2) expression and activity are reduced in HF (Lipskaia et al., 2014; Roe et al., 2015). Since intracellular Na+ is already higher at baseline and the NCX/SERCA2 balance is shifted in HF, a less pronounced NKA inhibition (e.g., by hypokalemia) could cause sufficient rise in Na+ to cause Ca2+ overload and spontaneous SR Ca2+ release. In addition, one study found pronounced reduction in the NKAα2 isoform expression and function despite only minor changes in the NKAα1 isoform (Swift et al., 2008). Increased expression and activity of CaMKII are also consistently observed in HF (Hoch et al., 1999; Kirchhefer et al., 1999; Zhang et al., 2003), and contribute to increased intracellular Na+ and Ca2+ by increasing INa−L (Anderson et al., 2011) and Ca2+ leak through RyRs (Ai et al., 2005). On the basis of these observations, we speculate that HF patients are “sensitized” and tolerate less changes in serum-[K+] compared to patients without cardiac diseases. This might help explain why HF patients with serum-[K+] <4 mM have increased risk of SCD and death (Macdonald and Struthers, 2004; Bowling et al., 2010), compared to the conventional cut-off at 3.5 mM (Unwin et al., 2011).
Spontaneous Ca2+ waves induce DADs by activating an inward current mainly consisting of INCX (Clusin, 2003). Since NCX typically is upregulated in HF, a given amount of spontaneously released Ca2+ generates more depolarizing inward current, reducing the threshold for DADs (Pogwizd and Bers, 2004). IK1 acts as a “safety valve” and counterbalances depolarizing inward currents caused by spontaneous Ca2+ release during the resting phase of the AP. IK1 is downregulated in HF (Fauconnier et al., 2005; Bers, 2006), thus increasing the likelihood that a spontaneous AP occurs through generation of EADs and/or DADs. In line with the typical observation of prolonged APD and reduced repolarization reserve (Wang and Hill, 2010), in addition to the earlier described factors that possibly contribute to Na+ and Ca2+ overload in HF, it is likely that there is a reduced threshold for hypokalemia-induced afterdepolarizations in HF.
No treatment that directly targets the underlying mechanism for hypokalemia-induced arrhythmias is currently available. The current treatment of hypokalemia in itself is potassium replacement (Cohn et al., 2000), and in patients with TDP, magnesium sulfate injection is used to prevent EADs and DADs through an unknown mechanism (Fazekas et al., 1993). TDP can also be treated with cardiac pacing or isoproterenol injection to shorten the APD (Banai and Tzivoni, 1993).
Ideally, new treatments against hypokalemia-induced arrhythmias should aim at (1) preventing EADs/DADs, (2) shortening the APD, and (3) directly targeting the underlying mechanism. On the basis of the model for hypokalemia-induced ventricular arrhythmias in Figure 2, we suggest CaMKII inhibition, NKA activation, and in particular NKAα2 activation, to be further investigated as future antiarrhythmic strategies. CaMKII inhibition prevents hypokalemia-induced EADs (Pezhouman et al., 2015), Ca2+ overload, and DADs in catecholaminergic polymorphic ventricular tachycardia (CPVT) (Di Pasquale et al., 2013) and in HF (Ai et al., 2005; Sag et al., 2009), in addition to hypokalemia-induced VT/VF (Pezhouman et al., 2015). CaMKII inhibition also shortens APD (Li et al., 2006; Bourgonje et al., 2012), although this effect might be species-dependent (Wagner et al., 2009). We are currently unaware of any drug that specifically activates the NKA. Theoretically, this could prove to be an effective antiarrhythmic strategy, as NKA inhibition causes Ca2+ overload, afterdepolarizations, and ventricular arrhythmias (Faggioni and Knollmann, 2015).
HF is also characterized by APD prolongation (Wang and Hill, 2010) and increased risk of afterdepolarizations (Pogwizd and Bers, 2004; Weiss et al., 2010), and we speculate that CaMKII inhibitors and NKA activators might be future antiarrhythmic options in HF even in the absence of hypokalemia.
Both authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Debbie Maizels (www.scientific-art.com) for the artwork included in this review and to Dr. Emma L. Robinson for valuable inputs.
(1982). Multiple risk factor intervention trial. Risk factor changes and mortality results. The Multiple Risk Factor Intervention Trial Research Group. JAMA 248, 1465–1477.
(1984). The effect of antihypertensive drug treatment on mortality in the presence of resting electrocardiographic abnormalities at baseline: the HDFP experience. The hypertension detection and Folow-up Program Cooperative Research Group. Circulation 70, 996–1003.
