 Divya Murthy
Divya Murthy Kuldeep S. Attri
Kuldeep S. Attri Pankaj K. Singh
Pankaj K. Singh
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 04 April 2018
Sec. Gastrointestinal Sciences
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.00335
This article is part of the Research Topic Inositide signaling targets in gastrointestinal diseases View all 7 articles
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive malignancy characterized by its sudden manifestation, rapid progression, poor prognosis, and limited therapeutic options. Genetic alterations in key signaling pathways found in early pancreatic lesions are pivotal for the development and progression of pancreatic intraepithelial neoplastic lesions into invasive carcinomas. More than 90% of PDAC tumors harbor driver mutations in K-Ras that activate various downstream effector-signaling pathways, including the phosphoinositide-3-kinase (PI3K) pathway. The PI3K pathway also responds to stimuli from various growth factor receptors present on the cancer cell surface that, in turn, modulate downstream signaling cascades. Thus, the inositide signaling acts as a central node in the complex cellular signaling networks to impact cancer cell growth, motility, metabolism, and survival. Also, recent publications highlight the importance of PI3K signaling in stromal cells, whereby PI3K signaling modifies the tumor microenvironment to dictate disease outcome. The high incidence of mutations in the PI3K signaling cascade, accompanied by activation of parallel signaling pathways, makes PI3K a promising candidate for drug therapy. In this review, we describe the role of PI3K signaling in pancreatic cancer development and progression. We also discuss the crosstalk between PI3K and other major cellular signaling cascades, and potential therapeutic opportunities for targeting pancreatic ductal adenocarcinoma.
Pancreatic cancer is the third-leading cause of cancer-related morbidity in the United States, owing largely to the short-term survival rates observed for pancreatic cancer patients despite a low incidence rate (Mazure et al., 1997; Siegel et al., 2016; Attri et al., 2017). Pancreatic adenocarcinoma (PDAC), characterized by a strong desmoplastic stromal formation around the cancerous tissues, accounts for more than 90% of pancreatic cancer cases (Mosdell and Doberneck, 1991). In advanced stages of pancreatic cancer development, the primary tumor reaches the surrounding lymph nodes and then disseminates into the distal metastatic organs like liver, lung, and diaphragm (Pour et al., 1991; Attri et al., 2017). The sudden onset of pancreatic cancer combined with rapid progression of the disease accounts for the lack of robust prognostic markers and early-stage diagnostic markers (Kaur et al., 2012; Le et al., 2016). The short-term survival of the pancreatic cancer patients limits the choice of therapeutic interventions available. Gemcitabine remains the standard care of therapy with success rate of 20–30% when used alone and has a greater efficacy when used in a combination therapy based on a multi-targeting approach. However, cancer tissues evolve rapidly and develop resistance to gemcitabine and other associated therapies (Chand et al., 2016). Specifically, cancer cells reprogram their metabolic machinery to confer gemcitabine resistance, making it extremely difficult to treat (Shukla et al., 2017). The current scenario not only demands a search of novel targets that can be utilized in co-therapeutic regimens but also indicates a need for alternative targets in gemcitabine-resistant and gemcitabine-unresponsive patients.
Pancreatic adenocarcinomas are genetically heterogeneous tumors marked by several genetic mutations found in cancer genomes. The oncogenic mutations can either be driver mutations that cause the onset of disease or passenger mutations that amplify the rate of tumor progression through different stages (Makohon-Moore and Iacobuzio-Donahue, 2016). K-Ras is the major driver mutation present in more than 90% of the adenocarcinoma patients (Lennerz and Stenzinger, 2015). The K-Ras mutations are found in early lesions and are involved in the progression of cancer to invasive metastatic PDAC (Eser et al., 2014). G12D and G12V are the most common K-Ras point mutations found in pancreatic cancer patients (Waddell et al., 2015). The genetically engineered mouse models expressing these oncogenic mutations result in constitutive activation of K-Ras, that regulates downstream signaling pathways involved in proliferation, migration, and metastasis of cancer cells (di Magliano and Logsdon, 2013). The passenger mutations frequently observed in tumor-suppressor genes CDKN2A, TP53, or SMAD4, and oncogenes ERBB2 and EGFR, accelerate the formation and progression of invasive pancreatic lesions (Waddell et al., 2015; Notta et al., 2016). The other commonly observed mutations in signaling pathway genes, metabolic genes, and other regulatory factors can also act as passenger mutations to aid in the rapid progression of the disease (Jones et al., 2008; Hardie et al., 2017). The convergent target of these genetic alterations is aberrant signaling observed in pancreatic cancer cells. Targeting these aberrant signaling pathways can provide alternative foci and thus help reduce the variability in the effects of pancreatic cancer treatment modalities.
Signaling pathways are critical for maintaining biological functions of all cellular subtypes in both healthy and cancer cells. Deregulation of these interconnected cellular networks results in a myriad of disease conditions, including cancer (Murthy et al., 2013; Creixell et al., 2015). Aberrant cellular signaling also regulates the other molecular hallmarks of cancer such as evasion of growth suppressors, resisting cell death, replicative immortality, angiogenesis, invasion and metastasis, tumor metabolism, and tumor-promoting immune modulation (Hanahan and Weinberg, 2000; Fouad and Aanei, 2017). The mutant K-Ras signaling in pancreatic cancer patients regulates the downstream inositide signaling pathway (Pirhonen et al., 1990; Jones et al., 2008). Phosphoinositide 3-kinase (PI3K) activity is important for cellular proliferation, protein synthesis, apoptosis, migration, metabolism, cytoskeletal rearrangement, response to growth factors, and malignant transformation (Yuan and Cantley, 2008). PI3K controls these functions by regulating a multitude of downstream signaling cascades such as the mechanistic target of rapamycin (mTOR), nuclear factor kB (NF-κB), glycogen synthase kinase 3 beta (GSK3β), p27 and Bad-Bax pathways (Arcaro and Guerreiro, 2007). In addition to K-Ras, the PI3K can be activated by a variety of oncogenic mutations and growth factor receptors present on the surface of cancer cells. While PI3K inhibitors alone have shown limited success in treating pancreatic cancer patients, the use of PI3K inhibitors in combination with other drugs has shown promising results and are currently in clinical trials.
Mutations present in PI3K pathway genes and other-associated regulated pathway genes contribute to tumorigenesis. The PI3KCA gene mutations present in 3–5% of pancreatic cancer patients can act as activating mutations to initiate pancreatic tumor formation and drive its progression and growth (Payne et al., 2015). In addition to their role in cancer cells, PI3K pathway also regulates key cellular functions of immune cells and plays a pivotal role in the interaction of tumor cells with immune cells. Thus, apart from their classical functions, PI3K also regulates metabolic attributes of cancer cell and tumor microenvironment-mediated regulation of tumor growth and survival (Landis and Shaw, 2014; Okkenhaug et al., 2016).
This review summarizes recent advances in the understanding of the regulation of the Class I PI3K signaling pathway (hereafter referred to as PI3K unless otherwise mentioned) in pancreatic cancer, with a particular emphasis on pancreatic cancer-specific regulators and mutations that modify PI3K-mediated functions. The recent literature also highlights an emergent role of the PI3K signaling network in the regulation of tumor metabolism and immune cell function. Further, this review discusses the scope and progress toward developing PI3K-targeted therapies for the treatment of pancreatic cancer.
The PI3K/Akt survival pathway is a key downstream target of the family of the rat sarcoma virus (Ras), which are proteins primarily involved in cell proliferation. It is estimated that at least 50% of all cancer patients and 60% of all PDAC patients have deregulation of the PI3K/Akt signaling pathway (Bondar et al., 2002; Schlieman et al., 2003; Yuan and Cantley, 2008; Schild et al., 2009). PI3Ks belong to the lipid kinase family that respond to signals from the Ras family, as well as the receptor tyrosine kinases, and regulate diverse cellular functions including cell transformation, proliferation, growth, motility, and survival (Cantley, 2002; Vanhaesebroeck et al., 2010) (Figure 1). Moreover, the PI3K pathway has been reported to inhibit cellular apoptosis to stimulate cell proliferation in cancer cells (Mao et al., 2016).
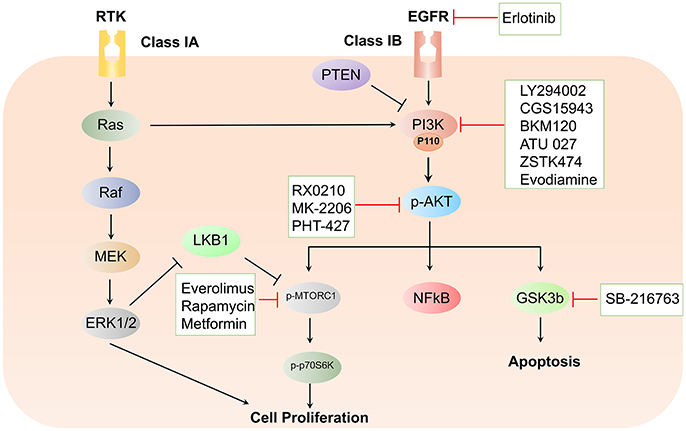
Figure 1. Schematic representation of the PI3K/Akt signaling cascade and targeted therapeutic interventions in pancreatic cancer.
The PI3K functions are mediated by diverse phosphorylated forms of phosphatidylinositols (PtdIns) that perform regulatory functions by binding and recruitment of various phosphoinositide binding domain-containing effector proteins. PI3Ks have been classified into three groups that are expressed in most pancreatic cancer cell lines (Edling et al., 2010). Two classes of PI3K class I have been described in this review, namely class IA and class IB. While class IA is composed of three isoforms of the catalytic subunit known as p110α, p110β, and p110δ, class IB encompasses only p110γ. In addition to differences in composition, there are variations in the cues essential for the activation of subclasses IA and IB. While subclass IA (PI3K α, β, and δ) is activated by receptor tyrosine kinases (RTKs), subclass IB (PI3Kγ) is activated by G-protein-coupled receptors (GCPRs). The PI3K signaling cascade is activated by stimulation of RTKs, which results in the recruitment of PI3Ks to the auto-phosphorylated tyrosine residues on RTKs. The interaction between the Src Homology 2 (SH2) domain in the adaptor subunit with PI3K propels the allosteric activation of the catalytic subunit. Activated Class I PI3Ks catalyze the phosphorylation of PtdIns moieties PtdIns(4,5)P2 (hereafter referred to as PIP2) at their 3′-OH position to generate phosphatidylinositol-3,4,5-triphosphate (hereafter referred to as PIP3). The resultant second messenger PIP3 further recruits pleckstrin homology-(PH) domain-containing signaling proteins to the cancer cell membrane. One such protein is the serine/threonine kinase, phosphoinositide-dependent kinase 1 (PDK1), which is a known activator of Akt/protein kinase B (PKB) (Falasca and Maffucci, 2007; Lien et al., 2017).
