
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 05 June 2018
Sec. Membrane Physiology and Membrane Biophysics
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.00682
 Ricardo Ramírez-Barrantes1
Ricardo Ramírez-Barrantes1 Claudio Córdova1
Claudio Córdova1 Sebastian Gatica2,3
Sebastian Gatica2,3 Belén Rodriguez1
Belén Rodriguez1 Carlo Lozano1
Carlo Lozano1 Ivanny Marchant1
Ivanny Marchant1 Cesar Echeverria4
Cesar Echeverria4 Felipe Simon2,3*
Felipe Simon2,3* Pablo Olivero1*
Pablo Olivero1*The transient receptor potential (TRP) ion channel family consists of a broad variety of non-selective cation channels that integrate environmental physicochemical signals for dynamic homeostatic control. Involved in a variety of cellular physiological processes, TRP channels are fundamental to the control of the cell life cycle. TRP channels from the vanilloid (TRPV) family have been directly implicated in cell death. TRPV1 is activated by pain-inducing stimuli, including inflammatory endovanilloids and pungent exovanilloids, such as capsaicin (CAP). TRPV1 activation by high doses of CAP (>10 μM) leads to necrosis, but also exhibits apoptotic characteristics. However, CAP dose–response studies are lacking in order to determine whether CAP-induced cell death occurs preferentially via necrosis or apoptosis. In addition, it is not known whether cytosolic Ca2+ and mitochondrial dysfunction participates in CAP-induced TRPV1-mediated cell death. By using TRPV1-transfected HeLa cells, we investigated the underlying mechanisms involved in CAP-induced TRPV1-mediated cell death, the dependence of CAP dose, and the participation of mitochondrial dysfunction and cytosolic Ca2+ increase. Together, our results contribute to elucidate the pathophysiological steps that follow after TRPV1 stimulation with CAP. Low concentrations of CAP (1 μM) induce cell death by a mechanism involving a TRPV1-mediated rapid and transient intracellular Ca2+ increase that stimulates plasma membrane depolarization, thereby compromising plasma membrane integrity and ultimately leading to cell death. Meanwhile, higher doses of CAP induce cell death via a TRPV1-independent mechanism, involving a slow and persistent intracellular Ca2+ increase that induces mitochondrial dysfunction, plasma membrane depolarization, plasma membrane loss of integrity, and ultimately, cell death.
Transient receptor potential (TRP) channels belong to a polymodal family of ion channels that act as molecular transducers and integrators of a variety of environmental physicochemical stimuli, such as temperature, osmotic pressure, mechanical stress, and exogenous and endogenous ligands (Ramsey et al., 2006; Latorre et al., 2007, 2009). TRP channels play an essential role in multiple physiological and pathological cellular processes, such as proliferation, differentiation, and death progression (Shimizu et al., 2004; Shirakawa et al., 2008; Carrasco et al., 2018). Deregulated activation of TRP channels from the vanilloid (TRPV) family has been directly implicated in cell death (Macho et al., 1999; Amantini et al., 2009; Chen et al., 2012). TRPV1 has been detected in a variety of organs, such as the brain, testes, lungs, and heart. (Hayes et al., 2000; Randhawa and Jaggi, 2018). TRPV1 is widely expressed in dorsal root ganglion (DRG) and trigeminal neurons.
TRPV1 is activated by pain-inducing stimuli, including inflammatory endovanilloids, TNF-α, TGF-β, heat (37–42°C), acids (pH < 6.3), and pungent exovanilloids, such as capsaicin (CAP) or resiniferatoxin (Caterina et al., 1997; Tominaga et al., 1998; Jordt et al., 2000; Olah et al., 2001, 2002; Latorre et al., 2007; Ma et al., 2011; Utreras et al., 2013; Rozas et al., 2016). TRPV1 activation by CAP is antagonized by the synthetic organic compound capsazepine (CPZ). At the cellular level, TRPV1 activation by high doses of CAP leads not only to necrotic processes with membrane bleb formation (Pecze et al., 2013; Wu et al., 2014) but also to apoptosis through caspase-3 activation and mitochondrial membrane potential attenuation (Ziglioli et al., 2009; Sun et al., 2014; Çiğ and Nazıroğlu, 2015). However, CAP dose–response studies are lacking in order to understand whether CAP-induced cell death occurs preferentially via necrosis or apoptosis. Thus, the relationship between CAP concentration and CAP-induced TRPV1-mediated cell death is not completely understood.
Increased TRPV1 activity induces high levels of cytosolic Ca2+, generating mitochondrial membrane depolarization and decreased cell viability (Thomas et al., 2007). Furthermore, TRPV1 activation triggers apoptotic cell death in neuron-rich cultures from rat cerebral cortex via Ca2+ channels opening, allowing Ca2+ influx (Shirakawa et al., 2008). However, it is not known whether cytosolic Ca2+ and mitochondrial dysfunction participate in CAP-induced TRPV1-mediated cell death. Thus, we focused on investigating the underlying mechanisms involved in CAP-induced TRPV1-mediated cell death, the dependence of CAP dose, and the participation of mitochondrial dysfunction and cytosolic Ca2+ increase.
Using an analytical three-state model (O’Neill et al., 2011) to describe the mechanistic sequential progression from a state of health to a state of death, we found that TRPV1 stimulation with 10 μM CAP significantly induces necrosis-like cell death characterized by extensive cell membrane damage but without affecting mitochondrial function. Interestingly, 100 μM CAP induced a different pattern for cell death, characterized by mitochondrial dysfunction and is independent of TRPV1 activity, resembling an apoptosis-like death pattern. Furthermore, we found that TRPV1 stimulation with 1 μM CAP induces a TRPV1-dependent fast and transient intracellular Ca2+ increase, while 10 μM CAP induces a fast and persistent increase, which can be explained by the combination of two intracellular Ca2+ signals, a TRPV1-dependent fast and transient increase that is inhibited by CPZ, and a slow, persistent, and TRPV1-independent rise of intracellular Ca2+. Finally, we demonstrated that 10 μM CAP induces plasma membrane depolarization via an influx of Ca2+ and Na+ from the extracellular space.
Our results show further mechanistical insights detailing how CAP induces TRPV1-dependent and independent cell death. Low concentrations of CAP (1 μM) induce a fast and transient increase in intracellular Ca2+, which leads to plasma membrane depolarization, thereby compromising plasma membrane integrity, and ultimately driving cell physiology to a state of death but without mitochondrial dysfunction. Meanwhile, 10 and 100 μM CAP induce a slow but persistent increase in intracellular Ca2+, which leads not only to plasma membrane depolarization but also to mitochondrial dysfunction, and ultimately cell death. Thus, CAP is shown to activate two independent pathways of Ca2+ homeostasis leading to cell death by necrosis or apoptosis.
HeLa cells were obtained from ATCC (Manassas, VA, United States). The culture medium used was Dulbecco’s Modified Eagle Medium/F12 supplemented with 10% fetal bovine serum and 50 U/ml penicillin–streptomycin. Cells were incubated in a conventional incubator at 37°C and a 95% air/5% CO2 atmosphere.
HeLa cells were cultured at 70–80% confluence and then were transfected with pcDNA3.1 containing the full length of rat TRPV1 (GenBankTM accession no. NM031982) using Lipofectamine (ThermoFisher). Transfected cells were selected using Geneticin (Sigma-Aldrich, St. Louis, MO, United States, 800 mg/mL) to generate a stable cell line encoding TRPV1. Stable TRPV1 expression was checked by RT-PCR and flow cytometry weekly (Supplementary Figure S1).
