 Rick Huisjes1
Rick Huisjes1 Anna Bogdanova2
Anna Bogdanova2 Wouter W. van Solinge1
Wouter W. van Solinge1 Raymond M. Schiffelers1
Raymond M. Schiffelers1 Lars Kaestner3,4
Lars Kaestner3,4 Richard van Wijk1*
Richard van Wijk1*- 1Department of Clinical Chemistry and Haematology, University Medical Center Utrecht, Utrecht University, Utrecht, Netherlands
- 2Red Blood Cell Research Group, Institute of Veterinary Physiology, Vetsuisse Faculty and the Zurich Center for Integrative Human Physiology (ZIHP), University of Zurich, Zürich, Switzerland
- 3Theoretical Medicine and Biosciences, Saarland University, Saarbrücken, Germany
- 4Experimental Physics, Saarland University, Saarbrücken, Germany
Deformability is an essential feature of blood cells (RBCs) that enables them to travel through even the smallest capillaries of the human body. Deformability is a function of (i) structural elements of cytoskeletal proteins, (ii) processes controlling intracellular ion and water handling and (iii) membrane surface-to-volume ratio. All these factors may be altered in various forms of hereditary hemolytic anemia, such as sickle cell disease, thalassemia, hereditary spherocytosis and hereditary xerocytosis. Although mutations are known as the primary causes of these congenital anemias, little is known about the resulting secondary processes that affect RBC deformability (such as secondary changes in RBC hydration, membrane protein phosphorylation, and RBC vesiculation). These secondary processes could, however, play an important role in the premature removal of the aberrant RBCs by the spleen. Altered RBC deformability could contribute to disease pathophysiology in various disorders of the RBC. Here we review the current knowledge on RBC deformability in different forms of hereditary hemolytic anemia and describe secondary mechanisms involved in RBC deformability.
Introduction
The primary function of RBCs is to enable respiration in tissues by providing oxygen and removing carbon dioxide via gas exchange in the lungs. During a typical 120 days lifespan of a RBC, it circulates through arteries, veins and small capillaries traveling –in total– a distance of 500 km (Lasch et al., 2000). RBC deformability, i.e., the ability of the RBC to change shape is essential for successful passage through these capillaries and splenic sinuses (Danielczok et al., 2017).
Deformability of RBC depends on the (i) structural properties of the “horizontal” cytoskeletal components such as spectrin (Burton and Bruce, 2011; Nans et al., 2011), (ii) vertical interaction of cytoskeleton and integral transmembrane complexes that is accomplished by ankyrin, 4.1 and 4.2 protein and the cytosolic domain of band 3 protein (also known as the anion exchanger 1 (AE1) or solute carrier family 4 member 1 (SLC4A1)) (Gallagher, 2004a) and (iii) resistance of the cytosolic pool (i.e., intracellular viscosity, RBC hydration state and surface-volume interaction) (Clark et al., 1983).
Deformability is also affected by metabolic processes controlling ATP levels and redox state. These factors control ion handling by pumps and passive transport pathways (Chu et al., 2012; Bogdanova et al., 2016), proteolytic activity of Ca2+-dependent protease calpain (Bogdanova et al., 2013), and mutations and structural integrity of each element of the membrane architecture (Gallagher, 2004b). Failure to sustain deformability results in shortening of RBC life span and, when not compensated by de novo RBC production, in hemolytic anemia.
Therefore, reliable estimation of RBC deformability and understanding of the processes in control of it are essential for evaluation of severity of patients’ state and choosing of the optimal therapeutic strategy. This particularly relates to the feasibility of splenectomy as an option to improve or worsen condition of patients with anemic state (Iolascon et al., 2017).
In this review, we provide an overview of the current knowledge on the primary and secondary mechanisms involved in regulation of RBC deformability in hereditary hemolytic anemia. We discuss methodologies that are currently used to assess RBC deformability in the clinical and research laboratories. We link different processes, such as ion channel activity, intracellular energy metabolism and phosphorylation of membrane proteins to RBC deformability and illustrate how these processes are affected in various RBC pathologies, such as sickle cell disease, thalassemia, HS and metabolic defects of RBCs. Finally, we describe the influence of shedding of nano-sized membrane vesicles from the RBC, the oxygenation state of hemoglobin and adaptive responses (such as exercise and high-altitude) on RBC deformability. Increased shedding of RBC vesicles, for example, is a feature of various RBC pathologies and vesicles are increasingly being considered to be a novel biomarker of RBC disorders (Pattanapanyasat et al., 2004; Nantakomol et al., 2012; Alaarg et al., 2013). They are considered to be involved in thrombosis and hemostasis (Biro et al., 2003; Livaja Koshiar et al., 2014) and associated with reduced RBC deformability (Waugh et al., 1992; Bosch et al., 1994).
RBC Deformability In Hereditary Hemolytic Anemia
Anemia is considered to be hemolytic when RBCs are prematurely cleared from the circulation. Hemolytic anemia can be further subdivided into intra- or extravascular hemolytic anemia, and the underlying cause can be either inherited or acquired. Intravascular hemolysis is, as the name suggests, lysis of RBC in the vasculature. The cause can be hereditary, as seen in sickle cell disease (Pauling and Itano, 1949; Kato et al., 2017), but intravascular hemolysis can also be initiated by certain drugs (Cappellini and Fiorelli, 2008), by mechanical stress (for example through shear forces generated by artificial heart valves), by cold-agglutination (Körmöczi et al., 2006) or as a result of exhaustive exercise (Jordan et al., 1998). Intravascular hemolysis causes the release of hemoglobin into the plasma. Free hemoglobin is toxic and can lead to various clinical manifestations, such as hemoglobinuria, renal dysfunction, pulmonary hypertension and platelet activation (Rother et al., 2005).
Extravascular hemolysis is directly related to reduced RBC deformability. RBCs with reduced deformability fail to pass the spleen, which acts as an RBC quality-control organ (Mebius and Kraal, 2005; Deplaine et al., 2010). The red pulp of the spleen contains narrow inter-endothelial slits (MacDonald et al., 1987). Failure to pass through these narrow slits (Mebius and Kraal, 2005) leads to the uptake and breakdown of RBCs by macrophages (Burger et al., 2012). A number of hereditary RBC disorders result in reduced RBC deformability, which, as a consequence, leads to premature removal of RBCs in the spleen. Removal of RBCs by the spleen is, however, not only dependent on reduced deformability, but also occurs after recognition by macrophages. Senescent RBCs can be recognized and phagocytized by macrophages in the spleen upon binding of autologous antibodies to band 3 (Kay et al., 1983; Kay, 1984), exposure of conformational altered CD47 (Burger et al., 2012) or exposure of PS (Boas et al., 1998).
Hereditary forms of hemolytic anemia can affect the RBC membrane (i.e., HS, elliptocytosis, and pyropoikilocytosis) (Gallagher, 2004a; Perrotta et al., 2008; Da Costa et al., 2013), its metabolism (i.e., enzymopathies) (Zanella and Bianchi, 2000; van Wijk and van Solinge, 2005; Koralkova et al., 2014), cell hemoglobin (i.e., sickle cell anemia, unstable hemoglobin variants) (Higgs et al., 2012; Ware et al., 2017), or cellular hydration (i.e., HS, hereditary xerocytosis or Gardos Channelopathy) (Vives Corrons and Besson, 2001; Albuisson et al., 2013; Andolfo et al., 2013, 2015; Beneteau et al., 2014; Faucherre et al., 2014; Glogowska et al., 2015; Fermo et al., 2017a). While the primary genetic causes of these disorders are often well determined, less is known about the factors triggering the actual hemolysis. Striking examples are sickle cell disease, thalassemia, HS and the metabolic disorders of the RBC. All have well-known and well-studied primary genetic and molecular defects. However, little is known about the secondary mechanisms that may decrease RBC deformability and thereby contribute to the premature removal of these affected RBCs.
For example, the homozygous single point mutation in the HBB gene, substituting glutamic acid for valine at position 6 leads to sickle cell disease (Ware et al., 2017). Although the discovery that these RBCs tend to sickle at low oxygen tension was already provided by Hahn and Gillespie (1927), Kenney et al. (1961), it took decades to unravel the processes that contribute to sickle cell dehydration (Lew and Bookchin, 2005; Ataga et al., 2008), decreased deformability and increased endothelial cell adhesion (Alapan et al., 2015, 2016). We still do not understand how does a point mutation in hemoglobin beta chain cause these secondary pathological alterations in density and adhesiveness and how all these changes in turn impact the disease symptoms of the patient. Other examples are the enzymopathies, which are caused by mutations in, for example, PK or HK and lead to a shortage of metabolic energy inside the RBC. Again, although the primary effect on decreased levels of ATP in these diseases is well understood (Zanella et al., 2007), secondary processes such as altered K+ fluxes (Nathan et al., 1965) and phosphorylation of intracellular proteins (Thali et al., 2010; Wang et al., 2010), need further elucidation.
Moreover, for a substantial number of patients with hereditary hemolytic anemia – the primary causes of disease remain unknown, or were only identified very recently. For these patients, a comprehensive understanding of both primary and secondary defects of the affected RBCs is still lacking. This is illustrated by the recently discovered mutations in patients with hereditary xerocytosis, also known as dehydrated stomatocytosis (Andolfo et al., 2013). In these patients, stomatocytes are typically found in peripheral blood smears. However, the primary molecular cause for the anemia and morphological change of the RBC remained unknown for a long time, let alone that the secondary processes that are involved in the disease process were elucidated. Only recently, it was shown that hereditary xerocytosis was caused by mutations in genes for the mechanosensitive PIEZO1 channel (Andolfo et al., 2013; Shmukler et al., 2014; Glogowska et al., 2017). Very recently, a mutation in the calcium-activated potassium channel subfamily N member KCNN4 (encoded by the human KCa3.1 gene, and also known as the Gardos channel in RBCs) (Andolfo et al., 2015; Glogowska et al., 2015; Rapetti-Mauss et al., 2015; Fermo et al., 2017a) was shown to cause ‘Gardos channelopathy’ a disease resembling yet at some points different from hereditary xerocytosis (Fermo et al., 2017a). Only then, the molecular diagnosis of these diseases was established in these patient groups and new insight on RBC deformability and RBC ion homeostasis with respect to ion channel function were obtained. The secondary processes that contribute to RBC clearance in these diseases are now being explored (Fermo et al., 2017a).
Techniques to Determine RBC Deformability
A currently well-established tool to probe RBC deformability is osmotic gradient ektacytometry, which is routinely used in the diagnosis of patients with hereditary hemolytic anemia. The technique is performed on a Laser-assisted Optical Rotational Cell Analyzer (Lorrca). It assesses RBC deformability, osmotic fragility and cellular hydration status (see Figure 1) (Clark et al., 1983; Da Costa et al., 2016; Lazarova et al., 2017; Llaudet-Planas et al., 2017). In osmotic gradient ektacytometry, the maximum RBC deformability is represented by the maximum elongation index (EImax). The Omin represents the osmotic value (mOsmol/kg H2O) where the elongation index (EI) is minimal, corresponding to the 50% lysis point as determined by the classical osmotical fragility test (Clark et al., 1983). The hydration status (or intracellular viscosity) is represented by Ohyper (mOsmol/kg H2O). The value corresponds with the hypertonic osmolarity where the EI is 50% of EImax. The value of these three parameters as well as the shape of the curve is used in the diagnosis of various disorders of the RBC membrane and hydration station (Figure 1B).
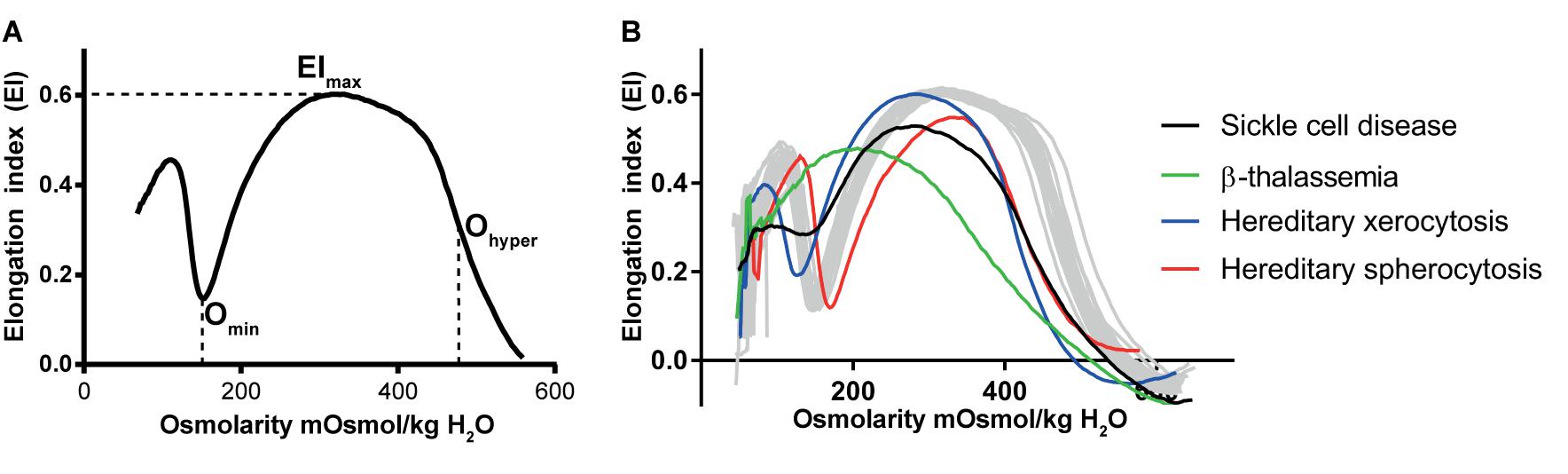
FIGURE 1. Osmotic gradient ektacytometry as a tool to measure red blood cell (RBC) deformability. The technique is discussed in detail by Clark et al. (1983); Lazarova et al. (2017), and Da Costa et al. (2016). Briefly, during osmotic gradient ektacytometry, the RBC is subjected to an osmotic gradient (range ≈50 mOsmol/kg H2O – 650 mOsmol/kg H2O) under constant shear stress, while the elongation index (EI) is measured. The EI corresponds to the deformability at various osmotic conditions. (A) the left graph depicts an osmotic gradient ektacytometry curve from a healthy control with various intersection points: the EImax reflects the maximal deformability of the RBC, Omin reflects the osmotic fragility and Ohyper reflects the (de)hydration state (or intracellular viscosity) of the RBC. (B) In the right graph osmotic gradient curves of patients with sickle cell disease (black line), β-thalassemia (green line), hereditary xerocytosis (blue line) and hereditary spherocytosis (red line) and several individual healthy controls (gray lines, n = 20) are depicted.
Ohyper correlates with the reciprocal function of the MCHC (Clark et al., 1983). In healthy individuals, during RBC aging increased MCHC is observed and this correlates with decreased RBC deformability (Clark et al., 1983; Bosch et al., 1994). A similar observation has been made for RBCs from patients with sickle cell disease and HS. Also here, a decrease in RBC hydration (i.e., decreased Ohyper and increased MCHC) of patient’s cells is associated with a decrease in the maximum RBC deformability (EImax) (Clark et al., 1983; Bunn, 1997).
Besides osmotic gradient ektacytometry, there are other methods to measure RBC deformability (Tomaiuolo, 2014). A number of techniques that are also used in diagnostics measures RBC deformability as a function of shear stress, for example the RheoScan-D (Shin et al., 2007), Automated Rheoscope and Cell Analyser (ARCA) (Dobbe et al., 2002) and the deformability measurement module on the Lorrca (Hardeman et al., 1987). The results of RheoScan-D and the deformability measurements (shear-stress module) on the Lorrca were observed to be comparable (Shin et al., 2007). However, RheoScan-D measures RBC deformability under microfluidic conditions after the application of shear stress, while shear-stress on RBCs in the deformability measurements on the Lorrca requires a larger set-up with a rotating cup (Hardeman et al., 1987; Shin et al., 2007). The ARCA measures RBC deformability with a microscope after the application of shear-stress, and therefore has the potential to measure the RBC deformability of subpopulations (Dobbe et al., 2002).
Most direct measurements of single cells include: micropipette (Waugh et al., 1992), atomic force spectroscopy and holographic optical tweezers (HOT) (Steffen et al., 2011; Kaestner et al., 2012). The mechanical probes and the optical approach are complementary since 30 pN is a lower force limit for the mechanical measurements and an upper limit for the HOT in the context of investigating RBCs (Minetti et al., 2013). Currently microfluidic approaches becoming increasingly popular to investigate RBC deformability (Cluitmans et al., 2014; Guo et al., 2014; Picot et al., 2015; Danielczok et al., 2017). While the latter method has the potential to become a routine diagnostic tool, the former single cell methods are mostly dedicated to basic research due to their instrumental complexity.
A further very simple method to measure RBC deformability is the readout of RBC filterability, e.g., by measuring RBC passage through cellulose columns (Oonishi et al., 1997). Unfortunately there is currently no standardized column/readout system on the market that would allow comparison of filterability between diagnostic laboratories. In addition, RBC passage through cellulose columns or filters is highly subjected to changes in cellular volume (such as MCV), which may affect results. A summary of available techniques to measure RBC deformability is depicted in Table 1.
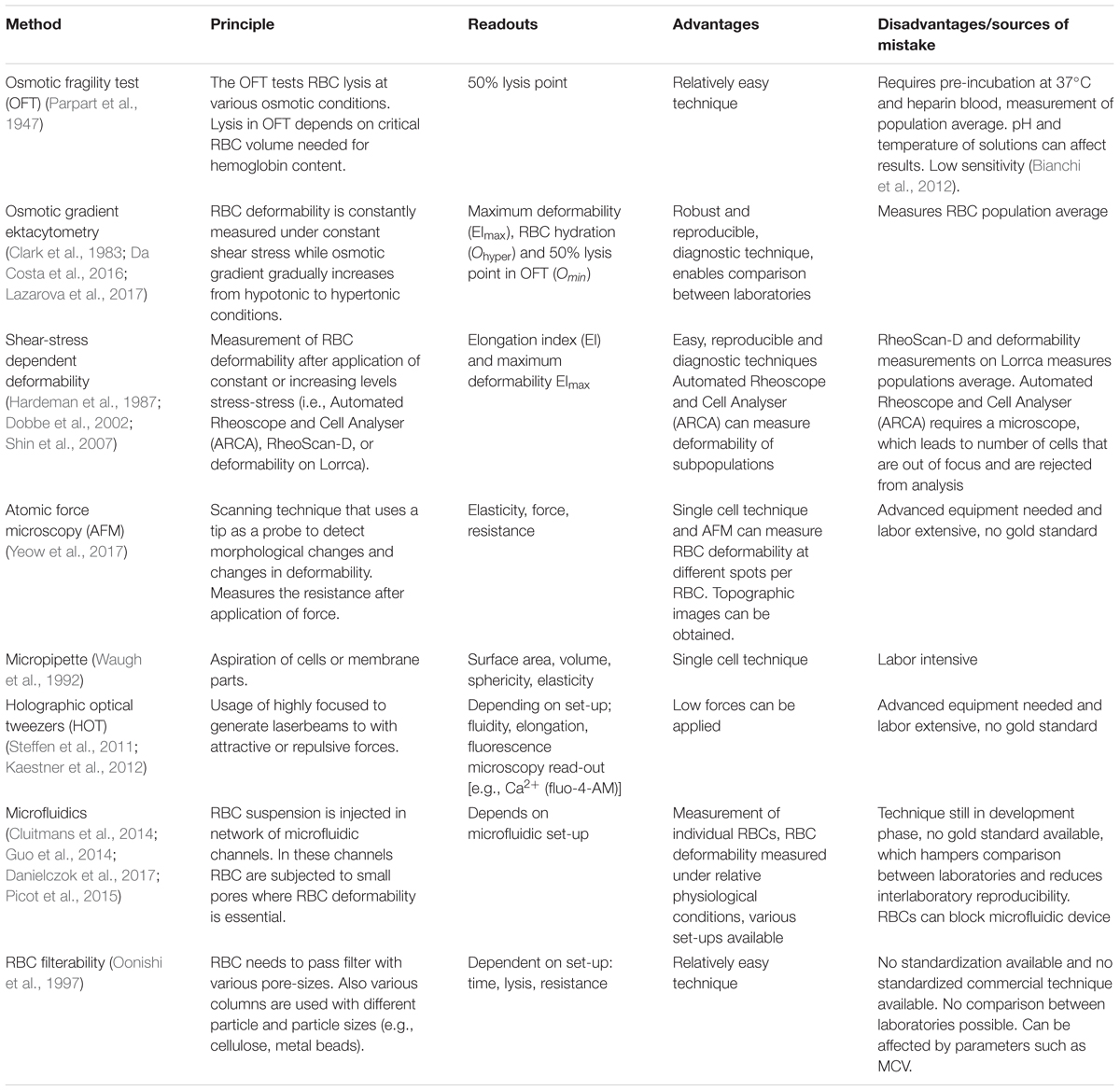
TABLE 1. Overview of techniques to measure RBC deformability.
