
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 26 July 2018
Sec. Vascular Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01018
This article is part of the Research Topic Arterial Aging and Age-Associated Arterial Diseases View all 18 articles
 Ali H. Eid1,2Ahmed F. El-Yazbi1,3†Fouad Zouein1†Abdelilah Arredouani4Allal Ouhtit2Md M. Rahman2Hatem Zayed5Gianfranco Pintus5Haissam Abou-Saleh2*
Ali H. Eid1,2Ahmed F. El-Yazbi1,3†Fouad Zouein1†Abdelilah Arredouani4Allal Ouhtit2Md M. Rahman2Hatem Zayed5Gianfranco Pintus5Haissam Abou-Saleh2*Chronic hypertension remains a major cause of global mortality and morbidity. It is a complex disease that is the clinical manifestation of multiple genetic, environmental, nutritional, hormonal, and aging-related disorders. Evidence supports a role for vascular aging in the development of hypertension involving an impairment in endothelial function together with an alteration in vascular smooth muscle cells (VSMCs) calcium homeostasis leading to increased myogenic tone. Changes in free intracellular calcium levels ([Ca2+]i) are mediated either by the influx of Ca2+ from the extracellular space or release of Ca2+ from intracellular stores, mainly the sarcoplasmic reticulum (SR). The influx of extracellular Ca2+ occurs primarily through voltage-gated Ca2+ channels (VGCCs), store-operated Ca2+ channels (SOC), and Ca2+ release-activated channels (CRAC), whereas SR-Ca2+ release occurs through inositol trisphosphate receptor (IP3R) and ryanodine receptors (RyRs). IP3R-mediated SR-Ca2+ release, in the form of Ca2+ waves, not only contributes to VSMC contraction and regulates VGCC function but is also intimately involved in structural remodeling of resistance arteries in hypertension. This involves a phenotypic switch of VSMCs as well as an alteration of cytoplasmic Ca2+ signaling machinery, a phenomena tightly related to the aging process. Several lines of evidence implicate changes in expression/function levels of IP3R isoforms in the development of hypertension, VSMC phenotypic switch, and vascular aging. The present review discusses the current knowledge of these mechanisms in an integrative approach and further suggests potential new targets for hypertension management and treatment.
Cardiovascular diseases (CVD) remain the leading cause of death worldwide, with hypertension being the number one cause of this high mortality (Forouzanfar et al., 2017). Nearly one-third of the yearly global mortality is due to CVD (Chen et al., 2013). At least half or more of ischemic stroke, hemorrhagic stroke, ischemic heart disease and other CVD such as cardiomyopathy, aortic aneurysms, or peripheral vascular disease are intimately attributed to elevated blood pressure (BP), or hypertension (Forouzanfar et al., 2017). This burden is on the rise, despite all therapeutic advances made in recent years, especially in elderly people (Gates et al., 2009; GBD 2013 Risk Factors Collaborators et al., 2015; Harvey et al., 2015; GBD 2015 Risk Factors Collaborators, 2016; Thijssen et al., 2016).
Hypertension is defined as a chronic and persistent elevation of systemic arterial pressure beyond normal values. Etiologically, hypertension is classified as primary and secondary. Primary hypertension, also known as essential hypertension, is the most prevalent form of high BP and constitutes around 90–95% of the cases with unknown etiology (Carretero and Oparil, 2000; Rossier et al., 2017). Secondary hypertension, on the other hand, constitutes around 5–10% of hypertensive cases and arises from known and identifiable causes such as kidney diseases, pregnancy, endocrine disorders, neurological diseases, and others (Chiong et al., 2008).
Chronic hypertension predisposes nearly 1.5 billion individuals in the world to CVD, including ventricular hypertrophy and heart failure, stroke, and renal damage (Chockalingam, 2008). A number of factors are known to increase the risk of high BP development including obesity, sedentary lifestyle, insulin resistance, high alcohol intake, high salt intake, smoking, and aging (Carretero and Oparil, 2000; Gates et al., 2009; Green et al., 2010). The development of essential hypertension involves multiple physiological mechanisms including cardiac output, peripheral resistance, renin–angiotensin–aldosterone system, autonomic nervous system, and vasoactive substances such as endothelin, bradykinin, natriuretic peptides, and others (Beevers et al., 2001; Cain and Khalil, 2002).
The etiology of hypertension is complex and results from the interaction of multiple genetic, neuronal, hormonal, environmental factors, and aging-associated diseases (Garbers and Dubois, 1999; Oparil et al., 2003; Chockalingam, 2008). In fact, with over 50 genes implicated in BP regulation, and other risk factors contributing to the pathogenesis of hypertension, it is rarely possible to determine the etiology of the disease. However, strong evidences support the role of “vascular aging” in the development of hypertension (Green et al., 2010; Fritze et al., 2012; van den Munckhof et al., 2012). In fact, progressive aging implies endothelial dysfunction, loss of nitric oxide (NO) bioavailability, impaired vasodilation, vascular remodeling, and increased arterial stiffness. In addition, the molecular and cellular mechanisms underlying vascular alterations are common and include impaired Ca2+ signaling, oxidative stress, and production of pro-inflammatory cytokines and pro-fibrotic growth factors.
Regardless of its etiology, a hallmark of all cases of hypertension is an increased vascular resistance that leads to elevated BP. Resistance arteries, with an internal diameter of less than 350 μm, are key elements in the control of peripheral vascular resistance. The major drop in hydrostatic pressure in the vascular tree occurs at the level of resistance arteries. As described by Poiseuille’s law, resistance to blood flow is inversely proportional to the vessel radius to the fourth power; therefore, small variations in the lumen of resistance arteries result in significant effects on peripheral resistance with a pronounced impact on BP. Hence, peripheral resistance is typically a function of the diameter of resistance arteries which, in turn, is intricately linked to the contractility state (vasomotor tone) of vascular smooth muscle cells (VSMCs) (Bosnjak, 1993; Hill et al., 2001). Indeed, it is these VSMCs in resistance arteries and arterioles that act as the main effectors in the continuous regulation of vascular resistance. By stretching VSMCs, BP activates a myriad of signaling events that eventually produces myogenic tone, a distinguishing feature of resistance arteries and arterioles (D’Angelo et al., 1997; Davis, 2012; Mufti et al., 2015; Kroetsch et al., 2017). Furthermore, this tone represents the baseline on which various primary messengers such as neurotransmitters, endothelium-derived vasoactive molecules, local metabolites, or hormones converge and act to modulate constriction and dilatation. Many membrane channels and receptors play a pivotal role in vasotone regulation. VSMCs of resistance arteries express several plasma membrane (PM) ion channels including K+ channels (Taguchi et al., 1994; Sobey et al., 1998; Tajada et al., 2012), Ca2+ channels, Cl− channels (Bulley et al., 2012; Dam et al., 2014; Heinze et al., 2014), transient receptor potential (TRP) family of ion channels, voltage-gated Ca2+ channels (VGCCs) (Hondeghem et al., 1986; Inoue et al., 2001; Liao et al., 2007), epithelial Na+/acid-sensing channel (ENaC) (Jernigan and Drummond, 2005; Drummond, 2009; Grifoni et al., 2010), and stretch-activated channel, also known as PIEZO1 (Allison, 2017). In addition to these channels, IP3R and ryanodine receptor (RyR), which are localized SR membrane play an important role in VSMC contractility and the development of hypertension (Long et al., 2007; Mufti et al., 2010; Lin et al., 2016).
It is important to note that with sustained hypertension, vessels undergo progressive alteration characterized by inflammatory responses, VSMC growth and migration, extracellular matrix synthesis and degradation, endothelial dysfunction that increases vascular stiffness and resistance, and decreases vascular elasticity (Shyu, 2009; Dharmashankar and Widlansky, 2010; Renna et al., 2013). Remodeled vessels heavily contribute to the pathophysiology of vascular diseases such as atherosclerosis, and are subsequently at high risk of blockage or rupture that could damage and fail the supplied organ (Renna et al., 2013). This review will highlight the role of alterations in inositol trisphosphate receptors (IP3R) expression/function in changes in vascular remodeling and vascular tone, and VSM contractility in response to chronic hypertension. A summary of the proposed model is presented in Figure 1.
