 Lars Kaestner
Lars Kaestner Xijia Wang3
Xijia Wang3 Laura Hertz
Laura Hertz Ingolf Bernhardt
Ingolf Bernhardt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Physiol., 27 April 2018
Sec. Membrane Physiology and Membrane Biophysics
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.00450
This article is part of the Research TopicThe red cell life-cycle from erythropoiesis to clearanceView all 22 articles
It is a well-known fact that voltage-activated ion channels are expressed in non-excitable cells (Jagannathan et al., 2002; Piskorowski et al., 2008; Sontheimer, 2008). However, their putative physiological functions and the regulation of their activity in non-excitable cells are controversial topics (Stokes et al., 2004; Badou et al., 2013). This also holds true for red blood cells (RBCs).
In the context of investigating ion transport across the membrane of RBCs, especially in low ionic strength media, the existence of ion transport dependent on the membrane potential was reported for the first time approximately 50 years ago (Donlon and Rothstein, 1969). In an investigation based on comparative physiology, it became evident that the low ionic strength-induced cation permeability in RBCs is not due to electrodiffusion but due to a transport protein-based process (Halperin et al., 1990; Bernhardt et al., 1991). Later, in addition to the K+(Na+)/H+ exchanger (Richter et al., 1997; Kummerow et al., 2000), the existence of a voltage-activated non-selective cation channel was functionally demonstrated utilizing the patch-clamp technique (Christophersen and Bennekou, 1991; Kaestner et al., 1999; Rodighiero et al., 2004). At present, the molecular identity of this particular channel remains unknown (Kaestner, 2011; Bouyer et al., 2012), and it has alternatively been proposed to reflect a conductance state of the voltage-dependent anion channel (VDAC) (Bouyer et al., 2011). On the other hand, evidence for the existence of a number of voltage-activated Ca2+ channels that are abundant in RBCs has been reported (Pinet et al., 2002; Romero et al., 2006), and the most convincing evidence is for CaV2.1, based on molecular biology data (Western blot) (Andrews et al., 2002) and, presumably, CaV2.1-specific pharmacological interactions (ω-agatoxinTK) (Andrews et al., 2002; Wagner-Britz et al., 2013). Nevertheless, so far, we and others have failed to obtain direct functional evidence for the existence of CaV2.1 or other voltage-activated Ca2+ channels in RBCs by patch-clamp techniques.
Although RBCs are undoubtedly non-excitable cells, sudden changes in membrane potential occur, when increased cation permeability is induced. This is the case because the resting membrane potential is determined by Cl− conductance (Hunter, 1977; Lassen et al., 1978). For example, when the Gardos channel (Gardos, 1958; Hoffman et al., 2003) is activated, the resting membrane potential changes from approximately −10 to −90 mV (Tiffert et al., 2003). The physiological function of the Gardos channel remained elusive for decades, until it was discovered that it is a major component of the suicidal process of RBCs (Kaestner and Bernhardt, 2002; Lang et al., 2003; Bogdanova et al., 2013) triggered by Ca2+ entry (Yang et al., 2000; Kaestner et al., 2004), resulting in cell shrinkage (Begenisich et al., 2004; Lew et al., 2005), and phosphatidylserine exposure (Chung et al., 2007; Nguyen et al., 2011). Only very recently was the interplay between the mechanosensitive channel Piezo1 and the Gardos channel established (Faucherre et al., 2013; Cahalan et al., 2015; Danielczok et al., 2017a), showing a Ca2+-mediated response when RBCs pass through constrictions such as small capillaries.
