
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 04 September 2018
Sec. Cardiac Electrophysiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01228
This article is part of the Research TopicCardiomyocyte Microdomains: An Emerging Concept of Local Regulation and RemodelingView all 18 articles
 Przemysław B. Radwański1,2,3,4*
Przemysław B. Radwański1,2,3,4* Christopher N. Johnson2,5
Christopher N. Johnson2,5 Sándor Györke1,2,3
Sándor Györke1,2,3 Rengasayee Veeraraghavan1,2,3,6*
Rengasayee Veeraraghavan1,2,3,6*A nanodomain is a collection of proteins localized within a specialized, nanoscale structural environment, which can serve as the functional unit of macroscopic physiologic processes. We are beginning to recognize the key roles of cardiomyocyte nanodomains in essential processes of cardiac physiology such as electrical impulse propagation and excitation–contraction coupling (ECC). There is growing appreciation of nanodomain dysfunction, i.e., nanopathy, as a mechanistic driver of life-threatening arrhythmias in a variety of pathologies. Here, we offer an overview of current research on the role of nanodomains in cardiac physiology with particular emphasis on: (1) sodium channel-rich nanodomains within the intercalated disk that participate in cell-to-cell electrical coupling and (2) dyadic nanodomains located along transverse tubules that participate in ECC. The beat to beat function of cardiomyocytes involves three phases: the action potential, the calcium transient, and mechanical contraction/relaxation. In all these phases, cell-wide function results from the aggregation of the stochastic function of individual proteins. While it has long been known that proteins that exist in close proximity influence each other’s function, it is increasingly appreciated that there exist nanoscale structures that act as functional units of cardiac biophysical phenomena. Termed nanodomains, these structures are collections of proteins, localized within specialized nanoscale structural environments. The nano-environments enable the generation of localized electrical and/or chemical gradients, thereby conferring unique functional properties to these units. Thus, the function of a nanodomain is determined by its protein constituents as well as their local structural environment, adding an additional layer of complexity to cardiac biology and biophysics. However, with the emergence of experimental techniques that allow direct investigation of structure and function at the nanoscale, our understanding of cardiac physiology and pathophysiology at these scales is rapidly advancing. Here, we will discuss the structure and functions of multiple cardiomyocyte nanodomains, and novel strategies that target them for the treatment of cardiac arrhythmias.
For over a century, it has been recognized that cardiac myocytes come into close contact at the intercalated disk (ID), where adjacent cells are only nanometers apart. By the mid-20th century, gap junctions (GJs) were identified as specialized structures within the ID, which afford electrochemical coupling between cells (Sjostrand and Andersson, 1954; Barr et al., 1965). The closest apposition between neighboring myocytes occurs within these GJs, with as little as 2 nm separating the cells. Despite this, GJs themselves do not constitute a functional nanodomain – they afford direct cytosolic continuity between coupled cells, and since neither cytosolic compartment is a restricted environment, the effects unique to nanoscale compartments do not come into play. Thus, cardiac conduction, the cell-to-cell spread of electrical excitation through the heart, was thought a relatively simple process with cardiac voltage-gated sodium (Na+) channels (NaV1.5) affording excitability, and GJs providing cell-to-cell electrical coupling (Kleber and Rudy, 2004).
This electrotonic view of cardiac conduction allowed the formalism of cable theory to be applied to cardiac conduction, and adequately explained experimental observations for several decades (Kleber and Rudy, 2004). However, findings have accumulated, that are not well-explained by this model. Transgenic mice with reduced Cx43 expression displayed significant conduction slowing in some studies (Guerrero et al., 1997; Eloff et al., 2001; Gutstein et al., 2001), but not in others (Morley et al., 1999; Vaidya et al., 2001; Thomas et al., 2003; Beauchamp et al., 2004; Danik et al., 2004; van Rijen et al., 2004; Stein et al., 2009). Likewise, GJ remodeling and reduced Cx43 expression correlated with arrhythmogenic conduction slowing in one pacing-induced canine heart failure model (Poelzing et al., 2004), but preceded it in another (Akar et al., 2007). Another discrepant finding stemmed from investigations of the electrophysiological impact of cardiac interstitial edema (fluid accumulation in the extracellular space): per the electrotonic model, an increase interstitial volume would lower extracellular resistance, and consequently, increase conduction velocity (Spach et al., 2004; Plonsey and Barr, 2007). However, experimental measurements in guinea pig ventricles revealed conduction slowing during to interstitial edema (Veeraraghavan et al., 2012, 2015, 2016) and edema was linked to reversible conduction block in patients undergoing ablation for the treatment of atrial fibrillation (AF) (Arujuna et al., 2012). These findings prompted speculation that non-electrotonic mechanisms of intercellular communication may play a role in the heart.
Theoretical studies had long raised the possibility of an alternate mode of intercellular coupling (Pertsov and Medvinskii, 1976; Sperelakis and Mann, 1977; Kucera et al., 2002; Sperelakis and McConnell, 2002; Copene and Keener, 2008; Mori et al., 2008). Dubbed ephaptic coupling, this mechanism envisions cells communicating via local extracellular electrochemical transients (ion accumulation/depletion). The aforementioned models suggest that a cardiac ephapse (a functional nanodomain capable of supporting ephaptic coupling) would require NaV1.5-rich membranes from neighboring cells, separated by a very narrow extracellular cleft (≤30 nm) (Veeraraghavan et al., 2014a,b). Whereas channels located at the lateral sarcolemma would face a large bulk of extracellular fluid with a practically inexhaustible supply of ions, channels facing narrow extracellular clefts would have a limited supply of ions. In the latter case, ion channel activity could mount local extracellular electrochemical transients, in turn altering the local transmembrane potential. In this context, early results demonstrating that NaV1.5 channels are enriched at the ID (Maier et al., 2004; Petitprez et al., 2011) prompted speculation that functional nanodomains capable of supporting ephaptic coupling, i.e., ephapses, may exist within the ID. Until recently, the 250–350 nm resolution limit imposed on confocal microscopy by diffraction had precluded precise localization of NaV1.5 within the ID. However, this restriction was removed by the advent of super-resolution microscopy techniques with resolutions extending down to 20 nm. Work conducted by the Gourdie (Rhett et al., 2012; Veeraraghavan et al., 2013, 2015; Veeraraghavan and Gourdie, 2016) and Delmar (Agullo-Pascual et al., 2013, 2014; Leo-Macias et al., 2016) labs using super-resolution techniques identified NaV1.5 enrichment within specific regions of the ID (Figure 1). One sub-population of ID-localized NaV1.5 was located at the perinexus, a specialized nanodomain located at the periphery of Cx43 GJs (Rhett et al., 2011; Rhett and Gourdie, 2012). A second localized to N-cadherin-rich plicate regions of the ID, where mechanical junctions are concentrated. Electron microscopy studies revealed disparate ultrastructural properties at these sites: within the perinexus, adjacent cell membranes were no more than 10–15 nm apart (Veeraraghavan et al., 2015), whereas at N-cadherin-rich mechanical junctions, intermembrane spacing was as high as 60–75 nm (Leo-Macias et al., 2015, 2016). The latter significantly exceeds the theoretically derived ≤ 30 nm intermembrane spacing limit for ephaptic coupling; however, the properties of the perinexus may enable it to function as an ephapse.
