 Yuxin Chen
Yuxin Chen Zhongyang Zhou
Zhongyang Zhou Wang Min
Wang Min- 1Department of Laboratory Medicine, Nanjing Drum Tower Hospital, Nanjing University Medical School, Nanjing, China
- 2Center for Translational Medicine, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, China
- 3Department of Pathology and the Vascular Biology and Therapeutics Program, Yale School of Medicine, New Haven, CT, United States
Canonical functions of mitochondria include the regulation of cellular survival, orchestration of anabolic and metabolic pathways, as well as reactive oxygen species (ROS) signaling. Recent discoveries, nevertheless, have demonstrated that mitochondria are also critical elements to stimulate innate immune signaling cascade that is able to intensify the inflammation upon cytotoxic stimuli beyond microbial infection. Here we review the expanding research field of mitochondria and oxidative stress in innate immune system to highlight the new mechanistic insights and discuss the pathological relevance of mitochondrial dysregulation induced aberrant innate immune responses in a growing list of sterile inflammatory diseases.
Introduction
Mitochondria are multifunctional organelles that collaborate with their host cells in biosynthesis, metabolism, and cell death or survival functions. Recent growing evidence has highlighted expanded roles of mitochondria in modulating oxidative stress as well as innate immune responses. Since mitochondria is the major resource of reactive oxygen species (ROS) that emerges as a convergent signaling hub that regulates diverse developmental, environmental, and pathological stimuli. Along with mitochondrial dysfunction and excessive ROS observed in various disease conditions including cardiovascular diseases, autoimmune diseases, metabolic syndromes as well as tumor, aberrant innate immune responses were also identified, which is considered to be a common factor which drives the inflammatory pathology of these conditions. In this review, we present a comprehensive landscape regarding the role of mitochondria in innate immunity. We also detail recent advances in understanding novel function of mitochondria in inflammasome machinery, highlighting their emerging roles in driving the sterile inflammatory responses upon injured or damaged tissues and cells.
Mitochondrial Function and ROS
Mitochondria are ubiquitous multifunctional organelles with a double-membrane structure, which are present in majority of mammalian cell types. They are dynamic, branched networks with continuous cycles of fission and fusion (Mishra and Chan, 2014). Mitochondria bear the residual genome (mitochondrial DNA, mtDNA) that is essential for their activity of oxidative phosphorylation (OXPHOS) and a protein library containing with 1,200 proteins (Calvo and Mootha, 2010) that are varied substantially between cell and tissue types (Scarpulla, 2008). It is believed that mitochondrial biogenesis and homeostasis, including mtDNAs, are under tight regulation of nucleus. This needs bi-directional signaling pathways that mediate crosstalk between the nucleus and mitochondria (Shadel and Horvath, 2015). Such mitochondrial retrograde signaling pathways are highly conserved from prokaryotic to eukaryotic cells which potentially can trigger both favorable and maladaptive responses. Therefore, the number, morphology, distribution, and activity of mitochondria are constantly altered in respond to physiological, developmental, and environmental stimuli.
As the sites of the tricarboxylic acid (TCA) cycle and OXPHOS, mitochondria produce substantial ATP using the electrochemical gradient by the electron transport chain (ETC), whereas mitochondria could also produce ROS predominately at the ETC complex I and complex III (Murphy, 2009). ROS were initially considered to exert damage-promoting, detrimental effects; however, recent studies have depicted it as an emerging central signaling molecule. Therefore, beyond the traditional roles of mitochondria in metabolism such as glucose oxidation as well as biosynthesis of fatty acid, amino acid and hormones, mitochondria are also actively involved in ROS signaling, apoptosis and innate immunity (Shadel and Horvath, 2015).
Given its intrinsic complicated nature of mitochondria, mitochondrial dysfunction can induce distinct stress signals. For example, reduced OXPHOS and ETC activity can result in disturbed mitochondrial ROS (mtROS) production, eliminated mitochondrial membrane potential, or reduced cellular adenosine 5′-triphosphate (ATP) or energy (Butow and Avadhani, 2004; Sena and Chandel, 2012). As the major generator of ROS, mitochondria are also prone to become the target of ROS followed by pathological consequences. For example, elevated free radicals associated with mtDNA oxidative damage trigger cell apoptosis by inducing mitochondrial stress and downstream signaling (Scheibye-Knudsen et al., 2015; West et al., 2015). Finally, dynamic morphology and distribution of mitochondria within cells can also elicit distinct forms of stress that are associated with mitochondrial elimination by mitophagy or autophagy (Labbe et al., 2014). However, mitochondria contain potent anti-oxidant systems that protect mitochondria from ROS-mediated damages, including mitochondrial superoxide dismutase (SOD2) and the thioredoxin system made of thioredoxin-2 (Trx2), thioredoxin reductase-2 and peroxidase 3.
One of the most studied mitochondrial protein is Trx2, a small redox protein with double redox-active sites (C90 and C93). Trx2 is ubiquitously presented in tissues with high metabolic activity, including liver, brain and heart. Trx2 is able to maintain cell in a reduced state by reversible oxidation to Cys disulfide (Trx-S2) via the transfer of reducing equivalents from the catalytic site Cys residue to a disulfide protein substrate (protein-S2). By such way, Trx2 regulates a large number of apoptosis related molecules and critical transcription factors, such as apoptosis signal-regulating kinase 1 (ASK1) (Zhang et al., 2004) and nuclear factor kappa B (NF-κB) (Hansen et al., 2006). Trx2 depletion leads to cytosolic release of cytochrome c from mitochondria and subsequently activation of caspase-3 and -9. Moreover, Trx2 is able to attenuate tumor necrosis factor (TNF)-dependent elevated mtROS and apoptosis (Zhang et al., 2004; Hansen et al., 2006), suggesting that Trx2 modulates TNF-dependent redox signaling in mitochondria (Benhar et al., 2008). Trx2 not only protects against oxidative stress in mitochondria, but also induces the cells insensitive in response to ROS-induced apoptosis. In femoral artery ligation model, mitochondrial Trx2 in the endothelium cells is able to inhibit ASK1-induced apoptosis by elimination of ROS to increase nitric oxide (NO) bioavailability and inhibition of ASK1 activity (Zhang et al., 2004, 2007; Dai et al., 2009). Moreover, a similar role of Trx2 in cardiomyocytes was also identified. The absence of Trx2 in cardiomyocytes exhibits the disorganized mitochondrial arrays and swelling as well as impaired ATP generation, whereas ASK1 is required for Trx2 deletion triggered apoptosis, dysfunctional mitochondria, excessive ROS production observed in cardiomyocytes. Our findings in small animal model were further also validated in the clinical samples (Huang et al., 2015). Reduced Trx2 expression, elevated levels of phosphorylated ASK1 and activated caspase-3 were found in cardiomyocytes of patients with dilated cardiomyopathy (DCM) compared to that of healthy organ donors, indicating human Trx2 is also required to inhibit ASK1-dependent apoptosis signaling. Taken together, as exemplified by Trx2, mitochondrial proteins are capable to modulate cellular activities by maintaining cellular redox state and limiting ROS production.