Since patch-clamp protocols for CaV2.1 in RBCs are lacking, imaging approaches based on the Ca2+ fluorophore Fluo-4 are the method of choice (Minetti et al., 2013). The sudden change in membrane potential following Gardos channel activation suggests that there is a link between Gardos channel activity and voltage-activated channels. Such an interplay was already demonstrated in a recent publication showing an 80% reduction in lysophosphatidic acid (LPA)-induced Ca2+ entry by the Gardos channel blocker charybdotoxin (Figure 4A in Wesseling et al., 2016). It would be nice to show CaV2.1 activity in direct response to Gardos channel activation. This is challenging because the activation stimulus for the Gardos channel (an increase in intracellular Ca2+) is the same parameter used to measure CaV2.1 activity. However, Gardos channel activity is increased in patients carrying a mutation that affects the calmodulin-binding site (R352H) (Rapetti-Mauss et al., 2015; Fermo et al., 2017). This could be used as a model to investigate the putative interplay between the Gardos channel and CaV2.1. One would expect an increase in CaV2.1 activity due to increased Gardos channel activity. Consequently, intracellular Ca2+ levels should be elevated in the RBCs of these patients, which indeed is the case (Figure 6 in Fermo et al., 2017). The finding that only a subpopulation of cells showed increased Ca2+ levels (Fermo et al., 2017) can probably be explained by the highly heterogeneous distribution of the participating channels, which is well-established for the Gardos channel (Grygorczyk et al., 1984; Lew et al., 2005) but is also likely to apply to other channels in RBCs (Kaestner, 2015).
Nevertheless, we are still left with one peculiarity: According to previous investigations, CaV2.1 activation is induced by depolarization (Catterall, 2011), not by hyperpolarization, which is the outcome of Gardos channel activity. However, hyperpolarization is a requirement to switch CaV2.1 channels from the inactivated state to the closed state, which is a prerequisite to subsequently transition to the open state (Catterall, 2000) (Figure 1A). Closing of the Gardos channels after their initial activation could well provide the necessary conditions for subsequent depolarisation to activate CaV2.1. Since the hypothetical switching behavior of the Gardos channel would be crucial for the activation of CaV2.1, we would like to discuss this aspect in more detail. We envision three principle modes by which this switching could occur:
(i) Because channel activity is a stochastic event and because the number of Gardos channels per RBC is rather low (in the single digit numbers; Grygorczyk et al., 1984; Wolff et al., 1988), depolarisation could be the result of stochastic Gardos channel closures. This hypothesis is supported by the rather sparse whole cell patch-clamp recordings of Gardos channel activity in human RBCs (Qadri et al., 2011; Kucherenko et al., 2012, 2013; Fermo et al., 2017). Whole cell current traces do not show a smooth appearance but rather a flickering pattern similar to that observed with single channel recordings, especially at higher (positive and negative) membrane potentials.
(ii) When looking at Gardos channel-induced changes in the membrane potential of cell populations, a gradual Ca2+ concentration-dependent effect can be seen (Baunbaek and Bennekou, 2008), i.e., the hyperpolarisation observed in RBC suspensions is a gradual Ca2+ concentration-dependent effect. However, the abovementioned study (Baunbaek and Bennekou, 2008) as well as another report (Seear and Lew, 2011) showed that the activation of the Gardos channel at the cellular level is an all-or-none response. This means that the gradual change in membrane potential would be the result of the summation of cells with open or closed Gardos channels. Taking into consideration that the Ca2+ pump (Schatzmann, 1973) continuously operates in response to any increase in intracellular Ca2+ levels, one would imagine that the state of the Gardos channels is exclusively modulated by variations in intracellular Ca2+ concentrations. Hence, the switching behavior of the Gardos channel would be the direct consequence of continuous variations in RBC intracellular Ca2+ concentrations.
(iii) Localized interactions between the Gardos channel and CaV2.1 in RBCs could occur in lipid rafts or nanodomains, as is the case with closely related ion transporters in other cell types, for example, within the fuzzy space or dyadic cleft in myocytes (Lines et al., 2006). Although RBCs do not possess membrane-constricted subspaces, there are indications for functional compartments in the immediate vicinity of the plasma membrane (Chu et al., 2012). Colocalization of ion channels is common in excitable cells (Rasband and Shrager, 2000; Bers, 2002). For RBCs, it is still unknown if the different ion channels colocalize or cluster to allow their interaction in nanodomains. However, in support of this idea is the observation that local activation of mechanosensitive channels (most likely Piezo 1) by patch-clamp micropipettes resulted in local activation (single-channel recordings) of the Gardos channel (Dyrda et al., 2010).