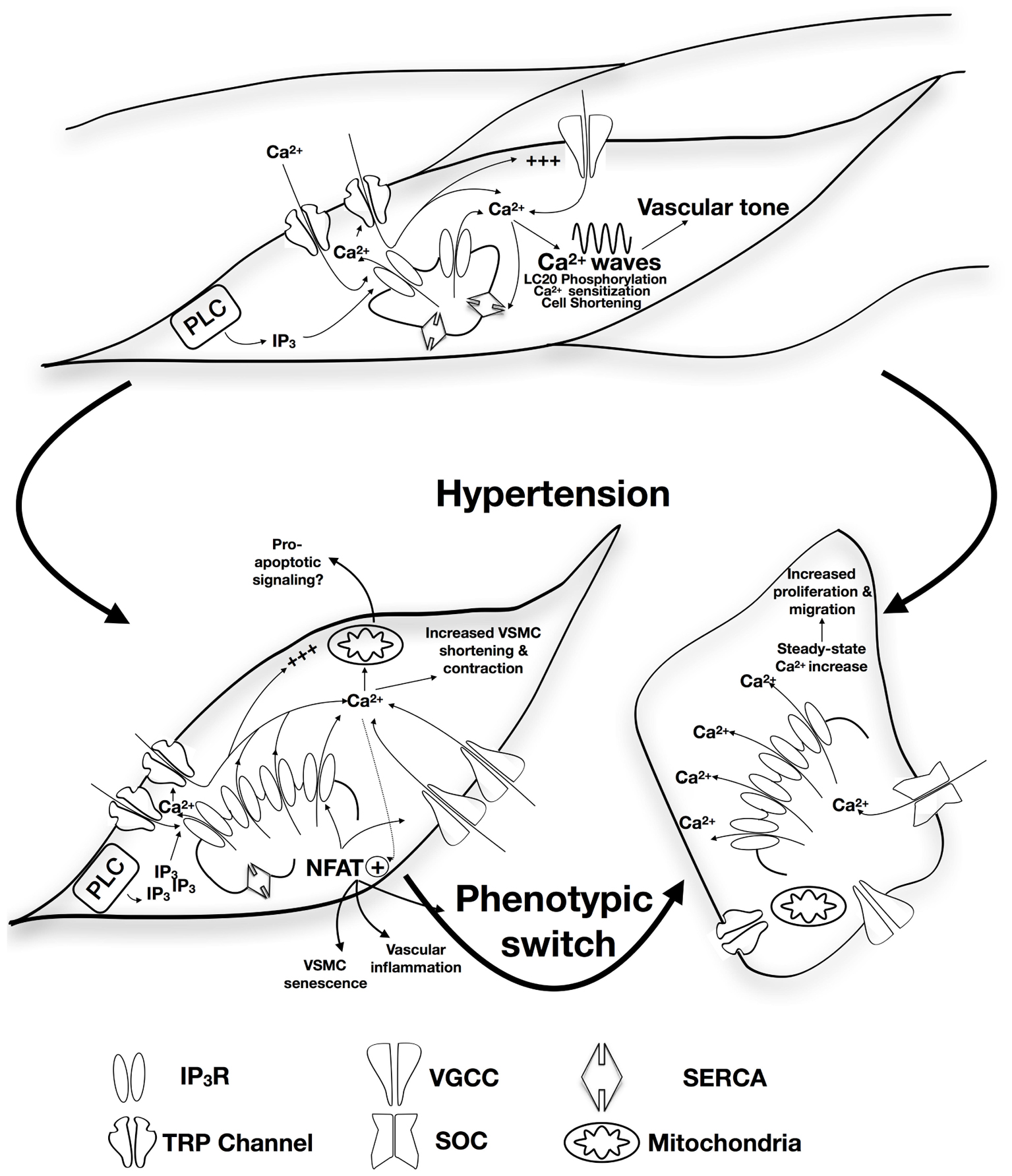
FIGURE 1. Proposed role of alteration in IP3R expression/function in the development of hypertension. Apart from contribution to vascular tone and VSM contractility via SR-Ca2+ release, calcium wave production, and induction of calcium sensitization, IP3R forms a mechanosensing complex with TRP channels that is proposed to initiate contraction in response to increased intraluminal pressure. Physiological patterns of IP3R-mediated Ca2+ release are affected by the expression of other SR membrane proteins including SERCA. Under circumstances of sustained increased blood pressure, IP3R expression increases together with increased sensitivity to IP3 and increased coupling with the TRP mechanosensing machinery resulting in increased intracellular Ca2+ release and increased VSM contraction. Interplay with NFAT-mediated signaling pathways might contribute pressure-induced changes in VSMC phenotype, vascular inflammation, and VSMC senescence. Increased VSMC proliferation and migration were reported to involve increased IP3R-mediated Ca2+ release with subsequent intracellular store depletion and increased store-operated Ca2+ entry. IP3R-mediated Ca2+ release can potentially relay apoptotic signaling to the mitochondria contributing to vascular aging.
The IP3R of VSMCs plays important roles in gene expression, cellular proliferation, and migration, as well as contractility (Wilkerson et al., 2006; Xi et al., 2008; Adebiyi et al., 2010). IP3R is a tetramer, with each subunit encompassing an amino terminus, six transmembrane domains, and a carboxy terminal tail (Michikawa et al., 1994; Yoshikawa et al., 1996). The amino terminus contains an IP3-binding domain, a suppressor domain that inhibits IP3 binding, and a regulatory domain (Yoshikawa et al., 1999). This regulatory domain contains binding sites for Ca2+ and ATP as well as consensus phosphorylation sites (Michikawa et al., 1994; Patel et al., 1999; Foskett et al., 2007). Within this regulatory domain, there is also a coupling motif that is important for physical interactions between IP3R and transient receptor potential canonical (TRPC) channels (Tang et al., 2001; Adebiyi et al., 2010; Zhao et al., 2017). The transmembrane and carboxy terminal domains are essential for tetramerization of IP3Rs (Mignery and Sudhof, 1990; Sayers et al., 1997).
The IP3R family comprises three subtypes (IP3R1, IP3R2, and IP3R3) that are encoded by Itpr1, Itpr2, and Itpr3, respectively. Almost all animal cells express IP3Rs (Prole and Taylor, 2016). The human isoforms share approximately 75% amino acid homology; however, their sensitivity toward IP3 or other regulatory factors is variable, thus adding a layer of complexity for their functions (Foskett et al., 2007; Mikoshiba, 2007). This complexity is further compounded by the existence of many splice variants as well as the possibility of tetramerization (Foskett et al., 2007; Mikoshiba, 2007).
Although most cells express more than one IP3R subtype, the different subtypes exhibit some tissue-specific pattern of expression, with one subtype being expressed at a higher level than the others (Vermassen et al., 2004; Ivanova et al., 2014). Moreover, the different subtypes exhibit marked difference in their affinity for their ligand IP3 (Ivanova et al., 2014; Vervloessem et al., 2015). They also differ in their regulation by Ca2+ and ATP as well as their phosphorylation by various kinases (Ivanova et al., 2014; Vervloessem et al., 2015). For instance, although all three isoforms exhibit a biphasic mode of IP3-induced Ca2+ release, isoform-specific characteristics of this response are observed (Miyakawa et al., 1999; Mak et al., 2001; Tu et al., 2005). Likewise, ATP regulates all three isoforms but with a clear differential effect on each. For example, IP3R2 is 10 times more sensitive to ATP than IP3R3, at least in pancreatic acinar cells (Park et al., 2008). Moreover, while all three isoforms are targets for many kinases such as Akt, PKA, and MAP kinases, isoform-specific regulation by these kinases are markedly noticed. For instance, ERK1/2 can recognize three phosphorylation resides (S436, T945, and S1765) in IP3R1 but not in IP3R2 or IP3R3 (Bosanac et al., 2004).
Vascular smooth muscle cells expresses all three subtypes, with IP3R1 being the predominant one in these cells (Islam et al., 1996; Wang et al., 2001; Grayson et al., 2004; Zhou et al., 2008). Levels of these proteins are determined by a well-regulated balance between transcription and degradation. For instance, while c-Myb stimulates its expression, retinoic acid and TGF-beta inhibit expression of IP3R1 (Sharma et al., 1997; Deelman et al., 1998; Afroze et al., 2007). Hydrogen peroxide Jak2 kinase, Herpud1, and vasopressin regulate levels of IP3R1 by modulating its degradation (Sipma et al., 1998; Wallace et al., 2005; Martin-Garrido et al., 2009; Torrealba et al., 2017).
The main location of IP3Rs in VSMCs is the SR, both central (perinuclear), and peripheral (beneath the PM) compartments (Nixon et al., 1994; Gordienko et al., 2008; Narayanan et al., 2012). Importantly, this localization may impart a functional effect. For instance, IP3Rs located around the nucleus are thought to regulate Ca2+-dependent gene expression without affecting the global intracellular pool of Ca2+. On the other hand, peripherally located SR allows their IP3Rs to be close enough to the PM for localized signaling to membrane proteins to be efficiently elicited (Adebiyi et al., 2010; Zhao et al., 2010). It is also important to note that in addition to their role in Ca2+ release, IP3Rs have other significant functions. For example, upon binding, IP3 causes IP3R–binding protein released with IP3 (IRBIT) to be released from the IP3-binding site. The now released IRBIT can then modulate other targets such as transporters, channels as well as ribonucleotide reductase (Ando et al., 2003; Arnaoutov and Dasso, 2014). Moreover, IP3Rs, independent of their Ca2+ release ability, may also regulate others proteins such as the opening of TRPC (Zhang et al., 2001). As such, cellular distribution of IP3Rs and their Ca2+-independent roles dictate the functions of these receptors, under both physiologic and pathophysiologic conditions as will be discussed below.
Under physiological conditions, individual components of the vascular system maintain a certain degree of spontaneous constriction constituting the vascular tone. This vascular property determines the dilatory capacity of the vascular bed and hence the organ, whereby a higher tone allows for a higher dilatory capacity as in the heart and skeletal muscles, and a lower tone leads to a limited dilatory capacity as in case of cerebral circulation (Klabunde, 2012). Indeed, vascular tone results from the integration of several competing stimuli that modulate the contractile state of VSMC. In isolated vessels, the myogenic response constitutes the fundamental form of vascular reactivity in response to increased intraluminal pressure (Uchida and Bohr, 1969). Extrinsic influences converge to modulate this intrinsic contractility. The overall vascular tone is set as a net outcome of the interaction of endothelial inputs activated by sheer stress (Koller et al., 1993), neuronal regulation (Fleming et al., 1987), humoral mediators (Waldemar and Paulson, 1989), tissue metabolic demand (Chovanes and Richards, 2012), and tubuloglomerular feedback (characteristic to the renal vascular bed) (Burke et al., 2014). The resultant level of constriction determines the extent of systemic vascular resistance and hence contributes to regulating BP, making the examination of alterations in vascular tone an attractive target in the study of hypertension.
Significantly, studies showed substantial alterations in vascular tone in hypertension. Whether it is a causative factor or adaptive consequence of hypertension, enhanced myogenic response was reported in humans and animal models of the disease (Henriksen et al., 1981; Sonoyama et al., 2007). Early studies on spontaneously hypertensive rats showed a reduced ability of cerebral arterioles to dilate increasing the cerebral blood flow in response to intraluminal pressure reduction (Waldemar and Paulson, 1989). Subsequent multiple reports on these animals described an enhanced myogenic constriction in response to intra-luminal pressure in different vascular beds including skeletal muscle arterioles (Falcone et al., 1993; Shibuya et al., 1998), mesenteric arteries (Matrougui et al., 2000), cerebral arterioles (Jarajapu and Knot, 2005), and renal afferent arterioles (Ren et al., 2010).
Among other factors, intracellular Ca2+ is known to play a pivotal role in the development and maintenance of vascular myogenic tone. The increased intraluminal pressure was shown to elicit an increased intracellular Ca2+ in a number of vessel preparations that develop myogenic response (Schubert and Mulvany, 1999). Knot and Nelson (1998) reported a strong correlation between vessel constriction in isolated pressurized rat cerebral arteries and intraluminal pressure increase, membrane depolarization, and increased intracellular Ca2+. Early studies using Ca2+-sensitive dyes and two-dimensional electrophoresis showed that the increase in intracellular Ca2+ levels ([Ca2+]i) in these vessels was associated with an increase in myosin light chain (LC20) phosphorylation (Zou et al., 1995). Further investigation of the temporal association between increased intraluminal pressure, increased [Ca2+]i, and LC20 phosphorylation showed close coincidence of the three events in vessels examined in pressure myography experiments (Zou et al., 2000). Upon activation by Ca2+/calmodulin, the myosin light-chain kinase (MLCK) specifically phosphorylates LC20 at serine-19 (Kamm and Stull, 2001), an event that is sufficient to activate the ATPase activity of actomyosin, cross-bridge cycling, and cell shortening and contraction (Walsh et al., 1982).