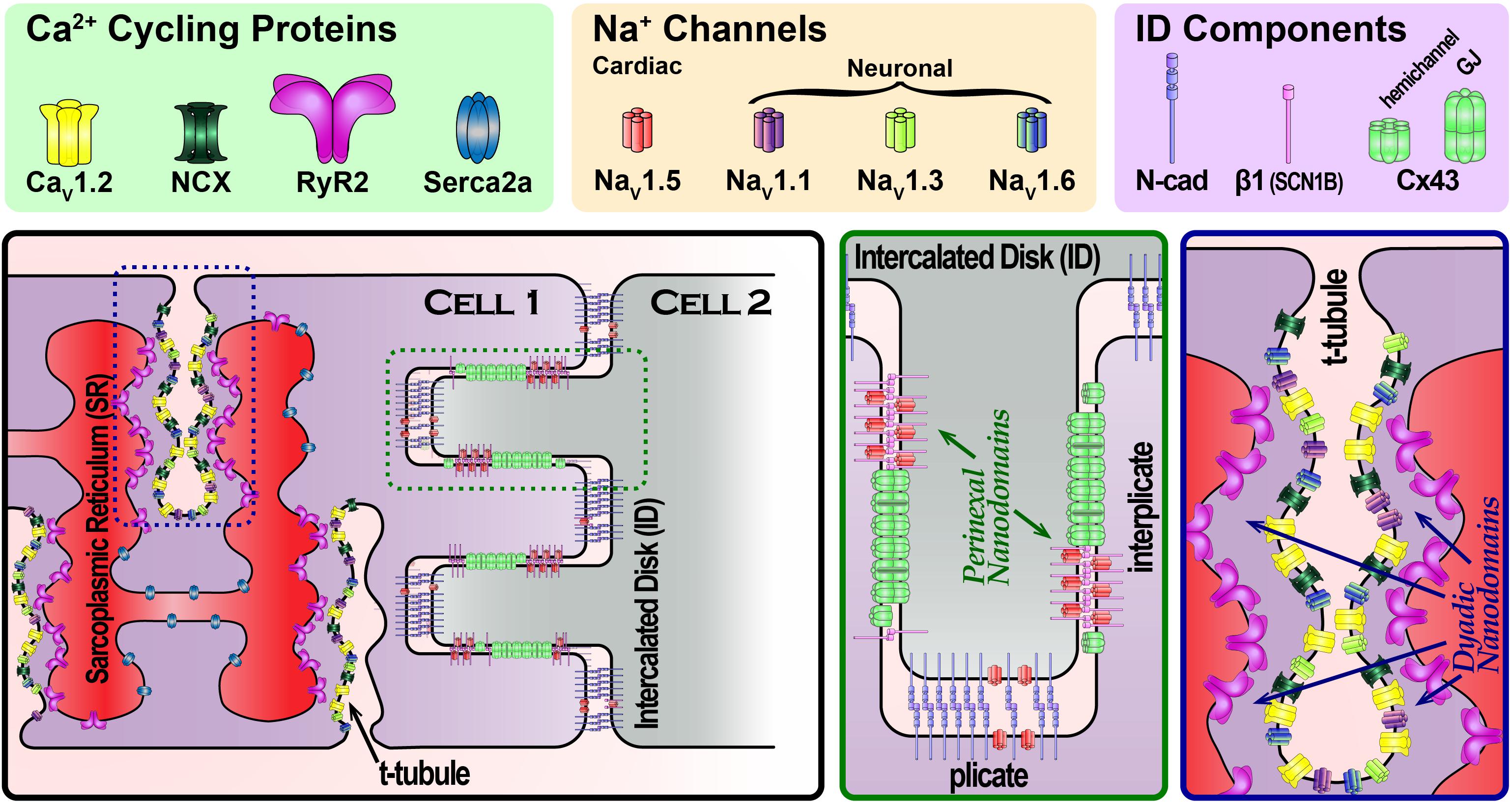
FIGURE 1. Schematic cartoon depicting the organization of Na+ channels in different parts of the cardiac myocyte, highlighting Na+ channel-rich nanodomains within the intercalated disk (green dashed box) and t-tubules (blue dashed box). Zoomed in views of these nanodomains are provided in the green and blue boxed panels.
Providing functional support for the ephaptic coupling hypothesis, the Poelzing and Gourdie groups demonstrated that acute interstitial edema selectively disrupted perinexal ultrastructure without altering other ID sites, and precipitated arrhythmogenic conduction slowing (Veeraraghavan et al., 2012, 2015, 2016). Additionally, perinexal disruption increased the dependence of conduction on GJ coupling and vice versa, suggesting that both modes of intercellular coupling operate in tandem (Veeraraghavan et al., 2012). Notably, in a setting of perinexal disruption, inhibition of channels enriched at perinexal sites – NaV1.5, and inward-rectifier potassium channels (Kir2.1) – resulted in anisotropic changes in conduction velocity, preferentially affecting transverse conduction (Veeraraghavan et al., 2015, 2016). In contrast, modulating ionic currents under normal conditions does not produce direction-specific impact. Keener and colleagues demonstrated that these experimental results could be predicted by a computational model incorporating both ephaptic and electrotonic coupling, but not by a purely electrotonic model (Lin and Keener, 2010, 2014; Veeraraghavan et al., 2015, 2016). Recently, Hichri and colleagues provided experimental and in silico evidence that the behavior of ID-localized NaV1.5 is modulated by their clustering, location within the ID (relative to the bulk interstitium), and the trans-alignment of these clusters (Hichri et al., 2017). In sum, these results illustrate macroscopic physiologic phenomena driven by the behavior of functional nanodomains, which can only be understood through knowledge of structure–function relationships at the nanodomain level.
In the larger context, the results highlighting the perinexus as a functional nanodomain involved in cardiac cell-to-cell communication pose the question: is the ID home to other functional nanodomains? The likelihood is high, given that the ID contains other sites enriched with NaV1.5 (Milstein et al., 2012; Veeraraghavan et al., 2015; Leo-Macias et al., 2016) and Kir2.1 (Milstein et al., 2012; Veeraraghavan et al., 2016; Ponce-Balbuena et al., 2018) as well as other ion channel species such as ATP-sensitive K+ channels (Kir6.2) (Hong et al., 2012). Notably, extracellular sodium has been shown to strongly modulate Kir2.1 function (Ishihara, 2018), hinting at the potential for complex interrelationships between ion channel species within ID nanodomains. Thus, ID-localized ion channels clearly merit further investigation, with particular emphasis on their protein neighbors and local structural environment, both within the cell and without. In this regard, available observations on the composition and subcellular location of NaV1.5-containing macromolecular complexes hint at significant complexity. NaV1.5 has been demonstrated to associate with ankyrin-G (Makara et al., 2014), SAP97 (Petitprez et al., 2011; Gillet et al., 2015), Kir2.1 and Kir2.2 (Milstein et al., 2012; Matamoros et al., 2016; Ponce-Balbuena et al., 2018), syntrophin and dystrophin (Petitprez et al., 2011), and CASK (Eichel et al., 2016). Both ankyrin-G and SAP97 preferentially associate with ID-localized NaV1.5. However, loss of ankyrin-G reduced functional NaV1.5 expression (Makara et al., 2014) whereas loss of SAP97 did not (Gillet et al., 2015). Similarly, NaV1.5 at the lateral sarcolemma associates with syntrophin, dystrophin, and CASK. Loss of dystrophin reduced NaV1.5 functional expression at this location (Petitprez et al., 2011). Conversely, loss of CASK increased NaV1.5 surface expression at the lateral sarcolemma (Eichel et al., 2016). These intriguing findings suggest that we have much to learn about the organization and regulation of sodium channel-rich nanodomains, and the functional implications thereof.
Pathological alterations in NaV1.5 function are a well-established cause of cardiac arrhythmias (Veerman et al., 2015; Sottas and Abriel, 2016). Likewise, GJ remodeling is widely recognized as a feature of multiple cardiac pathologies (Jongsma and Wilders, 2000; Stroemlund et al., 2015), and implicated as a contributor to arrhythmogenesis (De Vuyst et al., 2011; Ongstad et al., 2013). While the arrhythmogenic impacts of functional loss of INa and GJ coupling have long been subjects of intense inquiry, we are only beginning to recognize the effects of nanoscale organization of NaV1.5 and Cx43, various scaffolding proteins associated with them, and changes in their ultrastructural environment. An interesting development in this context is the growing appreciation of the non-channel functions of Cx43 and their role in pathophysiology (Agullo-Pascual and Delmar, 2012). Knockdown of Cx43 in mice was associated with concomitant reductions in NaV1.5 expression, and INa density (Jansen et al., 2012) but not with alterations in adherens junctions or desmosomes (Gutstein et al., 2003). Proximity ligation assays indicate that the association between Cx43, and NaV1.5 is concentrated within the perinexus (Rhett et al., 2012). This raises the possibility that the aforementioned loss of INa function may result from the loss of Cx43 to scaffold NaV1.5 within perinexal nanodomains, and thereby, preferentially impact these sites. Thus, nanopathy may underlie arrhythmias in the wide array of cardiac pathologies where Cx43 is remodeled (Akar et al., 2007; Desplantez et al., 2007; Dhein et al., 2011; Chkourko et al., 2012). Along similar lines, missense mutations in the desmosomal protein plakophilin-2 were demonstrated to reduce N-cadherin-associated NaV1.5 density at the ID, reduce INa, and result in a Brugada syndrome phenotype (Cerrone et al., 2014). Taken together, these results underscore the clinical relevance of understanding the molecular organization of functional nanodomains, and the specific role of nanopathy in disease.
In addition to direct alterations of NaV1.5 and/or its molecular partners, nanopathy could also result from pathological changes in nanodomain ultrastructure. In this context, the previously discussed effects of interstitial edema on the perinexal nanodomain take on interesting implications given the wide array of pathologies that are associated with both cardiac edema and arrhythmias (Mehlhorn et al., 1996, 2001; Boyle et al., 2007; Fernandez-Jimenez et al., 2015; Migliore et al., 2015). Additionally, in long QT syndrome type 3 (LQT3; a disorder stemming from pathological gain of NaV1.5 function), the Poelzing group recently demonstrated that transient depletion of extracellular Na+ within perinexal nanodomains may mitigate risk of premature beats, and that perinexal widening may unmask the latent arrhythmia risk (Greer-Short et al., 2017). More recently, they have demonstrated that wider perinexi associated with the occurrence of AF in human patients (Raisch et al., 2018). While these are early days yet, these results collectively support the view that ultrastructural alterations within NaV1.5-rich nanodomains may be a key determinant of arrhythmia risk, and therefore, a potential target for antiarrhythmic therapy.