The Class II PI3K (PI3KC2) subfamily comprises of three members (in vertebrates), namely, PI3KC2 α, β, and γ (Jean and Kiger, 2014). While the N terminal domain of PI3KC2 has been demonstrated to interact with adapter proteins that control distinct steps of vesicular trafficking processes such as endocytosis and vesicle recycling, the C terminal region binds to PIP2-containing membranes allowing membrane recruitment of PI3KC2α (Stahelin et al., 2006; Posor et al., 2013). The Class III PI3K, Vps34, catalyzes the synthesis of PtdIns(3)P at distinct intracellular membranes and plays a critical role in regulation of endosomal protein sorting, autophagosome formation, endosome-lysosome maturation, and cytokinesis (Sagona et al., 2010; Jaber et al., 2012; Ma et al., 2014). Among the different classes of the PI3K family, the Class I PI3Ks have been widely studied and implicated in pancreatic cancer, and henceforth, will be the focus of this review.
The serine-threonine kinase Akt belongs to the AGC kinase family and is the central signaling molecule that regulates diverse cellular processes critical for cell survival and cell cycle progression (Yamamoto et al., 2004). The Akt kinase family includes Akt1, Akt2, and Akt3, which display distinct features despite extensive structural homology. Interestingly, Akt was originally identified as a regulator of insulin receptor signaling. Akt is composed of the following three domains: (1) an N-terminal pleckstrin homology PH domain, (2) a C-terminal extension (EXT) domain containing a regulatory hydrophobic motif (HM), and (3) a central kinase catalytic subunit (CAT) domain (Kumar and Madison, 2005). Upon PI3K activation, Akt is recruited to the plasma membrane through the interaction of the pleckstrin homology domain with membrane lipids (James et al., 1996). The recruited Akt is phosphorylated by PDK1 at residues Thr308 and by mTORC2 at Ser473 present in the activation loop (Alessi et al., 1997; Sarbassov et al., 2005). The initiation of the Akt signaling cascade stimulates NF-kB, hypoxia-inducible factor-1α (HIF-1α), and Forkhead transcription factors (FoXO), in addition to regulation of the cell cycle and inhibition of apoptosis (Cardone et al., 1998; Ayabe, 1999; Brunet et al., 1999; Zhong et al., 2000). The Akt isoforms carry specific genetic aberrations in diverse types of tumors, which positively correlate to cancer aggressiveness and poor prognosis. Such genetic alterations underline the discrete functional role of Akt in pancreatic cancer development and progression.
The kinase mTOR, another serine/threonine kinase, functions downstream of PI3K signaling. Furthermore, mTOR regulates a wide array of growth-related functions by increasing cell proliferation and survival, protein degradation, and reorganization of the actin cytoskeleton (Foster and Fingar, 2010). While enhanced phosphorylation of ribosomal protein S6 kinase beta-1 (P70S6K) aids in protein synthesis, binding to eukaryotic translation initiation factor 4E (eIF4E) is abolished by phosphorylation of the eIF4E-binding proteins (4E-BPs) to relieve translational block, which leads to protein translation and cell growth (Gingras et al., 1998). Two distinct complexes of mTOR exist within the cells. While mTORC1, which is composed of mTOR, Gbl, and Raptor, transcends signals following PI3K-Akt activation, mTORC2, which is composed of mTOR, Gbl, and Rictor, is involved in full activation of Akt, thus promoting cell proliferation (Hay and Sonenberg, 2004). The activation of mTOR regulates translation of different proteins, including cyclin D1, which regulates cell cycle progression, and HIF-1α, which regulates expression of the pro-angiogenic vascular endothelial growth factor (VEGF) (Grewe et al., 1999). Because mTORC1 is highly sensitive to rapamycin, this family of molecules epitomizes the first-generation of mTOR inhibitors. Efforts are focused on inhibiting the mTORC1 complex, and little attention has been paid to mTORC2, which is largely insensitive to rapamycin. However, in a recent study by Driscoll et al. deletion of the obligate mTORC2 subunit Rictor delayed tumorigenesis. The study further demonstrated the utilization of combined inhibition of mTORC1/2 and PI3K as a potential therapeutic strategy to inhibit the progression of pancreatic cancer (Driscoll et al., 2016).
Being one of the most frequently disrupted tumor suppressors in cancer, a mutation in the phosphatase and tensin homolog deleted on chromosome 10 (PTEN), the natural antagonist of PI3K, relieves the repression of the PI3K/Akt signaling axis in PDAC. Investigators have observed that the development of pre-malignant PanIN in Pdx1-Cre; K-RasG12D/+ mice with conditional deletion of Pten was accelerated and accentuated the phenotype of acinar-to-ductal metaplasia (ADM) (Stanger et al., 2005; Hill et al., 2010). In principle, the PTEN phosphatase dephosphorylates PIP3 to PIP2 and reduces tumor cell growth and survival (Maehama and Dixon, 1998; Cantley and Neel, 1999; Di Cristofano and Pandolfi, 2000; Asano et al., 2004). Additional studies have shown that loss of PTEN expression in 25–70% of cases is concurrent with the short-term overall survival (Asano et al., 2004; Ying et al., 2011). Activation of the NF-κB pathway and its downstream cytokine network had been identified as a key altered pathway on combined oncogenic deletion of K-Ras and Pten. Aberrations in the PTEN/PI3K pathway are thus frequently observed in PDAC which results in activation of tumor-promoting stromal and immune cell populations that shape the PDAC tumor microenvironment (Ying et al., 2011). VEGF, predominantly known to promote tumor angiogenesis, is also inversely regulated by PTEN in pancreatic cancer cells (Ma et al., 2009).
K-Ras belongs to the Ras family of guanosine-5′-triphosphatases (GTPases). Activating K-Ras mutations, mainly in codon 12, are the first genetic changes detected during the progression of pancreatic cancer and are present in 75–90% of all pancreatic adenocarcinomas (Shibata et al., 1990; Dergham et al., 1997; Wang et al., 2002). Oncogenic K-Ras activates a plethora of signaling pathways associated with the survival of cancer cells. Such a characteristic suggests that K-Ras signaling is an ideal drug target to counteract the progression of pancreatic cancer. Classically, growth factor-mediated exogenous stimulation results in activation of Ras GTPases, which dimerize and further regulate downstream effector molecules. Attempts to identify critical Ras effectors in pancreatic duct epithelial cell systems have revealed a dependency of K-Ras on the PI3K/Akt signaling cascade. It is well-established that the PI3K/Akt pathway is activated in human PDAC as well as K-Ras-driven mouse models of pancreatic cancer (Jimeno et al., 2008; Kennedy et al., 2011; Eser et al., 2013). The various mouse models utilized for understanding the role of PI3K have been discussed in Table 1. A recent study, which utilized an in vivo genetic model, demonstrated a critical role of the K-Ras-PI3K-PDK1 axis in mediating ADM, PDAC formation, and maintenance. The enhanced ducts formed from the acinar cells further develop PanIN lesions (Baer et al., 2014). Activation of K-Ras by interaction with the protein-coding gene heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) is associated with upregulation of the mTOR signaling pathway and results in PDAC cell survival and tumor formation in mice (Barcelo et al., 2014). Other than directly activating the PI3K signaling cascade, increased interaction between the K-Ras 4B isoform with calmodulin via the hypervariable region indirectly modulates PI3K signaling (Nussinov et al., 2015). Reactive oxygen species (ROS) is an important determinant of pancreatic cancer pathogenesis. Oncogenic K-Ras-driven metabolic and signaling alterations regulate the production of ROS in pancreatic cancer (Wang et al., 2015; Storz, 2017). Furthermore, the membrane translocation and activation of ROS-producing family of enzymes, namely NADPH oxidases (NOX), is mediated by the PI3K signaling. NOX activation mediates the pro-survival effects of ROS by sustained phosphorylation of JAK2 and by suppressing apoptosis (Lee et al., 2007). Akt plays a direct role in the activation of NOX proteins through NFkB-mediated upregulation of the NOX subunit p22phox (Edderkaoui et al., 2013).
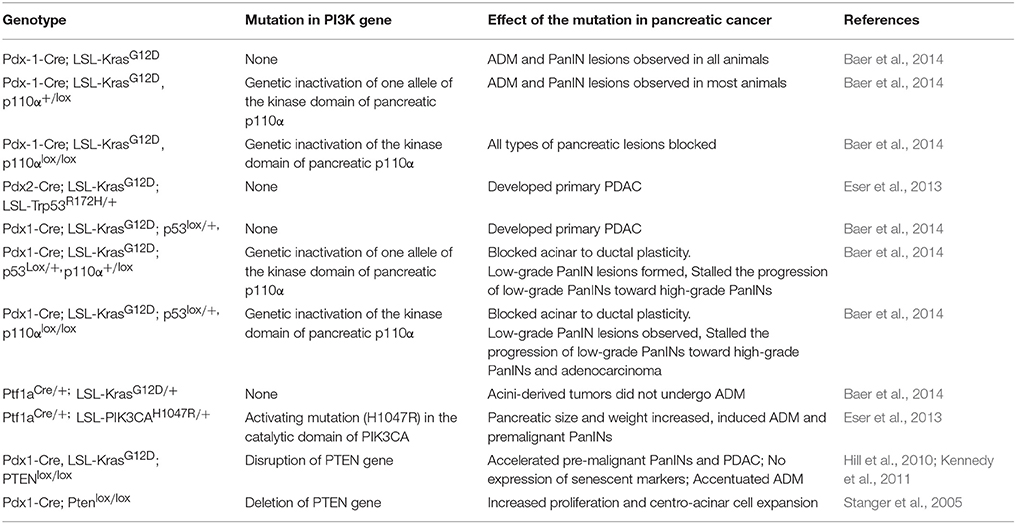
Table 1. Mouse models of pancreatic cancer utilized to understand the role of phosphoinositide signaling pathway in pancreatic cancer.
It is interesting to note that pharmacological inhibition of the protein kinase cascade pathway known as the mitogen-activated protein kinase/extracellular signal-regulated kinases (MAPK/ERK), one of the other well-established mediators of K-Ras-dependent cancer progression, remains ineffective in reducing the tumor burden in K-Ras-mutant cancers (Hayes et al., 2016). However, this recent study by Hayes et al. revealed dynamic reprogramming of signaling networks that resulted in ERK activation. Thus, ERK inhibition is a potential therapeutic approach in K-Ras-dependent pancreatic cancer. Further, the study by Hayes et al. also showed the regulatory roles of PI3K/Akt/mTOR signaling in the sensitivity of the ERK inhibitor. Therefore, co-targeting the ERK signaling pathway along with the PI3K signaling may bestow a distinct and advantageous therapeutic strategy for pancreatic cancer.
