 Andrea Gerbino
Andrea Gerbino Giuseppe Procino1
Giuseppe Procino1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 25 September 2018
Sec. Striated Muscle Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01356
This article is part of the Research TopicNuclear Envelope Function in Striated Muscle Health and DiseaseView all 6 articles
Nuclear lamin A/C are crucial components of the intricate protein mesh that underlies the inner nuclear membrane and confers mainly nuclear and cytosolic rigidity. However, throughout the years a number of other key physiological processes have been associated with lamins such as modulation of both genes expression and the activity of signaling mediators. To further solidify its importance in cell physiology, mutations in the lamin A/C gene (LMNA) have been associated to diverse pathological phenotypes with skeletal muscles and the heart being the most affected systems. When affected, the heart develops a wide array of phenotypes spanning from dilated cardiomyopathy with conduction defects to arrhythmogenic right ventricular cardiomyopathy. The surprising large number of cardiac phenotypes reflects the equally large number of specific mutations identified in the LMNA gene. In this review, we underlie how mutations in LMNA can impact the activity and the spatial/temporal organization of signaling mediators and transcription factors. We analyzed the ever-increasing amount of findings collected in LmnaH222P/H222P mice whose cardiomyopathy resemble the most important features of the disease in humans and a number of key evidences from other experimental models. With this mini review, we attempt to combine the newest insights regarding both the pathogenic effects of LMNA mutations in terms of signaling abnormalities and cardiac laminopathies.
A-type nuclear lamins are 3.5 nm diameter type V intermediate filament proteins expressed in the majority of differentiated mammalian somatic cells, including cardiomyocytes (Butin-Israeli et al., 2012). Lamin A and C are encoded by the same gene (LMNA, cytogenetic location: 1q22) thanks to alternative RNA splicing events (Lin and Worman, 1993) and targeted to the nucleus by a nuclear localization signal (NLS). Both proteins polymerize with B-type lamins to form the nuclear lamina (NL) a scaffold apposed to the inner nuclear membrane (INM) of metazoan cells. For more information regarding cellular and tissue expression of all lamin isoforms, their structure and assembly readers are referred to these reviews (Ho and Lammerding, 2012; Schreiber and Kennedy, 2013). Recently, Turgay et al. (2017), used cryo-electron tomography to gain insight about the architecture of the thin lamin meshwork within the NL. This mesh of proteins confers cellular and nuclear integrity against mechanical cues. Additionally, thanks to its interaction with over 100 cytosolic and intra nuclear membrane/nucleoplasmic proteins(Gerace and Tapia, 2018), the NL allows a wide number of other functions such as mechano-transduction, chromatin protection/organization, regulation of signaling, and gene expression (Carmosino et al., 2014; Swift and Discher, 2014; Gerace and Tapia, 2018). The unexpected involvement of the NL in a large array of physiological processes triggered the scientific interest and these proteins became the focus of a multidisciplinary field.
Lamin A/C gene is among the most common cardiomyopathy-causing gene (van Tintelen et al., 2007a). Hundreds of distinct LMNA mutations have been associated with 15 heritable and organ-specific or multisystem disease phenotypes such as accelerated aging or overlapping syndromes (Cattin et al., 2013b) which are usually identified as laminopathies (Scharner et al., 2018). Mostly, these diseases affect specifically the striated muscle with a recurrent involvement of the heart that develops different type of arrhythmogenic cardiomyopathies with high interfamilial heterogeneity being dilated cardiomyopathy with conduction defects (DCM-CD) the most prevalent. However, over the years, LMNA mutations have been also associated with a combination of morpho-functional phenotypes between DCM and arrhythmogenic right ventricular cardiomyopathy (ARVC) (Forleo et al., 2015), underlining the urge for a new (and wider) classification of cardiac laminopathies (Arbustini et al., 2013). Strikingly, in all the cardiac phenotypes reported, structural abnormalities and electrical instability always coexist being myocardial fibrosis a key player in the development of both types of cardiac impairment.
Cardiomyopathies induced by mutations in LMNA have a very aggressive and fast clinical course that could culminate with sudden cardiac death from malignant ventricular arrhythmias and end-stage heart failure occurring at earlier ages compared to other familial cardiomyopathies. Current therapies involve implantable pacemakers and defibrillators to manage arrhythmia and conduction defects in order to prevent sudden cardiac death (Peretto et al., 2018). The pharmacological interventions commonly focus on the symptoms of congestive heart failure. These therapies extend the survival rate of affected patients; however, they only improve cardiac function and decrease the complications and secondary features of the disease without focusing on the specific mutation causing the pathology. Unfortunately, the design of patient-specific therapeutic approaches is often hindered by the lack of insight regarding the underlying pathogenic mechanisms.
Two not competing hypothesis could explain the etiology of laminopathies. The mechanical stress hypothesis proposes that the reduced nuclear rigidity due to Lamin A/C mutations could increase cellular susceptibility against recurrent mechanical stress and reduce mechano-transduction, especially in cells subjected to mechanical forces such those of skeletal muscle and cardiomyocytes (Carmosino et al., 2014). On the other hand, the gene expression hypothesis proposes that mutation-induced defects in proteins of the nuclear envelope might lead to changes in signaling pathways and abnormal control of gene expression which, in turn, could be associated to skeletal muscle diseases (Chatzifrangkeskou et al., 2015) and cardiomyopathies (Worman, 2017).
Here we describe the newest insights regarding the mechanisms by which LMNA mutations impact diverse cardiac signaling pathways and intracellular mediators in an updated brief review.
