 Wenjia Li
Wenjia Li Lei Yin2
Lei Yin2 Junbo Ge
Junbo Ge Aijun Sun
Aijun Sun
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 09 October 2018
Sec. Cardiac Electrophysiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01372
The SCN5A gene encodes the alpha subunit of the main cardiac sodium channel Nav1.5. This channel predominates inward sodium current (INa) and plays a critical role in regulation of cardiac electrophysiological function. Since 1995, SCN5A variants have been found to be causatively associated with Brugada syndrome, long QT syndrome, cardiac conduction system dysfunction, dilated cardiomyopathy, etc. Previous genetic, electrophysiological, and molecular studies have identified the arrhythmic and cardiac structural characteristics induced by SCN5A variants. However, due to the variation of disease manifestations and genetic background, impact of environmental factors, as well as the presence of mixed phenotypes, the detailed and individualized physiological mechanisms in various SCN5A-related syndromes are not fully elucidated. This review summarizes the current knowledge of SCN5A genetic variations in different SCN5A-related cardiac disorders and the newly developed therapy strategies potentially useful to prevent and treat these disorders in clinical setting.
The SCN5A gene encodes the alpha subunit of the main cardiac sodium channel Nav1.5, which is known to be responsible for maintaining the normal function of inward sodium current (INa). INa current is the main component in fast depolarization phase after which the excitation–contraction coupling cascade and proper conduction of the electrical impulse is subsequently initiated within the heart (Aronsen et al., 2013).
Genetic variants of SCN5A are involved in a number of inherited cardiac channelopathies including Brugada syndrome (BrS), long QT syndrome (LQT3), and cardiac conduction system dysfunction. Meanwhile, SCN5A variants are also found to be correlated with myocardial contractile dysfunction, dilated cardiomyopathy, and heart failure. According to one of the most complete open database of on-line disease-related genetic variations “ClinVar” (ClinVar, 2018), more than 700 SCN5A variation locations are shown to be associated with cardiac disorders (Table 1) and about 90% variants account for non-synonymous variants, while the rest are due to deletion and duplication. With the help of patch clamp technique and transgenic animal technology, studies have clarified the major pathogenic mechanisms underlying sodium channelopathies due to SCN5A variants.
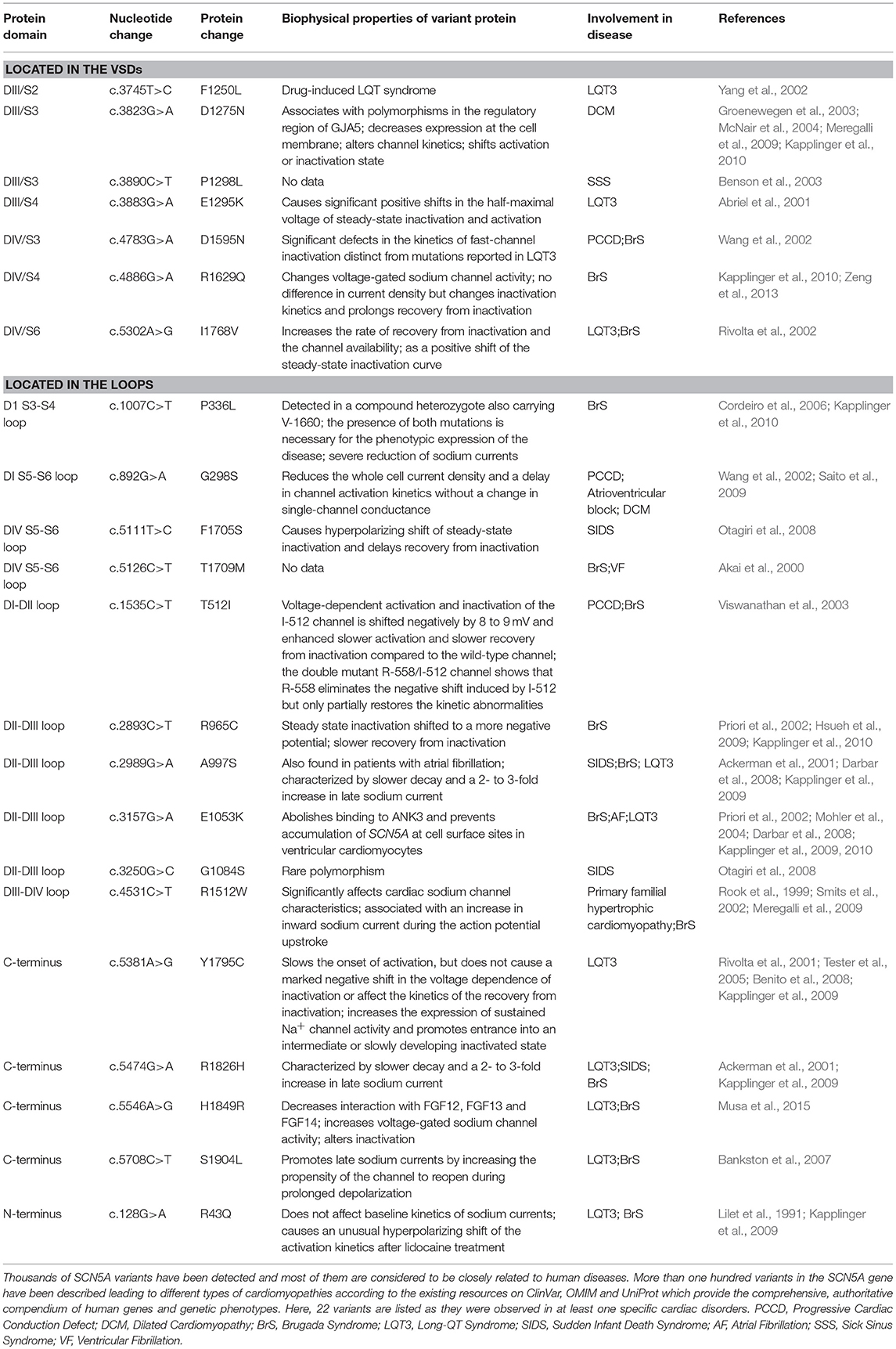
Table 1. Variants in SCN5A associated with cardiac disorders.
Recent research results indicated that the function and regulation of Nav1.5 was more complicated than traditionally assumed. For example, Nav1.5 has been found to be regulated by more than 20 interacting proteins in distinct membrane compartments (Shy et al., 2013). The dysfunction of Nav1.5 may not only be a cause but also a consequence in different pathophysiological procedures and various cardiac disorders. Undoubtedly the dysfunction of Nav1.5 contributes to arrhythmogenesis during pathophysiological conditions. However, sodium channel remodeling manifested with alterations in Nav1.5 clustering was also discovered within distinct cardiomyocyte microdomains in some pathophysiological procedures such as heart failure (Rivaud et al., 2017). We previously found that the SCN5A variant A1180V could directly lead to cardiac structural impairment without involvement of long-time arrhythmias (Shen et al., 2013). Similarly, the environmental and genetic factors may also play additional roles in pathogenesis of disorders linked to SCN5A variants.
In this review, we will summarize SCN5A variants-related arrhythmia syndromes, structural cardiac disorders and the underlying potential mechanisms. Then a brief description will also be given to introduce the current knowledge regarding the therapy strategies.
The SCN5A gene, located in chromosome 3p21 with 28 exons, is a member of the human voltage-gated sodium channel gene family and encodes alpha subunit of the main cardiac sodium channel Nav1.5. It was firstly identified by Georgè in 1995 with the help of fluorescence in situ hybridization (George et al., 1995). Nav1.5 is a large transmembrane protein (227 KDa) with four internally homologous domains (DI-DIV), each containing six transmembrane spanning segments (S1-S6) (Figure 1). S4 segment works as a critical voltage-sensor because of its ample positive charge residues. It is triggered to undergo transmembrane movement when the cell membrane depolarizes, allowing for sodium ion influx to generate sodium current. The P-loops between S5 and S6 segments constitute the central pore forming region, which determine the ion selectivity of the channel. The four domains are interconnected by intracellular peptide chains, with N-terminal and C-end both located in the intracellular side (Catterall, 2014; Chen-Izu et al., 2015). Previous studies suggested the SCN5A gene was mainly expressed in cardiomyocytes, however, recent researches found this gene also expressed and played fundamental roles in other tissues such as brain, gastrointestinal (GI) tract, and cancer tissues (Black and Waxman, 2013; Verstraelen et al., 2015).