Recent evidence has highlighted the essential role of mitochondria in activities of immune cells. Mitochondrial physiology, morphology, and metabolism tightly regulate immune cell fate during immune responses. For example, T cell development toward memory or effector phenotypes is tightly modulated by fission and fusion activities of mitochondria (Buck et al., 2016). OXPHOS and ROS production is required for T cell activation, while activated T cells can use either OXPHOS or glycolysis for proliferation. T cell mitochondrial dysfunction has been considered as a signature for infectious diseases and some autoimmune disease. During hepatitis B virus (HBV) infection, various cellular processes centered on mitochondrial activities and ROS were substantially downregulated in CD8 T cells, which might contribute to functional exhaustion of HBV CD8 T cells. Mitochondrion targeted antioxidants can elicit a notable improvement and restore anti-HBV CD8 function (Fisicaro et al., 2017). Mitochondrial hyperpolarization, ATP depletion and elevated ROS production has been observed from T cells from systemic lupus erythematosus (SLE) patients (Gergely et al., 2002), which might result from abnormal NO generation derived from monocytes (Nagy et al., 2003). CD4+ T cells from rheumatoid arthritis patients also exhibited elevated autophagy, ATP depletion, and impaired redox status (Yang et al., 2013, 2016). CD4+ T cells from multiple sclerosis patients displayed remarkable abundance in mitochondrial inner membrane lipid cardiolipin (Vergara et al., 2015). Potential therapeutic strategies include the inhibition of the mitochondrial oxidation and glycolytic rate to restore CD4+ T cell function in autoimmune diseases (Yin et al., 2015).
Beyond immune cells, mitochondrial dysfunction also extensively influences the function of non-immune cells. Taking endothelial cells for example, a representative mechanism that links to sterile inflammation is the so-called senescence associated secretory phenotype (SASP) (Tchkonia et al., 2013). It has been shown that upon mitochondrial dysfunction, endothelial cells secrete multiple pro-inflammatory cytokines includes interleukin (IL)-1, IL-6, and TNF-α, and upregulate intercellular adhesion molecule-1 (ICAM-1) expression which attracts monocyte activation and adhesion (Choi et al., 2018). Indeed, our recent unpublished data have also revealed that Trx2-deletion induced mitochondrial dysfunction also lead to endothelial cell senescence and SASP in vivo and in vitro, which might further recruit innate immune cells to augment inflammatory responses.
The Activation of Innate Immune Responses by Mitochondria-Derived ROS
Innate immunity provides a front line of host defense through direct engagement of pathogen or environmental insult, which further initiates the development of an adaptive immune response. However, the ability of innate immune system to resist a subsequent challenge with either the same or a different insult remains unaltered. Although the innate immune system lacks the fine specificity of adaptive immunity that is necessary to produce immunological memory, it can distinguish self from non-self by pattern recognition receptors (PRRs), germline-encoded receptors located at the cell surface and within endosomes (Kawai and Akira, 2006), serving as a sensor to monitor signs of infection or tissue injury. Such evolutionarily ancient PRRs sense a wide range of exogenous pathogen-associated molecular patterns (PAMPs) and endogenous danger-associated molecular patterns (DAMPs). DAMPs include ROS, heat shock proteins, oxidized lipoproteins, and cholesterol crystals. DAMPs could trigger sterile inflammation by binding with PPRs such as Toll-like receptors (TLRs), nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), retinoic acid–inducible gene (RIG)-I-like receptors (RLRs), and absent in Melanoma (AIM) 2-like receptors (ALRs) (Sellge and Kufer, 2015). Engagement of these receptors further recruit immune cells to the site of infection or injury in an attempt to clear pathogens or amply inflammatory responses within the host.
The innate immune receptors might also modulate OXPHOS, mitochondrial function (Figure 1), ROS and apoptosis present in tissue injury. For example, NLR family member X1 (NLRX1) is a designated innate immune receptor localized in mitochondria. During ischemia-reperfusion injury, NLRX1 is capable to protect against mortality by attenuating mitochondrial impairment and oxidative stress-induced apoptosis of epithelial cells (Stokman et al., 2017). By this way, the innate immune receptor could also control the metabolism of epithelial cells under cytotoxic stress.
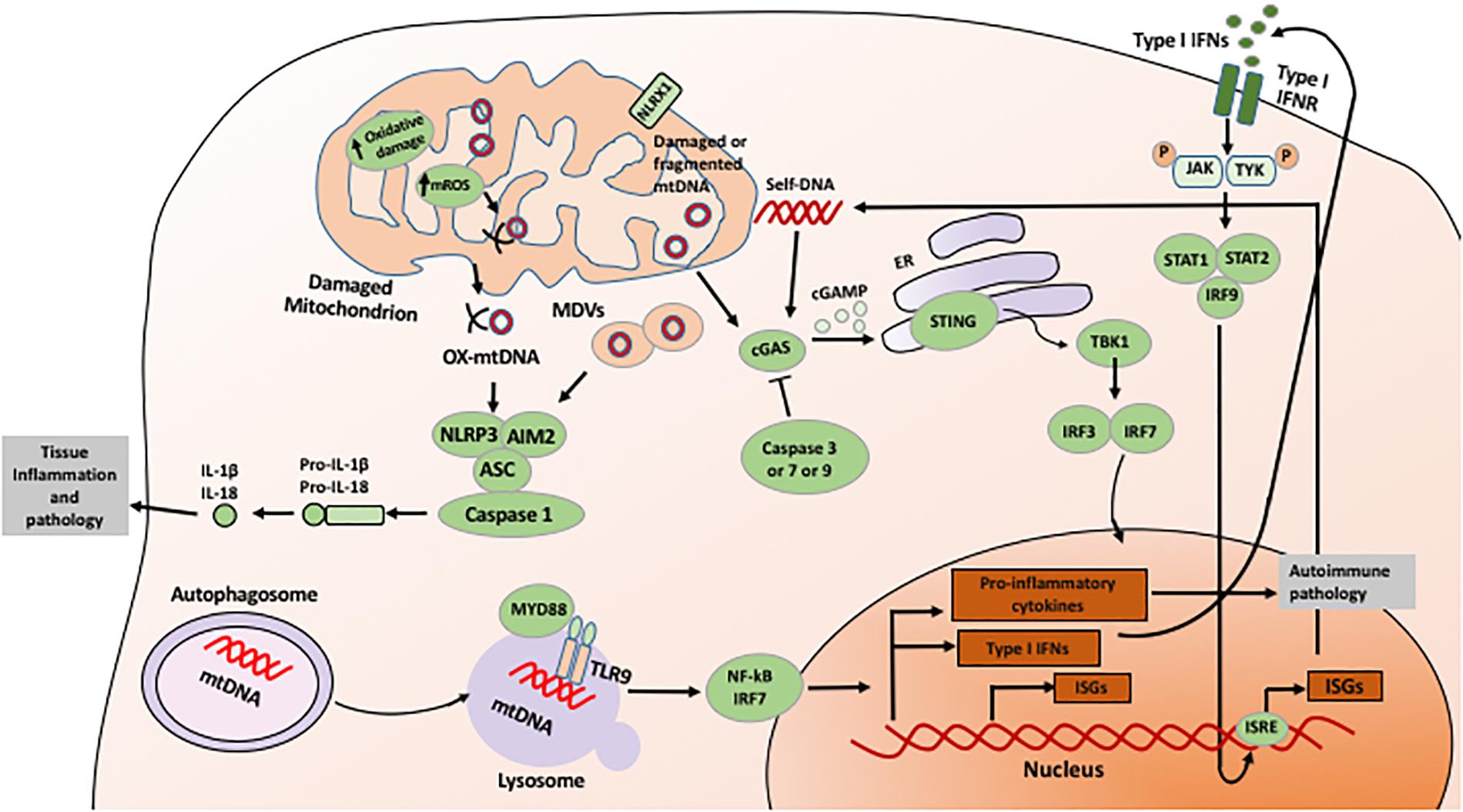
FIGURE 1. Mechanism of mitochondria-mediated signaling in innate immunity. Mitochondria-derived ROS and released mtDNA directly induces activation of innate immune responses, including activation of inflammasome, sGAS-STING, and NF-kB signaling pathways. See text for details.