Specific LMNA mutations can induce alterations in the interplay between lamins themselves or their interactors of the NL and core signaling factors potentially involved in transcriptional regulation in both cells and animal tissues (Worman, 2017). This may trigger diverse pathophysiological mechanisms that underpin the broad spectrum of clinical phenotypes described in the above paragraph. The lack of cardiac tissue samples from affected patients has limited our ability to identify the precise molecular mechanisms through which each lamin mutation functions, however, the generation of the first transgenic lamin A/C knockout mouse paved the road for overcoming this limitation (Sullivan et al., 1999). Since then, different transgenic mice, baring specific LMNA mutations, have been generated and reviewed in Brayson and Shanahan (2017). For instance, most of the evidences regarding all the signaling pathways and the specific targets modulated in the striated muscle by a single LMNA mutation rely on a single mouse model containing a knock-in mutation in lamin A/C gene (H222P, Figure 1) causing muscular dystrophy and DCM (Arimura et al., 2005). LmnaH222P/H222P mice recapitulate some of the most important features of cardiac laminopathies in humans such as left ventricular dilation and systolic dysfunction with male mice developing the disease with faster kinetics relative to female mice (Choi and Worman, 2014; Worman, 2017).
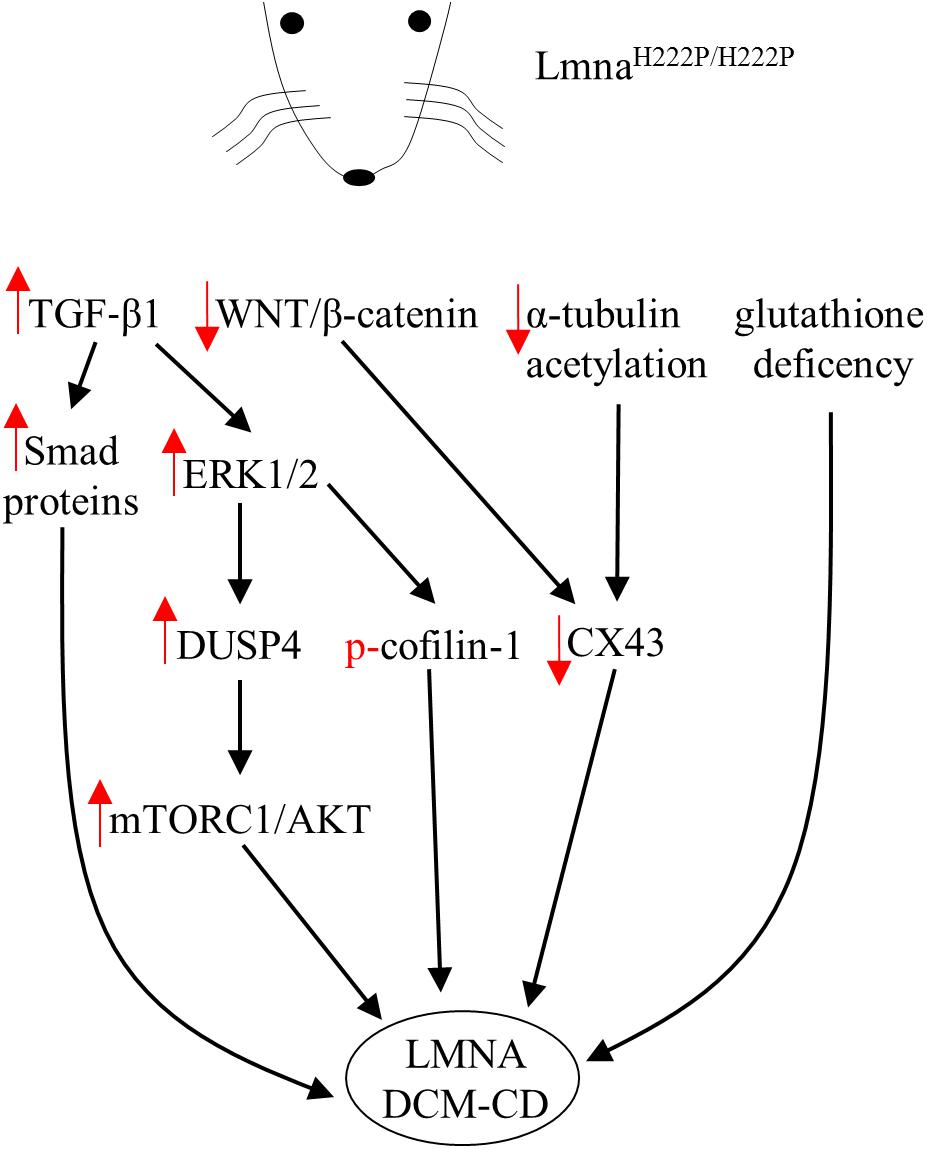
FIGURE 1. Diagram showing signaling mediators and cellular targets affected in hearts of LmnaH222P/H222P mice. Red arrows indicate the effects (increase/decrease) induced by the mutation in terms of expression level or activity.