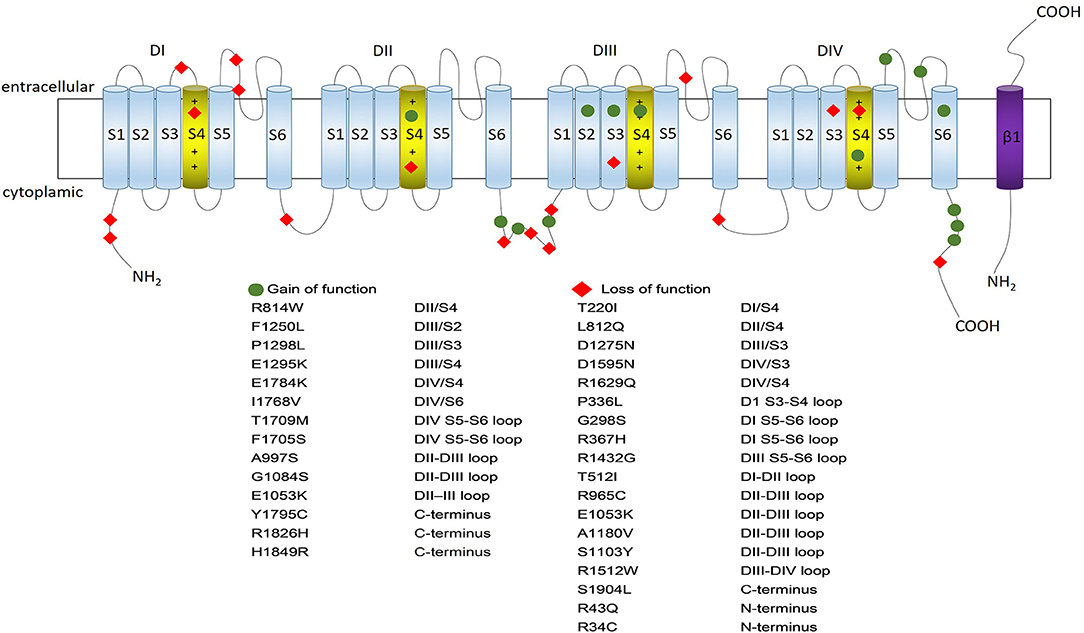
Figure 1. Nav1.5 protein structure and some typical variants associated with gain- or loss- of the sodium channel function. Nav1.5 is a large transmembrane protein with four internally homologous domains (DI-DIV), each containing six spanning segments (S1–S6) which are indicated by numbered cylinders. The four domains are interconnected by intracellular peptide chains, with N-terminal and C-end both located in the intracellular side. S4 segment works as a critical voltage-sensor because of its ample positive charge residues; S5 and S6 segments are lined by extracellular loops (P-loops) which are considered to determine the ion selectivity of the channel (S4 segments are depicted in yellow while the rest segments being blue). The transmembrane segment resembles one of the beta-subunits, which is depicted in purple. The location of the gain-of-function SCN5A variants are shown in blue circle while location of the loss-of-function SCN5A variants are shown in red rhombus.
There are two major SCN5A isoforms: “adult” isoform (SCN5A-003, NM_000335) and “neonatal” (or fetal) isoform (SCN5A-001, NM_001099404). The “adult” SCN5A isoform is abundantly expressed in adult human heart while the “neonatal” isoform is abundantly expressed in neonatal heart. The “adult” isoform differs from the “neonatal” isoform in exon 6 within a voltage-sensor domain (D1/S3-S4) (Murphy et al., 2012). In a mutually exclusive splicing manner, the “neonatal” isoform exhibited slower kinetic of activation and inactivation and a lower depolarized threshold of activation compared to the “adult” isoform (Onkal et al., 2008). The latest study found that both fetal (exon 6a) and adult (exon 6) isoforms of SCN5A were expressed after short-term culture of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) with one of SCN5A variants I230T. Interestingly, prolonged culture time increased the expression of adult sodium channel isoform, which was paralleled by a decrease in INa, representing a severe clinical phenotype in patients with recessive cardiac conduction disease (Veerman et al., 2017). This result partly explained the electrophysiological immaturity phenomenon of hiPSC-CMs and refined our understanding on pathophysiological mechanism of a recessive form of SCN5A-related conduction disorder. Apart from these two isoforms, SCN5A-014 (NM_198056) is also expressed in human heart and should be mentioned in some detail. SCN5A-014 includes an additional glutamine at position 1077 (1077Q) at the exon 17–18 splice boundary, resulting in smaller INa current and exhibiting functional differences with the previous standard isoforms (Makielski et al., 2003).
Sodium current participates in phase 0 and 2 of action potential (AP), which is an important determinant on the production of heart rhythms, normal conduction, and maintenance of excitement. Aberrant sodium channels resulting from SCN5A variants would potentially cause disorganization of the cardiac electrophysiological system, produce various arrhythmias and result in structural heart diseases. Inward and outward transmembrane currents are in dynamic balance physiologically, while variants in SCN5A could induce two states in the sodium channel: “gain-of-function state” or “loss-of-function state.” The former state appears as an increase in sodium influx (sodium current) and a delay in inactivation, while the latter one is manifested by a decrease in sodium influx and an acceleration of inactivation. Different variants-related phenotypes correspond to different state.
Sick sinus syndrome (SSS) is characterized by dysfunction of the sinus node. SSS patients exhibit inappropriate sinus bradycardia, sinus arrest, and reduced chronotropic response (Abe et al., 2014). Although the disease usually affects elderly patients with underlying structural cardiac abnormalities, such as fibrosis or ischemia, young hereditary SSS patients are also not rare (John and Kumar, 2016). One of the genes linked to hereditary SSS is SCN5A and 128 SCN5A variants were found to be causative of hereditary SSS by the complete sequencing of the human genome (ClinVar, 2018). Benson's study firstly discovered six compound heterozygous variants of SCN5A and defined SCN5A-related SSS was a “recessive” disorder often exhibiting compound heterozygosity for distinct SCN5A alleles which associated with dominant disorders of cardiac excitability (Benson et al., 2003). Current evidences indicated that cardiac excitability would decrease and sinus node became dysfunctional in case of both lost or enhanced sodium channel function induced by SCN5A variants. The most typical state of SCN5A variants is the loss-of-function property with reduced electric coupling between sinus node and surrounding atrial cells, resulting in the exit block, a common feature in SSS (Abe et al., 2014). SCN5A variants with gain-of-function feature could induce an increase in AP duration as a result of persistent current during the systolic phase. The prolongation of the sinus node AP would prolong the cycle length, thereby reduced the sinus rate (Wilders, 2018). The phenotypic and functional changes in sinus node caused by SCN5A variants have been carefully observed, however, the electrical activity and the underlying regulation mechanisms have not been extensively studied. Previous studies found that heterozygous SCN5A variant genotype was associated with inappropriate sinus bradycardia, impaired atrioventricular conduction, or increased ventricular refractoriness, while homozygous genotype resulted in intrauterine lethality (Papadatos et al., 2002). However, a recent case report challenged this viewpoint. Jenewein et al. (2017) studied a family with E1053K variant and found that the phenotypical expression in carriers of the same E1053K variant ranged from asymptomatic phenotype to sudden cardiac death (SCD), some asymptomatic individuals were even homozygous variant carriers. Moreover, there are some signaling pathways that regulate the expression and function of SCN5A. Notch signaling is a signaling pathway that can be transiently activated in response to various cardiomyocytes injuries. It was discovered that a transient Notch pulse could down-regulate SCN5A expression, which might result in a slowed atrial conduction velocity (Qiao et al., 2017). As SCN5A variants are associated with large phenotypic variability, future studies are warranted to understand the alternations of electrical activity in mutant pacemaker cells.
Atrial arrhythmias associated with SCN5A variants mostly include familial atrial standstill and atrial fibrillation. Atrial standstill (AS) is caused by atrial electrical and mechanical impairment, manifested by disappeared P wave, bradycardia, or borderline escape rhythm (Baskar et al., 2014). Missense variants such as R367H caused electrophysiological disorders with a non-functional Na+ channel, which could finally lead to AS and BrS (Takehara et al., 2004). Other variant types such as frameshift variants or compound heterozygous variants were also predicted to result in loss-of-function sodium channel and induce supraventricular arrhythmia in patients with AS (Baskar et al., 2014). Groenewegen et al. (2003) performed genetic screening of SCN5A and other atrial-specific genes in an apparently sporadic case of AS and found SCN5A D1275N variant in a familial AS pedigree. This variant resulted in a depolarizing shift in activation. Interestingly, D1275N was found to be closely linked to the atrial-specific gap junction protein connexin40 (Cx40). The decreased expression of Cx40 jointly impaired the electrical coupling between atrial cells. It was thus speculated that genetic variants could synergistically enhance the functional effect of each individual genetic change, consequently leading to a complete loss of electrical and mechanical activity.
Atrial fibrillation (AF) is the most common sustained arrhythmia characterized by rapid and irregular beating of the atria (Hucker et al., 2016). A Framingham study showed that the incidence of new-onset AF was 26.8% after a 9-year follow-up period. Besides, new-onset AF occurred more frequently among individuals with familial history than without familial AF history, indicating that the hereditary factors might play an important role in AF pathogenesis (Lubitz et al., 2010). To date, 32 genes including SCN5A have been shown to be associated with AF (Hayashi et al., 2017). A clinical study scanned SCN5A variants in 156 unrelated probands and AF was evidenced in 43% individuals with SCN5A variants (Olson et al., 2005). Another study also reported a high prevalence of SCN5A variants in familial AF patients (Darbar et al., 2008). Moreover, there was a high overlap between AF and other SCN5A variants related diseases i.e., LQT3, BrS, SSS, and conduction disease (Savio-Galimberti and Darbar, 2014; Ishikawa et al., 2017; Boddum et al., 2018). These emerging evidences suggested a possible link between variants in SCN5A and familial AF. Both gain-of-function and loss-of-function variants in SCN5A have been described to induce atrial arrhythmia. Gain-of-function variants such as D1275N promoted ectopic activity and increased atrial AP duration and excitability, while loss-of-function variants such as H558R brought a reduction in sodium current density and a shortening refractory period, then decreased conduction velocity in the atria and finally increased vulnerability to atrial arrhythmia.
Variants of SCN5A have been demonstrated to contribute to various kinds of ventricular arrhythmias including long QT syndrome (LQTS) type 3; Brugada syndrome (BrS) and idiopathic ventricular fibrillation (IVF), which greatly increase the risk of sudden death in young individuals with a structurally normal heart. Decades of researches have raised enormous insights into the role of SCN5A variants in ventricular arrhythmogenesis.