An essential member of downstream cytosolic surveillance is the inflammasome, a large multimolecular complex that intensifies the inflammation when sensing microbial components and endogenous danger signals (Kanneganti et al., 2007; Takeuchi and Akira, 2010). The activated inflammasome modulates the proteolytic enzyme caspase-1, result in the maturation of proinflammatory cytokines, IL-1ß and IL-18 (Martinon et al., 2002; Kayagaki et al., 2011). To date, at least two major types of inflammasomes were identified, NLR families and the ALR families (Lamkanfi et al., 2007). NLRs, including the NLRP1, NLRP3 and NLR family caspase recruitment domain (CARD) domain containing 4 (NLRC4) inflammasomes, are characterized by a central located NOD domain that is flanked at N-terminal CARD, pyrin domain (PYD), acidic transactivating domain, or baculovirus inhibitor repeat (BIR) (Franchi et al., 2009; Sutterwala et al., 2014; Tschope et al., 2017). These motifs are capable to bound NLRs with adapter proteins and effectors such as apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1. C-terminal of NLRs contain leucine-rich repeat (LRR) motifs that regulate NLR activity (Lamkanfi et al., 2007). AIM2 inflammasome, including AIM2 and interferon-gamma inducible factor 16 (IFI16), contains a prototypical DNA binding HIN200 domain and an amino-terminal pyrin motif, through which AIM2 recruits ASC and caspase-1.
The assembly and activation of these inflammasomes are dependent on specific pathogens and endogenous insults (Kanneganti, 2010). For example, the NLRC4 inflammasome is activated in response to bacterial infection such as Salmonella typhimurium (Zhao et al., 2011). The NLRP1b and NLRP12 inflammasomes are triggered by anthrax lethal toxin and Y. pestis infection, respectively (Levinsohn et al., 2012; Vladimer et al., 2012). NLRP6 is widely present in the intestine that is critical to maintain the intestinal homeostasis (Elinav et al., 2011), whereas NLPR7 inflammasome has been shown to recognize diacylated lipopeptides in human macrophages (Khare et al., 2012). Compared to the NLR family members mentioned above, NLRP3 inflammasome is the most well characterized inflammasome. NLRP3 inflammasome is activated by diverse stimuli including both RNA and DNA viruses, presence of cytosolic bacterial RNA during infection, injury-induced stress molecules such as ROS, ATP and the release of mitochondrial DNA, and harmful environmental substances such as silica and asbestos, K+ efflux, and lysosomal destabilization (Piccini et al., 2008; Martinon et al., 2009; Broz et al., 2010; Franchi et al., 2012). Activation of NLRP3 requires two-step signals: firstly priming with either TLR or NLR ligands to enhance NF-κB-driven transcriptional level of NLRP3, and subsequently exposing to microbial toxins and ionophores or endogenous alarmins to trigger inflammasome assembly (Tschopp and Schroder, 2010). Meanwhile, during microbial infection of macrophages, two-step signaling required by NLRP3 inflammasome activation may occur simultaneously (Tschopp and Schroder, 2010). Interestingly, although adenoviral DNA triggers assembly of the NLRP3 inflammasome activation, upon exposure with transfected non-viral cytosolic DNA, caspase-1 activation and IL-1β secretion, however, was dependent on ASC rather than NLRP3 (Muruve et al., 2008). Given such diverse stimuli NLRP3 recognizes, it is believed that the NLRP3 inflammasome was able to recognize cytosolic nucleic acids and other endogenous danger signals indirectly. Indeed, it has been suggested that NLRP3 activation might be triggered by perturbed cell membrane (Barlan et al., 2011).
Given diverse stimuli converge on the activation of NLRP3 including ROS, here we hypothesize that ROS might be the direct mediator to trigger NLRP3. Crystal structural analysis of NLRP3 has identified an unexpected, however, highly conserved disulfide bond between the PYD motif and the nucleotide-binding site motif, that is super sensitive to disturbed redox status (Bae and Park, 2011), indicating a crucial redox role for NLRP3. Indeed, ROS is identified as a potent endogenous ligand for NLRP3, since the early reports has demonstrated that the inhibition of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-induced ROS inhibited NLRP3 activation in macrophages treated with ATP (Cruz et al., 2007); meanwhile, the absence of the p22phox subunit within NADPH oxidase substantially attenuated IL-1β production when macrophages exposed with asbestos (Dostert et al., 2008). In addition to NADPH oxidase-derived ROS, mtROS has also demonstrated to elevate NLRP3 activation (Bulua et al., 2011; Zhou et al., 2011; Wu et al., 2013). Interestingly, NLRP3 is present in the cytosol and endoplasmic reticulum (ER) during rest state but is migrated to mitochondria upon activation, which is mediated by the association of mitochondrial anti-viral signaling protein (MAVS) (Subramanian et al., 2013). Specific inhibition of mtROS, instead of the inhibition of NADPH oxidase, was able to prevent inflammasome-dependent IL-1β expression triggered by cyclic stretch in alveolar macrophage (Wu et al., 2013).
The Activation of Innate Immune Responses by Mitochondria-Derived DNA
The innate immune system is ultrasensitive to dsDNA. Cellular sources of immune stimulatory self-DNA include nucleus DNA, mtDNA, and DNA from phagolysosomal compartments. Mitochondria in most cells have approximately 2–10 copies of mtDNA, characterized as circular, closed, dsDNA with size of 15,000 base pairs (Kukat and Larsson, 2013). Upon mitochondrial stress induced apoptosis, Bcl2 antagonist/killer (BAK) and Bcl2-associated X protein (BAX) induce permeabilization of mitochondrial outer-membrane (MOMP), leading to cytochrome c release and activation of apoptotic caspases. During this process, mitochondrial network is compromised and newly appeared BAK/BAX macropores allows mitochondrial matrix component containing mtDNA leakage into the cytosol. Similar to NLR inflammasomes, ALR inflammasomes also induce caspase-1 activation and IL-1β cytokine maturation. However, AIM2 inflammasome has been identified as a cytosolic receptor for DNA, including intracellular bacteria F. tularensis and mouse cytomegalovirus (Fernandes-Alnemri et al., 2010); while AIM-2 related inflammasome, IFI16 is in the nucleus, which has also implicated in forming an inflammasome complex during herpes simplex virus infection of endothelial cells (Kerur et al., 2011). Contrast with NLR inflammasomes, ALR inflammasomes is able to bind their ligand directly, dsDNA. Further, AIM2 and IFI16 lack CARD domains and recruitment of ASC is required for their activation. Of note, AIM2 is the first inflammasome where a direct receptor:ligand interaction has been formally demonstrated (Fernandes-Alnemri et al., 2009; Hornung et al., 2009). Intriguingly, AIM2 recognizes cytosolic DNA with a minimum length of 80 base pairs in a sequence-dependent fashion (Rathinam et al., 2012). When DNA binding with its C-terminal HIN200 domain, AIM2 undergoes oligomerization and further engages caspase-1 via ASC and secretes IL-1β. In Aim2-deficient mice, it has been shown that the AIM2 inflammasome plays a non-redundant role to induce defense responses against DNA viruses and intracellular bacterial infections (Fernandes-Alnemri et al., 2010). Moreover, AIM2 contributes to the adjuvanticity of DNA vaccines (Suschak et al., 2015) and enhance autoimmune disorders such as SLE via host DNA recognition (Panchanathan et al., 2011). During Nelfinavir treatment, an HIV aspartyl protease, AIM2 could also be activated upon DNA release from the nucleus with a compromised nuclear envelope integrity (Di Micco et al., 2016). Until recently, there is a few reports suggesting a role of AIM2 in caspase-1 activation trigger by mtDNA. The depletion of AIM2 in bone marrow-derived macrophages (BMDMs) leads to reduced IL-1β with mtDNA transfection compared to control BMDMs (Nakahira et al., 2011). A recent study elegantly demonstrated that cholesterol overload causes impaired mitochondrial metabolism and mtDNA release, which further triggers AIM2 inflammasome in activated BMDMs (Dang et al., 2017).