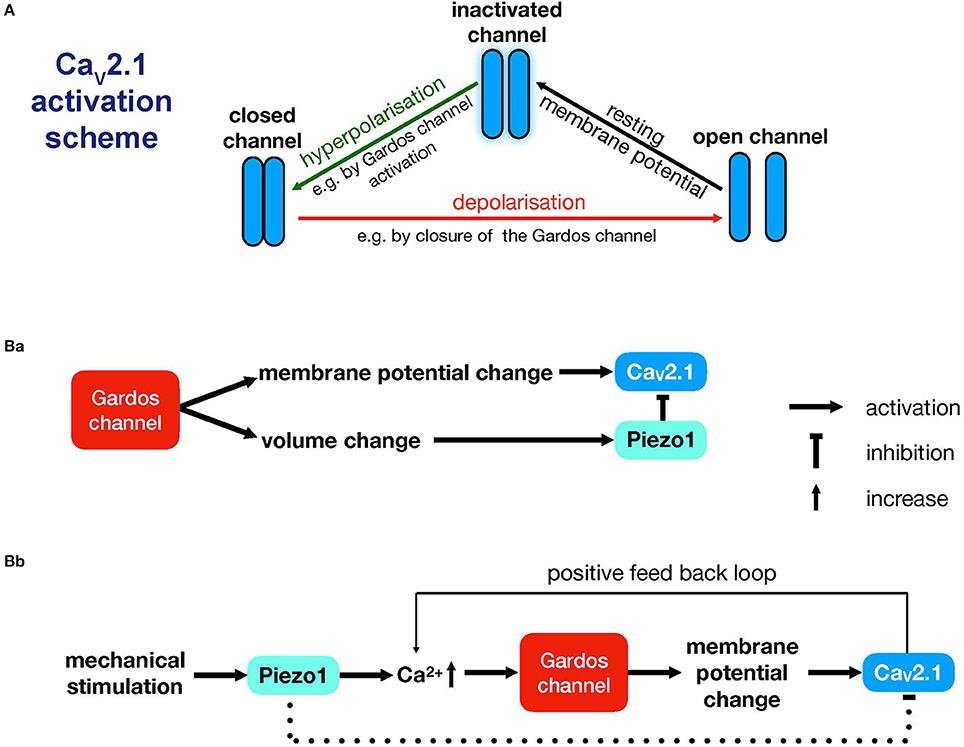
Figure 1. Interplay between CaV2.1, Piezo1 and the Gardos channel. (A) Activation scheme for the CaV2.1 channel modulated by underlying Gardos channel activity. (B) Proposed interactions between CaV2.1, the Gardos channel and Piezo1. (Ba) illustrates a mechanism that is compatible with the measurements presented in Wesseling et al. (2016) and that explains the pathophysiological events seen in Gardos channelopathy (Fermo et al., 2017). (Bb) illustrates a sequence of events that is more likely to be relevant to physiological mechanical stimulation conditions in the circulation (Faucherre et al., 2013; Cahalan et al., 2015; Danielczok et al., 2017a). Whether Piezo1 would exert an inhibitory effect in this scenario is an open question, and therefore, this is depicted with a dashed line. For more details, see the main text.
Although the previous three lines of argumentation are to some extent speculative and although we are unable to favor one over the others, we believe it is worthwhile to share our thoughts with both the RBC and CaV channel research communities in this opinion article. The concerted activity of channels is essential in numerous physiological mechanisms, such as the generation of action potentials in neurons (Rojas et al., 1970), excitation-contraction coupling in the heart (Bers, 2002), and during the formation of the immunological synapse (Quintana et al., 2006). In RBCs, there is evidence that the Gardos channel is activated in response to the opening of Piezo1 (Dyrda et al., 2010; Danielczok et al., 2017a), but the inverse process may also occur; activation of the Gardos channel may induce Piezo1 activity. Since Gardos channel activation is supposed to be associated with RBC volume changes, this effect is likely to activate the mechanosensitive channel Piezo1.
