
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 22 May 2018
Sec. Vascular Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.00579
 Julien Pottecher1,2*
Julien Pottecher1,2* Chris Adamopoulos1,3
Chris Adamopoulos1,3 Anne Lejay1,4
Anne Lejay1,4 Jamal Bouitbir1Anne-Laure Charles1,5
Jamal Bouitbir1Anne-Laure Charles1,5 Alain Meyer1,5Mervyn Singer6
Alain Meyer1,5Mervyn Singer6 Valerie Wolff1,7Pierre Diemunsch1,2
Valerie Wolff1,7Pierre Diemunsch1,2 Gilles Laverny8Daniel Metzger8
Gilles Laverny8Daniel Metzger8 Bernard Geny1,5
Bernard Geny1,5Objectives: Diabetic patients respond poorly to revascularization for peripheral arterial disease (PAD) but the underlying mechanisms are not well understood. We aimed to determine whether diabetes worsens ischemia-reperfusion (IR)-induced muscle dysfunction and the involvement of endogenous protective kinases in this process.
Materials and Methods: Streptozotocin-induced diabetic and non-diabetic rats were randomized to control or to IR injury (3 h of aortic cross-clamping and 2 h of reperfusion). Mitochondrial respiration, reactive oxygen species (ROS) production, protein levels of superoxide dismutase (SOD2) and endogenous protective kinases (RISK and SAFE pathways) were investigated in rat gastrocnemius, together with upstream (GSK-3β) and downstream (cleaved caspase-3) effectors of apoptosis.
Results: Although already impaired when compared to non-diabetic controls at baseline, the decline in mitochondrial respiration after IR was more severe in diabetic rats. In diabetic animals, IR-triggered oxidative stress (increased ROS production and reduced SOD2 levels) and effectors of apoptosis (reduced GSK-3β inactivation and higher cleaved caspase-3 levels) were increased to a higher level than in the non-diabetics. IR had no effect on the RISK pathway in non-diabetics and diabetic rats, but increased STAT 3 only in the latter.
Conclusion: Type 1 diabetes worsens IR-induced skeletal muscle injury, endogenous protective pathways not being efficiently stimulated.
Diabetes is a major risk factor for peripheral arterial disease (PAD) and in patients with diabetes, PAD is more severe and has a poorer response to revascularization (Jude et al., 2001; DeRubertis et al., 2008; Malmstedt et al., 2008). Yet, the mechanisms linking diabetes to worse outcomes in PAD are not well understood. Diabetes per se impairs skeletal muscle mitochondrial function (Kelley et al., 2002; Bonnard et al., 2008; Anderson et al., 2009). In type 1 diabetic patients the skeletal muscles present reduced mitochondrial oxidative phosphorylation, even without obvious vascular abnormalities (Karakelides et al., 2007) and the mitochondrial impairment may even precede hyperglycemia in type 2 diabetes (Petersen et al., 2004). These data suggest that mitochondrial dysfunction may be a common pathway of diabetes and PAD severity.
When PAD ischemia becomes critical, blood flow has to be reestablished by revascularization. However, the return in blood flow causes additional muscle damage. This paradoxical and detrimental effect is known as ischemia-reperfusion injury (IRI). Although the cause of PAD is occlusive arterial disease, muscle mitochondrial dysfunction is also critical for the severity of PAD. These abnormalities include impaired mitochondrial respiration and increased oxidative stress, and are present in both chronic PAD and after ischemia-reperfusion (IR) (Pipinos et al., 2006; Makris et al., 2007; Tran et al., 2012; Guillot et al., 2014; Lejay et al., 2014, 2017; Paradis et al., 2016). While it is generally assumed that muscle mitochondria are more susceptible to IRI in diabetic subjects, data supporting this assertion are lacking. Very little is known regarding the magnitude of the mitochondrial dysfunction and the mechanisms involved in this process.
It is well established that myocardial IR induces the reperfusion injury salvage kinase (RISK) and survivor activating factor enhancement (SAFE) protective pathways (Lecour, 2009; Rossello and Yellon, 2017). Decreased myocardial activation of RISK and SAFE increases the susceptibility of the diabetic heart to IRI (Tsang et al., 2005; Drenger et al., 2011). Acute activation of RISK and SAFE effectors appear to converge on the mitochondria and avert cell damage. In brief, RISK signaling involves protein kinase B (Akt) phosphorylation which, in turn, phosphorylates and inactivates glycogen synthase kinase 3β (GSK-3β). Inactivated GSK-3β cannot induce mitochondrial permeability transition pore (mPTP) opening anymore, thus preventing mitochondrial dysfunction and apoptosis (Juhaszova et al., 2004). SAFE activation involves phosphorylation and hence, activation of signal transducer and activator of transcription 3 (STAT3). Activated STAT3 stimulates mitochondrial respiration, inhibits mPTP opening and attenuates apoptosis. Yet, the involvement of these pathways in skeletal muscle IR is unknown.
The purpose of this study was to investigate the impact of type 1 diabetes on the skeletal muscle IRI and examine the activation of the RISK and SAFE pathways. We assessed mitochondrial respiration, oxidative stress, and effectors of apoptosis and of RISK and SAFE pathways, in streptozotocin-treated, type 1 diabetic rats, in comparison to non-diabetic controls.
Experiments were performed on 8-week-old male Wistar rats (Depré, Saint-Doulchard, France), either vehicle-treated or streptozotocin-treated to induce insulin-dependent type 1 diabetes. The study conformed to the “Principles of laboratory animal care” (NIH publication 85–23, revised 1985) and was approved by the Institutional Animal Care Committee (CREMEAS AL/02/10/06/2009).
Type 1 diabetes was induced in male rats by a single 65 mg/kg streptozotocin injection in the penile vein. Animals were considered diabetic when blood glucose was above 16.7 mmol/L, 8 days after induction of diabetes. Non-diabetic vehicle-treated animals received intravenous saline injection at the same time. Six weeks after diabetes induction, streptozotocin-treated and vehicle-treated rats were individually housed in metabolic cages for 2 days. After the first day (considered an acclimation period), blood glucose concentrations, food and water intake and urine output were recorded over a 24-h period.
Thirty days after diabetes induction, an oral glucose-tolerance test was performed in diabetic rats and non-diabetic animals to confirm the diabetic status of the formers. The test consisted in a 2 g/kg glucose loading given by oral gavage after a 12-h fasting period. Glucose concentrations were determined on tail vein blood at 20, 40, 60, 120, and 180 min after gavage.
As previously described (Mansour et al., 2012), after performing a midline laparotomy under isoflurane anesthesia, the infra-renal abdominal aorta was dissected and freed from adjacent adhesions. All arterial collaterals located between the renal arteries and the aortic bifurcation, were coagulated and sectioned using electrocautery (Geiger®, thermal cautery unit).