Long-QT syndrome (LQTS) is an inherited arrhythmogenic disease characterized by prolonged QT intervals. Among the 15 current forms of LQTS, long QT syndrome type 3 (LQT3) is caused by inherited variants of SCN5A accompanied with an increasing risk to sudden death during rest or sleep and electrocardiographically characterized by prolonged QT/QTc interval, accentuated QT dispersion, late onset of T wave and frequent prominent U wave. The first description of LQT3 variant was presented in 1995 with a deletion of amino acids 1,505–1,507 (ΔKPQ) in a patient with prolonged QT interval (Wang et al., 1995a). Up to now, over 300 SCN5A variants are known to be related to LQT3. Five to ten percentage of LQT3 patients possess gain-of-function variants in SCN5A such as E1784K, E1053K, I1768V etc. (Flaim et al., 2007; Remme, 2013; Veerman et al., 2015; Jenewein et al., 2017). These variants are mostly located in the intracellular junctions between DIII and DIV region, S4 segment and the C-terminal region, which could cause the channel inactivation disorders, persistent sodium channel opening state, inactivation of inward sodium current, and prolonged AP (Wang et al., 1995b). The current alternation disturbed the balance between ion influx and outflux in the plateau phase of the AP, which could result in a fast AP initiation and late inward Na+ current (INaL), thereby easily induced Torsades de pointes (TdP) and ventricular fibrillation (VF) (Fredj et al., 2006). In addition, intracellular calcium homeostasis and Ca2+ transient amplitude would influence window current or INaL, as a result, the mixed syndrome phenotype in LQT3 patients would ameliorated or exacerbated in the setting of a SCN5A variants (Rivaud et al., 2018). The predisposition to VF is also influenced by genetic modifiers. Ter Bekke and his colleagues phenotyped a 16-generation pedigree and found SCN5A-p.Phe1617del carriers were predominantly female family members, characterized with after arousal-evoked heart-rate acceleration and repolarization prolongation (Ter Bekke et al., 2017). Besides, many variants were associated with more than one arrhythmia phenotypes at the same time (Kimura et al., 2017). The most common SCN5A variant E1784K was not only underpinning both Brugada syndrome type 1 (BrS1) and LQT3, but being influenced by both elevated temperature and cytosolic calcium (Abdelsayed et al., 2018). The interrelationships and regulatory mechanisms of SCN5A variants are so complicated that there is still a long way to go to pinpoint various pathogenic mechanisms underlying the various arrhythmia syndrome.
Brugada syndrome (BrS) is a familial arrhythmia syndrome characterized by ventricular arrhythmias and SCD, in which the signature feature of type-I BrS ECG pattern is ST-elevations in the right precordial leads (Sieira and Brugada, 2017). It often occurs in healthy individuals at a relatively young age (< 40 years). In BrS, the first variant was identified in an α-subunit of the sodium channel gene, SCN5A in 1998 (Chen et al., 1998). A study examined the genotype-phenotype correlation of SCN5A variants in BrS with a 72 months followup period and found that BrS patients with SCN5A variants exhibited more conduction abnormalities and had higher risk for cardiac events (Yamagata et al., 2017). To date, nearly a quarter of BrS patients were found to be SCN5A variants carriers and over 300 SCN5A variants were found to be associated with BrS, which include missense variants, non-sense variants, nucleotide insertion/deletions, and splice site variants (Kapplinger et al., 2010). BrS-related SCN5A variants are usually loss-of-function variants and mainly located in the region between DI and DII, intracellular connection between DIII and DIV region, P ring and D-terminal of DIII region including SCN5A polymorphism H558R, R34C, S1103Y, L812Q, K817E, etc. (Kapplinger et al., 2010; Wang et al., 2015; Kinoshita et al., 2016; Matsumura et al., 2017). These variants disturbed the transmembrane ion flux balance at the end of the first phase of AP. As a result, there appeared notch on the epicardial AP, which presented ST segment elevation on ECG. When the current imbalance increased, the epicardial AP would become significantly shorter and the phase 2 bipolar state would become unbalanced with reentry, leading to the occurrence of ventricular tachycardia and ventricular fibrillation (Grant et al., 2002). In addition to the abnormal AP duration restitution properties, conduction delay in the right ventricle outflow tract were also observed in BrS patients. The depolarization disorder hypothesis mostly derived from clinical observations (Takami et al., 2003; Tukkie et al., 2004). The delayed and fragmented ventricular conduction was thought to be one of the potent arrhythmogenic substrates as strong predictors of ventricular arrhythmias in patients with BrS (Kanda et al., 2002; Nishii et al., 2010).
Idiopathic ventricular fibrillation (IVF) is characterized by spontaneous ventricular fibrillation and exclusion of any specific structural or functional cardiac diseases including structural cardiac diseases (i.e., coronary artery disease, valvular heart disease, myocarditis, hypertrophic, and dilated cardiomyopathy) and primary arrhythmia syndromes [i.e., BrS, long- and short-QT syndromes, catecholaminergic polymorphic ventricular tachycardia (CPVT), and early repolarization syndrome (ERS)] (Priori et al., 2013). Truncated, non-functional Na+ channel α-subunit resulting from SCN5A variants was found to be one of the underlying causes for IVF including missense variants such as R1432G, R1512W, A1924T, and insertion variants such as 1795insD (Chen et al., 1998). Some studies indicated that IVF variants of SCN5A reduced Na+ channel function and decreased the availability of Na+ conductance, however, the mutant phenotype became different with experimental conditions changes (e.g., presence of β1-subunits; temperature) as well as variable region location (Wan et al., 2001; Maltsev et al., 2009). The exact pathogenesis and pathophysiological mechanism of IVF are unknown. The origin might be mono- or polygenic variants and may be multifactorially influenced by particular environmental or discrete structural abnormalities.
Progressive cardiac conduction defect (PCCD), also called Lenegre syndrome or Lev disease, is one of the most common conduction disorders. It is characterized by progressive changes in the His-Purkinje system with either right or left bundle branch block and QRS complex broadening, eventually leading to complete atrioventricular block, syncope or sudden death. Variants in SCN5A as a cause of inherited PCCD were firstly described in 1999 (Schott et al., 1999). SCN5A variants led to a decrease in sodium channel density on cell membrane, as a result, the decreased current in phase 0 slowed down the myocardial conduction speed and eventually resulted in various conduction blocks. Considerable overlaps existed between PCCD and other clinical entities such as BrS and LQT3 (Glaaser et al., 2012; Park et al., 2015; Veltmann et al., 2016). Similar to BrS, loss-of-function SCN5A variants could decrease sodium channel availability and led to PCCD, while gain-of-function SCN5A variants underlying LQT3 could prolong AP and increased persistent inward sodium, which might also occur in PCCD. Some pathogenic variants of SCN5A associated with cardiac conduction disturbances might be a genetic marker and a non-negligible alarm signal associated with ventricular arrhythmia and SCD (Makarawate et al., 2017). Therefore, proper follow-up of SCN5A variants carriers with PCCD phenotype is warranted.
Dilated cardiomyopathy (DCM) is a myocardial disease characterized by left ventricular systolic dysfunction with dilatation of left ventricle or double ventricles. It is the most common type of primary cardiomyopathy and the most common reason for cardiac transplantation in adults and children (Sanbe, 2013). It was reported that 20–50% DCM patients had a family history, suggesting that genetic factors played an important role in their pathogenesis. Currently, more than 40 causative genes have been found in DCM, including SCN5A (Hershberger et al., 2013). In 2004, Mc Nair firstly identified a heterozygous G-to-A variant of SCN5A, which was co-segregated with DCM phenotype (McNair et al., 2004). Subsequently, SCN5A missense variants (T220I, R814W, D1595H) and truncation variants (2550-2551insTG) were successively discovered in DCM patients (Olson et al., 2005). Most variants are located in the voltage sensor domain (VSD) especially in S3 and S4 segment. As the electrophysiological properties of these variants are not fully clarified, the pathogenesis of DCM due to SCN5A variants remains inconclusive.
Systolic function of the heart was previously thought to be mainly associated with calcium channels. SCN5A variants were traditionally recognized as the cause of arrythmias in DCM patients, but did not directly affect the cardiac systolic function. A primary disruption of an ion channel through gene variants leading to DCM was explained that in one way, the ion channel disturbance caused dysfunction of cytoskeletal protein binding partners and resulted in a DCM phenotype. In another way, the electrical dysfunction caused by variants defects led to mechanical instability, and ultimately led to myocardial dilation (Towbin and Lorts, 2011). Furthermore, gain-of-function SCN5A variants were partly responsible for hyperexcitability of the fascicular-Purkinje system. The incomplete repolarization in Purkinje cells brought premature ventricular action potentials and developed DCM finally (Laurent et al., 2012).