NLRP3 inflammasome could also been directly activated by mtDNA. This was first identified by the observation that the autophagic proteins were able to regulate NLRP3-dependent inflammation by maintaining mitochondrial integrity (Nakahira et al., 2011). Upon stimulation of lipopolysaccharide (LPS) and ATP, the deficiency of autophagy proteins beclin 1 and LC3B in BMDMs leads to dysfunctional mitochondrial and cytosolic translocation of mtDNA. The later event was strictly relied on NALP3 inflammasome activation and mtROS, ultimately enhancing IL-1β and IL-18 secretion. A subsequent study by Shimada et al. (2012) suggested that the essential role of mROS during the process of NLRP3-dependent cytosolic release of mtDNA might be contributed by the intrinsic characteristics of NLRP3, which preferentially binds oxidized mtDNA and further stabilizes it in the cytoplasm after release.
Cyclic GMP-AMP synthase (cGAS) is a prominent cytosolic DNA sensor (Gao et al., 2013). The nucleotidyl transferase enzyme cGAS detects cytoplasmic DNA and produces the second-messenger, cyclic AMP-GMP (cGAMP), which is associated with and subsequently activates stimulator of interferon gene (STING) (Chen et al., 2016). Activated STING is able to recruit TANK-binding kinase 1 (TBK1), which phosphorylates interferon regulatory factor 3 (IRF3) to enhance its homodimerization and migrate into the nucleus, followed by induction of interferon β (IFNβ) and interferon-stimulated genes (ISGs) (Schoggins et al., 2014; Storek et al., 2015; Watson et al., 2015). Despite cGAS-STING signaling pathway has considered as a major defense mechanism against microbial infection, it is also essential to orchestrate type I interferon (IFN) and proinflammatory responses to self-DNA, driving type I IFN induction and ultimately resulting in autoimmune responses as well as antitumor immunity (Barber, 2015; Crow and Manel, 2015; Gray et al., 2015; Roers et al., 2016). Genetic studies in humans have demonstrated that mutations in DNA nucleases such as three prime repair exonuclease 1 (Trex1) lead to cytosolic DNA accumulation which activates the cGAS–STING pathway (Rice et al., 2015). With exposure of genotoxic stress, the collapsed micronuclear envelope help cGAS get access to the damaged nuclear DNA followed by activation of cGAS–STING pathway (Harding et al., 2017; Mackenzie et al., 2017). Interesting enough, it has been demonstrated that mtDNA leaked into the cytosol can activate the cGAS–STING pathway and type I IFN production in the absence of active caspases (West et al., 2015; McArthur et al., 2018).
Here comes an interesting question: since there are billions of cells that undergo apoptosis per day in our body, during which DNA is released into cytosol, how the immune system effectively keep immunologically silent from apoptosis triggered stimuli? A recent unexpected finding is that mitochondria and downstream caspases can determine the immunological status of cell death (Rongvaux et al., 2014). It has long been considered that the highly regulated caspase-dependent apoptosis is immunologically silent, in which the effector caspases including caspase-3, -7, and -9, were necessary to inhibit mtDNA triggered activation of STING (White et al., 2014; West et al., 2015). In contrast, cell death independent of caspase provokes an inflammatory response through releasing DAMPs into the local microenvironment. The activation of such DAMPs further recruits inflammatory cells such as granulocytes, monocytes, and macrophages. Specifically, without the presence of active caspases, Bax and Bak induced MOMP leads the activation of mitochondrial DNA-dependent cGAS–STING pathway followed by the potent induction of type I IFNs and a state of viral resistance. These unexpected mechanistic findings indicated an essential role of mitochondria and caspases, not only on the decision of the cell fate but also on the choice decease in an inflammatory or immunologically silent manner.
It worth mentioning that mtDNA uniquely affects the innate immunity distinct from NADPH oxidase-induced ROS and mtROS. NADPH oxidase-induced ROS serves as an alarm signal in the cytosol that induces efficient defense signal transduction pathways that use hydrogen peroxide as secondary messenger. Although NADPH oxidase-induced ROS could be partially scavenged by a versatile antioxidant system, it also results extensive cellular damage and necrosis. NADPH oxidase-induced ROS can exert oxidative damage on cellular proteins, lipids and nucleic acids, nevertheless, they are also crucial secondary messengers in innate immune responses. ROS is required for heightened sensing by innate immune receptors including TLRs, RLRs, and NLRP3 in an indirect manner via interaction with and modification of other molecules such as mtDNA (Banoth and Cassel, 2018). As reviewed above, mtROS directly contribute to inflammatory cytokine production and innate immune responses. mtROS can directly activate adenosine monophosphate-activated protein kinase (AMPK) and mitogen-activated protein kinase (MAPKs) (Bulua et al., 2011). Phosphorylation of their substrate can directly affect diverse metabolic pathways, regulation of gene expression by transcription factors, and direct activation or inhibition of specific target proteins including innate immune molecules such as RLRs (Tal et al., 2009) and NLRP3 (Zhou et al., 2011). In the absence of any mtROS, mtDNA upon releasing into cytoplasm, extracellular space or circulation during cell death or mitochondrial damage could be sensed by multiple PRRs including cGAS-STING, TLR9, NLRP3, NLRC4, and AIM2 in cell-type and context-dependent manners to trigger pro-inflammatory and type I IFN responses.
Mitochondria-Mediated Innate Immune Responses in the Context of Sterile Inflammation Related Diseases
Recent studies have illustrated a deleterious role of innate immunity in sterile inflammation, which we have summarized in Table 1. Tissue injury would result in cell death accompanied with nucleus and mitochondrial DNA damages. Recently, AIM2 has been shown to be involved in ionizing radiation induced hematopoietic syndrome and severe injury to gastrointestinal (GI) tract. During this process, AIM2 is capable to recognize nucleus DNA damage triggered by radiation and caspase-1 dependent death of bone marrow cells and intestinal epithelial cells (Hu et al., 2016). Blockage of AIM2 inflammasome activity might be an effective therapeutic regimen especially for cancer patients suffering from hematopoietic or GI toxicity. Similarly, using permanent coronary ligation after myocardial infarction (MI) in mice, King et al. (2017) reported a harmful role of IRF3-dependent innate immune response on ventricular remodeling, which supports the key molecules in innate immunity as a new target for protection against MI. Mechanistically, during MI a great number of dying ischemic cell from the heart catastrophically releases large quantities of DAMPs, such as self-DNA, that trigger the innate immune response. Phagocytic macrophages in the heart trigger a fatal response against MI by sensing DNA via cGAS, followed by IRF3-IFN signaling axis activation. Secreted type I IFNs is able to further amplify the response through ISGs by diffusing to the local microenvironment and signaling to cells with interferon-α/β receptor (IFNAR). Indeed, in contrast to wild type (WT) mice after MI, mice with genetic deficiency of cGAS, IRF3, or IFNAR suffer less ventricular dilation and rupture, greater contractile ability and improved survival time. Further, in cardiomyocytes, mitochondrial DNA that escapes degradation specifically activate TLR9-mediated inflammatory response, resulting in exacerbated heart failure (Oka et al., 2012). These studies elegantly demonstrate the molecular inflammation events during pathogenesis of myocardial disease.