Determinants of RBC Deformability
RBC Hydration
Regulation of RBC Hydration
Red blood cell deformability is highly influenced by RBC volume control and by ion content, both regulated by ion pumps, ion channels, symporters and antiporters (Gallagher, 2013) (see Figure 2). When ion channels are open, ions move following their electrochemical gradients, while ion pumps can actively move these ions against the gradient (Gadsby, 2009). Symporters and antiporters may also create secondary ion gradients, but require the pre-existing gradients for at least one type of ions as a driving force to transport the other ion types against the gradient. Symporters transport two (e.g., K+-Cl- cotransporter) or more (e.g., Na+-K+-2Cl- cotransporter) ions in the same direction using driving force for one of them, while antiporters (e.g., Na+/H+ exchanger or anion exchanger) exchange two ions that move in the opposite direction (Wolfersberger, 1994). These transporters, pumps and channels are crucial in resisting/adapting to local osmotic changes and maintaining RBC volume (Wieth, 1979; Liu et al., 2011; Thomas et al., 2011; Gallagher, 2013; Lew and Tiffert, 2017). RBC volume changes result from differences in osmotic pressure (Sugie et al., 2018), which results in water transport by aquaporins of which AQP1 and AQP3 are found in human RBCs (Yang et al., 2001). Cotransport of water is, however, also possible through the K+-Cl- cotransporter (KCC) (Zeuthen and Macaulay, 2012). Volume changes are also associated by in- and efflux of several amino acids and amino-acid derivates, such as taurine (Goldstein and Brill, 1991; Goldstein and Davis, 1994).
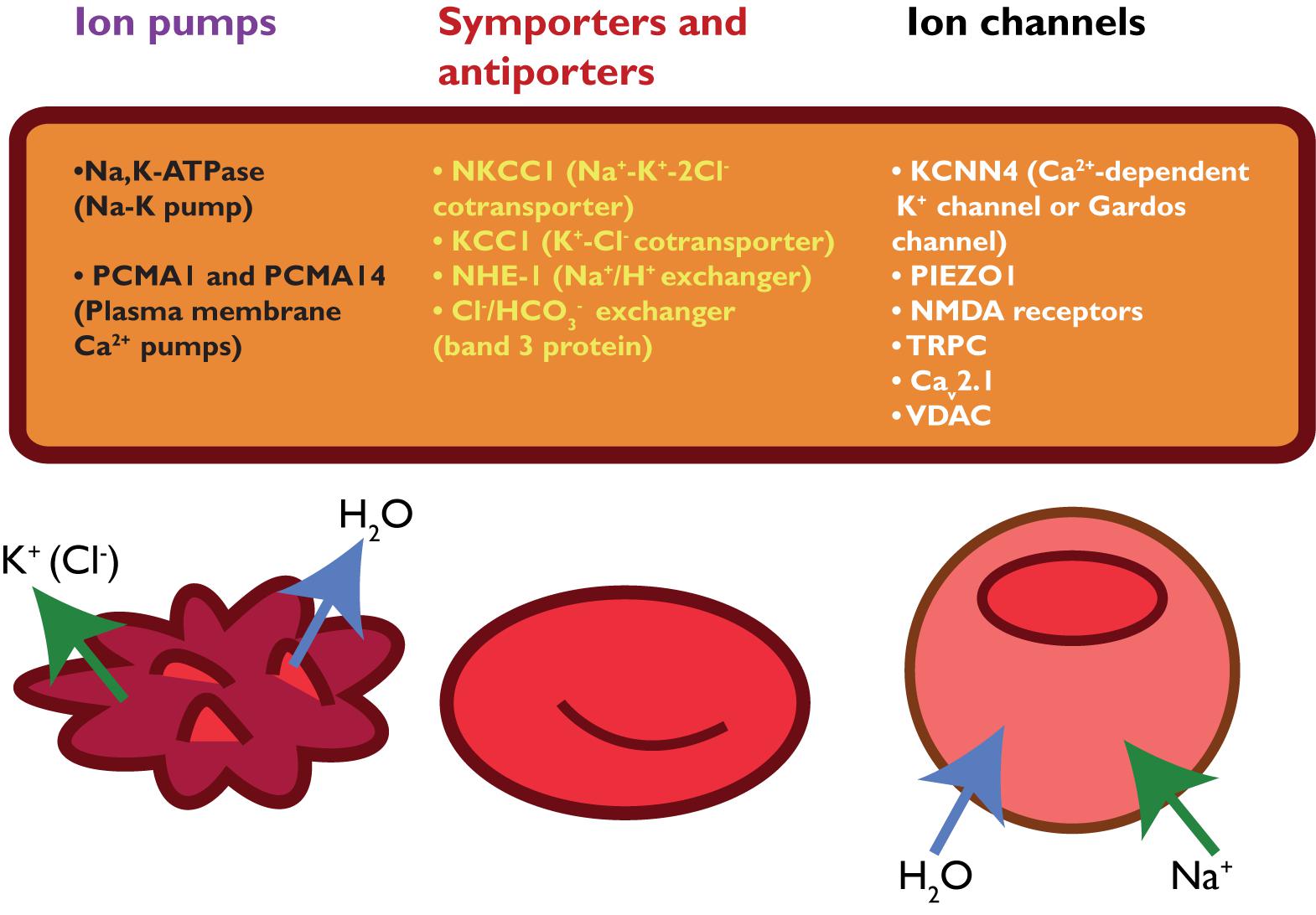
FIGURE 2. schematic overview of RBC ion pumps (Sachs, 2003; Tiffert et al., 2003), symporters and antiporters (Bernhardt et al., 1988; Wolfersberger, 1994) and ion channels (Bouyer et al., 2012; Kaestner, 2011, 2015).
As mentioned previously, MCHC is a parameter that reflects the RBC hydration state, and is dependent on the hemoglobin concentration in RBCs, RBC volume, RBC membrane loss and water content. Quick movements of ions and water result in acute changes in MCHC whereas long-term effects on MCHC are often associated with release of hemoglobin-free vesicles. RBC hydration may be quantified using the Ohyper value as obtained by osmotic gradient ektacytometry (see Figure 1 and see section “Techniques to Determine RBC Deformability”). Changes in RBC density detected using cell distribution within Percoll density gradients reflects both membrane loss and the changes in RBC surface-to-volume ratio and alterations in intracellular ion and water content. In-depth morphological analysis could be used as a complementary approach to discriminate between membrane and ion/water loss.
Primary Changes of RBC Hydration
In this section we shall address hereditary hemolytic anemias associated with the changes in the intracellular ion and secondary to the latter changes in water content. One of the players in this type of disorder is a mechano-sensitive PIEZO1 channel, a non-selective cation channel permeable for Ca2+ that in turn can activate the calcium activated K+ channel subfamily N member KCNN4 (or Gardos channel in RBCs) (Kaestner et al., 2018). Thus, hydration state is largely dependent on the shear stress intensity and the hydration state of RBCs from splenectomized and non-splenectomized patients with the same mutation may vary substantially. Furthermore, hydration state is a function of RBC age, young RBCs being more hydrated than the senescent cells (Clark et al., 1983; Lutz et al., 1992).
A RBC disorder characterized by primary hydration changes is hereditary xerocytosis (HX), also known as dehydrated stomatocytosis. HX is a form of hereditary hemolytic anemia, and patients often suffer from iron-overload (Zarychanski et al., 2012). In addition, in many RBC disorders splenectomy relieves symptoms and increases RBC survival, but in HX, splenectomy is contra-indicated as it drastically increases the risks for thrombotic events (Stewart et al., 1996; Jaïs et al., 2003; Iolascon et al., 2017). HX was related to mutations in the mechanosensitive PIEZO1 channel (Zarychanski et al., 2012; Albuisson et al., 2013; Li et al., 2014). PIEZO1 mutations lead to stabilization of the “active” state or destabilization of the “inactive” state of the PIEZO1 protein (Albuisson et al., 2013). These alterations in channel function lead to increased intracellular levels of Ca2+ and subsequent KCNN4 (Gardos channel) activation (Cahalan et al., 2015; Danielczok et al., 2017). KCNN4 activation leads to efflux of K+ and dehydration. The relatively low intracellular levels of K+ could therefore serve as a biomarker for HX (Zarychanski et al., 2012; Gallagher, 2017). Although PIEZO1 mutations leads to KCNN4 (or Gardos channel) activation, RBCs with mutations in KCNN4 (Gardos channelopathy) exhibit a different pathology (Fermo et al., 2017a). For example, HX caused by mutations in PIEZO1 lead to severe RBC dehydration, which is also shown by a left-shift of Ohyper in the osmotic gradient curve (see Figure 1B and see section “Techniques to Determine RBC Deformability”) while mutations in KCNN4 (or Gardos channel) may cause only slight alterations in the osmotic gradient curve (Fermo et al., 2017b). In addition, RBCs from patients with KCNN4 mutations were found to exhibit increased activity of Na-K-ATPase that is most likely triggered by the increase in the intracellular Na+ concentrations. The hyperactivated Na, K-pump compensates for the dissipation in Na/K gradients and pumps K+ back into the cells, but fails to restore normal K+ content completely (Fermo et al., 2017a). Currently, however, it is difficult to measure intracellular water content or RBC volume directly and accurately enough, which makes it challenging to discriminate between water loss and membrane loss.
Cytoskeletal Network
Primary Changes of the Cytoskeletal Network
In addition to the protein defects, described above, that directly disturb the membrane and thereby the ability of the RBC to deform there are also secondary causes affecting membrane (protein) function and, thereby, RBC deformability. Examples of these are membrane protein phosphorylation, RBC density and RBC vesiculation.
The RBC membrane is built from a basic triangular network of α- and β-spectrin molecules connected to band 3, ankyrin and protein 4.1 (Perrotta et al., 2008; Salomao et al., 2008). This network provides the RBC with a certain stability and simultaneously the ability to deform. HS (HS) is characterized by defects in proteins forming the cytoskeletal network or proteins forming ankyrin and junctional complexes. This usually concerns mutations in band 3, protein 4.2, α-spectrin, or ankyrin (Perrotta et al., 2008). These mutations lead to membrane instability, membrane loss through release of hemoglobin-free vesicles, with consequent decreased surface area-to-volume ratio, increase in MCHC and density and formation of spherocytes (Perrotta et al., 2008). Spherocytes show a characteristic loss of RBC deformability, leading to premature removal of these RBCs from the circulation (Perrotta et al., 2008). Hereditary elliptocytosis (HE) is a disorder characterized by disturbances of the horizontal cytoskeletal interactions and are usually caused by mutations in α-spectrin, β-spectrin and protein 4.1R. As the name suggests, the disturbed horizontal interactions lead to the formation of elliptocytes. The majority of HE patients are clinically asymptomatic, but few patients suffer from hemolysis, jaundice and splenomegaly (Da Costa et al., 2013).
Secondary Disturbances of the Cytoskeletal Network
The triangular spectrin network, connected through ankyrin and protein 4.1R to band 3 supports membrane stability and contributes to RBC flexibility and deformability. Tyrosine phosphorylation of in the individual proteins regulates interaction forces between the elements of this network. Tyrosine phosphorylation is regulated by phosphotyrosine kinases and by phosphotyrosine phosphatases. Under normal conditions, protein regulatory tyrosine residues are only phosphorylated to a limited degree (Terra et al., 1998; Zipser et al., 2002). In the RBC membrane, phosphorylation of protein 4.1R, for example, leads to a reduced membrane stability (Muravyov and Tikhomirova, 2013), and phosphorylation of ankyrin regulates its binding to band 3 (Muravyov and Tikhomirova, 2013). The dissociation constant (Kd) between ankyrin and band 3 influences RBC deformability without a loss in elasticity, underlining that cytoskeletal protein phosphorylation has implications on RBC deformability (Anong, 2006).
The effect of phosphorylation of band 3, the most abundant membrane protein, on RBC deformability is not entirely clear. The possible physiological relevance of band 3 protein Tyr phosphorylation state emerges from the fact that it is controlled by hormones such as insulin (Marques et al., 2000) and that it is altered in patients with hemoglobinopathies (Terra et al., 1998). Saldanha et al. (2007) showed that dephosphorylation of band 3 Tyr residues does not affect RBC deformability. It, therefore, appears that phosphorylation of band 3 does not affect RBC deformability directly, but may alter band 3 protein aggregation and its interaction with extracellular matrix components.
Lopes de Almeida et al. (2012) showed that high concentrations of fibrinogen in combination with dephosphorylated band 3, leads to mildly increased RBC deformability at low shear stress. In contrast to these findings, Reid et al. (1984) report decreased RBC deformability in diabetic patients with hyperfibrinogenemia in absence of dephosphorylating agents (Reid et al., 1984). The decreased RBC deformability in the absence of dephosphorylating agents seems to underline that phosphorylation is not involved. However, insulin is implicated in the activation of tyrosine kinases that in turn phosphorylate band 3. In diabetes types I and II, glucose and insulin homeostasis is disturbed leading to increased protein tyrosine phosphatase activities and aberrant band 3 phosphorylation patterns (Marques et al., 2000), potentially leading to reduced RBC deformability and increased RBC turnover. This same principle may also play a role in patients with sepsis, where investigators have shown that tyrosine phosphorylation of band 3 was increased in septic mice and that this increase was accompanied by reduced RBC deformability (Condon et al., 2007).
Band 3 phosphorylation was found to be increased by a rise in intracellular Ca2+-concentrations due to dissociation of phosphotyrosine phosphatase from band 3 (Zipser et al., 2002). Interestingly, sickle cell disease is characterized by an increase in both intracellular Ca2+ concentration (Bogdanova et al., 2013) and tyrosine phosphorylation of band 3 (Terra et al., 1998), which potentially could thus influence RBC membrane stability. The importance of band 3 phosphorylation for RBC shape and deformability is highlighted by a number of in vitro experiments. For example, morphological changes of RBCs were observed after Tyr-phosphorylation of band 3 mediated by pervanadate (Bordin et al., 1995). In contrast, Ser/Thr-phosphorylation of spectrin, which was selectively induced by okadaic acid did not result in morphological changes (Bordin et al., 1995). Unfortunately, the deformability of RBCs after pervanadate and okadaic treatment was not measured in these studies, but it seems reasonable to assume that RBC deformability is affected since both membrane protein phosphorylation and RBC morphology have been associated with altered RBC deformability (Reid et al., 1984; Chabanel et al., 1987; Hasegawa et al., 1993; Saldanha et al., 2007; Lopes de Almeida et al., 2012).
RBC Hydration Changes Associated With Cytoskeletal Defects
Although reduced RBC deformability in HS mainly results from cytoskeletal disturbances (Perrotta et al., 2008), spherocytes experience dehydration and increased leakage of K+ (De Franceschi et al., 1997; Gallagher, 2017). To compensate for this increased K+ leakage, spherocytes are also found to have increased activity of the Na-K pump and the NKCC1 (also known as the Na-K-2Cl cotransporter, coded by gene SLC12A2) (Vives Corrons and Besson, 2001). RBCs of transgenic mice deficient for ankyrin or spectrin show increased activity of the Na-K pump, but normal activities of the NKCC1 and K+-Cl- cotransporters (Peters et al., 1999). This contrasts with protein 4.2-deficient mice, where Na-K pump activity is normal, while activities of NKCC1 and K+-Cl- cotransporters are increased (Peters et al., 1999). These differences in pump activities between protein 4.2 deficient and spectrin- or ankyrin-deficient mice support the concept that membrane proteins interact with ion channels of the RBC.
The RBC dehydration in HS also involves increased intracellular Ca2+ levels (Hertz et al., 2017). Increased intracellular Ca2+ correlates with decreased RBC deformability and RBC membrane stiffening (Bogdanova et al., 2013). For example, after in vitro ATP-depletion of HS RBCs, Ca2+ influx is increased compared to healthy RBCs (Shimoda et al., 1984). The detailed mechanisms beyond this Ca2+ influx have up to now not been identified and may include PIEZO1 or NMDA receptors both of which are activated by shear stress in “stiff” less deformable cells.
In protein 4.2-/- mice, displaying the HS phenotype, KCNN4 (or known as Gardos channels in RBCs) were noted to be functionally up-regulated, or at least more KCNN4 activity was measured (Peters et al., 1999; De Franceschi et al., 2005). This can be considered as a protective mechanism that leads to an outward flux of K+ and water from RBCs. This mechanism may compensate for the decreased surface-to-area ratio. Indeed, increased lysis was observed when these RBCs were exposed to KCNN4 (or Gardos-channel) inhibitors (De Franceschi et al., 2005). These findings indicate that KCNN4 is activated in HS in order to facilitate RBC dehydration, which maintains hemoglobin in RBCs at the expense of water. Recent research confirms the finding that RBC dehydration and formation of denser RBCs may protect the RBC in HS from lysis and premature uptake. We observed that mild HS is accompanied with prolonged RBC life-span, relatively mild reductions in RBC deformability and denser RBCs when compared with RBCs from patients with moderate/severe HS (Huisjes et al., 2017a). Most likely, the mild reduction in RBC deformability in patients with mild HS avoids premature splenic uptake and thus may provide the HS RBC time to lose membrane and to become dense.
Red Cell Metabolism
Primary Changes of Red Cell Metabolism
Mature RBCs of healthy individuals lack mitochondria and are therefore entirely dependent on the glycolytic pathway for the production of energy in the form of ATP. In the glycolytic pathway, glucose is converted to lactate by several enzymatic steps (Figure 3) (van Wijk and van Solinge, 2005; Koralkova et al., 2014). Key regulatory enzymes in the glycolytic pathway are HK, phosphofructokinase and PK. Hereditary metabolic disorders, or enzymopathies, are disorders impairing cellular energy production and balance, in particular ATP production. ATP is essential in the regulation of RBC deformability, viability, regulatory cascades involving phosphorylation and activity of ion transport involving Na, K-ATPase, and Ca ATPase (Weed et al., 1969; Tuvia et al., 1998; Martinov et al., 2000; Fischer et al., 2003; Park et al., 2010; Makhro et al., 2016).