HeLa cells were exposed to different experimental conditions in Dulbecco’s Modified Eagle Medium/F12 supplemented with 1% bovine serum albumin instead of fetal bovine serum. Cellular death was determined as described in the literature (Darzynkiewicz et al., 1982) and analyzed according to a three-state model of cell death (O’Neill et al., 2011). Briefly, cell cultures were stained with 10 μM rhodamine 123 (Rho123, Invitrogen, Carlsbad, CA, United States) to assess mitochondrial membrane potential, and propidium iodide (PI, 10 μg/ml (Sigma-Aldrich, St. Louis, MO, United States) to assess plasma membrane integrity. Both measures were analyzed by flow cytometry (FACScalibur, BD Biosciences, CA, United States). A minimum of 10,000 cells/sample were analyzed to evaluate mitochondrial function and membrane permeability. Fluorescence intensity analysis was performed using FlowJo software (Tree Star, Inc., Ashland, OR, United States). Cell state was operationally defined with the following first-order rate process: Alive (A) ↔ Vulnerable (V) ↔ Dead (D). To determine probability for the A state (PA), data were normalized with respect to internal controls using the following equation: PA = X – CD/CA -CD, where X corresponds to living cells in each experimental condition, CD corresponds to living cells in the presence of 10% ethanol to induce cell death, and CA corresponds to living cells without treatment.
Cell cultures were loaded with Fura-2 AM (Molecular Probes, Eugene, OR, United States) for 30 min at room temperature in buffer solution [130 mM NaCl, 5.4 mM KCl, 2.5 mM CaCl2, 0.8 mM MgCl2, 5.6 mM glucose, 10 mM HEPES, pH 7.4 (adjusted with Tris base)], rinsed, and allowed to equilibrate for 5–10 min. Next, HeLa cells were cultured on 12-mm glass cover slips in a recording chamber mounted on an epifluorescence Olympus IX81 microscope (Olympus, Japan) equipped with a multiple-excitation filter wheel. CAP-induced activity was recorded for a minimum recording time of 2 s. Maximum resolution was obtained with a Plan Apo 40X 1.3 NA oil objective lens.
Cell cultures were equilibrated using DiBAC4(3) [Molecular Probes, Eugene, OR, United States] as described previously (Kunz et al., 2006). This anionic fluorescent dye is distributed across the plasma membrane relative to the membrane potential following Nernst’s equation (Olivero et al., 2008). DiBAC4(3) (200 nM) was applied extracellularly for approximately 20 min to ensure dye distribution across the cell membrane. Changes in fluorescence intensity were monitored by sampling every 10 s for 30 min with a 515 nm excitation filter and an emission filter of at least 600 nm. Fluorescence data were transformed to mV using a calibration curve from HeLa cells as described previously (Krasznai et al., 1995).
Total RNA from parental HeLa cells and cells transfected with TRPV1 was extracted with TRIzol (Invitrogen, Carlsbad, CA, United States), and reverse transcription was performed to create a cDNA library using reverse transcriptase M-MLV (Invitrogen, Carlsbad, CA, United States). An equal amount of RNA was used as template in each reaction. The PCR reactions were performed using GoTaq Master Mix (Promega Corp., Madison, WI, United States) following the manufacturer’s instructions.
HeLa cells were collected by trypsinization and fixed with 4% paraformaldehyde for 30 min. Next, the cells were blocked and permeabilized using a PBS solution with 5% bovine serum albumin (Merck KGaA, Darmstadt, Germany) and 2% Tween-20 (Merck KGaA, Darmstadt, Germany) for 1 h and then incubated with an anti-TRPV1 antibody (Santa Cruz Biotechnology, Inc., United States, 1:200) in blocking solution overnight at 4°C. After washing with PBS, the cells were incubated with anti-goat biotinylated secondary antibody (Jackson ImmunoResearch, United States, 1:500) for 1 h at 37°C. The cells were washed with PBS and incubated in the dark with streptavidin-Alexa Fluor 488 (Jackson ImmunoResearch, United States, 1:200) for 1 h at 37°C. The Alexa Fluor signal was measured with a 530/30 bandpass filter using an argon laser at 488 nm integrated into a FACScalibur flow cytometer (BD, Biosciences, CA, United States). Debris and duplets were excluded from the analysis, and a minimum of 10,000 cells were acquired in each experiment. Data were analyzed with FlowJo software (Tree Star, Inc., Ashland, OR, United States).
Cyanide-4-(trifluoromethoxy)phenylhydrazone, FCCP, ionomycin, rodamine123, and PI were obtained from Sigma-Aldrich (St. Louis, MO, United States). CAP and CPZ were obtained from Tocris Bioscience (Bristol, United Kingdom). Buffers, ethanol, and salts were purchased from Merck (Darmstadt, Germany).
All results are presented as the mean ± SD from at least three independent assays for each experimental condition. Fisher’s least significant difference test and an ANOVA test followed by the Bonferroni post hoc test were used to compare multiple groups using Statgraphics Plus 5.0 (GraphPad Software, Inc., San Diego, CA, United States). A p-value < 0.05 was used to indicate statistical significance.
The participation of TRPV1 expression and activity to sensitize cells to CAP-induced death was tested using a three-state cell death model. The three-state model [alive (A), vulnerable (V), and dead (D)] was established by means of flow cytometry dot plot analysis to determine cell death with or without mitochondrial involvement. Wild-type HeLa (wt-HeLa) cells were exposed to 10 μM FCCP (Figure 1A) for 0, 30, and 120 min, and 10% ethanol (Figure 1B) for 0, 1, and 12 h. The three-state cell death model displayed a pronounced progression from A (PIlowRho123hi), to V (PIlowRho123low, or PIhiRho123hi), to D (PIhiRho123low; Figures 1A,B). The induction of an intermediate PIlowRho123low V-phenotype indicates loss of mitochondrial membrane potential without plasma membrane disruption (Figure 1A), while PIhiRho123hi V-phenotype indicates plasma membrane disruption, but without loss of mitochondrial membrane potential (Figure 1B). Cell phenotype did not remain constant, and the proportion of cells in the three states varied over time. Cells achieved a full phenotype shift toward the D state after 3 h of exposure to FCCP (Figure 1C) and after 9 h of exposure to 10% ethanol (Figure 1D). Cells reached the intermediate V state at approximately 1 h of exposure to either FCCP or 10% ethanol, and PA was greater for cells exposed to FCCP than to 10% ethanol. Therefore, HeLa cell physiology appeared more sensitive to mitochondrial dysfunction than to plasma membrane disruption, as shown with PA progression curves.