Lamins are well-known molecular scaffolds that tether at the nuclear envelope mitogen-activated protein (MAP) kinase extracellular signal-regulated kinase (ERK) 1/2 and its downstream transcription factor c-Fos, a major regulator of important cellular processes such as cell proliferation, death, survival, and differentiation (Shaulian and Karin, 2002; Eferl and Wagner, 2003). Mutation-induced alterations in lamin A/C structure and or function might perturb the nuclear envelope geometry sufficiently to directly affect ERK1/2 activity and consequently its downstream signaling targets. In fact, ERK1/2 enhanced phosphorylation and nuclear sequestration were reported in the heart of patients with cardiolaminopathy (Muchir et al., 2007). In the hearts of LmnaH222P/H222P mice, hyper-activation of ERK1/2, JNK and P38α was reported before the onset of significant cardiac impairments (Muchir et al., 2007, 2012a) likely indicating that MAP kinases hyperactivation is not a consequence but rather a causative event of the cardiac disease. Strikingly, genetic or pharmacological approaches used to control ERK1/2 signaling in LmnaH222P/H222P mice had a positive outcome on cardiac symptoms led to survival prolongation (Muchir et al., 2012a; Wu et al., 2011, 2014, 2017). In addition, several downstream target genes were activated by MAPKs in hearts of LmnaH222P/H222P mice (Wu et al., 2011, 2017; Muchir et al., 2012a; Chatzifrangkeskou et al., 2016). All these targets might in turn, modulate the expression of additional genes encoding for proteins potentially involved in pathogenic mechanisms of cardiac diseases (Gillespie-Brown et al., 1995; Thorburn et al., 1995). The mechanisms by which expression of LmnaH222P leads to ERK1/2 signaling hyperactivation are under investigation. However, in the heart of LmnaH222P/H222P mice, ERK1/2 activation was functionally preceded by the elevation of both the transforming growth factor beta-1 (TGF-β1), an important mediator of fibrosis and extracellular matrix deposition (Rosenkranz, 2004), and its nuclear effectors, Smad proteins (Chatzifrangkeskou et al., 2016). Of note, treatment with a TGF-β receptor blocker lowered the amount of activated ERK1/2, indicating an increased TGF-β1 upstream ERK1/2 activation, as already reported for other cardiac pathophysiological states (Huang et al., 2015).
Recently, Chatzifrangkeskou et al. (2018) demonstrated that active cytosolic ERK1/2 can directly interact and phosphorylate cofilin-1, an F-actin depolymerizing factor. When phosphorylated, cofilin-1 impacts actin dynamics, which in turn leads defective actin organization in sarcomeres and alterations in contractile force generation of the left ventricular. Of note, left ventricular tissue from wild type mice injected with adeno-associated viruses expressing cofilin-1 showed myofibrillar disruption similar to that observed in LmnaH222P/H222P mice. Thus, pharmacological compounds able to correct defective actin dynamics could be used as new therapeutic approach to improve left ventricular dysfunction in LMNA-mediated cardiomyopathy (Chatzifrangkeskou et al., 2018).
One of the direct consequences of ERK1/2 hyperactivation and nuclear sequestration is the increased expression of the dual specific protein phosphatase 4 (DUSP4) together with left ventricular dilation markers in ventricular tissue of LmnaH222P/H222P (Choi et al., 2012b, 2018). DUSP4 is an ERK1/2-specific phosphatase predominantly located in the nucleus where it is involved in regulating the ERK1/2 cascade at nuclear level. Of note, genetic deletion of DUSP4 ameliorated heart function and prolonged survival in LmnaH222P/H222P mice (Choi et al., 2018). So far, this phosphatase is the leading molecular link between MAP kinases and the hyperactivation of protein kinase B/mammalian target of rapamycin complex 1 (mTORC1) signaling pathway in LmnaH222P/H222P mice (Choi et al., 2012b) and likely related to reduced autophagy, a key process for lifespan extension (Eisenberg et al., 2009; Morselli et al., 2010). Strikingly, treatment with temsirolimus, a specific inhibitor of mTOR, restores autophagy and prevents cardiac defects in LmnaH22P/H22P mice (Choi et al., 2012a).
Furthermore, LmnaH222P/H222P mice also showed reduced expression of WNT and β-catenin (Muchir et al., 2007, 2012b; Le Dour et al., 2017), two signaling mediators involved in several cellular mechanisms leading to proliferation, differentiation and apoptosis. β-catenin is usually phosphorylated, in the absence of WNT stimulation, by GSK-3β and degraded after ubiquitination by the proteasome. However, upon WNT activation, GSK-3β is inactivated and β-catenin firstly accumulates in the cytosol and then translocates to the nucleus where it forms a complex with transcription factors, which in turn activate target genes. In the heart β-catenin can interact with the gene encoding connexin 43 (CX43), an important cardiac connexin, to increase its transcription. In addition, β-catenin can also interact with CX43 itself, as part of a multiproteic complex within the intercalated disk, to stabilize its structure (Le Dour et al., 2017). Thus, as shown in LmnaH222P/H222P mice, the reduced CX43 expression seems tightly dependent by the deficient WNT/β-catenin signaling, since the pharmacological rescue of β-catenin obtained by blocking its specific inhibitor GSK-3β, restores the levels of CX43. Accordingly, mice lacking Lamin A have also decreased CX43 levels, while mutant mice homozygous for the Lmna N195K variation have similar conduction abnormalities induced by altered expression and distributions of both CX40 and CX43 and misregulation of HF1b/Sp4 a transcription factor of the Sp family (Mounkes et al., 2005). In addition, CX43 protein expression was reduced by about 40% in neonatal rat cardiomyocytes expressing Lmna E82K (Chen et al., 2013). Furthermore, alterations of the cytoskeleton through reduced acetylation of α-tubulin have to be taken into account when considering the abnormal distribution of CX43 in cardiomyocytes and cardiac tissue from LmnaH222P/H222P mice. Treatment with paclitaxel, in fact, stabilized the microtubule network via increased acetylation of α-tubulin and this, in turn, helped the correct localization of CX43 at intercalated disks improving conduction defects in LmnaH222P/H222P hearts (Macquart et al., 2018).
Finally, Rodriguez et al. (2018) showed that the cardiac phenotype associated with LmnaH222P/H222P underlines abnormal oxidative stress levels and glutathione deficiency. Of note, pharmacological glutathione replenishment lowered cardiac oxidative stress damage and mitigates contractile dysfunction in LmnaH222P/H222P mice (Rodriguez et al., 2018).