Seven weeks after vehicle or streptozotocin injection, rats (referred to as non-diabetic “n,” and diabetic “d,” respectively) were randomly assigned to the control (CON) or IR group. The control groups (nCON, dCON, 8 rats per group) underwent 5 h of isoflurane anesthesia and similar surgical manipulation to the IR groups, except for hindlimb ischemia (sham-operated). The ischemia–reperfusion groups (nIR and dIR, 10 rats per group) underwent 3 h of ischemia induced by infra-renal aortic occlusion and collateral vessel ligation, followed by 2 h of reperfusion. Ischemia was clinically characterized by cyanosis and lack of an arterial pulse distal to the clamp, and biochemically by an increase in capillary blood lactate measured in the right hindlimb (Lactate Pro device, LT1710; Arkray, KGK, Japan).
After reperfusion, gastrocnemius muscles, that are considered more sensitive to IR (Charles et al., 2017), were harvested and either analyzed immediately (mitochondrial respiration) or kept in ice or in liquid nitrogen-cooled isopentane. Animals were sacrificed by heart retrieval under deep anesthesia (5% isoflurane).
Mitochondrial respiration was studied in saponin-skinned fibers of white gastrocnemius muscle (glycolytic muscle), as previously described (Talha et al., 2013). Fibers were separated and subsequently permeabilized in a bath of solution S containing 50 μg/ml saponin for 30 min at 4°C, under shaking. Permeabilized fibers were washed for 10 min under shaking, to remove the saponin, and placed in a bath with the respiratory solution for 5 min twice, in order to remove any phosphates. Finally, oxygen consumption was measured polarographically with a Clark-type electrode in a 3 ml oxygraphic cell (Strathkelvin Instruments, Glasgow, Scotland) at 22.1°C in incubation buffer using 5 mM glutamate and 2.5 mM malate as substrates for complex I, or 25 mM succinate (in combination with 0.02 mM amytal to inhibit complex I) as substrate for complex II.
After recording of basal oxygen consumption (V0), maximal fiber respiration (VMax) rate was measured under continuous stirring in the presence of a saturating amount of ADP (2 mM) as a phosphate acceptor. Relative contributions of the respiratory chain complexes I, III and IV to the global mitochondrial respiratory rates were also determined. When VMax was recorded, the electron flow went through complexes I, III, and IV. For determining VSucc, complex I was blocked with amytal (0.02 mM) and complex II was stimulated with succinate (25 mM). Mitochondrial respiration in these conditions allowed to determine the contribution of complexes II, III, IV activities. Thereafter, N, N, N′, N′-tetramethyl-p-phenylenediamine dihydrochloride (TMPD, 0.5 mM) and ascorbate (0.5 mM) were added as an artificial electron donor to cytochrome c. In these conditions, the activity of cytochrome c oxidase (complex IV) was determined as an isolated step of the respiratory chain (VTMPD). In all cases, mitochondrial respiration assays in skinned fibers were performed immediately after harvesting. Fibers were then dried for 15 min at 150°C and respiration rates were expressed as μM O2/min/g dry weight.
As described previously (Pottecher et al., 2013), 10 μm-thick serial sections of white gastrocnemius muscle were prepared using a cryostat microtome and incubated with dihydroethidium (DHE) that produces red fluorescence when oxidized to ethidium bromide (EtBr) by ROS, including superoxide anion (Li and Jackson, 2002; Sheetz and King, 2002; Charles et al., 2011). To assess ROS production in each skeletal muscle section, mean fluorescence intensity (arbitrary units) was determined in 25 regions of interest under 20× epifluorescence magnification.
As ROS production by diabetic skeletal muscle mitochondria may be confounded by concurrent changes in respiration rates, this may leave raw, unadjusted ROS levels unchanged, while specific ROS production may increase, when considered relative to electron transport (Herlein et al., 2011). Consequently, the mean DHE fluorescence was divided by the mitochondrial respiration rate from skinned fibers.
This assessment is detailed below in the following paragraph.
Gastrocnemius muscles were grounded in a mortar at 4°C in RIPA buffer [50 mM Tris pH 7.5, 1% Nonident P40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 5 mM EDTA, 1 mM PMSF and phosphatase and protease inhibitor cocktails according to the manufacturer's protocol (PhosphoStop and Complete-Mini EDTA free, Roche)]. Homogenates (50 μg of protein, n = 3–6 per group) were electrophoresed on polyacrylamide gels. Proteins were electroblotted onto nitrocellulose membranes using a Trans-blot turbo transfer system (Biorad) and immunodetected using primary antibodies directed against SOD2 (SOD-110, StressGen), Akt (4691, Cell Signaling), phospho-Akt Ser473 (4060, Cell Signaling), phospho-Akt Thr308 (4056, Cell Signaling), GSK-3β (610201, BD Biosciences), phospho-GSK-3β Ser9 (5558, Cell Signaling), STAT3 (9132, Cell Signaling), phospho-STAT3 Tyr705 (9131, Cell Signaling), cleaved caspase-3 (9661, Cell Signaling), tubulin and GAPDH (MAB374, Millipore Upstate Chemicon), according the manufacturer's instructions. Proteins were revealed with secondary antibodies conjugated to horseradish peroxidase (Amersham Biosciences) using an enhanced chemiluminescence detection system (ECLplus, GE Healthcare) and an ImageQuant™ LAS 4000 biomolecular imager (GE Healthcare). Immunodetected proteins were quantified with the FIJI software and normalized to GAPDH levels (except cleaved caspase-3, which was normalized to tubulin levels).
All data are expressed as mean ± standard error of the mean (SEM), and were analyzed using Prism software (GraphPad Prism 5, Graph Pad Software, San Diego, USA). Comparisons between two groups were performed by Student two-tailed t-test. Two-way analysis of variance (ANOVA) was applied to test simultaneously the main effects and interaction for diabetes status and ischemia. Comparisons between more than two groups were performed using one-way ANOVA with Newman-Keuls post-hoc correction for multiple comparisons. Repeated-measures ANOVA was conducted when appropriate. In all cases, a p-value < 0.05 was considered statistically significant.
Compared to vehicle-treated animals, streptozotocin-treated rats displayed typical features of type 1 diabetes with hyperglycemia, polyuria, polydipsia, polyphagia and decreased body weight. Baseline heart rate was similar in both groups although streptozotocin-treated rats exhibited mild arterial hypertension (164 ± 5, mmHg vs. 140 ± 3; p < 0.001). The absolute and relative maximal increases in blood glucose were higher in streptozotocin-treated than in the non-diabetic rats (+319 ± 9 mg/dL vs. +73 ± 4 mg/dL and 309 ± 24% vs. 200 ± 4%, respectively; p < 0.001) (Figure 1). Hind limb capillary blood lactate levels pre-IR were similar between streptozotocin- and vehicle-treated animals (2.8 ± 0.3 and 2.6 ± 0.4 mmol/L respectively, p = NS), and were similarly increased at the end of the 3 h ischemia (17.7 ± 0.5 vs. 18.9 ± 0.9 mmol/L respectively, p = NS).