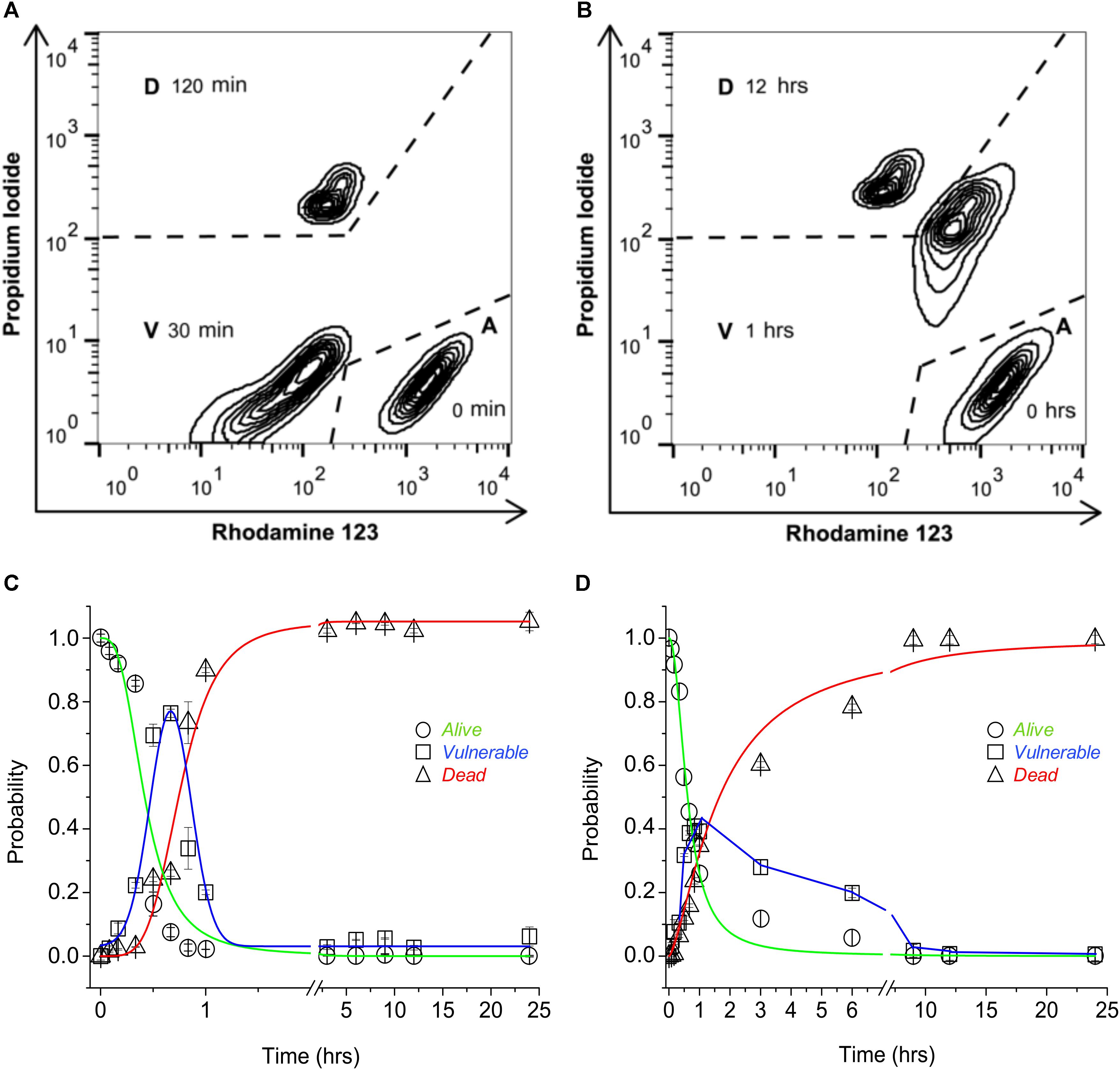
FIGURE 1. Three-state model of cell death. Representative flow cytometry dot plot output depicting 24-h exposure of non-transfected wt-HeLa cell phenotype exposed to 10 μM FCCP (A) and 10% ethanol (B). Cell death progression was evaluated using PI (to assess plasma membrane integrity) and Rho123 (to assess mitochondrial membrane potential). Cell state progression is symbolized as A (alive), V (vulnerable), and D (dead). Replicate experiments [from (A,B)] were performed for different times between 0 and 24 h, and were normalized against PA = 1 to determine the probability for cell state [(C,D) respectively]. Data are shown as mean ± SEM (n = 9).
Once the three-state cell death model was established, we investigated the effect of TRPV1 expression in HeLa cells stably transfected with TRPV1 (st-TRPV1). Transfection efficiency in generating the st-TRPV1 was confirmed at the mRNA level by RT-PCR and at the protein level by flow cytometry (Supplementary Figure S1). Flow cytometry analysis revealed that in the absence of CAP, wt-HeLa cells predominantly exhibited a phenotype consistent with the A state (PIlowRho123hi), without plasma membrane disruption or mitochondrial dysfunction (Figure 2, upper-left panel). Similar results were observed for st-TRPV1 HeLa cells in the absence (Figure 2, upper-middle panel) or presence of CPZ (Figure 2, upper-right panel), and in wt-HeLa cells exposed to 10 μM CAP (Figure 2, middle-left panel). However, st-TRPV1 HeLa cells exposed to 10 μM CAP showed a phenotype (PIlow-hiRho123hi) predominantly indicative of plasma membrane loss of integrity without mitochondrial dysfunction (Figure 2, middle-middle panel), a phenotype that resembles a necrosis-like cell death. Interestingly, st-TRPV1 HeLa cells pre-treated with 10 μM CPZ and then treated with 10 μM CAP exhibited a cell phenotype consistent with the A state (Figure 2, middle-right panel), suggesting that 10 μM CPZ is able to prevent cell death. Wt-HeLa cells exposed to 100 μM CAP showed a phenotype (PIhiRho123low) predominantly indicative of plasma membrane loss of integrity and severe mitochondrial dysfunction (Figure 2, lower-left panel). Similarly, st-TRPV1 HeLa cells exposed to 100 μM CAP exhibited a phenotype (PIhiRho123mid) consistent with the D state with both plasma membrane disruption and mitochondrial failure (Figure 2, lower-middle panel), indicative of an apoptosis-like cell death. Notably, pre-treatment with 10 μM CPZ of st-TRPV1 HeLa cells exposed to 100 μM CAP was not effective protecting the cells from CAP challenge, showing a phenotype (PIhiRho123mid) consistent with the D state (Figure 2, lower-right panel). Thus, these results indicate that 10 μM CAP induces TRPV1-mediated cell death without affecting mitochondrial physiology, while 100 μM CAP induces cell death in a TRPV1-independent fashion, characterized by mitochondrial potential dysfunction and plasma membrane disruption. Phase-contrast images highlighting the main morphological features of each condition for wt-HeLa and st-TRPV1 HeLa cells were documented and summarized in Supplementary Figure S2.
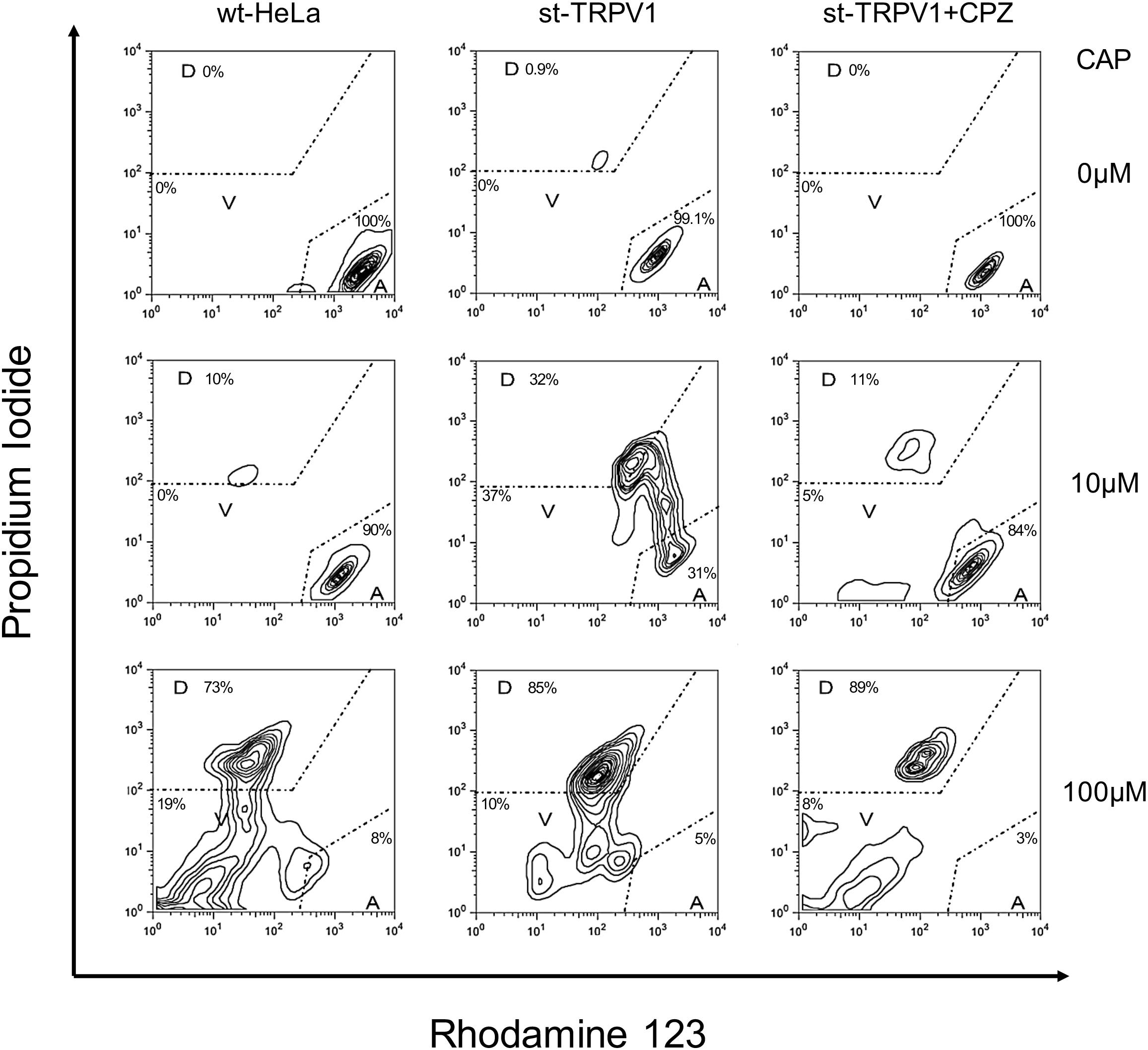
FIGURE 2. TRPV1 expression increases CAP-induced cell death. Representative flow cytometry dot plot output and quantification depicting wt-HeLa cells and st-TRPV1 HeLa cells exposed to 0, 10, and 100 μM CAP for 24 h in the presence or absence of 10 μM CPZ. Cell death progression was evaluated using PI (to assess plasma membrane integrity) and Rho123 (to assess mitochondrial membrane potential). Cell state progression is symbolized as A (alive), V (vulnerable), and D (dead). Data are shown as mean ± SEM (n = 6).