All the findings collected using LmnaH222P/H222P mice unveiled the unexpected impact that a single mutation of a structural gene may have in cell signaling. This animal model made us realize of the urge for the generation of new transgenic models in order to evaluate the impact of other mutations of lamin A/C. Hopefully, the collected information will help us discover the molecular mechanisms that underpin the surprising functional complexity of this protein.
Additional insights on the pathogenic mechanisms induced by LMNA mutations have been obtained using other experimental models. Specific LMNA mutations can significantly impact the dynamic equilibrium between cell death and survival in response to mechanical or metabolic cues. Cell death through apoptosis causes continuous loss of myocytes and leads to alterations of cardiac conduction and dispersion of refractoriness (Sen-Chowdhry and McKenna, 2012), potentially resulting in sudden cardiac death (James, 1994). In addition, reduction in cardiomyocytes content, likely compensated by additional fibrosis, may represent a substrate for conduction block and re-entrant arrhythmias (Basso et al., 2012; Sen-Chowdhry and McKenna, 2012). Indeed, previous reports have shown that LMNA mutation carriers with conduction defects and arrhythmias have histopathological signs of myocardial fibrosis that involves the cardiac conduction system (Fatkin et al., 1999; Arbustini et al., 2002; van Tintelen et al., 2007b).
Indeed, LmnaE82K/E82K transgenic mice were affected by DCM-CD, myocyte disarray and collagen accumulation. Lmna E82K was mislocalized in the nucleus with impaired nuclear envelope integrity, swollen mitochondria with reduced number of cristae. Of note, expression of Lmna E82K increased 9 fold the apoptosis rate, probably as consequence of first apoptosis signal receptor (FAS) overexpression, with activation of caspase-8/caspase-3 and caspase-9-dependent release of cytochrome c from mitochondria (Lu et al., 2010).
We identified in members of an Italian family a novel heterozygous lamin A/C variant consisting of a in-frame duplication of 21 nucleotides in the exon 2 of the LMNA gene (Forleo et al., 2015). This mutation co-segregates with arrhythmogenic cardiomyopathy of different phenotypes with high intra-familial variability. The in vitro characterization of this new LMNA variant showed a decreased nuclear stability and impaired nuclear-cytoskeletal coupling, resulting in a higher susceptibility to nuclear rupture and stress-induced cardiomyocyte apoptosis as the main pathogenic mechanism (Figure 2A).
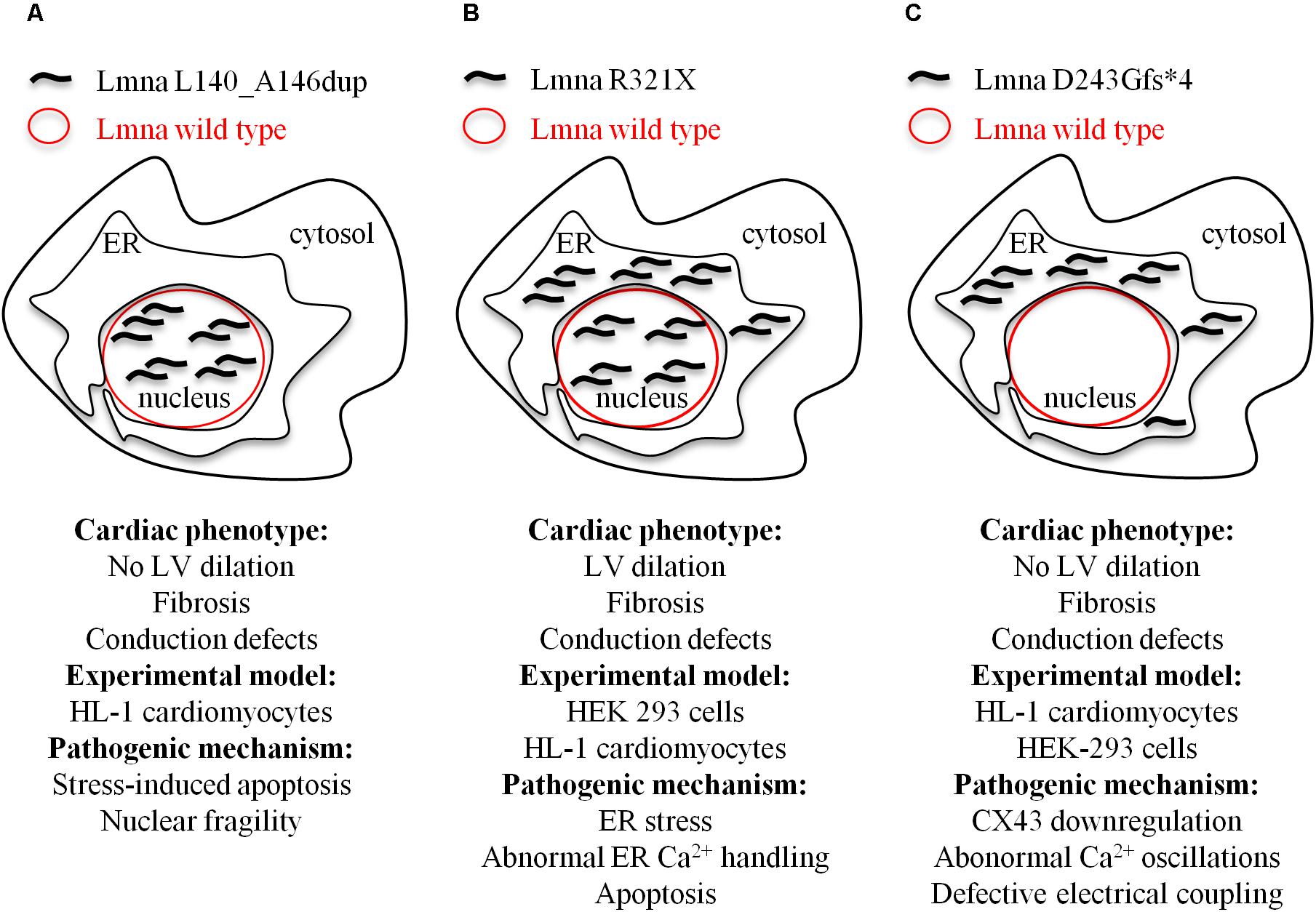
FIGURE 2. Model depicting the cellular mislocalization of the three mutants of lamin A in light of the cardiac phenotypes of the probands and the experimental models used for the evaluation of the pathogenic mechanisms at cellular level. (A) Lmna L140_A146dup; (B) Lmna R321X; and (C) D243Gfs∗4.