Through the electrophysiological analysis of A1180V, a novel SCN5A variant that was discovered by our team, we indicated that variant channel expressed a rate-dependent Na+ current reduction and a moderate increase in INaL, implying that A1180V caused DCM by disturbing cellular Na+ homeostasis. During the follow-up period of the affected pedigree, we found A1180V carriers exhibited a deterioration of cardiac function and progressed to DCM or atrioventricular block (AVB). These results suggested that A1180V was a risk factor for familiar DCM with preceding atrioventricular block (Ge et al., 2008; Shen et al., 2013). The variant might directly damage myocardium by affecting the intracellular sodium homeostasis, which in turn lead to cell membrane Na+/Ca2+- and Na+/H+-exchanger disorders and imbalance of intracellular calcium homeostasis. Apart from A1180V, we further explored three novel non-synonymous SCN5A variants in idiopathic DCM patients, including c.674G>A, c.677C>T, and c.4340T>A (Shen et al., 2017). Consistent with previous results, these newly defined idiopathic DCM related variants were mainly located in the S4 segment of domain I (DI-S4). By applying patch clamp technology, we found that R225Q (c.674G>A) predisposed electrical disorders by reducing peak sodium current density. Some Nav1.5 variants such as R222Q; R225W; R225P; R814W; and R219H were associated with an atypical phenotype combining several cardiac arrhythmias and DCM (McNair et al., 2011; Mann et al., 2012). Chahine's group investigated mutant channels and found the gating pore current generated by these variants with a cation leak through the typically non-conductive VSD. The presence of a proton-based leak current linked Nav1.5 VSD variants with human cardiac arrhythmias and dilatation of cardiac chambers (Gosselin-Badaroudine et al., 2012; Moreau et al., 2015a,b). As many of the molecular mechanisms between the dilated phenotype and DCM-related SCN5A variants have yet to be elucidated, future novel insights will be available depending on transgenic mice studies and large-scale clinical long-term follow-up studies.
Sudden infant death syndrome (SIDS) denominates cases of infant death, which mostly occurs during sleep without any preceding symptoms. It is the leading cause of post neonatal mortality (Matthews and Macdorman, 2013). Although many pathophysiological studies have been proposed, the specific pathogenic mechanisms remain unclear. SIDS is presumed to be co-regulated by multiple factors including neurotransmission, energy metabolism and genetic disorders. Ion channel genes such as KCNQ1, KCNH2, SCN5A, RYR2, and their molecular modifiers such as GPD1L and β-subunits account for ~10–15% of all SIDS cases, in which SCN5A variants account for approximately half of the channelopathic SIDS cases (Tan et al., 2010; Tester et al., 2018). Variants in SCN5A led to severe trafficking defects and a severely dysfunctional Nav1.5 channel in a structurally normal heart. The decreased current density was likely to be a major contributing factor to the lethal arrhythmia in SIDS victim (Gando et al., 2017). Given the potential risk of inherited cardiac conditions caused by ultra-rare gene variants, current experts has made consensus statement on post-mortem genetic testing (molecular autopsy) and clinical investigation of surviving blood relatives. However, under pressure from public moral principle and different physical environments, roll-out of this approach seems a long way away.
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiomyopathy characterized by myocardial atrophy with fibro-fatty replacement, eventually resulting in wall thinning and cardiac dilatation. The inflow tract, outflow tract and apex of the right ventricle constitute the so-called “triangle of dysplasia” (Oomen et al., 2018). ARVC manifests with myocardial inflammation, right ventricular aneurysms, and ventricular tachyarrhythmias, which represents one of the leading causes of sudden death in the young and in athletes (Basso et al., 2018). The estimated prevalence of ARVC is 1 per 5000 in the general population. Approximately >60% of patients have a pathogenic variant (Bhonsale et al., 2015; Calkins et al., 2017). Desmosomes variants was traditionally considered as the genetic basis of ARVC (Gandjbakhch et al., 2013). It was known that desmosomal protein desmoglein-2 and Nav1.5 interacted with each other (Rizzo et al., 2012). Decreased Na+ current density could slow down conduction and increase necrosis and fibrosis. An artificial loss of desmoplakin (DSP) expression induced an abnormal distribution of connexin43 (Cx43) and Nav1.5. As a result, the sodium current decreased and conduction velocity slowed down, which impaired the mechanical and electrical coupling, these factors also contributed to the pathogenesis of ARVC (Zhang et al., 2013). Beside these, Te Riele performed whole-exome sequencing in six ARVD/C patients without desmosomal variants and found a missense variant (R1898H) in SCN5A. Later, they expanded the study population and found almost 2% of ARVD/C patients had rare SCN5A variants (Te et al., 2017). These findings demonstrated that changes in sodium channel might interact with other molecules or form a tighter network of interactions and served as potential causal gene variants of ARVC.
Multiple SCN5A variants caused complex clinical phenotypes, which suggested that different arrhythmias might have the same genetic origin. The clinical and genetic overlap between these arrhythmias are considered overlap syndrome. 1795insD was the first SCN5A variant related to overlap syndrome, carriers not only presented with LQT3, but also with sinus bradycardia, PCCD, and BrS (Postema et al., 2009). Subsequently, similar findings were reported for other SCN5A variants such as DelK1500, E1784K, Q779X (Grant et al., 2002; Sumitomo, 2014; Aoki et al., 2017). Various SCN5A-related arrhythmia syndromes were not separated but closely linked clinical entities with numerous biophysical overlaps. Moreover, there was a strong genotype-phenotype correlation between arrhythmia syndromes and SCN5A variants, although some phenotypes presented during all decades of life, while others developed with increasing age. By observation in various heterologous expression systems, the biophysical characteristics underlying multiple phenotypes were found to delay fast inactivation, reduce peak sodium current density and increase late component of Ina (Remme and Wilde, 2008; Veltmann et al., 2016). The complex phenotype could be explained by the changes in membrane potential and current. Reduced penetrance and variable disease expression might limit the clinical and genetic diagnosis in SCN5A-related overlap syndromes. Even owning the same variant in SCN5A, reasons of the presence of different or even opposite phenotype are still unclear. The fact that the same SCN5A variant might have different phenotypes suggests the phenotype may have individual or family differences, or that certain regulatory genes or environmental factors may also affect the presence of phenotypes. In addition, clinical elements including age, gender, or medications may also modify disease expressivity. Large cohorts studies are thus needed to gain more insight into exact causal relationship between sodium channel function and various phenotypes.
For many years, researchers have been working on reversing of the pathogenic phenotypes caused by SCN5A variants. Although implantation of ICDs can prevent potentially fatal arrhythmias in patients, the main stream of therapy is the use of medications to prevent arrhythmias. Classical sodium channel blockers are classified as Class I anti-arrhythmic agents and furtherly subdivide into classes IA, IB and IC according to their effects on changes of cardiac AP length (Milne et al., 1984). The blockade of sodium channels can reduce the excitability of cardiomyocytes and stop reentrant wavefronts. However, the same mechanism may exert opposing effect at the same time (van Hoeijen et al., 2014). Toxicities include proarrhythmia are common. Whether the sodium channel blocker mexiletine can be used in clinic to shorten the QT interval without side effects among SCN5A variants carriers has long been controversial. As early as 2004, it was found to rescue the membrane expression of the voltage-gated sodium channel and assist in transporting proteins from the sarcoplasmic reticulum to plasma membrane (Valdivia et al., 2004). Later research queried mexiletine might facilitate trafficking of mutant proteins and exacerbating QT prolongation (Ruan et al., 2010). However, recent study demonstrated mexiletine could bring a major reduction of life-threatening arrhythmic events in LQT3 patients in a retrospective cohort study (Mazzanti et al., 2016). Even with so many conflicting studies, selective inhibition of INaL constitutes a promising pharmacological treatment for cardiac channelopathies. Many pharmacists are dedicated to finding a potent, selective inhibitor of INaL. For example, the INaL inhibitor GS967 was discovered to decrease repolarization abnormalities and had anti-arrhythmic effect without damnification on cardiac conduction (Portero et al., 2017). Other traditional drugs such as flecainide, lidocaine, or β-blockers have also been reported to have some clinical benefits in certain population or specific variants (Wilde et al., 2016; Anderson et al., 2017; Chorin et al., 2018). In general, the especially important issue in pharmacological treatment lays in rebranding of old medications and development of novel agents on the basis of ascertainment in the molecular derangements resulting from genetic variants (Varian and Tang, 2017).
In addition to pharmacological treatments and ICD implantation, some other new approaches are gradually emerging. Non-sense variant is one type of SCN5A pathogenic variants. Non-sense mutation readthrough is a new kind of gene-specific treatment to reduce or avoid deleterious consequences of non-sense variants which could make sure the ribosomes ignore a premature stop codon and produce a full-length protein (Lee and Dougherty, 2012). Some researchers applied readthrough-enhancing methods such as aminoglycosides, suppressor tRNAs or small interfering RNAs (siRNAs) to suppress variants (Roy et al., 2016; Baradaran-Heravi et al., 2017). The sodium currents were restored, however, the restored channels increased the risk of arrhythmia (Teng et al., 2017). Pre-implantation genetic diagnosis (PGD) is a diagnostic procedure by analyzing genotype of the preimplantation embryo, enabling selective transfer of unaffected embryos to the uterus. This procedure improves embryo selection and avoids pathogenesis-related genetic variants. A meta-analysis including four randomized controlled trials (RCTs) and seven cohort studies showed that comprehensive chromosome screening (CCS)-based PGD has better outcomes for women compared to classical morphological criteria (Chen et al., 2015). Although the prospect is raising difficult ethical considerations, PGD is one of the strategies for prevention of gene variants-related diseases somehow (Resnik, 2014; Vermeesch et al., 2016). It is known that gene works through mRNA level. Some studies attempted to regulate the expression of SCN5A mRNA through exogenous ingestion of the messenger RNA (mRNA) stabilizing protein ELAVL1/HuR and to verify whether it was beneficial to control arrhythmia. Researchers found injection of viral particles carrying HuR increased SCN5A expression and improved AP upstroke and conduction velocity, which reduced reentrant arrhythmia. Increasing mRNA stability to rescue decreased SCN5A expression may represent another new paradigm in antiarrhythmic therapy (Zhou et al., 2018). Abnormal bioelectric phenotype was discovered to be corrected in cystic fibrosis caused by variants of the cystic fibrosis transmembrane conductance regulator gene (CFTR) by administering replication-deficient, recombinant adenovirus vector containing a normal copy of the CFTR cDNA (AdCFTR) in a clinical trial (Hay et al., 1995). But arrhythmia genetic or epigenetic therapy investigations have been limited to short-term demonstrations of efficacy in pre-clinical pilot studies. For many arrhythmia applications, adequate delivery to the target tissue and proarrhythmic toxicity as well as safety risks still remain. With long-term efficacy and safety studies, integrated approaches of normalizing cardiac structure and function may represent the next generation of potential therapeutic strategies for cardiac disorders with gene variants.