To investigate the dose–response effect of CAP, st-TRPV1 HeLa cells were exposed to increasing doses of CAP for 24 h. The results showed that CAP has a sensitizing effect over st-TRPV1 HeLa cells, decreasing the PA when compared to wt-HeLa cells (PA50 from ∼3.5⋅10-5 to ∼4.5⋅10-6 μM CAP). Interestingly, addition of the TRPV1 competitive antagonist CPZ (which blocks CAP-induced Ca2+ uptake through TRPV1) to CAP treated st-TRPV1 HeLa cells completely overturned the original sensitizing effect of CAP, contributing to cell resistance to a level close to wt-HeLa cells response (Figure 3A). As a next step, we studied the time–response (0–24 h) of st-TRPV1 cells exposed to 10 μM CAP. The proportion of st-TRPV1 cells in the V state was maximal after 12 h of exposure to 10 μM CAP. The A state in st-TRPV1 cells reached 50% after ∼7 h of CAP exposure, while the D state increased steadily (Figure 3B). These results indicate that the CAP-induced cell-state transition is mediated by TRPV1.
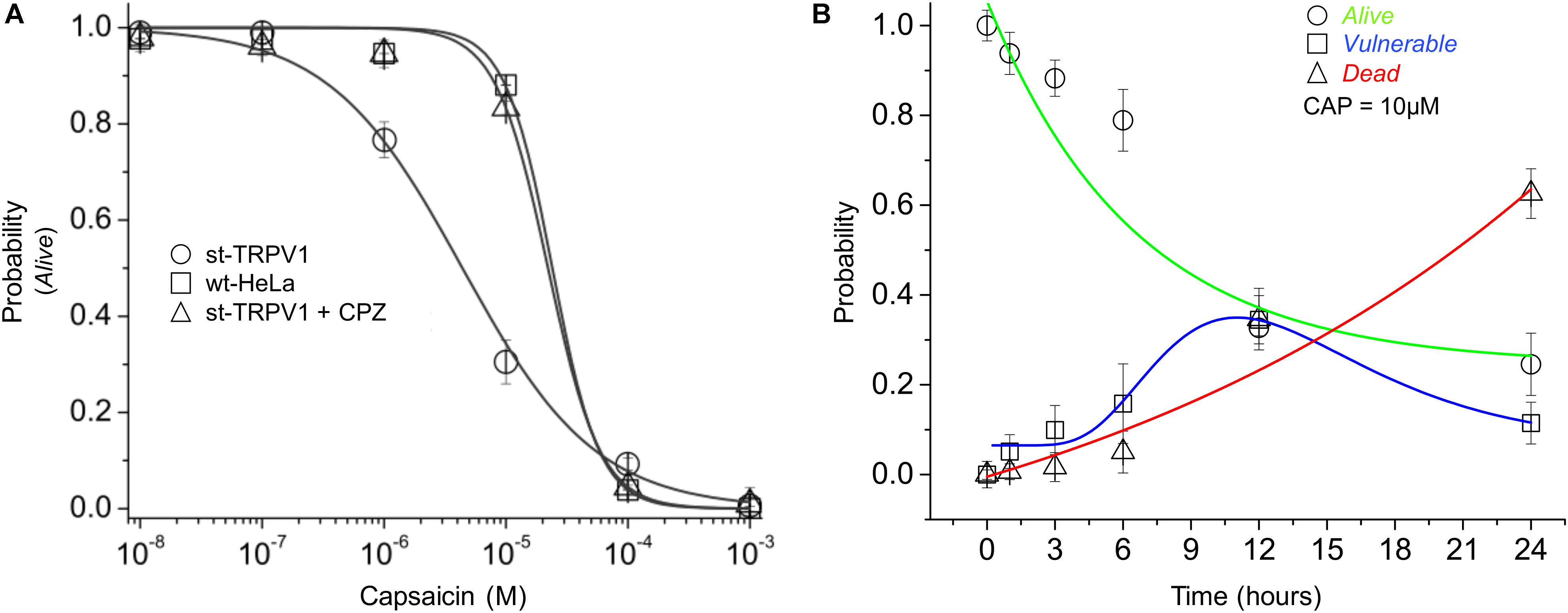
FIGURE 3. Dose- and time-response of CAP-induced cell death. (A) Probability of cell state A (PA) of wt-HeLa cells and st-TRPV1 HeLa cells in the presence or absence of 10 μM CPZ exposed to several concentration of CAP (1⋅10-3 to 1⋅10-8 M) for 24 h. (B) Probability of cell state progression of st-TRPV1 HeLa cells exposed to 10 μM CAP for 0, 1, 3, 6, 12, and 24 h. Data are shown as mean ± SEM (n = 5). ∗p < 0.01.
To investigate the intracellular effect of CAP-mediated TRPV1 stimulation, we measured Ca2+ dynamics with ratiometric assays. Measurements of intracellular Ca2+ levels showed that wt-HeLa cells were irresponsive to CAP stimulation. However, st-TRPV1 HeLa cells were able to respond to CAP treatment exhibiting transient and dose-dependent increases in intracellular calcium concentration ([Ca2+]I; Figure 4A). Addition of 1 μM CAP to st-TRPV1 HeLa cells showed a fast and transient rise in [Ca2+]I after 2 min of exposure to CAP reaching basal levels shortly before 10 min (Figure 4B). However, st-TRPV1 HeLa cells treated with 10 μM CAP showed a fast and persistent increase in [Ca2+]I, without return to basal levels after 10 min. Interestingly, when the latter cells were simultaneously exposed to 10 μM CPZ, the fast increase in [Ca2+]I was prevented, showing a slow and constant increase in [Ca2+]I reaching levels similar to those observed in the absence of CPZ (Figure 4B). Thus, 10 μM CAP elicits two Ca2+ signals combined, a TRPV1-dependent fast and transient increase and another slow and persistent Ca2+ increase, without participation of TRPV1. As a positive control, cells were responsive to ionomycin (Figures 4A,B).