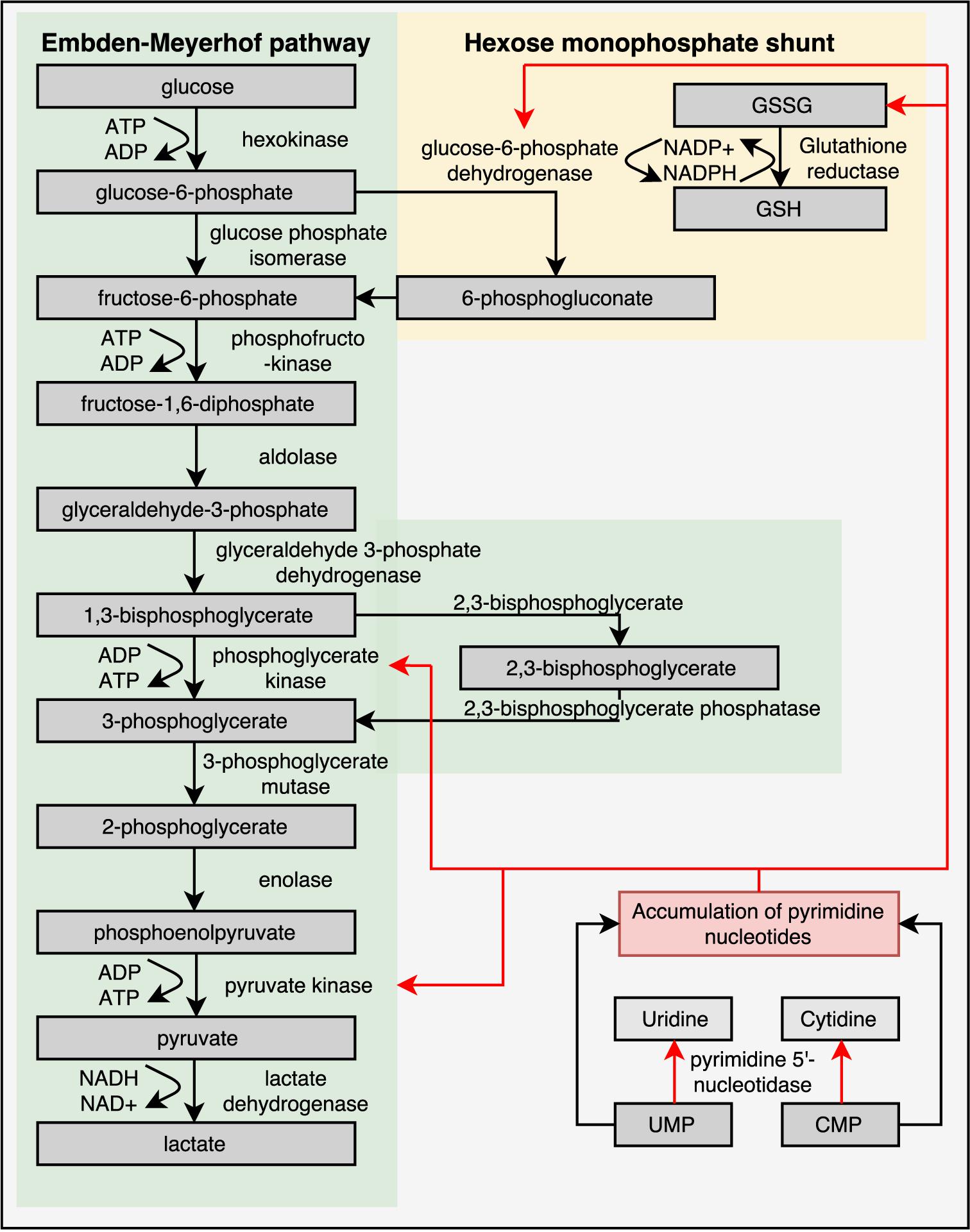
FIGURE 3. Glycolytic pathway of RBCs is essential for energy metabolism as RBCs lack mitochondria and a nucleus. Enzyme deficiencies can lead to decreased intracellular ATP levels, which possibly reduce RBC deformability. Depicted here are the Embden–Meyerhof pathway, hexose monophosphate shunt and the influence of a pyrimidine 5′-nucleotidase deficiency. Red arrows illustrate negative feedback or reduced production. This illustration is a merged figure adapted from both van Wijk and van Solinge (2005) and Zanella et al. (2006).
Adenosine triphosphate plays a key role in maintenance of ion gradients and should impact cytoskeletal structure and RBC shape (Tuvia et al., 1998; Park et al., 2010). However, glycolytic enzymopathies usually do not show altered RBC morphology (Koralkova et al., 2014; Huisjes et al., 2017b). For reasons that are not yet well understood, metabolically defective RBCs are prematurely removed by the spleen, causing chronic Hereditary Non-Spherocytic Hemolytic Anemia (HNSHA) (Koralkova et al., 2014).
Several observations point to mechanisms that may be involved in the premature clearance. RBCs from a patient with pyruvate kinase deficiency (PKD) were found to leak K+ more rapidly and were found to consume ATP at an accelerated rate (Nathan et al., 1965). This most likely contributes to the decreased RBC viability in this disease and could result in reduced RBC deformability (Leblond et al., 1978). A possible explanation for increased clearance of RBCs with metabolic defects is the imbalance in Ca2+ uptake and extrusion (Bogdanova et al., 2013; Hertz et al., 2017). In agreement with the hypothesis that ATP deprivation will cause Ca2+ overload due to the inability to actively extrude Ca2+ from the cells by the Ca2+ pumps, decreased ATP levels in RBCs with metabolic defects were associated with abnormally high intracellular Ca2+ and exposure of PS, an important cellular ‘eat-me’ signal (de Back et al., 2014). Alternatively, defective metabolism may impact the redox state of RBCs making them more susceptible to oxidative stress (Cappellini and Fiorelli, 2008).
Despite the intuitive feeling that intracellular ATP levels should correlate with cellular deformability, evidence for this is so far inconclusive. This may be related to the techniques used. For example, research performed by Karger et al. (2012) did not show any correlation between intracellular ATP and deformability in RBC-concentrates intended for transfusion when assessing deformability by increasing shear stress. On the other hand, studies using viscosity and filterability assays did find correlations between intracellular ATP and deformability and showed that deformability is indeed dependent on intracellular ATP levels (Weed et al., 1969; Fischer et al., 2003). Atomic force microscopy studies also revealed increased membrane stiffness after ATP depletion due to reduced spectrin phosphorylation (Picas et al., 2013). We hypothesize that the different results from these experiments are possibly caused by the fact that ATP is not required for RBCs to elongate after the application of external forces, such as shear stress but ATP might be essential in the process of RBC deformation toward different shapes, as seen in filterability measurements.
Secondary Changes of Metabolism
Cyclic AMP (cAMP) is a second messenger and is derived from ATP. In nucleated human cells, cyclic AMP (cAMP) is synthesized from ATP by adenylate cyclase (AC) upon stimulation by G-protein coupled receptors (GPCR) and there is evidence that this process also occurs in human RBCs (Oonishi et al., 1997; Tuvia et al., 1999; Sprague et al., 2006; Kostova et al., 2015) (see Figure 4). Examples of GPCRs on RBCs are the erythrocyte β2-adrenergic receptor (Harrison et al., 2003), the lysophosphatidic acid (LPA) receptor (Wang et al., 2013) and the purinergic (P2Y) receptor (Kostova et al., 2015). The activation of the erythrocyte β2-adrenergic receptor by catecholamines such as epinephrine (or adrenaline) affects RBC deformability (Tuvia et al., 1999). In vitro experiments with increased concentration of adrenaline, lead to increased membrane fluctuations and increased RBC filterability (Tuvia et al., 1999).
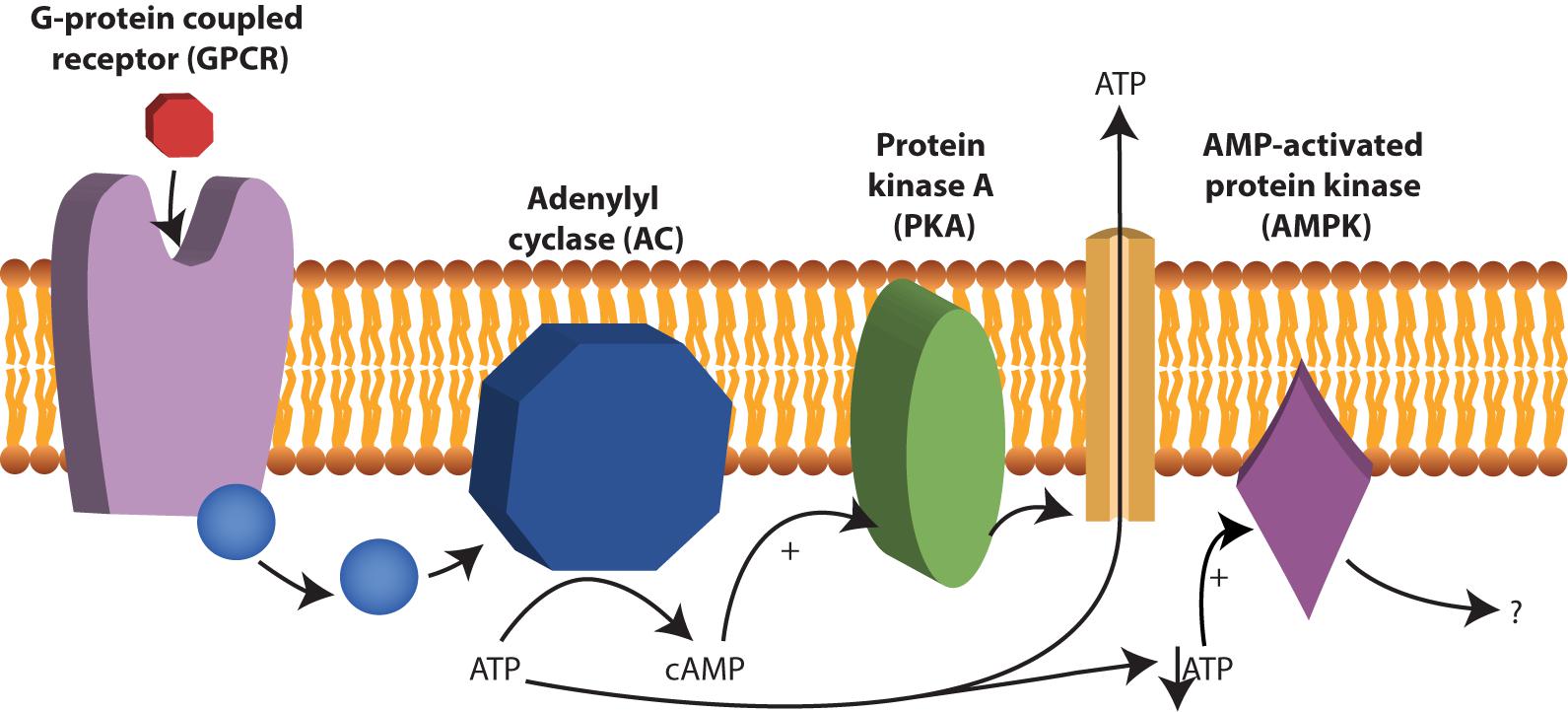
FIGURE 4. cAMP production by adenylyl cyclase (AC). AC is activated after stimulation by G-protein coupled receptors (GPCR) and converts ATP to cAMP. Examples of GPCRs 2-adrenergic receptor, the lysophosphatidic β in RBCs are the erythrocyte acid (LPA) receptor and the purinergic (P2Y) receptor. cAMP acts as a second messenger in RBCs and among other things stimulate protein kinase A (PKA). Under low ATP concentrations, AMP-activated protein kinase (AMPK) becomes active and can inactivate or activate yet not fully understood processes in RBCs.
Intracellular mechanisms that sense and control cellular energy status in RBCs
Phosphodiesterases tightly regulate levels of cAMP in RBCs (Adderley et al., 2011). PDEs catabolize the phosphodiesters in cyclic nucleotides, such as cAMP to AMP or cGMP to GMP. By inhibiting these PDEs, cyclic nucleotides remain intact and active. The reports of cAMP effects on RBC deformability are, however, ambivalent. Recently it was found that sildenafil, which is a PDE inhibitor and well-known for its function in the treatment of erectile dysfunction and pulmonary hypertension, has beneficial effects on RBC deformability in sickle cell disease at low concentrations in vitro. Increase in nitric monoxide bioavailability was suggested to improve RBC deformability (Grau et al., 2013; Kuhn et al., 2017). However, increased concentrations of sildenafil impaired sickle RBC filterability (Hagley et al., 2011). Typically, inhibition of cGMP-specific PDE type 5 (PDE-5) by sildenafil reduces the degradation of the second messenger cGMP to GMP and increases smooth muscles relaxation. In RBCs, this PDE-5 inhibition increases not only intracellular levels of cGMP but also preserves intracellular levels of cAMP via the inhibitor activity of cGMP on PDE-3 (see Figure 5) (Adderley et al., 2010; Knebel et al., 2013). The effect of increased cAMP levels was also studied using a spleen-like microfiltration system. Increased retention in the microfiltration system, reflecting decreased deformability, was observed after incubation of RBCs with sildenafil (Ramdani et al., 2015). Moreover, the production of cGMP from GTP is synthesized by guanylate cyclase (Derbyshire and Marletta, 2012). Guanylate cyclase is activated by nitric oxide (NO) and the addition of sodium nitroprusside to RBCs as an artificial nitric oxide donor does prevent Ca2+ influx and Ca2+-mediated stiffness of RBCs (Barodka et al., 2014).
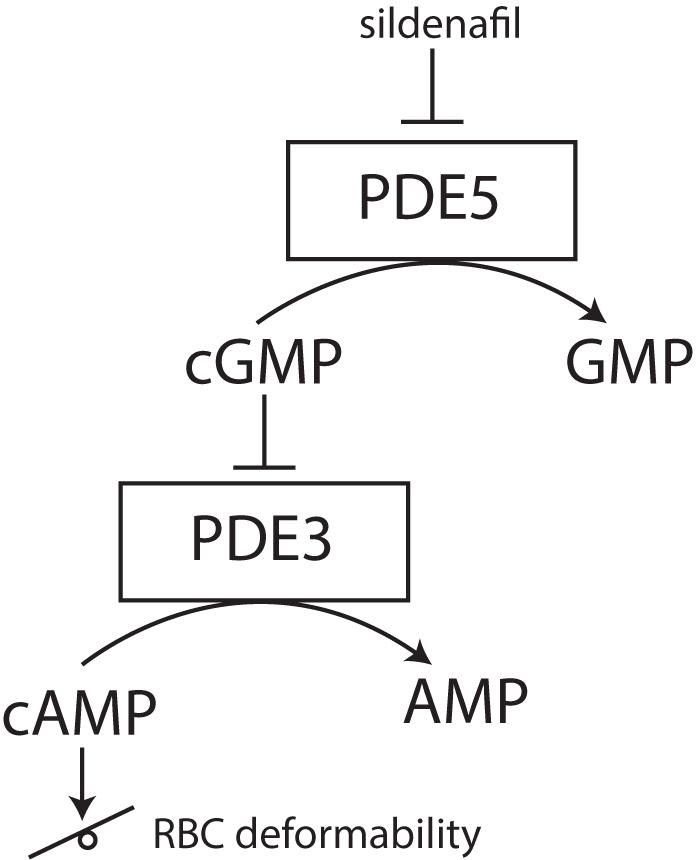
FIGURE 5. effect of PDE-5 inhibitors, such as sildenafil, on RBC deformability. PDE-5 inhibitors elevate intracellular concentrations of cGMP, thereby indirectly inhibiting PDE3. Inhibition of PDE3 does raise the intracellular concentration of cAMP. Depending on the degree of this intracellular cAMP elevation, RBC deformability can be either increased or decreased.
In addition, the effect of cAMP on the release of ATP is still under debate. A role for cAMP in the release of ATP to its extracellular environment via PKA was proposed (Sprague et al., 2007; Adderley et al., 2011; Knebel et al., 2013) (see Figure 4). Others, however, have attributed the release of ATP solely to hemolysis of the RBC (Sikora et al., 2014) or to release via transporters such as Pannexin 1 (Locovei et al., 2006) or PIEZO1 (Cinar et al., 2015).
Recently AMPK was also found to be present in RBCs (Thali et al., 2010). The exact function of AMPK in RBCs is unknown, however, in other cells, AMPK is essential to balance ATP consumption and ATP production. In muscles and fat AMPK regulates glucose uptake (Mihaylova and Shaw, 2011). Likely, AMPK also regulates these processes in RBCs, as RBCs from AMPK-/- mice were less viable after in vitro glucose depletion (Föller et al., 2009). Interestingly, these AMPK-/- mice had increased reticulocytes count, decreased RBC deformability, increased osmotic fragility, increased spleen size and decreased RBC lifespan (Foretz et al., 2010; Wang et al., 2010). This indicates a significant contribution of AMPK in maintaining RBC function and viability. AMPK is an energy-sensing enzyme, which is activated by low AMP: ATP ratios and inhibited by high AMP: ATP ratios (Wang et al., 2003). Under low ATP concentrations, AMP and ADP can bind to AMPK and change its conformation, thereby preventing the protein from activation by phosphorylation (Mihaylova and Shaw, 2011). Chemical agents that increase intracellular levels of cAMP are forskolin and isobutyl-methyl-xanthine (IBMX). Forskolin activates AC and thus leads to increased formation cAMP from ATP, while IBMX inhibits PDEs and maintains cAMP levels. Both forskolin and IBMX lead to AMPK inhibition, which reveals a molecular interaction between cAMP and AMPK activity (Hurley et al., 2006). This interaction could play a role in altered deformability in patients suffering from glycolytic enzymopathies. The low RBC ATP concentrations in these patients would lead to low levels of cAMP and increased intracellular levels of non-phosphorylated (inactive) AMPK.
Phosphorylation of AMPK may also be involved in G6PD-deficiency. G6PD deficiency is the most common RBC enzyme deficiency with over 400 million affected people (Cappellini and Fiorelli, 2008). Deficiency of this enzyme is associated with decreased ability of the RBC to withstand oxidative stress (Cappellini and Fiorelli, 2008; Mangat et al., 2014; Tang et al., 2015). Exposure of normal and G6PD-deficient RBCs to the oxidant diamide leads to a decrease in RBC deformability of the G6PD-deficient RBCs due to the inability of the RBC to maintain the intracellular glutathione-pool (Tang et al., 2015). Depletion of glutathione also leads to activation of AMPK by phosphorylation in G6PD-deficient RBCs. The activation of AMPK in G6PD-deficient RBCs after exposure to oxidative stress is most likely a consequence of ATP-consuming compensation mechanisms after excessive glutathione loss (Tang et al., 2015).
Protein kinase C alpha (PKCα) possibly also regulates RBC deformability. PKCα is involved in the phosphorylation of proteins in the RBC, such as adducin, protein 4.1R and protein 4.9 (George et al., 2010). Moreover, PKCα is involved in stimulation of glucose uptake by phosphorylation of the glucose transporter and participates in the activity of calcium channels (Kanno et al., 1995; George et al., 2010; Wagner-Britz et al., 2013). Consequently, phosphorylation and dephosphorylation of these proteins can be altered upon depletion of glucose. Glucose depletion was found to activate PKCα and this is accompanied by an increase in intracellular Ca2+ and, consequently, exposure of the membrane phospholipid PS (Klarl et al., 2006). The phosphorylation of adducin and protein 4.1R by PKCα could change RBC behavior and could influence RBC deformability (George et al., 2010) in a way similar to what is seen, for example, after phosphorylation of band 3 (Saldanha et al., 2007). Interestingly, homology analysis between murine, rat and human showed a conserved region in both liver- and RBC-specific PK for a PKCα binding site (Kanno et al., 1995). PKCα regulates RBC membrane stability and this conserved region for PK indicates a possible role for PK in this process (Kanno et al., 1995). In addition, PK converts phosphoenolpyruvate (PEP) to pyruvate (Figure 3) and previously it has been recognized that phosphorylation of PK by cAMP-dependent protein kinases leads to a reduced affinity of PEP for PK, which leads to a reduced production of ATP (Marie et al., 1980). The decreased cAMP and ATP production in metabolic disorders of the RBC, such as PK-deficiency, could subsequently result in disturbed RBC deformability (Leblond et al., 1978).