A characteristic feature that defines pancreatic cancer is the aberrant overexpression of a high molecular weight glycoproteins, mucins (MUC). Mucins, such as MUC4, have been demonstrated to be the targets of K-RasG12D mutant, which is regulated by a transcriptional or post-transcriptional mechanisms (Vasseur et al., 2015). Furthermore, PI3K signaling is regulated by MUC1, a type I transmembrane glycoprotein that regulates aggressiveness in PDAC by inducing metabolic and signaling alterations (Chaika et al., 2012a; Liu et al., 2014; Mehla and Singh, 2014; Gebregiworgis et al., 2017; King et al., 2017). MUC1 regulates the expression and signaling of multiple receptor tyrosine kinases, including PDGFR, EGFR, and c-Met that signals through the PI3K signaling cascade to drive cellular processes such as proliferation, migration, and survival (Singh and Hollingsworth, 2006; Singh et al., 2007, 2008; Engel et al., 2016). Also, MUC1 mediates the nuclear localization of EGFR to influence the interaction between EGFR and transcriptionally active promoter regions (Bitler et al., 2010). Interestingly, an intimate link between MUC1 cytoplasmic tail expression and the activation of the PI3K-Akt pathway has been observed previously in fibroblasts and thyroid cancer cells (Raina et al., 2004). In accordance with these previous findings, a recent study on non-small cell lung cancer also showed that the interaction between the cytoplasmic domain of MUC1 with the SH2 domain of PI3K p85 subunit is critical for the activation of the PI3K-Akt-mTOR pathway (Kato et al., 2007). Additionally, multidrug resistance is a prominent phenomenon in pancreatic cancer that is modulated by the upregulation of transporters of the ATP-binding cassette (Nath et al., 2013). MUC1 has been further shown to regulate the expression of the multidrug-resistance genes by Akt-dependent and -independent pathways conferring the multidrug-resistance phenotype in pancreatic cancer cells (Nath et al., 2013). It is also pertinent that MUC1-mediated resistance to radiotherapy and chemotherapy may be governed by PI3K signaling (Gunda et al., 2017; Shukla et al., 2017). Therefore, targeting the regulatory axis of MUC-PI3K signaling could be a promising therapeutic strategy for pancreatic cancer.
PI3K and its downstream effectors are constitutively activated in K-Ras-driven pancreatic cancer. p110α, the PI3K class IA subunit, is encoded by PIK3CA and encompasses hotspot mutations in the helical (E542K and E545K) and catalytic domains (H1047R). Such oncogenic mutations result in constitutive activation of the PI3K signaling, as reported in breast and lung cancers (Bader et al., 2005; Liu et al., 2009). Despite the sparse occurrence of activating mutations in p110α in PDAC, the enhanced expression of activated p110α mimics mutated K-Ras-mediated oncogenesis (Schonleben et al., 2006; Jones et al., 2008; Biankin et al., 2012; Eser et al., 2013). When expressed specifically in the pancreas, p110αH1047R induces PI3K activation, leading to enhanced ADM and PanIN formation. Overexpression of p110αH1047R phenocopies mutant K-Ras driven PDAC and is independent of cross-activation of K-Ras (Engelman et al., 2008; Adams et al., 2011; Liu et al., 2011). A subsequent study showed the activation of Akt and GSK3 in a KC mice model (Eser et al., 2013). Along these lines, pancreas-specific expression of kinase-dead p110α isoform inhibited initiation of pancreatic pre-neoplastic lesions. In contrast, the PI3K activity of pancreatic p110β is dispensable for oncogenic K-Ras-induced cancer formation (Baer et al., 2014). In opposition to these findings, Collisson et al. reported the inability of p110αH1047R to induce PanIN and PDAC formation using a Pdx1-CreER mouse line, an alternate murine model utilized to study pancreatic cancer formation and progression. In addition to studies on p110α, a report by Edling et al. highlighted the role of p110γ in oncogenic transformation of pancreatic cells. This study shows increased expression of p110γ in PDAC tissue compared with normal ducts. Further, depletion of p110γ resulted in reduced cell proliferation, emphasizing the participation of p110γ in pancreatic cancer progression (Edling et al., 2010).
Constitutive activation of PI3K-effector Akt is an indicator of the aggressiveness of pancreatic cancer (Edling et al., 2010; Massihnia et al., 2017). In addition to activated Akt in general, Akt2 amplification has been observed in 10–32% of pancreatic adenocarcinomas and contributes to the malignant phenotype in a subset of human PDAC patients (Cheng et al., 1996; Altomare and Testa, 2005). Moreover, amplification of Akt2 has been identified in studies that apply comparative genomic hybridization (array CGH; Liang et al., 2014). Likewise, Akt2 has long been implicated in signaling pathways downstream of various mitogenic growth factors critical in the development of pancreatic cancer (Ruggeri et al., 1998). Interestingly, PDAC hetero-transplants that possess mutant K-Ras and Akt2 amplification are extremely responsive to co-treatment with dactolisib (BEZ235) and panobinostat, resulting in the inhibition of tumor growth (Venkannagari et al., 2012). Mutations and amplification of Akt are thus instrumental in determining the oncogenic landscape of pancreatic cancer.
In addition to amplified Akt, also observed in pancreatic tumor cell lines is decreased PTEN expression accompanied by an elevation in PI3K/Akt signaling. The poor expression of PTEN is shown to be a result of promoter methylation (Asano et al., 2004). Previous studies show the significantly low frequency of deletion or loss-of-function mutations targeting PTEN in human PDAC (Asano et al., 2004). However, recent human PDAC genome analyses, along with mouse genetic studies, have revealed frequent deletion of the PTEN gene in pancreatic tumor specimens, leading to the activation of NF-κB and its downstream cytokine pathway, which is associated with shaping the tumor microenvironment in PDAC (Ying et al., 2011). Further, a multiplatform-based survey was performed to study alterations in the PI3K/Akt/mTOR pathways in over 1,000 cancer cases. Analyses revealed that around 1% of the patients harbored non-silent somatic mutations in the components of the PI3K/Akt/mTOR pathways, < 1% encompassed copy number loss, and 2–5% contained amplification of Akt2, which is consistent with a previous finding (Zhang et al., 2017). Taken together, it is evident that the PI3K/Akt/PTEN signaling loop is a critical signaling hub, which is altered during PDAC initiation and progression.
Pancreatic cancer is characterized by altered metabolic pathways rewired predominantly by the mutant K-Ras. One of the key metabolic changes driven by oncogenic K-Ras involves elevated glucose uptake (Ying et al., 2012; Kerr et al., 2016). In addition to enhanced glycolysis, mutant K-Ras is responsible for the shuttling of glycolytic intermediates to various anabolic pathways such as pentose phosphate pathway and hexosamine biosynthetic pathway, which are critical for the genesis, proliferation, and progression of pancreatic cancer (Ying et al., 2012). The reprogramming of glutamine metabolism and the dependence of PDAC cells on this non-canonical pathway for supporting pancreatic cancer growth is also contingent on oncogenic K-Ras (Son et al., 2013). In addition to its role in regulating anabolic glucose metabolism, K-Ras drives transcriptional reprogramming to elevate the expression of autophagic and macropinocytosis-associated genes in order to meet the metabolic requirements of the cell (Yang et al., 2011; Commisso et al., 2013).
The engagement of K-Ras with various metabolic pathways is primarily mediated by PI3K/Akt and MAPK (Deprez et al., 1997; Barthel et al., 1999). Such regulatory alterations of key metabolic factors are strongly associated with the Akt signaling pathway. Oncogenic K-Ras can enhance the activity of the metabolic enzyme ATP citrate lyase (ACLY) in an Akt-dependent manner, resulting in increased histone acetylation (Lee et al., 2014). Altered histone acetylation can impinge upon cellular metabolism by altering the cellular acetyl-CoA pool, expression patterns of genes, response to DNA damage in cancer cells, and DNA replication (Unnikrishnan et al., 2010; Sulli et al., 2012). The role of PI3K/Akt has been well established in lung adenocarcinoma and hepatoma cells wherein PI3K-mediated enhanced expression levels and membrane localization of GLUT1 were observed (Barthel et al., 1999; Makinoshima et al., 2015). In the case of pancreatic cancer, treatment with the PI3K inhibitors LY294002 and wortmannin led to a significant decrease in the expression of GLUT1 and subsequently, a reduction in the glucose uptake by cells (Melstrom et al., 2008). While stimulation of phosphofructokinase enzymatic activity by Akt has been observed, Akt mediates regulation of the mitochondrial localization of the glycolytic enzymes Hexokinase (HK)1 and HK2 in Rat1a fibroblasts (Majewski et al., 2004).
The PI3K/Akt signaling pathway also regulates the HIF-1α, one of the master regulators of PDAC metabolism (Chaika et al., 2012a; Kang et al., 2014). Notably, it has been shown that inhibition of PI3K/Akt pathway results in a significant decrease in the expression and the DNA-binding ability of HIF-1α (Kilic-Eren et al., 2013). The strong suppressive effects of the downregulation of K-Ras and downstream signaling in the glycolytic pathway via HIF-1α is evident in diverse tumor cells (Mazure et al., 1997; Fukuda et al., 2002; Gao et al., 2002).
MUC16, known to facilitate PDAC progression, regulates the activity of mTOR and consequently, its downstream target c-myc, which is crucial for PDAC growth and metabolism. Reduction in mTOR activity and c-myc expression in MUC16-knockdown cells results in global metabolic alterations that significantly reduce the cellular glycolytic and nucleotide metabolite pools (Shukla et al., 2015). Apart from metabolic regulation of c-myc, PI3K signaling also controls c-myc protein abundance in a GSK3-dependent fashion in PDAC cells (Schild et al., 2009). Although available studies do not adequately demonstrate the direct role of PI3K signaling in commencing metabolic reprogramming of PDAC cells, future studies directed toward elucidating the interplay between this tumorigenic metabolic pathway with PDAC metabolism will potentially identify new test models and therapeutic options for pancreatic cancer.
PDAC is a very unusual tumor where in the stromal cells outnumber the tumor cells (Erkan et al., 2008). The thick desmoplasia makes the tumor microenvironment hypoxic, acidic, and impermeable to drugs, thus creating a barrier to treatment options (Pandol et al., 2009). Studies performed in this arena have shown that manipulating the tumor microenvironment impacts tumor growth kinetics (Whatcott et al., 2015; Abrego et al., 2017). The pancreatic tumor microenvironment is a heterogeneous compartment, majorly consisting of cancer-associated fibroblasts, different immune cells, stellate cells, endothelial cells, and the extracellular matrix. The dynamicity of the tumor microenvironment is mediated through various signaling factors secreted during tumor and accessory cell crosstalk (Feig et al., 2012). Such a heterocellular process of oncogenic cross-signaling results in increased pancreatic cancer proliferation, metastasis, and altered apoptosis. For instance, the interaction between stromal cells and pancreatic cancer cells has been shown to alter intracellular signaling and metabolic pathways in pancreatic cancer cells (Bailey et al., 2008; Hwang et al., 2008; Behrens et al., 2010; Chaika et al., 2012b; Tape et al., 2016; Rucki et al., 2017). Interestingly, PI3K signaling is also regulated by the cross-exchange of such signaling stimuli (Yuan and Cantley, 2008). While the role of PI3K signaling is well-known in the development and function of different immune cells, the significance of PI3K signaling in the pancreatic cancer tumor microenvironment is currently being examined.