Our understanding of excitation–contraction coupling (ECC) has followed a similar trajectory to the one outlined for the electrical excitation. Early studies first identified invaginations in the membrane called transverse (T)-tubules (Lindner, 1957) and demonstrated that an activating current localized to those domains prompts contraction (Huxley and Taylor, 1955, 1958). Subsequent work demonstrated that Ca2+ entry through the sarcolemma, via L-type Ca2+ channels (LTCC), results in calcium-induced calcium release from the sarcoplasmic reticulum (SR) via ryanodine receptor channels (RyR2) to couple electrical excitation with mechanical contraction (Fabiato and Fabiato, 1977). Examination of the structural underpinnings of cardiac ECC led to the identification of a restricted space dubbed the “fuzzy” dyadic space (Lederer et al., 1990) where the sarcolemma and the terminal cisternae of the SR are separated by only ∼12 nm (Forbes and Sperelakis, 1983) (Figure 1). These results led to the realization that local protein function and electrochemical fluxes within nanodomains govern ECC rather than bulk effects across larger scales. Indeed, both systolic and diastolic Ca2+ concentrations in the dyadic cleft have been found to exceed that of the bulk cytosol (Despa et al., 2014; Popescu et al., 2016), and differences in Na+ concentrations have been reported as well (Despa and Bers, 2003).
An early clue to the complexity of the dyadic nanodomain function came with the demonstration by Leblanc and Hume that Na+ influx may regulate Ca2+ release (Leblanc and Hume, 1990). Specifically, they provided evidence that Na+ influx during the action potential upstroke was linked with Ca2+ cycling through the action of the Na+–Ca2+ exchanger (NCX). Interestingly, a component of INa persists during the plateau phases, dubbed late INa, and is tetrodotoxin (TTX) – sensitive (Conforti et al., 1993). Further investigation along these intriguing lines by the Bridge and Goldhaber groups suggested the involvement of neuronal Na+ channel (nNaV) isoforms (Torres et al., 2010). This findings are consistent with aforementioned structural results: (1) NaV1.5 localizes to the ID and the lateral sarcolemma and (2) nNaVs localize to t-tubules (Dhar Malhotra et al., 2001; Maier et al., 2002, 2004; Westenbroek et al., 2013; Radwański et al., 2015) (Figure 1). Indeed, up to 50% of the late INa in canine cardiac myocytes has been reported to be TTX-sensitive (Biet et al., 2012). These results raised the possibility that nNaVs, and NCX may constitute essential components of dyadic nanodomains along with LTCCs and RYR2. Since then, multiple groups have provided evidence for co-compartmentation of Na+ and Ca2+ handling proteins within dyadic nanodomains (Scriven et al., 2000; Jayasinghe et al., 2009). In recent years, evidence has mounted that the high gain system of cardiac Ca2+ cycling is tightly regulated by nNaVs, and NCX localized within the cleft (Radwański and Poelzing, 2011; Radwański et al., 2013, 2015, 2016; Veeraraghavan et al., 2017). However, the molecular stoichiometry of Na+/Ca2+ handling machinery (NCX, RyR, NaVs) and inter-species differences remain to be elucidated.
An additional layer of complexity in dyadic nanodomain function derives from its structure. It encompasses three spatial compartments – the extracellular space within the t-tubule, the cytosolic subspace, and the terminal cisternae of the junctional SR. In contrast, Na+ channel-rich nanodomains at the ID only consist of two compartments, extracellular and intracellular. While much of the inquiry into dyadic nanodomain function has focused on dynamics within the cytosolic subspace, emerging evidence is drawing attention to the complexity of the t-tubular network. Electron microscopy has revealed microfolds along t-tubules (Lavorato et al., 2015), thought to be generated by the action of the protein Bridging integrator 1 (BIN1) (Hong et al., 2014; Fu and Hong, 2016). Experimental observations also suggest that diffusion within t-tubules is significantly slowed in comparison to bulk interstitial space (Blatter and Niggli, 1998; Shepherd and McDonough, 1998; Pasek et al., 2006; Swift et al., 2006). The Lopatin group recently demonstrated that the presence of expansions and constrictions along t-tubules, the result of microfolds, likely accounts for this slow diffusion (Uchida and Lopatin, 2018). This slowed diffusion within t-tubules enables the development of local electrochemical transients within t-tubular nanodomains, paralleling ID nanodomains. This phenomenon has important consequences for cardiac electrophysiology as well as ECC. Likewise local dynamics in luminal Ca2+ within the junctional SR would strongly modulate dyadic nanodomain behavior (particularly RYR2 gating) and merit further investigation (Gyorke et al., 2017). While there has been much progress in understanding dyadic nanodomains as the structural and functional units of cardiac ECC, further investigation is needed to understand how structure–function relationships at the nanoscale determine physiology at cellular, tissue, and organ levels.
Dysregulated Ca2+ cycling is widely recognized as the cause of arrhythmias in multiple pathological states. Aberrant Ca2+ release from the SR can be prompted by SR Ca2+ overload, RyR2 dysfunction, or mistimed Ca2+ entry across the sarcolemma. This in turn can activate NCX, depolarizing the membrane, and prompting premature beats that can trigger arrhythmias (Berlin et al., 1989; Venetucci et al., 2008). Intriguingly, a recent report suggests that aberrant cytosolic Ca2+ levels may also produce arrhythmogenic conditions by facilitating untimely NaV re-opening (Johnson et al., 2018). Overall, the behavior of individual Na+ and Ca2+ -cycling proteins is fairly well-characterized, yet the interplay between Na+ and Ca2+ within dyadic nanodomains is very much the subject of active inquiry.
Modeling studies suggest that enhanced late Na+ entry into dyadic nanodomains can prompt reversal of NCX, resulting in Ca2+ entry into the subspace, and thereby, contribute to arrhythmogenic aberrance Ca2+ release (Armoundas et al., 2003; Radwański and Poelzing, 2011). Late INa carried by nNaVs has been implicated in inherited arrhythmia disorders such as catecholaminergic polymorphic ventricular tachycardia (CPVT) (Radwański et al., 2015, 2016). Specifically, Ca2+/calmodulin – dependent kinase II (CaMKII) -dependent enhancement of nNaV activity during β-adrenergic stimulation contributes to diastolic Ca2+ release and consequent arrhythmias in vivo via an NCX-mediated mechanism. Additionally, this pro-arrhythmic mechanism was consistent regardless of whether the CPVT resulted from “leaky” RyRs or from SR Ca2+ overload. In the broader context, these results point to pathological overload of cytosolic Na+ and Ca2+ being inextricably linked, particularly within the dyadic subspace.
Consistent with this hypothesis, aberrant Ca2+ release is the principal mechanism of arrhythmogenesis in disorders characterized by pathological gain of Na+ channel function. Multiple studies have linked pathological enhancement of nNaV function with arrhythmogenic diastolic Ca2+ release. These include mice lacking the Na+ channel auxiliary subunit β1 (SCN1B) which exhibit enhanced NaV1.3 expression (Lin et al., 2014), a rat pilocarpine-induced status epilepticus model where NaV1.1 expression is elevated (Biet et al., 2015), and a murine model of epileptic encephalopathy resulting from a gain of function mutation in NaV1.6 (Frasier et al., 2016). While these cases involve direct enhancement of Na+ entry into the dyadic nanodomain via nNaVs, LQT3 is characterized by gain of NaV1.5 function, resulting in global Na+ overload throughout the myocyte. Nonetheless, recent evidence indicates that Na+ entry into the dyadic subspace via nNaVs is still a key element of arrhythmogenesis in LQT3 (Koleske et al., 2018). Paralleling the aforementioned results in inherited arrhythmic syndromes, are studies implicating pathological enhancement of late INa in arrhythmias in acquired forms of ECC dysfunction such as heart failure (Valdivia et al., 2005; Undrovinas and Maltsev, 2008; Undrovinas et al., 2010; Sossalla and Maier, 2012; Antzelevitch et al., 2014; Makielski, 2016). Specifically, augmentation of Na+ influx via NaV1.1 (Mishra et al., 2014) as well as of NCX function (Pogwizd and Bers, 2002) in failing hearts have been shown to contribute to arrhythmias.