Role of purines and pyrimidines in RBC deformability
It has been suggested that pools of ATP are intracellular enclosed by the membrane proteins ankyrin, β-spectrin, band 3, and GAPDH, serving as substrates for both the Na+, K+-, and Ca2+ -pumps (Chu et al., 2012). This indicates an interplay between membrane proteins, energy stores and ion channel function of the RBC. Interestingly, the chemotherapeutic agent 5-FU was shown to induce changes in RBC rigidity, morphology and ion balance, most likely by altering ATP levels in the RBC (Spasojević et al., 2005). 5-FU is a pyrimidine antagonist (Longley et al., 2003) and since purine and pyrimidines play key roles in cellular metabolism and energy homeostasis. 5-FU could, therefore, influence RBC deformability. The activating and feedback mechanisms of purines and pyrimidines on glycolytic enzymes have previously been described by Seider and Kim (1979) and Tomoda et al. (1982). For example, in bovine RBCs incubated with glucose, adenosine stimulated ATP production compared to incubation with glucose alone. Most likely, adenine (i.e., the nucleobase of adenosine) stimulates HK activity (Seider and Kim, 1979). The effect of pyrimidines in RBCs has also been studied in two patients with a pyrimidine 5′-nucleotidase (P5N) deficiency, which lack this enzyme involved in clearance of pyrimidines from the RBC. P5N deficiency increases intracellular concentrations of pyrimidine nucleotides, which eventually leads to hemolytic anemia (Vives L Corrons, 2000). P5N-deficient patients were also found to have increased levels of reduced glutathione (Tomoda et al., 1982). At the same time, pyrimidine 5′-nucleotides (such as cytidine mono-, di-, and triphosphate (CMP, CDP, CTP) or uridine mono-, di-, and triphosphate (UMP, UDP, UTP)) decreased the activity of glucose-6-phosphate dehydrogenase (G6PD, Figure 3). As mentioned earlier G6PD activity is crucial in the anti-oxidative defense of RBCs. Indeed, RBCs from P5N-deficient patients were found to be more susceptible to oxidative stress, as reflected by increased formation of Heinz bodies (Tomoda et al., 1982), even despite increased reduced glutathione concentrations. Besides their role as a morphological marker of oxidative stress, Heinz bodies can lead to a decreased deformability by themselves (Hasegawa et al., 1993).
Hemoglobin
Primary Changes of Hemoglobin
Hemoglobin is the main component of the RBC and responsible for the delivery and removal of oxygen and carbon dioxide to and from the tissues, respectively. Intracellular hemoglobin concentrations and its state (polymerization, crystallization, degradation, and oxidation) also defines cytosolic viscosity making up 19.9–22.3 mmol/L in cells of healthy humans. Hemoglobin is composed of two α- and β-hemoglobin molecules together composing a heterotetramer. Disorders of hemoglobin can be subdivided into hemoglobinopathies (e.g., sickle cell anemia) and thalassemias (α- and β-thalassemia) (Galanello and Origa, 2010; Harteveld and Higgs, 2010; Higgs et al., 2012; Ware et al., 2017).
Sickle cell anemia is caused by a single point mutation in the HBB gene at position 6 substituting glutamic acid to valine (HbS). This substitution causes formation of HbS polymers of deoxygenated hemoglobin. This transition is usually rapidly reversed upon reoxygenation, but induces progressive damage of membrane driving dehydration to the extreme to the state when HbS polymers do not dissociate as its concentration exceeds it solubility threshold. Destabilization of the membrane and decrease in deformability leads to intravascular hemolysis and to vaso-occlusive events (Ware et al., 2017). Sickle RBCs are poorly deformable (Alapan et al., 2015, 2016) and this is, in part, due to changes in hydration status of the RBC.
In α- and β-thalassemia, α- and β-hemoglobin chains are affected, respectively, which leads to an imbalance in the synthesis of globin chains and to an inability to form sufficient quantities of hemoglobin heterotetramers. The imbalanced synthesis in thalassemia leads to the formation of hemoglobin precipitates, so-called Heinz bodies. RBCs in α-thalassemia show increased hydration (Bunyaratvej et al., 1994; Chui et al., 2003), whereas the hydration of RBCs in β-thalassemia is either decreased or increased (Bunyaratvej et al., 1994; Brugnara and Mohandas, 2013). Both Heinz bodies and the altered hydration state of the RBC are known to impair RBC deformability (Clark et al., 1983; Losco et al., 2001).
Oxygenation of Hemoglobin
The main function of RBCs is to transport oxygen to the tissues in the human body. The oxygenation state of hemoglobin is known to influence various processes of the RBC (Gibson et al., 2000; Stefanovic et al., 2013). Several ion transporters are oxygen sensitive, such as K+-Cl- cotransporter (KCC1) and the NKCC1 (Na-K-2Cl cotransporter) (Bogdanova et al., 2009). The function of band 3 seems, however, unaltered upon deoxygenation and oxygenation (Gibson et al., 2000). On the other hand, hemoglobin binds the cytosolic domain of band 3 and this binding is regulated by pH (Eisinger et al., 1982; Chétrite and Cassoly, 1985). Deoxygenation reduces the binding of ankyrin to band 3 and dissociation leads to more freely diffusible band 3 (Stefanovic et al., 2013). In addition, the binding between ankyrin and band 3 is also regulated by 2,3-disphosphoglycerate (2,3-DPG) (Moriyama et al., 1993). 2,3-DPG promotes the dissociation of oxygen from hemoglobin and promotes the release of oxygen from the RBC to the tissues (van Wijk and van Solinge, 2005). In the oxygenated state 2,3-DPG is not bound to hemoglobin and leads to less deformable and more fragile RBCs, while in the deoxygenated state 2,3-DPG is bound to hemoglobin and leads to an increase in RBC deformability and less fragile RBCs (Moriyama et al., 1993). The exact role of the reduced binding between ankyrin and band 3 after deoxygenation and binding of 2,3-DPG to hemoglobin is currently unknown, although it is hypothesized that a mild membrane weakening is beneficial in deoxygenated capillaries to facilitate smooth traveling of the RBC (Stefanovic et al., 2013).
RBC Hydration Changes Associated With Hemoglobin Defects
RBC dehydration in sickle cell anemia
The RBC dehydration in sickle cell anemia is caused by facilitated K+ loss through hyperactivated KCNN4 (also known as the Gardos Channel in RBCs) and K+-Cl- cotransporter that is not compensated by uptake of equal amounts of Na+ (Clark et al., 1982; Izumo et al., 1987). Regardless of the ion transport pathway involved, RBC dehydration raises the HbS concentration, thereby affecting the equilibrium of HbS-polymerization and depolymerization in favor of the polymerized version (Eaton and Hofrichter, 1990; Brugnara, 1993). Na/K/ATPase was found to be more active in RBCs from sickle cell disease patients and can contribute to RBC dehydration (Izumo et al., 1987). Dehydration of RBCs in sickle cell disease contributes significantly to the decreased deformability since dehydration promotes the probability of polymerization of HbS by 20–40-fold (Eaton and Hofrichter, 1990) and result in prolonged polymerization of hemoglobin and thus prolonged sickling of RBCs (Brugnara, 1993). Sickled RBCs are far less deformable.
Two mechanisms involved in abnormally high K+ loss are presented in Figure 6. The first mechanism of K+ loss involves the ubiquitously expressed K-Cl co-transporter (KCC), of which the isoforms KCC1, KCC3, and KCC4 are expressed in human RBCs (Crable et al., 2005).
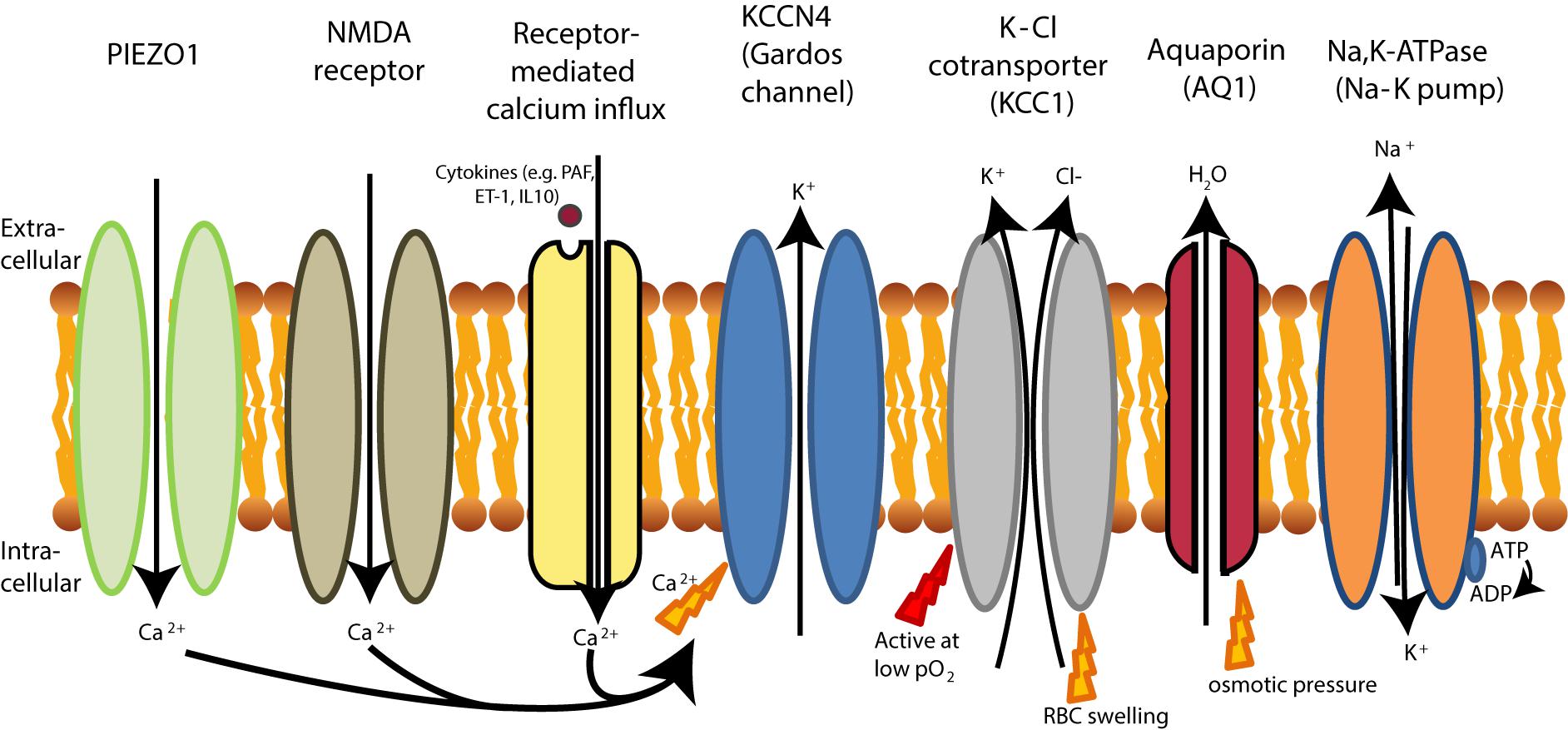
FIGURE 6. RBC dehydration in sickle cell disease. The Na-K pump (Na, K-ATPase) is more active in sickle RBCs, leading to dehydration as extrusion of 3 Na+ ions leads to the influx of 2 K+ ions. Dehydration of sickle cell is initiated by PIEZO1 activation, likely because PIEZO1 is stretch-activation after deoxygenation (Ma et al., 2012). Dehydration in sickle cell disease is also initiated by the upregulation of the NMDA receptor. Both the NMDA receptor and PIEZO1 activation result in Ca2+ influx that leads to KCNN4 activation (KCCN4 in RBCs is better known as the Gardos channel). Gardos channel activation in sickle cell disease is also achieved by signaling cascades which respond to increased levels of cytokines. This leads to K+ and water efflux by aquaporins (AQ1). In sickle cell disease, the K-Cl cotransporter is also activated at low oxygen tensions, causing efflux of K+ and subsequently loss of water by aquaporins (AQ1), while under normal conditions KCC is only active at normal pO2. Although water can be cotransported through KCC1 (Zeuthen and Macaulay, 2012), water efflux in RBCs mainly driven by aquaporins (AQ1).
KCC1, is most often studied in RBCs and referred to as KCC. KCC in RBCs are known to be regulated by intracellular pH, Mg2+ concentrations as well as volume and oxygenation states of hemoglobin (Adragna et al., 2004; Khan et al., 2004). Under physiological conditions, KCC activation restores cell volume after swelling, but KCC activation may also respond to the changes in the RBC redox state (Adragna et al., 2004). In healthy RBCs, KCC is oxygen sensitive but only activated at high oxygen tension (pO2). At low oxygen tension, KCC becomes sensitive to other stimuli (Gibson et al., 1998, 2001; Muzyamba et al., 2006). In sickle cell disease, in contrast, KCC is also activated at low oxygen tension, most likely due to increased phosphorylation of KCC in sickle RBCs (Muzyamba et al., 2006). In addition, KCC activity is intrinsically higher, even in older sickle RBCs (Bize et al., 2003).
The loss of deformability caused by the formation of HbS-polymers at low oxygen tensions (Odièvre et al., 2011) is enhanced by the KCC activation and leads to additional cell shrinkage. The clinical relevance of KCC activity is emphasized by observations in individuals with mild sickle cell disease. In these patients, an increased K-Cl cotransport activity is associated with increased likelihood of hospitalization, because of acute vaso-occlusive problems caused by less deformable dehydrated RBCs (Rees et al., 2015).
One more trigger of K+ leak from RBCs is the increase in the intracellular Ca2+ levels and the activation of Ca2+-sensitive K+ channels (KCNN4 or known as the Gardos channel in RBCs) (Gallagher, 2017). These channels driving K+ loss and dehydration are hyperactivated in RBCs of sickle cell disease patients secondary to the high intracellular Ca2+ levels (Bookchin and Lew, 1980; Bogdanova et al., 2013) (see Figure 6). KCNN4 is expressed in various cell types (Gallagher, 2013) and are inhibited by imidazole antimycotics, such as clotrimazole (McNaughton-Smith et al., 2008). In sickle cell disease, KCNN4 (or known as the Gardos channel in RBCs) is activated by two pathways. Firstly, KCNN4 can be activated by a signaling cascade initially triggered by several cytokines, such as PAF, interleukin-10 (IL-10) and endothelin 1 (ET-1) (Rivera, 2002) also involving activation of PKCα (Rivera et al., 1999; Wagner-Britz et al., 2013). In addition, PAF and ET-1 concentrations are increased in plasma from patients with sickle cell disease and are assumed to contribute to the adhesion of sickle RBCs to endothelium that is responsible for the vaso-occlusive events (Rivera, 2002). Also, these cytokines generate denser RBCs through RBC dehydration after an oxygenation/deoxygenation cycle (Rivera, 2002). Secondly, sickle cell disease patients have increased expression of the NMDA-receptor on their RBC membranes, causing pathological influx of Ca2+ after stimulation with receptor agonists (such as glycine and homocysteic acid) (Hänggi et al., 2014) which could contribute to dehydration mediated by KCNN4. Currently, a clinical trial using memantine as a NMDA-receptor antagonist is being tested in sickle cell disease patients (Bogdanova et al., 2017). In addition, the KCNN4 (or Gardos channel) blocker senicapoc (ICA-17043) has recently been tested in sickle cell disease. Senicapoc increased hemoglobin concentrations and hematocrit, and improved RBC hydration. Despite these encouraging findings, this study has been terminated prematurely because senicapoc did not meet its primary efficacy endpoint, defined as a decrease in painful crises (Ataga et al., 2011). From the clinical results obtained with senicapoc it can be hypothesized that the RBC dehydration and changes in RBC hydration after treatment with senicapoc are not causally related to the vaso-occlusive events which are often observed in sickle cell anemia.
Another pathway that increases intracellular Ca2+ levels in sickle cell disease is the mechanosensitive PIEZO1 channel (also described at section “Primary Changes of RBC Hydration”). PIEZO1 channels are widely expressed in vertebrates across various cell types and in RBC these channels are responsible for regulation of volume homeostasis upon mechanical signals (Zarychanski et al., 2012; Ge et al., 2015). Sickle RBCs show increased permeability for Ca2+ and Mg2+ upon deoxygenation. This increased permeability results in an increased deoxygenation-induced cation conductance in sickle cell disease, which can be entirely blocked by GsMTx4 (Ma et al., 2012; Cahalan et al., 2015). The gating modifier GsMTx4 blocks the mechanically sensitive part of PIEZO1 that supports a closed state of this ion channel (Bae et al., 2011), preventing Ca2+ influx (Jacques-Fricke et al., 2006).
RBC hydration changes in thalassemia
Three increasingly severe phenotypes can be distinguished in β-thalassemia, i.e., β-thalassemia minor, intermedia and major (Higgs et al., 2012). Intracellular concentrations of Na+ in RBCs from patients with β-thalassemia minor and β-thalassemia major are comparable with those in RBCs from healthy adults, but K+ levels are slightly increased (Cividalli et al., 1971). Upon in vitro incubation at 37°C, RBCs from patients with β-thalassemia major show increased leakage of K+ and elevation of intracellular Na+ levels when compared with healthy RBCs. In RBCs from β-thalassemia minor patients, leakage of K+ and intracellular Na+ levels are comparable to the healthy RBCs after in vitro incubation at 37°C (Cividalli et al., 1971).
Leakage of K+ from RBCs in thalassemia is possibly linked to the precipitation of excess α- or β-globin chains inside the RBCs, which is a characteristic feature of this disease. The precipitated globin chains in RBCs can lead to oxidative stress and can affect transport of Na+ and K+ in thalassemia RBCs (Olivieri et al., 1994). In both β-thalassemia and α-thalassemia, RBCs show increased efflux of K+ (Olivieri et al., 1994). Moreover, the amount of hemoglobin aggregates in RBCs is correlated with K+ efflux (Nathan and Gunn, 1966). The presence of these aggregates reduces RBC deformability (Lubin and Desforges, 1972; Hasegawa et al., 1993) thereby revealing correlation between K+ fluxes, Heinz body formation, and reduced RBC deformability. The precipitation of globin chains and leakage of K+ from RBCs in thalassemia is also linked, or may even be augmented, by overload of Ca2+ (Shalev et al., 1984; Bookchin et al., 1988).
Another important factor in K+ loss from RBCs in β-thalassemia involves the K+-Cl- cotransporter, which can be affected by oxidative stress (Olivieri et al., 1994). This K+ loss by the K+-Cl- cotransporter is inhibited by increased intracellular concentrations of Mg2+ (De Franceschi et al., 1998; Adragna et al., 2004). In a trial with β-thalassemia intermedia patients, dietary Mg2+ supplementation did lead to increased intracellular concentrations of Mg2+ and to a reduction in the activities for the K+-Cl- cotransporter and Na-K pump (Na/K/ATPase). However, Mg2+ did not influence the activities of NKCC1 (also known as the Na-K-Cl cotransporter). Although dietary Mg2+ supplementation in β-thalassemia patients does not influence the hemoglobin concentration in blood, it does lead to a significant decrease in the absolute reticulocyte number. This is possibly due to improved RBC survival time (De Franceschi et al., 1998).
Little research has been performed on KCNN4 (or also known as the Gardos channel in RBCs) in thalassemia. An exception is the work on a mouse model with a homozygous deletion in the β-globin chain (de Franceschi et al., 1996). These mice were treated with clotrimazole, an antifungal imidazole derivate with KCNN4 blocking properties. During treatment with clotrimazole, hemoglobin concentrations in blood remained constant. In addition, it caused a decrease in MCHC whereas hematocrit and intracellular K+ levels increased indicating that hydration state of the RBC was restored. Combination of clotrimazole with erythropoietin administration did increase hemoglobin levels in these mice to a higher extent than erythropoietin alone, possibly by promoting proliferation and differentiation during erythropoiesis (de Franceschi et al., 1996).
RBC Vesiculation
Intracellular levels of Ca2+ tightly regulate RBC vesiculation and elevated levels of intracellular Ca2+ are known to induce PS exposure and RBC vesiculation (Bevers et al., 1992; Nguyen et al., 2011; Fens et al., 2012; Alaarg et al., 2013) (Figure 7).