Cancer-associated fibroblasts are specialized fibroblasts that constitute the majority of the cells present in the tumor microenvironment (Apte et al., 2013). These fibroblasts are mostly derived from stellate cells in pancreatic cancer (Ohlund et al., 2017) and play a secretory role in the tumor microenvironment by releasing a variety of factors like collagen, proteoglycans, glycoproteins, and other components that comprise the extracellular matrix (Shan et al., 2017). Cancer-associated fibroblasts have been shown to protect cancer cells from chemotherapeutic agents, and increase their proliferation and migration in vitro and in vivo (Xing et al., 2010; Shiga et al., 2015). Similarly, the platelet-derived growth factor secreted by immune cells regulate migration and proliferation of cancer cells by the activation of PI3K signaling pathway (Figure 2; Cho et al., 2016). Another mitogen, cholecystokinin, binds to the receptors present on stellate cells to activate PI3K pathway for regulation of collagen production and fibrosis (Berna et al., 2010; Smith et al., 2014). Pancreatic cancer cells and cancer-associated fibroblasts show a reciprocal release of mitogens from both the cell types that regulate activation of PI3K signaling. This positive loop for PI3K activation is critical for pancreatic cancer progression (Figure 2; Bussard et al., 2016).
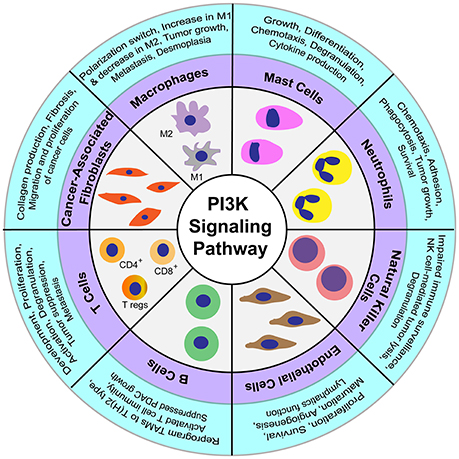
Figure 2. Regulatory functions of PI3K signaling in diverse cellular components constituting the tumor microenvironment.
Macrophages are phagocytic immune cells involved primarily in the defense against pathogens and in the wound-healing process (Ginhoux et al., 2016). Macrophages can exist as either classically activated—M1 macrophages involved in defense against pathogens or alternatively activated—M2 macrophages involved in wound healing and tissue repair (Yoshikawa et al., 2012; Barros et al., 2013). These two polarized subtypes of macrophages play an antagonistic function in the regulation of pancreatic tumor cell growth. While M1 macrophages are pro-tumorigenic, M2 macrophages are anti-tumorigenic (Karnevi et al., 2014). Notably, the survival, adhesion, metabolism, polarization, and motility of macrophages have been shown to be regulated by PI3K signaling (Luyendyk et al., 2008; Xie et al., 2014; Covarrubias et al., 2016). The PI3K signaling pathway also activates Akt1 and Akt2 kinases, thereby regulating the macrophage polarization switch. Furthermore, Akt1-deficient macrophages were shown to produce M1 macrophages that secrete pro-inflammatory cytokines, and Akt2-deficient macrophages were shown to produce M2 polarized macrophages that express Arg1, Fizz1, and interleukin-10 (IL-10) (Chaudhuri, 2014). Another marker associated with PDAC tumor-associated macrophages is PI3Kγ, a class I PI3K lipid kinase isoform specifically expressed in myeloid cells. Inhibition of PI3Kγ in PDAC-associated macrophages suppresses the expression of M2 macrophage markers, while increasing expression of M1 macrophage markers, thereby leading to the suppression of CD8+ T-cell-mediated immunity. Additionally, it has been shown that inhibition of PI3Kγ can suppress tumor growth, invasion, metastasis, and desmoplasia in pancreatic cancer (Figure 2; Kaneda et al., 2016a). In another study, therapeutic targeting of PI3Kγ along with checkpoint inhibitor treatment resulted in tumor suppression and increased survival (Kaneda et al., 2016b). Macrophages also interact with pancreatic stellate cells to regulate fibrosis. Lipopolysaccharide-activated macrophages regulate stellate cell activity through the signaling of transforming growth factor beta (TGF-β) to induce secretion of Th2 cytokines, which in turn polarizes macrophages to the M2 subtype (Schmid-Kotsas et al., 1999; Xue et al., 2015). Moreover, the crosstalk between M1 macrophages and stellate cells, through cytokine signaling, regulates the suppression of pancreatic tumors (Shi C. et al., 2014).
Neutrophils are primary cells recruited to the site of inflammation caused by bacterial infections. These cells migrate to the inflammation sites by sensing chemotactic signals, where the neutrophils release cytokines, chemokines, and proteinases that further recruit macrophages and dendritic cells (Kolaczkowska and Kubes, 2013). Pancreatic cancer cells attract neutrophils into the vicinity of tumor cells and the surrounding stromal compartment (Reid et al., 2011; Chao et al., 2016). The neutrophil influx in tumors correlates with tumor growth and poor prognosis in PDAC patients (Reid et al., 2011; Ino et al., 2013). Moreover, tumor-associated neutrophils promote cancer progression by reactive oxygen species-mediated DNA damage and modulate migration of tumor and other immune cells by secretion of various pro-inflammatory cytokines and chemokines (Scapini et al., 2000; Mangerich et al., 2012). The PI3K signaling pathway in neutrophils is known to regulate survival, growth, adhesion, phagocytosis, and chemotaxis (Figure 2; Engelman et al., 2006; Pinho et al., 2007). However, the exact mechanism and significance of PI3K signaling with respect to neutrophils in PDAC remains elusive.
Mast cells primarily associated with an inflammatory allergic reaction, are also found in the pancreatic cancer tumor microenvironment (Ma et al., 2013). Comparison of human pancreatic cancer samples with the normal pancreas showed an elevated number of mast cells, which were associated with reduced patient survival (Strouch et al., 2010). In a study conducted on spontaneous PDAC K-Ras (G12V) mice, the deficiency of mast cells was shown to suppress pancreatic tumor cell growth (Chang et al., 2011). Importantly, the PI3K pathway is critical in regulating growth, differentiation, chemotaxis, degranulation, and cytokine production in mast cells (Figure 2; Kim et al., 2008).
Natural killer (NK) cells are effector lymphocytic cells of the innate immune system that are known to migrate early to the sites of inflammation during infection. The role of NK cells in cancer is recently being realized, in addition to their classical role in host defense during viral infection (Cerwenka and Lanier, 2016). The cytotoxic activity of NK cells is not only regulated by the extent of receptors present on the cell surface, but also by sensing the availability of ligands in the modified microenvironment (Long et al., 2013). NK cells can elicit their effector function without being educated on the epitopes present in the altered cells or undergoing any clonal selection (Cerwenka and Lanier, 2016). Activating killer receptors NKp30 and NKG2D are two important receptors present on the NK cell surface that are primarily involved in tumor cell recognition and subsequent killing (Peng et al., 2013). Cancer-associated fibroblasts, macrophages, T cells, and other accessory cells present in the tumor microenvironment are also known to modulate expression of the NKG2D receptor and production of interferon gamma (IFNγ) in NK cells (Vitale et al., 2014). Additionally, inhibition of the PI3K signaling pathway in NK cells impairs their function in immune surveillance by preventing their degranulation activity (Jiang et al., 2000). The activity of NK cells is reduced in pancreatic cancers, and the decreased expression of NKG2D, NKp30, and NKp46 correlates with tumor progression in pancreatic cancer patients (Figure 2; Peng et al., 2013, 2014). Furthermore, expression of the receptors CD226 and CD96 in NK cells is associated with cell dysfunction-mediated immune escape and the progression of pancreatic cancer (Peng et al., 2016). In contrast to the tumoricidal function of the receptor NKG2D in NK cells, its expression in cancer cells regulates proliferation and metastasis by activation of PI3K signaling (Benitez et al., 2011; El-Gazzar et al., 2014). Moreover, treatment of pancreatic cancer cells by valproic acid, a histone deacetylase inhibitor, has been shown to promote NK cell-mediated lysis of tumors by activation of PI3K/Akt signaling pathway (Figure 2; Shi P. et al., 2014). Taken together, these studies suggest that PI3K signaling pathway in NK cells is also critical for regulating the pancreatic tumor biology.
Adaptive immune cells are responsible for a range of functions, from long-term memory and pathogenic infections to complex diseases like cancer (Grivennikov et al., 2010). Adaptive immune responses are mostly suppressed in pancreatic cancer due to the evolution of immune escape mechanisms, immunoediting, and the development of mechanisms of immune resistance (Gajewski et al., 2013). CD3+ T cells are the key adaptive immune cells known to invade the stroma of tumor cells (Emmrich et al., 1998). CD3+ T lymphocytes mainly consist of CD4+ helper T cells, CD8+ cytotoxic T cells, and the regulatory T cells (Tregs). Interestingly, increased migration of Tregs in tumors and blood circulation is associated with a decrease in tumor progression (Hiraoka et al., 2006). The infiltration of Tregs in pancreatic tumors is mediated by binding of tumor-secreted chemokines to receptors on the Treg cell surfaces (Tan et al., 2009). In addition to Tregs, T helper cells play key roles in tumor cell growth or inhibition. While the response of type 2 T helper cells (Th2) is critical for imparting tolerance to tumors, the immune response of CD4+ T-cell-mediated type 1 T helper (Th1) is known to promote the death of pancreatic cancer cells (Tassi et al., 2008). Notably, in pancreatic cancer patients, an increased level of tumor-infiltrating CD4+ T cells is associated with improved survival (Ino et al., 2013). However, pancreatic cancer cells inhibit the proliferation and migration of CD4+ T cells (Fogar et al., 2011). Similarly, targeting the plasticity in Th1/2 subtype switch has shown promise as a candidate for cancer immunotherapy (Tassi et al., 2009). On the other hand, CD8+T cells comprise the most abundant population of T cells in pancreatic cancer, and a reduction in this T cell population is observed during the progression of pancreatic cancer (Ene-Obong et al., 2013; Shibuya et al., 2014). Specifically, pancreatic cancer cells inhibit the cytotoxic action of T cells by multiple mechanisms including inhibition of perforin and granzyme secretion, lack of expression of major histocompatibility complex (MHC) class I molecules, and expression of the programmed death-ligand 1 (PD-L1) by tumor cells that bind to the programmed cell death-1 (PD-1) to suppress the function of CD8+T cells (Dong et al., 2002; Ryschich et al., 2005; Thomas and Massague, 2005). Critical for the development of T cells is the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) receptor expressed on CD4+T and CD8+ T cells. Activation of the CTLA-4 receptor is required for suppression of the immune response by Treg cells, resulting in a decrease in cytotoxic T cells. Blockage of CTLA-4 mounts an effective immune response mediated by an increased cytotoxic T cell population and a decreased Treg population to suppress tumor progression (Johansson et al., 2016). Notably, the PI3K pathway is critical for the development, proliferation, and activation of T cells. For example, disruption of the PI3Kδ gene by a D910A-inactivating point mutation in mice impaired T and B cell receptor signaling, leading to a suppressed immune response (Okkenhaug et al., 2002). Furthermore, PI3K inhibitor treatment in a KPC mouse model of PDAC reduced disease pathology and metastasis, and prolonged survival (Figure 2). This immunoprotective effect in pancreatic lesions correlated with a decrease in Treg cells concomitant with an increase in CD44highCD8+ T lymphocyte population (Ali et al., 2014). PI3K-mTOR pathway has also been implicated in regulating the release of granzyme B by Treg cells. Deletion of PI3Kδ affected the secretory perforin-granzyme pathway leading to degranulation of cytotoxic T lymphocytes to impact tumor surveillance (Figure 2; Putz et al., 2012). Thus, targeting pathways of the immune response, with a particular focus on T cells, holds promise as a potential treatment strategy.