Several receptors and transporters contribute to [Ca2+]i dynamics in VSM, but the two primary pathways for Ca2+ influx are the PM L-type VGCC and the ER membrane IP3R (Hill et al., 2001). On the one hand, depolarization of the PM activates α1C, the pore-forming subunit of the VGCC, causing a rapid Ca2+ entry from extracellular space and thus leading to VSMC contraction. On the other hand, triggering of IP3R by IP3 induces Ca2+ release from the ER Ca2+ stores. The fundamental role of these two Ca2+ signaling pathways in the clinical management of hypertension is demonstrated by the fact that pharmacological blockers of the L-type VGCC or α-adrenergic receptors are effective in lowering BP (Oparil et al., 2003). In contractile VSMC, VGCCs are the major determinants of [Ca2+]i and vascular tone. Indeed, it is mainly via through these channels that Ca2+ enters the cell. Nevertheless, studies also implicated RyR-mediated SR Ca2+ release not only as a potential contributor to the generation of myogenic tone (Mufti et al., 2010), but also via feedback regulation of VSMC depolarization through activation of large conductance Ca2+-dependent potassium channels (Krishnamoorthy et al., 2014).
The idea of Ca2+ influx through VGCC contributing to the development of myogenic response stemmed from early results demonstrating a complete loss of myogenic response following extracellular Ca2+ removal in a variety of vessel preparations (Schubert and Mulvany, 1999) and later corroborated by the close association between membrane potential, intracellular Ca2+ level, and myogenic contractility (Knot and Nelson, 1998). Voltage-associated Ca2+ currents were shown to occur following membrane stretching in cerebral artery (McCarron et al., 1997) and blockade of VGCC, while not affecting the depolarization produced by the increase in intraluminal pressure, inhibited the increase in vessel wall Ca2+ and the myogenic response (Knot and Nelson, 1998). Models proposed for this mechanotransduction process spanned the involvement of membrane integrins activating downstream Ca2+-sensitive and insensitive contractile pathways to a role for stretch sensitive channels (Colinas et al., 2015; Mufti et al., 2015).
Out of the several members of the VGCC family, the L-type Ca2+ channels received the most and earliest attention as the mediator of the extracellular Ca2+ influx in myogenic response. Certainly, L-type Ca2+ channels are broadly expressed in VSMC (Abd El-Rahman et al., 2013), and interference with Ca2+ influx through these channels with selective blockers was shown to preclude the myogenic response, at least partially, in many vessel preparations in early studies (McCarron et al., 1997; Knot and Nelson, 1998). On the other hand, interventions that increased L-type Ca2+ channel expression were associated with an increased myogenic tone (Narayanan et al., 2010). Interestingly, earlier studies of spontaneously hypertensive rats implicated increased Ca2+ influx via VGCC in the observed augmentation of myogenic contractility (Ren et al., 2010). It is now widely accepted that an upregulation of VGCC expression and/or function occurs in the context of hypertension (Joseph et al., 2013; Tajada et al., 2013). Several signaling proteins are implicated in this process including protein kinase C (PKC) (Joseph et al., 2013) and PI3K (Carnevale and Lembo, 2012), providing a mechanistic context for the contribution of humoral mediators such as angiotensin in increased vascular resistance.
Of interest, a model was proposed implicating a role for IP3R in regulating extracellular Ca2+ influx in VSMCs. IP3R activation synergistically enhanced TRP channels mediated stretch-induced depolarization (Gonzales et al., 2014). IP3R organizes in a signaling complex with TRPC and TRPM channels whereby stretch activates a phospholipase C isoform in addition to Ca2+ influx through TRPC channels. The resultant IP3 sensitizes IP3R to Ca2+ entering through TRPC leading to an increased SR Ca2+ release activating TRPM currents establishing VSMC depolarization. Significantly, the physical coupling between IP3R and TRP channels increased in resistance arteriole myocytes from animal models of genetic hypertension leading to an enhanced IP3-dependent cationic current and depolarization (Adebiyi et al., 2012).
In addition to the extracellular Ca2+ influx, it is well documented that Ca2+ release from the SR in the form of Ca2+ waves is involved in arterial constriction (Boittin et al., 1999; Jaggar and Nelson, 2000; Lee et al., 2005). Specifically, during the myogenic response, both the number of active cells that display Ca2+ waves and the frequency of these waves in a given VSMC dramatically increased upon raising the intraluminal pressure from 20 to 40 mmHg (Mufti et al., 2010). The incidence of Ca2+ waves at high pressure was not affected by L-type Ca2+ channel blockade but was rather sensitive to interference with SR Ca2+ release. SR Ca2+ depletion precluded Ca2+ wave production, LC20 phosphorylation, and myogenic response generation. Specifically, direct inhibition of IP3R was associated with impaired Ca2+ wave generation and interference with the myogenic tone production (Mufti et al., 2015). Similar effects of IP3R inhibition on micro-vessel contractility and Ca2+ wave production were recently observed in human tissues (Navarro-Dorado et al., 2014). Importantly, the expression of several SR and PM-associated Ca2+ handling proteins, including IP3R, sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), and Na+/Ca2+ exchanger, was upregulated in different hypertensive animal models. Together with an increased SR Ca2+ release, the upregulation of these proteins leads to enhanced basal and evoked vascular constriction (Linde et al., 2012; Abou-Saleh et al., 2013). Specifically, IP3R expression was shown to be higher in VSMC from spontaneously hypertensive rats compared to non-hypertensive controls (Bernier and Guillemette, 1993). Moreover, in rat models of genetic hypertension, both IP3 production and IP3/IP3R-binding affinity are increased (Wu and de Champlain, 1996) together with an increased global [Ca2+]i (Jarajapu and Knot, 2005).
Yet, Osol et al. (2002) showed that increased force production in a myogenic vessel preparation in the pressure range that is associated with myogenic contractility (60–140 mmHg) was not associated with appreciable increases in membrane depolarization or intracellular Ca2+ concentration. As well, a number of early studies raised the possibility of the involvement of Ca2+-independent force generation mechanisms in myogenic contractility. Calcium–tone relationships were shown to be fivefold more sensitive during pressure-induced activation (VanBavel et al., 1998), and myogenic contractility persisted in situations with reduced extracellular Ca2+ levels (McCarron et al., 1997) or where membrane potential was clamped in a depolarized state with high extracellular potassium precluding further Ca2+ entry (McCarron et al., 1997; Lagaud et al., 2002). Multiple lines of evidence implicated signaling pathways involving activation of G proteins, PKC, and Rho-associated protein kinase (ROK) in the generation of Ca2+-independent contraction within the context of the vascular tone development (McCarron et al., 1997; VanBavel et al., 2001; Wesselman et al., 2001; Lagaud et al., 2002). Interestingly, a growing body of evidence suggests that enhanced Ca2+ sensitization contributes to augmented vascular tone in models of hypertension (Uehata et al., 1997; Jarajapu and Knot, 2005; Zicha et al., 2014; Behuliak et al., 2017). However, despite the direct observations that a sustained arteriolar constriction could be obtained via enhanced actin cytoskeleton reorganization triggered by a seemingly Ca2+-independent signaling pathways (ROK- and PKC-mediated pathways) (Moreno-Domínguez et al., 2013, 2014; Colinas et al., 2015; El-Yazbi et al., 2015). These studies clearly demonstrated the obligate dependence of the generation of arteriolar vascular tone on Ca2+. Not only interferences with extracellular Ca2+ levels affected the myogenic response and the mechanisms of all force generation including the Ca2+-independent pathways; specific inhibition of IP3R precluded a pressure-dependent increase in Ca2+ sensitization (Mufti et al., 2015).
Apart from mechanisms contributing to vascular tone through regulation of the contractile machinery, an additional interesting factor is the alteration in structural properties of the vessel wall, referred to as vascular remodeling, a phenomenon strongly associated with age (Baek and Kim, 2011). Initially, vascular remodeling constituted an adaptive response of VSMC to hemodynamic changes that can be sensed by vascular cells, both endothelial and SMCs and translated into structural alteration within the vessel wall. On the long run, however, these adaptations lead to increased media thickness, reduced luminal diameter, and extracellular matrix reorganization (Mulvany et al., 1996; Touyz, 2005; Lemarie et al., 2010; Rizzoni and Agabiti-Rosei, 2012). Furthermore, vascular injury induced by disruption of atheromatous plaque or balloon angioplasty triggers a reparative response that includes inflammation, migration and proliferation of VSMC, and intimal hyperplasia. Ultimately, due to changes in vessel architecture and geometry, this leads to a negative constrictive remodeling of the arterial wall (Gibbons and Dzau, 1994; Faxon et al., 1997).
It is now accepted that structural remodeling in resistance arteries is closely related to the development of hypertension (Lemarie et al., 2010; Rizzoni and Agabiti-Rosei, 2012). In this perspective, smooth muscle cells display a significant degree of phenotypic plasticity and, unlike most other differentiated cells, can change their phenotype even at the differentiated state (Yoshida and Owens, 2005; Matchkov et al., 2012). This involves a phenotypic switch from a contractile to a proliferative, migrating, and or/synthetic phenotype and is associated with gene regulation and alteration of cytoplasmic Ca2+ signaling machinery (House et al., 2008; Matchkov et al., 2012). While vascular remodeling in aging has been partially investigated (Wang et al., 2005, 2006, 2007), the molecular mechanisms involved in the remodeling of Ca2+ signaling pathways observed in hypertension is still poorly understood.