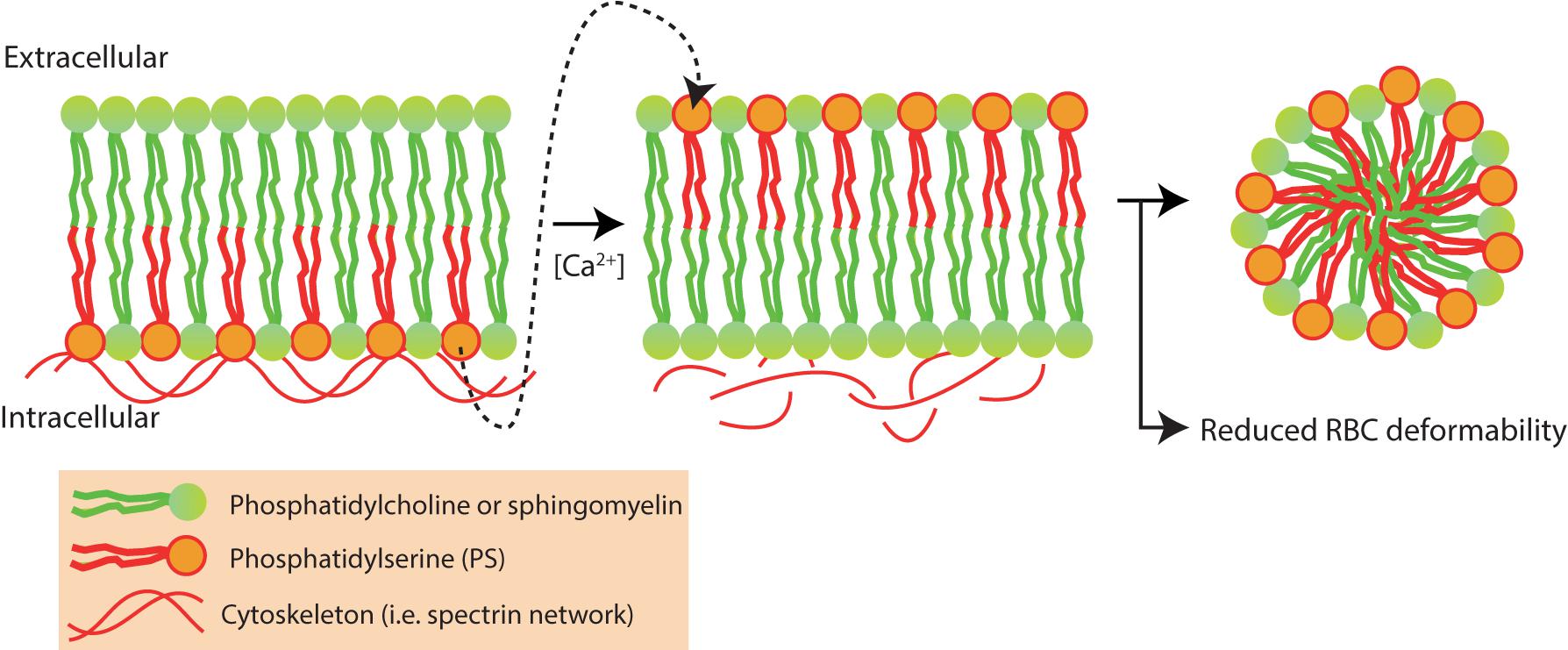
FIGURE 7. Red blood cell vesiculation is driven by increased concentrations of intracellular Ca2+. Phosphatidylserine (PS) is normally presented in the inner-leaflet of the RBC. Elevated intracellular Ca2+ levels activate scramblase and inhibit flippase, leading to phosphatidylserine (PS) externalization and redistribution of membrane and cytoskeletal proteins. PS externalization and protein redistribution leads to vesicle formation and loss of RBC deformability (Alaarg et al., 2013).
The RBC-vesiculation is a physiological process and leads to reduced RBC deformability. During its 120-day life span, RBC lose membrane surface and hemoglobin content through vesiculation (Werre et al., 2004; Ciana et al., 2017) a process in particular prominent in the youngest RBCs (Greenwalt and Dumaswala, 1988). As membrane is shed, this leads to a decreased surface-to-volume ratio in the RBC. During this RBC aging, hemoglobin-free vesicles are shed which leads to RBC dehydration and older RBCs with increased MCHC (Bosch et al., 1994; Bosman, 2013). Altogether, this leads to a reduction in membrane elasticity and RBC deformability (Linderkamp et al., 1993; Bosch et al., 1994). This implies that there is an important role for vesiculation in RBC deformability.
While RBC-vesiculation is considered a physiological process, RBC vesiculation is increased in various forms of hereditary hemolytic anemia, such as HS and sickle cell disease (Perrotta et al., 2008; Alaarg et al., 2013). In HS, differences in deformability between spectrin/ankyrin-deficient and band 3-deficient RBCs were observed after splenectomy. While splenectomy prevented premature removal of young RBCs from the circulation in both groups, the loss of deformability during RBC-aging was delayed after splenectomy in spectrin/ankyrin-deficient RBCs. This contrasts with band 3-deficient RBCs, where splenectomy did not lead to a delay in the deformability decrease during RBC-aging. This could be explained by the fact that spectrin/ankyrin-deficient RBCs are more prone to shed band 3-containing vesicles. Clustered band 3 is known to induce binding of autologous IgG, which facilitates removal of RBC by macrophages (Kay et al., 1983; Kay, 1984; Willekens et al., 2008). Shedding of band 3-containing vesicles from spectrin/ankyrin-deficient RBCs, therefore, would avoid IgG-opsonisation and clearance of the RBC (Reliene et al., 2002).
Red blood cell vesiculation is suggested as a mechanism to protect the cell from removal from the circulation. By vesiculation, the RBC can shed “eat-me” signals such as PS and specific band 3 cleavage products that react with senescent antigens (Willekens et al., 2008). By shedding these “eat-me” signals, the RBC may escape clearance until the reduction in RBC deformability causes trapping in the spleen. In addition, the lipid bilayer of the RBC is a complex system with various microdomains. These microdomains of the RBC, or so called ‘lipid rafts’ are specific membrane parts with high concentrations of cholesterol, sphingomyelin and gangliosides (Pike, 2003). These specific lipid rafts have a high abundance of the membrane proteins stomatin, flotillin-1, flottilin-2 (Salzer and Prohaska, 2001) and band 3 (Cai et al., 2012). RBC vesicles shed during storage are enriched in the lipid raft marker stomatin (Salzer et al., 2008) and indicates that specific lipid domains are shed during RBC vesiculation (Leonard et al., 2017).
The relationship between vesiculation and deformability is nicely illustrated by an experiment where chlorpromazine is added to RBCs to stabilize the RBC membrane. During overnight incubation with chlorpromazine at 37°C in a glucose-free buffer, vesiculation is inhibited and RBC deformability is maintained. Without chlorpromazine strong vesiculation of RBCs under these conditions occur as PS is exposed on the RBC surface. The activity of chlorpromazine may be explained by the amphiphilic properties of the molecule directly affecting the RBC membrane (Bütikofer et al., 1989).
High-throughput screening with chemical compound libraries also revealed that RBC-vesiculation can be driven by drugs and chemical compounds. For example, vesiculation of RBCs is driven by certain kinase pathways, including Jak-STAT and protein kinase B. Increased RBC vesiculation is also observed after addition of paclitaxel to blood, probably through its formulation excipient containing Cremophor. Not only RBC vesiculation is influenced by the vehicle Cremophor, but the compound also increases whole blood viscosity and transforms RBC to stomatocytes (Vader et al., 2013).
Adaptive Responses
Exercise leads to increased heart rate as a response to provide tissues with sufficient amount of oxygen. Exercise is accompanied with several stress factors that may affect RBC deformability, such as shear stress, hyperthermia, and glucose consumption (Carlson and Mawdsley, 1986; Szygula, 1990; Smith, 1995; Mairbäurl, 2013). These stress factors can lead to mechanical rupture, stimulated erythropoiesis and can decrease the average RBC age in athletes (Mairbäurl, 2013).
In a study with 24 trained cyclists, RBC deformability was decreased directly after exercise (Oostenbrug et al., 1997). The biology beyond the decrease in RBC deformability is unknown, although increased blood flow may be involved. The change in deformability after exercise can be the results of cytoskeletal changes, such as membrane loss. In addition, decreased levels of haptoglobin and increased levels of bilirubin were observed after marathon races (Jordan et al., 1998; Simpson et al., 2006). On the other hand, RBC deformability seems to be generally increased in well-trained athletes (Mairbäurl, 2013). A study comparing RBC membrane fluidity with the RBC membrane composition in controls and runners observed increased RBC membrane fluidity in endurance runners and sprinters. Although the intake of nutrients was comparable between the running groups and the control group, endurance running was accompanied with reduced concentrations of saturated fatty acids in RBC membranes (Kamada et al., 1993). These results indicate that the RBC can adapt upon exposure to exercise and that these adapted RBCs facilitate exercise and proper delivery of oxygen to the tissues. Interestingly, Smith et al. (1999) observed a higher maximal RBC deformability in elite cyclists when compared with sedentary healthy controls. Also, RBC populations with lower RBC densities were observed in the elite cyclists, which could indicate increased RBC turnover (Smith et al., 1999). A possible explanation for the increased RBC deformability in elite cyclist could be that extreme exercise leads to membrane loss and early RBC uptake, which is likely to be compensated with relatively young and good deformable RBCs. Whether increased RBC deformability would lead to better sports performances is unknown.
Autologous erythropoietin production is stimulated upon exposure to high altitudes. Acute exposure to high altitude, however, does not affect RBC deformability and RBC rheology (Reinhart et al., 1991). The effects of chronic exposure to high altitude seems largely unexplored. RBC deformability related processes were investigated in RBC concentrates obtained from Tibetans living at high altitude and from Tibetans at living at lowland. RBC viscosity was increased, and RBCs were more osmotic fragile in Tibetians living at low altitude when compared to RBC concentrates obtained from their lowland residents (Zhong et al., 2015). The increased viscosity can, however, be affected by increased hemoglobin levels and the decreased osmotic fragility can be affected by the decreased MCV values in Tibetans living at high altitude. The effects of exercise under hypoxia on RBC properties were investigated by Mao et al. (2011). Exercise under hypoxic conditions was found to decrease KCNN4 (or Gardos-channel) modulated deformability and KCNN4 modulated volume and down-regulated the senescence markers CD47 and CD147 (Mao et al., 2011). These results indicate RBC dysfunction after exercise under hypoxia. The dysfunction of RBCs under hypoxic conditions in combination with the erythropoietin-driven increase in RBC production leads to RBCs with relative low densities (Samaja et al., 1993), possibly caused by both increased RBC turnover and increased RBC production.
Summary
Deformability is an important parameter that regulates RBC rheology, its longevity, and efficacy of O2 transport. Altered deformability is a characteristic feature of multiple forms of hereditary hemolytic anemias and is likely related to the severity of the disease. Factors regulating deformability at the cellular level are dehydration, membrane protein phosphorylation, cytoskeletal integrity, metabolism and the integrity of hemoglobin. Interaction of these factors makes RBCs more or less deformable. Measuring RBC deformability in research and diagnostic laboratories can be challenging as there are many techniques available with all their specific advantages and disadvantages.
Measuring RBC deformability is important from both a diagnostic and research point of view. RBC deformability (i) could provide information about the patient’s disease and clinical severity, and (ii) could be a target for pharmacological intervention or predict the toxicity of drugs for patients with hemolytic anemias.
Reduced RBC deformability leads to an inability of the RBC to pass the splenic circulation and leads to premature removal of RBCs from the blood. Altered RBC deformability can be attributed to primary and secondary changes of RBC deformability. Primary changes of RBC deformability are directly related to the disease, such as the membrane weakening in HS or the formation of poorly deformable sickle cells in sickle cell disease. Secondary changes of RBC deformability, such as altered ion fluxes, aberrant membrane protein phosphorylation or RBC vesiculation, are not directly related to the cause of disease. Thus, both primary and secondary causes of RBC deformability can contribute to premature uptake of RBCs in the spleen. In this review and in Table 2, we have summarized the current knowledge on primary and secondary mechanisms of RBC deformability in sickle cell anemia, thalassemia, HS, hereditary xerocytosis, and a number of metabolic disorders of the RBC. We have addressed and discussed the effects of ion regulation, ion channels and RBC (de)hydration on RBC deformability. In addition, we discuss the role and current knowledge of the adaptive responses on RBC deformability, intracellular energy-sensing molecules, membrane protein phosphorylation, hemoglobin deoxygenation and RBC vesiculation on RBC deformability. Knowledge about these processes in the RBC will lead to better understanding of the secondary processes involved in premature removal, and could lead to the discovery of new targets for pharmacological treatment. Furthermore, we postulate that a number of currently undiagnosed, patients with hereditary hemolytic anemia may have (genetic) defects in the here discussed secondary pathways or secondary processes that regulate RBC deformability. This review helps to understand the molecular mechanisms that maintain RBC deformability in healthy and diseased individuals, and enumerates the molecular mechanisms that are altered in RBC disorders leading to hereditary hemolytic anemia.
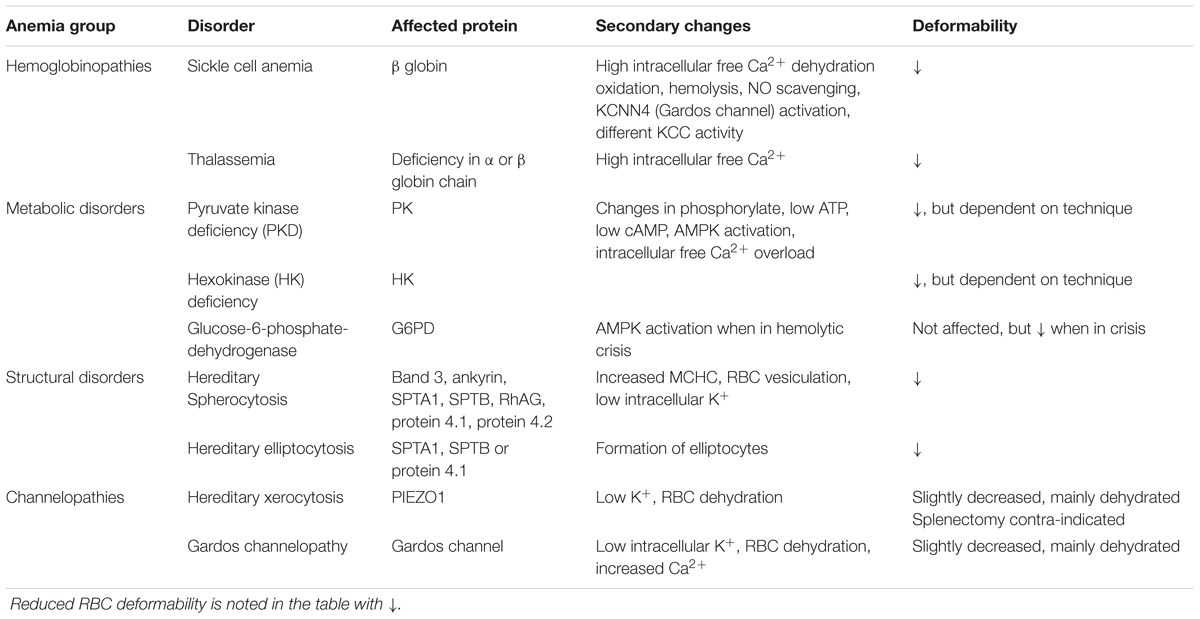
TABLE 2. Summary of primary and secondary changes that lead to reduced RBC deformability in hereditary hemolytic anemia.
Author Contributions
RvW, RS, and WvS encouraged RH to investigate the properties of RBC deformability. RH and RvW wrote the manuscript with support from RS, AB, LK, and WvS.
Funding
This research has received funding from the European Seventh Framework Program under grant agreement number 602121 (CoMMiTMenT).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
5-FU, 5-fluorouracil; AC, adenyl cyclase; AMPK, AMP-activated protein kinase; ATP, adenosine triphosphate; Ca2+, calcium; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; Cl-, chloride; DNA, deoxyribonucleic acid; ET-1, endothelin-1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HbS, hemoglobin S; HK, hexokinase; HS, hereditary spherocytosis; IL-10, interleukine-10; K+, potassium; KCC = K+-Cl- cotransporter; KCNN4, calcium-activated potassium channel or Gardos channel; MCHC, mean corpuscular hemoglobin concentration; Mg2+, magnesium; Na+, sodium; PAF, platelet-activating factor; PDE, phosphodiesterase; PK, pyruvate kinase; PKA, protein kinase A; PKCα, protein kinase C alpha; PS, phosphatidylserine; RBCs, red blood cells; RNA, ribonucleic acid.
References
Adderley, S. P., Sprague, R. S., Stephenson, A. H., and Hanson, M. S. (2010). Regulation of cAMP by phosphodiesterases in erythrocytes. Pharmacol. Rep. 62, 475–482. doi: 10.1016/s1734-1140(10)70303-0
Adderley, S. P., Thuet, K. M., Sridharan, M., Bowles, E. A., Stephenson, A. H., Ellsworth, M. L., et al. (2011). Identification of cytosolic phosphodiesterases in the erythrocyte: a possible role for PDE5. Med. Sci. Monit. 17, CR241–CR247. doi: 10.12659/msm.881763
Adragna, N. C., Di Fulvio, M., and Lauf, P. K. (2004). Regulation of K-Cl cotransport: from function to genes. J. Membr. Biol. 201, 109–137. doi: 10.1007/s00232-004-0695-6
Alaarg, A., Schiffelers, R. M., van Solinge, W. W., and van Wijk, R. (2013). Red blood cell vesiculation in hereditary hemolytic anemia. Front. Physiol. 4:365. doi: 10.3389/fphys.2013.00365
Alapan, Y., Little, J. A., and Gurkan, U. A. (2015). Heterogeneous red blood cell adhesion and deformability in sickle cell disease. Sci. Rep. 4:7173. doi: 10.1038/srep07173
Alapan, Y., Matsuyama, Y., Little, J. A., and Gurkan, U. A. (2016). Dynamic deformability of sickle red blood cells in microphysiological flow. Technology 4, 71–79. doi: 10.1142/S2339547816400045
Albuisson, J., Murthy, S. E., Bandell, M., Coste, B., Louis-Dit-Picard, H., Mathur, J., et al. (2013). Dehydrated hereditary stomatocytosis linked to gain-of-function mutations in mechanically activated PIEZO1 ion channels. Nat. Commun. 4:1884. doi: 10.1038/ncomms2899
Andolfo, I., Alper, S. L., De Franceschi, L., Auriemma, C., Russo, R., De Falco, L., et al. (2013). Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood 121, S1–S12. doi: 10.1182/blood-2013-02-482489
Andolfo, I., Russo, R., Manna, F., Shmukler, B. E., Gambale, A., Vitiello, G., et al. (2015). Novel Gardos channel mutations linked to dehydrated hereditary stomatocytosis (xerocytosis). Am. J. Hematol. 90, 921–926. doi: 10.1002/ajh.24117
Anong, W. A. (2006). Rate of rupture and reattachment of the band 3-ankyrin bridge on the human erythrocyte membrane. J. Biol. Chem. 281, 22360–22366. doi: 10.1074/jbc.M513839200
Ataga, K. I., Reid, M., Ballas, S. K., Yasin, Z., Bigelow, C., James, L. S., et al. (2011). Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: a phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapo. Br. J. Haematol. 153, 92–104. doi: 10.1111/j.1365-2141.2010.08520.x
Ataga, K. I., Smith, W. R., De Castro, L. M., Swerdlow, P., Saunthararajah, Y., Castro, O., et al. (2008). Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood 111, 3991–3997. doi: 10.1182/blood-2007-08-110098
Bae, C., Sachs, F., and Gottlieb, P. A. (2011). The mechanosensitive ion channel Piezo1 is inhibited by the peptide GsMTx4. Biochemistry 50, 6295–6300. doi: 10.1021/bi200770q
Barodka, V., Mohanty, J. G., Mustafa, A. K., Santhanam, L., Nyhan, A., Bhunia, A. K., et al. (2014). Nitroprusside inhibits calcium-induced impairment of red blood cell deformability. Transfusion 54, 434–444. doi: 10.1111/trf.12291
Beneteau, C., Thierry, G., Blesson, S., Le Vaillant, C., Picard, V., Béné, M. C., et al. (2014). Recurrent mutation in the PIEZO1 gene in two families of hereditary xerocytosis with fetal hydrops. Clin. Genet. 85, 293–295. doi: 10.1111/cge.12147
Bernhardt, I., Hall, A. C., and Ellory, J. C. (1988). Transport pathways for monovalent cations through erythrocyte membranes. Stud. Biophys 126, 5–21.