B cells are known to infiltrate tumor cells during PDAC progression and support the growth of cancer cells by suppressing CD8+ T cells and tumor-associated macrophages (TAMs). Likewise, inhibition or genetic deletion of B cells leads to the activation of CD8+ T cells that further inhibit tumor cell growth (Roghanian et al., 2016). A study conducted in a mouse model of PDAC showed that the stroma in PanIN lesions secretes the chemokine ligand 13 (CXCL13), a chemokine that attracts B cells to the tumor periphery. These recruited B cells promote the proliferation of transformed epithelial cells via interleukin-35 (IL-35)-mediated paracrine signaling (Pylayeva-Gupta et al., 2016). Further, HIF-1α deletion in the KC mouse model resulted in enhanced secretion of B cell chemoattractants that increased the population of the B cell subclass B1b in early pancreatic neoplasia. Notably, depletion of B cells has been shown to result in reduced progression of PanINs and tumorigenesis in the mice (Lee et al., 2016). In a study on Bruton tyrosine kinase (BTK) expressed in B cells and macrophages, this enzyme was shown to play a critical role in suppressing PDAC growth. Specifically, the B cell-macrophage crosstalk was demonstrated to reprogram TAMs to Th2 type via the activation of BTK in a PI3Kγ-dependent manner. Inhibition of either BTK or PI3Kγ was found to reprogram macrophages to Th1 type leading to activation of T cell-mediated immunity to control pancreatic cancer growth (Figure 2; Gunderson et al., 2016). Thus, the interconnected signaling pathways of B cell and TAMS are critical for regulating tumor growth.
Endothelial cells present in the tumor microenvironment are critical for regulating angiogenesis and maintaining vasculature inside tumors. Remodeling of the local tumor vasculature is critical for the exchange of nutrients and signaling molecules between the hypoxic core and the peripheral regions (Folkman, 2007). Pancreatic tumor cells secrete high levels of VEGF that in turn bind to VEGF receptors present on the endothelial cells (Yamazaki et al., 2008). This interaction activates PI3K signaling crucial for proliferation, survival, and maturation of endothelial cells (Figure 2; Luo et al., 2001). Apart from VEGF receptor, endothelial cells secrete various other receptors like TIE, PDGFR, FGR, and ERBB receptor tyrosine kinases, whose activation triggers PI3K signaling (Hofer and Schweighofer, 2007). Hence, PI3K signaling acts as a key regulator of angiogenesis and lymphatic vessel formation (Hamada et al., 2005).
Constitutively activated mutant K-Ras signals via the MAPK, PI3K-Akt, NF-κB, WNT–β-catenin, Notch, and SMAD pathways. The existence of complex cross-signaling and feedback loops between these pathways remains one of the major factors in determining the development of resistance to therapeutic drug regimens. In addition to K-Ras-driven PI3K activation, aberrant expression of the PTEN protein results in constitutive activation of the PI3K and Akt signaling pathways in pancreatic cancer. In light of such dysregulation of the PI3K signaling cascade, intense research has been directed toward the development of inhibitors that target this critical node. Various classes of inhibitors have been developed that specifically target PI3K, Akt, and mTOR signaling pathways (Figure 1). Interestingly, many of these inhibitors are under clinical trials, thereby paving a new path for improved therapeutic approaches pertinent to the treatment of PDAC.
Multiple generations of PI3K inhibitors have been developed, many of which are under clinical trials (Table 2). Historically, the poor pharmacokinetic properties of various Pan PI3K inhibitors, such as Wortmannin and LY294002, has led to the evolution of next-generation PI3K inhibitors that include small molecule drugs, such as BKM120, GDC0941, GSK2126458, and RNA interfering (RNAi) agent, ATU027 (Khan et al., 2013). Despite the success of many of these drugs, one of the major challenges that contribute to the suboptimal response to monotherapies is the development of drug resistance. Multiple mechanisms underlying such resistance phenotypes have been reported which encompass activation of alternative signaling pathways, mutations in the secondary target, and amplification of downstream signaling moieties within the same pathway (Zahreddine and Borden, 2013). Under such scenarios, the identification of new targeted combination therapies is indispensable for developing a superior response to therapies to treat pancreatic cancer.
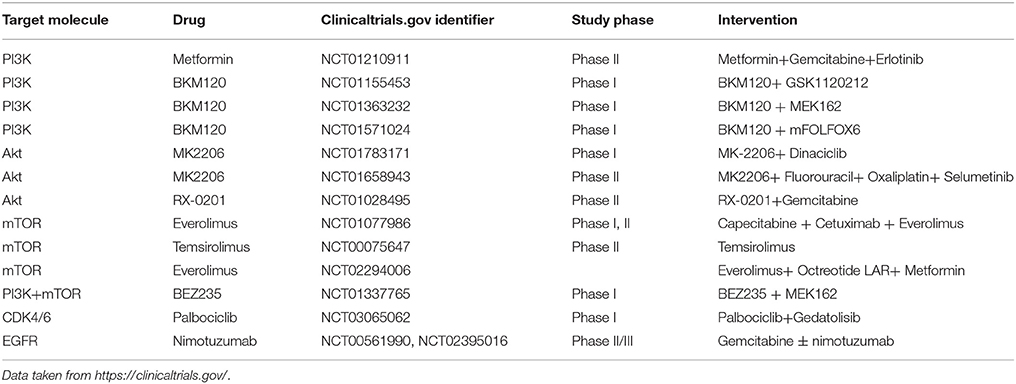
Table 2. Current ongoing clinical trials targeting the phosphoinositide signaling cascade for the treatment of pancreatic cancer.
While LY294002 monotherapy has low efficacy against tumors, the desired efficacy of this drug may be achieved by combination with the NSAID sulindac or gemcitabine, which improved the growth inhibitory effects of LY294002 by reducing the apoptotic threshold in PDAC cells (Yip-Schneider et al., 2003). A recent study has demonstrated that the suppression of the PI3K/Akt /mTOR pathway results in a compensatory activation of the MAPK/MEK pathway. The authors further established a reduction in the viability of pancreatic cancer cell lines by application of a dual-acting agent using the PI3K inhibitor, ZSTK474, and the Raf/MEK inhibitor, RO5126766 (Van Dort et al., 2015). Interestingly, MEK inhibition by AZD6244 alone remained cytostatic until it was combined with the PI3K-inhibitor BKM120 or erlotinib, which delayed tumor formation and improved overall survival compared with single-agent therapy (Alagesan et al., 2015; NCT01222689). A phase I study of BKM120 in combination with mFOLFOX6 in patients with metastatic pancreatic cancer is ongoing (NCT01571024). Gemcitabine has been the standard first-line treatment for patients with advanced or metastatic pancreatic cancer (Von Hoff et al., 2013). The therapeutic potential of gemcitabine can be improved by evodiamine, which negatively regulates the NF-κB signaling by targeting the PI3K/Akt pathway (Wei et al., 2012). Thus, targeting PI3K pathways by a combination of agents may be crucial for achieving the desired efficacy against tumors.
Akt signaling is pertinent to the activation of anti-apoptotic pathways that support cancer cell survival. Akt inhibitors have been classified as ATP-competitive, allosteric, and Akt-protein-substrate-binding-site inhibitors. However, due to the extensive similarity of the ATP binding pocket of the Akt kinases to the AGC family of kinases, it has been particularly difficult to develop ATP-competitive inhibitors that target Akt kinases (Scheid and Woodgett, 2001). A recent report by Yap et al. assessed the effect of MK-2206, an oral allosteric inhibitor of all Akt isoforms, for anti-tumor activity in preclinical models. Their study showed a reduction in tumor size and a decrease in cancer antigen levels in a PDAC patient treated with 60 mg of the drug on alternate days (Yap et al., 2011). The drug has further been demonstrated to function along with dinaciclib, a cyclin-dependent kinase inhibitor, to abolish tumor growth in pancreatic cancer models (Hu et al., 2015). In addition to small molecule inhibitors, the emergence of antisense nucleotides that target key signaling molecules is promising. RX-0201, an Akt antisense oligonucleotide, has been tested in combination with gemcitabine in metastatic pancreatic cancer and is in phase II trials (NCT01028495).
The mTOR kinase, one of the key downstream effectors of K-Ras and the PI3K pathway, coordinates distal metabolic features, such as the presence of growth factors, to cell survival, growth, and proliferation by regulating various transcriptional and translational regulatory programs (Utomo et al., 2014). Rapamycin (sirolimus), one of the most long-standing inhibitors of mTOR, has shown broad anticancer activity (Douros and Suffness, 1981; Garber, 2001; Utomo et al., 2014). In the recent years, multiple rapamycin analogs have been developed, including everolimus, temsirolimus, and deforolimus (Hudes et al., 2007; Motzer et al., 2008). The oral mTOR-inhibitor everolimus is in phase 2 trials to treat advanced pancreatic neuroendocrine tumors and has shown antitumor activity by prolonging progression-free survival in patients (Yao et al., 2011). Meanwhile, researchers have developed inhibitors to target EGFR, which is frequently overexpressed in PDAC patients. The EGFR-inhibitor erlotinib tested in combination with rapamycin significantly disrupted the PI3K-Akt-mTOR signaling cascade producing a synergistic effect on cell growth inhibition (Buck et al., 2006).
Taken together, previous in vitro screens reveal that PDAC cell lines are relatively resistant to single-agent therapies and thus, targeting multiple nodes of the PI3K-Akt-mTOR signaling cascade using combination drug therapies might reinforce the response to individual drug therapies. The PI3K pathway status may serve as genetic determinants of therapeutic response in clinical trials.
Aberrant signaling pathways are an important hallmark of cancer and essential for development and progression of tumors. Signaling networks are activated in cancer cells upon sensing the molecular cues from outside the cells to regulate cellular functions (Jones et al., 2008). Cancer cells and surrounding cells present in the tumor microenvironment continuously interact by modifying the signaling cues during the different stages of cancer evolution. These dynamic cues are perceived by cellular signaling networks to impart adaptation to the tumor cells during the disease progression (Liu et al., 2017). K-Ras and PI3K are key regulated nodes in the complex, inter-connected signaling networks altered during pancreatic cancer (Jones et al., 2008). K-Ras is the key driver mutation in pancreatic adenocarcinomas and is required for initiation, progression, and maintenance of the disease (Bryant et al., 2014; Cancer Genome Atlas Research Network Electronic Address and Cancer Genome Atlas Research, 2017). PI3K mutations, although rarely found in pancreatic cancer compared to other forms of cancer, can contribute to tumorigenesis in a small subset of pancreatic cancer patients (Schonleben et al., 2006; Payne et al., 2015). Targeting PI3K in this small cohort of patients can be of great significance in this lethal malignancy. Because PI3K can be regulated by a myriad of growth factor receptors and pathways including K-Ras, it dispenses its effector functions by differentially regulating a multitude of downstream signaling cascades. PI3K signaling is directly activated by mutations in the K-Ras gene in pancreatic cancer (Eser et al., 2013). Despite the lack of significant genomic alterations in the PI3K pathway, it is still considered a key node in the signaling network considering that it is interconnected with other signaling pathways at multiple levels. Co-targeting the PI3K pathway with multiple other signaling pathways and known drugs has seen limited success, but it remains a viable strategy given that multiple PI3K inhibitors are in human clinical trials (Bowles and Jimeno, 2011). It would be beneficial for future research to be targeted to understand PI3K-inhibitor unresponsiveness and resistance, and eventually design novel combinatorial drug strategies using resistance-pathway inhibitors and other targets.