Given that late INa is central to aberrant Ca2+ cycling and arrhythmogenesis in multiple pathologies, it should come as no surprise that late INa inhibition by drugs such as ranolazine has demonstrated efficacy as an antiarrhythmic therapy (Burashnikov, 2017). However, emerging research suggests that selective targeting of nNaV-mediated Na+ entry into dyadic nanodomains may hold even greater promise (Radwański et al., 2015, 2016; Koleske et al., 2018). Additional impetus for pursuing this strategy comes from the dire negative consequences resulting from the off-target effects of non-isoform-selective Na+ channel inhibition: although non-selective INa inhibition suppressed triggered activity following myocardial infarction (The Cardiac Arrhythmia Pilot Study, 1986), the concomitant reduction in excitability precipitated reentrant arrhythmias, thereby increasing mortality (Echt et al., 1991; Starmer et al., 1991). Selective inhibition of nNaV would reduce Na+ entry into dyadic nanodomains, thereby ameliorating triggered arrhythmia incidence, without any attendant adverse impact on excitability. Indeed, selective inhibition of nNaVs, and of NaV1.6 in particular, whether using TTX analogs or a clinically relevant drug, riluzole, has been demonstrated to effectively suppress arrhythmias in murine models of CPVT (Radwański et al., 2015, 2016) and LQT3 (Radwański et al., 2013; Koleske et al., 2018).
Based on this logic, and available evidence, we posit the following requirements for arrhythmogenesis in pathologies directly driven by Ca2+ cycling defects (i.e., CPVT), and those arising from QT prolongation: (1) abnormal RyR2 function, whether genetic or acquired, (2) increased dyadic subspace Ca2+ levels, and (3) augmentation of nanodomain Na+ entry via nNaVs (Radwański et al., 2010, 2016; Cheng et al., 2011; Radwański and Poelzing, 2011; Terentyev et al., 2014). Any individual factor in the absence of the other two is unlikely to cause arrhythmia. For instance, genetic defects in the RyR2 complex alone are insufficient to induce triggered activity (Rios and Györke, 2009; Radwański et al., 2016); augmentation of Na+ entry via nNaV, and SR Ca2+ load, secondary to β-adrenergic receptor stimulation, is necessary for arrhythmogenesis (Radwański et al., 2016). On the other hand, LQT may promote CaMKII activity, which in turn modifies the components of the dyadic nanodomain (Terentyev et al., 2014; Viatchenko-Karpinski et al., 2014; Radwański et al., 2016). Given that augmentation of nNaV-mediated Na+ influx into dyadic nanodomains is a common element of both arrhythmogenic processes, it follows that late INa inhibition should prove antiarrhythmic in a broad range of pathologies including CPVT (Radwański et al., 2015, 2016), LQT3 (Koleske et al., 2018), LQT type 7 (Andersen-Tawil Syndrome; resulting from loss of repolarization reserve) (Radwański et al., 2013; Janson et al., 2014), and LQT type 8 (Timothy syndrome; resulting from pathological gain of LTCC function) (Gao et al., 2013).
While NaV isoform-selectivity is one focus in developing novel antiarrhythmic drugs, we note that the pharmacological mode of action (i.e., use-dependence vs. tonic block) will also determine therapeutic success. Use-dependent NaV inhibitors can effectively ameliorate triggered arrhythmias (The Cardiac Arrhythmia Pilot Study, 1986). However, under conditions such as elevated heart rates, they can suppress excitability and thereby, exacerbate conduction slowing. This, in turn, precipitates reentrant arrhythmias and increases mortality (Echt et al., 1991; Starmer et al., 1991). In contrast, tonic NaV inhibitors function independently of heart rate, thereby avoiding this adverse effect. Therefore, we postulate that tonic blockade may hold greater potential for delivering efficacy with safety. Intriguingly, a recent report by Buyan et al. (2018) provides clues to fundamental properties (inhibitor protonation state determines binding site) that may lead to development of novel NaV blockers with tailored modes of action. This highlights the need for mechanistically-driven drug development research grounded in understanding of atomic level NaV structure.
As with ID nanodomains, the behavior of dyadic nanodomains is determined not only by the function of their Na+/Ca2+ cycling protein constituents but also by their local ultrastructure. An example of this is found in failing hearts where β-adrenergic stimulation fails to effectively enhance ECC, despite augmented nNaV and NCX function (Viatchenko-Karpinski et al., 2005). This is likely a result of t-tubules being severely disrupted in failing hearts (Li et al., 2015), compromising cell-wide coordination of individual nanodomains. In addition, the remaining t-tubules in these hearts contain abnormal dyadic nanodomains consisting of nNaVs, NCX, and hypersensitized RyRs which are prone to arrhythmogenic aberrant Ca2+ release (Belevych et al., 2012). Additionally, loss of BIN1 has been linked to compromised LTCC trafficking as well as loss of t-tubule microfolds in heart failure (Caldwell et al., 2014; Hong et al., 2014; Laury-Kleintop et al., 2015). Both experimental and modeling studies suggest that the latter effect could compromise the previously discussed slowing of diffusion within t-tubules, thereby dysregulating electrophysiology and ECC. Indeed, the Sachse and Bridge groups have demonstrated that the arrhythmia burden in failing hearts is reduced secondary to restoration of the t-tubule network following cardiac resynchronization therapy (Sachse et al., 2012; Lichter et al., 2014; Li et al., 2015). Thus, available evidence points to dyadic nanodomains as promising targets for the prevention of arrhythmias resulting from aberrant Ca2+ cycling.
Current research, propelled by emerging technologies capable of assessing structure/function at the nanoscale, suggests that nanodomains located at the ID and the t-tubule may respectively constitute the functional units of cardiac electrical excitation and ECC. Thus, we are beginning to appreciate nanodomain dysfunction, i.e., nanopathy, as a key mechanistic driver of cardiac disease and arrhythmogenesis. It follows therefore that antiarrhythmic treatments should be designed to correct underlying nanopathies, and indeed, such therapies currently under investigation show a great deal of promise. In the broader context, the emerging understanding of how nanoscale biophysics and biochemistry determine cardiovascular physiology and pathophysiology across protein, cell, tissue, and organ scales may represent a paradigm shift on par with the advent of molecular biology. It should drive multiple avenues of scientific and medical research including (1) new diagnostic approaches that can non-invasively interrogate functional nanodomains within patients, (2) new methods to assess nanodomain alterations during autopsies, and (3) new therapeutic approaches designed to restore nanodomain structure/function.
RV, CNJ, SG, and PR drafted the work or revised it critically for important intellectual content.
This work was supported by American Heart Association (Grant No. 16SDG29870007 to RV) and NIH (Grant Nos. R01-HL074045, R01-HL063043, and R01-HL138579 to SG; R00-HL127299 to PR).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Agullo-Pascual, E., and Delmar, M. (2012). The noncanonical functions of Cx43 in the heart. J. Membr. Biol. 245, 477–482. doi: 10.1007/s00232-012-9466-y
Agullo-Pascual, E., Lin, X., Leo-Macias, A., Zhang, M., Liang, F. X., Li, Z., et al. (2014). Super-resolution imaging reveals that loss of the C-terminus of connexin43 limits microtubule plus-end capture and NaV1.5 localization at the intercalated disc. Cardiovasc. Res. 104, 371–381. doi: 10.1093/cvr/cvu195
Agullo-Pascual, E., Lin, X., Pfenniger, A., Lubkemeier, I., Willecke, K., Rothenberg, E., et al. (2013). A novel noncanonical role of cx43 in the heart: ensuring the arrival of nav1.5 to the intercalated disk. Heart Rhythm. 10:1742. doi: 10.1016/j.hrthm.2013.09.016