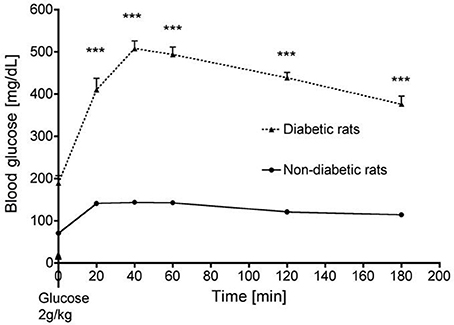
Figure 1. Time course of blood glucose in streptozotocin-treated (diabetic) and non-diabetic rats during an oral glucose tolerance test. ***p < 0.001.
Heart rate changes were also not statistically different between diabetic and non- diabetic animals at baseline, at the end of ischemia and after 2 h of reperfusion (data not shown). These results suggest that streptozotocin- and vehicle-treated animals were subjected to a similar ischemic insult.
Among controls, oxygen consumption was halved in streptozotocin-treated rats as compared to non-diabetic rats (Figure 2A). Ischemia-reperfusion decreased VMax in both diabetic and non-diabetic animals (Figure 2A). However, VMax was significantly more reduced after IR in type 1 diabetic rats compared to the non-diabetics (−57 ± 14% vs. −23 ± 7%; p < 0.05, Figure 2B). While IR did not alter VSucc in non-diabetic animals (−11 ± 6%; p = NS), it induced a significant decrease in VSucc in diabetic rats (−60 ± 7%; p < 0.001 compared to non-diabetic animals). IR did neither alter VTMPD/Asc in streptozotocin- nor in non-diabetic animals (Figure 2B). These results show that, after IR, mitochondrial respiration is more impaired in diabetic than in non-diabetic animals.
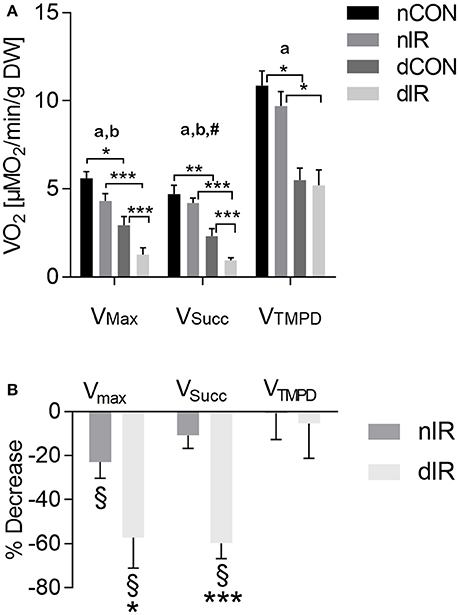
Figure 2. Mitochondrial respiration in gastrocnemius muscles from vehicle-treated and streptozotocin-treated rats with and without hindlimb ischemia-reperfusion. (A) Mitochondrial respiration was determined in gastrocnemius muscles from sham-operated, vehicle-treated (nCON) or streptozotocin-treated rats (dCON), and after ischemia reperfusion in vehicle-treated (nIR) and streptozotocin-treated rats (dIR). Maximal fiber respiration (VMax) rate, combined complexes II, III, IV activities (VSucc) and isolated complex IV activity (VTMPD) were measured. Results are expressed as mean ± SEM [μM O2/min/g dry weight tissue]. *p < 0.05; **p < 0.01; ***p < 0.001, ap < 0.05 for diabetes effect; bp < 0.05 for IR effect; #p < 0.05 for interaction. (B) Variations in mitochondrial respiration in gastrocnemius muscles obtained after ischemia-reperfusion vs. sham operation in vehicle-treated (“nIR”) and streptozotocin- treated rats (“dIR”). Results show a variation in oxygen consumption after ischemia- reperfusion vs. sham operation, expressed as percent decrease ± SEM. §indicates that the variation is significantly different from zero, while *p < 0.05 and ***p < 0.001 indicate that the variation is significantly different between diabetic and non-diabetic rats.
After IR, we observed a 2-fold increase in raw DHE fluorescence in vehicle-treated rats (p < 0.001). In contrast, DHE staining, which was already enhanced in the diabetic rats, was not further increased after IR in this group (Figures 3A,B). However, ROS levels normalized either to VMax (DHE/VMax) or VSucc (DHE/VSucc) were significantly increased after IR in both diabetic and non-diabetic rats, but to a larger extent in diabetic animals (Figure 3B).
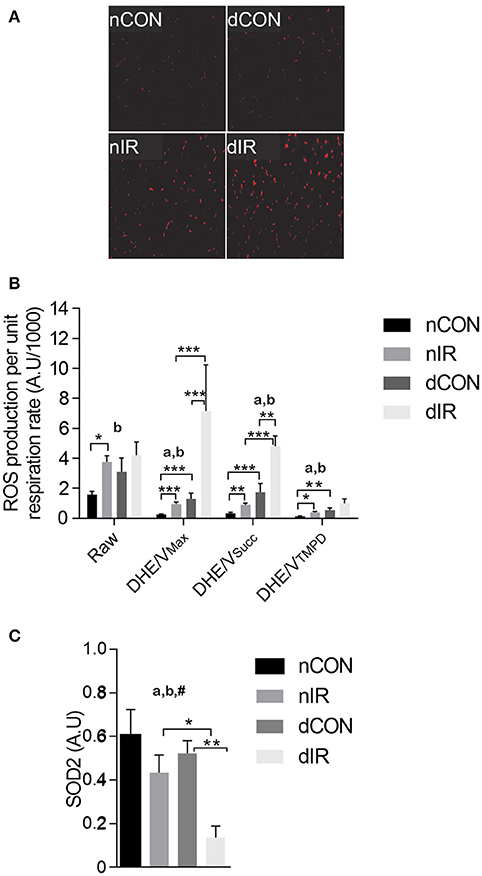
Figure 3. Reactive oxygen species production and antioxidant protein level. (A) Representative histological sections after dihydroethidium (DHE) staining in sham- operated, vehicle-treated (nCON) or streptozotocin-treated rats (dCON), and after ischemia reperfusion in vehicle-treated (nIR) and streptozotocin-treated rats (dIR). (B) Raw and specific (normalized to respiration rate) reactive oxygen species (ROS) production in gastrocnemius muscles from sham-operated, vehicle-treated (nCON) or streptozotocin-treated rats (dCON), and after ischemia reperfusion in vehicle-treated (nIR) and streptozotocin-treated rats (dIR). Data are expressed as mean ± SEM [Arbitrary Unit/μM O2/min/g dry weight]. *p < 0.05; **p < 0.01; ***p < 0.001, ap < 0.05 for diabetes effect; bp < 0.05 for IR effect. (C) Quantification of mitochondrial superoxide dismutase (SOD2) protein level by Western blotting in gastrocnemius muscles from sham-operated, vehicle-treated (nCON) or streptozotocin-treated rats (dCON), and after ischemia reperfusion in vehicle-treated (nIR) and streptozotocin-treated rats (dIR). Data are expressed as the mean ± SEM of the intensity of the SOD2 bands reported to the intensity of the internal control (glyceraldehyde-3-phosphate dehydrogenase, GAPDH). *p < 0.05; **p < 0.01, ap < 0.05 for diabetes effect; bp < 0.05 for IR effect; #p < 0.05 for interaction.