A second critical aspect of pancreatic cancer research is the interaction of tumor cells with the surrounding cells present in the tumor microenvironment. Recent studies not only highlight the importance of PI3K signaling in development and maintenance of immune and stromal cell functions but also in modifying signaling cues between tumor and accessory cells during the progression of cancer (Koyasu, 2003; Polo et al., 2015; Gunderson et al., 2016). It is now well-established that both tumor cells and the tumor microenvironment continuously interact to modify the outcome during tumor development and changing the properties of one component modifies the other (Whatcott et al., 2015). While the genome of a cancer cell continues to evolve, thus making it hard to target, the stromal or immune cells, on the other hand, harbor a robust and stable genome. Therefore, targeting signaling pathways in these accessory cells can yield notable results in cancer prevention and disease control. PI3K has been examined in the tumor microenvironment to a certain extent; a comprehensive understanding of all the signaling pathways that include PI3K will need to be elucidated in the future. Hence, understanding the oncogenic mechanisms during tumor-stromal and tumor-immune crosstalk will be of paramount importance to dissect the pathways critical for pancreatic cancer pathogenesis. This will aid in designing multi-drug, multi-target combination therapies and will prove critical in systematically screening patient biopsies for both driver mutations and other downstream-activated signaling pathways, like PI3K, in order to design personalized therapies.
DM and KA: made figures; DM, KA, and PS: wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by funding from the National Institutes of Health grant (R01 CA163649, R01 CA210439, and R01 CA216853, NCI) to PS and the Specialized Programs for Research Excellence (SPORE, 2P50 CA127297, NCI) to PS. We would also like to acknowledge the Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727, NCI) for supporting shared resources.
4E-BP, eIF4E-binding proteins; ACLY, ATP citrate lyase; ADM, acinar-to-ductal metaplasia; BTK, Bruton tyrosine kinase; CAT, Catalytic domain; CTLA, Cytotoxic T-lymphocyte-associated protein; CXCL13, Chemokine ligand 13; eIF4E, eukaryotic translation initiation factor 4E; ERK, Extracellular signal-regulated kinases; EXT, Extension domain; FoXO, Forkhead transcription factors; GCPR, G-protein-coupled receptors; GSK3β), glycogen synthase kinase 3 beta; GTPases, Guanosine-5′-triphosphatases; HIF-1α, Hypoxia-inducible factor 1- α; HK, Hexokinase; HM, Hydrophobic motif; HNRNPA2B1, Heterogeneous nuclear ribonucleoprotein A2/B1; IFNγ, Interferon gamma; MAPK, Mitogen-activated protein kinase; MHC, Major histocompatibility complex; mTOR, Mechanistic target of Rapamycin; MUC, Mucin; NF-κB, Nuclear factor kB; NK, Natural killer; P70S6K, ribosomal protein S6 kinase beta-1; PanINs, Pancreatic intraepithelial neoplastic lesions; PD-1, Programmed cell death-1; PD-L1, Programmed death-ligand 1; PDAC, Pancreatic ductal adenocarcinoma; PDK1, Phosphoinositide-dependent kinase 1; PH, Pleckstrin homology; PI3K, Phosphoinositide-3-kinase; PKB, Protein kinase B; PtdIns, Phosphatidylinositols; PTEN, Phosphatase and tensin homolog deleted on chromosome 10; Ras, Rat sarcoma virus; ROS, Reactive oxygen species; RTKs, Receptor tyrosine kinases; SH2, Src Homology 2; TAM, Tumor-associated macrophages; TGF-β, Transforming growth factor beta; Th, T helper; Treg, Regulatory T cells; VEGF, Vascular endothelial growth factor.
Abrego, J., Gunda, V., Vernucci, E., Shukla, S. K., King, R. J., Dasgupta, A., et al. (2017). GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett. 400, 37–46. doi: 10.1016/j.canlet.2017.04.029
Adams, J. R., Xu, K., Liu, J. C., Agamez, N. M., Loch, A. J., Wong, R. G., et al. (2011). Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer Res. 71, 2706–2717. doi: 10.1158/0008-5472.CAN-10-0738
Alagesan, B., Contino, G., Guimaraes, A. R., Corcoran, R. B., Deshpande, V., Wojtkiewicz, G. R., et al. (2015). Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin. Cancer Res. 21, 396–404. doi: 10.1158/1078-0432.CCR-14-1591
Alessi, D. R., James, S. R., Downes, C. P., Holmes, A. B., Gaffney, P. R., Reese, C. B., et al. (1997). Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 7, 261–269. doi: 10.1016/S0960-9822(06)00122-9
Ali, K., Soond, D. R., Pineiro, R., Hagemann, T., Pearce, W., Lim, E. L., et al. (2014). Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 510, 407–411. doi: 10.1038/nature13444
Altomare, D. A., and Testa, J. R. (2005). Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464. doi: 10.1038/sj.onc.1209085
Apte, M. V., Wilson, J. S., Lugea, A., and Pandol, S. J. (2013). A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 144, 1210–1219. doi: 10.1053/j.gastro.2012.11.037
Arcaro, A., and Guerreiro, A. S. (2007). The phosphoinositide 3-kinase pathway in human cancer: genetic alterations and therapeutic implications. Curr. Genomics 8, 271–306. doi: 10.2174/138920207782446160
Asano, T., Yao, Y., Zhu, J., Li, D., Abbruzzese, J. L., and Reddy, S. A. (2004). The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene 23, 8571–8580. doi: 10.1038/sj.onc.1207902
Attri, K. S., Murthy, D., and Singh, P. K. (2017). Racial disparity in metabolic regulation of cancer. Front. Biosci. 22, 1221–1246. doi: 10.2741/4543
Ayabe, K. (1999). [Trends in adjuvant therapy after surgery of lung cancer]. Kyobu Geka 52, 707–713.
Bader, A. G., Kang, S., Zhao, L., and Vogt, P. K. (2005). Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer 5, 921–929. doi: 10.1038/nrc1753
Baer, R., Cintas, C., Dufresne, M., Cassant-Sourdy, S., Schonhuber, N., Planque, L., et al. (2014). Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110α. Genes Dev. 28, 2621–2635. doi: 10.1101/gad.249409.114
Bailey, J. M., Swanson, B. J., Hamada, T., Eggers, J. P., Singh, P. K., Caffery, T., et al. (2008). Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 14, 5995–6004. doi: 10.1158/1078-0432.CCR-08-0291
Barcelo, C., Etchin, J., Mansour, M. R., Sanda, T., Ginesta, M. M., Sanchez-Arevalo Lobo, V. J., et al. (2014). Ribonucleoprotein HNRNPA2B1 interacts with and regulates oncogenic KRAS in pancreatic ductal adenocarcinoma cells. Gastroenterology 147, 882–892 e888. doi: 10.1053/j.gastro.2014.06.041
Barros, M. H., Hauck, F., Dreyer, J. H., Kempkes, B., and Niedobitek, G. (2013). Macrophage polarisation: an immunohistochemical approach for identifying M1 and M2 macrophages. PLoS ONE 8:e80908. doi: 10.1371/journal.pone.0080908
Barthel, A., Okino, S. T., Liao, J., Nakatani, K., Li, J., Whitlock, J. P. Jr., et al. (1999). Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J. Biol. Chem. 274, 20281–20286. doi: 10.1074/jbc.274.29.20281
Behrens, M. E., Grandgenett, P. M., Bailey, J. M., Singh, P. K., Yi, C. H., Yu, F., et al. (2010). The reactive tumor microenvironment: MUC1 signaling directly reprograms transcription of CTGF. Oncogene 29, 5667–5677. doi: 10.1038/onc.2010.327
Benitez, A. C., Dai, Z., Mann, H. H., Reeves, R. S., Margineantu, D. H., Gooley, T. A., et al. (2011). Expression, signaling proficiency, and stimulatory function of the NKG2D lymphocyte receptor in human cancer cells. Proc. Natl. Acad. Sci. U.S.A. 108, 4081–4086. doi: 10.1073/pnas.1018603108
Berna, M. J., Seiz, O., Nast, J. F., Benten, D., Blaker, M., Koch, J., et al. (2010). CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J. Biol. Chem. 285, 38905–38914. doi: 10.1074/jbc.M110.125534
Biankin, A. V., Waddell, N., Kassahn, K. S., Gingras, M. C., Muthuswamy, L. B., Johns, A. L., et al. (2012). Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405. doi: 10.1038/nature11547
Bitler, B. G., Goverdhan, A., and Schroeder, J. A. (2010). MUC1 regulates nuclear localization and function of the epidermal growth factor receptor. J. Cell Sci. 123, 1716–1723. doi: 10.1242/jcs.062661
Bondar, V. M., Sweeney-Gotsch, B., Andreeff, M., Mills, G. B., and Mcconkey, D. J. (2002). Inhibition of the phosphatidylinositol 3'-kinase-AKT pathway induces apoptosis in pancreatic carcinoma cells in vitro and in vivo. Mol. Cancer Ther. 1, 989–997.