In VSMCs, resting [Ca2+]i is slightly higher than in other cells, allowing the vessel to be in a constant state of partial contraction. In the synthetic phenotype, however, this turns to be less important or even voltage-independent. In contrast to the role proposed for VGCC and IP3R in VSM contraction, it has been suggested that regulation of [Ca2+]i in synthetic VSMC occurs via alternative pathways including store-operated channels (SOCs) and receptor-operated channels (ROCs) (Berra-Romani et al., 2008; Baryshnikov et al., 2009). SOCs are activated by depletion of internal Ca2+ stores mainly through IP3-mediated Ca2+ release (Trebak, 2012), whereas ROCs activation involves different components of the PLC signaling cascade including IP3 (House et al., 2008). It is beyond the scope of this review to discuss these two pathways in more detail. Of note, however, the expression level of all three IP3R isoforms increase during VSMC switch from contractile to synthetic phenotype (Berra-Romani et al., 2008). Additionally, IP3R-mediated Ca2+ release increases in proliferating VSMC offering a possible explanation for the observed increased in SOC Ca2+ entry (Moses et al., 2001; Wilkerson et al., 2006). Selective inhibition of IP3R not only reduced VSMC proliferation (Wang et al., 2001; Wilkerson et al., 2006) but also inhibited in vitro pressure-induced increase in VSMC migration (Tada et al., 2008). Evidence in synthetic human VSMCs point to an altered mode of Ca2+ release via IP3R (Bobe et al., 2011). IP3-mediated release in these cells occurs in a steady state followed by store-operated calcium entry. This pattern was restored to the oscillatory Ca2+ release pattern characteristic to contractile VSMCs upon upregulation of SERCA pump expression. This switch reduced nuclear factor of activated T cells (NFAT) signaling. In the context of hypertension, we have previously shown that L-type Ca2+ channels and IP3R are specifically and concomitantly upregulated in an angiotensin-induced hypertension model through a NFAT-dependent pathway (Abou-Saleh et al., 2013). Functionally, this was associated with enhancement and sensitization of IP3-dependent Ca2+ release, thereby resulting in higher basal Ca2+ levels and increased VSM contraction. In addition to hypertension, upregulated NFAT signaling in the vasculature was implicated in a number of age-related disorders including post-injury restenosis (Bonnet et al., 2009), vascular inflammation, and aggravation of atherosclerosis in diabetes (Nilsson-Berglund et al., 2010; Zetterqvist et al., 2014), as well as vascular smooth muscle senescence (Min et al., 2009). This latter observation taken together with the evidence regarding the association of increased IP3R expression/activity, NFAT signaling, and VSMC phenotypic switch may add novel insights into the role of IP3R in VSMC molecular remodeling as a part of the aging process. Specifically, studies in several cell types demonstrated that different IP3R isoforms occur in close proximity to the mitochondria and transmit pro-apoptotic Ca2+ signals (Simpson et al., 1998; Szalai et al., 1999; Mendes et al., 2005). Yet, it is worth mentioning that the role of IP3R in aging is far from being clear. Whereas IP3 content was shown to increase in rat brain (Igwe and Ning, 1993), IP3R expression and IP3 binding were shown to be decreased (Igwe and Filla, 1997).
In addition to NFAT, other Ca2+ sensitive transcription factors such as serum response factor (SRF), c-response element binding (CREB) seem to play an important role in switching VSMC from a contractile to a synthetic phenotype (Matchkov et al., 2012). Future studies on the role of IP3R in this process need to be conducted in IP3R-deficient mice. In this regard, the role of IP3R in VSMC contractility in vivo was recently highlighted in a conditional triple knockout mouse, where the agonist-mediated vascular constriction was attenuated together with a lack of development of hypertension in response to chronic angiotensin infusion (Lin et al., 2016). However, the effect of the conditional knockout on VSMC phenotypic switch in response to hypertension has not been addressed so far.
Hypertension is a complex disease that arises from the interaction of multiple genetic, environmental, nutritional, hormonal, and age-related pathological conditions. The etiology of “essential hypertension,” which accounts for more than 90% of clinical hypertension, comprises an increased vascular resistance and is associated with structural alterations in the wall of resistance arteries. Modulation of [Ca2+]i in VSMC allows small arteries and arterioles to establish vasomotor tone and regulate blood flow, and determine peripheral vascular resistance and BP. These changes require a phenotypic switch of VSMC from a contractile quiescent to a versatile proliferative phenotype, a phenomenon widely observed in age-associated vascular remodeling. As described above, IP3R activity was shown to be essential in almost every cellular mechanism involved in setting vascular tone level. Additionally, modulation of IP3-dependent Ca2+ signaling may represent an essential stimulus for VSMC shift from quiescent to the proliferative state. KT-362 was an investigational drug targeting IP3R-mediated Ca2+ release that showed a clinically relevant antihypertensive action (Hester and Shibata, 1990). However, clinical trials were discontinued at phase II. In addition to known targets for antihypertensive therapy, novel interventions within the PLC–IP3R pathway constitute attractive therapeutic targets for future research given their ubiquitous involvement in cellular processes leading to hypertension.
AE and HA-S designed and wrote the first draft of the manuscript. AE-Y designed the graphical abstract and reviewed the manuscript. FZ, AA, AO, and MR reviewed the manuscript. HZ, GP, and HA-S proofread and revised the manuscript.
This publication was made possible by an MPP fund (#320133) from the American University of Beirut to AE.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abd El-Rahman, R. R., Harraz, O. F., Brett, S. E., Anfinogenova, Y., Mufti, R. E., Goldman, D., et al. (2013). Identification of L- and T-type Ca2+ channels in rat cerebral arteries: role in myogenic tone development. Am. J. Physiol. Heart Circ. Physiol. 304, H58–H71. doi: 10.1152/ajpheart.00476.2012
Abou-Saleh, H., Pathan, A. R., Daalis, A., Hubrack, S., Abou-Jassoum, H., Al-Naeimi, H., et al. (2013). Inositol 1,4,5-trisphosphate (IP3) receptor up-regulation in hypertension is associated with sensitization of Ca2+ release and vascular smooth muscle contractility. J. Biol. Chem. 288, 32941–32951. doi: 10.1074/jbc.M113.496802
Adebiyi, A., Thomas-Gatewood, C. M., Leo, M. D., Kidd, M. W., Neeb, Z. P., and Jaggar, J. H. (2012). An elevation in physical coupling of type 1 inositol 1,4,5-trisphosphate (IP3) receptors to transient receptor potential 3 (TRPC3) channels constricts mesenteric arteries in genetic hypertension. Hypertension 60, 1213–1219. doi: 10.1161/hypertensionaha.112.198820
Adebiyi, A., Zhao, G., Narayanan, D., Thomas-Gatewood, C. M., Bannister, J. P., and Jaggar, J. H. (2010). Isoform-selective physical coupling of TRPC3 channels to IP3 receptors in smooth muscle cells regulates arterial contractility. Circ. Res. 106, 1603–1612. doi: 10.1161/CIRCRESAHA.110.216804
Afroze, T., Sadi, A. M., Momen, M. A., Gu, S., Heximer, S., and Husain, M. (2007). c-Myb-dependent inositol 1,4,5-trisphosphate receptor type-1 expression in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 27, 1305–1311. doi: 10.1161/ATVBAHA.107.142059
Allison, S. J. (2017). Hypertension: mechanosensation by PIEZO1 in blood pressure control. Nat. Rev. Nephrol. 13:3. doi: 10.1038/nrneph.2016.165
Ando, H., Mizutani, A., Matsu-ura, T., and Mikoshiba, K. (2003). IRBIT, a novel inositol 1,4,5-trisphosphate (IP3) receptor-binding protein, is released from the IP3 receptor upon IP3 binding to the receptor. J. Biol. Chem. 278, 10602–10612. doi: 10.1074/jbc.M210119200
Arnaoutov, A., and Dasso, M. (2014). Enzyme regulation. IRBIT is a novel regulator of ribonucleotide reductase in higher eukaryotes. Science 345, 1512–1515. doi: 10.1126/science.1251550
Baek, E. B., and Kim, S. J. (2011). Mechanisms of myogenic response: Ca2+-dependent and -independent signaling. J. Smooth Muscle Res. 47, 55–65. doi: 10.1540/jsmr.47.55
Baryshnikov, S. G., Pulina, M. V., Zulian, A., Linde, C. I., and Golovina, V. A. (2009). Orai1, a critical component of store-operated Ca2+ entry, is functionally associated with Na+/Ca2+ exchanger and plasma membrane Ca2+ pump in proliferating human arterial myocytes. Am. J. Physiol. Cell Physiol. 297, C1103–C1112. doi: 10.1152/ajpcell.00283.2009
Beevers, G., Lip, G. Y., and O’Brien, E. (2001). ABC of hypertension: the pathophysiology of hypertension. BMJ 322, 912–916. doi: 10.1136/bmj.322.7291.912
Behuliak, M., Bencze, M., Vaneckova, I., Kunes, J., and Zicha, J. (2017). Basal and activated calcium sensitization mediated by RhoA/Rho kinase pathway in rats with genetic and salt hypertension. Biomed Res. Int. 2017:8029728. doi: 10.1155/2017/8029728
Bernier, S., and Guillemette, G. (1993). Increased inositol 1,4,5-trisphosphate binding capacity in vascular smooth muscle of spontaneously hypertensive rats. Am. J. Hypertens. 6(3 Pt 1), 217–225.