Cardiolaminopathies are not only the result of missense mutations. Mutations like nonsense or splice-site or insertions/deletions are more rare (van Rijsingen et al., 2012; Kawakami et al., 2018) but result in more severe cardiac events. We characterized at cellular level two different nonsense mutations, Lmna R321X (Carmosino et al., 2016) and Lmna D243Gfs∗4 (Gerbino et al., 2017), both segregating with cardiomyopathies with conduction defects (Figures 2B,C). Both mutations generated a premature termination codon before the NLS. When exogenously expressed in HEK293 or HL-1 cardiomyocytes both mutants mislocalized and accumulated within the endoplasmic reticulum (ER). Of note, Lmna R321X ER-accumulation led to unfolded protein response (UPR), an adaptive mechanism involving a complex transcriptional program that induces the hyperphosphorylation of PERK, a kinase localized on the ER membrane (Oslowski and Urano, 2011). PERK phosphorylates eIF2 leading to the expression of transcription factors such as CHOP, ultimately promoting the expression of proapoptotic genes. In our study, we found increased expression of both p-PERK and CHOP in cells expressing Lmna R321X and increased levels of p-PERK in samples of the heart explanted from a mutation carrier. If not compensated, UPR leads to apoptosis since the cellular expression of Lmna R321X itself was able to significantly increase the apoptotic rate when compared with cells expressing the wild type Lamin A. Thus, the fibro-fatty replacement of the cardiac tissue could well explain the conduction defect diagnosed in patients carrying this specific mutation (Carmosino et al., 2016). An alternative pathogenic mechanism that leads to DCM in LmnaΔK32/+ mice was showed by Cattin et al. (2013a). These authors proposed a scenario in which haploinsufficiency induced by ΔK32 degradation caused cardiac remodeling which, in turn, impaired UPR. Consequently, cardiac function worsened leading to DCM due to toxic accumulation of both lamin wild type and ΔK32 (Cattin et al., 2013a).
When we functionally characterized the second nonsense LMNA mutation, we found that ER Lmna D243Gfs∗4 accumulation did not induce neither UPR/ER stress nor apoptosis (Gerbino et al., 2017). The fact that the Lmna D243Gfs∗4 mutant was silent in terms of UPR suggested its aggregation in a more tolerated unfolded state likely because of the lack of the whole coil 2b region, which is involved in the polymeric head-to-coil assembly of Lamin monomers (Strelkov et al., 2004). While performing experiments to evaluate the impact of ER Lmna D243Gfs∗4 accumulation on intracellular Ca2+ dynamics we measured a significant impairment of Ca2+ oscillations both in the cardiomyocyte carrying the mutation and in the cardiomyocyte directly coupled with it, indicating defective electrical coupling between these cardiac cells. Under this scenario, we showed that HL-1 cardiomyocytes expressing Lmna D243Gfs∗4 had a significant reduction in CX43 expression level that was, however, independent of both β-catenin expression level and cytoskeleton polymerization (measured as RhoA activity). Of note, partial restoration of CX43 activity in Lmna D243Gfs∗4-expressing cells by lithium improved cell-to-cell signal propagation suggesting CX43 as potential pharmaceutical target for this form of cardiolaminopathy.
During the functional characterization of both truncated mutants we uncovered critical changes in intracellular Ca2+ dynamics. On one hand, Lmna R321X ER accumulation led to an abnormal Ca2+ handling by the cell; ER Ca2+ content was significantly reduced as well as the Ca2+-mediated agonist responses in both the cytosol and the nucleus. On the other hand, Lmna D243Gfs∗4 expression affected the regular Ca2+ oscillatory pattern of cardiomyocytes through a CX-43 dysfunction. Thus, the effect of LMNA mutations on Ca2+ changes might represent a new and under-investigated event in the pathogenesis of LMNA-mediated cardiomyopathies. Of note, Arimura et al. (2010) showed that SCH00013, a Ca2+ sensitizer able to increase muscular Ca2+ sensitivity without increasing cytosolic Ca2+ levels, ameliorated systolic dysfunction, reduced cardiac interstitial fibrosis and modulated the expression of genes involved in cardiac remodeling when administrated in LmnaH222P/H222P mice. Nuclear and cytosolic Ca2+ changes can activate Ca2+-dependent PKC isoforms either resident within the nucleus or translocated from the cytosol, respectively. Interestingly, lamin A has at least two consensus sites for PKC and others for kinases that can be modulated by Ca2+ (Machowska et al., 2015).
Overall, we believe that the exact dissection of all the cellular pathogenic mechanisms induced by mutations in lamin A/C gene will pave the road toward personalized treatment for these cardiomyopathies. Patients carrying different mutations with the same cardiac phenotype should not be therapeutically approached with the same strategy. For instance, cardiac fibrosis is not only induced by apoptosis but also by abnormal CX43 expression triggers due to enhanced activity of non-excitable cardiac fibroblasts (Jansen et al., 2012). Of note, normalization of CX43 expression might prevent fibrosis and reduce the susceptibility to fatal arrhythmia. Thus, a complete palette of all the pathogenic mechanisms involved in LMNA-mediated cardiomyopathies will help cardiologists to design more precise and effective therapeutic protocols.