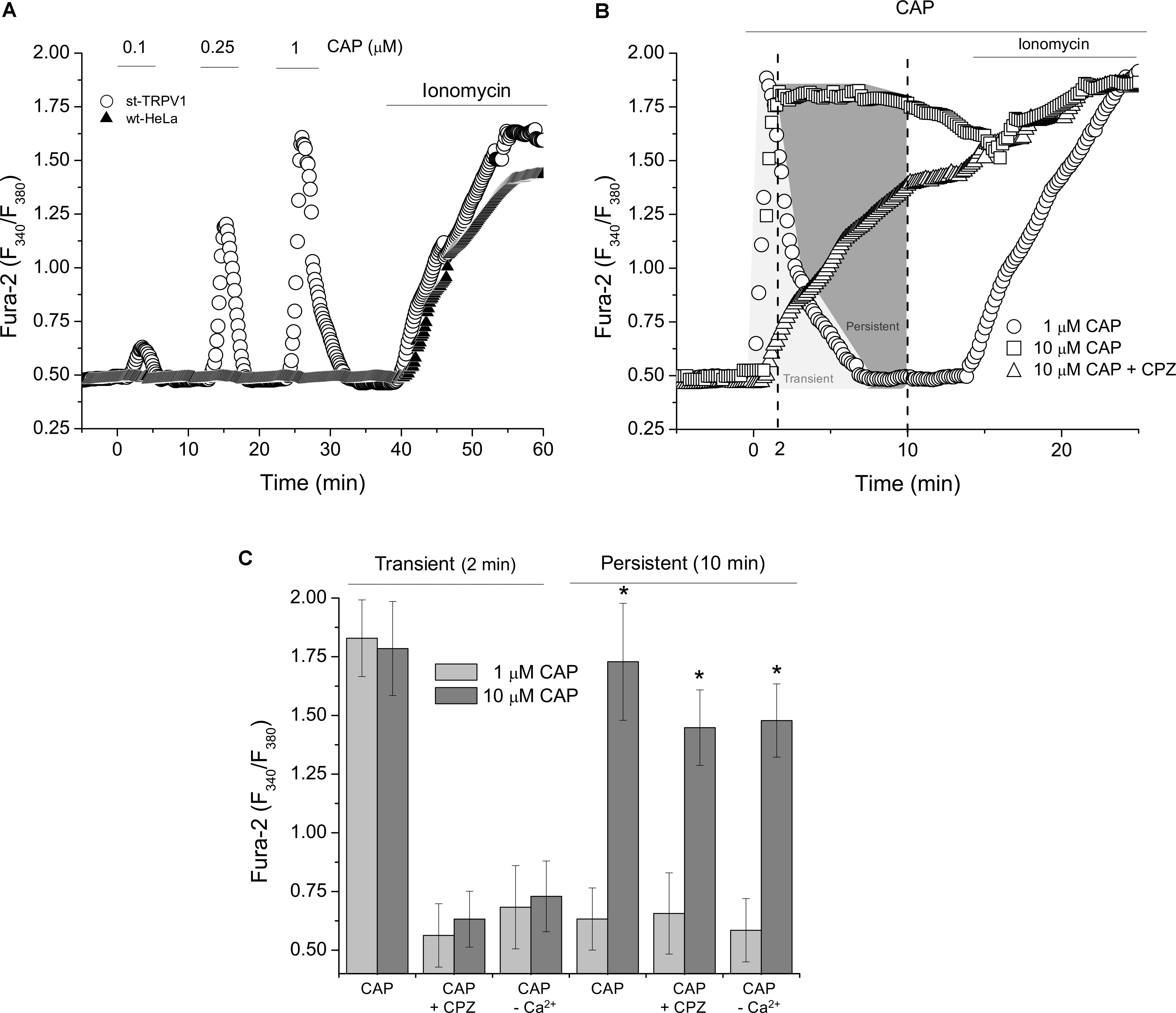
FIGURE 4. TRPV1 expression increases intracellular calcium level in response to CAP. (A) Dose–response curve of ratiometric assay recordings for [Ca2+]I in wt-HeLa cells and st-TRPV1 HeLa cells exposed to 0.1, 0.25, and 1 μM CAP for 60 min. Representative experiments from five independent experiments. (B) Dose–response curve of ratiometric assay recordings for [Ca2+]I in st-TRPV1 HeLa cells exposed to 1 and 10 μM CAP in the presence or absence of 10 μM CPZ for 20 min. Representative experiments from five independent experiments. 10 μM ionomycin was added at the end of experiments (A) and (B) as positive control. (C) Cross-sectional ratiometric [Ca2+]I in st-TRPV1 HeLa cells exposed to 1 and 10 μM CAP at times 2 and 10 min after CAP exposition, in the presence or absence of 10 μM CPZ or Ca2+-free condition. Data are shown as mean ± SEM (n = 15). ∗p < 0.01.
Whether the increase in intracellular Ca2+ levels was mediated by TRPV1, we were prompted to determine the participation of extracellular Ca2+ on the CAP-induced increase on intracellular Ca2+ levels. Figure 4C depicts both the transient and the persistent increases in [Ca2+]I from experiments as shown in Figure 4B. St-TRPV1 HeLa cells exposed to 1 μM CAP showed a transient, but not persistent increase in [Ca2+]I, while cells exposed to 10 μM CAP showed a persistent increase in [Ca2+]I. The addition of 10 μM CPZ prevented both the transient increase in [Ca2+]I independent of CAP concentration, but CPZ failed into inhibit the persistent increase in [Ca2+]I when 10 μM CAP was present. Interestingly, after removal of extracellular Ca2+, transient Ca2+ increases were prevented for both 1 and 10 μM CAP treatments, suggesting that CAP-induced transient increases in [Ca2+]I are dependent on extracellular Ca2+ and considering that it is CPZ-sensitive, this influx is mediated TRPV1. However, after extracellular Ca2+ depletion, 10 μM CAP was still able to stimulate an increase in [Ca2+]I, potentially due to a secondary mechanism involving Ca2+ mobilization from an intracellular storage compartment (Figure 4C).
Considering that stimulation of st-TRPV1 with CAP generated a fast increase in [Ca2+]I (Figure 4B) and that CAP induced a disruption in membrane integrity but not mitochondrial function (Figure 2, middle panel), we determined whether CAP challenge could depolarize plasma membrane potential. St-TRPV1 HeLa cells exposed to 10 μM CAP for 30 min increased the fluorescence of the membrane potential fluorescent indicator DIBAC4(3), suggesting that exposure to 10 μM CAP induced a significant depolarization of the plasma membrane from approximately –80 mV in non-treated st-TRPV1 HeLa cells to –35 mV (Figure 5). Interestingly, selective inactivation of TRPV1 with 10 μM CPZ prevented the CAP-induced plasma membrane depolarization, suggesting that TRPV1 participates in plasma membrane depolarization induced by CAP.