Berra-Romani, R., Mazzocco-Spezzia, A., Pulina, M. V., and Golovina, V. A. (2008). Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am. J. Physiol. Cell Physiol. 295, C779–C790. doi: 10.1152/ajpcell.00173.2008
Bobe, R., Hadri, L., Lopez, J. J., Sassi, Y., Atassi, F., Karakikes, I., et al. (2011). SERCA2a controls the mode of agonist-induced intracellular Ca2+ signal, transcription factor NFAT and proliferation in human vascular smooth muscle cells. J. Mol. Cell Cardiol. 50, 621–633. doi: 10.1016/j.yjmcc.2010.12.016
Boittin, F. X., Macrez, N., Halet, G., and Mironneau, J. (1999). Norepinephrine-induced Ca2+ waves depend on InsP(3) and ryanodine receptor activation in vascular myocytes. Am. J. Physiol. 277(1 Pt 1), C139–C151. doi: 10.1152/ajpcell.1999.277.1.C139
Bonnet, S., Paulin, R., Sutendra, G., Dromparis, P., Roy, M., Watson, K. O., et al. (2009). Dehydroepiandrosterone reverses systemic vascular remodeling through the inhibition of the Akt/GSK3-beta/NFAT Axis. Circulation 120, 1231–1240. doi: 10.1161/Circulationaha.109.848911
Bosanac, I., Michikawa, T., Mikoshiba, K., and Ikura, M. (2004). Structural insights into the regulatory mechanism of IP3 receptor. Biochim. Biophys. Acta 1742, 89–102. doi: 10.1016/j.bbamcr.2004.09.016
Bosnjak, Z. J. (1993). Ion channels in vascular smooth muscle. Physiology and pharmacology. Anesthesiology 79, 1392–1401. doi: 10.1097/00000542-199312000-00031
Bulley, S., Neeb, Z. P., Burris, S. K., Bannister, J. P., Thomas-Gatewood, C. M., Jangsangthong, W., et al. (2012). TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ. Res. 111, 1027–1036. doi: 10.1161/CIRCRESAHA.112.277145
Burke, M., Pabbidi, M. R., Farley, J., and Roman, R. J. (2014). Molecular mechanisms of renal blood flow autoregulation. Curr. Vasc. Pharmacol. 12, 845–858. doi: 10.2174/15701611113116660149
Cain, A. E., and Khalil, R. A. (2002). Pathophysiology of essential hypertension: role of the pump, the vessel, and the kidney. Semin. Nephrol. 22, 3–16.
Carnevale, D., and Lembo, G. (2012). PI3Kγ in hypertension: a novel therapeutic target controlling vascular myogenic tone and target organ damage. Cardiovasc. Res. 95, 403–408. doi: 10.1093/cvr/cvs166
Carretero, O. A., and Oparil, S. (2000). Essential hypertension. Part I: definition and etiology. Circulation 101, 329–335. doi: 10.1161/01.CIR.101.3.329
Chen, R., Dharmarajan, K., Kulkarni, V. T., Punnanithinont, N., Gupta, A., Bikdeli, B., et al. (2013). Most important outcomes research papers on hypertension. Circ. Cardiovasc. Qual. Outcomes 6, e26–e35. doi: 10.1161/CIRCOUTCOMES.113.000424
Chiong, J. R., Aronow, W. S., Khan, I. A., Nair, C. K., Vijayaraghavan, K., Dart, R. A., et al. (2008). Secondary hypertension: current diagnosis and treatment. Int. J. Cardiol. 124, 6–21. doi: 10.1016/j.ijcard.2007.01.119
Chockalingam, A. (2008). World Hypertension Day and global awareness. Can. J. Cardiol. 24, 441–444. doi: 10.1016/S0828-282X(08)70617-2
Chovanes, G. I., and Richards, R. M. (2012). The predominance of metabolic regulation of cerebral blood flow and the lack of “Classic” autoregulation curves in the viable brain. Surg. Neurol. Int. 3:12. doi: 10.4103/2152-7806.92185
Colinas, O., Moreno-Dominguez, A., Zhu, H. L., Walsh, E. J., Perez-Garcia, M. T., Walsh, M. P., et al. (2015). alpha5-Integrin-mediated cellular signaling contributes to the myogenic response of cerebral resistance arteries. Biochem. Pharmacol. 97, 281–291. doi: 10.1016/j.bcp.2015.08.088
Dam, V. S., Boedtkjer, D. M., Nyvad, J., Aalkjaer, C., and Matchkov, V. (2014). TMEM16A knockdown abrogates two different Ca2+-activated Cl− currents and contractility of smooth muscle in rat mesenteric small arteries. Pflugers Arch. 466, 1391–1409. doi: 10.1007/s00424-013-1382-1
D’Angelo, G., Davis, M. J., and Meininger, G. A. (1997). Calcium and mechanotransduction of the myogenic response. Am. J. Physiol. 273(1 Pt 2), H175–H182. doi: 10.1152/ajpheart.1997.273.1.H175
Davis, M. J. (2012). Perspective: physiological role(s) of the vascular myogenic response. Microcirculation 19, 99–114. doi: 10.1111/j.1549-8719.2011.00131.x
Deelman, L. E., Jonk, L. J., and Henning, R. H. (1998). The isolation and characterization of the promoter of the human type 1 inositol 1,4,5-trisphosphate receptor. Gene 207, 219–225. doi: 10.1016/S0378-1119(97)00630-6
Dharmashankar, K., and Widlansky, M. E. (2010). Vascular endothelial function and hypertension: insights and directions. Curr. Hypertens. Rep. 12, 448–455. doi: 10.1007/s11906-010-0150-2
Drummond, H. A. (2009). Yes, no, maybe so ENaC proteins as mediators of renal myogenic constriction. Hypertension 54, 962–963. doi: 10.1161/Hypertensionaha.109.139014
El-Yazbi, A. F., Abd-Elrahman, K. S., and Moreno-Dominguez, A. (2015). PKC-mediated cerebral vasoconstriction: Role of myosin light chain phosphorylation versus actin cytoskeleton reorganization. Biochem. Pharmacol. 95, 263–278. doi: 10.1016/j.bcp.2015.04.011
Falcone, J. C., Granger, H. J., and Meininger, G. A. (1993). Enhanced myogenic activation in skeletal muscle arterioles from spontaneously hypertensive rats. Am. J. Physiol. 265(6 Pt 2), H1847–H1855. doi: 10.1152/ajpheart.1993.265.6.H1847
Faxon, D. P., Coats, W., and Currier, J. (1997). Remodeling of the coronary artery after vascular injury. Prog. Cardiovasc. Dis. 40, 129–140. doi: 10.1016/S0033-0620(97)80005-9
Fleming, B. P., Barron, K. W., Howes, T. W., and Smith, J. K. (1987). Response of the microcirculation in rat cremaster muscle to peripheral and central sympathetic stimulation. Circ. Res. 61(5 Pt 2), Ii26–Ii31.
Forouzanfar, M. H., Liu, P., Roth, G. A., Ng, M., Biryukov, S., Marczak, L., et al. (2017). global burden of hypertension and systolic blood pressure of at least 110 to 115 mm Hg, 1990-2015. JAMA 317, 165–182. doi: 10.1001/jama.2016.19043
Foskett, J. K., White, C., Cheung, K. H., and Mak, D. O. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658. doi: 10.1152/physrev.00035.2006
Fritze, O., Romero, B., Schleicher, M., Jacob, M. P., Oh, D. Y., Starcher, B., et al. (2012). Age-related changes in the elastic tissue of the human aorta. J. Vasc. Res. 49, 77–86. doi: 10.1159/000331278
Garbers, D. L., and Dubois, S. K. (1999). The molecular basis of hypertension. Annu. Rev. Biochem. 68, 127–155. doi: 10.1146/annurev.biochem.68.1.127
Gates, P. E., Strain, W. D., and Shore, A. C. (2009). Human endothelial function and microvascular ageing. Exp. Physiol. 94, 311–316. doi: 10.1113/expphysiol.2008.043349
GBD 2013 Risk Factors Collaborators, Forouzanfar, M. H., Alexander, L., Anderson, H. R., Bachman, V. F., Biryukov, S., et al. (2015). Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 386, 2287–2323. doi: 10.1016/S0140-6736(15)00128-2
GBD 2015 Risk Factors Collaborators (2016). Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388, 1659–1724.
Gibbons, G. H., and Dzau, V. J. (1994). The emerging concept of vascular remodeling. N. Engl. J. Med. 330, 1431–1438. doi: 10.1056/NEJM199405193302008
Gonzales, A. L., Yang, Y., Sullivan, M. N., Sanders, L., Dabertrand, F., Hill-Eubanks, D. C., et al. (2014). A PLCgamma1-dependent, force-sensitive signaling network in the myogenic constriction of cerebral arteries. Sci. Signal. 7:ra49. doi: 10.1126/scisignal.2004732
Gordienko, D. V., Harhun, M. I., Kustov, M. V., Pucovsky, V., and Bolton, T. B. (2008). Sub-plasmalemmal [Ca2+]i upstroke in myocytes of the guinea-pig small intestine evoked by muscarinic stimulation: IP3R-mediated Ca2+ release induced by voltage-gated Ca2+ entry. Cell Calcium 43, 122–141. doi: 10.1016/j.ceca.2007.04.012
Grayson, T. H., Haddock, R. E., Murray, T. P., Wojcikiewicz, R. J. H., and Hill, C. E. (2004). Inositol 1,4,5-trisphosphate receptor subtypes are differentially distributed between smooth muscle and endothelial layers of rat arteries. Cell Calcium 36, 447–458. doi: 10.1016/j.ceca.2004.04.005
Green, D. J., Swart, A., Exterkate, A., Naylor, L. H., Black, M. A., Cable, N. T., et al. (2010). Impact of age, sex and exercise on brachial and popliteal artery remodelling in humans. Atherosclerosis 210, 525–530. doi: 10.1016/j.atherosclerosis.2010.01.048
Grifoni, S. C., Chiposi, R., McKey, S. E., Ryan, M. J., and Drummond, H. A. (2010). Altered whole kidney blood flow autoregulation in a mouse model of reduced beta-ENaC. Am. J. Physiol. Renal Physiol. 298, F285–F292. doi: 10.1152/ajprenal.00496.2009
Harvey, A., Montezano, A. C., and Touyz, R. M. (2015). Vascular biology of ageing—Implications in hypertension. J. Mol. Cell Cardiol. 83, 112–121. doi: 10.1016/j.yjmcc.2015.04.011
Heinze, C., Seniuk, A., Sokolov, M. V., Huebner, A. K., Klementowicz, A. E., Szijarto, I. A., et al. (2014). Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J. Clin. Invest. 124, 675–686. doi: 10.1172/JCI70025
Henriksen, O., Skagen, K., Amtorp, O., and Hartling, O. (1981). Augmented vasoconstrictor response to changes in vascular transmural pressure in patients with essential arterial hypertension. Acta Physiol. Scand. 112, 323–329. doi: 10.1111/j.1748-1716.1981.tb06823.x
Hester, R. K., and Shibata, S. (1990). KT-362 related effects on intracellular calcium release and associated clinical potential: arrhythmias, myocardial ischemia, and hypertension. Cardiovasc. Drugs Ther. 4, 1345–1354. doi: 10.1007/BF02018262
Hill, M. A., Zou, H., Potocnik, S. J., Meininger, G. A., and Davis, M. J. (2001). Arteriolar smooth muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic reactivity. J. Appl. Physiol. 91, 973–983. doi: 10.1152/jappl.2001.91.2.973
Hondeghem, L. M., Ayad, M. J., and Robertson, R. M. (1986). Verapamil, diltiazem and nifedipine block the depolarization-induced potentiation of norepinephrine contractions in rabbit aorta and porcine coronary arteries. J. Pharmacol. Exp. Ther. 239, 808–813.