AG conceived the idea of this article. AG and MC wrote and edited the manuscript. Both GP and MS reviewed and critically edited the content of the manuscript.
This work was supported by the Master in “Structural Osteopathy” and by “Fondi per la Ricerca di interesse Locale” both of the University of Basilicata to MC. This research was also supported by funding from the CLUSTER TECNOLOGICO REGIONALE “DICLIMAX” (project # MTJU9H8) to MS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DB and handling Editor declared their shared affiliation at the time of the review.
We thank K. Lefkimmiatis for critically reading the manuscript and valuable feedback.
Arbustini, E., Narula, N., Dec, G. W., Reddy, K. S., Greenberg, B., Kushwaha, S., et al. (2013). The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J. Am. Coll. Cardiol. 62, 2046–2072. doi: 10.1016/j.jacc.2013.08.1644
Arbustini, E., Pilotto, A., Repetto, A., Grasso, M., Negri, A., Diegoli, M., et al. (2002). Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J. Am. Coll. Cardiol. 39, 981–990.
Arimura, T., Helbling-Leclerc, A., Massart, C., Varnous, S., Niel, F., Lacène, E., et al. (2005). Mouse model carrying H222P- Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 14, 155–169. doi: 10.1093/hmg/ddi017
Arimura, T., Sato, R., Machida, N., Bando, H., Zhan, D.-Y., Morimoto, S., et al. (2010). Improvement of left ventricular dysfunction and of survival prognosis of dilated cardiomyopathy by administration of calcium sensitizer SCH00013 in a mouse model. J. Am. Coll. Cardiol. 55, 1503–1505. doi: 10.1016/j.jacc.2009.10.065
Basso, C., Bauce, B., Corrado, D., and Thiene, G. (2012). Pathophysiology of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 9, 223–233. doi: 10.1038/nrcardio.2011.173
Brayson, D., and Shanahan, C. M. (2017). Current insights into LMNA cardiomyopathies: existing models and missing LINCs. Nucleus 8, 17–33. doi: 10.1080/19491034.2016.1260798
Butin-Israeli, V., Adam, S. A., Goldman, A. E., and Goldman, R. D. (2012). Nuclear lamin functions and disease. Trends Genet. 28, 464–471. doi: 10.1016/j.tig.2012.06.001
Carmosino, M., Gerbino, A., Schena, G., Procino, G., Miglionico, R., Forleo, C., et al. (2016). The expression of Lamin A mutant R321X leads to endoplasmic reticulum stress with aberrant Ca2+ handling. J. Cell. Mol. Med. 20, 2194–2207. doi: 10.1111/jcmm.12926
Carmosino, M., Torretta, S., Procino, G., Gerbino, A., Forleo, C., Favale, S., et al. (2014). Role of nuclear Lamin A/C in cardiomyocyte functions. Biol. Cell 106, 346–358. doi: 10.1111/boc.201400033
Cattin, M.-E., Bertrand, A. T., Schlossarek, S., Le Bihan, M.-C., Skov Jensen, S., Neuber, C., et al. (2013a). Heterozygous LmnadelK32 mice develop dilated cardiomyopathy through a combined pathomechanism of haploinsufficiency and peptide toxicity. Hum. Mol. Genet. 22, 3152–3164. doi: 10.1093/hmg/ddt172
Cattin, M.-E., Muchir, A., and Bonne, G. (2013b). “State-of-the-heart” of cardiac laminopathies. Curr. Opin. Cardiol. 28, 297–304. doi: 10.1097/HCO.0b013e32835f0c79
Chatzifrangkeskou, M., Bonne, G., and Muchir, A. (2015). Nuclear envelope and striated muscle diseases. Curr. Opin. Cell Biol. 32, 1–6. doi: 10.1016/j.ceb.2014.09.007
Chatzifrangkeskou, M., Le Dour, C., Wu, W., Morrow, J. P., Joseph, L. C., Beuvin, M., et al. (2016). ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum. Mol. Genet. 25, 2220–2233. doi: 10.1093/hmg/ddw090
Chatzifrangkeskou, M., Yadin, D., Marais, T., Chardonnet, S., Cohen-Tannoudji, M., Mougenot, N., et al. (2018). Cofilin-1 phosphorylation catalyzed by ERK1/2 alters cardiac actin dynamics in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 27, 3060–3078. doi: 10.1093/hmg/ddy215
Chen, S. C., Kennedy, B. K., and Lampe, P. D. (2013). Phosphorylation of connexin43 on S279/282 may contribute to laminopathy-associated conduction defects. Exp. Cell Res. 319, 888–896. doi: 10.1016/j.yexcr.2012.12.014
Choi, J. C., Muchir, A., Wu, W., Iwata, S., Homma, S., Morrow, J. P., et al. (2012a). Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 4:144ra102. doi: 10.1126/scitranslmed.3003875
Choi, J. C., Wu, W., Muchir, A., Iwata, S., Homma, S., and Worman, H. J. (2012b). Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J. Biol. Chem. 287, 40513–40524. doi: 10.1074/jbc.M112.404541
Choi, J. C., and Worman, H. J. (2014). “Nuclear envelope regulation of signaling cascades,” in Cancer Biology and the Nuclear Envelope Advances in Experimental Medicine and Biology, eds E. C. Schirmer and J. I. de las Heras (New York, NY: Springer), 187–206. doi: 10.1007/978-1-4899-8032-8_9
Choi, J. C., Wu, W., Phillips, E., Plevin, R., Sera, F., Homma, S., et al. (2018). Elevated dual specificity protein phosphatase 4 in cardiomyopathy caused by lamin A/C gene mutation is primarily ERK1/2-dependent and its depletion improves cardiac function and survival. Hum. Mol. Genet. 27, 2290–2305. doi: 10.1093/hmg/ddy134