Cardiac sodium channel plays a central role in cardiomyocyte excitability and proper conduction of cardiac electrical impulses. The function and mechanisms of sodium channel are complicate and remain a lot of controversies. Even within families, the observed phenotypes carrying the same SCN5A variant are highly diverse. Besides, environmental and epigenetic alterations also determine variable disease severity. Continuous exploration and novel insight of this issue will contribute to clarify detailed mechanisms and ultimately enable improved diagnosis, risk stratification, and development of more effective treatment strategies.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication. In particular, WL, LY, and CS: drafting of the manuscript; KH, JG, and AS: revising the manuscript for important intellectual content critically and for final approval of the manuscript.
This work was supported by the National Science Fund for Distinguished Young Scholars to AS (81725002) and a grant of Innovative Research Groups of the National Natural Science Foundation of China (81521001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdelsayed, M., Ruprai, M., and Ruben, P. C. (2018). The efficacy of Ranolazine on E1784K is altered by temperature and calcium. Sci. Rep. 8:3643. doi: 10.1038/s41598-018-22033-1
Abe, K., Machida, T., Sumitomo, N., Yamamoto, H., Ohkubo, K., Watanabe, I., et al. (2014). Sodium channelopathy underlying familial sick sinus syndrome with early onset and predominantly male characteristics. Circ. Arrhythm. Electrophysiol. 7, 511–517. doi: 10.1161/CIRCEP.113.001340
Abriel, H., Cabo, C., Wehrens, X. H., Rivolta, I., Motoike, H. K., Memmi, M., et al. (2001). Novel arrhythmogenic mechanism revealed by a long-QT syndrome mutation in the cardiac Na(+) channel. Circ. Res. 88, 740–745. doi: 10.1161/hh0701.089668
Ackerman, M. J., Siu, B. L., Sturner, W. Q., Tester, D. J., Valdivia, C. R., Makielski, J. C., et al. (2001). Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA 286, 2264–2269. doi: 10.1001/jama.286.18.2264
Akai, J., Makita, N., Sakurada, H., Shirai, N., Ueda, K., Kitabatake, A., et al. (2000). A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 479, 29–34. doi: 10.1016/S0014-5793(00)01875-5
Anderson, H. N., Bos, J. M., Kapplinger, J. D., Meskill, J. M., Ye, D., and Ackerman, M. J. (2017). Lidocaine attenuation testing: an in vivo investigation of putative LQT3-associated variants in the SCN5A-encoded sodium channel. Heart Rhythm 14, 1173–1179. doi: 10.1016/j.hrthm.2017.04.020
Aoki, H., Nakamura, Y., Ohno, S., Makiyama, T., and Horie, M. (2017). Cardiac conduction defects and Brugada syndrome: a family with overlap syndrome carrying a nonsense SCN5A mutation. J Arrhythm 33, 35–39. doi: 10.1016/j.joa.2016.05.007
Aronsen, J. M., Swift, F., and Sejersted, O. M. (2013). Cardiac sodium transport and excitation-contraction coupling. J. Mol. Cell. Cardiol. 61, 11–19. doi: 10.1016/j.yjmcc.2013.06.003
Bankston, J. R., Sampson, K. J., Kateriya, S., Glaaser, I. W., Malito, D. L., Chung, W. K., et al. (2007). A novel LQT-3 mutation disrupts an inactivation gate complex with distinct rate-dependent phenotypic consequences. Channels 1, 273–280. doi: 10.4161/chan.4956
Baradaran-Heravi, A., Niesser, J., Balgi, A. D., Choi, K., Zimmerman, C., South, A. P., et al. (2017). Gentamicin B1 is a minor gentamicin component with major nonsense mutation suppression activity. Proc. Natl. Acad. Sci. U.S.A. 114, 3479–3484. doi: 10.1073/pnas.1620982114
Baskar, S., Ackerman, M. J., Clements, D., Mayuga, K. A., and Aziz, P. F. (2014). Compound heterozygous mutations in the SCN5A-encoded Nav1.5 cardiac sodium channel resulting in atrial standstill and His-Purkinje system disease. J. Pediatr. 165, 1050–1052. doi: 10.1016/j.jpeds.2014.07.036
Basso, C., Pilichou, K., Bauce, B., Corrado, D., and Thiene, G. (2018). Diagnostic criteria, genetics, and molecular basis of arrhythmogenic cardiomyopathy. Heart Fail. Clin. 14, 201–213. doi: 10.1016/j.hfc.2018.01.002
Benito, B., Brugada, R., Perich, R. M., Lizotte, E., Cinca, J., Mont, L., et al. (2008). A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm 5, 1434–1440. doi: 10.1016/j.hrthm.2008.07.013
Benson, D. W., Wang, D. W., Dyment, M., Knilans, T. K., Fish, F. A., Strieper, M. J., et al. (2003). Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 112, 1019–1028. doi: 10.1172/JCI200318062
Bhonsale, A., Groeneweg, J. A., James, C. A., Dooijes, D., Tichnell, C., Jongbloed, J. D., et al. (2015). Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 36, 847–855. doi: 10.1093/eurheartj/ehu509
Black, J. A., and Waxman, S. G. (2013). Noncanonical roles of voltage-gated sodium channels. Neuron 80, 280–291. doi: 10.1016/j.neuron.2013.09.012
Boddum, K., Saljic, A., Jespersen, T., and Christensen, A. H. (2018). A novel SCN5A variant associated with abnormal repolarization, atrial fibrillation, and reversible cardiomyopathy. Cardiology 140, 8–13. doi: 10.1159/000487475
Calkins, H., Corrado, D., and Marcus, F. (2017). Risk stratification in arrhythmogenic right ventricular cardiomyopathy. Circulation 136, 2068–2082. doi: 10.1161/CIRCULATIONAHA.117.030792
Catterall, W. A. (2014). Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu. Rev. Pharmacol. Toxicol. 54, 317–338. doi: 10.1146/annurev-pharmtox-011112-140232
Chen, M., Wei, S., Hu, J., and Quan, S. (2015). Can comprehensive chromosome screening technology improve IVF/ICSI outcomes? A meta-analysis. PLoS ONE 10:e140779. doi: 10.1371/journal.pone.0140779
Chen, Q., Kirsch, G. E., Zhang, D., Brugada, R., Brugada, J., Brugada, P., et al. (1998). Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296. doi: 10.1038/32675
Chen-Izu, Y., Shaw, R. M., Pitt, G. S., Yarov-Yarovoy, V., Sack, J. T., Abriel, H., et al. (2015). Na+ channel function, regulation, structure, trafficking and sequestration. J. Physiol. 593, 1347–1360. doi: 10.1113/jphysiol.2014.281428
Chorin, E., Taub, R., Medina, A., Flint, N., Viskin, S., and Benhorin, J. (2018). Long-term flecainide therapy in type 3 long QT syndrome. Europace 20, 370–376. doi: 10.1093/europace/euw439
ClinVar [Z]. (2018). Available online at: https://www.ncbi.nlm.nih.gov/clinvar/?term=SCN5A%5Bgene%5D.