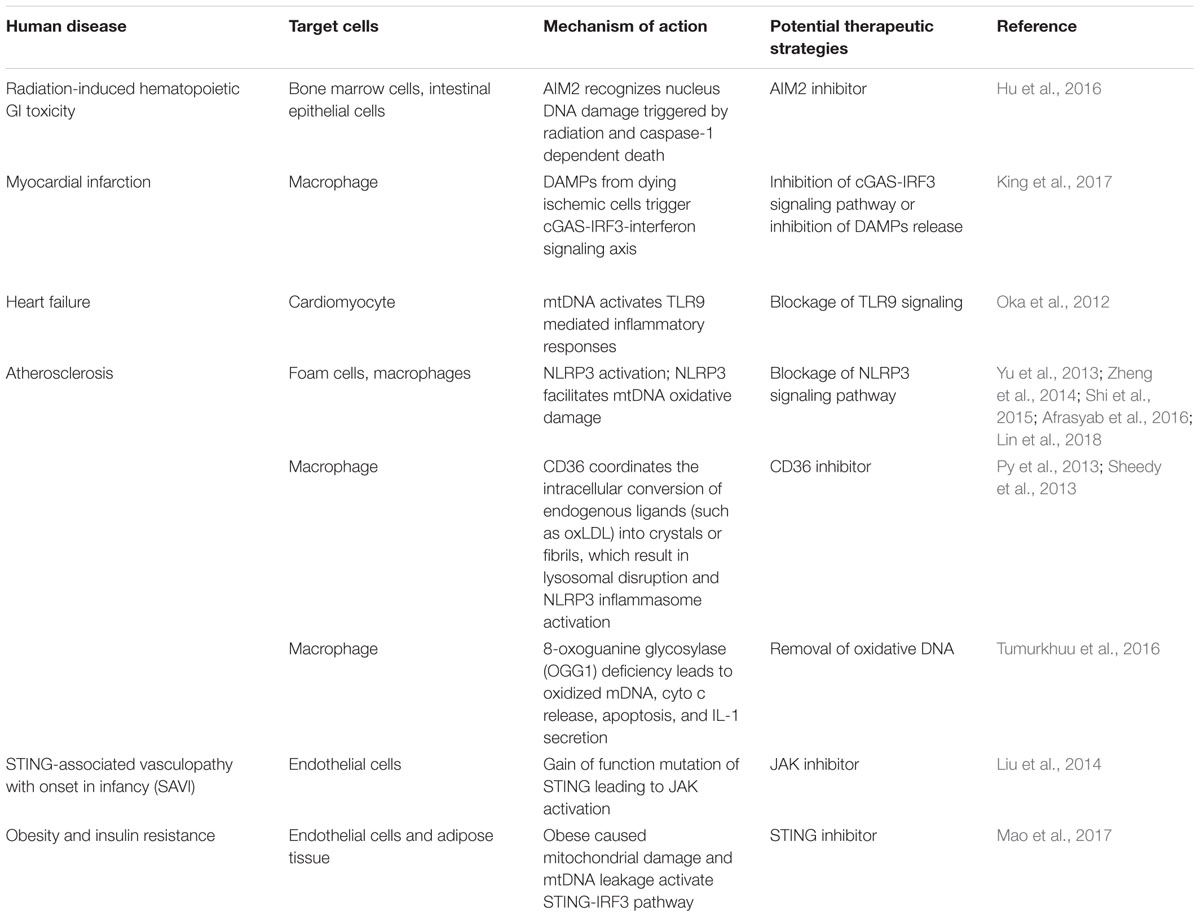
TABLE 1. Summary of sterile inflammation diseases triggered by mitochondrial dysregulation-induced aberrant innate immune responses and potential therapeutic strategies.
During early atherosclerosis, endothelial cells are activated by the elevation of ATP synthesis-uncoupled but proton leak-coupled mtROS without causing mitochondrial damage and EC death (Li et al., 2016, 2017), highlighting a novel regulatory network triggered by mtROS among mitochondrial metabolism, physiological EC activation, patrolling cell migration, and pathological inflammation. This suggests that mitochondrial antioxidants are promising therapies for vascular inflammation and cardiovascular diseases. Indeed, during the progression of atherosclerosis, excessive oxidative stress, dysfunctional mitochondria (Victor et al., 2009), ER stress (Chistiakov et al., 2014), and lysosome rupture (Yuan et al., 2000) are considered as one of the critical driver responsible for inflammasome activation. Clinical evidence has shown that the level of NLRP3 from peripheral blood monocyte is highly associated with the severity of coronary atherosclerosis in patients (Afrasyab et al., 2016), especially NLRP3 inflammasome has been observed to localize in the cytoplasm of foam cells and macrophages (Shi et al., 2015). Blockage of NLRP3 signaling inhibits the progression of atherosclerosis in apolipoprotein E-deficient (ApoE−/−) mice treated with a high-fat diet. NLRP3 knockdown also reduces macrophages and lipids while increases smooth muscle cells and collagen deposition of the plaque, contributing to plaque stabilization (Zheng et al., 2014). Interestingly, hydrogen sulfide which has shown to have anti-oxidative properties is able to also attenuate oxidative stress induced NLRP3 inflammasome activation via S-sulfhydrating c-Jun in macrophages (Lin et al., 2018). The priming event of NLRP3 is initiated by the cooperation of CD36 and a heterodimer of TLR4–TLR6 to convert intracellular ligands to crystals or fibrils followed by lysosomal disruption and NLRP3 activation (Sheedy et al., 2013). CD36 is an archetypal PRR which has been involved in the pathogenesis of atherosclerosis via modified endogenous danger signals such as oxidized low density lipoprotein (LDL) (oxLDL). CD36−/−ApoE−/− mice had a reduced serological IL-1β and plaque cholesterol crystal accumulation. TLR and ROS signaling can increase the levels of NLRP3 via BRCA1/BRCA2-containing complex subunit 3 (BRCC3) which mediates deubiquitination of the LRR domain of NLRP3 and such modification is essential for its activation (Py et al., 2013). NLRP3 might also facilitate atherosclerosis via sensing oxidative mtDNA (Tumurkhuu et al., 2016). Increased mtDNA damage has been indicated in human atherosclerotic plaques compared to normal vessels. Further, leukocyte mtDNA damage was associated with higher-risk plaques in humans (Yu et al., 2013). 8-oxoguanine glycosylase (OGG1) is one of the major DNA glycosylase that is responsible for removing oxidative DNA. Ogg1 deficiency in atherosclerotic mice induces larger plaque and greater amount of lipid content, while both knockout of Ogg1 and NLRP3 rescue the enhanced atherosclerosis observed in Ogg1−/− mice. Specifically, Ogg1−/− macrophages showed increased oxidized mtDNA, cytochrome c, apoptosis, and IL-1 secretion. All these studies highlight NLRP3 as a potential therapeutic target for atherosclerosis (Tumurkhuu et al., 2016).
Chronic inflammation of endothelial cells initiates cardiovascular disease (Liao, 2013). Mitochondrial damage induced by palmitic acid and mitochondrial DNA release activates the STING–IRF3 pathway, which further trigger ICAM-1 expression and endothelial inflammation. Clinical evidence also identified STING-associated vasculopathy with onset in infancy (SAVI), an autoinflammatory disease result from gain-of-function mutations of STING (Liu et al., 2014). With the identified STING expression, endothelial cells exposed with cGAMP, the STING ligand, result in cellular activation indicated by elevated inducible nitric oxide synthase (iNOS) and E-selectin followed by apoptosis. Since mutant STING increased level of phosphorylated signal transducer and activator of transcription 1 (STAT1), the elucidation of such mechanism also suggests Janus kinase (JAK) inhibitor as a potential therapeutic strategy for SAVI, which is currently under clinical assessment. STING pathway is also involved in adipose tissue inflammation and insulin resistance. In obese mice triggered by high-fat diet, STING was found in adipose tissue and was actively involved in tissue inflammation and insulin resistance (Mao et al., 2017).