Akar, F. G., Nass, R. D., Hahn, S., Cingolani, E., Shah, M., Hesketh, G. G., et al. (2007). Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 293, H1223–H1230. doi: 10.1152/ajpheart.00079.2007
Antzelevitch, C., Nesterenko, V., Shryock, J. C., Rajamani, S., Song, Y., and Belardinelli, L. (2014). The role of late I Na in development of cardiac arrhythmias. Handb. Exp. Pharmacol. 221, 137–168. doi: 10.1007/978-3-642-41588-3_7
Armoundas, A. A., Hobai, I. A., Tomaselli, G. F., Winslow, R. L., and O’Rourke, B. (2003). Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ. Res. 93, 46–53. doi: 10.1161/01.RES.0000080932.98903.D8
Arujuna, A., Karim, R., Caulfield, D., Knowles, B., Rhode, K., Schaeffter, T., et al. (2012). Acute pulmonary vein isolation is achieved by a combination of reversible and irreversible atrial injury after catheter ablation: evidence from magnetic resonance imaging. Circ. Arrhythm. Electrophysiol. 5, 691–700. doi: 10.1161/CIRCEP.111.966523
Barr, L., Dewey, M. M., and Berger, W. (1965). Propagation of action potentials and the structure of the nexus in cardiac muscle. J. Gen. Physiol. 48, 797–823. doi: 10.1085/jgp.48.5.797
Beauchamp, P., Choby, C., Desplantez, T., de Peyer, K., Green, K., Yamada, K. A., et al. (2004). Electrical propagation in synthetic ventricular myocyte strands from germline connexin43 knockout mice. Circ. Res. 95, 170–178. doi: 10.1161/01.RES.0000134923.05174.2f
Belevych, A. E., Terentyev, D., Terentyeva, R., Ho, H. T., Gyorke, I., Bonilla, I. M., et al. (2012). Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ. Res. 110, 569–577. doi: 10.1161/CIRCRESAHA.111.260455
Berlin, J. R., Cannell, M. B., and Lederer, W. J. (1989). Cellular origins of the transient inward current in cardiac myocytes. Role of fluctuations and waves of elevated intracellular calcium. Circ. Res. 65, 115–126. doi: 10.1161/01.RES.65.1.115
Biet, M., Barajas-Martinez, H., Ton, A. T., Delabre, J. F., Morin, N., and Dumaine, R. (2012). About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na(+) channels. J. Mol. Cell Cardiol. 53, 593–598. doi: 10.1016/j.yjmcc.2012.06.012
Biet, M., Morin, N., Lessard-Beaudoin, M., Graham, R. K., Duss, S., Gagne, J., et al. (2015). Prolongation of action potential duration and QT interval during epilepsy linked to increased contribution of neuronal sodium channels to cardiac late Na+ current: potential mechanism for sudden death in epilepsy. Circ. Arrhythm. Electrophysiol. 8, 912–920. doi: 10.1161/CIRCEP.114.002693
Blatter, L. A., and Niggli, E. (1998). Confocal near-membrane detection of calcium in cardiac myocytes. Cell Calcium 23, 269–279. doi: 10.1016/S0143-4160(98)90023-9
Boyle, A., Maurer, M. S., and Sobotka, P. A. (2007). Myocellular and interstitial edema and circulating volume expansion as a cause of morbidity and mortality in heart failure. J. Card. Fail. 13, 133–136. doi: 10.1016/j.cardfail.2006.10.015
Burashnikov, A. (2017). Late INa inhibition as an antiarrhythmic strategy. J. Cardiovasc. Pharmacol. 70, 159–167. doi: 10.1097/FJC.0000000000000510
Buyan, A., Sun, D., and Corry, B. (2018). Protonation state of inhibitors determines interaction sites within voltage-gated sodium channels. Proc. Natl. Acad. Sci. U.S.A. 115, E3135–E3144. doi: 10.1073/pnas.1714131115
Caldwell, J. L., Smith, C. E., Taylor, R. F., Kitmitto, A., Eisner, D. A., Dibb, K. M., et al. (2014). Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1). Circ. Res. 115, 986–996. doi: 10.1161/CIRCRESAHA.116.303448
Cerrone, M., Lin, X., Zhang, M., Agullo-Pascual, E., Pfenniger, A., Chkourko Gusky, H., et al. (2014). Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 129, 1092–1103. doi: 10.1161/CIRCULATIONAHA.113.003077
Cheng, E. P., Yuan, C., Navedo, M. F., Dixon, R. E., Nieves-Cintron, M., Scott, J. D., et al. (2011). Restoration of normal L-type Ca2+ channel function during timothy syndrome by ablation of an anchoring protein. Circ. Res. 109, 255–261. doi: 10.1161/CIRCRESAHA.111.248252
Chkourko, H. S., Guerrero-Serna, G., Lin, X., Darwish, N., Pohlmann, J. R., Cook, K. E., et al. (2012). Remodeling of mechanical junctions and of microtubule-associated proteins accompany cardiac connexin43 lateralization. Heart Rhythm. 9, 1133.e6–1140.e6. doi: 10.1016/j.hrthm.2012.03.003
Conforti, L., Tohse, N., and Sperelakis, N. (1993). Tetrodotoxin-sensitive sodium current in rat fetal ventricular myocytes–contribution to the plateau phase of action potential. J. Mol. Cell Cardiol. 25, 159–173. doi: 10.1006/jmcc.1993.1019
Copene, E. D., and Keener, J. P. (2008). Ephaptic coupling of cardiac cells through the junctional electric potential. J. Math. Biol. 57, 265–284. doi: 10.1007/s00285-008-0157-3
Danik, S. B., Liu, F., Zhang, J., Suk, H. J., Morley, G. E., Fishman, G. I., et al. (2004). Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ. Res. 95, 1035–1041. doi: 10.1161/01.RES.0000148664.33695.2a
De Vuyst, E., Boengler, K., Antoons, G., Sipido, K. R., Schulz, R., and Leybaert, L. (2011). Pharmacological modulation of connexin-formed channels in cardiac pathophysiology. Br. J. Pharmacol. 163, 469–483. doi: 10.1111/j.1476-5381.2011.01244.x
Despa, S., and Bers, D. M. (2003). Na/K pump current and [Na](i) in rabbit ventricular myocytes: local [Na](i) depletion and Na buffering. Biophys. J. 84, 4157–4166. doi: 10.1016/S0006-3495(03)75140-6
Despa, S., Shui, B., Bossuyt, J., Lang, D., Kotlikoff, M. I., and Bers, D. M. (2014). Junctional cleft [Ca(2)(+)]i measurements using novel cleft-targeted Ca(2)(+) sensors. Circ. Res. 115, 339–347. doi: 10.1161/CIRCRESAHA.115.303582
Desplantez, T., Dupont, E., Severs, N., and Weingart, R. (2007). Gap junction channels and cardiac impulse propagation. J. Membr. Biol. 218, 13–28. doi: 10.1007/s00232-007-9046-8
Dhar Malhotra, J., Chen, C., Rivolta, I., Abriel, H., Malhotra, R., Mattei, L. N., et al. (2001). Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation 103, 1303–1310. doi: 10.1161/01.CIR.103.9.1303
Dhein, S., Rothe, S., Busch, A., Rojas Gomez, D. M., Boldt, A., Reutemann, A., et al. (2011). Effects of metoprolol therapy on cardiac gap junction remodelling and conduction in human chronic atrial fibrillation. Br. J. Pharmacol. 164, 607–616. doi: 10.1111/j.1476-5381.2011.01460.x
Echt, D. S., Liebson, P. R., Mitchell, L. B., Peters, R. W., Obias-Manno, D., Barker, A. H., et al. (1991). Mortality and morbidity in patients receiving encainide, flecainide, or placebo, the cardiac arrhythmia suppression trial. N. Engl. J. Med. 324, 781–788. doi: 10.1056/NEJM199103213241201
Eichel, C. A., Beuriot, A., Chevalier, M. Y., Rougier, J. S., Louault, F., Dilanian, G., et al. (2016). Lateral membrane-specific MAGUK CASK down-regulates NaV1.5 channel in cardiac myocytes. Circ. Res. 119, 544–556. doi: 10.1161/CIRCRESAHA.116.309254
Eloff, B. C., Lerner, D. L., Yamada, K. A., Schuessler, R. B., Saffitz, J. E., and Rosenbaum, D. S. (2001). High resolution optical mapping reveals conduction slowing in connexin43 deficient mice. Cardiovasc. Res. 51, 681–690. doi: 10.1016/S0008-6363(01)00341-8
Fabiato, A., and Fabiato, F. (1977). Calcium release from the sarcoplasmic reticulum. Circ. Res. 40, 119–129. doi: 10.1161/01.RES.40.2.119
Fernandez-Jimenez, R., Sanchez-Gonzalez, J., Aguero, J., Garcia-Prieto, J., Lopez-Martin, G. J., Garcia-Ruiz, J. M., et al. (2015). Myocardial edema after ischemia/reperfusion is not stable and follows a bimodal pattern: imaging and histological tissue characterization. J. Am. Coll. Cardiol. 65, 315–323. doi: 10.1016/j.jacc.2014.11.004
Forbes, M. S., and Sperelakis, N. (1983). The membrane systems and cytoskeletal elements of mammalian myocardial cells. Cell Muscle Motil. 3, 89–155. doi: 10.1007/978-1-4615-9296-9_5
Frasier, C. R., Wagnon, J. L., Bao, Y. O., McVeigh, L. G., Lopez-Santiago, L. F., Meisler, M. H., et al. (2016). Cardiac arrhythmia in a mouse model of sodium channel SCN8A epileptic encephalopathy. Proc. Natl. Acad. Sci. U.S.A. doi: 10.1073/pnas.1612746113 [Epub ahead of print].