Bevers, E. M., Wiedmer, T., Comfurius, P., Shattil, S. J., Weiss, H. J., Zwaal, R. F., et al. (1992). Defective Ca2+-induced microvesiculation and deficient expression of procoagulant activity in erythrocytes from a patient with a bleeding disorder: a study of the red blood cells of Scott syndrome. Blood 79, 380–388.
Bianchi, P., Fermo, E., Vercellati, C., Marcello, A. P., Porretti, L., Cortelezzi, A., et al. (2012). Diagnostic power of laboratory tests for hereditary spherocytosis: a comparison study in 150 patients grouped according to molecular and clinical characteristics. Haematologica 97, 516–523. doi: 10.3324/haematol.2011.052845
Biro, E., Sturk-Maquelin, K. N., Vogel, G. M. T., Meuleman, D. G., Smit, M. J., Hack, C. E., et al. (2003). Human cell-derived microparticles promote thrombus formation in vivo in a tissue factor-dependent manner. J. Thromb. Haemost. 1, 2561–2568. doi: 10.1046/j.1538-7836.2003.00456.x
Bize, I., Taher, S., and Brugnara, C. (2003). Regulation of K-Cl cotransport during reticulocyte maturation and erythrocyte aging in normal and sickle erythrocytes. Am. J. Physiol. Cell Physiol. 285, C31–C38. doi: 10.1152/ajpcell.00447.2002
Boas, F. E., Forman, L., and Beutler, E. (1998). Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc. Natl. Acad. Sci. U.S.A. 95, 3077–3081. doi: 10.1073/pnas.95.6.3077
Bogdanova, A., Berenbrink, M., and Nikinmaa, M. (2009). Oxygen-dependent ion transport in erythrocytes. Acta Physiol. 195, 305–319. doi: 10.1111/j.1748-1716.2008.01934.x
Bogdanova, A., Makhro, A., Seiler, E., Max Gass, A., Hegemann, I., and Muller, R. (2017). “N-methyl D-glutamate receptor as a pharmacological target for treatment of sickle cell disease: effect of memantine on red cells of patients on a pilot clinical trail MemSID,” in Proceedings of the 21th Meeting of the Red Cell Society, Heidelberg.
Bogdanova, A., Makhro, A., Wang, J., Lipp, P., and Kaestner, L. (2013). Calcium in red blood cells-a perilous balance. Int. J. Mol. Sci. 14, 9848–9872. doi: 10.3390/ijms14059848
Bogdanova, A., Petrushanko, I. Y., Hernansanz-Agustín, P., and Martínez-Ruiz, A. (2016). “Oxygen Sensing” by Na,K-ATPase: these miraculous thiols. Front. Physiol. 7:314. doi: 10.3389/fphys.2016.00314
Bookchin, R. M., and Lew, V. L. (1980). Progressive inhibition of the Ca pump and Ca:Ca exchange in sickle red cells. Nature 284, 561–563. doi: 10.1038/284561a0
Bookchin, R. M., Ortiz, O. E., Shalev, O., Tsurel, S., Rachmilewitz, E. A., Hockaday, A., et al. (1988). Calcium transport and ultrastructure of red cells in beta-thalassemia intermedia. Blood 72, 1602–1607.
Bordin, L., Clari, G., Moro, I., Dalla Vecchia, F., and Moret, V. (1995). Functional link between phosphorylation state of membrane proteins and morphological changes of human erythrocytes. Biochem. Biophys. Res. Commun. 213, 249–257. doi: 10.1006/bbrc.1995.2123
Bosch, F. H., Werre, J. M., Schipper, L., Roerdinkholder-Stoelwinder, B., Huls, T., Willekens, F. L., et al. (1994). Determinants of red blood cell deformability in relation to cell age. Eur. J. Haematol. 52, 35–41. doi: 10.1111/j.1600-0609.1994.tb01282.x
Bosman, G. J. (2013). Survival of red blood cells after transfusion: processes and consequences. Front. Physiol. 4:376. doi: 10.3389/fphys.2013.00376
Bouyer, G., Thomas, S., and Ege, S. (2012). Patch-Clamp Analysis of Membrane Transport in Erythrocytes, Patch Clamp Technique. Available at: https://www.intechopen.com/books/patch-clamp-technique/patch-clamp-analysis-of-membrane-transport-in-erythrocytes
Brugnara, C. (1993). Membrane transport of Na and K and cell dehydration in sickle erythrocytes. Experientia 49, 100–109. doi: 10.1007/bf01989413
Brugnara, C., and Mohandas, N. (2013). Red cell indices in classification and treatment of anemias: from M.M. Wintrobes’s original 1934 classification to the third millennium. Curr. Opin. Hematol. 20, 222–230. doi: 10.1097/MOH.0b013e32835f5933
Bunn, H. F. (1997). Pathogenesis and treatment of sickle cell disease. N. Engl. J. Med. 337, 762–769. doi: 10.1056/NEJM199709113371107
Bunyaratvej, A., Fucharoen, S., Greenbaum, A., and Mohandas, N. (1994). Hydration of red cells in alpha and beta thalassemias differs. A useful approach to distinguish between these red cell phenotypes. Am. J. Clin. Pathol. 102, 217–222. doi: 10.1093/ajcp/102.2.217
Burger, P., Hilarius-Stokman, P., de Korte, D., van den Berg, T. K., and van Bruggen, R. (2012). CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood 119, 5512–5521. doi: 10.1182/blood-2011-10-386805
Burton, N. M., and Bruce, L. J. (2011). Modelling the structure of the red cell membrane. Biochem. Cell Biol. 89, 200–215. doi: 10.1139/o10-154
Bütikofer, P., Lin, Z. W., Kuypers, F. A., Scott, M. D., Xu, C. M., Wagner, G. M., et al. (1989). Chlorpromazine inhibits vesiculation, alters phosphoinositide turnover and changes deformability of ATP-depleted RBCs. Blood 73, 1699–1704. doi: 10.1128/EC.00038-15
Cahalan, S. M., Lukacs, V., Ranade, S. S., Chien, S., Bandell, M., and Patapoutian, A. (2015). Piezo1 links mechanical forces to red blood cell volume. eLife 4:e07370. doi: 10.7554/eLife.07370
Cai, M., Zhao, W., Shang, X., Jiang, J., Ji, H., Tang, Z., et al. (2012). Direct evidence of lipid rafts by in situ atomic force microscopy. Small 8, 1243–1250. doi: 10.1002/smll.201102183
Cappellini, M. D., and Fiorelli, G. (2008). Glucose-6-phosphate dehydrogenase deficiency. Lancet 371, 64–74. doi: 10.1016/S0140-6736(08)60073-2
Carlson, D. L., and Mawdsley, R. H. (1986). Sports anemia: a review of the literature. Am. J. Sports Med. 14, 109–112. doi: 10.1177/036354658601400202
Chabanel, A., Reinhart, W., and Chien, S. (1987). Increased resistance to membrane deformation of shape-transformed human red blood cells. Blood 69, 739–743.
Chétrite, G., and Cassoly, R. (1985). Affinity of hemoglobin for the cytoplasmic fragment of human erythrocyte membrane band 3. Equilibrium measurements at physiological pH using matrix-bound proteins: the effects of ionic strength, deoxygenation and of 2,3-diphosphoglycerate. J. Mol. Biol. 185, 639–644. doi: 10.1016/0022-2836(85)90076-2
Chu, H., Puchulu-Campanella, E., Galan, J. A., Tao, W. A., Low, P. S., and Hoffman, J. F. (2012). Identification of cytoskeletal elements enclosing the ATP pools that fuel human red blood cell membrane cation pumps. Proc. Natl. Acad. Sci. U.S.A. 109, 12794–12799. doi: 10.1073/pnas.1209014109
Chui, D. H., Fucharoen, S., and Chan, V. (2003). Hemoglobin H disease: not necessarily a benign disorder. Blood 101, 791–800. doi: 10.1182/blood-2002-07-1975
Ciana, A., Achilli, C., Gaur, A., and Minetti, G. (2017). Membrane remodelling and vesicle formation during ageing of human red blood cells. Cell. Physiol. Biochem. 42, 1127–1138. doi: 10.1159/000478768
Cinar, E., Zhou, S., DeCourcey, J., Wang, Y., Waugh, R. E., and Wan, J. (2015). Piezo1 regulates mechanotransductive release of ATP from human RBCs. Proc. Natl. Acad. Sci. U.S.A. 112, 11783–11788. doi: 10.1073/pnas.1507309112
Cividalli, G., Locker, H., and Russell, A. (1971). Increased permeability of erythrocyte membrane in thalassemia. Blood 37, 716–724.
Clark, M. R., Mohandas, N., and Shohet, S. B. (1982). Hydration of sickle cells using the sodium ionophore Monensin. A model for therapy. J. Clin. Invest. 70, 1074–1080. doi: 10.1172/jci110695
Clark, M. R., Mohandas, N., and Shohet, S. B. (1983). Osmotic gradient ektacytometry: comprehensive characterization of red cell volume and surface maintenance. Blood 61, 899–910.
Cluitmans, J. C. A., Chokkalingam, V., Janssen, A. M., Brock, R., Huck, W. T. S., and Bosman, G. J. C. G. M. (2014). Alterations in red blood cell deformability during storage: a microfluidic approach. Biomed Res. Int. 2014:764268. doi: 10.1155/2014/764268
Condon, M. R., Feketova, E., Machiedo, G. W., Deitch, E. A., and Spolarics, Z. (2007). Augmented erythrocyte band-3 phosphorylation in septic mice. Biochim. Biophys. Acta 1772, 580–586. doi: 10.1016/j.bbadis.2007.02.004
Crable, S. C., Hammond, S. M., Papes, R., Rettig, R. K., Zhou, G.-P., Gallagher, P. G., et al. (2005). Multiple isoforms of the KC1 cotransporter are expressed in sickle and normal erythroid cells. Exp. Hematol. 33, 624–631. doi: 10.1016/j.exphem.2005.02.006
Da Costa, L., Galimand, J., Fenneteau, O., and Mohandas, N. (2013). Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 27, 167–178. doi: 10.1016/j.blre.2013.04.003
Da Costa, L., Suner, L., Galimand, J., Bonnel, A., Pascreau, T., Couque, N., et al. (2016). Diagnostic tool for red blood cell membrane disorders: assessment of a new generation ektacytometer. Blood Cells Mol. Dis. 56, 9–22. doi: 10.1016/j.bcmd.2015.09.001
Danielczok, J. G., Terriac, E., Hertz, L., Petkova-Kirova, P., Lautenschläger, F., Laschke, M. W., et al. (2017). Red blood cell passage of small capillaries is associated with transient Ca2+-mediated adaptations. Front. Physiol. 8:979. doi: 10.3389/fphys.2017.00979
de Back, D. Z., Kostova, E. B., van Kraaij, M., van den Berg, T. K., and van Bruggen, R. (2014). Of macrophages and red blood cells; a complex love story. Front. Physiol. 5:9. doi: 10.3389/fphys.2014.00009
De Franceschi, L., Cappellini, M. D., Graziadei, G., Manzato, F., Olivieri, O., Corrocher, R., et al. (1998). The effect of dietary magnesium supplementation on the cellular abnormalities of erythrocytes in patients with beta thalassemia intermedia. Haematologica 83, 118–125.
De Franceschi, L., Olivieri, O., Miraglia del Giudice, E., Perrotta, S., Sabato, V., Corrocher, R., et al. (1997). Membrane cation and anion transport activities in erythrocytes of hereditary spherocytosis: effects of different membrane protein defects. Am. J. Hematol. 55, 121–128. doi: 10.1002/(SICI)1096-8652(199707)55:3<121::AID-AJH1>3.0.CO;2-U
De Franceschi, L., Rivera, A., Fleming, M. D., Honczarenko, M., Peters, L. L., Gascard, P., et al. (2005). Evidence for a protective role of the Gardos channel against hemolysis in murine spherocytosis. Blood 106, 1454–1459. doi: 10.1182/blood-2005-01-0368
de Franceschi, L., Rouyer-Fessard, P., Alper, S. L., Jouault, H., Brugnara, C., and Beuzard, Y. (1996). Combination therapy of erythropoietin, hydroxyurea, and clotrimazole in a beta thalassemic mouse: a model for human therapy. Blood 87, 1188–1195.
Deplaine, G., Safeukui, I., Jeddi, F., Lacoste, F., Brousse, V., Perrot, S., et al. (2010). The sensing of poorly deformable red blood cells by the human spleen can be mimicked in vitro. Blood 117, e88–e95. doi: 10.1182/blood-2010-10-312801
Derbyshire, E. R., and Marletta, M. A. (2012). Structure and regulation of soluble guanylate cyclase. Annu. Rev. Biochem. 81, 533–559. doi: 10.1146/annurev-biochem-050410-100030
Dobbe, J. G., Streekstra, G. J., Hardeman, M. R., Ince, C., and Grimbergen, C. A. (2002). Measurement of the distribution of red blood cell deformability using an automated rheoscope. Cytometry 50, 313–325. doi: 10.1002/cyto.10171
Eaton, W. A., and Hofrichter, J. (1990). Sickle cell hemoglobin polymerization. Adv. Protein Chem. 40, 63–279. doi: 10.1016/S0065-3233(08)60287-9
Eisinger, J., Flores, J., and Salhany, J. M. (1982). Association of cytosol hemoglobin with the membrane in intact erythrocytes. Proc. Natl. Acad. Sci. U.S.A. 79, 408–412. doi: 10.1073/pnas.79.2.408
Faucherre, A., Kissa, K., Nargeot, J., Mangoni, M. E., and Jopling, C. (2014). Piezo1 plays a role in erythrocyte volume homeostasis. Haematologica 99, 70–75. doi: 10.3324/haematol.2013.086090
Fens, M. H., van Wijk, R., Andringa, G., van Rooijen, K. L., Dijstelbloem, H. M., Rasmussen, J. T., et al. (2012). A role for activated endothelial cells in red blood cell clearance: implications for vasopathology. Haematologica 97, 500–508. doi: 10.3324/haematol.2011.048694
Fermo, E., Bogdanova, A., Petkova-Kirova, P., Zaninoni, A., Marcello, A. P., Makhro, A., et al. (2017a). “Gardos Channelopathy”: a variant of hereditary Stomatocytosis with complex molecular regulation. Sci. Rep. 7:1744. doi: 10.1038/s41598-017-01591-w
Fermo, E., Vercellati, C., Marcello, A. P., Zaninoni, A., van Wijk, R., Mirra, N., et al. (2017b). Hereditary xerocytosis due to mutations in PIEZO1 gene associated with heterozygous pyruvate kinase deficiency and beta-thalassemia trait in two unrelated families. Case Rep. Hematol. 2017:2769570. doi: 10.1155/2017/2769570
Fischer, D. J., Torrence, N. J., Sprung, R. J., and Spence, D. M. (2003). Determination of erythrocyte deformability and its correlation to cellular ATP release using microbore tubing with diameters that approximate resistance vessels in vivo. Analyst 128, 1163–1168. doi: 10.1039/b308225n
Föller, M., Sopjani, M., Koka, S., Gu, S., Mahmud, H., Wang, K., et al. (2009). Regulation of erythrocyte survival by AMP-activated protein kinase. FASEB J. 23, 1072–1080. doi: 10.1096/fj.08-121772
Foretz, M., Guihard, S., Leclerc, J., Fauveau, V., Couty, J.-P., Andris, F., et al. (2010). Maintenance of red blood cell integrity by AMP-activated protein kinase alpha1 catalytic subunit. FEBS Lett. 584, 3667–3671. doi: 10.1016/j.febslet.2010.07.041
Gadsby, D. C. (2009). Ion channels versus ion pumps: the principal difference, in principle. Nat. Rev. Mol. Cell Biol. 10, 344–352. doi: 10.1038/nrm2668
Galanello, R., and Origa, R. (2010). Beta-thalassemia. Orphanet J. Rare Dis. 5, 11. doi: 10.1186/1750-1172-5-11
Gallagher, P. G. (2004a). Hereditary elliptocytosis: spectrin and protein 4.1R. Semin. Hematol. 41, 142–164. doi: 10.1053/j.seminhematol.2004.01.003
Gallagher, P. G. (2004b). Update on the clinical spectrum and genetics of red blood cell membrane disorders. Curr. Hematol. Rep. 3, 85–91.
Gallagher, P. G. (2013). Disorders of red cell volume regulation. Curr. Opin. Hematol. 20, 201–207. doi: 10.1097/MOH.0b013e32835f6870
Gallagher, P. G. (2017). Disorders of erythrocyte hydration. Blood 130, 2699–2708. doi: 10.1182/blood-2017-04-590810
Ge, J., Li, W., Zhao, Q., Li, N., Chen, M., Zhi, P., et al. (2015). Architecture of the mammalian mechanosensitive Piezo1 channel. Nature 527, 64–69. doi: 10.1038/nature15247
George, A., Pushkaran, S., Li, L., An, X., Zheng, Y., Mohandas, N., et al. (2010). Altered phosphorylation of cytoskeleton proteins in sickle red blood cells: the role of protein kinase C. Rac GTPases, and reactive oxygen species. Blood Cells Mol. Dis. 45, 41–45. doi: 10.1016/j.bcmd.2010.02.006
Gibson, J. S., Cossins, A. R., and Ellory, J. C. (2000). Oxygen-sensitive membrane transporters in vertebrate red cells. J. Exp. Biol. 203, 1395–1407.
Gibson, J. S., Khan, A., Speake, P. F., and Ellory, J. C. (2001). O2 dependence of K+ transport in sickle cells: the effect of different cell populations and the substituted benzaldehyde 12C79. FASEB J. 15, 823–832. doi: 10.1096/fj.00-0177com
Gibson, J. S., Speake, P. F., and Ellory, J. C. (1998). Differential oxygen sensitivity of the K+-Cl- cotransporter in normal and sickle human red blood cells. J. Physiol. 511(Pt 1), 225–234. doi: 10.1111/j.1469-7793.1998.225bi.x
Glogowska, E., Lezon-Geyda, K., Maksimova, Y., Schulz, V. P., and Gallagher, P. G. (2015). Mutations in the Gardos channel (KCNN4) are associated with hereditary xerocytosis. Blood 126, 1281–1284. doi: 10.1182/blood-2015-07-657957
Glogowska, E., Schneider, E. R., Maksimova, Y., Schulz, V. P., Lezon-Geyda, K., Wu, J., et al. (2017). Novel mechanisms of PIEZO1 dysfunction in hereditary xerocytosis. Blood 130, 1845–1856. doi: 10.1182/blood-2017-05-786004
Goldstein, L., and Brill, S. R. (1991). Volume-activated taurine efflux from skate erythrocytes: possible band 3 involvement. Am. J. Physiol. 260, R1014–R1020. doi: 10.1152/ajpregu.1991.260.5.R1014
Goldstein, L., and Davis, E. M. (1994). Taurine, betaine, and inositol share a volume-sensitive transporter in skate erythrocyte cell membrane. Am. J. Physiol. 267, R426–R431. doi: 10.1152/ajpregu.1994.267.2.R426
Grau, M., Pauly, S., Ali, J., Walpurgis, K., Thevis, M., Bloch, W., et al. (2013). RBC-NOS-dependent S-nitrosylation of cytoskeletal proteins improves RBC deformability. PLoS One 8:e56759. doi: 10.1371/journal.pone.0056759
Greenwalt, T. J., and Dumaswala, U. J. (1988). Effect of red cell age on vesiculation in vitro. Br. J. Haematol. 68, 465–467. doi: 10.1111/j.1365-2141.1988.tb04237.x
Guo, Q., Duffy, S. P., Matthews, K., Santoso, A. T., Scott, M. D., and Ma, H. (2014). Microfluidic analysis of red blood cell deformability. J. Biomech. 47, 1767–1776. doi: 10.1016/j.jbiomech.2014.03.038
Hagley, A. C., Younger-Coleman, N. O., Richards, A. A., Nwokocha, C. R., and Pepple, D. J. (2011). The effect of sildenafil on the elasticity of erythrocytes in homozygous sickle cell disease. Indian J. Med. Sci. 65, 243–249. doi: 10.4103/0019-5359.107024
Hahn, E. V., and Gillespie, E. B. (1927). Sickle cell anemia. Arch. Intern. Med. 39, 233–254. doi: 10.1001/archinte.1927.00130020072006
Hänggi, P., Makhro, A., Gassmann, M., Schmugge, M., Goede, J. S., Speer, O., et al. (2014). Red blood cells of sickle cell disease patients exhibit abnormally high abundance of N-methyl D-aspartate receptors mediating excessive calcium uptake. Br. J. Haematol. 167, 252–264. doi: 10.1111/bjh.13028
Hardeman, M. R., Goedhart, P., and Breederveld, D. (1987). Laser diffraction ellipsometry of erythrocytes under controlled shear stress using a rotational viscosimeter. Clin. Chim. Acta 165, 227–234. doi: 10.1016/0009-8981(87)90166-5
Harrison, T., Samuel, B. U., Akompong, T., Hamm, H., Mohandas, N., Lomasney, J. W., et al. (2003). Erythrocyte G protein-coupled receptor signaling in malarial infection. Science 301, 1734–1736. doi: 10.1126/science.1089324
Harteveld, C. L., and Higgs, D. R. (2010). Alpha-thalassaemia. Orphanet J. Rare Dis. 5:13. doi: 10.1186/1750-1172-5-13
Hasegawa, S., Rodgers, G. P., Shio, H., Schechter, A. N., and Uyesaka, N. (1993). Impaired deformability of Heinz body-forming red cells. Biorheology 30, 275–286. doi: 10.3233/BIR-1993-303-412
Hertz, L., Huisjes, R., Llaudet-Planas, E., Petkova-Kirova, P., Makhro, A., Danielczok, J. G., et al. (2017). Is increased intracellular calcium in red blood cells a common component in the molecular mechanism causing anemia? Front. Physiol. 8:673. doi: 10.3389/FPHYS.2017.00673
Higgs, D. R., Engel, J. D., and Stamatoyannopoulos, G. (2012). Thalassaemia. Lancet 379, 373–383. doi: 10.1016/S0140-6736(11)60283-3
Huisjes, R., Petkova-Kirova, P., Makhro, A., Llaudet-Planas, E., Hertz, L., Verhagen, L., et al. (2017a). “In-depth characterization of the red blood cell in hereditary spherocytosis,” in Proceedings of the 21th Meeting of the Red Cell Society, Heidelberg.