Ahmed, A., Zannad, F., Love, T. E., Tallaj, J., Gheorghiade, M., Ekundayo, O. J., et al. (2007). A propensity-matched study of the association of low serum potassium levels and mortality in chronic heart failure. Eur. Heart J. 28, 1334–1343. doi: 10.1093/eurheartj/ehm091
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., and Pogwizd, S. M. (2005). Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97, 1314–1322. doi: 10.1161/01.RES.0000194329.41863.89
Aldahl, M., Jensen, A. C., Davidsen, L., Eriksen, M. A., Moller Hansen, S., Nielsen, B. J., et al. (2017). Associations of serum potassium levels with mortality in chronic heart failure patients. Eur. Heart J. 38, 2890–2896. doi: 10.1093/eurheartj/ehx460
Anderson, M. E., Brown, J. H., and Bers, D. M. (2011). CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 51, 468–473. doi: 10.1016/j.yjmcc.2011.01.012
Antzelevitch, C., and Burashnikov, A. (2011). Overview of basic mechanisms of cardiac arrhythmia. Card. Electrophysiol. Clin. 3, 23–45. doi: 10.1016/j.ccep.2010.10.012
Arnold, J. M. O., Liu, P., Howlett, J., Ignaszewski, A., Leblanc, M. H., Kaan, A., et al. (2013). Ten year survival by NYHA functional class in heart failure outpatients referred to specialized multidisciplinary heart failure clinics 1999 to 2011. Eur. Heart J. 34, 291–291. doi: 10.1093/eurheartj/eht308.P1505
Aronsen, J. M., Skogestad, J., Lewalle, A., Louch, W. E., Hougen, K., Stokke, M. K., et al. (2015). Hypokalaemia induces Ca2+ overload and Ca2+ waves in ventricular myocytes by reducing Na+,K+-ATPase α2 activity. J. Physiol. 593, 1509–1521. doi: 10.1113/jphysiol.2014.279893
Aronsen, J. M., Swift, F., and Sejersted, O. M. (2013). Cardiac sodium transport and excitation-contraction coupling. J. Mol. Cell. Cardiol. 61, 11–19. doi: 10.1016/j.yjmcc.2013.06.003
Azuma, K. K., Hensley, C. B., Putnam, D. S., and McDonough, A. A. (1991). Hypokalemia decreases Na+-K+-ATPase α2- but not alpha 1-isoform abundance in heart, muscle, and brain. Am. J. Physiol. 260(5 Pt 1), C958–C964.
Banai, S., and Tzivoni, D. (1993). Drug therapy for torsade de pointes. J. Cardiovasc. Electrophysiol. 4, 206–210. doi: 10.1111/j.1540-8167.1993.tb01224.x
Berry, R. G., Despa, S., Fuller, W., Bers, D. M., and Shattock, M. J. (2007). Differential distribution and regulation of mouse cardiac Na+/K+-ATPase α1 and α2 subunits in T-tubule and surface sarcolemmal membranes. Cardiovasc. Res. 73, 92–100. doi: 10.1016/j.cardiores.2006.11.006
Bers, D. M. (2001). Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd Edn. Dordrecht; Boston, MA: Kluwer Academic Publishers.
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
Bers, D. M. (2006). Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 21, 380–387. doi: 10.1152/physiol.00019.2006
Bers, D. M., and Despa, S. (2009). Na+ transport in cardiac myocytes; Implications for excitation-contraction coupling. IUBMB Life 61, 215–221. doi: 10.1002/iub.163
Boguslavskyi, A., Pavlovic, D., Aughton, K., Clark, J. E., Howie, J., Fuller, W., et al. (2014). Cardiac hypertrophy in mice expressing unphosphorylatable phospholemman. Cardiovasc. Res. 104, 72–82. doi: 10.1093/cvr/cvu182
Bossuyt, J., Ai, X., Moorman, J. R., Pogwizd, S. M., and Bers, D. M. (2005). Expression and phosphorylation of the na-pump regulatory subunit phospholemman in heart failure. Circ. Res. 97, 558–565. doi: 10.1161/01.RES.0000181172.27931.c3
Bouchard, R., Clark, R. B., Juhasz, A. E., and Giles, W. R. (2004). Changes in extracellular K+ concentration modulate contractility of rat and rabbit cardiac myocytes via the inward rectifier K+ current IK1. J. Physiol. 556(Pt 3), 773–790. doi: 10.1113/jphysiol.2003.058248
Bourgonje, V. J., Schoenmakers, M., Beekman, J. D., van der Nagel, R., Houtman, M. J., Miedema, L. F., et al. (2012). Relevance of calmodulin/CaMKII activation for arrhythmogenesis in the AV block dog. Heart Rhythm. 9, 1875–1883. doi: 10.1016/j.hrthm.2012.07.023
Bowling, C. B., Pitt, B., Ahmed, M. I., Aban, I. B., Sanders, P. W., Mujib, M., et al. (2010). Hypokalemia and outcomes in patients with chronic heart failure and chronic kidney disease: findings from propensity-matched studies. Circ. Heart Fail. 3, 253–260. doi: 10.1161/CIRCHEARTFAILURE.109.899526