Fu, Y., and Hong, T. (2016). BIN1 regulates dynamic t-tubule membrane. Biochim. Biophys. Acta 1863(7Pt B), 1839–1847. doi: 10.1016/j.bbamcr.2015.11.004
Gao, Y., Xue, X., Hu, D., Liu, W., Yuan, Y., Sun, H., et al. (2013). Inhibition of late sodium current by mexiletine: a novel pharmotherapeutical approach in timothy syndrome. Circ. Arrhythm. Electrophysiol. 6, 614–622. doi: 10.1161/CIRCEP.113.000092
Gillet, L., Rougier, J. S., Shy, D., Sonntag, S., Mougenot, N., Essers, M., et al. (2015). Cardiac-specific ablation of synapse-associated protein SAP97 in mice decreases potassium currents but not sodium current. Heart Rhythm. 12, 181–192. doi: 10.1016/j.hrthm.2014.09.057
Greer-Short, A., George, S. A., Poelzing, S., and Weinberg, S. H. (2017). Revealing the concealed nature of long-QT type 3 syndrome. Circ. Arrhythm. Electrophysiol. 10:e004400. doi: 10.1161/CIRCEP.116.004400
Guerrero, P. A., Schuessler, R. B., Davis, L. M., Beyer, E. C., Johnson, C. M., Yamada, K. A., et al. (1997). Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J. Clin. Invest. 99, 1991–1998. doi: 10.1172/JCI119367
Gutstein, D. E., Liu, F. Y., Meyers, M. B., Choo, A., and Fishman, G. I. (2003). The organization of adherens junctions and desmosomes at the cardiac intercalated disc is independent of gap junctions. J. Cell Sci. 116(Pt 5), 875–885. doi: 10.1242/jcs.00258
Gutstein, D. E., Morley, G. E., Tamaddon, H., Vaidya, D., Schneider, M. D., Chen, J., et al. (2001). Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ. Res. 88, 333–339. doi: 10.1161/01.RES.88.3.333
Gyorke, S., Belevych, A. E., Liu, B., Kubasov, I. V., Carnes, C. A., and Radwański, P. B. (2017). The role of luminal Ca regulation in Ca signaling refractoriness and cardiac arrhythmogenesis. J. Gen. Physiol. 149, 877–888. doi: 10.1085/jgp.201711808
Hichri, E., Abriel, H., and Kucera, J. P. (2017). Distribution of cardiac sodium channels in clusters potentiates ephaptic interactions in the intercalated disc. J. Physiol. 596, 563–589. doi: 10.1113/JP275351
Hong, M., Bao, L., Kefaloyianni, E., Agullo-Pascual, E., Chkourko, H., Foster, M., et al. (2012). Heterogeneity of ATP-sensitive K+ channels in cardiac myocytes: enrichment at the intercalated disk. J. Biol. Chem. 287, 41258–41267. doi: 10.1074/jbc.M112.412122
Hong, T., Yang, H., Zhang, S. S., Cho, H. C., Kalashnikova, M., Sun, B., et al. (2014). Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat. Med. 20, 624–632. doi: 10.1038/nm.3543
Huxley, A. F., and Taylor, R. E. (1955). Function of Krause’s membrane. Nature 176:1068. doi: 10.1038/1761068a0
Huxley, A. F., and Taylor, R. E. (1958). Local activation of striated muscle fibres. J. Physiol. 144, 426–441. doi: 10.1113/jphysiol.1958.sp006111
Ishihara, K. (2018). External K(+) dependence of strong inward rectifier K(+) channel conductance is caused not by K(+) but by competitive pore blockade by external Na. J. Gen. Physiol. 150, 977–989. doi: 10.1085/jgp.201711936
Jansen, J. A., Noorman, M., Musa, H., Stein, M., de Jong, S., van der Nagel, R., et al. (2012). Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm. 9, 600–607. doi: 10.1016/j.hrthm.2011.11.025
Janson, C. M., Poelzing, S., and Shah, M. J. (2014). Combined inhibition of Na(+) and Ca(2)(+) channels: a novel paradigm for the treatment of incessant ventricular arrhythmias in Andersen-Tawil syndrome. Heart Rhythm. 11, 318–320. doi: 10.1016/j.hrthm.2013.11.003
Jayasinghe, I. D., Cannell, M. B., and Soeller, C. (2009). Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys. J. 97, 2664–2673. doi: 10.1016/j.bpj.2009.08.036
Johnson, C. N., Potet, F., Thompson, M. K., Kroncke, B. M., Glazer, A. M., Voehler, M. W., et al. (2018). A mechanism of calmodulin modulation of the human cardiac sodium channel. Structure 26, 683.e3–694.e3. doi: 10.1016/j.str.2018.03.005
Jongsma, H. J., and Wilders, R. (2000). Gap junctions in cardiovascular disease. Circ. Res. 86, 1193–1197. doi: 10.1161/01.RES.86.12.1193
Kleber, A. G., and Rudy, Y. (2004). Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 84, 431–488. doi: 10.1152/physrev.00025.2003
Koleske, M., Bonilla, I., Thomas, J., Zaman, N., Baine, S., Knollmann, B. C., et al. (2018). Tetrodotoxin-sensitive Navs contribute to early and delayed afterdepolarizations in long QT arrhythmia models. J. Gen. Physiol. 150, 991–1002. doi: 10.1085/jgp.201711909
Kucera, J. P., Rohr, S., and Rudy, Y. (2002). Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ. Res. 91, 1176–1182. doi: 10.1161/01.RES.0000046237.54156.0A
Laury-Kleintop, L. D., Mulgrew, J. R., Heletz, I., Nedelcoviciu, R. A., Chang, M. Y., Harris, D. M., et al. (2015). Cardiac-specific disruption of Bin1 in mice enables a model of stress- and age-associated dilated cardiomyopathy. J. Cell Biochem. 116, 2541–2551. doi: 10.1002/jcb.25198
Lavorato, M., Huang, T. Q., Iyer, V. R., Perni, S., Meissner, G., and Franzini-Armstrong, C. (2015). Dyad content is reduced in cardiac myocytes of mice with impaired calmodulin regulation of RyR2. J. Muscle Res. Cell Motil. 36, 205–214. doi: 10.1007/s10974-015-9405-5
Leblanc, N., and Hume, J. R. (1990). Sodium current-induced release of calcium from cardiac sarcoplasmic reticulum. Science 248, 372–376. doi: 10.1126/science.2158146
Lederer, W. J., Niggli, E., and Hadley, R. W. (1990). Sodium-calcium exchange in excitable cells: fuzzy space. Science 248:283. doi: 10.1126/science.2326638
Leo-Macias, A., Agullo-Pascual, E., Sanchez-Alonso, J. L., Keegan, S., Lin, X., Arcos, T., et al. (2016). Nanoscale visualization of functional adhesion/excitability nodes at the intercalated disc. Nat. Commun. 7:10342. doi: 10.1038/ncomms10342
Leo-Macias, A., Liang, F. X., and Delmar, M. (2015). Ultrastructure of the intercellular space in adult murine ventricle revealed by quantitative tomographic electron microscopy. Cardiovasc. Res. 107, 442–452. doi: 10.1093/cvr/cvv182
Li, H., Lichter, J. G., Seidel, T., Tomaselli, G. F., Bridge, J. H., and Sachse, F. B. (2015). Cardiac resynchronization therapy reduces subcellular heterogeneity of ryanodine receptors, T-Tubules, and Ca2+ sparks produced by dyssynchronous heart failure. Circ. Heart Fail. 8, 1105–1114. doi: 10.1161/CIRCHEARTFAILURE.115.002352
Lichter, J. G., Carruth, E., Mitchell, C., Barth, A. S., Aiba, T., Kass, D. A., et al. (2014). Remodeling of the sarcomeric cytoskeleton in cardiac ventricular myocytes during heart failure and after cardiac resynchronization therapy. J. Mol. Cell Cardiol. 72, 186–195. doi: 10.1016/j.yjmcc.2014.03.012
Lin, J., and Keener, J. P. (2010). Modeling electrical activity of myocardial cells incorporating the effects of ephaptic coupling. Proc. Natl. Acad. Sci. U.S.A. 107, 20935–20940. doi: 10.1073/pnas.1010154107
Lin, J., and Keener, J. P. (2014). Microdomain effects on transverse cardiac propagation. Biophys. J. 106, 925–931. doi: 10.1016/j.bpj.2013.11.1117
Lin, X., O’Malley, H., Chen, C., Auerbach, D., Foster, M., Shekhar, A., et al. (2014). Scn1b deletion leads to increased tetrodotoxin-sensitive sodium current, altered intracellular calcium homeostasis and arrhythmias in murine hearts. J Physiol. 593, 1389–1407. doi: 10.1113/jphysiol.2014.277699
Lindner, E. (1957). [Submicroscopic morphology of the cardiac muscle]. Z. Zellforsch. Mikrosk. Anat. 45, 702–746.