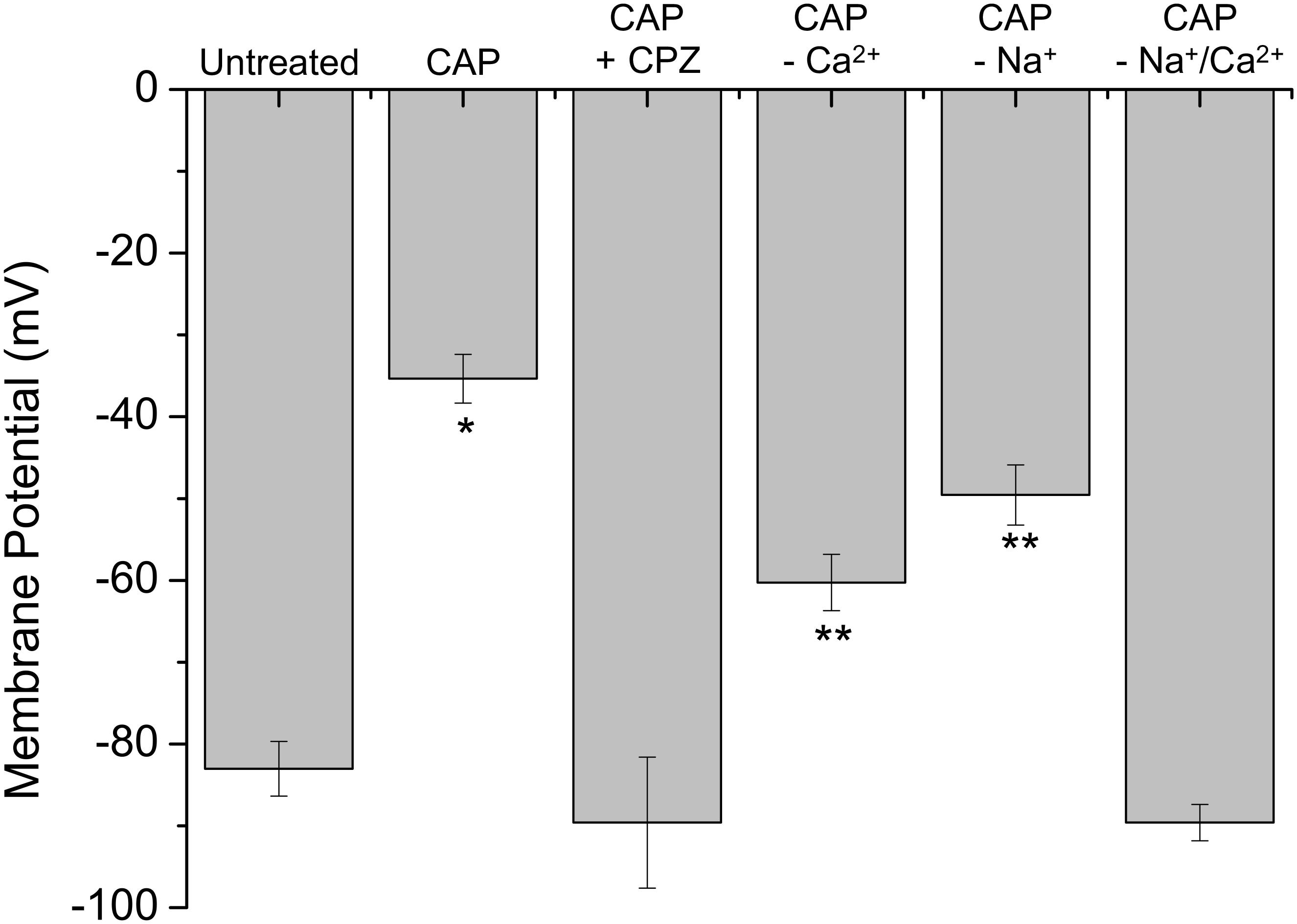
FIGURE 5. TRPV1 expression induces plasma membrane depolarization in response to CAP. Membrane potential measured by the change in DiBAC4(3) fluorescence intensity in st-TRPV1 HeLa cells exposed to 10 μM CAP for 30 min, in the presence or absence of 10 μM CPZ, Ca2+-free condition, or Na+-free condition. Data are shown as mean ± SEM (n = 5). ∗p < 0.01 and ∗∗p < 0.05.
To investigate whether CAP-induced plasma membrane depolarization requires external Ca2+, st-TRPV1 HeLa cells were exposed to 10 μM CAP in the absence of extracellular Ca2+. These results showed that in the absence of external Ca2+, CAP-induced plasma membrane depolarization was significantly decreased. Often, intracellular Ca2+ increases are followed by increases in intracellular Na+, which severely contribute to plasma membrane depolarization (Figure 5; Castro et al., 2006). To test this possibility, st-TRPV1 HeLa cells were exposed to 10 μM CAP in the presence of a culture medium depleted of Na+ by means of replacing Na+ with the non-permeable cation NMDG+, thereby maintaining osmolarity and tonicity constant. These results showed that in the absence of external Na+, the CAP-induced plasma membrane depolarization was significantly decreased. Remarkably, CAP-induced plasma membrane depolarization was completely prevented when both Ca2+ and Na+ were absent from the extracellular medium (Figure 5). Thus, CAP-induced plasma membrane depolarization appears to be an additive effect of Ca2+ and Na+ influxes. DIBAC4(3) efficiency to measure plasma membrane potential was validated using valinomycin and gramicidin (Supplementary Figure S3).
This study highlights how TRPV1 activity is required to induce cell death. Here, we suggest that TRPV1 stimulation with CAP induces necrotic-like cell death without mitochondrial dysfunction, in a mechanism that involves a fast and transient increase in intracellular Ca2+, leading to plasma membrane depolarization and a loss of plasma membrane integrity. Interestingly, 100 μM CAP generates mitochondrial dysfunction and TRPV1-independent apoptotic-like cell death.
TRPV1 expression exhibits dose-dependent cytotoxic effects, including mitochondrial store-dependent Ca2+ overload (Lam et al., 2007; Hu et al., 2008; Davies et al., 2010), membrane bleb formation (Pecze et al., 2013; Wu et al., 2014), pyknotic nuclear fragmentation, cytochrome c release from mitochondria, caspase-3 activation (Davies et al., 2010), and cell viability (Maeno et al., 2000; Bortner and Cidlowski, 2002; Stutzin and Hoffmann, 2006; Lam et al., 2007; Panayiotidis et al., 2010). Indeed, TRPV1 activation promises therapeutic use by rapidly and selectively inducing necrosis in TRPV1-expressing nociceptive neurons (Olah et al., 2001), thereby inducing analgesia most likely via Ca2+-mediated cytotoxicity (Marsch et al., 2007; Gunthorpe and Chizh, 2009; Lambert, 2009). Furthermore, the analgesic effects of CAP – via TRPV1 activation – are associated with the inhibition of hyperpolarization-activated cation currents (Ih), which depend on intracellular Ca2+ mobilization (Kwak, 2012). Optimal mitochondrial physiology maintains a low cytoplasmic Ca2+ concentration through mitochondrial refilling and/or ATP-dependent Ca2+ compartmentalization processes (Varikmaa et al., 2013). Thus, CAP can induce sustained Ca2+ increases, likely via the release of Ca2+ stores caused by mitochondrial failure or mitochondrial fission.
Stable TRPV1 expression and stimulation induces membrane depolarization through an increase in intracellular Ca2+. Although transient increases in Ca2+ did not induce a change in cell state, heavy stimulation of TRPV1 with 100 μM CAP was able to trigger a toxic Ca2+ overload, likely due to intracellular mitochondrial Ca2+ release. Mitochondrial Ca2+ exchange with the cytoplasm has been previously reported (Malli et al., 2003) and Ca2+ uptake has been proposed to generate microdomains of low Ca2+ concentrations across the cytoplasm (Youle and van der Bliek, 2012). In fact, mitochondrial function has been found to be finely regulated by Ca2+-dependent ion channels, capable of regulating the electrochemical gradients required to mobilize Ca2+ into intracellular stores (Malli et al., 2003). Thus, the loss of membrane potential is likely induced by the activation of TRPV1 and a massive Ca2+ influx, leading to CAP-induced cell death.
Endogenous TRPV1 expression in many tissues, such as the brain, heart, skin, and testis, has been reported to play a role in cell death (Kunert-Keil et al., 2006; Marsch et al., 2007). For example, TRPV1-mediated neurotoxicity has been reported in a large spectrum of assays involving primary cultures, as well as tissues, organs, and in toto experiments (Shin et al., 2003; Cernak et al., 2004; Shirakawa et al., 2008). In fact, direct application of CAP to the substantia nigra can induce the depletion of dopaminergic neurons (Kim et al., 2005). Moreover, deregulated TRPV1 activation by endogenous agonists induces the loss of hippocampal neurons and an impairment of cognitive functions (Cernak et al., 2004). These findings, along with observations by other authors, raise the possibility that TRPV1 receptors may be inducing cell death via oxidative stress (Macho et al., 1999; Movsesyan et al., 2004; Lam et al., 2007; Shirakawa et al., 2008), mitochondrial disruption (Macho et al., 1999; Shin et al., 2003), and intracellular Ca2+ overload (Chard et al., 1995; Cernak et al., 2004; Lam et al., 2007; Shirakawa et al., 2008). Nevertheless, this conjecture falls beyond the scope of this work and warrants further investigation in neuronal cells.
Taken together, the results shown in this study suggest that the expression and specific activation of TRPV1 can induce TRPV1-dependent Ca2+ signaling modifications that lead to a plasma membrane potential depolarization contributing to cell death.
RR-B, CC, SG, BR, CL, IM, CE, FS, and PO critically revised and edited this manuscript. RR-B, FS, and PO participated in the research design. RR-B, CC, BR, CL, IM, and PO conducted the experiments and performed data analyses. R-RB, SG, FS, and PO contributed to the figure design. RR-B, SG, IM, CE, FS, and PO wrote the paper.