Camors, E., Mohler, P. J., Bers, D. M., and Despa, S. (2012). Ankyrin-B reduction enhances Ca spark-mediated SR Ca release promoting cardiac myocyte arrhythmic activity. J. Mol. Cell. Cardiol. 52, 1240–1248. doi: 10.1016/j.yjmcc.2012.02.010
Chah, Q. T., Braly, G., Bouzouita, K., and Faucon, G. (1982). Effects of hypokalemia on the various parts of the conduction system of the dog heart in situ. Naunyn Schmiedebergs. Arch. Pharmacol. 319, 178–183. doi: 10.1007/BF00503934
Chan, Y. H., Tsai, W. C., Ko, J. S., Yin, D., Chang, P. C., Rubart, M., Chen, P. S., et al. (2015). Small-conductance calcium-activated potassium current is activated during hypokalemia and masks short-term cardiac memory induced by ventricular pacing. Circulation 132, 1377–1386. doi: 10.1161/CIRCULATIONAHA.114.015125
Cleland, J. G., Dargie, H. J., and Ford, I. (1987). Mortality in heart failure: clinical variables of prognostic value. Br. Heart J. 58, 572–582. doi: 10.1136/hrt.58.6.572
Clusin, W. T. (2003). Calcium and cardiac arrhythmias: DADs, EADs, and alternans. Crit. Rev. Clin. Lab. Sci. 40, 337–375. doi: 10.1080/713609356
Cohn, J. N., Kowey, P. R., Whelton, P. K., and Prisant, L. M. (2000). New guidelines for potassium replacement in clinical practice: a contemporary review by the National Council on Potassium in Clinical Practice. Arch. Intern. Med. 160, 2429–2436. doi: 10.1001/archinte.160.16.2429
Collins, A. J., Pitt, B., Reaven, N., Funk, S., McGaughey, K., Wilson, D., et al. (2017). Association of serum potassium with all-cause mortality in patients with and without heart failure, chronic kidney disease, and/or diabetes. Am. J. Nephrol. 46, 213–221. doi: 10.1159/000479802
Colombo, M. G., Kirchberger, I., Amann, U., Dinser, L., and Meisinger, C. (2018). Association of serum potassium concentration with mortality and ventricular arrhythmias in patients with acute myocardial infarction: a systematic review and meta-analysis. Eur. J. Prev. Cardiol. 25, 576–595. doi: 10.1177/2047487318759694
Cooper, H. A., Dries, D. L., Davis, C. E., Shen, Y. L., and Domanski, M. J. (1999). Diuretics and risk of arrhythmic death in patients with left ventricular dysfunction. Circulation 100, 1311–1315. doi: 10.1161/01.CIR.100.12.1311
Cunha, S. R., and Mohler, P. J. (2009). Ankyrin protein networks in membrane formation and stabilization. J. Cell. Mol. Med. 13, 4364–4376. doi: 10.1111/j.1582-4934.2009.00943.x
Despa, S., and Bers, D. M. (2007). Functional analysis of Na+/K+-ATPase isoform distribution in rat ventricular myocytes. Am. J. Physiol. Cell Physiol. 293, C321–C327. doi: 10.1152/ajpcell.00597.2006
Despa, S., and Bers, D. M. (2013). Na+ transport in the normal and failing heart-remember the balance. J. Mol. Cell. Cardiol. 61, 2–10. doi: 10.1016/j.yjmcc.2013.04.011
Despa, S., Bossuyt, J., Han, F., Ginsburg, K. S., Jia, L. G., Kutchai, H., et al. (2005). Phospholemman-phosphorylation mediates the beta-adrenergic effects on Na/K pump function in cardiac myocytes. Circ. Res. 97, 252–259. doi: 10.1161/01.RES.0000176532.97731.e5
Despa, S., Islam, M. A., Weber, C. R., Pogwizd, S. M., and Bers, D. M. (2002). Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 105, 2543–2548. doi: 10.1161/01.CIR.0000016701.85760.97
Despa, S., Lingrel, J. B., and Bers, D. M. (2012). Na+/K+-ATPase α2-isoform preferentially modulates Ca2+ transients and sarcoplasmic reticulum Ca2+ release in cardiac myocytes. Cardiovasc. Res. 95, 480–486. doi: 10.1093/cvr/cvs213
Despa, S., Tucker, A. L., and Bers, D. M. (2008). Phospholemman-mediated activation of Na/K-ATPase limits [Na]i and inotropic state during beta-adrenergic stimulation in mouse ventricular myocytes. Circulation 117, 1849–1855. doi: 10.1161/CIRCULATIONAHA.107.754051
Di Pasquale, E., Lodola, F., Miragoli, M., Denegri, M., Avelino-Cruz, J. E., Buonocore, M., et al. (2013). CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 4:e843. doi: 10.1038/cddis.2013.369
Ebinger, M. W., Krishnan, S., and Schuger, C. D. (2005). Mechanisms of ventricular arrhythmias in heart failure. Curr. Heart Fail. Rep. 2, 111–117. doi: 10.1007/s11897-005-0018-y
Eisner, D. A., and Lederer, W. J. (1979). The role of the sodium pump in the effects of potassium-depleted solutions on mammalian cardiac muscle. J. Physiol. 294, 279–301.