Maier, S. K., Westenbroek, R. E., McCormick, K. A., Curtis, R., Scheuer, T., and Catterall, W. A. (2004). Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation 109, 1421–1427. doi: 10.1161/01.CIR.0000121421.61896.24
Maier, S. K., Westenbroek, R. E., Schenkman, K. A., Feigl, E. O., Scheuer, T., and Catterall, W. A. (2002). An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc. Natl. Acad. Sci. U.S.A. 99, 4073–4078. doi: 10.1073/pnas.261705699
Makara, M. A., Curran, J., Little, S. C., Musa, H., Polina, I., Smith, S. A., et al. (2014). Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ. Res. 115, 929–938. doi: 10.1161/CIRCRESAHA.115.305154
Makielski, J. C. (2016). Late sodium current: a mechanism for angina, heart failure, and arrhythmia. Trends Cardiovasc. Med. 26, 115–122. doi: 10.1016/j.tcm.2015.05.006
Matamoros, M., Perez-Hernandez, M., Guerrero-Serna, G., Amoros, I., Barana, A., Nunez, M., et al. (2016). Nav1.5 N-terminal domain binding to alpha1-syntrophin increases membrane density of human Kir2.1, Kir2.2 and Nav1.5 channels. Cardiovasc. Res. 110, 279–290. doi: 10.1093/cvr/cvw009
Mehlhorn, U., Davis, K. L., Laine, G. A., Geissler, H. J., and Allen, S. J. (1996). Myocardial fluid balance in acute hypertension. Microcirculation 3, 371–378. doi: 10.3109/10739689609148309
Mehlhorn, U., Geissler, H. J., Laine, G. A., and Allen, S. J. (2001). Myocardial fluid balance. Eur. J. Cardiothorac. Surg. 20, 1220–1230. doi: 10.1016/S1010-7940(01)01031-4
Migliore, F., Zorzi, A., Perazzolo Marra, M., Iliceto, S., and Corrado, D. (2015). Myocardial edema as a substrate of electrocardiographic abnormalities and life-threatening arrhythmias in reversible ventricular dysfunction of takotsubo cardiomyopathy: imaging evidence, presumed mechanisms, and implications for therapy. Heart Rhythm. 12, 1867–1877. doi: 10.1016/j.hrthm.2015.04.041
Milstein, M. L., Musa, H., Balbuena, D. P., Anumonwo, J. M., Auerbach, D. S., Furspan, P. B., et al. (2012). Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc. Natl. Acad. Sci. U.S.A. 109, E2134–E2143. doi: 10.1073/pnas.1109370109
Mishra, S., Reznikov, V., Maltsev, V. A., Undrovinas, N. A., Sabbah, H. N., and Undrovinas, A. (2014). Contribution of sodium channel neuronal isoform Nav1.1 to late sodium current in ventricular myocytes from failing hearts. J. Physiol. 593, 1409–1427. doi: 10.1113/jphysiol.2014.278259
Mori, Y., Fishman, G. I., and Peskin, C. S. (2008). Ephaptic conduction in a cardiac strand model with 3D electrodiffusion. Proc. Natl. Acad. Sci. U.S.A. 105, 6463–6468. doi: 10.1073/pnas.0801089105
Morley, G. E., Vaidya, D., Samie, F. H., Lo, C., Delmar, M., and Jalife, J. (1999). Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J. Cardiovasc. Electrophysiol. 10, 1361–1375. doi: 10.1111/j.1540-8167.1999.tb00192.x
Ongstad, E. L., O’Quinn, M. P., Ghatnekar, G. S., Yost, M. J., and Gourdie, R. G. (2013). A connexin43 mimetic peptide promotes regenerative healing and improves mechanical properties in skin and heart. Adv. Wound Care (New Rochelle) 2, 55–62. doi: 10.1089/wound.2011.0341
Pasek, M., Simurda, J., and Christe, G. (2006). The functional role of cardiac T-tubules explored in a model of rat ventricular myocytes. Philos. Trans. A Math Phys. Eng. Sci. 364, 1187–1206. doi: 10.1098/rsta.2006.1764
Pertsov, A. M., and Medvinskii, A. B. (1976). [Electric coupling in cells without highly permeable cell contacts]. Biofizika 21, 698–670.
Petitprez, S., Zmoos, A. F., Ogrodnik, J., Balse, E., Raad, N., El-Haou, S., et al. (2011). SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ. Res. 108, 294–304. doi: 10.1161/CIRCRESAHA.110.228312
Poelzing, S., Akar, F. G., Baron, E., and Rosenbaum, D. S. (2004). Heterogeneous connexin43 expression produces electrophysiological heterogeneities across ventricular wall. Am. J. Physiol. Heart Circ. Physiol. 286, H2001–H2009. doi: 10.1152/ajpheart.00987.2003
Pogwizd, S. M., and Bers, D. M. (2002). Na/Ca exchange in heart failure: contractile dysfunction and arrhythmogenesis. Ann. N. Y. Acad. Sci. 976, 454–465. doi: 10.1111/j.1749-6632.2002.tb04775.x
Ponce-Balbuena, D., Guerrero-Serna, G., Valdivia, C. R., Caballero, R., Diez-Guerra, F. J., Jimenez-Vazquez, E. N., et al. (2018). Cardiac Kir2.1 and NaV1.5 channels traffic together to the sarcolemma to control excitability. Circ Res. 122, 1501–1516. doi: 10.1161/CIRCRESAHA.117.311872
Popescu, I., Galice, S., Mohler, P. J., and Despa, S. (2016). Elevated local [Ca2+] and CaMKII promote spontaneous Ca2+ release in ankyrin-B-deficient hearts. Cardiovasc. Res. 111, 287–294. doi: 10.1093/cvr/cvw093
Radwański, P. B., Brunello, L., Veeraraghavan, R., Ho, H. T., Lou, Q., Makara, M. A., et al. (2015). Neuronal Na+ channel blockade suppresses arrhythmogenic diastolic Ca2+ release. Cardiovasc. Res. 106, 143–152. doi: 10.1093/cvr/cvu262
Radwański, P. B., Greer-Short, A., and Poelzing, S. (2013). Inhibition of Na(+) channels ameliorates arrhythmias in a drug-induced model of Andersen-Tawil syndrome. Heart Rhythm. 10, 255–263. doi: 10.1016/j.hrthm.2012.10.005
Radwański, P. B., Ho, H.-T., Veeraraghavan, R., Brunello, L., Liu, B., Belevych, A. E., et al. (2016). Neuronal Na+ channels are integral components of pro-arrhythmic Na+/Ca2+ signaling nanodomain that promotes cardiac arrhythmias during β-adrenergic stimulation. JACC Basic Transl. Sci. 1, 251–266. doi: 10.1016/j.jacbts.2016.04.004
Radwański, P. B., and Poelzing, S. (2011). NCX is an important determinant for premature ventricular activity in a drug-induced model of Andersen-Tawil syndrome. Cardiovasc. Res. 92, 57–66. doi: 10.1093/cvr/cvr180
Radwański, P. B., Veeraraghavan, R., and Poelzing, S. (2010). Cytosolic calcium accumulation and delayed repolarization associated with ventricular arrhythmias in a guinea pig model of Andersen-Tawil syndrome. Heart Rhythm. 7, 1428–1435. doi: 10.1016/j.hrthm.2010.03.044
Raisch, T. B., Yanoff, M. S., Larsen, T. R., Farooqui, M. A., King, D. A., Veeraraghavan, R., et al. (2018). Intercalated disc extracellular nanodomain expansion in patients with atrial fibrillation. Front. Physiol. 9:398. doi: 10.3389/fphys.2018.00398
Rhett, J. M., and Gourdie, R. G. (2012). The perinexus: a new feature of Cx43 gap junction organization. Heart Rhythm. 9, 619–623. doi: 10.1016/j.hrthm.2011.10.003
Rhett, J. M., Jourdan, J., and Gourdie, R. G. (2011). Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 22, 1516–1528. doi: 10.1091/mbc.e10-06-0548
Rhett, J. M., Ongstad, E. L., Jourdan, J., and Gourdie, R. G. (2012). Cx43 associates with Na(v)1.5 in the cardiomyocyte perinexus. J. Membr. Biol. 245, 411–422. doi: 10.1007/s00232-012-9465-z
Rios, E., and Györke, S. (2009) Calsequestrin, triadin and more: the molecules that modulate calcium release in cardiac and skeletal muscle. J. Physiol. 587, 3069–3070. doi: 10.1113/jphysiol.2009.175083
Sachse, F. B., Torres, N. S., Savio-Galimberti, E., Aiba, T., Kass, D. A., Tomaselli, G. F., et al. (2012). Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ. Res. 110, 588–597. doi: 10.1161/CIRCRESAHA.111.257428
Scriven, D. R., Dan, P., and Moore, E. D. (2000). Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys. J. 79, 2682–2691. doi: 10.1016/S0006-3495(00)76506-4
Shepherd, N., and McDonough, H. B. (1998). Ionic diffusion in transverse tubules of cardiac ventricular myocytes. Am. J. Physiol. 275(3 Pt 2), H852–H860. doi: 10.1152/ajpheart.1998.275.3.H852
Sjostrand, F. S., and Andersson, E. (1954). Electron microscopy of the intercalated discs of cardiac muscle tissue. Experientia 10, 369–370. doi: 10.1007/BF02160542