Huisjes, R., van Solinge, W. W., Levin, M. D., van Wijk, R., and Riedl, J. A. (2017b). Digital microscopy as a screening tool for the diagnosis of hereditary hemolytic anemia. Int. J. Lab. Hematol. 40, 159–168. doi: 10.1111/ijlh.12758
Hurley, R. L., Barré, L. K., Wood, S. D., Anderson, K. A., Kemp, B. E., Means, A. R., et al. (2006). Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J. Biol. Chem. 281, 36662–36672. doi: 10.1074/jbc.M606676200
Iolascon, A., Andolfo, I., Barcellini, W., Corcione, F., Garçon, L., De Franceschi, L., et al. (2017). Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica 102, 1304–1313. doi: 10.3324/haematol.2016.161166
Izumo, H., Lear, S., Williams, M., Rosa, R., and Epstein, F. H. (1987). Sodium-potassium pump, ion fluxes, and cellular dehydration in sickle cell anemia. J. Clin. Invest. 79, 1621–1628. doi: 10.1172/JCI112998
Jacques-Fricke, B. T., Seow, Y., Gottlieb, P. A., Sachs, F., and Gomez, T. M. (2006). Ca2+ influx through mechanosensitive channels inhibits neurite outgrowth in opposition to other influx pathways and release from intracellular stores. J. Neurosci. 26, 5656–5664. doi: 10.1523/JNEUROSCI.0675-06.2006
Jaïs, X., Till, S. J., Cynober, T., Ioos, V., Garcia, G., Tchernia, G., et al. (2003). An extreme consequence of splenectomy in dehydrated hereditary stomatocytosis: gradual thrombo-embolic pulmonary hypertension and lung-heart transplantation. Hemoglobin 27, 139–147. doi: 10.1081/HEM-120023377
Jordan, J., Kiernan, W., Merker, H., Wenzel, M., and Beneke, R. (1998). Red cell membrane skeletal changes in marathon runners. Int. J. Sports Med. 19, 16–19. doi: 10.1055/s-2007-971873
Kaestner, L. (2011) Cation Channels in Erythrocytes - Historical and Future Perspective. Open Biol. J. 4, 27–34.
Kaestner, L. (2015). Channelizing the red blood cell: molecular biology competes with patch-clamp. Front. Mol. Biosci. 2:46. doi: 10.3389/fmolb.2015.00046
Kaestner, L., Steffen, P., Nguyen, D. B., Wang, J., Wagner-Britz, L., Jung, A., et al. (2012). Lysophosphatidic acid induced red blood cell aggregation in vitro. Bioelectrochemistry 87, 89–95. doi: 10.1016/j.bioelechem.2011.08.004
Kaestner, L., Wang, X., Hertz, L., and Bernhardt, I. (2018). Voltage-activated ion channels in non-excitable cells — a viewpoint regarding their physiological justification. Front Physiol. 9:450. doi: 10.3389/fphys.2018.00450
Kamada, T., Tokuda, S., Aozaki, S., and Otsuji, S. (1993). Higher levels of erythrocyte membrane fluidity in sprinters and long-distance runners. J. Appl. Physiol. 74, 354–358. doi: 10.1152/jappl.1993.74.1.354
Kanno, H., Morimoto, M., Fujii, H., Tsujimura, T., Asai, H., Noguchi, T., et al. (1995). Primary structure of murine red blood cell-type pyruvate kinase (PK) and molecular characterization of PK deficiency identified in the CBA strain. Blood 86, 3205–3210.
Karger, R., Lukow, C., and Kretschmer, V. (2012). Deformability of red blood cells and correlation with ATP content during storage as leukocyte-depleted whole blood. Transfus. Med. Hemother. 39, 277–282. doi: 10.1159/000339809
Kato, G. J., Steinberg, M. H., and Gladwin, M. T. (2017). Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Invest. 127, 750–760. doi: 10.1172/JCI89741
Kay, M. M. (1984). Localization of senescent cell antigen on band 3. Proc. Natl. Acad. Sci. U.S.A. 81, 5753–5757. doi: 10.1073/pnas.81.18.5753
Kay, M. M., Goodman, S. R., Sorensen, K., Whitfield, C. F., Wong, P., Zaki, L., et al. (1983). Senescent cell antigen is immunologically related to band 3. Proc. Natl. Acad. Sci. U.S.A. 80, 1631–1635. doi: 10.1073/pnas.80.6.1631
Kenney, H. W., Smoot, R. T., Trader, J. D., Dowe, C. R., Allain, C. C., and Campbell, T. M. (1961). The sickle cell disease: current status and preliminary research observations. J. Natl. Med. Assoc. 53, 137–143.
Khan, A. I., Drew, C., Ball, S. E., Ball, V., Ellory, J. C., and Gibson, J. S. (2004). Oxygen dependence of K+-Cl- cotransport in human red cell ghosts and sickle cells. Bioelectrochemistry 62, 141–146. doi: 10.1016/j.bioelechem.2003.07.005
Klarl, B. A., Lang, P. A., Kempe, D. S., Niemoeller, O. M., Akel, A., Sobiesiak, M., et al. (2006). Protein kinase C mediates erythrocyte “programmed cell death” following glucose depletion. Am. J. Physiol. Cell Physiol. 290, C244–C253. doi: 10.1152/ajpcell.00283.2005
Knebel, S. M., Elrick, M. M., Bowles, E. A., Zdanovec, A. K., Stephenson, A. H., Ellsworth, M. L., et al. (2013). Synergistic effects of prostacyclin analogs and phosphodiesterase inhibitors on cyclic adenosine 3′,5′ monophosphate accumulation and adenosine 3′5′ triphosphate release from human erythrocytes. Exp. Biol. Med. 238, 1069–1074. doi: 10.1177/1535370213498981
Koralkova, P., van Solinge, W. W., and van Wijk, R. (2014). Rare hereditary red blood cell enzymopathies associated with hemolytic anemia - pathophysiology, clinical aspects, and laboratory diagnosis. Int. J. Lab. Hematol. 36, 388–397. doi: 10.1111/ijlh.12223
Körmöczi, G. F., Säemann, M. D., Buchta, C., Peck-Radosavljevic, M., Mayr, W. R., Schwartz, D. W. M., et al. (2006). Influence of clinical factors on the haemolysis marker haptoglobin. Eur. J. Clin. Invest. 36, 202–209. doi: 10.1111/j.1365-2362.2006.01617.x
Kostova, E. B., Beuger, B. M., Klei, T. R. L., Halonen, P., Lieftink, C., Beijersbergen, R., et al. (2015). Identification of signalling cascades involved in red blood cell shrinkage and vesiculation. Biosci. Rep. 35:e00187. doi: 10.1042/BSR20150019
Kuhn, V., Diederich, L., Keller, T. C. S., Kramer, C. M., Lückstädt, W., Panknin, C., et al. (2017). Red blood cell function and dysfunction: redox regulation, nitric oxide metabolism, anemia. Antioxid. Redox Signal. 26, 718–742. doi: 10.1089/ars.2016.6954
Lasch, J., Küllertz, G., and Opalka, J. R. (2000). Separation of erythrocytes into age-related fractions by density or size? Counterflow centrifugation. Clin. Chem. Lab. Med. 38, 629–632. doi: 10.1515/CCLM.2000.092
Lazarova, E., Gulbis, B., Oirschot, B., and van Wijk, R. (2017). Next-generation osmotic gradient ektacytometry for the diagnosis of hereditary spherocytosis: interlaboratory method validation and experience. Clin. Chem. Lab. Med. 55, 394–402. doi: 10.1515/cclm-2016-0290
Leblond, P. F., Coulombe, L., and Lyonnais, J. (1978). Erythrocyte populations in pyruvate kinase deficiency anaemia following splenectomy. II. Cell deformability. Br. J. Haematol. 39, 63–70. doi: 10.1111/j.1365-2141.1978.tb07128.x
Leonard, C., Conrard, L., Guthmann, M., Pollet, H., Carquin, M., Vermylen, C., et al. (2017). Contribution of plasma membrane lipid domains to red blood cell (re)shaping. Sci. Rep. 7:4264. doi: 10.1038/s41598-017-04388-z
Lew, V. L., and Bookchin, R. M. (2005). Ion transport pathology in the mechanism of sickle cell dehydration. Physiol. Rev. 85, 179–200. doi: 10.1152/physrev.00052.2003
Lew, V. L., and Tiffert, T. (2017). On the mechanism of human red blood cell longevity: roles of calcium, the sodium pump, PIEZO1 and Gardos channels. Front. Physiol. 8:977. doi: 10.3389/FPHYS.2017.00977
Li, J., Hou, B., Tumova, S., Muraki, K., Bruns, A., Ludlow, M. J., et al. (2014). Piezo1 integration of vascular architecture with physiological force. Nature 515, 279–282. doi: 10.1038/nature13701
Linderkamp, O., Friederichs, E., Boehler, T., and Ludwig, A. (1993). Age dependency of red blood cell deformability and density: studies in transient erythroblastopenia of childhood. Br. J. Haematol. 83, 125–129. doi: 10.1111/j.1365-2141.1993.tb04642.x
Liu, L., Lei, T., Bankir, L., Zhao, D., Gai, X., Zhao, X., et al. (2011). Erythrocyte permeability to urea and water: comparative study in rodents, ruminants, carnivores, humans, and birds. J. Comp. Physiol. B 181, 65–72. doi: 10.1007/s00360-010-0515-5
Livaja Koshiar, R., Somajo, S., Norström, E., Dahlbäck, B., and McMahon, C. (2014). Erythrocyte-derived microparticles supporting activated protein C-mediated regulation of blood coagulation. PLoS One 9:e104200. doi: 10.1371/journal.pone.0104200
Llaudet-Planas, E., Vives-Corrons, J. L., Rizzuto, V., Gómez-Ramírez, P., Sevilla Navarro, J., Coll Sibina, M. T., et al. (2017). Osmotic gradient ektacytometry: a valuable screening test for hereditary spherocytosis and other red blood cell membrane disorders. Int. J. Lab. Hematol. 40, 94–102. doi: 10.1111/ijlh.12746
Locovei, S., Bao, L., and Dahl, G. (2006). Pannexin 1 in erythrocytes: function without a gap. Proc. Natl. Acad. Sci. U.S.A. 103, 7655–7659. doi: 10.1073/pnas.0601037103
Longley, D. B., Harkin, D. P., and Johnston, P. G. (2003). 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338. doi: 10.1038/nrc1074
Lopes de Almeida, J. P., Freitas-Santos, T., and Saldanha, C. (2012). Erythrocyte deformability dependence on band 3 protein in an in-vitro model of hyperfibrinogenemia. Clin. Hemorheol. Microcirc. 50, 213–219. doi: 10.3233/CH-2010-1433
Losco, P., Nash, G., Stone, P., and Ventre, J. (2001). Comparison of the effects of radiographic contrast media on dehydration and filterability of red blood cells from donors homozygous for hemoglobin A or hemoglobin S. Am. J. Hematol. 68, 149–158. doi: 10.1002/ajh.1171
Lubin, A., and Desforges, J. F. (1972). Effect of Heinz bodies on red cell deformability. Blood 39, 658–665.
Lutz, H. U., Stammler, P., Fasler, S., Ingold, M., and Fehr, J. (1992). Density separation of human red blood cells on self forming Percoll gradients: correlation with cell age. Biochim. Biophys. Acta 1116, 1–10. doi: 10.1016/0304-4165(92)90120-J
Ma, Y.-L., Rees, D. C., Gibson, J. S., and Ellory, J. C. (2012). The conductance of red blood cells from sickle cell patients: ion selectivity and inhibitors. J. Physiol. 590, 2095–2105. doi: 10.1113/jphysiol.2012.229609
MacDonald, I. C., Ragan, D. M., Schmidt, E. E., and Groom, A. C. (1987). Kinetics of red blood cell passage through interendothelial slits into venous sinuses in rat spleen, analyzed by in vivo microscopy. Microvasc. Res. 33, 118–134. doi: 10.1016/0026-2862(87)90011-2
Mairbäurl, H. (2013). Red blood cells in sports: effects of exercise and training on oxygen supply by red blood cells. Front. Physiol. 4:332. doi: 10.3389/fphys.2013.00332
Makhro, A., Huisjes, R., Verhagen, L. P., Mañú-Pereira, M., Llaudet-Planas, E., Petkova-Kirova, P., et al. (2016). Red cell properties after different modes of blood transportation. Front. Physiol. 7:288. doi: 10.3389/fphys.2016.00288
Mangat, C., Inoue, S., Saah, E., and Sharman, M. (2014). Acute haemolytic anaemia and myolysis due to G6PD deficiency. BMJ Case Rep. 2014:bcr2014203631. doi: 10.1136/bcr-2014-203631
Mao, T.-Y., Fu, L.-L., and Wang, J.-S. (2011). Hypoxic exercise training causes erythrocyte senescence and rheological dysfunction by depressed Gardos channel activity. J. Appl. Physiol. 111, 382–391. doi: 10.1152/japplphysiol.00096.2011
Marie, J., Buc, H., Simon, M. P., and Kahn, A. (1980). Phosphorylation of human erythrocyte pyruvate kinase by soluble cyclic-AMP-dependent protein kinases. Comparison with human liver L-type enzyme. Eur. J. Biochem. 108, 251–260. doi: 10.1111/j.1432-1033.1980.tb04718.x
Marques, F., Crespo, M. E., Silva, Z. I., and Bicho, M. (2000). Insulin and high glucose modulation of phosphatase and reductase enzymes in the human erythrocytes: a comparative analysis in normal and diabetic states. Diabetes Res. Clin. Pract. 47, 191–198. doi: 10.1016/S0168-8227(99)00128-X
Martinov, M. V., Plotnikov, A. G., Vitvitsky, V. M., and Ataullakhanov, F. I. (2000). Deficiencies of glycolytic enzymes as a possible cause of hemolytic anemia. Biochim. Biophys. Acta 1474, 75–87. doi: 10.1016/S0304-4165(99)00218-4
McNaughton-Smith, G. A., Burns, J. F., Stocker, J. W., Rigdon, G. C., Creech, C., Arrington, S., et al. (2008). Novel inhibitors of the Gardos channel for the treatment of sickle cell disease. J. Med. Chem. 51, 976–982. doi: 10.1021/jm070663s
Mebius, R. E., and Kraal, G. (2005). Structure and function of the spleen. Nat. Rev. Immunol. 5, 606–616. doi: 10.1038/nri1669
Mihaylova, M. M., and Shaw, R. J. (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023. doi: 10.1038/ncb2329
Minetti, G., Egée, S., Mörsdorf, D., Steffen, P., Makhro, A., Achilli, C., et al. (2013). Red cell investigations: art and artefacts. Blood Rev. 27, 91–101. doi: 10.1016/j.blre.2013.02.002
Moriyama, R., Lombardo, C. R., Workman, R. F., and Low, P. S. (1993). Regulation of linkages between the erythrocyte membrane and its skeleton by 2,3-diphosphoglycerate. J. Biol. Chem. 268, 10990–10996.
Muravyov, A. V., and Tikhomirova, I. A. (2013). Role molecular signaling pathways in changes of red blood cell deformability. Clin. Hemorheol. Microcirc. 53, 45–59. doi: 10.3233/CH-2012-1575
Muzyamba, M. C., Campbell, E. H., and Gibson, J. S. (2006). Effect of intracellular magnesium and oxygen tension on K+-Cl- cotransport in normal and sickle human red cells. Cell. Physiol. Biochem. 17, 121–128. doi: 10.1159/000092073
Nans, A., Mohandas, N., and Stokes, D. L. (2011). Native ultrastructure of the red cell cytoskeleton by cryo-electron tomography. Biophys. J. 101, 2341–2350. doi: 10.1016/j.bpj.2011.09.050
Nantakomol, D., Palasuwan, A., Chaowanathikhom, M., Soogarun, S., and Imwong, M. (2012). Red cell and platelet-derived microparticles are increased in G6PD-deficient subjects. Eur. J. Haematol. 89, 423–429. doi: 10.1111/ejh.12010
Nathan, D. G., and Gunn, R. B. (1966). Thalassemia: the consequences of unbalanced hemoglobin synthesis. Am. J. Med. 41, 815–830. doi: 10.1016/0002-9343(66)90039-8
Nathan, D. G., Oski, F. A., Sidel, V. W., and Diamond, L. K. (1965). Extreme hemolysis and red-cell distortion in erythrocyte pyruvate kinase deficiency. N. Engl. J. Med. 272, 118–123. doi: 10.1056/NEJM196501212720302
Nguyen, D. B., Wagner-Britz, L., Maia, S., Steffen, P., Wagner, C., Kaestner, L., et al. (2011). Regulation of phosphatidylserine exposure in red blood cells. Cell. Physiol. Biochem. 28, 847–856. doi: 10.1159/000335798
Odièvre, M.-H., Verger, E., Silva-Pinto, A. C., and Elion, J. (2011). Pathophysiological insights in sickle cell disease. Indian J. Med. Res. 134, 532–537.
Olivieri, O., De Franceschi, L., Capellini, M. D., Girelli, D., Corrocher, R., and Brugnara, C. (1994). Oxidative damage and erythrocyte membrane transport abnormalities in thalassemias. Blood 84, 315–320.