To imagine what may happen when Piezo1 is activated, we need to consider the membrane potential. Activation of Piezo1, which is a non-selective cation channel, would lead to a disruption of the hyperpolarisation induced by the Gardos channel, thus preventing voltage activation of CaV2.1. Therefore, if Piezo1 is closed, CaV2.1 activation would be facilitated, resulting in increased intracellular Ca2+ compared to control conditions. We propose two scenarios explaining the interplay between the Gardos channel, CaV2.1 and Piezo1 (Figure 1B).
The first scenario takes into account the RBCs of patients carrying the R352H mutation (Rapetti-Mauss et al., 2015; Fermo et al., 2017) or the V282M/E mutation (Andolfo et al., 2015; Glogowska et al., 2015; Rapetti-Mauss et al., 2015). The RBCs of these patients show increased baseline Gardos channel activity, which is schematically sketched in Figure 1Ba. This sequence of events can be initiated by the abovementioned mutations or by an increase in intracellular Ca2+ independent of mechanical stress, e.g., by NMDA receptor activity (Makhro et al., 2013), TRPC channel openings (Danielczok et al., 2017b), or VDAC activity (Bouyer et al., 2011).
The second scenario envisions an alternative, independent sequence of events. Piezo1 may indirectly modulate CaV2.1 activity, as outlined in Figure 1Bb. If Piezo1 is the source of the increase in intracellular Ca2+, then subsequent Gardos channel activity would induce the opening of CaV2.1 channels, while Piezo1 channels might still be in an inactivated state. It is likely that Piezo1 channels would remain inactive. After mechanical stimulation, inactivation occurs within 100 ms (Wu et al., 2017), and channel reopening would require a new (repetitive; not lasting) mechanical stimulation (Lewis et al., 2017). Whether the volume change induced by mechanical stress is sufficient for repetitive activation remains unclear, and therefore, the inhibitory effect by Piezo1 is indicated by a dashed line in Figure 1Bb. This mechanism (Figure 1Bb) might explain the long-lasting Ca2+ signal seen after mechanical stimulation and reported in this Research Topic (Danielczok et al., 2017a).
In summary, here, we propose several reasonable mechanisms (Figure 1) to explain how voltage-activated (Ca2+) channels could fulfill a physiological function in non-excitable RBCs. We hope to initiate a discussion on this topic and to encourage further investigations beyond the content of this paper.
LK designed the experimental approach. XW and LH performed and analyzed the experiments. IB and LK supervised the experiments. LK wrote the manuscript. LH and IB revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The research leading to this opinion has received funding from the European Seventh Framework Program under grant agreement number 602121 (CoMMiTMenT) and the European Framework Horizon 2020 under grant agreement number 675115 (RELEVANCE).
Andolfo, I., Russo, R., Manna, F., Shmukler, B. E., Gambale, A., Vitiello, G., et al. (2015). Novel Gardos channel mutations linked to dehydrated hereditary stomatocytosis (xerocytosis). Am. J. Hematol. 90, 921–926. doi: 10.1002/ajh.24117
Andrews, D. A., Yang, L., and Low, P. S. (2002). Phorbol ester stimulates a protein kinase C-mediated agatoxin-TK-sensitive calcium permeability pathway in human red blood cells. Blood 100, 3392–3399. doi: 10.1182/blood.V100.9.3392
Badou, A., Jha, M. K., Matza, D., and Flavell, R. A. (2013). Emerging roles of L-type voltage-gated and other calcium channels in T lymphocytes. Front. Immunol. 4:243. doi: 10.3389/fimmu.2013.00243
Baunbaek, M., and Bennekou, P. (2008). Evidence for a random entry of Ca2+ into human red cells. Bioelectrochemistry 73, 145–150. doi: 10.1016/j.bioelechem.2008.04.006
Begenisich, T., Nakamoto, T., Ovitt, C. E., Nehrke, K., Brugnara, C., Alper, S. L., et al. (2004). Physiological roles of the intermediate conductance, Ca2+-activated potassium channel Kcnn4. J. Biol. Chem. 279, 47681–47687. doi: 10.1074/jbc.M409627200
Bernhardt, I., Hall, A. C., and Ellory, J. C. (1991). Effects of low ionic strength media on passive human red cell monovalent cation transport. J. Physiol. 434, 489–506.