The antioxidant SOD2 level was conversely 3-fold lower in diabetic rats after IR (Figure 3C). Thus, diabetic muscles produced more ROS and had reduced antioxidant defenses after IR.
To test whether the alterations on mitochondrial function and oxidative stress after IR were associated with activation of the endogenous protective kinases, we investigated the effector proteins of RISK (Akt) and SAFE (STAT3) pathways.
Among controls (no IR), the activated Akt (phosphorylated at the threonine 308 and the serine 473 residues) was higher in the non-diabetic rats (p < 0.01). After IR, the phosphorylated Akt levels did neither change in diabetic, nor in non-diabetic rats, indicating no RISK activation. However, both diabetic groups had consistently lower levels of activated Akt than their non-diabetic counterparts (p < 0.01; Figures 4A,B).
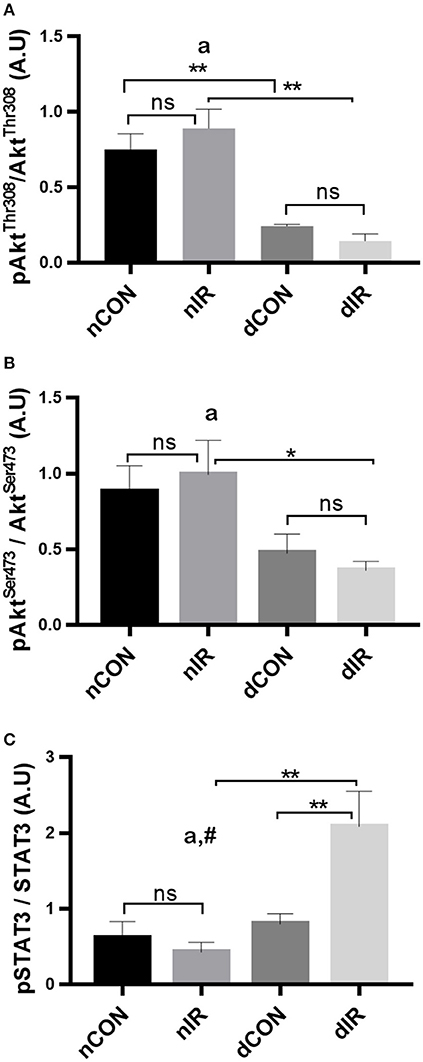
Figure 4. Protein levels of key components of the endogenous protective RISK and SAFE pathways. Quantification of the levels of effector proteins from the RISK pathway [Akt, phospho-Akt/Total Akt Thr308 (A), phospho-Akt/Total Akt Ser473 (B)] and the SAFE pathway [STAT3, phospho-STAT3/Total STAT3 tyr705 (C)] in gastrocnemius muscle from sham-operated, vehicle-treated (nCON) or streptozotocin-treated rats (dCON), and after ischemia reperfusion in vehicle-treated (nIR) and streptozotocin-treated rats (dIR). GAPDH was used as an internal control for all proteins. Data are expressed as the mean ± SEM of the intensity of the bands reported to the intensity of the internal control. *p < 0.05; **p < 0.01, ap < 0.05 for diabetes effect; #p < 0.05 for interaction.
Regarding SAFE pathway, the active, phosphorylated form of STAT3 was not different between diabetic and non-diabetic rats who did not undergo IR. After IR, the phosphorylated STAT3 levels showed a significant 2-fold increase in the diabetic animals only (p < 0.01) (Figure 4C). Therefore, one effector of the SAFE pathway is activated by IR in diabetic but not in in the non-diabetic rats.
While the phosphorylated GSK-3β levels were not altered after IR in non-diabetic animals, they dropped markedly in diabetic rats (p < 0.01), resulting in a 2.5-fold decrease in phospho-GSK-3β/GSK-3β ratio (Figure 5A). Thus, after IR, the protective, phosphorylated GSK-3β was markedly lower in the diabetic than in the non-diabetic animals and was even lower compared to the diabetic rats that did not undergo IR (p < 0.05 and p < 0.01 respectively). Cleaved caspase-3 is a strong downstream effector of mitochondrial-mediated apoptosis and characterized as the “key executioner” of apoptosis.
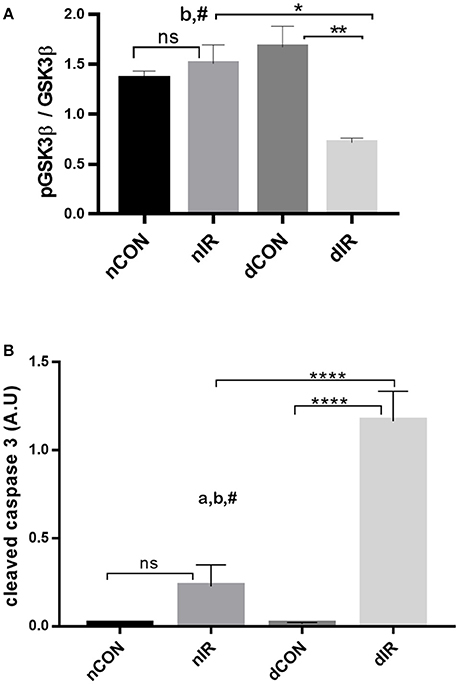
Figure 5. Protein levels of GSK-3β and cleaved caspase-3. Quantification of the levels of (A) GSK-3β, (GSK-3β, phospho-GSK-3β/Total GSK-3β Ser9) and (B) caspase-3 effector of apoptosis in gastrocnemius muscle from sham-operated, vehicle-treated (nCON) or streptozotocin-treated rats (dCON), and after ischemia reperfusion in vehicle-treated (nIR) and streptozotocin-treated rats (dIR). GAPDH was used as an internal control except cleaved caspase-3 for which tubulin was used. Data are expressed as the mean ± SEM of the intensity of the bands reported to the intensity of the internal control. *p < 0.05, **p < 0.001, ****p < 0.0001, ap < 0.05 for diabetes effect; bp < 0.05 for IR effect; #p < 0.05 for interaction.