Conclusion
Mitochondria are not only house machineries that support cellular essential activities, but also important sources of endogenous DAMPs including ROS as well as necessary triggers for inflammasome signaling. A large number of evidence has emerged linking dysfunctional mitochondria to aberrant innate immune responses. Nevertheless, our understanding of precise roles the inflammasomes in response to mitochondrial malfunction and ROS are still lacking. Many significant questions regarding the molecular machineries which initiate inflammasome activation upon mitochondria disorder and ROS remain to be addressed. Further elucidation of the interplay of ROS, mitochondrial function and inflammasome pathways might open up a new horizon for the development of immunotherapeutic strategy for chronic inflammation diseases such as cardiovascular diseases.
Author Contributions
YC, ZZ, and WM wrote the paper.
Funding
This work was supported by National Natural Science Foundation of China (Nos. 91539110, U1601219, and 81600201). This work was also supported by NIH grants R01 HL136507, HL109420, and HL115148.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Afrasyab, A., Qu, P., Zhao, Y., Peng, K., Wang, H., Lou, D., et al. (2016). Correlation of NLRP3 with severity and prognosis of coronary atherosclerosis in acute coronary syndrome patients. Heart Vessels 31, 1218–1229. doi: 10.1007/s00380-015-0723-8
Bae, J. Y., and Park, H. H. (2011). Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J. Biol. Chem. 286, 39528–39536. doi: 10.1074/jbc.M111.278812
Banoth, B., and Cassel, S. L. (2018). Mitochondria in innate immune signaling. Transl. Res. doi: 10.1016/j.trsl.2018.07.014 [Epub ahead of print].
Barber, G. N. (2015). STING: infection, inflammation and cancer. Nat. Rev. Immunol. 15, 760–770. doi: 10.1038/nri3921
Barlan, A. U., Griffin, T. M., McGuire, K. A., and Wiethoff, C. M. (2011). Adenovirus membrane penetration activates the NLRP3 inflammasome. J. Virol. 85, 146–155. doi: 10.1128/JVI.01265-10
Benhar, M., Forrester, M. T., Hess, D. T., and Stamler, J. S. (2008). Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 320, 1050–1054. doi: 10.1126/science.1158265
Broz, P., Newton, K., Lamkanfi, M., Mariathasan, S., Dixit, V. M., and Monack, D. M. (2010). Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 207, 1745–1755. doi: 10.1084/jem.20100257
Buck, M. D., O’Sullivan, D., Klein Geltink, R. I., Curtis, J. D., Chang, C. H., Sanin, D. E., et al. (2016). Mitochondrial dynamics controls T Cell fate through metabolic programming. Cell 166, 63–76. doi: 10.1016/j.cell.2016.05.035
Bulua, A. C., Simon, A., Maddipati, R., Pelletier, M., Park, H., Kim, K. Y., et al. (2011). Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208, 519–533. doi: 10.1084/jem.20102049
Butow, R. A., and Avadhani, N. G. (2004). Mitochondrial signaling: the retrograde response. Mol. Cell. 14, 1–15. doi: 10.1016/S1097-2765(04)00179-0
Calvo, S. E., and Mootha, V. K. (2010). The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 11, 25–44. doi: 10.1146/annurev-genom-082509-141720
Chen, Q., Sun, L., and Chen, Z. J. (2016). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149. doi: 10.1038/ni.3558
Chistiakov, D. A., Sobenin, I. A., Orekhov, A. N., and Bobryshev, Y. V. (2014). Role of endoplasmic reticulum stress in atherosclerosis and diabetic macrovascular complications. Biomed. Res. Int. 2014:610140. doi: 10.1155/2014/610140
Choi, S. J., Piao, S., Nagar, H., Jung, S. B., Kim, S., Lee, I., et al. (2018). Isocitrate dehydrogenase 2 deficiency induces endothelial inflammation via p66sh-mediated mitochondrial oxidative stress. Biochem. Biophys. Res. Commun. 503, 1805–1811. doi: 10.1016/j.bbrc.2018.07.117
Crow, Y. J., and Manel, N. (2015). Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol. 15, 429–440. doi: 10.1038/nri3850
Cruz, C. M., Rinna, A., Forman, H. J., Ventura, A. L., Persechini, P. M., and Ojcius, D. M. (2007). ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 282, 2871–2879. doi: 10.1074/jbc.M608083200
Dai, S., He, Y., Zhang, H., Yu, L., Wan, T., Xu, Z., et al. (2009). Endothelial-specific expression of mitochondrial thioredoxin promotes ischemia-mediated arteriogenesis and angiogenesis. Arterioscler. Thromb Vasc. Biol. 29, 495–502. doi: 10.1161/ATVBAHA.108.180349
Dang, E. V., McDonald, J. G., Russell, D. W., and Cyster, J. G. (2017). Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell 171, 1057.e11–1071.e11. doi: 10.1016/j.cell.2017.09.029
Di Micco, A., Frera, G., Lugrin, J., Jamilloux, Y., Hsu, E. T., Tardivel, A., et al. (2016). AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc. Natl. Acad. Sci. U.S.A. 113, E4671–E4680. doi: 10.1073/pnas.1602419113
Dostert, C., Petrilli, V., Van Bruggen, R., Steele, C., Mossman, B. T., and Tschopp, J. (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677. doi: 10.1126/science.1156995
Elinav, E., Strowig, T., Kau, A. L., Henao-Mejia, J., Thaiss, C. A., Booth, C. J., et al. (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757. doi: 10.1016/j.cell.2011.04.022
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J., and Alnemri, E. S. (2009). AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513. doi: 10.1038/nature07710
Fernandes-Alnemri, T., Yu, J. W., Juliana, C., Solorzano, L., Kang, S., Wu, J., et al. (2010). The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393. doi: 10.1038/ni.1859
Fisicaro, P., Barili, V., Montanini, B., Acerbi, G., Ferracin, M., Guerrieri, F., et al. (2017). Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat Med. 23, 327–336. doi: 10.1038/nm.4275
Franchi, L., Munoz-Planillo, R., and Nunez, G. (2012). Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332. doi: 10.1038/ni.2231
Franchi, L., Warner, N., Viani, K., and Nunez, G. (2009). Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227, 106–128. doi: 10.1111/j.1600-065X.2008.00734.x
Gao, D., Wu, J., Wu, Y. T., Du, F., Aroh, C., Yan, N., et al. (2013). Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906. doi: 10.1126/science.1240933
Gergely, P. Jr., Niland, B., Gonchoroff, N., Pullmann, R. Jr., Phillips, P. E., and Perl, A. (2002). Persistent mitochondrial hyperpolarization, increased reactive oxygen intermediate production, and cytoplasmic alkalinization characterize altered IL-10 signaling in patients with systemic lupus erythematosus. J. Immunol. 169, 1092–1101. doi: 10.4049/jimmunol.169.2.1092
Gray, E. E., Treuting, P. M., Woodward, J. J., and Stetson, D. B. (2015). Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of aicardi-goutieres syndrome. J. Immunol. 195, 1939–1943. doi: 10.4049/jimmunol.1500969
Hansen, J. M., Zhang, H., and Jones, D. P. (2006). Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol. Sci. 91, 643–650. doi: 10.1093/toxsci/kfj175
Harding, S. M., Benci, J. L., Irianto, J., Discher, D. E., Minn, A. J., and Greenberg, R. A. (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. doi: 10.1038/nature23470