Eferl, R., and Wagner, E. F. (2003). AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868. doi: 10.1038/nrc1209
Eisenberg, T., Knauer, H., Schauer, A., Büttner, S., Ruckenstuhl, C., Carmona-Gutierrez, D., et al. (2009). Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314. doi: 10.1038/ncb1975
Fatkin, D., MacRae, C., Sasaki, T., Wolff, M. R., Porcu, M., Frenneaux, M., et al. (1999). Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 341, 1715–1724. doi: 10.1056/NEJM199912023412302
Forleo, C., Carmosino, M., Resta, N., Rampazzo, A., Valecce, R., Sorrentino, S., et al. (2015). Clinical and functional characterization of a novel mutation in lamin a/c gene in a multigenerational family with arrhythmogenic cardiac laminopathy. PLoS One 10:e0121723. doi: 10.1371/journal.pone.0121723
Gerace, L., and Tapia, O. (2018). Messages from the voices within: regulation of signaling by proteins of the nuclear lamina. Curr. Opin. Cell Biol. 52, 14–21. doi: 10.1016/j.ceb.2017.12.009
Gerbino, A., Bottillo, I., Milano, S., Lipari, M., Zio, R. D., Morlino, S., et al. (2017). Functional characterization of a novel truncating mutation in lamin A/C gene in a family with a severe cardiomyopathy with conduction defects. CPB 44, 1559–1577. doi: 10.1159/000485651
Gillespie-Brown, J., Fuller, S. J., Bogoyevitch, M. A., Cowley, S., and Sugden, P. H. (1995). The mitogen-activated protein kinase kinase MEK1 stimulates a pattern of gene expression typical of the hypertrophic phenotype in rat ventricular cardiomyocytes. J. Biol. Chem. 270, 28092–28096.
Ho, C. Y., and Lammerding, J. (2012). Lamins at a glance. J. Cell Sci. 125, 2087–2093. doi: 10.1242/jcs.087288
Huang, H., Tang, Y., Wu, G., Mei, Y., Liu, W., Liu, X., et al. (2015). ALK7 protects against pathological cardiac hypertrophy in mice. Cardiovasc. Res. 108, 50–61. doi: 10.1093/cvr/cvv206
James, T. N. (1994). Normal and abnormal consequences of apoptosis in the human heart. From postnatal morphogenesis to paroxysmal arrhythmias. Circulation 90, 556–573. doi: 10.1161/01.CIR.90.1.556
Jansen, J. A., van Veen, T. A. B., de Jong, S., van der Nagel, R., van Stuijvenberg, L., Driessen, H., et al. (2012). Reduced Cx43 expression triggers increased fibrosis due to enhanced fibroblast activity. Circ. Arrhythm. Electrophysiol. 5, 380–390. doi: 10.1161/CIRCEP.111.966580
Kawakami, H., Ogimoto, A., Tokunaga, N., Nishimura, K., Kawakami, H., Higashi, H., et al. (2018). A novel truncating LMNA mutation in patients with cardiac conduction disorders and dilated cardiomyopathy. Int. Heart J. 59, 531–541. doi: 10.1536/ihj.17-377
Le Dour, C., Macquart, C., Sera, F., Homma, S., Bonne, G., Morrow, J. P., et al. (2017). Decreased WNT/β-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/C gene. Hum. Mol. Genet. 26, 333–343. doi: 10.1093/hmg/ddw389
Lin, F., and Worman, H. J. (1993). Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 268, 16321–16326.
Lu, D., Lian, H., Zhang, X., Shao, H., Huang, L., Qin, C., et al. (2010). LMNA E82K mutation activates FAS and mitochondrial pathways of apoptosis in heart tissue specific transgenic mice. PLoS One 5:e15167. doi: 10.1371/journal.pone.0015167
Machowska, M., Piekarowicz, K., and Rzepecki, R. (2015). Regulation of lamin properties and functions: does phosphorylation do it all? Open Biol. 5:150094. doi: 10.1098/rsob.150094
Macquart, C., Jüttner, R., Le Dour, C., Chatzifrangkeskou, M., Schmitt, A., Gotthardt, M., et al. (2018). Microtubule cytoskeleton regulates connexin 43 localization and cardiac conduction in cardiomyopathy caused by mutation in A-type lamins gene. Hum. Mol. Genet. doi: 10.1093/hmg/ddy227 [Epub ahead of print].
Morselli, E., Maiuri, M. C., Markaki, M., Megalou, E., Pasparaki, A., Palikaras, K., et al. (2010). Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 1:e10. doi: 10.1038/cddis.2009.8
Mounkes, L. C., Kozlov, S. V., Rottman, J. N., and Stewart, C. L. (2005). Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum. Mol. Genet. 14, 2167–2180. doi: 10.1093/hmg/ddi221
Muchir, A., Pavlidis, P., Decostre, V., Herron, A. J., Arimura, T., Bonne, G., et al. (2007). Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Invest. 117, 1282–1293. doi: 10.1172/JCI29042
Muchir, A., Reilly, S. A., Wu, W., Iwata, S., Homma, S., Bonne, G., et al. (2012a). Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc. Res. 93, 311–319. doi: 10.1093/cvr/cvr301
Muchir, A., Wu, W., Choi, J. C., Iwata, S., Morrow, J., Homma, S., et al. (2012b). Abnormal p38α mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 21, 4325–4333. doi: 10.1093/hmg/dds265
Oslowski, C. M., and Urano, F. (2011). Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 490, 71–92. doi: 10.1016/B978-0-12-385114-7.00004-0
Peretto, G., Sala, S., Benedetti, S., Resta, C. D., Gigli, L., Ferrari, M., et al. (2018). Updated clinical overview on cardiac laminopathies: an electrical and mechanical disease. Nucleus doi: 10.1080/19491034.2018.1489195 [Epub ahead of print].