IR was followed by an increased cleaved caspase-3 expression in gastrocnemius, which was non-significant in non-diabetic animals and highly significant in diabetic rats. Moreover, the increased expression of cleaved caspase 3 was 6-fold larger in the muscles of diabetic animals than in the muscles of their non-diabetic counterparts (p < 0.0001) (Figure 5B).
Taken together, the decreased levels of inactivated GSK-3β and the increased levels of cleaved caspase-3 in the diabetic animals after IR, denote a reduced protection against IRI and a strong trend for apoptosis (presumably through mPTP opening) and cell damage. Representative Western blots of the effector proteins (RISK and SAFE pathways, together with cleaved caspase 3) are provided in Figure 6.
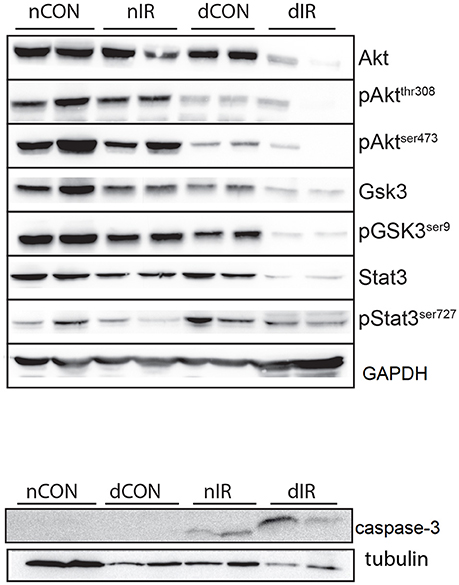
Figure 6. Representative Western blots of the effector proteins. (Upper panel) Effector proteins of RISK and SAFE pathways. (Lower panel) Cleaved caspase-3. GAPDH was used as an internal control except cleaved caspase-3 for which tubulin was used.
Histological gastrocnemius samples from the diabetic-IR rats invariably showed totally distorted anatomy with extensive necrosis and cell lysis, compared to the non-diabetic-IR animals that presented much less severe cell injury (Figure 7).
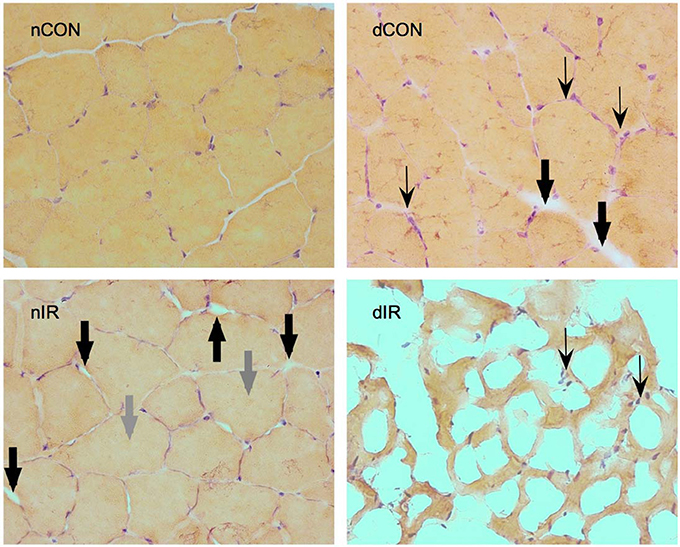
Figure 7. Hematoxylin-eosin staining of gastrocnemius muscles from sham-operated, vehicle-treated (nCON) and streptozotocin-treated rats (dCON), and after ischemia reperfusion in vehicle-treated (nIR) and streptozotocin-treated rats (dIR). Magnification ×400. Compared to nCON, gastrocnemius muscles from dCON rats showed reduced muscle fiber diameter, increased number of inflammatory cells invading the intercellular space (thin black, swept arrowheads) and intercellular edema (large black, solid arrowheads). After IR, gastrocnemius muscles from the nIR group displayed both increased intercellular and intracellular edema (large gray, solid arrowheads). After IR, gastrocnemius from the dIR group showed totally distorted anatomy with extensive necrosis and cell lysis surrounded by clusters of inflammatory cells (thin black, swept arrowheads).
The main finding of this study is that the impact of skeletal muscle IRI is worse in subjects with type 1 diabetes phenotype. Before ischemia, mitochondrial respiration and oxidative stress were more impaired in animals with diabetes than in those without, and these differences became more prominent after IR. Despite activation of one effector of the SAFE pathway, apoptosis and cell damage also appear to be more severe in diabetic subjects after IR. To the best of our knowledge, this is the first study that examined the combined effect of IR and diabetes on skeletal muscle.
Impaired skeletal muscle mitochondrial function and increased oxidative stress are the key elements of PAD-related IR injury (Tran et al., 2011; Guillot et al., 2014). Increased ROS production appears to precede (Guillot et al., 2014) and mediate mitochondrial dysfunction in skeletal muscles. In turn, dysfunctional mitochondria increase ROS production in a vicious cycle (Zorov et al., 2006; Chouchani et al., 2014). Type 1 and type 2 diabetes are also independently associated with increased oxidative stress and defective mitochondrial function, in both humans and experimental models (Kelley et al., 2002; Petersen et al., 2004; Karakelides et al., 2007; Bonnard et al., 2008; Anderson et al., 2009). Put in context, our results indicate that, after IR, diabetes further compromises mitochondrial function and oxidative stress in skeletal muscles, and suggest a cumulative or synergistic deleterious effect of both pathologies. A plausible mechanistic explanation could be that the diabetes-induced compromised mitochondrial function may be predisposing the limb muscles to respond poorly to IRI.
In the same line of evidence, Ryan et al., investigating the impact of type 2 diabetes on mice with acute limb ischemia, also reported exacerbation of skeletal muscle mitochondriopathy, oxidative stress and necrosis in diabetic animals. Although they did not extend their observations to the reperfusion period, they reported a reversal of these disturbances, by overexpression of catalase-mediated antioxidant defenses (Ryan et al., 2016). Bonnard et al. observed mitochondrial alterations that were similar in both type-1 and type-2 diabetic mice and associated with ROS production. Interestingly, the authors found that normalization of glycemia (by insulin) or antioxidant treatment restored mitochondrial integrity and reversed the proapoptotic process in the type-1 diabetic animals (Bonnard et al., 2008). Other studies, focusing on diabetic myocardium, have also shown that diabetes was associated with increased susceptibility to IRI (Whittington et al., 2012). Most of these studies suggested that the hyperglycemia-induced elevated levels of ROS and depletion of antioxidants were the key players in diabetic IRI (Ceriello et al., 2000; Nishikawa et al., 2000; Song et al., 2007).