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., et al. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518. doi: 10.1038/nature07725
Hu, B., Jin, C., Li, H. B., Tong, J., Ouyang, X., Cetinbas, N. M., et al. (2016). The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 354, 765–768. doi: 10.1126/science.aaf7532
Huang, Q., Zhou, H. J., Zhang, H., Huang, Y., Hinojosa-Kirschenbaum, F., Fan, P., et al. (2015). Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation 131, 1082–1097. doi: 10.1161/CIRCULATIONAHA.114.012725
Kanneganti, T. D. (2010). Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 10, 688–698. doi: 10.1038/nri2851
Kanneganti, T. D., Lamkanfi, M., and Nunez, G. (2007). Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559. doi: 10.1016/j.immuni.2007.10.002
Kawai, T., and Akira, S. (2006). TLR signaling. Cell Death Differ. 13, 816–825. doi: 10.1038/sj.cdd.4401850
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. doi: 10.1038/nature10558
Kerur, N., Veettil, M. V., Sharma-Walia, N., Bottero, V., Sadagopan, S., Otageri, P., et al. (2011). IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375. doi: 10.1016/j.chom.2011.04.008
Khare, S., Dorfleutner, A., Bryan, N. B., Yun, C., Radian, A. D., de Almeida, L., et al. (2012). An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 36, 464–476. doi: 10.1016/j.immuni.2012.02.001
King, K. R., Aguirre, A. D., Ye, Y. X., Sun, Y., Roh, J. D., Ng, R. P., et al. (2017). IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23, 1481–1487. doi: 10.1038/nm.4428
Kukat, C., and Larsson, N. G. (2013). mtDNA makes a U-turn for the mitochondrial nucleoid. Trends Cell Biol. 23, 457–463. doi: 10.1016/j.tcb.2013.04.009
Labbe, K., Murley, A., and Nunnari, J. (2014). Determinants and functions of mitochondrial behavior. Annu. Rev. Cell Dev. Biol. 30, 357–391. doi: 10.1146/annurev-cellbio-101011-155756
Lamkanfi, M., Kanneganti, T. D., Franchi, L., and Nunez, G. (2007). Caspase-1 inflammasomes in infection and inflammation. J. Leukoc. Biol. 82, 220–225. doi: 10.1189/jlb.1206756
Levinsohn, J. L., Newman, Z. L., Hellmich, K. A., Fattah, R., Getz, M. A., Liu, S., et al. (2012). Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 8:e1002638. doi: 10.1371/journal.ppat.1002638
Li, X., Fang, P., Li, Y., Kuo, Y. M., Andrews, A. J., Nanayakkara, G., et al. (2016). Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arterioscler. Thromb. Vasc. Biol. 36, 1090–1100. doi: 10.1161/ATVBAHA.115.306964
Li, X., Fang, P., Yang, W. Y., Chan, K., Lavallee, M., Xu, K., et al. (2017). Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells. Can. J. Physiol. Pharmacol. 95, 247–252. doi: 10.1139/cjpp-2016-0515
Liao, J. K. (2013). Linking endothelial dysfunction with endothelial cell activation. J. Clin. Invest. 123, 540–541. doi: 10.1172/JCI66843
Lin, Z., Altaf, N., Li, C., Chen, M., Pan, L., Wang, D., et al. (2018). Hydrogen sulfide attenuates oxidative stress-induced NLRP3 inflammasome activation via S-sulfhydrating c-Jun at Cys269 in macrophages. Biochim. Biophys. Acta 1864(9 Pt B), 2890–2900. doi: 10.1016/j.bbadis.2018.05.023
Liu, Y., Jesus, A. A., Marrero, B., Yang, D., Ramsey, S. E., Sanchez, G. A. M., et al. (2014). Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 371, 507–518. doi: 10.1056/NEJMoa1312625
Mackenzie, K. J., Carroll, P., Martin, C. A., Murina, O., Fluteau, A., Simpson, D. J., et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. doi: 10.1038/nature23449
Mao, Y., Luo, W., Zhang, L., Wu, W., Yuan, L., Xu, H., et al. (2017). STING-IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler. Thromb Vasc. Biol. 37, 920–929. doi: 10.1161/ATVBAHA.117.309017
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 10, 417–426. doi: 10.1016/S1097-2765(02)00599-3
Martinon, F., Mayor, A., and Tschopp, J. (2009). The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265. doi: 10.1146/annurev.immunol.021908.132715
McArthur, K., Whitehead, L. W., Heddleston, J. M., Li, L., Padman, B. S., Oorschot, V., et al. (2018). BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359:eaao6047. doi: 10.1126/science.aao6047
Mishra, P., and Chan, D. C. (2014). Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 15, 634–646. doi: 10.1038/nrm3877
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Muruve, D. A., Petrilli, V., Zaiss, A. K., White, L. R., Clark, S. A., Ross, P. J., et al. (2008). The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452, 103–107. doi: 10.1038/nature06664
Nagy, G., Koncz, A., and Perl, A. (2003). T cell activation-induced mitochondrial hyperpolarization is mediated by Ca2 + - and redox-dependent production of nitric oxide. J. Immunol. 171, 5188–5197. doi: 10.4049/jimmunol.171.10.5188
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. doi: 10.1038/ni.1980
Oka, T., Hikoso, S., Yamaguchi, O., Taneike, M., Takeda, T., Tamai, T., et al. (2012). Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255. doi: 10.1038/nature10992
Panchanathan, R., Duan, X., Arumugam, M., Shen, H., Liu, H., and Choubey, D. (2011). Cell type and gender-dependent differential regulation of the p202 and Aim2 proteins: implications for the regulation of innate immune responses in SLE. Mol. Immunol. 49, 273–280. doi: 10.1016/j.molimm.2011.08.022
Piccini, A., Carta, S., Tassi, S., Lasiglie, D., Fossati, G., and Rubartelli, A. (2008). ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc. Natl. Acad. Sci. U.S.A. 105, 8067–8072. doi: 10.1073/pnas.0709684105
Py, B. F., Kim, M. S., Vakifahmetoglu-Norberg, H., and Yuan, J. (2013). Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell. 49, 331–338. doi: 10.1016/j.molcel.2012.11.009
Rathinam, V. A., Vanaja, S. K., and Fitzgerald, K. A. (2012). Regulation of inflammasome signaling. Nat. Immunol. 13, 333–342. doi: 10.1038/ni.2237
Rice, G. I., Rodero, M. P., and Crow, Y. J. (2015). Human disease phenotypes associated with mutations in TREX1. J. Clin. Immunol. 35, 235–243. doi: 10.1007/s10875-015-0147-3
Roers, A., Hiller, B., and Hornung, V. (2016). Recognition of endogenous nucleic acids by the innate immune system. Immunity 44, 739–754. doi: 10.1016/j.immuni.2016.04.002
Rongvaux, A., Jackson, R., Harman, C. C., Li, T., West, A. P., de Zoete, M. R., et al. (2014). Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577. doi: 10.1016/j.cell.2014.11.037
Scarpulla, R. C. (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88, 611–638. doi: 10.1152/physrev.00025.2007
Scheibye-Knudsen, M., Fang, E. F., Croteau, D. L., Wilson, D. M. III, and Bohr, V. A. (2015). Protecting the mitochondrial powerhouse. Trends Cell Biol. 25, 158–170. doi: 10.1016/j.tcb.2014.11.002
Schoggins, J. W., MacDuff, D. A., Imanaka, N., Gainey, M. D., Shrestha, B., Eitson, J. L., et al. (2014). Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695. doi: 10.1038/nature12862