Faggioni, M., and Knollmann, B. C. (2015). Arrhythmia protection in hypokalemia: a novel role of Ca2+-Activated K+ currents in the ventricle. Circulation 132, 1371–1373. doi: 10.1161/CIRCULATIONAHA.115.018874
Fauconnier, J., Lacampagne, A., Rauzier, J. M., Vassort, G., and Richard, S. (2005). Ca2+-dependent reduction of IK1 in rat ventricular cells: a novel paradigm for arrhythmia in heart failure? Cardiovasc. Res. 68, 204–212. doi: 10.1016/j.cardiores.2005.05.024
Fazekas, T., Scherlag, B. J., Vos, M., Wellens, H. J., and Lazzara, R. (1993). Magnesium and the heart: antiarrhythmic therapy with magnesium. Clin. Cardiol. 16, 768–774. doi: 10.1002/clc.4960161105
Fedak, P. W., Verma, S., Weisel, R. D., and Li, R. K. (2005). Cardiac remodeling and failure: from molecules to man (Part I). Cardiovasc. Pathol. 14, 1–11. doi: 10.1016/j.carpath.2004.12.002
Figtree, G. A., Liu, C. C., Bibert, S., Hamilton, E. J., Garcia, A., White, C. N., et al. (2009). Reversible oxidative modification: a key mechanism of Na+-K+ pump regulation. Circ. Res. 105, 185–193. doi: 10.1161/CIRCRESAHA.109.199547
Glitsch, H. G. (2001). Electrophysiology of the sodium-potassium-ATPase in cardiac cells. Physiol. Rev. 81, 1791–1826. doi: 10.1152/physrev.2001.81.4.1791
Gonano, L. A., Sepulveda, M., Rico, Y., Kaetzel, M., Valverde, C. A., Dedman, J., et al. (2011). Calcium-calmodulin kinase II mediates digitalis-induced arrhythmias. Circ. Arrhythm. Electrophysiol. 4, 947–957. doi: 10.1161/CIRCEP.111.964908
Goyal, A., Spertus, J. A., Gosch, K., Venkitachalam, L., Jones, P. G., Van den Berghe, G., et al. (2012). Serum potassium levels and mortality in acute myocardial infarction. JAMA 307, 157–164. doi: 10.1001/jama.2011.1967
Guo, H., Lee, J. D., Ueda, T., Wang, J., Lu, D., He, H., et al. (2005). Different clinical features, biochemical profiles, echocardiographic and elctrocardiographic findings in older and younger patients with idiopathic dilated cardiomyopathy. Acta Cardiol. 60, 27–31. doi: 10.2143/AC.60.1.2005045
Han, F., Tucker, A. L., Lingrel, J. B., Despa, S., and Bers, D. M. (2009). Extracellular potassium dependence of the Na+-K+-ATPase in cardiac myocytes: isoform specificity and effect of phospholemman. Am. J. Physiol,. Cell Physiol. 297, C699–C705. doi: 10.1152/ajpcell.00063.2009
Hoch, B., Meyer, R., Hetzer, R., Krause, E. G., and Karczewski, P. (1999). Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ. Res. 84, 713–721. doi: 10.1161/01.RES.84.6.713
Hoppe, L. K., Muhlack, D. C., Koenig, W., Carr, P. R., Brenner, H., and Schottker, B. (2018). Association of abnormal serum potassium levels with arrhythmias and cardiovascular mortality: a systematic review and meta-analysis of observational studies. Cardiovasc. Drugs Ther. 32, 197–212. doi: 10.1007/s10557-018-6783-0
James, P. F., Grupp, I. L., Grupp, G., Woo, A. L., Askew, G. R., Croyle, M. L., et al. (1999). Identification of a specific role for the Na,K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol. Cell 3, 555–563. doi: 10.1016/S1097-2765(00)80349-4
Jin, H., Lyon, A. R., and Akar, F. G. (2008). Arrhythmia mechanisms in the failing heart. Pacing Clin. Electrophysiol. 31, 1048–1056. doi: 10.1111/j.1540-8159.2008.01134.x
Kaplan, N. M. (1984). Our appropriate concern about hypokalemia. Am. J. Med. 77, 1–4. doi: 10.1016/0002-9343(84)90427-3
Kehat, I., and Molkentin, J. D. (2010). Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 122, 2727–2735. doi: 10.1161/CIRCULATIONAHA.110.942268
Khafaga, M., Bossuyt, J., Mamikonian, L., Li, J. C., Lee, L. L., Yarov-Yarovoy, V., et al. (2012). Na(+)/K(+)-ATPase E960 and phospholemman F28 are critical for their functional interaction. Proc. Natl. Acad. Sci. U.S.A. 109, 20756–20761. doi: 10.1073/pnas.1207866109
Killeen, M. J., Gurung, I. S., Thomas, G., Stokoe, K. S., Grace, A. A., and Huang, C. L. (2007). Separation of early afterdepolarizations from arrhythmogenic substrate in the isolated perfused hypokalaemic murine heart through modifiers of calcium homeostasis. Acta Physiol. (Oxf). 191, 43–58. doi: 10.1111/j.1748-1716.2007.01715.x
Kirchhefer, U., Schmitz, W., Scholz, H., and Neumann, J. (1999). Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc. Res. 42, 254–261. doi: 10.1016/S0008-6363(98)00296-X