Bowles, D. W., and Jimeno, A. (2011). New phosphatidylinositol 3-kinase inhibitors for cancer. Expert Opin. Invest. Drugs 20, 507–518. doi: 10.1517/13543784.2011.562192
Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., et al. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. doi: 10.1016/S0092-8674(00)80595-4
Bryant, K. L., Mancias, J. D., Kimmelman, A. C., and Der, C. J. (2014). KRAS: feeding pancreatic cancer proliferation. Trends Biochem. Sci. 39, 91–100. doi: 10.1016/j.tibs.2013.12.004
Buck, E., Eyzaguirre, A., Brown, E., Petti, F., Mccormack, S., Haley, J. D., et al. (2006). Rapamycin synergizes with the epidermal growth factor receptor inhibitor erlotinib in non-small-cell lung, pancreatic, colon, and breast tumors. Mol. Cancer Ther. 5, 2676–2684. doi: 10.1158/1535-7163.MCT-06-0166
Bussard, K. M., Mutkus, L., Stumpf, K., Gomez-Manzano, C., and Marini, F. C. (2016). Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 18:84. doi: 10.1186/s13058-016-0740-2
Cancer Genome Atlas Research Network Electronic Address A.a.D.H.E. and Cancer Genome Atlas Research Network (2017). Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32, 185–203.e113. doi: 10.1016/j.ccell.2017.07.007
Cantley, L. C. (2002). The phosphoinositide 3-kinase pathway. Science 296, 1655–1657. doi: 10.1126/science.296.5573.1655
Cantley, L. C., and Neel, B. G. (1999). New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 4240–4245. doi: 10.1073/pnas.96.8.4240
Cardone, M. H., Roy, N., Stennicke, H. R., Salvesen, G. S., Franke, T. F., Stanbridge, E., et al. (1998). Regulation of cell death protease caspase-9 by phosphorylation. Science 282, 1318–1321. doi: 10.1126/science.282.5392.1318
Cerwenka, A., and Lanier, L. L. (2016). Natural killer cell memory in infection, inflammation and cancer. Nat. Rev. Immunol. 16, 112–123. doi: 10.1038/nri.2015.9
Chaika, N. V., Gebregiworgis, T., Lewallen, M. E., Purohit, V., Radhakrishnan, P., Liu, X., et al. (2012a). MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc. Natl. Acad. Sci. U.S.A. 109, 13787–13792. doi: 10.1073/pnas.1203339109
Chaika, N. V., Yu, F., Purohit, V., Mehla, K., Lazenby, A. J., Dimaio, D., et al. (2012b). Differential expression of metabolic genes in tumor and stromal components of primary and metastatic loci in pancreatic adenocarcinoma. PLoS ONE 7:e32996. doi: 10.1371/journal.pone.0032996
Chand, S., O'hayer, K., Blanco, F. F., Winter, J. M., and Brody, J. R. (2016). The landscape of pancreatic cancer therapeutic resistance mechanisms. Int. J. Biol. Sci. 12, 273–282. doi: 10.7150/ijbs.14951
Chang, D. Z., Ma, Y., Ji, B., Wang, H., Deng, D., Liu, Y., et al. (2011). Mast cells in tumor microenvironment promotes the in vivo growth of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 17, 7015–7023. doi: 10.1158/1078-0432.CCR-11-0607
Chao, T., Furth, E. E., and Vonderheide, R. H. (2016). CXCR2-dependent accumulation of tumor-associated neutrophils regulates T-cell immunity in pancreatic ductal adenocarcinoma. Cancer Immunol. Res. 4, 968–982. doi: 10.1158/2326-6066.CIR-16-0188
Chaudhuri, A. (2014). Regulation of macrophage polarization by RON receptor tyrosine kinase signaling. Front. Immunol. 5:546. doi: 10.3389/fimmu.2014.00546
Cheng, J. Q., Ruggeri, B., Klein, W. M., Sonoda, G., Altomare, D. A., Watson, D. K., et al. (1996). Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc. Natl. Acad. Sci. U.S.A. 93, 3636–3641. doi: 10.1073/pnas.93.8.3636
Cho, Y., Cho, E. J., Lee, J. H., Yu, S. J., Kim, Y. J., Kim, C. Y., et al. (2016). Hypoxia enhances tumor-stroma crosstalk that drives the progression of hepatocellular carcinoma. Dig. Dis. Sci. 61, 2568–2577. doi: 10.1007/s10620-016-4158-6
Commisso, C., Davidson, S. M., Soydaner-Azeloglu, R. G., Parker, S. J., Kamphorst, J. J., Hackett, S., et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. doi: 10.1038/nature12138
Covarrubias, A. J., Aksoylar, H. I., Yu, J., Snyder, N. W., Worth, A. J., Iyer, S. S., et al. (2016). Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife 5:e11612. doi: 10.7554/eLife.11612
Creixell, P., Reimand, J., Haider, S., Wu, G., Shibata, T., Vazquez, M., et al. (2015). Pathway and network analysis of cancer genomes. Nat. Methods 12, 615–621. doi: 10.1038/nmeth.3440
Deprez, J., Vertommen, D., Alessi, D. R., Hue, L., and Rider, M. H. (1997). Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J. Biol. Chem. 272, 17269–17275. doi: 10.1074/jbc.272.28.17269
Dergham, S. T., Dugan, M. C., Kucway, R., Du, W., Kamarauskiene, D. S., Vaitkevicius, V. K., et al. (1997). Prevalence and clinical significance of combined K-ras mutation and p53 aberration in pancreatic adenocarcinoma. Int. J. Pancreatol. 21, 127–143.
Di Cristofano, A., and Pandolfi, P. P. (2000). The multiple roles of PTEN in tumor suppression. Cell 100, 387–390. doi: 10.1016/S0092-8674(00)80674-1
di Magliano, M. P., and Logsdon, C. D. (2013). Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 144, 1220–1229. doi: 10.1053/j.gastro.2013.01.071
Dong, H., Strome, S. E., Salomao, D. R., Tamura, H., Hirano, F., Flies, D. B., et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8, 793–800. doi: 10.1038/nm730
Douros, J., and Suffness, M. (1981). New antitumor substances of natural origin. Cancer Treat. Rev. 8, 63–87. doi: 10.1016/S0305-7372(81)80006-0
Driscoll, D. R., Karim, S. A., Sano, M., Gay, D. M., Jacob, W., Yu, J., et al. (2016). mTORC2 signaling drives the development and progression of pancreatic cancer. Cancer Res. 76, 6911–6923. doi: 10.1158/0008-5472.CAN-16-0810
Edderkaoui, M., Nitsche, C., Zheng, L., Pandol, S. J., Gukovsky, I., and Gukovskaya, A. S. (2013). NADPH oxidase activation in pancreatic cancer cells is mediated through Akt-dependent up-regulation of p22phox. J. Biol. Chem. 288:36259. doi: 10.1074/jbc.A113.200063
Edling, C. E., Selvaggi, F., Buus, R., Maffucci, T., Di Sebastiano, P., Friess, H., et al. (2010). Key role of phosphoinositide 3-kinase class IB in pancreatic cancer. Clin. Cancer Res. 16, 4928–4937. doi: 10.1158/1078-0432.CCR-10-1210
El-Gazzar, A., Cai, X., Reeves, R. S., Dai, Z., Caballero-Benitez, A., Mcdonald, D. L., et al. (2014). Effects on tumor development and metastatic dissemination by the NKG2D lymphocyte receptor expressed on cancer cells. Oncogene 33, 4932–4940. doi: 10.1038/onc.2013.435
Emmrich, J., Weber, I., Nausch, M., Sparmann, G., Koch, K., Seyfarth, M., et al. (1998). Immunohistochemical characterization of the pancreatic cellular infiltrate in normal pancreas, chronic pancreatitis and pancreatic carcinoma. Digestion 59, 192–198. doi: 10.1159/000007488
Ene-Obong, A., Clear, A. J., Watt, J., Wang, J., Fatah, R., Riches, J. C., et al. (2013). Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 145, 1121–1132. doi: 10.1053/j.gastro.2013.07.025
Engel, B. J., Bowser, J. L., Broaddus, R. R., and Carson, D. D. (2016). MUC1 stimulates EGFR expression and function in endometrial cancer. Oncotarget 7, 32796–32809. doi: 10.18632/oncotarget.8743
Engelman, J. A., Chen, L., Tan, X., Crosby, K., Guimaraes, A. R., Upadhyay, R., et al. (2008). Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 14, 1351–1356. doi: 10.1038/nm.1890
Engelman, J. A., Luo, J., and Cantley, L. C. (2006). The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619. doi: 10.1038/nrg1879
Erkan, M., Michalski, C. W., Rieder, S., Reiser-Erkan, C., Abiatari, I., Kolb, A., et al. (2008). The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 6, 1155–1161. doi: 10.1016/j.cgh.2008.05.006
Eser, S., Reiff, N., Messer, M., Seidler, B., Gottschalk, K., Dobler, M., et al. (2013). Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 23, 406–420. doi: 10.1016/j.ccr.2013.01.023
Eser, S., Schnieke, A., Schneider, G., and Saur, D. (2014). Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 111, 817–822. doi: 10.1038/bjc.2014.215
Falasca, M., and Maffucci, T. (2007). Role of class II phosphoinositide 3-kinase in cell signalling. Biochem. Soc. Trans. 35, 211–214. doi: 10.1042/BST0350211
Feig, C., Gopinathan, A., Neesse, A., Chan, D. S., Cook, N., and Tuveson, D. A. (2012). The pancreas cancer microenvironment. Clin. Cancer Res. 18, 4266–4276. doi: 10.1158/1078-0432.CCR-11-3114
Fogar, P., Basso, D., Fadi, E., Greco, E., Pantano, G., Padoan, A., et al. (2011). Pancreatic cancer alters human CD4+ T lymphocyte function: a piece in the immune evasion puzzle. Pancreas 40, 1131–1137. doi: 10.1097/MPA.0b013e31822077b8
Folkman, J. (2007). Angiogenesis: an organizing principle for drug discovery? Nat. Rev. Drug Discov. 6, 273–286. doi: 10.1038/nrd2115
Foster, K. G., and Fingar, D. C. (2010). Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J. Biol. Chem. 285, 14071–14077. doi: 10.1074/jbc.R109.094003
Fouad, Y. A., and Aanei, C. (2017). Revisiting the hallmarks of cancer. Am. J. Cancer Res. 7, 1016–1036.
Fukuda, R., Hirota, K., Fan, F., Jung, Y. D., Ellis, L. M., and Semenza, G. L. (2002). Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J. Biol. Chem. 277, 38205–38211. doi: 10.1074/jbc.M203781200
Gajewski, T. F., Schreiber, H., and Fu, Y. X. (2013). Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022. doi: 10.1038/ni.2703
Gao, N., Ding, M., Zheng, J. Z., Zhang, Z., Leonard, S. S., Liu, K. J., et al. (2002). Vanadate-induced expression of hypoxia-inducible factor 1 alpha and vascular endothelial growth factor through phosphatidylinositol 3-kinase/Akt pathway and reactive oxygen species. J. Biol. Chem. 277, 31963–31971. doi: 10.1074/jbc.M200082200
Garber, K. (2001). Rapamycin's resurrection: a new way to target the cancer cell cycle. J. Natl. Cancer Inst. 93, 1517–1519. doi: 10.1093/jnci/93.20.1517
Gebregiworgis, T., Purohit, V., Shukla, S. K., Tadros, S., Chaika, N. V., Abrego, J., et al. (2017). Glucose limitation alters glutamine metabolism in MUC1-overexpressing pancreatic cancer cells. J. Proteome Res. 16, 3536–3546. doi: 10.1021/acs.jproteome.7b00246
Gingras, A. C., Kennedy, S. G., O'leary, M. A., Sonenberg, N., and Hay, N. (1998). 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 12, 502–513. doi: 10.1101/gad.12.4.502
Ginhoux, F., Schultze, J. L., Murray, P. J., Ochando, J., and Biswas, S. K. (2016). New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 17, 34–40. doi: 10.1038/ni.3324
Grewe, M., Gansauge, F., Schmid, R. M., Adler, G., and Seufferlein, T. (1999). Regulation of cell growth and cyclin D1 expression by the constitutively active FRAP-p70s6K pathway in human pancreatic cancer cells. Cancer Res. 59, 3581–3587.