Although present in skeletal muscles (Sandri, 2008), RISK and SAFE pathways activation has not been previously examined before in skeletal muscle IR. In this study, reduced Akt activation in diabetic animals indicates that the RISK pathway is not activated, and is consistent with the low inhibition of GSK-3β. GSK-3β is the downstream point of convergence of the RISK pathway and of several other prosurvival pathways and when phosphorylated (and thus inhibited) prevents mPTP opening (Juhaszova et al., 2004). Therefore, the lower levels of inactive GSK-3β in diabetic rats after IR may suggest failure of RISK and other protective pathways, ultimately resulting in mPTP opening, apoptosis and cell death. Consistently, downstream to GSK-3β, the proapoptotic cleaved caspase-3 levels were several times higher in diabetic rats. Several cardiac studies in diabetes also reported that hyperglycemia, insulin deficiency and insulin resistance were associated with loss of the conditioning-mediated cardioprotection and that this effect was driven by impaired signaling to Akt and GSK3β of the RISK pathway (Tsang et al., 2005; Gross et al., 2007; Song et al., 2007). In cardiomyocytes, upon IR, Akt activation appeared to be maximal after 10 min, remained elevated until 60 min of reperfusion, and returned toward basal levels by 2 h (Mockridge et al., 2000). Notwithstanding the lack of RISK activation at 2 h of reperfusion in our experiment, activation of RISK at an earlier timepoint cannot be excluded.
In contrast to RISK, SAFE pathway appeared to be activated in our diabetic animals after IR, as we observed an increased activation of STAT3. As a part of the SAFE pathway, STAT3 is involved in ischemic conditioning. Through its localization in the mitochondria of several organs, STAT3 modulates mitochondrial respiration and attenuates ROS production, mPTP opening and apoptosis (Wegrzyn et al., 2009). SAFE activation in diabetic animals should have been protective and yet, we found much higher cleaved caspase-3 levels in this group. These seemingly contradictory results may have several explanations. SAFE and RISK activation protects ischemic myocardium when triggered acutely, at the very first minutes of reperfusion, by specific conditioning maneuvers or pharmacological agents (Rossello and Yellon, 2017). In our study, STAT3 activation was spontaneous and hence, may not be sufficient or not activated early enough, to completely counteract the brunt of the increased oxidative stress. While spontaneous STAT3 activation has been shown to occur after myocardial ischemia and was further increased by reperfusion, it failed to reduce the infarct size (Hausenloy et al., 2011). Interestingly, STAT3 activation, despite its acute protective myocardial effects after IR, has also been strongly implied in atrophying skeletal muscle in experimental models of cancer cachexia or renal failure (Piccirillo and Giavazzi, 2015). Contrary to many chronic conditions, STAT3 activation usually triggers protective mechanisms in acute situations (like IR), probably accounting for the widespread “SAFE” label. In some situations, like in our study, one should probably not use the “SAFE” acronym, because STAT3 is not protective.
In our study, activated cleaved caspase-3 was several times higher in diabetic than in the non-diabetic subjects after IR, suggesting a strong trend toward apoptosis and cell death. Our histological findings, although not formally quantified, strongly corroborated this assertion. Chowdhry et al. reported comparable results in diabetic human myocardium. They demonstrated that diabetes increased both necrosis and apoptosis after IR, and that this effect was mediated by caspase-3 (Chowdhry et al., 2007). Other studies have also shown that increased activation of caspase-3 increased myocardial infarct size and that inhibiting caspases at the time of reperfusion, limited the infarct size (Mocanu et al., 2000; Condorelli et al., 2001). In skeletal muscles, caspases also mediate apoptosis after IR and trigger accelerated proteolysis in catabolic conditions (Du et al., 2004; Tran et al., 2012).
Several limitations of this study need to be acknowledged. Although the general limitations of the preclinical models may also apply to our study, we used a well-characterized pharmacological model of type 1 diabetes. Our data are descriptive and do not test specific mechanisms. That these pathways and mitochondrial function are altered in specific ways, simply make a series of observations and causality cannot be established. RISK and SAFE in skeletal muscle are investigated for the first time in this IR study and the analyses were performed at a single timepoint, after 3 h of ischemia and 2 h of reperfusion. In this regard, further studies are needed to determine the kinetics and the mechanisms of the different effectors. Executioner caspase-3 alone, although strongly inferring apoptosis and cell damage, cannot formally be interpreted as evidence of ultimate cell death. However, the histological findings seem to confirm a more extensive cell damage in the diabetic group.
In conclusion, our experimental data demonstrate that mitochondrial dysfunction, oxidative stress and apoptosis are enhanced in type 1 diabetes, after lower-limb IR. The so-called RISK and SAFE protective pathways appeared not to be activated in diabetic animals after IR, despite STAT 3 enhancement, potentially explaining the increased susceptibility to apoptosis and cell damage. These disturbances may contribute toward the high failure rate of revascularization in diabetic subjects with PAD. More studies are needed to further elucidate the mechanisms of IR mitochondriopathy in diabetes, determine the impact of specific anti-diabetic treatments on mitochondrial IR dysfunction and clarify the kinetics and the role of the protective pathways. Therapeutic approaches targeting mitochondria may reduce lower-limb IR damage and improve local and general prognosis in patients with PAD.
Conception or design of the work JP, AL, JB, A-LC, GL, BG. Acquisition, analysis JP, CA, AL, JB, A-LC, AM, MS, VW, PD, GL, DM, BG. Interpretation of data for the work JP, CA, AL, JB, A-LC, AM, MS, VW, PD, GL, DM, BG. Drafting or revising the work: JP, CA, AL, AM, GL, BG. Final approval JP, CA, AL, JB, A-LC, AM, MS, VW, PD, GL, DM, BG. Agreement to be accountable of all aspects of the work JP, CA, AL, JB, A-LC, AM, MS, VW, PD, GL, DM, BG.
This work was supported by institutional funds only: Bourse des Hôpitaux Universitaires de Strasbourg, Programme Hospitalier de Recherche Clinique, Fondation pour la Recherche Médicale.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are greatly indebted to Fabienne Goupilleau and Isabelle Bentz for their invaluable technical assistance, to Thien-Nga Chamaraux-Tran and Max Guillot for participation in data acquisition and to Christiane Damge for the gift of diabetic rats.
Akt, protein kinase B; DHE, dihydroethidium; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GSK-3β, glycogen synthase kinase 3β; IR, ischemia-reperfusion; IRI, ischemia-reperfusion injury; mPTP, mitochondrial permeability transition pore; PAD, peripheral arterial disease; RISK, reperfusion injury salvage kinases; ROS, reactive oxygen species; SAFE, survivor activating factor enhancement; SOD2, mitochondrial manganese superoxide dismutase; STAT3, signal transducer and activator of transcription 3; TMPD, N, N, N', N'-tetramethyl-p-phenylenediamine dihydrochloride.