This work was supported by research grants from Fondo Nacional de Desarrollo Científico y Tecnológico – FONDECYT 1140693, 11100047 (PO), 1111039 (IM), 11170840 (CE), and 1161288 (FS), Millennium Institute on Immunology and Immunotherapy P09-016-F (FS), and Comisión Nacional de Investigación Científica y Tecnológica (CONICYT) Ph.D. scholarship 21171566 (SG) and UNAB DI-741-15/N (FS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2018.00682/full#supplementary-material
Amantini, C., Ballarini, P., Caprodossi, S., Nabissi, M., Morelli, M. B., Lucciarini, R., et al. (2009). Triggering of transient receptor potential vanilloid type 1 (TRPV1) by capsaicin induces Fas/CD95-mediated apoptosis of urothelial cancer cells in an ATM-dependent manner. Carcinogenesis 30, 1320–1329. doi: 10.1093/carcin/bgp138
Bortner, C. D., and Cidlowski, J. A. (2002). Apoptotic volume decrease and the incredible shrinking cell. Cell Death Differ. 9, 1307–1310. doi: 10.1038/sj.cdd.4401126
Carrasco, C., Naziroĝlu, M., Rodriìguez, A., and Pariente, J. (2018). Neuropathic pain: delving into the oxidative origin and the possible implication of transient receptor potential channels. Front. Physiol. 9:95. doi: 10.3389/fphys.2018.00095
Castro, J., Ruminot, I., Porras, O. H., Flores, C. M., Hermosilla, T., Verdugo, E., et al. (2006). ATP steal between cation pumps: a mechanism linking Na+ influx to the onset of necrotic Ca2+ overload. Cell Death Differ. 13, 1675–1685. doi: 10.1038/sj.cdd.4401852
Caterina, M. J., Schumacher, M. A., Tominaga, M., Rosen, T. A., Levine, J. D., and Julius, D. (1997). The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824. doi: 10.1038/39807
Cernak, I., Vink, R., Natale, J., Stoica, B., Lea, P. M., Movsesyan, V., et al. (2004). The “dark side” of endocannabinoids: a neurotoxic role for anandamide. J. Cereb. Blood Flow Metab. 24, 564–578. doi: 10.1097/00004647-200405000-00011
Chard, P. S., Bleakman, D., Savidge, J. R., and Miller, R. J. (1995). Capsaicin-induced neurotoxicity in cultured dorsal root ganglion neurons: involvement of calcium-activated proteases. Neuroscience 65, 1099–1108. doi: 10.1016/0306-4522(94)00548-J
Chen, D., Yang, Z., Wang, Y., Zhu, G., and Wang, X. (2012). Capsaicin induces cycle arrest by inhibiting cyclin-dependent-kinase in bladder carcinoma cells. Int. J. Urol. 19, 662–668. doi: 10.1111/j.1442-2042.2012.02981.x
Çiğ, B., and Nazıroğlu, M. (2015). Investigation of the effects of distance from sources on apoptosis, oxidative stress and cytosolic calcium accumulation via TRPV1 channels induced by mobile phones and Wi-Fi in breast cancer cells. Biochim. Biophys. Acta 1848, 2756–2765. doi: 10.1016/j.bbamem.2015.02.013
Darzynkiewicz, Z., Traganos, F., Staiano-Coico, L., Kapuscinski, J., and Melamed, M. R. (1982). Interaction of rhodamine 123 with living cells studied by flow cytometry. Cancer Res. 42, 799–806.
Davies, J. W., Hainsworth, A. H., Guerin, C. J., and Lambert, D. G. (2010). Pharmacology of capsaicin-, anandamide-, and N-arachidonoyl-dopamine-evoked cell death in a homogeneous transient receptor potential vanilloid subtype 1 receptor population. Br. J. Anaesth. 104, 596–602. doi: 10.1093/bja/aeq067
Gunthorpe, M. J., and Chizh, B. A. (2009). Clinical development of TRPV1 antagonists: targeting a pivotal point in the pain pathway. Drug Discov. Today 14, 56–67. doi: 10.1016/j.drudis.2008.11.005
Hayes, P., Meadows, H. J., Gunthorpe, M. J., Harries, M. H., Duckworth, D. M., Cairns, W., et al. (2000). Cloning and functional expression of a human orthologue of rat vanilloid receptor-1. Pain 88, 205–215. doi: 10.1016/S0304-3959(00)00353-5
Hu, F., Sun, W. W., Zhao, X. T., Cui, Z. J., and Yang, W. X. (2008). TRPV1 mediates cell death in rat synovial fibroblasts through calcium entry-dependent ROS production and mitochondrial depolarization. Biochem. Biophys. Res. Commun. 369, 989–993. doi: 10.1016/j.bbrc.2008.02.155
Jordt, S. E., Tominaga, M., and Julius, D. (2000). Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc. Natl. Acad. Sci. U.S.A. 97, 8134–8139. doi: 10.1073/pnas.100129497
Kim, S. R., Lee, D. Y., Chung, E. S., Oh, U. T., Kim, S. U., and Jin, B. K. (2005). Transient receptor potential vanilloid subtype 1 mediates cell death of mesencephalic dopaminergic neurons in vivo and in vitro. J. Neurosci. 25, 662–671. doi: 10.1523/jneurosci.4166-04.2005
Krasznai, Z., Márián, T., Balkay, L., Emri, M., and Trón, L. (1995). Flow cytometric determination of absolute membrane potential of cells. J. Photochem. Photobiol. B Biol. 28, 93–99. doi: 10.1016/1011-1344(94)07099-A
Kunert-Keil, C., Bisping, F., Krüger, J., and Brinkmeier, H. (2006). Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genomics 7:159. doi: 10.1186/1471-2164-7-159
Kunz, L., Richter, J. S., and Mayerhofer, A. (2006). The adenosine 5’-triphosphate-sensitive potassium channel in endocrine cells of the human ovary: role in membrane potential generation and steroidogenesis. J. Clin. Endocrinol. Metab. 91, 1950–1955. doi: 10.1210/jc.2005-2269
Kwak, J. (2012). Capsaicin blocks the hyperpolarization-activated inward currents via TRPV1 in the rat dorsal root ganglion neurons. Exp. Neurobiol. 21, 75–82. doi: 10.5607/en.2012.21.2.75
Lam, P. M., Hainsworth, A. H., Smith, G. D., Owen, D. E., Davies, J., and Lambert, D. G. (2007). Activation of recombinant human TRPV1 receptors expressed in SH-SY5Y human neuroblastoma cells increases [Ca2+](i), initiates neurotransmitter release and promotes delayed cell death. J. Neurochem. 102, 801–811. doi: 10.1111/j.1471-4159.2007.04569.x
Lambert, D. G. (2009). Capsaicin receptor antagonists: a promising new addition to the pain clinic. Br. J. Anaesth. 102, 153–155. doi: 10.1093/bja/aen354
Latorre, R., Brauchi, S., Orta, G., Zaelzer, C., and Vargas, G. (2007). ThermoTRP channels as modular proteins with allosteric gating. Cell Calcium 42, 427–438. doi: 10.1016/j.ceca.2007.04.004
Latorre, R., Zaelzer, C., and Brauchi, S. (2009). Structure-functional intimacies of transient receptor potential channels. Q. Rev. Biophys. 42, 201–246. doi: 10.1017/S0033583509990072
Ma, L., Zhong, J., Zhao, Z., Luo, Z., Ma, S., Sun, J., et al. (2011). Activation of TRPV1 reduces vascular lipid accumulation and attenuates atherosclerosis. Cardiovasc. Res. 92, 504–513. doi: 10.1093/cvr/cvr245
Macho, A., Calzado, M. A., Muñoz-Blanco, J., Gómez-Díaz, C., Gajate, C., Mollinedo, F., et al. (1999). Selective induction of apoptosis by capsaicin in transformed cells: the role of reactive oxygen species and calcium. Cell Death Differ. 6, 155–165. doi: 10.1038/sj.cdd.4400465