Sossalla, S., and Maier, L. S. (2012). Role of ranolazine in angina, heart failure, arrhythmias, and diabetes. Pharmacol. Ther. 133, 311–323. doi: 10.1016/j.pharmthera.2011.11.003
Sottas, V., and Abriel, H. (2016). Negative-dominance phenomenon with genetic variants of the cardiac sodium channel Nav1.5. Biochim. Biophys. Acta 1863(7 Pt B), 1791–1798. doi: 10.1016/j.bbamcr.2016.02.013
Spach, M. S., Heidlage, J. F., Barr, R. C., and Dolber, P. C. (2004). Cell size and communication: role in structural and electrical development and remodeling of the heart. Heart Rhythm. 1, 500–515. doi: 10.1016/j.hrthm.2004.06.010
Sperelakis, N., and Mann, J. E. J. (1977). Evaluation of electric field changes in the cleft between excitable cells. J. Theor. Biol. 64, 71–96. doi: 10.1016/0022-5193(77)90114-X
Sperelakis, N., and McConnell, K. (2002). Electric field interactions between closely abutting excitable cells. IEEE Eng. Med. Biol. Mag. 21, 77–89. doi: 10.1109/51.993199
Starmer, C. F., Lastra, A. A., Nesterenko, V. V., and Grant, A. O. (1991). Proarrhythmic response to sodium channel blockade, theoretical model and numerical experiments. Circulation 84, 1364–1377. doi: 10.1161/01.CIR.84.3.1364
Stein, M., van Veen, T. A., Remme, C. A., Boulaksil, M., Noorman, M., van Stuijvenberg, L., et al. (2009). Combined reduction of intercellular coupling and membrane excitability differentially affects transverse and longitudinal cardiac conduction. Cardiovasc. Res. 83, 52–60. doi: 10.1093/cvr/cvp124
Stroemlund, L. W., Jensen, C. F., Qvortrup, K., Delmar, M., and Nielsen, M. S. (2015). Gap junctions – Guards of excitability. Biochem. Soc. Trans. 43, 508–512. doi: 10.1042/BST20150059
Swift, F., Stromme, T. A., Amundsen, B., Sejersted, O. M., and Sjaastad, I. (2006). Slow diffusion of K+ in the T tubules of rat cardiomyocytes. J. Appl. Physiol. 101, 1170–1176. doi: 10.1152/japplphysiol.00297.2006
Terentyev, D., Rees, C. M., Li, W., Cooper, L. L., Jindal, H. K., Peng, X., et al. (2014). Hyperphosphorylation of RyRs underlies triggered activity in transgenic rabbit model of LQT2 syndrome. Circ. Res. 115, 919–928. doi: 10.1161/CIRCRESAHA.115.305146
Thomas, S. P., Kucera, J. P., Bircher-Lehmann, L., Rudy, Y., Saffitz, J. E., and Kleber, A. G. (2003). Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ. Res. 92, 1209–1216. doi: 10.1161/01.RES.0000074916.41221.EA
Torres, N. S., Larbig, R., Rock, A., Goldhaber, J. I., and Bridge, J. H. (2010). Na+ currents are required for efficient excitation-contraction coupling in rabbit ventricular myocytes: a possible contribution of neuronal Na+ channels. J. Physiol. 588(Pt 21), 4249–4260. doi: 10.1113/jphysiol.2010.194688
Uchida, K., and Lopatin, A. N. (2018). Diffusional and electrical properties of T-tubules are governed by their constrictions and dilations. Biophys. J. 114, 437–449. doi: 10.1016/j.bpj.2017.11.3742
Undrovinas, A., and Maltsev, V. A. (2008). Late sodium current is a new therapeutic target to improve contractility and rhythm in failing heart. Cardiovasc. Hematol. Agents Med. Chem. 6, 348–359. doi: 10.2174/187152508785909447
Undrovinas, N. A., Maltsev, V. A., Belardinelli, L., Sabbah, H. N., and Undrovinas, A. (2010). Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J. Physiol. Sci. 60, 245–257. doi: 10.1007/s12576-010-0092-0
Vaidya, D., Tamaddon, H. S., Lo, C. W., Taffet, S. M., Delmar, M., Morley, G. E., et al. (2001). Null mutation of connexin43 causes slow propagation of ventricular activation in the late stages of mouse embryonic development. Circ. Res. 88, 1196–1202. doi: 10.1161/hh1101.091107
Valdivia, C. R., Chu, W. W., Pu, J., Foell, J. D., Haworth, R. A., Wolff, M. R., et al. (2005). Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell Cardiol. 38, 475–483. doi: 10.1016/j.yjmcc.2004.12.012
van Rijen, H. V., Eckardt, D., Degen, J., Theis, M., Ott, T., Willecke, K., et al. (2004). Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation 109, 1048–1055. doi: 10.1161/01.CIR.0000117402.70689.75
Veeraraghavan, R., and Gourdie, R. (2016). Stochastic optical reconstruction microscopy-based relative localization analysis (STORM-RLA) for quantitative nanoscale assessment of spatial protein organization. Mol. Biol. Cell. 27, 3583–3590. doi: 10.1091/mbc.E16-02-0125
Veeraraghavan, R., Gourdie, R., and Poelzing, S. (2014a). Mechanisms of cardiac conduction: a history of revisions. Am. J. Physiol. Heart Circ. Physiol. 306, H619–H627. doi: 10.1152/ajpheart.00760.2013
Veeraraghavan, R., Poelzing, S., and Gourdie, R. G. (2014b). Intercellular electrical communication in the heart: a new, active role for the intercalated Disk. Cell Commun. Adhes. 21, 161–167. doi: 10.3109/15419061.2014.905932
Veeraraghavan, R., Gyorke, S., and Radwański, P. B. (2017). Neuronal sodium channels: emerging components of the nano-machinery of cardiac calcium cycling. J. Physiol. 595, 3823–3834. doi: 10.1113/JP273058
Veeraraghavan, R., Larsen, A. P., Torres, N. S., Grunnet, M., and Poelzing, S. (2013). Potassium channel activators differentially modulate the effect of sodium channel blockade on cardiac conduction. Acta Physiol. (Oxf.) 207, 280–289. doi: 10.1111/j.1748-1716.2012.02481.x
Veeraraghavan, R., Lin, J., Hoeker, G. S., Keener, J. P., Gourdie, R. G., and Poelzing, S. (2015). Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study. Pflugers Arch. 467, 2093–2105. doi: 10.1007/s00424-014-1675-z
Veeraraghavan, R., Lin, J., Keener, J. P., Gourdie, R., and Poelzing, S. (2016). Potassium channels in the Cx43 gap junction perinexus modulate ephaptic coupling: an experimental and modeling study. Pflugers. Arch. 468, 1651–1661. doi: 10.1007/s00424-016-1861-2
Veeraraghavan, R., Salama, M. E., and Poelzing, S. (2012). Interstitial volume modulates the conduction velocity- gap junction relationship. Am. J. Physiol. Heart Circ. Physiol. 302, H278–H286. doi: 10.1152/ajpheart.00868.2011
Veerman, C. C., Wilde, A. A., and Lodder, E. M. (2015). The cardiac sodium channel gene SCN5A and its gene product NaV1.5: role in physiology and pathophysiology. Gene 573, 177–187. doi: 10.1016/j.gene.2015.08.062
Venetucci, L. A., Trafford, A. W., O’Neill, S. C., and Eisner, D. A. (2008). The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 77, 285–292. doi: 10.1093/cvr/cvm009
Viatchenko-Karpinski, S., Kornyeyev, D., El-Bizri, N., Budas, G., Fan, P., Jiang, Z., et al. (2014). Intracellular Na+ overload causes oxidation of CaMKII and leads to Ca2+ mishandling in isolated ventricular myocytes. J. Mol. Cell Cardiol. 76, 247–256. doi: 10.1016/j.yjmcc.2014.09.009
Viatchenko-Karpinski, S., Terentyev, D., Jenkins, L. A., Lutherer, L. O., and Gyorke, S. (2005). Synergistic interactions between Ca2+ entries through L-type Ca2+ channels and Na+-Ca2+ exchanger in normal and failing rat heart. J. Physiol. 567(Pt 2), 493–504. doi: 10.1113/jphysiol.2005.091280
Keywords: nanopathies, arrhythmias, cardiac, sodium channels, sodium calcium exchanger, RyR2
Citation: Radwański PB, Johnson CN, Györke S and Veeraraghavan R (2018) Cardiac Arrhythmias as Manifestations of Nanopathies: An Emerging View. Front. Physiol. 9:1228. doi: 10.3389/fphys.2018.01228
Received: 01 June 2018; Accepted: 14 August 2018;
Published: 04 September 2018.
Edited by:
Alexey V. Glukhov, University of Wisconsin System, United StatesReviewed by:
Yohannes Castro Shiferaw, California State University, Northridge, United StatesCopyright © 2018 Radwański, Johnson, Györke and Veeraraghavan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Przemysław B. Radwański, UHJ6ZW15c2xhdy5SYWR3YW5za2lAb3N1bWMuZWR1 Rengasayee Veeraraghavan, VmVlcmFyYWdoYXZhbi4xMkBvc3UuZWR1; U2FpdkB2dC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.