Rodriguez, B. M., Khouzami, L., Decostre, V., Varnous, S., Pekovic-Vaughan, V., Hutchison, C. J., et al. (2018). N-acetyl cysteine alleviates oxidative stress and protects mice from dilated cardiomyopathy caused by mutations in nuclear A-type lamins gene. Hum. Mol. Genet. doi: 10.1093/hmg/ddy243 [Epub ahead of print].
Rosenkranz, S. (2004). TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc. Res. 63, 423–432. doi: 10.1016/j.cardiores.2004.04.030
Scharner, J., Brown, C. A., Bower, M., Iannaccone, S. T., Khatri, I. A., Escolar, D., et al. (2018). Novel LMNA mutations in patients with Emery-Dreifuss muscular dystrophy and functional characterization of four LMNA mutations. Hum. Mutat. 32, 152–167. doi: 10.1002/humu.21361
Schreiber, K. H., and Kennedy, B. K. (2013). When lamins go bad: nuclear structure and disease. Cell 152, 1365–1375. doi: 10.1016/j.cell.2013.02.015
Sen-Chowdhry, S., and McKenna, W. J. (2012). Sudden death from genetic and acquired cardiomyopathies. Circulation 125, 1563–1576. doi: 10.1161/CIRCULATIONAHA.111.025528
Shaulian, E., and Karin, M. (2002). AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136. doi: 10.1038/ncb0502-e131
Strelkov, S. V., Schumacher, J., Burkhard, P., Aebi, U., and Herrmann, H. (2004). Crystal structure of the human lamin A coil 2B dimer: implications for the head-to-tail association of nuclear lamins. J. Mol. Biol. 343, 1067–1080. doi: 10.1016/j.jmb.2004.08.093
Sullivan, T., Escalante-Alcalde, D., Bhatt, H., Anver, M., Bhat, N., Nagashima, K., et al. (1999). Loss of a-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147, 913–920.
Swift, J., and Discher, D. E. (2014). The nuclear lamina is meccano-responsive to ECM elasticity in mature tissue. J. Cell Sci. 127, 3005–3015. doi: 10.1242/jcs.149203
Thorburn, J., Carlson, M., Mansour, S. J., Chien, K. R., Ahn, N. G., and Thorburn, A. (1995). Inhibition of a signaling pathway in cardiac muscle cells by active mitogen-activated protein kinase kinase. Mol. Biol. Cell 6, 1479–1490.
Turgay, Y., Eibauer, M., Goldman, A. E., Shimi, T., Khayat, M., Ben-Harush, K., et al. (2017). The molecular architecture of lamins in somatic cells. Nature 543, 261–264. doi: 10.1038/nature21382
van Rijsingen, I. A. W., Arbustini, E., Elliott, P. M., Mogensen, J., Hermans-van Ast, J. F., van der Kooi, A. J., et al. (2012). Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers: a European cohort study. J. Am. Coll. Cardiol. 59, 493–500. doi: 10.1016/j.jacc.2011.08.078
van Tintelen, J. P., Hofstra, R. M. W., Katerberg, H., Rossenbacker, T., Wiesfeld, A. C. P., du Marchie Sarvaas, G. J., et al. (2007a). High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am. Heart J. 154, 1130–1139. doi: 10.1016/j.ahj.2007.07.038
van Tintelen, J. P., Tio, R. A., Kerstjens-Frederikse, W. S., van Berlo, J. H., Boven, L. G., Suurmeijer, A. J. H., et al. (2007b). Severe myocardial fibrosis caused by a deletion of the 5’ end of the lamin A/C gene. J. Am. Coll. Cardiol. 49, 2430–2439. doi: 10.1016/j.jacc.2007.02.063
Worman, H. J. (2017). Cell signaling abnormalities in cardiomyopathy caused by lamin A/C gene mutations. Biochem. Soc. Transac. 46, 37–42. doi: 10.1042/BST20170236
Wu, W., Chordia, M. D., Hart, B. P., Kumarasinghe, E. S., Ji, M. K., Bhargava, A., et al. (2017). Macrocyclic MEK1/2 inhibitor with efficacy in a mouse model of cardiomyopathy caused by lamin A/C gene mutation. Bioorg. Med. Chem. 25, 1004–1013. doi: 10.1016/j.bmc.2016.12.014
Wu, W., Iwata, S., Homma, S., Worman, H. J., and Muchir, A. (2014). Depletion of extracellular signal-regulated kinase 1 in mice with cardiomyopathy caused by lamin A/C gene mutation partially prevents pathology before isoenzyme activation. Hum. Mol. Genet. 23, 1–11. doi: 10.1093/hmg/ddt387
Keywords: nucleus, lamin A/C gene, nuclear lamina, nuclear envelope, gene expression, signaling pathways, Ca2+ signaling, cardiac pathophysiology
Citation: Gerbino A, Procino G, Svelto M and Carmosino M (2018) Role of Lamin A/C Gene Mutations in the Signaling Defects Leading to Cardiomyopathies. Front. Physiol. 9:1356. doi: 10.3389/fphys.2018.01356
Received: 10 July 2018; Accepted: 07 September 2018;
Published: 25 September 2018.
Edited by:
Julien Ochala, King’s College London, United KingdomReviewed by:
Han-Zhong Feng, Wayne State University School of Medicine, United StatesCopyright © 2018 Gerbino, Procino, Svelto and Carmosino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Gerbino, YW5kcmVhLmdlcmJpbm9AdW5pYmEuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.