Cordeiro, J. M., Barajas-Martinez, H., Hong, K., Burashnikov, E., Pfeiffer, R., Orsino, A. M., et al. (2006). Compound heterozygous mutations P336L and I1660V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation 114, 2026–2033. doi: 10.1161/CIRCULATIONAHA.106.627489
Darbar, D., Kannankeril, P. J., Donahue, B. S., Kucera, G., Stubblefield, T., Haines, J. L., et al. (2008). Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 117, 1927–1935. doi: 10.1161/CIRCULATIONAHA.107.757955
Flaim, S. N., Giles, W. R., and Mcculloch, A. D. (2007). Arrhythmogenic consequences of Na+ channel mutations in the transmurally heterogeneous mammalian left ventricle: analysis of the I1768V SCN5A mutation. Heart Rhythm 4, 768–778. doi: 10.1016/j.hrthm.2007.02.009
Fredj, S., Lindegger, N., Sampson, K. J., Carmeliet, P., and Kass, R. S. (2006). Altered Na+ channels promote pause-induced spontaneous diastolic activity in long QT syndrome type 3 myocytes. Circ. Res. 99, 1225–1232. doi: 10.1161/01.RES.0000251305.25604.b0
Gandjbakhch, E., Vite, A., Gary, F., Fressart, V., Donal, E., Simon, F., et al. (2013). Screening of genes encoding junctional candidates in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Europace 15, 1522–1525. doi: 10.1093/europace/eut224
Gando, I., Morganstein, J., Jana, K., McDonald, T. V., Tang, Y., and Coetzee, W. A. (2017). Infant sudden death: mutations responsible for impaired Nav1.5 channel trafficking and function. Pacing Clin. Electrophysiol. 40, 703–712. doi: 10.1111/pace.13087
Ge, J., Sun, A., Paajanen, V., Wang, S., Su, C., Yang, Z., et al. (2008). Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ. Arrhythm. Electrophysiol. 1, 83–92. doi: 10.1161/CIRCEP.107.750752
George, A. L., Varkony, T. A., Drabkin, H. A., Han, J., Knops, J. F., Finley, W. H., et al. (1995). Assignment of the human heart tetrodotoxin-resistant voltage-gated Na+ channel alpha-subunit gene (SCN5A) to band 3p21. Cytogenet. Cell Genet. 68, 67–70. doi: 10.1159/000133892
Glaaser, I. W., Osteen, J. D., Puckerin, A., Sampson, K. J., Jin, X., and Kass, R. S. (2012). Perturbation of sodium channel structure by an inherited Long QT Syndrome mutation. Nat. Commun. 3:706. doi: 10.1038/ncomms1717
Gosselin-Badaroudine, P., Keller, D. I., Huang, H., Pouliot, V., Chatelier, A., Osswald, S., et al. (2012). A proton leak current through the cardiac sodium channel is linked to mixed arrhythmia and the dilated cardiomyopathy phenotype. PLoS ONE 7:e38331. doi: 10.1371/journal.pone.0038331
Grant, A. O., Carboni, M. P., Neplioueva, V., Starmer, C. F., Memmi, M., Napolitano, C., et al. (2002). Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J. Clin. Invest. 110, 1201–1209. doi: 10.1172/JCI0215570
Groenewegen, W. A., Firouzi, M., Bezzina, C. R., Vliex, S., van Langen, I. M., Sandkuijl, L., et al. (2003). A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ. Res. 92, 14–22. doi: 10.1161/01.RES.0000050585.07097.D7
Hay, J. G., McElvaney, N. G., Herena, J., and Crystal, R. G. (1995). Modification of nasal epithelial potential differences of individuals with cystic fibrosis consequent to local administration of a normal CFTR cDNA adenovirus gene transfer vector. Hum. Gene Ther. 6, 1487–1496. doi: 10.1089/hum.1995.6.11-1487
Hayashi, K., Tada, H., and Yamagishi, M. (2017). The genetics of atrial fibrillation. Curr. Opin. Cardiol. 32, 10–16. doi: 10.1097/HCO.0000000000000356
Hershberger, R. E., Hedges, D. J., and Morales, A. (2013). Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531–547. doi: 10.1038/nrcardio.2013.105
Hsueh, C. H., Chen, W. P., Lin, J. L., Tsai, C. T., Liu, Y. B., Juang, J. M., et al. (2009). Distinct functional defect of three novel Brugada syndrome related cardiac sodium channel mutations. J. Biomed. Sci. 16:23. doi: 10.1186/1423-0127-16-23
Hucker, W. J., Saini, H., Lubitz, S. A., and Ellinor, P. T. (2016). Atrial fibrillation genetics: is there a practical clinical value now or in the future? Can. J. Cardiol. 32, 1300–1305. doi: 10.1016/j.cjca.2016.02.032
Ishikawa, T., Ohno, S., Murakami, T., Yoshida, K., Mishima, H., Fukuoka, T., et al. (2017). Sick sinus syndrome with HCN4 mutations shows early onset and frequent association with atrial fibrillation and left ventricular noncompaction. Heart Rhythm 14, 717–724. doi: 10.1016/j.hrthm.2017.01.020
Jenewein, T., Beckmann, B. M., Rose, S., Osterhues, H. H., Schmidt, U., Wolpert, C., et al. (2017). Genotype-phenotype dilemma in a case of sudden cardiac death with the E1053K mutation and a deletion in the SCN5A gene. Forensic Sci. Int. 275, 187–194. doi: 10.1016/j.forsciint.2017.02.038
John, R. M., and Kumar, S. (2016). Sinus node and atrial arrhythmias. Circulation 133, 1892–1900. doi: 10.1161/CIRCULATIONAHA.116.018011
Kanda, M., Shimizu, W., Matsuo, K., Nagaya, N., Taguchi, A., Suyama, K., et al. (2002). Electrophysiologic characteristics and implications of induced ventricular fibrillation in symptomatic patients with Brugada syndrome. J. Am. Coll. Cardiol. 39, 1799–1805. doi: 10.1016/S0735-1097(02)01867-3
Kapplinger, J. D., Tester, D. J., Alders, M., Benito, B., Berthet, M., Brugada, J., et al. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7, 33–46. doi: 10.1016/j.hrthm.2009.09.069
Kapplinger, J. D., Tester, D. J., Salisbury, B. A., Carr, J. L., Harris-Kerr, C., Pollevick, G. D., et al. (2009). Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 6, 1297–1303. doi: 10.1016/j.hrthm.2009.05.021
Kimura, M., Kohno, T., Aizawa, Y., Inohara, T., Shiraishi, Y., Katsumata, Y., et al. (2017). A novel SCN5A mutation found in a familial case of long QT syndrome complicated by severe left ventricular dysfunction. Can. J. Cardiol. 33, 554–555. doi: 10.1016/j.cjca.2016.10.010
Kinoshita, K., Takahashi, H., Hata, Y., Nishide, K., Kato, M., Fujita, H., et al. (2016). SCN5A(K817E), a novel Brugada syndrome-associated mutation that alters the activation gating of NaV1.5 channel. Heart Rhythm 13, 1113–1120. doi: 10.1016/j.hrthm.2016.01.008
Laurent, G., Saal, S., Amarouch, M. Y., Beziau, D. M., Marsman, R. F., Faivre, L., et al. (2012). Multifocal ectopic Purkinje-related premature contractions: a new SCN5A-related cardiac channelopathy. J. Am. Coll. Cardiol. 60, 144–156. doi: 10.1016/j.jacc.2012.02.052
Lee, H. L., and Dougherty, J. P. (2012). Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol. Ther. 136, 227–266. doi: 10.1016/j.pharmthera.2012.07.007
Lilet, H., Krzentowski, G., Bodson, A., Scheen, A. J., and Lefebvre, P. J. (1991). [Lack of benefit from the intermittent administration of insulin in treatment using subcutaneous perfusion pump in type 1 diabetes]. Diabete Metab. 17, 363–372.
Lubitz, S. A., Yin, X., Fontes, J. D., Magnani, J. W., Rienstra, M., Pai, M., et al. (2010). Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA 304, 2263–2269. doi: 10.1001/jama.2010.1690
Makarawate, P., Chaosuwannakit, N., Vannaprasaht, S., Sahasthas, D., Koo, S. H., Lee, E., et al. (2017). SCN5A genetic polymorphisms associated with increased defibrillator shocks in Brugada syndrome. J. Am. Heart Assoc. 6:e005009. doi: 10.1161/JAHA.116.005009
Makielski, J. C., Ye, B., Valdivia, C. R., Pagel, M. D., Pu, J., Tester, D. J., et al. (2003). A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ. Res. 93, 821–828. doi: 10.1161/01.RES.0000096652.14509.96
Maltsev, V. A., Kyle, J. W., and Undrovinas, A. (2009). Late Na+ current produced by human cardiac Na+ channel isoform Nav1.5 is modulated by its beta1 subunit. J. Physiol. Sci. 59, 217–225. doi: 10.1007/s12576-009-0029-7
Mann, S. A., Castro, M. L., Ohanian, M., Guo, G., Zodgekar, P., Sheu, A., et al. (2012). R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J. Am. Coll. Cardiol. 60, 1566–1573. doi: 10.1016/j.jacc.2012.05.050
Matsumura, H., Nakano, Y., Ochi, H., Onohara, Y., Sairaku, A., Tokuyama, T., et al. (2017). H558R, a common SCN5A polymorphism, modifies the clinical phenotype of Brugada syndrome by modulating DNA methylation of SCN5A promoters. J. Biomed. Sci. 24:91. doi: 10.1186/s12929-017-0397-x
Matthews, T. J., and Macdorman, M. F. (2013). Infant mortality statistics from the 2010 period linked birth/infant death data set. Natl. Vital Stat. Rep. 62, 1–26.