Leier, C. V., Dei Cas, L., and Metra, M. (1994). Clinical relevance and management of the major electrolyte abnormalities in congestive heart failure: hyponatremia, hypokalemia, and hypomagnesemia. Am. Heart J. 128, 564–574. doi: 10.1016/0002-8703(94)90633-5
Li, J., Marionneau, C., Zhang, R., Shah, V., Hell, J. W., Nerbonne, J. M., et al. (2006). Calmodulin kinase II inhibition shortens action potential duration by upregulation of K+ currents. Circ. Res. 99, 1092–1099. doi: 10.1161/01.RES.0000249369.71709.5c
Lindinger, M. I. (1995). Potassium regulation during exercise and recovery in humans: implications for skeletal and cardiac muscle. J. Mol. Cell. Cardiol. 27, 1011–1022. doi: 10.1016/0022-2828(95)90070-5
Lines, G. T., Sande, J. B., Louch, W. E., Mork, H. K., Grottum, P., and Sejersted, O. M. (2006). Contribution of the Na+/Ca2+ exchanger to rapid Ca2+ release in cardiomyocytes. Biophys. J. 91, 779–792. doi: 10.1529/biophysj.105.072447
Lipskaia, L., Keuylian, Z., Blirando, K., Mougenot, N., Jacquet, A., Rouxel, C., et al. (2014). Expression of sarco (endo) plasmic reticulum calcium ATPase (SERCA) system in normal mouse cardiovascular tissues, heart failure and atherosclerosis. Biochim. Biophys. Acta 1843, 2705–2718. doi: 10.1016/j.bbamcr.2014.08.002
Lucchesi, P. A., and Sweadner, K. J. (1991). Postnatal changes in Na,K-ATPase isoform expression in rat cardiac ventricle. Conservation of biphasic ouabain affinity. J. Biol. Chem. 266, 9327–9331.
Macdonald, J. E., and Struthers, A. D. (2004). What is the optimal serum potassium level in cardiovascular patients? J. Am. Coll. Cardiol. 43, 155–161. doi: 10.1016/j.jacc.2003.06.021
McDonough, A. A., Zhang, Y., Shin, V., and Frank, J. S. (1996). Subcellular distribution of sodium pump isoform subunits in mammalian cardiac myocytes. Am. J. Physiol. 270(4 Pt 1), C1221–C1227.
Medbo, J. I., and Sejersted, O. M. (1990). Plasma potassium changes with high intensity exercise. J. Physiol. 421, 105–122.
Mohler, P. J., Davis, J. Q., and Bennett, V. (2005). Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 3:e423. doi: 10.1371/journal.pbio.0030423
Mohler, P. J., Schott, J. J., Gramolini, A. O., Dilly, K. W., Guatimosim, S., duBell, W. H., et al. (2003). Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421, 634–639. doi: 10.1038/nature01335
Moreno, J. D., and Clancy, C. E. (2012). Pathophysiology of the cardiac late Na current and its potential as a drug target. J. Mol. Cell. Cardiol. 52, 608–619. doi: 10.1016/j.yjmcc.2011.12.003
Motloch, L. J., Ishikawa, K., Xie, C., Hu, J., Aguero, J., Fish, K. M., et al. (2017). Increased afterload following myocardial infarction promotes conduction-dependent arrhythmias that are unmasked by hypokalemia. JACC Basic Trans. Sci. 2, 258–269. doi: 10.1016/j.jacbts.2017.02.002
Nolan, J., Batin, P. D., Andrews, R., Lindsay, S. J., Brooksby, P., Mullen, M., et al. (1998). Prospective study of heart rate variability and mortality in chronic heart failure: results of the United Kingdom heart failure evaluation and assessment of risk trial (UK-heart). Circulation 98, 1510–1516. doi: 10.1161/01.CIR.98.15.1510
Nordrehaug, J. E., Johannessen, K. A., and von der Lippe, G. (1985). Serum potassium concentration as a risk factor of ventricular arrhythmias early in acute myocardial infarction. Circulation 71, 645–649. doi: 10.1161/01.CIR.71.4.645
Nunez, J., Bayes-Genis, A., Zannad, F., Rossignol, P., Nunez, E., Bodi, V., et al. (2018). Long-term potassium monitoring and dynamics in heart failure and risk of mortality. Circulation 137, 1320–1330. doi: 10.1161/CIRCULATIONAHA.117.030576
Officers, A., Coordinators for the, A. C. R. G. T. A., and Lipid-Lowering Treatment to Prevent Heart Attack, T. (2002). Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs. diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 288, 2981–2997. doi: 10.1001/jama.288.23.2981
Osadchii, O. E. (2010). Mechanisms of hypokalemia-induced ventricular arrhythmogenicity. Fundam. Clin. Pharmacol. 24, 547–559. doi: 10.1111/j.1472-8206.2010.00835.x
Packer, M. (1990). Potential role of potassium as a determinant of morbidity and mortality in patients with systemic hypertension and congestive heart-failure. Am. J. Cardiol. 65, E45–E51. doi: 10.1016/0002-9149(90)90251-U
Papademetriou, V. (2006). Diuretics, hypokalemia, and cardiac arrhythmia: a 20-year controversy. J. Clin. Hypertens. (Greenwich) 8, 86–92. doi: 10.1111/j.1524-6175.2005.04722.x