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
Bogdanova, A., Makhro, A., Wang, J., Lipp, P., and Kaestner, L. (2013). Calcium in red blood cells-a perilous balance. Int. J. Mol. Sci. 14, 9848–9872. doi: 10.3390/ijms14059848
Bouyer, G., Cueff, A., Egée, S., Kmiecik, J., Maksimova, Y., Glogowska, E., et al. (2011). Erythrocyte peripheral type benzodiazepine receptor/voltage-dependent anion channels are upregulated by Plasmodium falciparum. Blood 118, 2305–2312. doi: 10.1182/blood-2011-01-329300
Bouyer, G., Thomas, S., and Egée, S. (2012). “Patch-clamp analysis of membrane transport in erythrocytes,” in Patch Clamp Technique (Rijeka: InTech), 171–202.
Cahalan, S. M., Lukacs, V., Ranade, S. S., Chien, S., Bandell, M., and Patapoutian, A. (2015). Piezo1 links mechanical forces to red blood cell volume. Elife 4:e07370. doi: 10.7554/eLife.07370
Catterall, W. A. (2000). Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16, 521–555. doi: 10.1146/annurev.cellbio.16.1.521
Catterall, W. A. (2011). Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 3:a003947. doi: 10.1101/cshperspect.a003947
Christophersen, P., and Bennekou, P. (1991). Evidence for a voltage-gated, non-selective cation channel in the human red cell membrane. Biochim. Biophys. Acta 1065, 103–106.
Chu, H., Puchulu-Campanella, E., Galan, J. A., Tao, W. A., Low, P. S., and Hoffman, J. F. (2012). Identification of cytoskeletal elements enclosing the ATP pools that fuel human red blood cell membrane cation pumps. Proc. Natl. Acad. Sci. U.S.A. 109, 12794–12799. doi: 10.1073/pnas.1209014109
Chung, S. M., Bae, O. N., Lim, K. M., Noh, J. Y., Lee, M. Y., Jung, Y. S., et al. (2007). Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler. Thromb. Vasc. Biol. 27, 414–421. doi: 10.1161/01.ATV.0000252898.48084.6a
Danielczok, J. G., Terriac, E., Hertz, L., Petkova-Kirova, P., Lautenschläger, F., Laschke, M. W., et al. (2017a). Red blood cell passage of small capillaries is associated with transient Ca2+-mediated adaptations. Front. Physiol. 8:979. doi: 10.3389/fphys.2017.00979
Danielczok, J., Hertz, L., Ruppenthal, S., Kaiser, E., Petkova-Kirova, P., Bogdanova, A., et al. (2017b). Does erythropoietin regulate TRPC channels in red blood cells? Cell. Physiol. Biochem. 41, 1219–1228. doi: 10.1159/000464384
Donlon, J. A., and Rothstein, A. (1969). The cation permeability of erythrocytes in low ionic strength media of various tinicities. J. Membr. Biol. 1, 37–52.
Dyrda, A., Cytlak, U., Ciuraszkiewicz, A., Lipinska, A., Cueff, A., Bouyer, G., et al. (2010). Local membrane deformations activate Ca2+-dependent K+ and anionic currents in intact human red blood cells. PLoS ONE 5:e9447. doi: 10.1371/journal.pone.0009447
Faucherre, A., Kissa, K., Nargeot, J., Mangoni, M. E., and Jopling, C. (2013). Piezo1 plays a role in erythrocyte volume homeostasis. Haematologica 99, 70–75. doi: 10.3324/haematol.2013.086090
Fermo, E., Bogdanova, A., Petkova-Kirova, P., Zaninoni, A., Marcello, A. P., Makhro, A., et al. (2017). “Gardos Channelopathy”: a variant of hereditary Stomatocytosis with complex molecular regulation. Sci. Rep. 7:1744. doi: 10.1038/s41598-017-01591-w
Gardos, G. (1958). The function of calcium in the potassium permeability of human erythrocytes. Biochim. Biophys. Acta. 30, 653–654.