House, S. J., Potier, M., Bisaillon, J., Singer, H. A., and Trebak, M. (2008). The non-excitable smooth muscle: calcium signaling and phenotypic switching during vascular disease. Pflugers Arch. 456, 769–785. doi: 10.1007/s00424-008-0491-8
Igwe, O. J., and Filla, M. B. (1997). Aging-related regulation of myo-inositol 1,4,5-trisphosphate signal transduction pathway in the rat striatum. Brain Res. Mol. Brain Res. 46, 39–53. doi: 10.1016/S0169-328X(96)00269-0
Igwe, O. J., and Ning, L. (1993). Inositol 1,4,5-trisphosphate arm of the phosphatidylinositide signal transduction pathway in the rat cerebellum during aging. Neurosci. Lett. 164, 167–170. doi: 10.1016/0304-3940(93)90883-M
Inoue, R., Okada, T., Onoue, H., Hara, Y., Shimizu, S., Naitoh, S., et al. (2001). The transient receptor potential protein homologue TRP6 is the essential component of vascular alpha(1)-adrenoceptor-activated Ca2+-permeable cation channel. Circ. Res. 88, 325–332. doi: 10.1161/01.RES.88.3.325
Islam, M. O., Yoshida, Y., Koga, T., Kojima, M., Kangawa, K., and Imai, S. (1996). Isolation and characterization of vascular smooth muscle inositol 1,4,5-trisphosphate receptor. Biochem. J. 316 (Pt 1), 295–302. doi: 10.1042/bj3160295
Ivanova, H., Vervliet, T., Missiaen, L., Parys, J. B., De Smedt, H., and Bultynck, G. (2014). Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim. Biophys. Acta 1843, 2164–2183. doi: 10.1016/j.bbamcr.2014.03.007
Jaggar, J. H., and Nelson, M. T. (2000). Differential regulation of Ca2+ sparks and Ca2+ waves by UTP in rat cerebral artery smooth muscle cells. Am. J. Physiol. Cell Physiol. 279, C1528–C1539. doi: 10.1152/ajpcell.2000.279.5.C1528
Jarajapu, Y. P., and Knot, H. J. (2005). Relative contribution of Rho kinase and protein kinase C to myogenic tone in rat cerebral arteries in hypertension. Am. J. Physiol. Heart Circ. Physiol. 289, H1917–1922. doi: 10.1152/ajpheart.01012.2004
Jernigan, N. L., and Drummond, H. A. (2005). Vascular ENaC proteins are required for renal myogenic constriction. Am. J. Physiol. Renal Physiol. 289, F891–F901. doi: 10.1152/ajprenal.00019.2005
Joseph, B. K., Thakali, K. M., Moore, C. L., and Rhee, S. W. (2013). Ion channel remodeling in vascular smooth muscle during hypertension: implications for novel therapeutic approaches. Pharmacol. Res. 70, 126–138. doi: 10.1016/j.phrs.2013.01.008
Kamm, K. E., and Stull, J. T. (2001). Dedicated myosin light chain kinases with diverse cellular functions. J. Biol. Chem. 276, 4527–4530. doi: 10.1074/jbc.R000028200
Klabunde, R. E. (2012). Cardiovascular Physiology Concepts. Philadelphia, PA: Lippincott Williams & Wilkins.
Knot, H. J., and Nelson, M. T. (1998). Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J. Physiol. 508 (Pt 1), 199–209. doi: 10.1111/j.1469-7793.1998.199br.x
Koller, A., Sun, D., and Kaley, G. (1993). Role of shear stress and endothelial prostaglandins in flow- and viscosity-induced dilation of arterioles in vitro. Circ. Res. 72, 1276–1284. doi: 10.1161/01.RES.72.6.1276
Krishnamoorthy, G., Sonkusare, S. K., Heppner, T. J., and Nelson, M. T. (2014). Opposing roles of smooth muscle BK channels and ryanodine receptors in the regulation of nerve-evoked constriction of mesenteric resistance arteries. Am. J. Physiol. Heart Circ. Physiol. 306, H981–H988. doi: 10.1152/ajpheart.00866.2013
Kroetsch, J. T., Levy, A. S., Zhang, H., Aschar-Sobbi, R., Lidington, D., Offermanns, S., et al. (2017). Constitutive smooth muscle tumour necrosis factor regulates microvascular myogenic responsiveness and systemic blood pressure. Nat. Commun. 8:14805. doi: 10.1038/ncomms14805
Lagaud, G., Gaudreault, N., Moore, E. D., Van Breemen, C., and Laher, I. (2002). Pressure-dependent myogenic constriction of cerebral arteries occurs independently of voltage-dependent activation. Am. J. Physiol. Heart Circ. Physiol. 283, H2187–H2195. doi: 10.1152/ajpheart.00554.2002
Lee, C.-H., Kuo, K.-H., Dai, J., and van Breemen, C. (2005). Asynchronous calcium waves in smooth muscle cells. Can. J. Physiol. Pharmacol. 83, 733–741. doi: 10.1139/y05-083
Lemarie, C. A., Tharaux, P. L., and Lehoux, S. (2010). Extracellular matrix alterations in hypertensive vascular remodeling. J. Mol. Cell. Cardiol. 48, 433–439. doi: 10.1016/j.yjmcc.2009.09.018
Liao, P., Yu, D., Li, G., Yong, T. F., Soon, J. L., Chua, Y. L., et al. (2007). A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. J. Biol. Chem. 282, 35133–35142. doi: 10.1074/jbc.M705478200
Lin, Q., Zhao, G., Fang, X., Peng, X., Tang, H., Wang, H., et al. (2016). IP3 receptors regulate vascular smooth muscle contractility and hypertension. JCI Insight 1:e89402. doi: 10.1172/jci.insight.89402
Linde, C. I., Karashima, E., Raina, H., Zulian, A., Wier, W. G., Hamlyn, J. M., et al. (2012). Increased arterial smooth muscle Ca2+ signaling, vasoconstriction, and myogenic reactivity in Milan hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 302, H611–H620. doi: 10.1152/ajpheart.00950.2011
Long, C., Cook, L. G., Hamilton, S. L., Wu, G. Y., and Mitchell, B. M. (2007). FK506 binding protein 12/12.6 depletion increases endothelial nitric oxide synthase threonine 495 phosphorylation and blood pressure. Hypertension 49, 569–576. doi: 10.1161/01.hyp.0000257914.80918.72
Mak, D. O., McBride, S., and Foskett, J. K. (2001). Regulation by Ca2+ and inositol 1,4,5-trisphosphate (InsP3) of single recombinant type 3 InsP3 receptor channels. Ca2+ activation uniquely distinguishes types 1 and 3 insp3 receptors. J. Gen. Physiol. 117, 435–446. doi: 10.1085/jgp.117.5.435
Martin-Garrido, A., Boyano-Adanez, M. C., Alique, M., Calleros, L., Serrano, I., Griera, M., et al. (2009). Hydrogen peroxide down-regulates inositol 1,4,5-trisphosphate receptor content through proteasome activation. Free Radic. Biol. Med. 47, 1362–1370. doi: 10.1016/j.freeradbiomed.2009.07.006
Matchkov, V. V., Kudryavtseva, O., and Aalkjaer, C. (2012). Intracellular Ca2+ signalling and phenotype of vascular smooth muscle cells. Basic Clin. Pharmacol. Toxicol. 110, 42–48. doi: 10.1111/j.1742-7843.2011.00818.x
Matrougui, K., Levy, B. I., and Henrion, D. (2000). Tissue angiotensin II and endothelin-1 modulate differently the response to flow in mesenteric resistance arteries of normotensive and spontaneously hypertensive rats. Br. J. Pharmacol. 130, 521–526. doi: 10.1038/sj.bjp.0703371
McCarron, J. G., Crichton, C. A., Langton, P. D., MacKenzie, A., and Smith, G. L. (1997). Myogenic contraction by modulation of voltage-dependent calcium currents in isolated rat cerebral arteries. J. Physiol. 498 (Pt 2), 371–379. doi: 10.1113/jphysiol.1997.sp021864
Mendes, C. C. P., Gomes, D. A., Thompson, M., Souto, N. C., Goes, T. S., Goes, A. M., et al. (2005). The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem. 280, 40892–40900. doi: 10.1074/jbc.M506623200
Michikawa, T., Hamanaka, H., Otsu, H., Yamamoto, A., Miyawaki, A., Furuichi, T., et al. (1994). Transmembrane topology and sites of N-glycosylation of inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 269, 9184–9189.
Mignery, G. A., and Sudhof, T. C. (1990). The ligand binding site and transduction mechanism in the inositol-1,4,5-triphosphate receptor. EMBO J. 9, 3893–3898.