Maeno, E., Ishizaki, Y., Kanaseki, T., Hazama, A., and Okada, Y. (2000). Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. U.S.A. 97, 9487–9492. doi: 10.1073/pnas.140216197
Malli, R., Frieden, M., Osibow, K., and Graier, W. F. (2003). Mitochondria efficiently buffer subplasmalemmal Ca2+ elevation during agonist stimulation. J. Biol. Chem. 278, 10807–10815. doi: 10.1074/jbc.M212971200
Marsch, R., Foeller, E., Rammes, G., Bunck, M., Kössl, M., Holsboer, F., et al. (2007). Reduced anxiety, conditioned fear, and hippocampal long-term potentiation in transient receptor potential vanilloid type 1 receptor-deficient mice. J. Neurosci. 27, 832–839. doi: 10.1523/jneurosci.3303-06.2007
Movsesyan, V. A., Stoica, B. A., Yakovlev, A. G., Knoblach, S. M., Lea, P. M., Cernak, I., et al. (2004). Anandamide-induced cell death in primary neuronal cultures: role of calpain and caspase pathways. Cell Death Differ. 11, 1121–1132. doi: 10.1038/sj.cdd.4401442
Olah, Z., Karai, L., and Iadarola, M. J. (2002). Protein kinase C(alpha) is required for vanilloid receptor 1 activation. Evidence for multiple signaling pathways. J. Biol. Chem. 277, 35752–35759. doi: 10.1074/jbc.M201551200
Olah, Z., Szabo, T., Karai, L., Hough, C., Fields, R. D., Caudle, R. M., et al. (2001). Ligand-induced dynamic membrane changes and cell deletion conferred by vanilloid receptor 1. J. Biol. Chem. 276, 11021–11030. doi: 10.1074/jbc.M008392200
Olivero, P., Leiva-Salcedo, E., Devoto, L., and Stutzin, A. (2008). Activation of Cl- channels by human chorionic gonadotropin in luteinized granulosa cells of the human ovary modulates progesterone biosynthesis. Endocrinology 149, 4680–4687. doi: 10.1210/en.2008-0303
O’Neill, D. P., Peng, T., Stiegler, P., Mayrhauser, U., Koestenbauer, S., Tscheliessnigg, K., et al. (2011). A three-state mathematical model of hyperthermic cell death. Ann. Biomed. Eng. 39, 570–579. doi: 10.1007/s10439-010-0177-1
Panayiotidis, M. I., Franco, R., Bortner, C. D., and Cidlowski, J. A. (2010). Ouabain-induced perturbations in intracellular ionic homeostasis regulate death receptor-mediated apoptosis. Apoptosis 15, 834–849. doi: 10.1007/s10495-010-0494-8
Pecze, L., Blum, W., and Schwaller, B. (2013). Mechanism of capsaicin receptor TRPV1-mediated toxicity in pain-sensing neurons focusing on the effects of Na+/Ca2+ fluxes and the Ca2+-binding protein calretinin. Biochim. Biophys. Acta 1833, 1680–1691. doi: 10.1016/j.bbamcr.2012.08.018
Ramsey, I. S., Delling, M., and Clapham, D. E. (2006). An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647. doi: 10.1146/annurev.physiol.68.040204.100431
Randhawa, P. K., and Jaggi, A. S. (2018). A review on potential involvement of TRPV1 channels in ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. Ther. 23, 38–45. doi: 10.1177/1074248417707050
Rozas, P., Lazcano, P., Piña, R., Cho, A., Terse, A., Pertusa, M., et al. (2016). Targeted overexpression of tumor necrosis factor-α increases cyclin-dependent kinase 5 activity and TRPV1-dependent Ca2+ influx in trigeminal neurons. Pain 157, 1346–1362. doi: 10.1097/j.pain.0000000000000527
Shimizu, T., Numata, T., and Okada, Y. (2004). A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl- channel. Proc. Natl. Acad. Sci. U.S.A. 101, 6770–6773. doi: 10.1073/pnas.0401604101
Shin, C. Y., Shin, J., Kim, B.-M., Wang, M.-H., Jang, J.-H., Surh, Y.-J., et al. (2003). Essential role of mitochondrial permeability transition in vanilloid receptor 1-dependent cell death of sensory neurons. Mol. Cell. Neurosci. 24, 57–68. doi: 10.1016/S1044-7431(03)00121-0
Shirakawa, H., Yamaoka, T., Sanpei, K., Sasaoka, H., Nakagawa, T., and Kaneko, S. (2008). TRPV1 stimulation triggers apoptotic cell death of rat cortical neurons. Biochem. Biophys. Res. Commun. 377, 1211–1215. doi: 10.1016/j.bbrc.2008.10.152
Stutzin, A., and Hoffmann, E. K. (2006). Swelling-activated ion channels: functional regulation in cell-swelling, proliferation and apoptosis. Acta Physiol. 187, 27–42. doi: 10.1111/j.1748-1716.2006.01537.x
Sun, Z., Han, J., Zhao, W., Zhang, Y., Wang, S., Ye, L., et al. (2014). TRPV1 activation exacerbates hypoxia/reoxygenation-induced apoptosis in H9C2 cells via calcium overload and mitochondrial dysfunction. Int. J. Mol. Sci. 15, 18362–18380. doi: 10.3390/ijms151018362
Thomas, K. C., Sabnis, A. S., Johansen, M. E., Lanza, D. L., Moos, P. J., Yost, G. S., et al. (2007). Transient receptor potential vanilloid 1 agonists cause endoplasmic reticulum stress and cell death in human lung cells. J. Pharmacol. Exp. Ther. 321, 830–838. doi: 10.1124/jpet.107.119412
Tominaga, M., Caterina, M. J., Malmberg, A. B., Rosen, T. A., Gilbert, H., Skinner, K., et al. (1998). The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543. doi: 10.1016/S0896-6273(00)80564-4
Utreras, E., Prochazkova, M., Terse, A., Gross, J., Keller, J., Iadarola, M. J., et al. (2013). TGF-β1 sensitizes TRPV1 through Cdk5 signaling in odontoblast-like cells. Mol. Pain 9:24. doi: 10.1186/1744-8069-9-24
Varikmaa, M., Guzun, R., Grichine, A., Gonzalez-Granillo, M., Usson, Y., Boucher, F., et al. (2013). Matters of the heart in bioenergetics: mitochondrial fusion into continuous reticulum is not needed for maximal respiratory activity. J. Bioenerg. Biomembr. 45, 319–331. doi: 10.1007/s10863-012-9494-4
Wu, T. T., Peters, A. A., Tan, P. T., Roberts-Thomson, S. J., and Monteith, G. R. (2014). Consequences of activating the calcium-permeable ion channel TRPV1 in breast cancer cells with regulated TRPV1 expression. Cell Calcium 56, 59–67. doi: 10.1016/j.ceca.2014.04.006
Youle, R. J., and van der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. doi: 10.1126/science.1219855
Keywords: TRPV1, capsaicin, cell death, mitochondria, necrosis, calcium
Citation: Ramírez-Barrantes R, Córdova C, Gatica S, Rodriguez B, Lozano C, Marchant I, Echeverria C, Simon F and Olivero P (2018) Transient Receptor Potential Vanilloid 1 Expression Mediates Capsaicin-Induced Cell Death. Front. Physiol. 9:682. doi: 10.3389/fphys.2018.00682
Received: 06 February 2018; Accepted: 16 May 2018;
Published: 05 June 2018.
Edited by:
Mauricio Antonio Retamal, Universidad del Desarrollo, ChileReviewed by:
Felipe Yasser Eltit, The University of British Columbia, CanadaCopyright © 2018 Ramírez-Barrantes, Córdova, Gatica, Rodriguez, Lozano, Marchant, Echeverria, Simon and Olivero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Felipe Simon, ZnNpbW9uQHVuYWIuY2w= Pablo Olivero, cGFibG8ub2xpdmVyb0B1di5jbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.