Glogowska, E., Lezon-Geyda, K., Maksimova, Y., Schulz, V. P., and Gallagher, P. G. (2015). Mutations in the Gardos channel (KCNN4) are associated with hereditary xerocytosis. Blood 126, 1281–1284. doi: 10.1182/blood-2015-07-657957
Grygorczyk, R., Schwarz, W., and Passow, H. (1984). Ca2+-activated K+ channels in human red cells. Comparison of single-channel currents with ion fluxes. Biophys. J. 45, 693–698. doi: 10.1016/S0006-3495(84)84211-3
Halperin, J. A., Brugnara, C., Van Ha, T., and Tosteson, D. C. (1990). Voltage-activated cation permeability in high-potassium but not low-potassium red blood cells. Am. J. Physiol. Cell Physiol. 258, 1169–1172.
Hoffman, J. F., Joiner, W., Nehrke, K., Potapova, O., Foye, K., and Wickrema, A. (2003). The hSK4 (KCNN4) isoform is the Ca2+-activated K+ channel (Gardos channel) in human red blood cells. Proc. Natl. Acad. Sci. U.S.A. 100, 7366–7371. doi: 10.1073/pnas.1232342100
Hunter, M. J. (1977). Human erythrocyte anion permeabilities measured under conditions of net charge transfer. J. Physiol. 268, 35–49. doi: 10.1111/(ISSN)1469-7793
Jagannathan, S., Publicover, S. J., and Barratt, C. L. (2002). Voltage-operated calcium channels in male germ cells. Reproduction 123, 203–215. doi: 10.1530/rep.0.1230203
Kaestner, L. (2011). Cation channels in erythrocytes - historical and future perspective. Open Biol. J. 4, 27–34. doi: 10.2174/1874196701104010027
Kaestner, L. (2015). Channelizing the red blood cell: molecular biology competes with patch-clamp. Front. Mol. Biosci. 2:46. doi: 10.3389/fmolb.2015.00046
Kaestner, L., and Bernhardt, I. (2002). Ion channels in the human red blood cell membrane: their further investigation and physiological relevance. Bioelectrochemistry 55, 71–74. doi: 10.1016/S1567-5394(01)00164-5
Kaestner, L., Bollensdorff, C., and Bernhardt, I. (1999). Non-selective voltage-activated cation channel in the human red blood cell membrane. Biochim. Biophys. Acta 1417, 9–15.
Kaestner, L., Tabellion, W., Lipp, P., and Bernhardt, I. (2004). Prostaglandin E2 activates channel-mediated calcium entry in human erythrocytes: an indication for a blood clot formation supporting process. Thromb. Haemostasis 92, 1269–1272. doi: 10.1267/THRO04061269
Kucherenko, Y. V., Wagner-Britz, L., Bernhardt, I., and Lang, F. (2013). Effect of chloride channel inhibitors on cytosolic Ca2+ levels and Ca2+-activated K+ (Gardos) channel activity in human red blood cells. J. Membr. Biol. 246, 315–326. doi: 10.1007/s00232-013-9532-0
Kucherenko, Y., Zelenak, C., Eberhard, M., Qadri, S. M., and Lang, F. (2012). Effect of casein kinase 1α activator pyrvinium pamoate on erythrocyte ion channels. Cell. Physiol. Biochem. 30, 407–417. doi: 10.1159/000339034
Kummerow, D., Hamann, J., Browning, J. A., Wilkins, R., Ellory, J. C., and Bernhardt, I. (2000). Variations of intracellular pH in human erythrocytes via K+(Na+)/H+ exchange under low ionic strength conditions. J. Membr. Biol. 176, 207–216. doi: 10.1007/s00232001089
Lang, K. S., Duranton, C., Poehlmann, H., Myssina, S., Bauer, C., Lang, F., et al. (2003). Cation channels trigger apoptotic death of erythrocytes. Cell Death Differ. 10, 249–256. doi: 10.1038/sj.cdd.4401144
Lassen, U. V., Pape, L., and Vestergaard-Bogind, B. (1978). Chloride conductance of the amphiuma red cell membrane. J. Membr. Biol. 39, 27–48.