Mazzanti, A., Maragna, R., Faragli, A., Monteforte, N., Bloise, R., Memmi, M., et al. (2016). Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome Type 3. J. Am. Coll. Cardiol. 67, 1053–1058. doi: 10.1016/j.jacc.2015.12.033
McNair, W. P., Ku, L., Taylor, M. R., Fain, P. R., Dao, D., Wolfel, E., et al. (2004). SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 110, 2163–2167. doi: 10.1161/01.CIR.0000144458.58660.BB
McNair, W. P., Sinagra, G., Taylor, M. R., Di Lenarda, A., Ferguson, D. A., Salcedo, E. E., et al. (2011). SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J. Am. Coll. Cardiol. 57, 2160–2168. doi: 10.1016/j.jacc.2010.09.084
Meregalli, P. G., Tan, H. L., Probst, V., Koopmann, T. T., Tanck, M. W., Bhuiyan, Z. A., et al. (2009). Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 6, 341–348. doi: 10.1016/j.hrthm.2008.11.009
Milne, J. R., Hellestrand, K. J., Bexton, R. S., Burnett, P. J., Debbas, N. M., and Camm, A. J. (1984). Class 1 antiarrhythmic drugs–characteristic electrocardiographic differences when assessed by atrial and ventricular pacing. Eur. Heart J. 5, 99–107. doi: 10.1093/oxfordjournals.eurheartj.a061633
Mohler, P. J., Rivolta, I., Napolitano, C., LeMaillet, G., Lambert, S., Priori, S. G., et al. (2004). Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 101, 17533–17538. doi: 10.1073/pnas.0403711101
Moreau, A., Gosselin-Badaroudine, P., Boutjdir, M., and Chahine, M. (2015a). Mutations in the voltage sensors of domains I and II of Nav1.5 that are associated with arrhythmias and dilated cardiomyopathy generate gating pore currents. Front. Pharmacol. 6:301. doi: 10.3389/fphar.2015.00301
Moreau, A., Gosselin-Badaroudine, P., Delemotte, L., Klein, M. L., and Chahine, M. (2015b). Gating pore currents are defects in common with two Nav1.5 mutations in patients with mixed arrhythmias and dilated cardiomyopathy. J. Gen. Physiol. 145, 93–106. doi: 10.1085/jgp.201411304
Murphy, L. L., Moon-Grady, A. J., Cuneo, B. F., Wakai, R. T., Yu, S., Kunic, J. D., et al. (2012). Developmentally regulated SCN5A splice variant potentiates dysfunction of a novel mutation associated with severe fetal arrhythmia. Heart Rhythm 9, 590–597. doi: 10.1016/j.hrthm.2011.11.006
Musa, H., Kline, C. F., Sturm, A. C., Murphy, N., Adelman, S., Wang, C., et al. (2015). SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc. Natl. Acad. Sci. U.S.A. 112, 12528–12533. doi: 10.1073/pnas.1516430112
Nishii, N., Nagase, S., Morita, H., Kusano, K. F., Namba, T., Miura, D., et al. (2010). Abnormal restitution property of action potential duration and conduction delay in Brugada syndrome: both repolarization and depolarization abnormalities. Europace 12, 544–552. doi: 10.1093/europace/eup432
Olson, T. M., Michels, V. V., Ballew, J. D., Reyna, S. P., Karst, M. L., Herron, K. J., et al. (2005). Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 293, 447–454. doi: 10.1001/jama.293.4.447
Onkal, R., Mattis, J. H., Fraser, S. P., Diss, J. K., Shao, D., Okuse, K., et al. (2008). Alternative splicing of Nav1.5: an electrophysiological comparison of 'neonatal' and 'adult' isoforms and critical involvement of a lysine residue. J. Cell. Physiol. 216, 716–726. doi: 10.1002/jcp.21451
Oomen, A. W. G. J., Semsarian, C., Puranik, R., and Sy, R. W. (2018). Diagnosis of arrhythmogenic right ventricular cardiomyopathy: progress and pitfalls. Heart Lung Circ. 27, 1310–1317. doi: 10.1016/j.hlc.2018.03.023
Otagiri, T., Kijima, K., Osawa, M., Ishii, K., Makita, N., Matoba, R., et al. (2008). Cardiac ion channel gene mutations in sudden infant death syndrome. Pediatr. Res. 64, 482–487. doi: 10.1203/PDR.0b013e3181841eca
Papadatos, G. A., Wallerstein, P. M., Head, C. E., Ratcliff, R., Brady, P. A., Benndorf, K., et al. (2002). Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl. Acad. Sci. U.S.A. 99, 6210–6215. doi: 10.1073/pnas.082121299
Park, D. S., Cerrone, M., Morley, G., Vasquez, C., Fowler, S., Liu, N., et al. (2015). Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J. Clin. Invest. 125, 403–412. doi: 10.1172/JCI76919
Portero, V., Casini, S., Hoekstra, M., Verkerk, A. O., Mengarelli, I., Belardinelli, L., et al. (2017). Anti-arrhythmic potential of the late sodium current inhibitor GS-458967 in murine Scn5a-1798insD+/- and human SCN5A-1795insD+/- iPSC-derived cardiomyocytes. Cardiovasc. Res. 113, 829–838. doi: 10.1093/cvr/cvx077
Postema, P. G., Van den Berg, M., Van Tintelen, J. P., Van den Heuvel, F., Grundeken, M., Hofman, N., et al. (2009). Founder mutations in the Netherlands: SCN5a 1795insD, the first described arrhythmia overlap syndrome and one of the largest and best characterised families worldwide. Neth. Heart J. 17, 422–428. doi: 10.1007/BF03086296
Priori, S. G., Napolitano, C., Gasparini, M., Pappone, C., Della, B. P., Giordano, U., et al. (2002). Natural history of Brugada syndrome: insights for risk stratification and management. Circulation 105, 1342–1347. doi: 10.1161/hc1102.105288
Priori, S. G., Wilde, A. A., Horie, M., Cho, Y., Behr, E. R., Berul, C., et al. (2013). HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 10, 1932–1963. doi: 10.1016/j.hrthm.2013.05.014
Qiao, Y., Lipovsky, C., Hicks, S., Bhatnagar, S., Li, G., Khandekar, A., et al. (2017). Transient notch activation induces long-term gene expression changes leading to sick sinus syndrome in mice. Circ. Res. 121, 549–563. doi: 10.1161/CIRCRESAHA.116.310396
Remme, C. A. (2013). Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J. Physiol. 591, 4099–4116. doi: 10.1113/jphysiol.2013.256461
Remme, C. A., and Wilde, A. A. (2008). SCN5A overlap syndromes: no end to disease complexity? Europace 10, 1253–1255. doi: 10.1093/europace/eun267
Resnik, D. B. (2014). The morality of patents on pre-implantation genetic diagnosis. Nat. Biotechnol. 32, 319–320. doi: 10.1038/nbt.2865
Rivaud, M. R., Agullo-Pascual, E., Lin, X., Leo-Macias, A., Zhang, M., Rothenberg, E., et al. (2017). Sodium channel remodeling in subcellular microdomains of murine failing cardiomyocytes. J. Am. Heart Assoc. 6:e007622. doi: 10.1161/JAHA.117.007622
Rivaud, M. R., Baartscheer, A., Verkerk, A. O., Beekman, L., Rajamani, S., Belardinelli, L., et al. (2018). Enhanced late sodium current underlies pro-arrhythmic intracellular sodium and calcium dysregulation in murine sodium channelopathy. Int. J. Cardiol. 263, 54–62. doi: 10.1016/j.ijcard.2018.03.044
Rivolta, I., Abriel, H., Tateyama, M., Liu, H., Memmi, M., Vardas, P., et al. (2001). Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. J. Biol. Chem. 276, 30623–30630. doi: 10.1074/jbc.M104471200
Rivolta, I., Clancy, C. E., Tateyama, M., Liu, H., Priori, S. G., and Kass, R. S. (2002). A novel SCN5A mutation associated with long QT-3: altered inactivation kinetics and channel dysfunction. Physiol. Genomics 10, 191–197. doi: 10.1152/physiolgenomics.00039.2002
Rizzo, S., Lodder, E. M., Verkerk, A. O., Wolswinkel, R., Beekman, L., Pilichou, K., et al. (2012). Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc. Res. 95, 409–418. doi: 10.1093/cvr/cvs219
Rook, M. B., Bezzina, A. C., Groenewegen, W. A., van Gelder, I. C., van Ginneken, A. C., Jongsma, H. J., et al. (1999). Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc. Res. 44, 507–517. doi: 10.1016/S0008-6363(99)00350-8
Roy, B., Friesen, W. J., Tomizawa, Y., Leszyk, J. D., Zhuo, J., Johnson, B., et al. (2016). Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad. Sci. U.S.A. 113, 12508–12513. doi: 10.1073/pnas.1605336113
Ruan, Y., Denegri, M., Liu, N., Bachetti, T., Seregni, M., Morotti, S., et al. (2010). Trafficking defects and gating abnormalities of a novel SCN5A mutation question gene-specific therapy in long QT syndrome type 3. Circ. Res. 106, 1374–1383. doi: 10.1161/CIRCRESAHA.110.218891
Saito, Y. A., Strege, P. R., Tester, D. J., Locke, G. R., Talley, N. J., Bernard, C. E., et al. (2009). Sodium channel mutation in irritable bowel syndrome: evidence for an ion channelopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G211–G218. doi: 10.1152/ajpgi.90571.2008
Sanbe, A. (2013). Dilated cardiomyopathy: a disease of the myocardium. Biol. Pharm. Bull. 36, 18–22. doi: 10.1248/bpb.b212023
Savio-Galimberti, E., and Darbar, D. (2014). Atrial fibrillation and SCN5A variants. Card. Electrophysiol. Clin. 6, 741–748. doi: 10.1016/j.ccep.2014.07.006
Schott, J. J., Alshinawi, C., Kyndt, F., Probst, V., Hoorntje, T. M., Hulsbeek, M., et al. (1999). Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 23, 20–21. doi: 10.1038/12618