Anderson, E. J., Lustig, M. E., Boyle, K. E., Woodlief, T. L., Kane, D. A., Lin, C. T., et al. (2009). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 119, 573–581. doi: 10.1172/JCI37048
Bonnard, C., Durand, A., Peyrol, S., Chanseaume, E., Chauvin, M. A., Morio, B., et al. (2008). Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Invest. 118, 789–800. doi: 10.1172/JCI32601
Ceriello, A., Morocutti, A., Mercuri, F., Quagliaro, L., Moro, M., Damante, G., et al. (2000). Defective intracellular antioxidant enzyme production in type 1 diabetic patients with nephropathy. Diabetes 49, 2170–2177. doi: 10.2337/diabetes.49.12.2170
Charles, A. L., Guilbert, A. S., Bouitbir, J., Goette-Di Marco, P., Enache, I., Zoll, J., et al. (2011). Effect of postconditioning on mitochondrial dysfunction in experimental aortic cross-clamping. Br. J. Surg. 98, 511–516. doi: 10.1002/bjs.7384
Charles, A. L., Guilbert, A.-S., Guillot, M., Talha, S., Lejay, A., Meyer, A., et al. (2017). Muscles susceptibility to ischemia-reperfusion injuries depends on fiber type specific antioxidant level. Front. Physiol. 8:52. doi: 10.3389/fphys.2017.00052
Chouchani, E. T., Pell, V. R., Gaude, E., Aksentijević, D., Sundier, S. Y., Robb, E. L., et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. doi: 10.1038/nature13909
Chowdhry, M. F., Vohra, H. A., and Galiñanes, M. (2007). Diabetes increases apoptosis and necrosis in both ischemic and nonischemic human myocardium: role of caspases and poly-adenosine diphosphate-ribose polymerase. J. Thorac. Cardiovasc. Surg. 134, 124–131, 131.e121–123. doi: 10.1016/j.jtcvs.2006.12.059
Condorelli, G., Roncarati, R., Ross, J. Jr., Pisani, A., Stassi, G., Todaro, M., et al. (2001). Heart-targeted overexpression of caspase3 in mice increases infarct size and depresses cardiac function. Proc. Natl. Acad. Sci. U.S.A. 98, 9977–9982. doi: 10.1073/pnas.161120198
DeRubertis, B. G., Pierce, M., Ryer, E. J., Trocciola, S., Kent, K. C., and Faries, P. L. (2008). Reduced primary patency rate in diabetic patients after percutaneous intervention results from more frequent presentation with limb-threatening ischemia. J. Vasc. Surg. 47, 101–108. doi: 10.1016/j.jvs.2007.09.018
Drenger, B., Ostrovsky, I. A., Barak, M., Nechemia-Arbely, Y., Ziv, E., and Axelrod, J. H. (2011). Diabetes blockade of sevoflurane postconditioning is not restored by insulin in the rat heart: phosphorylated signal transducer and activator of transcription 3- and phosphatidylinositol 3-kinase-mediated inhibition. Anesthesiology 114, 1364–1372. doi: 10.1097/ALN.0b013e31820efafd
Du, J., Wang, X., Miereles, C., Bailey, J. L., Debigare, R., Zheng, B., et al. (2004). Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Invest. 113, 115–123. doi: 10.1172/JCI18330
Gross, E. R., Hsu, A. K., and Gross, G. J. (2007). Diabetes abolishes morphine-induced cardioprotection via multiple pathways upstream of glycogen synthase kinase-3beta. Diabetes 56, 127–136. doi: 10.2337/db06-0907
Guillot, M., Charles, A. L., Chamaraux-Tran, T. N., Bouitbir, J., Meyer, A., Zoll, J., et al. (2014). Oxidative stress precedes skeletal muscle mitochondrial dysfunction during experimental aortic cross-clamping but is not associated with early lung, heart, brain, liver, or kidney mitochondrial impairment. J. Vasc. Surg. 60, 1043–1051 e1045. doi: 10.1016/j.jvs.2013.07.100
Hausenloy, D. J., Lecour, S., and Yellon, D. M. (2011). Reperfusion injury salvage kinase and survivor activating factor enhancement prosurvival signaling pathways in ischemic postconditioning: two sides of the same coin. Antioxid. Redox Signal. 14, 893–907. doi: 10.1089/ars.2010.3360
Herlein, J. A., Fink, B. D., Henry, D. M., Yorek, M. A., Teesch, L. M., and Sivitz, W. I. (2011). Mitochondrial superoxide and coenzyme Q in insulin-deficient rats: increased electron leak. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1616–R1624. doi: 10.1152/ajpregu.00395.2011
Jude, E. B., Oyibo, S. O., Chalmers, N., and Boulton, A. J. (2001). Peripheral arterial disease in diabetic and nondiabetic patients: a comparison of severity and outcome. Diabetes Care 24, 1433–1437. doi: 10.2337/diacare.24.8.1433
Juhaszova, M., Zorov, D. B., Kim, S. H., Pepe, S., Fu, Q., Fishbein, K. W., et al. (2004). Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest. 113, 1535–1549. doi: 10.1172/JCI19906
Karakelides, H., Asmann, Y. W., Bigelow, M. L., Short, K. R., Dhatariya, K., Coenen-Schimke, J., et al. (2007). Effect of insulin deprivation on muscle mitochondrial ATP production and gene transcript levels in type 1 diabetic subjects. Diabetes 56, 2683–2689. doi: 10.2337/db07-0378
Kelley, D. E., He, J., Menshikova, E. V., and Ritov, V. B. (2002). Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51, 2944–2950. doi: 10.2337/diabetes.51.10.2944
Lecour, S. (2009). Multiple protective pathways against reperfusion injury: a SAFE path without Aktion? J. Mol. Cell. Cardiol. 46, 607–609. doi: 10.1016/j.yjmcc.2009.01.003
Lejay, A., Laverny, G., Paradis, S., Schlagowski, A. I., Charles, A. L., Singh, F., et al. (2017). Moderate exercise allows for shorter recovery time in critical limb ischemia. Front. Physiol. 8:523. doi: 10.3389/fphys.2017.00523
Lejay, A., Meyer, A., Schlagowski, A. I., Charles, A. L., Singh, F., Bouitbir, J., et al. (2014). Mitochondria: mitochondrial participation in ischemia-reperfusion injury in skeletal muscle. Int. J. Biochem. Cell Biol. 50, 101–105. doi: 10.1016/j.biocel.2014.02.013
Li, C., and Jackson, R. M. (2002). Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Cell Physiol. 282, C227–C241. doi: 10.1152/ajpcell.00112.2001
Makris, K. I., Nella, A. A., Zhu, Z., Swanson, S. A., Casale, G. P., Gutti, T. L., et al. (2007). Mitochondriopathy of peripheral arterial disease. Vascular 15, 336–343. doi: 10.2310/6670.2007.00054