Oonishi, T., Sakashita, K., and Uyesaka, N. (1997). Regulation of red blood cell filterability by Ca2+ influx and cAMP-mediated signaling pathways. Am. J. Physiol. 273, C1828–C1834. doi: 10.1152/ajpcell.1997.273.6.C1828
Oostenbrug, G. S., Mensink, R. P., Hardeman, M. R., De Vries, T., Brouns, F., and Hornstra, G. (1997). Exercise performance, red blood cell deformability, and lipid peroxidation: effects of fish oil and vitamin E. J. Appl. Physiol. 83, 746–752. doi: 10.1152/jappl.1997.83.3.746
Park, Y., Best, C. A., Auth, T., Gov, N. S., Safran, S. A., Popescu, G., et al. (2010). Metabolic remodeling of the human red blood cell membrane. Proc. Natl. Acad. Sci. U.S.A. 107, 1289–1294. doi: 10.1073/pnas.0910785107
Parpart, A. K., Lorenz, P. B., Parpart, E. R., Gregg, J. R., and Chase, A. M. (1947). The osmotic resistance (fragility) of human red cells. J. Clin. Invest. 26, 636–640. doi: 10.1172/JCI101847
Pattanapanyasat, K., Noulsri, E., Fucharoen, S., Lerdwana, S., Lamchiagdhase, P., Siritanaratkul, N., et al. (2004). Flow cytometric quantitation of red blood cell vesicles in thalassemia. Cytometry B Clin. Cytom. 57, 23–31. doi: 10.1002/cyto.b.10064
Pauling, L., and Itano, H. A. (1949). Sickle cell anemia a molecular disease. Science 110, 543–548. doi: 10.1126/science.110.2865.543
Perrotta, S., Gallagher, P. G., and Mohandas, N. (2008). Hereditary spherocytosis. Lancet 372, 1411–1426. doi: 10.1016/S0140-6736(08)61588-3
Peters, L. L., Jindel, H. K., Gwynn, B., Korsgren, C., John, K. M., Lux, S. E., et al. (1999). Mild spherocytosis and altered red cell ion transport in protein 4. 2-null mice. J. Clin. Invest. 103, 1527–1537. doi: 10.1172/JCI5766
Picas, L., Rico, F., Deforet, M., and Scheuring, S. (2013). Structural and mechanical heterogeneity of the erythrocyte membrane reveals hallmarks of membrane stability. ACS Nano 7, 1054–1063. doi: 10.1021/nn303824j
Picot, J., Ndour, P. A., Lefevre, S. D., El Nemer, W., Tawfik, H., Galimand, J., et al. (2015). A biomimetic microfluidic chip to study the circulation and mechanical retention of red blood cells in the spleen. Am. J. Hematol. 90, 339–345. doi: 10.1002/ajh.23941
Ramdani, G., Naissant, B., Thompson, E., Breil, F., Lorthiois, A., Dupuy, F., et al. (2015). cAMP-signalling regulates gametocyte-infected erythrocyte deformability required for malaria parasite transmission. PLoS Pathog. 11:e1004815. doi: 10.1371/journal.ppat.1004815
Rapetti-Mauss, R., Lacoste, C., Picard, V. V., Guitton, C., Lombard, E., Loosveld, M., et al. (2015). A mutation in the Gardos channel is associated with hereditary xerocytosis. Blood 126, 1273–1280. doi: 10.1182/blood-2015-04-642496
Rees, D. C., Thein, S. L., Osei, A., Drasar, E., Tewari, S., Hannemann, A., et al. (2015). The clinical significance of K-Cl cotransport activity in red cells of patients with HbSC disease. Haematologica 100, 595–600. doi: 10.3324/haematol.2014.120402
Reid, H. L., Obi, G. O., and Oli, J. M. (1984). Reduced erythrocyte deformability and hyperfibrinogenemia in Nigerian diabetics with hemoglobin genotype HbAA. Acta Diabetol. Lat. 21, 105–114. doi: 10.1007/BF02591099
Reinhart, W. H., Kayser, B., Singh, A., Waber, U., Oelz, O., and Bärtsch, P. (1991). Blood rheology in acute mountain sickness and high-altitude pulmonary edema. J. Appl. Physiol. 71, 934–938. doi: 10.1152/jappl.1991.71.3.934
Reliene, R., Mariani, M., Zanella, A., Reinhart, W. H., Ribeiro, M. L., del Giudice, E. M., et al. (2002). Splenectomy prolongs in vivo survival of erythrocytes differently in spectrin/ankyrin- and band 3-deficient hereditary spherocytosis. Blood 100, 2208–2215.
Rivera, A. (2002). Modulation of Gardos channel activity by cytokines in sickle erythrocytes. Blood 99, 357–363. doi: 10.1182/blood.V99.1.357
Rivera, A., Rotter, M. A., and Brugnara, C. (1999). Endothelins activate Ca2+-gated K+ channels via endothelin B receptors in CD-1 mouse erythrocytes. Am. J. Physiol. 277, C746–C754. doi: 10.1152/ajpcell.1999.277.4.C746
Rother, R. P., Bell, L., Hillmen, P., and Gladwin, M. T. (2005). The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA 293, 1653–1662. doi: 10.1001/jama.293.13.1653
Sachs, J. R. (2003). “Na+/K+ Pump,” in Red Cell Membrane Transport in Health and Disease, eds I. Bernhardt, and J. C. Ellory (Berlin: Springer), 111–137. doi: 10.1007/978-3-662-05181-8_5
Saldanha, C., Silva, A. S., Gonçalves, S., and Martins-Silva, J. (2007). Modulation of erythrocyte hemorheological properties by band 3 phosphorylation and dephosphorylation. Clin. Hemorheol. Microcirc. 36, 183–194.
Salomao, M., Zhang, X., Yang, Y., Lee, S., Hartwig, J. H., Chasis, J. A., et al. (2008). Protein 4.1R-dependent multiprotein complex: New insights into the structural organization of the red blood cell membrane. Proc. Natl. Acad. Sci. U.S.A. 105, 8026–8031. doi: 10.1073/pnas.0803225105
Salzer, U., and Prohaska, R. (2001). Stomatin, flotillin-1, and flotillin-2 are major integral proteins of erythrocyte lipid rafts. Blood 97, 1141–1143. doi: 10.1182/blood.v97.4.1141
Salzer, U., Zhu, R., Luten, M., Isobe, H., Pastushenko, V., Perkmann, T., et al. (2008). Vesicles generated during storage of red cells are rich in the lipid raft marker stomatin. Transfusion 48, 451–462. doi: 10.1111/j.1537-2995.2007.01549.x
Samaja, M., Brenna, L., Allibardi, S., and Cerretelli, P. (1993). Human red blood cell aging at 5,050-m altitude: a role during adaptation to hypoxia. J. Appl. Physiol. 75, 1696–1701. doi: 10.1152/jappl.1993.75.4.1696
Seider, M. J., and Kim, H. D. (1979). Cow red blood cells. I. Effect of purines, pyrimidines, and nucleosides in bovine red cell glycolysis. Am. J. Physiol. 236, C255–C261.
Shalev, O., Mogilner, S., Shinar, E., Rachmilewitz, E. A., and Schrier, S. L. (1984). Impaired erythrocyte calcium homeostasis in beta-thalassemia. Blood 64, 564–566.
Shimoda, M., Miyashima, K., and Yawata, Y. (1984). Increased calcium uptake in the red cells of unsplenectomized patients with hereditary spherocytosis: significant contribution of reticulocytosis. Clin. Chim. Acta 142, 183–192. doi: 10.1016/0009-8981(84)90378-4
Shin, S., Hou, J. X., Suh, J. S., and Singh, M. (2007). Validation and application of a microfluidic ektacytometer (RheoScan-D) in measuring erythrocyte deformability. Clin. Hemorheol. Microcirc. 37, 319–328.
Shmukler, B. E., Vandorpe, D. H., Rivera, A., Auerbach, M., Brugnara, C., and Alper, S. L. (2014). Dehydrated stomatocytic anemia due to the heterozygous mutation R2456H in the mechanosensitive cation channel PIEZO1: a case report. Blood Cells Mol. Dis. 52, 53–54. doi: 10.1016/j.bcmd.2013.07.015
Sikora, J., Orlov, S. N., Furuya, K., and Grygorczyk, R. (2014). Hemolysis is a primary ATP-release mechanism in human erythrocytes. Blood 124, 2150–2157. doi: 10.1182/blood-2014-05-572024
Simpson, R. J., Florida-James, G. D., Whyte, G. P., Middleton, N., Shave, R., George, K., et al. (2006). The effects of marathon running on expression of the complement regulatory proteins CD55 (DAF) and CD59 (MACIF) on red blood cells. Eur. J. Appl. Physiol. 99, 201–204. doi: 10.1007/s00421-006-0326-2
Smith, J. A. (1995). Exercise, training and red blood cell turnover. Sports Med. 19, 9–31. doi: 10.2165/00007256-199519010-00002
Smith, J. A., Martin, D. T., Telford, R. D., Ballas, S. K., and John, A. (1999). Greater erythrocyte deformability in world-class endurance athletes Greater erythrocyte deformability in world-class endurance athletes. Am. J. Physiol. 276, H2188–H2193.
Spasojević, I., Maksimović, V., Zakrzewska, J., and Bacić, G. (2005). Effects of 5-fluorouracil on erythrocytes in relation to its cardiotoxicity: membrane structure and functioning. J. Chem. Inf. Model. 45, 1680–1685. doi: 10.1021/ci0501746
Sprague, R. S., Stephenson, A. H., Bowles, E. A., Stumpf, M. S., and Lonigro, A. J. (2006). Reduced expression of Gi in erythrocytes of humans with type 2 diabetes is associated with impairment of both cAMP generation and ATP release. Diabetes 55, 3588–3593. doi: 10.2337/db06-0555
Sprague, R. S., Stephenson, A. H., and Ellsworth, M. L. (2007). Red not dead: signaling in and from erythrocytes. Trends Endocrinol. Metab. 18, 350–355. doi: 10.1016/j.tem.2007.08.008
Stefanovic, M., Puchulu-Campanella, E., Kodippili, G., and Low, P. S. (2013). Oxygen regulates the band 3-ankyrin bridge in the human erythrocyte membrane. Biochem. J. 449, 143–150. doi: 10.1042/BJ20120869
Steffen, P., Jung, A., Nguyen, D. B., Müller, T., Bernhardt, I., Kaestner, L., et al. (2011). Stimulation of human red blood cells leads to Ca2+-mediated intercellular adhesion. Cell Calcium 50, 54–61. doi: 10.1016/j.ceca.2011.05.002
Stewart, G. W., Amess, J. A., Eber, S. W., Kingswood, C., Lane, P. A., Smith, B. D., et al. (1996). Thrombo-embolic disease after splenectomy for hereditary stomatocytosis. Br. J. Haematol. 93, 303–310. doi: 10.1046/j.1365-2141.1996.4881033.x
Sugie, J., Intaglietta, M., and Sung, L. A. (2018). Water transport and homeostasis as a major function of erythrocytes. Am. J. Physiol. Circ. Physiol. 314, H1098–H1107. doi: 10.1152/ajpheart.00263.2017
Szygula, Z. (1990). Erythrocytic system under the influence of physical exercise and training. Sports Med. 10, 181–197. doi: 10.2165/00007256-199010030-00004
Tang, H.-Y., Ho, H.-Y., Wu, P.-R., Chen, S.-H., Kuypers, F. A., Cheng, M.-L., et al. (2015). Inability to maintain GSH pool in G6PD-deficient red cells causes futile AMPK activation and irreversible metabolic disturbance. Antioxid. Redox Signal. 22, 744–759. doi: 10.1089/ars.2014.6142
Terra, H. T., Saad, M. J., Carvalho, C. R., Vicentin, D. L., Costa, F. F., and Saad, S. T. (1998). Increased tyrosine phosphorylation of band 3 in hemoglobinopathies. Am. J. Hematol. 58, 224–230. doi: 10.1002/(SICI)1096-8652(199807)58:3<224::AID-AJH11>3.0.CO;2-9
Thali, R. F., Tuerk, R. D., Scholz, R., Yoho-Auchli, Y., Brunisholz, R. A., and Neumann, D. (2010). Novel candidate substrates of AMP-activated protein kinase identified in red blood cell lysates. Biochem. Biophys. Res. Commun. 398, 296–301. doi: 10.1016/j.bbrc.2010.06.084
Thomas, S. L. Y., Bouyer, G., Cueff, A., Egée, S., Glogowska, E., and Ollivaux, C. (2011). Ion channels in human red blood cell membrane: actors or relics? Blood Cells Mol. Dis. 46, 261–265. doi: 10.1016/j.bcmd.2011.02.007
Tiffert, T., Bookchin, R. M., and Lew, V. L. (2003). “Calcium homeostasis in normal and abnormal human red cells,” in Red Cell Membrane Transport in Health and Disease, eds I. Bernhardt, and J. C. Ellory (Berlin: Springer), 373–405. doi: 10.1007/978-3-662-05181-8_15
Tomaiuolo, G. (2014). Biomechanical properties of red blood cells in health and disease towards microfluidics. Biomicrofluidics 8:051501. doi: 10.1063/1.4895755
Tomoda, A., Noble, N. A., Lachant, N. A., and Tanaka, K. R. (1982). Hemolytic anemia in hereditary pyrimidine 5′-nucleotidase deficiency: nucleotide inhibition of G6PD and the pentose phosphate shunt. Blood 60, 1212–1218.
Tuvia, S., Levin, S., Bitler, A., and Korenstein, R. (1998). Mechanical fluctuations of the membrane-skeleton are dependent on F-actin ATPase in human erythrocytes. J. Cell Biol. 141, 1551–1561. doi: 10.1083/jcb.141.7.1551
Tuvia, S., Moses, A., Gulayev, N., Levin, S., and Korenstein, R. (1999). Beta-adrenergic agonists regulate cell membrane fluctuations of human erythrocytes. J. Physiol. 516(Pt 3), 781–792. doi: 10.1111/j.1469-7793.1999.0781u.x
Vader, P., Fens, M. H., Sachini, N., van Oirschot, B. A., Andringa, G., Egberts, A. C., et al. (2013). Taxol(®)-induced phosphatidylserine exposure and microvesicle formation in red blood cells is mediated by its vehicle Cremophor(®) EL. Nanomedicine 8, 1127–1135. doi: 10.2217/nnm.12.163
van Wijk, R., and van Solinge, W. W. (2005). The energy-less red blood cell is lost: erythrocyte enzyme abnormalities of glycolysis. Blood 106, 4034–4042. doi: 10.1182/blood-2005-04-1622
Vives Corrons, J. L., and Besson, I. (2001). Red cell membrane Na+ transport systems in hereditary spherocytosis: relevance to understanding the increased Na+ permeability. Ann. Hematol. 80, 535–539. doi: 10.1007/s002770100342
Vives L Corrons, J. L. (2000). Chronic non-spherocytic haemolytic anaemia due to congenital pyrimidine 5’ nucleotidase deficiency: 25 years later. Best Pract. Res. Clin. Haematol. 13, 103–118. doi: 10.1053/beha.1999.0060
Wagner-Britz, L., Wang, J., Kaestner, L., and Bernhardt, I. (2013). Protein kinase Ca and P-Type Ca2+ Channel CaV2.1 in red blood cell calcium signalling. Cell. Physiol. Biochem. 31, 883–891. doi: 10.1159/000350106
Wang, J., Wagner-Britz, L., Bogdanova, A., Ruppenthal, S., Wiesen, K., Kaiser, E., et al. (2013). Morphologically homogeneous red blood cells present a heterogeneous response to hormonal stimulation. PLoS One 8:e67697. doi: 10.1371/journal.pone.0067697
Wang, S., Dale, G. L., Song, P., Viollet, B., and Zou, M.-H. (2010). AMPKalpha1 deletion shortens erythrocyte life span in mice: role of oxidative stress. J. Biol. Chem. 285, 19976–19985. doi: 10.1074/jbc.M110.102467
Wang, W., Yang, X., López de Silanes, I., Carling, D., and Gorospe, M. (2003). Increased AMP:ATP ratio and AMP-activated protein kinase activity during cellular senescence linked to reduced HuR function. J. Biol. Chem. 278, 27016–27023. doi: 10.1074/jbc.M300318200
Ware, R. E., de Montalembert, M., Tshilolo, L., and Abboud, M. R. (2017). Sickle cell disease. Lancet 390, 311–323. doi: 10.1016/S0140-6736(17)30193-9
Waugh, R. E., Narla, M., Jackson, C. W., Mueller, T. J., Suzuki, T., and Dale, G. L. (1992). Rheologic properties of senescent erythrocytes: loss of surface area and volume with red blood cell age. Blood 79, 1351–1358.
Weed, R. I., LaCelle, P. L., and Merrill, E. W. (1969). Metabolic dependence of red cell deformability. J. Clin. Invest. 48, 795–809. doi: 10.1172/JCI106038
Werre, J. M., Willekens, F. L., Bosch, F. H., de Haans, L. D., van der Vegt, S. G., van den Bos, A. G., et al. (2004). The red cell revisited–matters of life and death. Cell. Mol. Biol. 50, 139–145.
Wieth, J. O. (1979). Bicarbonate exchange through the human red cell membrane determined with [14C] bicarbonate. J. Physiol. 294, 521–539. doi: 10.1113/jphysiol.1979.sp012944
Willekens, F. L., Werre, J. M., Groenen-Döpp, Y. A., Roerdinkholder-Stoelwinder, B., de Pauw, B., and Bosman, G. J. (2008). Erythrocyte vesiculation: a self-protective mechanism? Br. J. Haematol. 141, 549–556. doi: 10.1111/j.1365-2141.2008.07055.x
Yang, B., Ma, T., and Verkman, A. S. (2001). Erythrocyte water permeability and renal function in double knock out mice lacking aquaporin-1 and aquaporin-3. J. Biol. Chem. 276, 624–628. doi: 10.1074/jbc.M008664200
Yeow, N., Tabor, R. F., and Garnier, G. (2017). Atomic force microscopy: from red blood cells to immunohaematology. Adv. Colloid Interface Sci. 249, 149–162. doi: 10.1016/j.cis.2017.05.011
Zanella, A., and Bianchi, P. (2000). Red cell pyruvate kinase deficiency: from genetics to clinical manifestations. Best Pract. Res. Clin. Haematol. 13, 57–81. doi: 10.1053/beha.1999.0057
Zanella, A., Bianchi, P., Fermo, E., and Valentini, G. (2006). Hereditary pyrimidine 5 ’-nucleotidase deficiency: from genetics to clinical manifestations. Br. J. Haematol. 133, 113–123. doi: 10.1111/j.1365-2141.2006.05992.x
Zanella, A., Fermo, E., Bianchi, P., Chiarelli, L. R., and Valentini, G. (2007). Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev. 21, 217–231. doi: 10.1016/j.blre.2007.01.001
Zarychanski, R., Schulz, V. P., Houston, B. L., Maksimova, Y., Houston, D. S., Smith, B., et al. (2012). Mutations in the mechanotransduction protein PIEZO1 are associated with hereditary xerocytosis. Blood 120, 1908–1915. doi: 10.1182/blood-2012-04-422253
Zeuthen, T., and Macaulay, N. (2012). Transport of water against its concentration gradient: fact or fiction? WIREs Membr. Transp. Signal. 1, 373–381. doi: 10.1002/wmts.54
Zhong, R., Liu, H., Wang, H., Li, X., He, Z., Gangla, M., et al. (2015). Adaption to high altitude: an evaluation of the storage quality of suspended red blood cells prepared from the whole blood of Tibetan plateau migrants. PLoS One 10:e0144201. doi: 10.1371/journal.pone.0144201
Keywords: deformability, vesiculation, sickle cell anemia, thalassemia, hereditary spherocytosis, enzymopathies, hydration, hemolysis
Citation: Huisjes R, Bogdanova A, van Solinge WW, Schiffelers RM, Kaestner L and van Wijk R (2018) Squeezing for Life – Properties of Red Blood Cell Deformability. Front. Physiol. 9:656. doi: 10.3389/fphys.2018.00656
Received: 29 January 2018; Accepted: 14 May 2018;
Published: 01 June 2018.
Edited by:
Francisco Javier Alvarez-Leefmans, Wright State University, United StatesReviewed by:
Joy G. Mohanty, National Institute on Aging (NIA), United StatesHector Rasgado-Flores, Rosalind Franklin University of Medicine and Science, United States
Copyright © 2018 Huisjes, Bogdanova, van Solinge, Schiffelers, Kaestner and van Wijk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard van Wijk, ci52YW53aWprQHVtY3V0cmVjaHQubmw=