Sellge, G., and Kufer, T. A. (2015). PRR-signaling pathways: Learning from microbial tactics. Semin. Immunol. 27, 75–84. doi: 10.1016/j.smim.2015.03.009
Sena, L. A., and Chandel, N. S. (2012). Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 48, 158–167. doi: 10.1016/j.molcel.2012.09.025
Shadel, G. S., and Horvath, T. L. (2015). Mitochondrial ROS signaling in organismal homeostasis. Cell 163, 560–569. doi: 10.1016/j.cell.2015.10.001
Sheedy, F. J., Grebe, A., Rayner, K. J., Kalantari, P., Ramkhelawon, B., Carpenter, S. B., et al. (2013). CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820. doi: 10.1038/ni.2639
Shi, X., Xie, W. L., Kong, W. W., Chen, D., and Qu, P. (2015). Expression of the nlrp3 inflammasome in carotid atherosclerosis. J. Stroke Cerebrovas. Dis. 24, 2455–2466. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.024
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414. doi: 10.1016/j.immuni.2012.01.009
Stokman, G., Kors, L., Bakker, P. J., Rampanelli, E., Claessen, N., Teske, G. J. D., et al. (2017). NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J. Exp Med. 214, 2405–2420. doi: 10.1084/jem.20161031
Storek, K. M., Gertsvolf, N. A., Ohlson, M. B., and Monack, D. M. (2015). cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J. Immunol. 194, 3236–3245. doi: 10.4049/jimmunol.1402764
Subramanian, N., Natarajan, K., Clatworthy, M. R., Wang, Z., and Germain, R. N. (2013). The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361. doi: 10.1016/j.cell.2013.02.054
Suschak, J. J., Wang, S., Fitzgerald, K. A., and Lu, S. (2015). Identification of Aim2 as a sensor for DNA vaccines. J. Immunol. 194, 630–636. doi: 10.4049/jimmunol.1402530
Sutterwala, F. S., Haasken, S., and Cassel, S. L. (2014). Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 1319, 82–95. doi: 10.1111/nyas.12458
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. doi: 10.1016/j.cell.2010.01.022
Tal, M. C., Sasai, M., Lee, H. K., Yordy, B., Shadel, G. S., and Iwasaki, A. (2009). Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 2770–2775. doi: 10.1073/pnas.0807694106
Tchkonia, T., Zhu, Y., van Deursen, J., Campisi, J., and Kirkland, J. L. (2013). Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 123, 966–972. doi: 10.1172/JCI64098
Tschope, C., Muller, I., Xia, Y., Savvatis, K., Pappritz, K., Pinkert, S., et al. (2017). NOD2 (Nucleotide-binding oligomerization domain 2) is a major pathogenic mediator of coxsackievirus B3-Induced Myocarditis. Circ. Heart Fail. 10:e003870. doi: 10.1161/CIRCHEARTFAILURE.117.003870
Tschopp, J., and Schroder, K. (2010). NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 10, 210–215. doi: 10.1038/nri2725
Tumurkhuu, G., Shimada, K., Dagvadorj, J., Crother, T. R., Zhang, W., Luthringer, D., et al. (2016). Ogg1-dependent DNA repair regulates NLRP3 inflammasome and prevents atherosclerosis. Circ. Res. 119, e76–e90. doi: 10.1161/CIRCRESAHA.116.308362
Vergara, D., D’Alessandro, M., Rizzello, A., De Riccardis, L., Lunetti, P., Del Boccio, P., et al. (2015). A lipidomic approach to the study of human CD4( + ) T lymphocytes in multiple sclerosis. BMC Neurosci. 16:46. doi: 10.1186/s12868-015-0183-1
Victor, V. M., Apostolova, N., Herance, R., Hernandez-Mijares, A., and Rocha, M. (2009). Oxidative stress and mitochondrial dysfunction in atherosclerosis: mitochondria-targeted antioxidants as potential therapy. Curr. Med. Chem. 16, 4654–4667. doi: 10.2174/092986709789878265
Vladimer, G. I., Weng, D., Paquette, S. W., Vanaja, S. K., Rathinam, V. A., Aune, M. H., et al. (2012). The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37, 96–107. doi: 10.1016/j.immuni.2012.07.006
Watson, R. O., Bell, S. L., MacDuff, D. A., Kimmey, J. M., Diner, E. J., Olivas, J., et al. (2015). The cytosolic sensor cGAS Detects Mycobacterium tuberculosis DNA to induce Type I interferons and activate autophagy. Cell Host Microbe. 17, 811–819. doi: 10.1016/j.chom.2015.05.004
West, A. P., Khoury-Hanold, W., Staron, M., Tal, M. C., Pineda, C. M., Lang, S. M., et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. doi: 10.1038/nature14156
White, M. J., McArthur, K., Metcalf, D., Lane, R. M., Cambier, J. C., Herold, M. J., et al. (2014). Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562. doi: 10.1016/j.cell.2014.11.036
Wu, J., Yan, Z., Schwartz, D. E., Yu, J., Malik, A. B., and Hu, G. (2013). Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J. Immunol. 190, 3590–3599. doi: 10.4049/jimmunol.1200860
Yang, Z., Fujii, H., Mohan, S. V., Goronzy, J. J., and Weyand, C. M. (2013). Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J. Exp. Med. 210, 2119–2134. doi: 10.1084/jem.20130252
Yang, Z., Shen, Y., Oishi, H., Matteson, E. L., Tian, L., Goronzy, J. J., et al. (2016). Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci. Transl. Med. 8:331ra338. doi: 10.1126/scitranslmed.aad7151
Yin, Y., Choi, S. C., Xu, Z., Perry, D. J., Seay, H., Croker, B. P., et al. (2015). Normalization of CD4 + T cell metabolism reverses lupus. Sci. Transl. Med. 7:274ra218. doi: 10.1126/scitranslmed.aaa0835
Yu, E., Calvert, P. A., Mercer, J. R., Harrison, J., Baker, L., Figg, N. L., et al. (2013). Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 128, 702–712. doi: 10.1161/CIRCULATIONAHA.113.002271
Yuan, X. M., Li, W., Brunk, U. T., Dalen, H., Chang, Y. H., and Sevanian, A. (2000). Lysosomal destabilization during macrophage damage induced by cholesterol oxidation products. Free Radic. Biol. Med. 28, 208–218. doi: 10.1016/S0891-5849(99)00220-8
Zhang, H., Luo, Y., Zhang, W., He, Y., Dai, S., Zhang, R., et al. (2007). Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am. J. Pathol. 170, 1108–1120. doi: 10.2353/ajpath.2007.060960
Zhang, R., Al-Lamki, R., Bai, L., Streb, J. W., Miano, J. M., Bradley, J., et al. (2004). Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ. Res. 94, 1483–1491. doi: 10.1161/01.RES.0000130525.37646.a7
Zhao, Y., Yang, J., Shi, J., Gong, Y. N., Lu, Q., Xu, H., et al. (2011). The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600. doi: 10.1038/nature10510
Zheng, F., Xing, S., Gong, Z., Mu, W., and Xing, Q. (2014). Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflamm. 2014:507208. doi: 10.1155/2014/507208
Keywords: mitochondria, innate and adaptive immune response, inflammasome, ROS, thioredoxin
Citation: Chen Y, Zhou Z and Min W (2018) Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 9:1487. doi: 10.3389/fphys.2018.01487
Received: 24 June 2018; Accepted: 02 October 2018;
Published: 18 October 2018.
Edited by:
Magnus Gram, Lund University, SwedenReviewed by:
Xiao-feng Yang, Temple University, United StatesHouzao Chen, Chinese Academy of Medical Sciences, China
Copyright © 2018 Chen, Zhou and Min. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuxin Chen, eXV4aW5fY2hlbjIwMTVAMTYzLmNvbQ== Wang Min, d2FuZy5taW5AeWFsZS5lZHU=