Lew, V. L., Tiffert, T., Etzion, Z., Perdomo, D., Daw, N., Macdonald, L., et al. (2005). Distribution of dehydration rates generated by maximal Gardos-channel activation in normal and sickle red blood cells. Blood 105, 361–367. doi: 10.1182/blood-2004-01-0125
Lewis, A. H., Cui, A. F., McDonald, M. F., and Grandl, J. (2017). Transduction of repetitive mechanical stimuli by Piezo1 and Piezo2 ion channels. Cell Rep. 19, 2572–2585. doi: 10.1016/j.celrep.2017.05.079
Lines, G. T., Sande, J. B., Louch, W. E., Mørk, H. K., Grøttum, P., and Sejersted, O. M. (2006). Contribution of the Na+/Ca2+ exchanger to rapid Ca2+ release in cardiomyocytes. Biophys. J. 91, 779–792. doi: 10.1529/biophysj.105.072447
Makhro, A., Hänggi, P., Goede, J. S., Wang, J., Brüggemann, A., Gassmann, M., et al. (2013). N-methyl D-aspartate (NMDA) receptors in human erythroid precursor cells and in circulating red blood cells contribute to the intracellular calcium regulation. Am. J. Physiol. Cell Physiol. 305, C1123–C1138. doi: 10.1152/ajpcell.00031.2013
Minetti, G., Egée, S., Mörsdorf, D., Steffen, P., Makhro, A., Achilli, C., et al. (2013). Red cell investigations: art and artefacts. Blood Rev. 27, 91–101. doi: 10.1016/j.blre.2013.02.002
Nguyen, D. B., Wagner-Britz, L., Maia, S., Steffen, P., Wagner, C., Kaestner, L., et al. (2011). Regulation of phosphatidylserine exposure in red blood cells. Cell. Physiol. Biochem. 28, 847–856. doi: 10.1159/000335798
Pinet, C., Antoine, S., Filoteo, A. G., Penniston, J. T., and Coulombe, A. (2002). Reincorporated plasma membrane Ca2+−ATPase can mediate B-Type Ca2+ channels observed in native membrane of human red blood cells. J. Membr. Biol. 187, 185–201. doi: 10.1007/s00232-001-0163-5
Piskorowski, R., Haeberle, H., Panditrao, M. V., and Lumpkin, E. A. (2008). Voltage-activated ion channels and Ca(2+)-induced Ca (2+) release shape Ca (2+) signaling in Merkel cells. Pflugers Arch. 457, 197–209. doi: 10.1007/s00424-008-0496-3
Qadri, S. M., Kucherenko, Y., and Lang, F. (2011). Beauvericin induced erythrocyte cell membrane scrambling. Toxicology 283, 24–31. doi: 10.1016/j.tox.2011.01.023
Quintana, A., Schwarz, E. C., Schwindling, C., Lipp, P., Kaestner, L., and Hoth, M. (2006). Sustained activity of CRAC channels requires translocation of mitochondria to the plasma membrane. J. Biol. Chem. 281, 40302–40309. doi: 10.1074/jbc.M607896200
Rapetti-Mauss, R., Lacoste, C., Picard, V., Guitton, C., Lombard, E., Loosveld, M., et al. (2015). A mutation in the Gardos channel is associated with hereditary xerocytosis. Blood 126, 1273–1280. doi: 10.1182/blood-2015-04-642496
Rasband, M. N., and Shrager, P. (2000). Ion channel sequestration in central nervous system axons. J. Physiol. 525 (Pt 1), 63–73. doi: 10.1111/j.1469-7793.2000.00063.x
Richter, S., Hamann, J., Kummerow, D., and Bernhardt, I. (1997). The monovalent cation “leak” transport in human erythrocytes: an electroneutral exchange process. Biophys. J. 73, 733–745.