Pavlovic, D., Fuller, W., and Shattock, M. J. (2007). The intracellular region of FXYD1 is sufficient to regulate cardiac Na/K ATPase. FASEB J. 21, 1539–1546. doi: 10.1096/fj.06-7269com
Pavlovic, D., Fuller, W., and Shattock, M. J. (2013a). Novel regulation of cardiac Na pump via phospholemman. J. Mol. Cell. Cardiol. 61:83–93. doi: 10.1016/j.yjmcc.2013.05.002
Pavlovic, D., Hall, A. R., Kennington, E. J., Aughton, K., Boguslavskyi, A., Fuller, W., et al. (2013b). Nitric oxide regulates cardiac intracellular Na+ and Ca2+ by modulating Na/K ATPase via PKCepsilon and phospholemman-dependent mechanism. J. Mol. Cell. Cardiol. 61, 164–171. doi: 10.1016/j.yjmcc.2013.04.013
Pezhouman, A., Singh, N., Song, Z., Nivala, M., Eskandari, A., Cao, H., et al. (2015). Molecular basis of hypokalemia-induced ventricular fibrillation. Circulation 132, 1528–1537. doi: 10.1161/CIRCULATIONAHA.115.016217
Pieske, B., Maier, L. S., Piacentino, V. III., Weisser, J., Hasenfuss, G., and Houser, S. (2002). Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation 106, 447–453. doi: 10.1161/01.CIR.0000023042.50192.F4
Poelzing, S., and Veeraraghavan, R. (2007). Heterogeneous ventricular chamber response to hypokalemia and inward rectifier potassium channel blockade underlies bifurcated T wave in guinea pig. Am. J. Physiol. Heart Circ. Physiol. 292, H3043–H3051. doi: 10.1152/ajpheart.01312.2006
Pogwizd, S. M., and Bers, D. M. (2004). Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc. Med. 14, 61–66. doi: 10.1016/j.tcm.2003.12.002
Pogwizd, S. M., Hoyt, R. H., Saffitz, J. E., Corr, P. B., Cox, J. L., and Cain, M. E. (1992). Reentrant and focal mechanisms underlying ventricular tachycardia in the human heart. Circulation 86, 1872–1887. doi: 10.1161/01.CIR.86.6.1872
Pogwizd, S. M., Qi, M., Yuan, W., Samarel, A. M., and Bers, D. M. (1999). Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ. Res. 85, 1009–1019. doi: 10.1161/01.RES.85.11.1009
Pogwizd, S. M., Sipido, K. R., Verdonck, F., and Bers, D. M. (2003). Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc. Res. 57, 887–896. doi: 10.1016/S0008-6363(02)00735-6
Popescu, I., Galice, S., Mohler, P. J., and Despa, S. (2016). Elevated local [Ca2+] and CaMKII promote spontaneous Ca2+ release in ankyrin-B-deficient hearts. Cardiovasc. Res. 111, 287–294. doi: 10.1093/cvr/cvw093
Poulsen, H., Morth, P., Egebjerg, J., and Nissen, P. (2010). Phosphorylation of the Na+,K+-ATPase and the H+,K+-ATPase. FEBS Lett. 584, 2589–2595. doi: 10.1016/j.febslet.2010.04.035
Reinecke, H., Studer, R., Vetter, R., Holtz, J., and Drexler, H. (1996). Cardiac Na+/Ca2+ exchange activity in patients with end-stage heart failure. Cardiovasc. Res. 31, 48–54. doi: 10.1016/S0008-6363(95)00176-X
Roe, A. T., Frisk, M., and Louch, W. E. (2015). Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr. Pharm. Des. 21, 431–448. doi: 10.2174/138161282104141204124129
Sag, C. M., Wadsack, D. P., Khabbazzadeh, S., Abesser, M., Grefe, C., Neumann, K., et al. (2009). Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ. Heart Fail. 2, 664–675. doi: 10.1161/CIRCHEARTFAILURE.109.865279
Sejersted, O. M., and Sjogaard, G. (2000). Dynamics and consequences of potassium shifts in skeletal muscle and heart during exercise. Physiol. Rev. 80, 1411–1481. doi: 10.1152/physrev.2000.80.4.1411
Semb, S. O., Lunde, P. K., Holt, E., Tonnessen, T., Christensen, G., and Sejersted, O. M. (1998). Reduced myocardial Na+, K+-pump capacity in congestive heart failure following myocardial infarction in rats. J. Mol. Cell. Cardiol. 30, 1311–1328. doi: 10.1006/jmcc.1998.0696
Shamraj, O. I., Grupp, I. L., Grupp, G., Melvin, D., Gradoux, N., Kremers, W., et al. (1993). Characterisation of Na/K-ATPase, its isoforms, and the inotropic response to ouabain in isolated failing human hearts. Cardiovasc. Res. 27, 2229–2237. doi: 10.1093/cvr/27.12.2229
Siegel, D., Hulley, S. B., Black, D. M., Cheitlin, M. D., Sebastian, A., Seeley, D. G., et al. (1992). Diuretics, serum and intracellular electrolyte levels, and ventricular arrhythmias in hypertensive men. JAMA 267, 1083–1089. doi: 10.1001/jama.1992.03480080053026
Singh, S. N., Carson, P. E., and Fisher, S. G. (1997). Nonsustained ventricular tachycardia in severe heart failure. Circulation 96, 3794–3795.