Mikoshiba, K. (2007). IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J. Neurochem. 102, 1426–1446. doi: 10.1111/j.1471-4159.2007.04825.x
Min, L. J., Mogi, M., Tamura, K., Iwanami, J., Sakata, A., Fujita, T., et al. (2009). Angiotensin II type 1 receptor-associated protein prevents vascular smooth muscle cell senescence via inactivation of calcineurin/nuclear factor of activated T cells pathway. J. Mol. Cell. Cardiol. 47, 798–809. doi: 10.1016/j.yjmcc.2009.09.006
Miyakawa, T., Maeda, A., Yamazawa, T., Hirose, K., Kurosaki, T., and Iino, M. (1999). Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 18, 1303–1308. doi: 10.1093/emboj/18.5.1303
Moreno-Domínguez, A., Colinas, O., El-Yazbi, A., Walsh, E. J., Hill, M. A., Walsh, M. P., et al. (2013). Ca2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J. Physiol. 591, 1235–1250. doi: 10.1113/jphysiol.2012.243576
Moreno-Domínguez, A., El-Yazbi, A. F., Zhu, H. L., Colinas, O., Zhong, X. Z., Walsh, E. J., et al. (2014). Cytoskeletal reorganization evoked by Rho-associated kinase- and protein kinase C-catalyzed phosphorylation of cofilin and heat shock protein 27, respectively, contributes to myogenic constriction of rat cerebral arteries. J. Biol. Chem. 289, 20939–20952. doi: 10.1074/jbc.M114.553743
Moses, S., Dreja, K., Lindqvist, A., Lovdahl, C., Hellstrand, P., and Hultgardh-Nilsson, A. (2001). Smooth muscle cell response to mechanical injury involves intracellular calcium release and ERK1/ERK2 phosphorylation. Exp. Cell Res. 269, 88–96. doi: 10.1006/excr.2001.5308
Mufti, R. E., Brett, S. E., Tran, C. H. T., Abd El-Rahman, R., Anfinogenova, Y., El-Yazbi, A., et al. (2010). Intravascular pressure augments cerebral arterial constriction by inducing voltage-insensitive Ca2+ waves. J. Physiol. 588(Pt 20), 3983–4005. doi: 10.1113/jphysiol.2010.193300
Mufti, R. E., Zechariah, A., Sancho, M., Mazumdar, N., Brett, S. E., and Welsh, D. G. (2015). Implications of alphavbeta3 Integrin Signaling in the Regulation of Ca2+ Waves and Myogenic Tone in Cerebral Arteries. Arterioscler. Thromb. Vasc. Biol. 35, 2571–2578. doi: 10.1161/atvbaha.115.305619
Mulvany, M. J., Baumbach, G. L., Aalkjaer, C., Heagerty, A. M., Korsgaard, N., Schiffrin, E. L., et al. (1996). Vascular remodeling. Hypertension 28, 505–506.
Narayanan, D., Adebiyi, A., and Jaggar, J. H. (2012). Inositol trisphosphate receptors in smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 302, H2190–H2210. doi: 10.1152/ajpheart.01146.2011
Narayanan, D., Xi, Q., Pfeffer, L. M., and Jaggar, J. H. (2010). Mitochondria control functional CaV1.2 expression in smooth muscle cells of cerebral arteries. Circ. Res. 107, 631–641. doi: 10.1161/circresaha.110.224345
Navarro-Dorado, J., Garcia-Alonso, M., van Breemen, C., Tejerina, T., and Fameli, N. (2014). Calcium oscillations in human mesenteric vascular smooth muscle. Biochem. Biophys. Res. Commun. 445, 84–88. doi: 10.1016/j.bbrc.2014.01.150
Nilsson-Berglund, L. M., Zetterqvist, A. V., Nilsson-Ohman, J., Sigvardsson, M., Gonzalez Bosc, L. V., Smith, M. L., et al. (2010). Nuclear factor of activated T cells regulates osteopontin expression in arterial smooth muscle in response to diabetes-induced hyperglycemia. Arterioscler. Thromb. Vasc. Biol. 30, 218–224. doi: 10.1161/ATVBAHA.109.199299
Nixon, G. F., Mignery, G. A., and Somlyo, A. V. (1994). Immunogold localization of inositol 1,4,5-trisphosphate receptors and characterization of ultrastructural features of the sarcoplasmic reticulum in phasic and tonic smooth muscle. J. Muscle Res. Cell Motil. 15, 682–700. doi: 10.1007/BF00121075
Oparil, S., Zaman, M. A., and Calhoun, D. A. (2003). Pathogenesis of hypertension. Ann. Intern. Med. 139, 761–776. doi: 10.7326/0003-4819-139-9-200311040-00011
Osol, G., Brekke, J. F., McElroy-Yaggy, K., and Gokina, N. I. (2002). Myogenic tone, reactivity, and forced dilatation: a three-phase model of in vitro arterial myogenic behavior. Am. J. Physiol. Heart Circ. Physiol. 283, H2260–H2267. doi: 10.1152/ajpheart.00634.2002
Park, H. S., Betzenhauser, M. J., Won, J. H., Chen, J., and Yule, D. I. (2008). The type 2 inositol (1,4,5)-trisphosphate (InsP3) receptor determines the sensitivity of InsP3-induced Ca2+ release to ATP in pancreatic acinar cells. J. Biol. Chem. 283, 26081–26088. doi: 10.1074/jbc.M804184200
Patel, S., Joseph, S. K., and Thomas, A. P. (1999). Molecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium 25, 247–264. doi: 10.1054/ceca.1999.0021
Prole, D. L., and Taylor, C. W. (2016). Inositol 1,4,5-trisphosphate receptors and their protein partners as signalling hubs. J. Physiol. 594, 2849–2866. doi: 10.1113/JP271139
Ren, Y., D’Ambrosio, M. A., Liu, R., Pagano, P. J., Garvin, J. L., and Carretero, O. A. (2010). Enhanced myogenic response in the afferent arteriole of spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 298, H1769–H1775. doi: 10.1152/ajpheart.00537.2009
Renna, N. F., de Las Heras, N., and Miatello, R. M. (2013). Pathophysiology of vascular remodeling in hypertension. Int. J. Hypertens. 2013:808353. doi: 10.1155/2013/808353
Rizzoni, D., and Agabiti-Rosei, E. (2012). Structural abnormalities of small resistance arteries in essential hypertension. Intern. Emerg. Med. 7, 205–212. doi: 10.1007/s11739-011-0548-0
Rossier, B. C., Bochud, M., and Devuyst, O. (2017). The hypertension pandemic: an evolutionary perspective. Physiology 32, 112–125. doi: 10.1152/physiol.00026.2016
Sayers, L. G., Miyawaki, A., Muto, A., Takeshita, H., Yamamoto, A., Michikawa, T., et al. (1997). Intracellular targeting and homotetramer formation of a truncated inositol 1,4,5-trisphosphate receptor-green fluorescent protein chimera in Xenopus laevis oocytes: evidence for the involvement of the transmembrane spanning domain in endoplasmic reticulum targeting and homotetramer complex formation. Biochem. J. 323 (Pt 1), 273–280. doi: 10.1042/bj3230273
Schubert, R., and Mulvany, M. J. (1999). The myogenic response: established facts and attractive hypotheses. Clin. Sci. 96, 313–326. doi: 10.1042/cs0960313
Sharma, K., Wang, L., Zhu, Y., Bokkala, S., and Joseph, S. K. (1997). Transforming growth factor-beta1 inhibits type I inositol 1,4,5-trisphosphate receptor expression and enhances its phosphorylation in mesangial cells. J. Biol. Chem. 272, 14617–14623. doi: 10.1074/jbc.272.23.14617
Shibuya, J., Ohyanagi, M., and Iwasaki, T. (1998). Enhanced myogenic response in resistance small arteries from spontaneously hypertensive rats: relationship to the voltage-dependent calcium channel. Am. J. Hypertens. 11, 767–773. doi: 10.1016/S0895-7061(98)00055-7
Shyu, K. G. (2009). Cellular and molecular effects of mechanical stretch on vascular cells and cardiac myocytes. Clin. Sci. 116, 377–389. doi: 10.1042/CS20080163
Simpson, P. B., Mehotra, S., Langley, D., Sheppard, C. A., and Russell, J. T. (1998). Specialized distributions of mitochondria and endoplasmic reticulum proteins define Ca2+ wave amplification sites in cultured astrocytes. J. Neurosci. Res. 52, 672–683. doi: 10.1002/(SICI)1097-4547(19980615)52:6<672::AID-JNR6>3.0.CO;2-5
Sipma, H., Deelman, L., Smedt, H. D., Missiaen, L., Parys, J. B., Vanlingen, S., et al. (1998). Agonist-induced down-regulation of type 1 and type 3 inositol 1,4,5-trisphosphate receptors in A7r5 and DDT1 MF-2 smooth muscle cells. Cell Calcium 23, 11–21. doi: 10.1016/S0143-4160(98)90070-7
Sobey, C. G., Heistad, D. D., and Faraci, F. M. (1998). Potassium channels mediate dilatation of cerebral arterioles in response to arachidonate. Am. J. Physiol. 275(5 Pt 2), H1606–H1612. doi: 10.1152/ajpheart.1998.275.5.H1606
Sonoyama, K., Greenstein, A., Price, A., Khavandi, K., and Heagerty, T. (2007). Vascular remodeling: implications for small artery function and target organ damage. Ther. Adv. Cardiovasc. Dis. 1, 129–137. doi: 10.1177/1753944707086358
Szalai, G., Krishnamurthy, R., and Hajnoczky, G. (1999). Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 18, 6349–6361. doi: 10.1093/emboj/18.22.6349
Tada, T., Nawata, J., Wang, H., Onoue, N., Zhulanqiqige, D., Ito, K., et al. (2008). Enhanced pulsatile pressure accelerates vascular smooth muscle migration: implications for atherogenesis of hypertension. Cardiovasc. Res. 80, 346–353. doi: 10.1093/cvr/cvn211
Taguchi, H., Heistad, D. D., Kitazono, T., and Faraci, F. M. (1994). ATP-sensitive K+ channels mediate dilatation of cerebral arterioles during hypoxia. Circ. Res. 74, 1005–1008. doi: 10.1161/01.RES.74.5.1005
Tajada, S., Cidad, P., Colinas, O., Santana, L. F., Lopez-Lopez, J. R., and Perez-Garcia, M. T. (2013). Down-regulation of CaV1.2 channels during hypertension: how fewer CaV1.2 channels allow more Ca2+ into hypertensive arterial smooth muscle. J. Physiol. 591, 6175–6191. doi: 10.1113/jphysiol.2013.265751
Tajada, S., Cidad, P., Moreno-Dominguez, A., Perez-Garcia, M. T., and Lopez-Lopez, J. R. (2012). High blood pressure associates with the remodelling of inward rectifier K+ channels in mice mesenteric vascular smooth muscle cells. J. Physiol. 590, 6075–6091. doi: 10.1113/jphysiol.2012.236190