Rodighiero, S., De Simoni, A., and Formenti, A. (2004). The voltage-dependent nonselective cation current in human red blood cells studied by means of whole-cell and nystatin-perforated patch-clamp techniques. Biochim. Biophys. Acta 1660, 164–170. doi: 10.1016/j.bbamem.2003.11.011
Rojas, E., Bezanilla, F., and Taylor, R. E. (1970). Demonstration of sodium and potassium conductance changes during a nerve action potential. Nature 225, 747–748.
Romero, P. J., Romero, E. A., Mateu, D., Hernández, C., and Fernández, I. (2006). Voltage-dependent calcium channels in young and old human red cells. Cell Biochem. Biophys. 46, 265–276. doi: 10.1385/CBB:46:3:265
Schatzmann, H. J. (1973). Dependence on calcium concentration and stoichiometry of the calcium pump in human red cells. J. Physiol. 235, 551–569.
Seear, R. V., and Lew, V. L. (2011). IKCa agonist (NS309)-elicited all-or-none dehydration response of human red blood cells is cell-age dependent. Cell Calcium 50, 444–448. doi: 10.1016/j.ceca.2011.07.005
Sontheimer, H. (2008). An unexpected role for ion channels in brain tumor metastasis. Exp. Biol. Med. 233, 779–791. doi: 10.3181/0711-MR-308
Stokes, L., Gordon, J., and Grafton, G. (2004). Non-voltage-gated L-type Ca2+ channels in human T cells: pharmacology and molecular characterization of the major alpha pore-forming and auxiliary beta-subunits. J. Biol. Chem. 279, 19566–19573. doi: 10.1074/jbc.M401481200
Tiffert, T., Bookchin, R. M., and Lew, V. L. (2003). “Calcium homeostasis in normal and abnormal human red cells,” in Red Cell Membrane Transport in Health and Disease, eds I. Bernhardt and C. Ellory (Berlin; Heidelberg; New York, NY: Springer Verlag), 373–405.
Wagner-Britz, L., Wang, J., Kaestner, L., and Bernhardt, I. (2013). Protein kinase Cα and P-type Ca channel CaV2.1 in red blood cell calcium signalling. Cell. Physiol. Biochem. 31, 883–891. doi: 10.1159/000350106
Wesseling, M. C., Wagner-Britz, L., Nguyen, D. B., Asanidze, S., Mutua, J., Mohamed, N., et al. (2016). Novel insights in the regulation of phosphatidylserine exposure in human red blood cells. Cell. Physiol. Biochem. 39, 1941–1954. doi: 10.1159/000447891
Wolff, D., Cecchi, X., Spalvins, A., and Canessa, M. (1988). Charybdotoxin blocks with high affinity the Ca-activated K+ channel of Hb A and Hb S red cells: individual differences in the number of channels. J. Membr. Biol. 106, 243–252.
Wu, J., Young, M., Lewis, A. H., Martfeld, A. N., Kalmeta, B., and Grandl, J. (2017). Inactivation of mechanically activated Piezo1 ion channels is determined by the C-terminal extracellular domain and the inner pore helix. Cell Rep. 21, 2357–2366. doi: 10.1016/j.celrep.2017.10.120
Yang, L., Andrews, D. A., and Low, P. S. (2000). Lysophosphatidic acid opens a Ca(++) channel in human erythrocytes. Blood 95, 2420–2425. Available online at: http://www.bloodjournal.org/content/95/7/2420/tab-article-info
Keywords: red blood cells, CaV2.1, Piezo1, hSK4 (KCNN4), Calcium signaling, Gardos-channelopathy
Citation: Kaestner L, Wang X, Hertz L and Bernhardt I (2018) Voltage-Activated Ion Channels in Non-excitable Cells—A Viewpoint Regarding Their Physiological Justification. Front. Physiol. 9:450. doi: 10.3389/fphys.2018.00450
Received: 17 January 2018; Accepted: 10 April 2018;
Published: 27 April 2018.
Edited by:
Ali Mobasheri, University of Surrey, United KingdomReviewed by:
Agnieszka Zdzisława Robaszkiewicz, University of Łódz, PolandCopyright © 2018 Kaestner, Wang, Hertz and Bernhardt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lars Kaestner, bGFyc19rYWVzdG5lckBtZS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.