Sipido, K. R., Volders, P. G., de Groot, S. H., Verdonck, F., Van de Werf, F., Wellens, H. J., et al. (2000). Enhanced Ca2+ release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation 102, 2137–2144. doi: 10.1161/01.CIR.102.17.2137
Siscovick, D. S., Raghunathan, T. E., Psaty, B. M., Koepsell, T. D., Wicklund, K. G., Lin, X., et al. (1994). Diuretic therapy for hypertension and the risk of primary cardiac arrest. N. Engl. J. Med. 330, 1852–1857. doi: 10.1056/NEJM199406303302603
Siscovick, D. S., Weiss, N. S., Fletcher, R. H., and Lasky, T. (1984). The incidence of primary cardiac arrest during vigorous exercise. N. Engl. J. Med. 311, 874–877. doi: 10.1056/NEJM198410043111402
Smeets, J. L., Allessie, M. A., Lammers, W. J., Bonke, F. I., and Hollen, J. (1986). The wavelength of the cardiac impulse and reentrant arrhythmias in isolated rabbit atrium. The role of heart rate, autonomic transmitters, temperature, and potassium. Circ Res 58, 96–108. doi: 10.1161/01.RES.58.1.96
Stanley, C. M., Gagnon, D. G., Bernal, A., Meyer, D. J., Rosenthal, J. J., and Artigas, P. (2015). Importance of the voltage dependence of cardiac Na/K ATPase isozymes. Biophys. J. 109, 1852–1862. doi: 10.1016/j.bpj.2015.09.015
Steiness, E., and Olesen, K. H. (1976). Cardiac arrhythmias induced by hypokalaemia and potassium loss during maintenance digoxin therapy. Br. Heart J. 38, 167–172. doi: 10.1136/hrt.38.2.167
Studer, R., Reinecke, H., Bilger, J., Eschenhagen, T., Bohm, M., Hasenfuss, G., et al. (1994). Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ. Res. 75, 443–453. doi: 10.1161/01.RES.75.3.443
Sweadner, K. J., Herrera, V. L., Amato, S., Moellmann, A., Gibbons, D. K., and Repke, K. R. (1994). Immunologic identification of Na+,K+-ATPase isoforms in myocardium. Isoform change in deoxycorticosterone acetate-salt hypertension. Circ. Res. 74, 669–678. doi: 10.1161/01.RES.74.4.669
Swift, F., Birkeland, J. A., Tovsrud, N., Enger, U. H., Aronsen, J. M., Louch, W. E., et al. (2008). Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase α2-isoform in heart failure. Cardiovasc. Res. 78, 71–78. doi: 10.1093/cvr/cvn013
Swift, F., Tovsrud, N., Enger, U. H., Sjaastad, I., and Sejersted, O. M. (2007). The Na+/K+-ATPase α2-isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc. Res. 75, 109–117. doi: 10.1016/j.cardiores.2007.03.017
Swift, F., Tovsrud, N., Sjaastad, I., Sejersted, O. M., Niggli, E., and Egger, M. (2010). Functional coupling of α2-isoform Na+/K+-ATPase and Ca2+ extrusion through the Na+/Ca2+-exchanger in cardiomyocytes. Cell Calcium. 48, 54–60. doi: 10.1016/j.ceca.2010.06.006
Teerlink, J. R., Jalaluddin, M., Anderson, S., Kukin, M. L., Eichhorn, E. J., Francis, G., et al. (2000). Ambulatory ventricular arrhythmias in patients with heart failure do not specifically predict an increased risk of sudden death. PROMISE (Prospective Randomized Milrinone Survival Evaluation) Investigators. Circulation 101, 40–46. doi: 10.1161/01.CIR.101.1.40
Thompson, P. D., Franklin, B. A., Balady, G. J., Blair, S. N., Corrado, D., Estes, N. A. III., et al. (2007). Exercise and acute cardiovascular events placing the risks into perspective: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism and the Council on Clinical Cardiology. Circulation 115, 2358–2368. doi: 10.1161/CIRCULATIONAHA.107.181485
Tomaselli, G. F., and Zipes, D. P. (2004). What causes sudden death in heart failure? Circ. Res. 95, 754–763. doi: 10.1161/01.RES.0000145047.14691.db
Undrovinas, A. I., Maltsev, V. A., and Sabbah, H. N. (1999). Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell. Mol. Life Sci. 55, 494–505. doi: 10.1007/s000180050306
Unwin, R. J., Luft, F. C., and Shirley, D. G. (2011). Pathophysiology and management of hypokalemia: a clinical perspective. Nat. Rev. Nephrol. 7, 75–84. doi: 10.1038/nrneph.2010.175
Urso, C., Brucculeri, S., and Caimi, G. (2015). Acid-base and electrolyte abnormalities in heart failure: pathophysiology and implications. Heart Fail. Rev. 20, 493–503. doi: 10.1007/s10741-015-9482-y