Tang, J., Lin, Y., Zhang, Z., Tikunova, S., Birnbaumer, L., and Zhu, M. X. (2001). Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J. Biol. Chem. 276, 21303–21310. doi: 10.1074/jbc.M102316200
Thijssen, D. H., Carter, S. E., and Green, D. J. (2016). Arterial structure and function in vascular ageing: are you as old as your arteries? J. Physiol. 594, 2275–2284. doi: 10.1113/JP270597
Torrealba, N., Navarro-Marquez, M., Garrido, V., Pedrozo, Z., Romero, D., Eura, Y., et al. (2017). Herpud1 negatively regulates pathological cardiac hypertrophy by inducing IP3 receptor degradation. Sci. Rep. 7:13402. doi: 10.1038/s41598-017-13797-z
Touyz, R. M. (2005). Intracellular mechanisms involved in vascular remodelling of resistance arteries in hypertension: role of angiotensin II. Exp. Physiol. 90, 449–455. doi: 10.1113/expphysiol.2005.030080
Trebak, M. (2012). STIM/Orai signalling complexes in vascular smooth muscle. J. Physiol. 590, 4201–4208. doi: 10.1113/jphysiol.2012.233353
Tu, H., Wang, Z., and Bezprozvanny, I. (2005). Modulation of mammalian inositol 1,4,5-trisphosphate receptor isoforms by calcium: a role of calcium sensor region. Biophys. J. 88, 1056–1069. doi: 10.1529/biophysj.104.049601
Uchida, E., and Bohr, D. F. (1969). Myogenic tone in isolated perfused vessels. Occurrence among vascular beds and along vascular trees. Circ. Res. 25, 549–555. doi: 10.1161/01.RES.25.5.549
Uehata, M., Ishizaki, T., Satoh, H., Ono, T., Kawahara, T., Morishita, T., et al. (1997). Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389, 990–994. doi: 10.1038/40187
van den Munckhof, I., Scholten, R., Cable, N. T., Hopman, M. T., Green, D. J., and Thijssen, D. H. (2012). Impact of age and sex on carotid and peripheral arterial wall thickness in humans. Acta Physiol. 206, 220–228. doi: 10.1111/j.1748-1716.2012.02457.x
VanBavel, E., van der Meulen, E. T., and Spaan, J. A. (2001). Role of Rho-associated protein kinase in tone and calcium sensitivity of cannulated rat mesenteric small arteries. Exp. Physiol. 86, 585–592. doi: 10.1113/eph8602217
VanBavel, E., Wesselman, J. P., and Spaan, J. A. (1998). Myogenic activation and calcium sensitivity of cannulated rat mesenteric small arteries. Circ. Res. 82, 210–220. doi: 10.1161/01.RES.82.2.210
Vermassen, E., Parys, J. B., and Mauger, J. P. (2004). Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: functional relevance and molecular determinants. Biol. Cell 96, 3–17. doi: 10.1016/j.biolcel.2003.11.004
Vervloessem, T., Yule, D. I., Bultynck, G., and Parys, J. B. (2015). The type 2 inositol 1,4,5-trisphosphate receptor, emerging functions for an intriguing Ca2+-release channel. Biochim. Biophys. Acta 1853, 1992–2005. doi: 10.1016/j.bbamcr.2014.12.006
Waldemar, G., and Paulson, O. B. (1989). Angiotensin converting enzyme inhibition and cerebral circulation–a review. Br. J. Clin. Pharmacol. 28(Suppl. 2), 177s–182s. doi: 10.1111/j.1365-2125.1989.tb03593.x
Wallace, T. A., Xia, S. L., and Sayeski, P. P. (2005). Jak2 tyrosine kinase prevents angiotensin II-mediated inositol 1,4,5 trisphosphate receptor degradation. Vascul. Pharmacol. 43, 336–345. doi: 10.1016/j.vph.2005.08.022
Walsh, M. P., Bridenbaugh, R., Hartshorne, D. J., and Kerrick, W. G. (1982). Phosphorylation-dependent activated tension in skinned gizzard muscle fibers in the absence of Ca2+. J. Biol. Chem. 257, 5987–5990.
Wang, M., Zhang, J., Jiang, L.-Q., Spinetti, G., Pintus, G., Monticone, R., et al. (2007). Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension 50, 219–227. doi: 10.1161/HYPERTENSIONAHA.107.089409
Wang, M., Zhang, J., Spinetti, G., Jiang, L.-Q., Monticone, R., Zhao, D., et al. (2005). Angiotensin II activates matrix metalloproteinase type II and mimics age-associated carotid arterial remodeling in young rats. Am. J. Pathol. 167, 1429–1442. doi: 10.1016/S0002-9440(10)61229-1
Wang, M., Zhao, D., Spinetti, G., Zhang, J., Jiang, L.-Q., Pintus, G., et al. (2006). Matrix metalloproteinase 2 activation of transforming growth factor-β1 (TGF-β1) and TGF-β1–Type II receptor signaling within the aged arterial wall. Arterioscler. Thromb. Vasc. Biol. 26, 1503–1509. doi: 10.1161/01.ATV.0000225777.58488.f2
Wang, Y., Chen, J., Wang, Y., Taylor, C. W., Hirata, Y., Hagiwara, H., et al. (2001). Crucial role of type 1, but not type 3, inositol 1,4,5-trisphosphate (IP(3)) receptors in IP(3)-induced Ca2+ release, capacitative Ca2+ entry, and proliferation of A7r5 vascular smooth muscle cells. Circ. Res. 88, 202–209. doi: 10.1161/01.RES.88.2.202
Wesselman, J. P., Spaan, J. A., van der Meulen, E. T., and VanBavel, E. (2001). Role of protein kinase C in myogenic calcium-contraction coupling of rat cannulated mesenteric small arteries. Clin. Exp. Pharmacol. Physiol. 28, 848–855. doi: 10.1046/j.1440-1681.2001.03534.x
Wilkerson, M. K., Heppner, T. J., Bonev, A. D., and Nelson, M. T. (2006). Inositol trisphosphate receptor calcium release is required for cerebral artery smooth muscle cell proliferation. Am. J. Physiol. Heart Circ. Physiol. 290, H240–H247. doi: 10.1152/ajpheart.01191.2004
Wu, L., and de Champlain, J. (1996). Inhibition by cyclic AMP of basal and induced inositol phosphate production in cultured aortic smooth muscle cells from Wistar-Kyoto and spontaneously hypertensive rats. J. Hypertens. 14, 593–599. doi: 10.1097/00004872-199605000-00008
Xi, Q., Adebiyi, A., Zhao, G., Chapman, K. E., Waters, C. M., Hassid, A., et al. (2008). IP3 constricts cerebral arteries via IP3 receptor-mediated TRPC3 channel activation and independently of sarcoplasmic reticulum Ca2+ release. Circ. Res. 102, 1118–1126. doi: 10.1161/CIRCRESAHA.108.173948
Yoshida, T., and Owens, G. K. (2005). Molecular determinants of vascular smooth muscle cell diversity. Circ. Res. 96, 280–291. doi: 10.1161/01.RES.0000155951.62152.2e
Yoshikawa, F., Iwasaki, H., Michikawa, T., Furuichi, T., and Mikoshiba, K. (1999). Cooperative formation of the ligand-binding site of the inositol 1,4, 5-trisphosphate receptor by two separable domains. J. Biol. Chem. 274, 328–334. doi: 10.1074/jbc.274.1.328
Yoshikawa, F., Morita, M., Monkawa, T., Michikawa, T., Furuichi, T., and Mikoshiba, K. (1996). Mutational analysis of the ligand binding site of the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 271, 18277–18284. doi: 10.1074/jbc.271.30.18277
Zetterqvist, A. V., Berglund, L. M., Blanco, F., Garcia-Vaz, E., Wigren, M., Duner, P., et al. (2014). Inhibition of nuclear factor of activated T-cells (NFAT) suppresses accelerated atherosclerosis in diabetic mice. PLoS One 8, e65020. doi: 10.1371/journal.pone.0065020
Zhang, Z., Tang, J., Tikunova, S., Johnson, J. D., Chen, Z., Qin, N., et al. (2001). Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc. Natl. Acad. Sci. U.S.A. 98, 3168–3173. doi: 10.1073/pnas.051632698
Zhao, G., Neeb, Z. P., Leo, M. D., Pachuau, J., Adebiyi, A., Ouyang, K., et al. (2010). Type 1 IP3 receptors activate BKCa channels via local molecular coupling in arterial smooth muscle cells. J. Gen. Physiol. 136, 283–291. doi: 10.1085/jgp.201010453
Zhao, M., Jia, H. H., Liu, L. Z., Bi, X. Y., Xu, M., Yu, X. J., et al. (2017). Acetylcholine attenuated TNF-alpha-induced intracellular Ca2+ overload by inhibiting the formation of the NCX1-TRPC3-IP3R1 complex in human umbilical vein endothelial cells. J. Mol. Cell. Cardiol. 107, 1–12. doi: 10.1016/j.yjmcc.2017.04.001
Zhou, H., Nakamura, T., Matsumoto, N., Hisatsune, C., Mizutani, A., Iesaki, T., et al. (2008). Predominant role of type 1 IP3 receptor in aortic vascular muscle contraction. Biochem. Biophys. Res. Commun. 369, 213–219. doi: 10.1016/j.bbrc.2007.12.194
Zicha, J., Behuliak, M., Pinterova, M., Bencze, M., Kunes, J., and Vaneckova, I. (2014). The interaction of calcium entry and calcium sensitization in the control of vascular tone and blood pressure of normotensive and hypertensive rats. Physiol. Res. 63(Suppl. 1), S19–S27.
Zou, H., Ratz, P. H., and Hill, M. A. (1995). Role of myosin phosphorylation and [Ca2+]i in myogenic reactivity and arteriolar tone. Am. J. Physiol. 269(5 Pt 2), H1590–H1596. doi: 10.1152/ajpheart.1995.269.5.H1590
Keywords: aging, hypertension, Ca2+, IP3R, VSMC
Citation: Eid AH, El-Yazbi AF, Zouein F, Arredouani A, Ouhtit A, Rahman MM, Zayed H, Pintus G and Abou-Saleh H (2018) Inositol 1,4,5-Trisphosphate Receptors in Hypertension. Front. Physiol. 9:1018. doi: 10.3389/fphys.2018.01018
Received: 22 December 2017; Accepted: 09 July 2018;
Published: 26 July 2018.
Edited by:
Lacolley Patrick, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Angélica Rueda, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional (CINVESTAV-IPN), MexicoCopyright © 2018 Eid, El-Yazbi, Zouein, Arredouani, Ouhtit, Rahman, Zayed, Pintus and Abou-Saleh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haissam Abou-Saleh, hasaleh@qu.edu.qa
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.