Malmstedt, J., Leander, K., Wahlberg, E., Karlstrom, L., Alfredsson, L., and Swedenborg, J. (2008). Outcome after leg bypass surgery for critical limb ischemia is poor in patients with diabetes: a population-based cohort study. Diabetes Care 31, 887–892. doi: 10.2337/dc07-2424
Mansour, Z., Bouitbir, J., Charles, A. L., Talha, S., Kindo, M., Pottecher, J., et al. (2012). Remote and local ischemic preconditioning equivalently protects rat skeletal muscle mitochondrial function during experimental aortic cross-clamping. J. Vasc. Surg. 55, 497–505 e491. doi: 10.1016/j.jvs.2011.07.084
Mocanu, M. M., Baxter, G. F., and Yellon, D. M. (2000). Caspase inhibition and limitation of myocardial infarct size: protection against lethal reperfusion injury. Br. J. Pharmacol. 130, 197–200. doi: 10.1038/sj.bjp.0703336
Mockridge, J. W., Marber, M. S., and Heads, R. J. (2000). Activation of Akt during simulated ischemia/reperfusion in cardiac myocytes. Biochem. Biophys. Res. Commun. 270, 947–952. doi: 10.1006/bbrc.2000.2522
Nishikawa, T., Edelstein, D., Du, X. L., Yamagishi, S., Matsumura, T., Kaneda, Y., et al. (2000). Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404, 787–790. doi: 10.1038/35008121
Paradis, S., Charles, A. L., Meyer, A., Lejay, A., Scholey, J. W., Chakfé, N., et al. (2016). Chronology of mitochondrial and cellular events during skeletal muscle ischemia-reperfusion. Am. J. Physiol,. Cell Physiol. 310, C968–C982. doi: 10.1152/ajpcell.00356.2015
Petersen, K. F., Dufour, S., Befroy, D., Garcia, R., and Shulman, G. I. (2004). Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 350, 664–671. doi: 10.1056/NEJMoa031314
Piccirillo, R., and Giavazzi, R. (2015). Inactivating STAT3: bad for tumor, good for muscle. Cell Cycle 14, 939–940. doi: 10.1080/15384101.2015.1010979
Pipinos, I. I., Judge, A. R., Zhu, Z., Selsby, J. T., Swanson, S. A., Johanning, J. M., et al. (2006). Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic. Biol. Med. 41, 262–269. doi: 10.1016/j.freeradbiomed.2006.04.003
Pottecher, J., Guillot, M., Belaidi, E., Charles, A. L., Lejay, A., Gharib, A., et al. (2013). Cyclosporine A normalizes mitochondrial coupling, reactive oxygen species production, and inflammation and partially restores skeletal muscle maximal oxidative capacity in experimental aortic cross-clamping. J. Vasc. Surg. 57, 1100–1108 e1102. doi: 10.1016/j.jvs.2012.09.020
Rossello, X., and Yellon, D. M. (2017). The RISK pathway and beyond. Basic Res. Cardiol. 113:2. doi: 10.1007/s00395-017-0662-x
Ryan, T. E., Schmidt, C. A., Green, T. D., Spangenburg, E. E., Neufer, P. D., and McClung, J. M. (2016). Targeted expression of catalase to mitochondria protects against ischemic myopathy in high-fat diet-fed mice. Diabetes 65, 2553–2568. doi: 10.2337/db16-0387
Sandri, M. (2008). Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 23, 160–170. doi: 10.1152/physiol.00041.2007
Sheetz, M. J., and King, G. L. (2002). Molecular understanding of hyperglycemia's adverse effects for diabetic complications. JAMA 288, 2579–2588. doi: 10.1001/jama.288.20.2579
Song, P., Wu, Y., Xu, J., Xie, Z., Dong, Y., Zhang, M., et al. (2007). Reactive nitrogen species induced by hyperglycemia suppresses Akt signaling and triggers apoptosis by upregulating phosphatase PTEN (phosphatase and tensin homologue deleted on chromosome 10) in an LKB1-dependent manner. Circulation 116, 1585–1595. doi: 10.1161/CIRCULATIONAHA.107.716498
Talha, S., Bouitbir, J., Charles, A. L., Zoll, J., Goette-Di Marco, P., Meziani, F., et al. (2013). Pretreatment with brain natriuretic peptide reduces skeletal muscle mitochondrial dysfunction and oxidative stress after ischemia-reperfusion. J. Appl. Physiol. (1985) 114, 172–179. doi: 10.1152/japplphysiol.00239.2012
Tran, T. P., Tu, H., Liu, J., Muelleman, R. L., and Li, Y. L. (2012). Mitochondria-derived superoxide links to tourniquet-induced apoptosis in mouse skeletal muscle. PLoS ONE 7:e43410. doi: 10.1371/journal.pone.0043410
Tran, T. P., Tu, H., Pipinos, I. I., Muelleman, R. L., Albadawi, H., and Li, Y. L. (2011). Tourniquet-induced acute ischemia-reperfusion injury in mouse skeletal muscles: Involvement of superoxide. Eur. J. Pharmacol. 650, 328–334. doi: 10.1016/j.ejphar.2010.10.037
Tsang, A., Hausenloy, D. J., Mocanu, M. M., Carr, R. D., and Yellon, D. M. (2005). Preconditioning the diabetic heart: the importance of Akt phosphorylation. Diabetes 54, 2360–2364. doi: 10.2337/diabetes.54.8.2360
Wegrzyn, J., Potla, R., Chwae, Y. J., Sepuri, N. B., Zhang, Q., Koeck, T., et al. (2009). Function of mitochondrial Stat3 in cellular respiration. Science 323, 793–797. doi: 10.1126/science.1164551
Whittington, H. J., Babu, G. G., Mocanu, M. M., Yellon, D. M., and Hausenloy, D. J. (2012). The diabetic heart: too sweet for its own good? Cardiol. Res. Pract. 2012:845698. doi: 10.1155/2012/845698
Keywords: diabetes, ischemia-reperfusion, peripheral arterial disease, protective kinases, muscles, mitochondria
Citation: Pottecher J, Adamopoulos C, Lejay A, Bouitbir J, Charles A-L, Meyer A, Singer M, Wolff V, Diemunsch P, Laverny G, Metzger D and Geny B (2018) Diabetes Worsens Skeletal Muscle Mitochondrial Function, Oxidative Stress, and Apoptosis After Lower-Limb Ischemia-Reperfusion: Implication of the RISK and SAFE Pathways? Front. Physiol. 9:579. doi: 10.3389/fphys.2018.00579
Received: 02 February 2018; Accepted: 01 May 2018;
Published: 22 May 2018.
Edited by:
Agustín Guerrero-Hernández, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional (CINVESTAV-IPN), MexicoReviewed by:
Adam Salmon, University of Texas Health Science Center San Antonio, United StatesCopyright © 2018 Pottecher, Adamopoulos, Lejay, Bouitbir, Charles, Meyer, Singer, Wolff, Diemunsch, Laverny, Metzger and Geny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julien Pottecher, anVsaWVuLnBvdHRlY2hlckBjaHJ1LXN0cmFzYm91cmcuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.