Venetucci, L. A., Trafford, A. W., O'Neill, S. C., and Eisner, D. A. (2008). The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 77, 285–292. doi: 10.1093/cvr/cvm009
Verdonck, F., Volders, P. G., Vos, M. A., and Sipido, K. R. (2003). Increased Na+ concentration and altered Na/K pump activity in hypertrophied canine ventricular cells. Cardiovasc. Res. 57, 1035–1043. doi: 10.1016/S0008-6363(02)00734-4
Volders, P. G., Vos, M. A., Szabo, B., Sipido, K. R., de Groot, S. H., Gorgels, A. P., et al. (2000). Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovasc. Res. 46, 376–392. doi: 10.1016/S0008-6363(00)00022-5
Wagner, S., Hacker, E., Grandi, E., Weber, S. L., Dybkova, N., Sossalla, S., et al. (2009). Ca/calmodulin kinase II differentially modulates potassium currents. Circ. Arrhythm. Electrophysiol. 2, 285–294. doi: 10.1161/CIRCEP.108.842799
Wang, Y., and Hill, J. A. (2010). Electrophysiological remodeling in heart failure. J. Mol. Cell. Cardiol. 48, 619–632. doi: 10.1016/j.yjmcc.2010.01.009
Wasserstrom, J. A., and Aistrup, G. L. (2005). Digitalis: new actions for an old drug. Am. J. Physiol. Heart Circ. Physiol. 289, H1781–H1793. doi: 10.1152/ajpheart.00707.2004
Weiss, J. N., Garfinkel, A., Karagueuzian, H. S., Chen, P. S., and Qu, Z. (2010). Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm 7, 1891–1899. doi: 10.1016/j.hrthm.2010.09.017
Weiss, J. N., Qu, Z., and Shivkumar, K. (2017). Electrophysiology of hypokalemia and hyperkalemia. Circ. Arrhythm. Electrophysiol. 10:e004667. doi: 10.1161/CIRCEP.116.004667
Wester, P. O., and Dyckner, T. (1986). Intracellular electrolytes in cardiac failure. Acta Med. Scand. Suppl. 707, 33–36.
Williams, M. E., Gervino, E. V., Rosa, R. M., Landsberg, L., Young, J. B., Silva, P., et al. (1985). Catecholamine modulation of rapid potassium shifts during exercise. N. Engl. J. Med. 312, 823–827. doi: 10.1056/NEJM198503283121304
Wolk, R., Kane, K. A., Cobbe, S. M., and Hicks, M. N. (1998). Regional electrophysiological effects of hypokalaemia, hypomagnesaemia and hyponatraemia in isolated rabbit hearts in normal and ischaemic conditions. Cardiovasc. Res. 40, 492–501. doi: 10.1016/S0008-6363(98)00200-4
Yancy, C. W., Jessup, M., Bozkurt, B., Butler, J., Casey, D. E. Jr., Drazner, M. H., et al. (2013). 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 128, 1810–1852. doi: 10.1161/CIR.0b013e31829e8807
Yang, T., Snyders, D. J., and Roden, D. M. (1997). Rapid inactivation determines the rectification and [K+]o dependence of the rapid component of the delayed rectifier K+ current in cardiac cells. Circ. Res. 80, 782–789. doi: 10.1161/01.RES.80.6.782
Young, D. Z., Lampert, S., Graboys, T. B., and Lown, B. (1984). Safety of maximal exercise testing in patients at high risk for ventricular arrhythmia. Circulation 70, 184–191. doi: 10.1161/01.CIR.70.2.184
Yuen, G. K., Galice, S., and Bers, D. M. (2017). Subcellular localization of Na/K-ATPase isoforms in ventricular myocytes. J. Mol. Cell. Cardiol. 108:158–169. doi: 10.1016/j.yjmcc.2017.05.013
Zhang, T., Maier, L. S., Dalton, N. D., Miyamoto, S., Ross, J. Jr., Bers, D. M., et al. (2003). The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 92, 912–919. doi: 10.1161/01.RES.0000069686.31472.C5
Keywords: calcium, arrhythmia (heart rhythm disorders), Na+ - K+-ATPase, hypokalaemia, heart failure
Citation: Skogestad J and Aronsen JM (2018) Hypokalemia-Induced Arrhythmias and Heart Failure: New Insights and Implications for Therapy. Front. Physiol. 9:1500. doi: 10.3389/fphys.2018.01500
Received: 06 July 2018; Accepted: 05 October 2018;
Published: 07 November 2018.
Edited by:
Daniel M. Johnson, University of Birmingham, United KingdomReviewed by:
Fadi G. Akar, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2018 Skogestad and Aronsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonas Skogestad, am9uYXMuc2tvZ2VzdGFkQG1lZGlzaW4udWlvLm5v
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.