Shen, C., Xu, L., Han, S., Dong, Z., Zhao, X., Wang, S., et al. (2017). Novel idiopathic DCM-related SCN5A variants localised in DI-S4 predispose electrical disorders by reducing peak sodium current density. J. Med. Genet. 54:762–770. doi: 10.1136/jmedgenet-2017-104780
Shen, C., Xu, L., Yang, Z., Zou, Y., Hu, K., Fan, Z., et al. (2013). A1180V of cardiac sodium channel gene (SCN5A): is it a risk factor for dilated cardiomyopathy or just a common variant in Han Chinese? Dis. Markers 35, 531–535. doi: 10.1155/2013/659528
Shy, D., Gillet, L., and Abriel, H. (2013). Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim. Biophys. Acta 1833, 886–894. doi: 10.1016/j.bbamcr.2012.10.026
Sieira, J., and Brugada, P. (2017). The definition of the Brugada syndrome. Eur. Heart J. 38, 3029–3034. doi: 10.1093/eurheartj/ehx490
Smits, J. P., Eckardt, L., Probst, V., Bezzina, C. R., Schott, J. J., Remme, C. A., et al. (2002). Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J. Am. Coll. Cardiol. 40, 350–356. doi: 10.1016/S0735-1097(02)01962-9
Sumitomo, N. (2014). E1784K mutation in SCN5A and overlap syndrome. Circ. J. 78, 1839–1840. doi: 10.1253/circj.CJ-14-0564
Takami, M., Ikeda, T., Enjoji, Y., and Sugi, K. (2003). Relationship between ST-segment morphology and conduction disturbances detected by signal-averaged electrocardiography in Brugada syndrome. Ann. Noninvasive Electrocardiol. 8, 30–36. doi: 10.1046/j.1542-474X.2003.08106.x
Takehara, N., Makita, N., Kawabe, J., Sato, N., Kawamura, Y., Kitabatake, A., et al. (2004). A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J. Intern. Med. 255, 137–142. doi: 10.1046/j.0954-6820.2003.01247.x
Tan, B. H., Pundi, K. N., Van Norstrand, D. W., Valdivia, C. R., Tester, D. J., Medeiros-Domingo, A., et al. (2010). Sudden infant death syndrome-associated mutations in the sodium channel beta subunits. Heart Rhythm 7, 771–778. doi: 10.1016/j.hrthm.2010.01.032
Te, R. A., Agullo-Pascual, E., James, C. A., Leo-Macias, A., Cerrone, M., Zhang, M., et al. (2017). Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 113, 102–111. doi: 10.1093/cvr/cvw234
Teng, S., Huang, J., Gao, Z., Hao, J., Yang, Y., Zhang, S., et al. (2017). Readthrough of SCN5A nonsense mutations p.R1623X and p.S1812X questions gene-therapy in Brugada syndrome. Curr. Gene Ther. 17, 50–58. doi: 10.2174/1566523217666170529074758
Ter Bekke, R. M. A., Isaacs, A., Barysenka, A., Hoos, M. B., Jongbloed, J., Hoorntje, J., et al. (2017). Heritability in a SCN5A-mutation founder population with increased female susceptibility to non-nocturnal ventricular tachyarrhythmia and sudden cardiac death. Heart Rhythm 14, 1873–1881. doi: 10.1016/j.hrthm.2017.07.036
Tester, D. J., Will, M. L., Haglund, C. M., and Ackerman, M. J. (2005). Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2, 507–517. doi: 10.1016/j.hrthm.2005.01.020
Tester, D. J., Wong, L., Chanana, P., Jaye, A., Evans, J. M., FitzPatrick, D. R., et al. (2018). Cardiac genetic predisposition in sudden infant death syndrome. J. Am. Coll. Cardiol. 71, 1217–1227. doi: 10.1016/j.jacc.2018.01.030
Towbin, J. A., and Lorts, A. (2011). Arrhythmias and dilated cardiomyopathy common pathogenetic pathways? J. Am. Coll. Cardiol. 57, 2169–2171. doi: 10.1016/j.jacc.2010.11.061
Tukkie, R., Sogaard, P., Vleugels, J., de Groot, I. K., Wilde, A. A., and Tan, H. L. (2004). Delay in right ventricular activation contributes to Brugada syndrome. Circulation 109, 1272–1277. doi: 10.1161/01.CIR.0000118467.53182.D1
Valdivia, C. R., Tester, D. J., Rok, B. A., Porter, C. B., Munger, T. M., Jahangir, A., et al. (2004). A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc. Res. 62, 53–62. doi: 10.1016/j.cardiores.2004.01.022
van Hoeijen, D. A., Blom, M. T., and Tan, H. L. (2014). Cardiac sodium channels and inherited electrophysiological disorders: an update on the pharmacotherapy. Expert Opin. Pharmacother. 15, 1875–1887. doi: 10.1517/14656566.2014.936380
Varian, K., and Tang, W. (2017). Therapeutic strategies targeting inherited cardiomyopathies. Curr. Heart Fail. Rep. 14, 321–330. doi: 10.1007/s11897-017-0346-8
Veerman, C. C., Mengarelli, I., Lodder, E. M., Kosmidis, G., Bellin, M., Zhang, M., et al. (2017). Switch from fetal to adult SCN5A isoform in human induced pluripotent stem cell-derived cardiomyocytes unmasks the cellular phenotype of a conduction disease-causing mutation. J. Am. Heart Assoc. 6:e005135. doi: 10.1161/JAHA.116.005135
Veerman, C. C., Wilde, A. A., and Lodder, E. M. (2015). The cardiac sodium channel gene SCN5A and its gene product NaV1.5: role in physiology and pathophysiology. Gene 573, 177–187. doi: 10.1016/j.gene.2015.08.062
Veltmann, C., Barajas-Martinez, H., Wolpert, C., Borggrefe, M., Schimpf, R., Pfeiffer, R., et al. (2016). Further insights in the most common SCN5A mutation causing overlapping phenotype of long QT syndrome, Brugada syndrome, and conduction defect. J. Am. Heart Assoc. 5:e003379. doi: 10.1161/JAHA.116.003379
Vermeesch, J. R., Voet, T., and Devriendt, K. (2016). Prenatal and pre-implantation genetic diagnosis. Nat. Rev. Genet. 17, 643–656. doi: 10.1038/nrg.2016.97
Verstraelen, T. E., Ter Bekke, R. M., Volders, P. G., Masclee, A. A., and Kruimel, J. W. (2015). The role of the SCN5A-encoded channelopathy in irritable bowel syndrome and other gastrointestinal disorders. Neurogastroenterol. Motil. 27, 906–913. doi: 10.1111/nmo.12569
Viswanathan, P. C., Benson, D. W., and Balser, J. R. (2003). A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J. Clin. Invest. 111, 341–346. doi: 10.1172/JCI200316879
Wan, X., Chen, S., Sadeghpour, A., Wang, Q., and Kirsch, G. E. (2001). Accelerated inactivation in a mutant Na(+) channel associated with idiopathic ventricular fibrillation. Am. J. Physiol. Heart Circ. Physiol. 280, H354–H360. doi: 10.1152/ajpheart.2001.280.1.H354
Wang, D. W., Viswanathan, P. C., Balser, J. R., George, A. J., and Benson, D. W. (2002). Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction block. Circulation 105, 341–346. doi: 10.1161/hc0302.102592
Wang, L., Meng, X., Yuchi, Z., Zhao, Z., Xu, D., Fedida, D., et al. (2015). De novo mutation in the SCN5A gene associated with Brugada syndrome. Cell. Physiol. Biochem. 36, 2250–2262. doi: 10.1159/000430189
Wang, Q., Shen, J., Li, Z., Timothy, K., Vincent, G. M., Priori, S. G., et al. (1995a). Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum. Mol. Genet. 4, 1603–1607. doi: 10.1093/hmg/4.9.1603
Wang, Q., Shen, J., Splawski, I., Atkinson, D., Li, Z., Robinson, J. L., et al. (1995b). SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80, 805–811. doi: 10.1016/0092-8674(95)90359-3
Wilde, A. A., Moss, A. J., Kaufman, E. S., Shimizu, W., Peterson, D. R., Benhorin, J., et al. (2016). Clinical aspects of type 3 long-QT syndrome: an international multicenter study. Circulation 134, 872–882. doi: 10.1161/CIRCULATIONAHA.116.021823
Wilders, R. (2018). Sinus bradycardia in carriers of the SCN5A-1795insD mutation: unraveling the mechanism through computer simulations. Int. J. Mol. Sci. 19:634. doi: 10.3390/ijms19020634
Yamagata, K., Horie, M., Aiba, T., Ogawa, S., Aizawa, Y., Ohe, T., et al. (2017). Genotype-phenotype correlation of SCN5A mutation for the clinical and electrocardiographic characteristics of probands with Brugada syndrome: a Japanese multicenter registry. Circulation 135, 2255–2270. doi: 10.1161/CIRCULATIONAHA.117.027983
Yang, P., Kanki, H., Drolet, B., Yang, T., Wei, J., Viswanathan, P. C., et al. (2002). Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation 105, 1943–1948. doi: 10.1161/01.CIR.0000014448.19052.4C
Zeng, Z., Zhou, J., Hou, Y., Liang, X., Zhang, Z., Xu, X., et al. (2013). Electrophysiological characteristics of a SCN5A voltage sensors mutation R1629Q associated with Brugada syndrome. PLoS ONE 8:e78382. doi: 10.1371/journal.pone.0078382
Zhang, Q., Deng, C., Rao, F., Modi, R. M., Zhu, J., Liu, X., et al. (2013). Silencing of desmoplakin decreases connexin43/Nav1.5 expression and sodium current in HL1 cardiomyocytes. Mol. Med. Rep. 8, 780–786. doi: 10.3892/mmr.2013.1594
Keywords: SCN5A, Nav1.5, cardiac disorders, cardiac sodium channelopathy, therapeutic potential
Citation: Li W, Yin L, Shen C, Hu K, Ge J and Sun A (2018) SCN5A Variants: Association With Cardiac Disorders. Front. Physiol. 9:1372. doi: 10.3389/fphys.2018.01372
Received: 24 June 2018; Accepted: 10 September 2018;
Published: 09 October 2018.
Edited by:
Jiashin Wu, University of South Florida, United StatesReviewed by:
Dan Hu, Renmin Hospital, Wuhan University, ChinaCopyright © 2018 Li, Yin, Shen, Hu, Ge and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aijun Sun, c3VuLmFpanVuQHpzLWhvc3BpdGFsLnNoLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.