Grivennikov, S. I., Greten, F. R., and Karin, M. (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. doi: 10.1016/j.cell.2010.01.025
Gunda, V., Souchek, J., Abrego, J., Shukla, S. K., Goode, G. D., Vernucci, E., et al. (2017). MUC1-mediated metabolic alterations regulate response to radiotherapy in pancreatic cancer. Clin. Cancer Res. 23, 5881–5891. doi: 10.1158/1078-0432.CCR-17-1151
Gunderson, A. J., Kaneda, M. M., Tsujikawa, T., Nguyen, A. V., Affara, N. I., Ruffell, B., et al. (2016). Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. 6, 270–285. doi: 10.1158/2159-8290.CD-15-0827
Hamada, K., Sasaki, T., Koni, P. A., Natsui, M., Kishimoto, H., Sasaki, J., et al. (2005). The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 19, 2054–2065. doi: 10.1101/gad.1308805
Hanahan, D., and Weinberg, R. A. (2000). The hallmarks of cancer. Cell 100, 57–70. doi: 10.1016/S0092-8674(00)81683-9
Hardie, R. A., Van Dam, E., Cowley, M., Han, T. L., Balaban, S., Pajic, M., et al. (2017). Mitochondrial mutations and metabolic adaptation in pancreatic cancer. Cancer Metab. 5:2. doi: 10.1186/s40170-017-0164-1
Hay, N., and Sonenberg, N. (2004). Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945. doi: 10.1101/gad.1212704
Hayes, T. K., Neel, N. F., Hu, C., Gautam, P., Chenard, M., Long, B., et al. (2016). Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell 29, 75–89. doi: 10.1016/j.ccell.2015.11.011
Hill, R., Calvopina, J. H., Kim, C., Wang, Y., Dawson, D. W., Donahue, T. R., et al. (2010). PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res. 70, 7114–7124. doi: 10.1158/0008-5472.CAN-10-1649
Hiraoka, N., Onozato, K., Kosuge, T., and Hirohashi, S. (2006). Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 12, 5423–5434. doi: 10.1158/1078-0432.CCR-06-0369
Hofer, E., and Schweighofer, B. (2007). Signal transduction induced in endothelial cells by growth factor receptors involved in angiogenesis. Thromb. Haemost. 97, 355–363. doi: 10.1160/TH06-08-0470
Hu, C., Dadon, T., Chenna, V., Yabuuchi, S., Bannerji, R., Booher, R., et al. (2015). Combined inhibition of cyclin-dependent kinases (Dinaciclib) and AKT (MK-2206) blocks pancreatic tumor growth and metastases in patient-derived xenograft models. Mol. Cancer Ther. 14, 1532–1539. doi: 10.1158/1535-7163.MCT-15-0028
Hudes, G., Carducci, M., Tomczak, P., Dutcher, J., Figlin, R., Kapoor, A., et al. (2007). Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 356, 2271–2281. doi: 10.1056/NEJMoa066838
Hwang, R. F., Moore, T., Arumugam, T., Ramachandran, V., Amos, K. D., Rivera, A., et al. (2008). Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 68, 918–926. doi: 10.1158/0008-5472.CAN-07-5714
Ino, Y., Yamazaki-Itoh, R., Shimada, K., Iwasaki, M., Kosuge, T., Kanai, Y., et al. (2013). Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 108, 914–923. doi: 10.1038/bjc.2013.32
Jaber, N., Dou, Z., Chen, J. S., Catanzaro, J., Jiang, Y. P., Ballou, L. M., et al. (2012). Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. U.S.A. 109, 2003–2008. doi: 10.1073/pnas.1112848109
James, S. R., Downes, C. P., Gigg, R., Grove, S. J., Holmes, A. B., and Alessi, D. R. (1996). Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem. J. 315(Pt 3), 709–713. doi: 10.1042/bj3150709
Jean, S., and Kiger, A. A. (2014). Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 127, 923–928. doi: 10.1242/jcs.093773
Jiang, K., Zhong, B., Gilvary, D. L., Corliss, B. C., Hong-Geller, E., Wei, S., et al. (2000). Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat. Immunol. 1, 419–425. doi: 10.1038/80859
Jimeno, A., Tan, A. C., Coffa, J., Rajeshkumar, N. V., Kulesza, P., Rubio-Viqueira, B., et al. (2008). Coordinated epidermal growth factor receptor pathway gene overexpression predicts epidermal growth factor receptor inhibitor sensitivity in pancreatic cancer. Cancer Res. 68, 2841–2849. doi: 10.1158/0008-5472.CAN-07-5200
Johansson, H., Andersson, R., Bauden, M., Hammes, S., Holdenrieder, S., and Ansari, D. (2016). Immune checkpoint therapy for pancreatic cancer. World J. Gastroenterol. 22, 9457–9476. doi: 10.3748/wjg.v22.i43.9457
Jones, S., Zhang, X., Parsons, D. W., Lin, J. C., Leary, R. J., Angenendt, P., et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. doi: 10.1126/science.1164368
Kaneda, M. M., Cappello, P., Nguyen, A. V., Ralainirina, N., Hardamon, C. R., Foubert, P., et al. (2016a). Macrophage PI3Kγ drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 6, 870–885. doi: 10.1158/2159-8290.CD-15-1346
Kaneda, M. M., Messer, K. S., Ralainirina, N., Li, H., Leem, C. J., Gorjestani, S., et al. (2016b). PI3Kγ is a molecular switch that controls immune suppression. Nature 539, 437–442. doi: 10.1038/nature19834
Kang, R., Hou, W., Zhang, Q., Chen, R., Lee, Y. J., Bartlett, D. L., et al. (2014). RAGE is essential for oncogenic KRAS-mediated hypoxic signaling in pancreatic cancer. Cell Death Dis. 5:e1480. doi: 10.1038/cddis.2014.445
Karnevi, E., Andersson, R., and Rosendahl, A. H. (2014). Tumour-educated macrophages display a mixed polarisation and enhance pancreatic cancer cell invasion. Immunol. Cell Biol. 92, 543–552. doi: 10.1038/icb.2014.22
Kato, K., Lu, W., Kai, H., and Kim, K. C. (2007). Phosphoinositide 3-kinase is activated by MUC1 but not responsible for MUC1-induced suppression of Toll-like receptor 5 signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L686–L692. doi: 10.1152/ajplung.00423.2006
Kaur, S., Baine, M. J., Jain, M., Sasson, A. R., and Batra, S. K. (2012). Early diagnosis of pancreatic cancer: challenges and new developments. Biomark. Med. 6, 597–612. doi: 10.2217/bmm.12.69
Kennedy, A. L., Morton, J. P., Manoharan, I., Nelson, D. M., Jamieson, N. B., Pawlikowski, J. S., et al. (2011). Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol. Cell 42, 36–49. doi: 10.1016/j.molcel.2011.02.020
Kerr, E. M., Gaude, E., Turrell, F. K., Frezza, C., and Martins, C. P. (2016). Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 531, 110–113. doi: 10.1038/nature16967
Khan, K. H., Yap, T. A., Yan, L., and Cunningham, D. (2013). Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin. J. Cancer 32, 253–265. doi: 10.5732/cjc.013.10057
Kilic-Eren, M., Boylu, T., and Tabor, V. (2013). Targeting PI3K/Akt represses Hypoxia inducible factor-1α activation and sensitizes Rhabdomyosarcoma and Ewing's sarcoma cells for apoptosis. Cancer Cell Int. 13:36. doi: 10.1186/1475-2867-13-36
Kim, M. S., Radinger, M., and Gilfillan, A. M. (2008). The multiple roles of phosphoinositide 3-kinase in mast cell biology. Trends Immunol. 29, 493–501. doi: 10.1016/j.it.2008.07.004
King, R. J., Yu, F., and Singh, P. K. (2017). Genomic alterations in mucins across cancers. Oncotarget 8, 67152–67168. doi: 10.18632/oncotarget.17934
Kolaczkowska, E., and Kubes, P. (2013). Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175. doi: 10.1038/nri3399
Koyasu, S. (2003). The role of PI3K in immune cells. Nat. Immunol. 4, 313–319. doi: 10.1038/ni0403-313
Kumar, C. C., and Madison, V. (2005). AKT crystal structure and AKT-specific inhibitors. Oncogene 24, 7493–7501. doi: 10.1038/sj.onc.1209087
Landis, J., and Shaw, L. M. (2014). Insulin receptor substrate 2-mediated phosphatidylinositol 3-kinase signaling selectively inhibits glycogen synthase kinase 3beta to regulate aerobic glycolysis. J. Biol. Chem. 289, 18603–18613. doi: 10.1074/jbc.M114.564070
Le, N., Sund, M., and Vinci, A. (2016). Prognostic and predictive markers in pancreatic adenocarcinoma. Dig. Liver Dis. 48, 223–230. doi: 10.1016/j.dld.2015.11.001
Lee, J. K., Edderkaoui, M., Truong, P., Ohno, I., Jang, K. T., Berti, A., et al. (2007). NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterology 133, 1637–1648. doi: 10.1053/j.gastro.2007.08.022
Lee, J. V., Carrer, A., Shah, S., Snyder, N. W., Wei, S., Venneti, S., et al. (2014). Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 20, 306–319. doi: 10.1016/j.cmet.2014.06.004
Lee, K. E., Spata, M., Bayne, L. J., Buza, E. L., Durham, A. C., Allman, D., et al. (2016). Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Discov. 6, 256–269. doi: 10.1158/2159-8290.CD-15-0822
Lennerz, J. K., and Stenzinger, A. (2015). Allelic ratio of KRAS mutations in pancreatic cancer. Oncologist 20, e8–e9. doi: 10.1634/theoncologist.2014-0408
Liang, J. W., Shi, Z. Z., Shen, T. Y., Che, X., Wang, Z., Shi, S. S., et al. (2014). Identification of genomic alterations in pancreatic cancer using array-based comparative genomic hybridization. PLoS ONE 9:e114616. doi: 10.1371/journal.pone.0114616
Lien, E. C., Dibble, C. C., and Toker, A. (2017). PI3K signaling in cancer: beyond AKT. Curr. Opin. Cell Biol. 45, 62–71. doi: 10.1016/j.ceb.2017.02.007
Liu, P., Cheng, H., Roberts, T. M., and Zhao, J. J. (2009). Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8, 627–644. doi: 10.1038/nrd2926
Liu, P., Cheng, H., Santiago, S., Raeder, M., Zhang, F., Isabella, A., et al. (2011). Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat. Med. 17, 1116–1120. doi: 10.1038/nm.2402
Liu, Q., Liao, Q., and Zhao, Y. (2017). Chemotherapy and tumor microenvironment of pancreatic cancer. Cancer Cell Int. 17:68. doi: 10.1186/s12935-017-0437-3
Liu, X., Caffrey, T. C., Steele, M. M., Mohr, A., Singh, P. K., Radhakrishnan, P., et al. (2014). MUC1 regulates cyclin D1 gene expression through p120 catenin and beta-catenin. Oncogenesis 3:e107. doi: 10.1038/oncsis.2014.19
Long, E. O., Kim, H. S., Liu, D., Peterson, M. E., and Rajagopalan, S. (2013). Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol. 31, 227–258. doi: 10.1146/annurev-immunol-020711-075005
Luo, J., Guo, P., Matsuda, K., Truong, N., Lee, A., Chun, C., et al. (2001). Pancreatic cancer cell-derived vascular endothelial growth factor is biologically active in vitro and enhances tumorigenicity in vivo. Int. J. Cancer 92, 361–369. doi: 10.1002/ijc.1202
Luyendyk, J. P., Schabbauer, G. A., Tencati, M., Holscher, T., Pawlinski, R., and Mackman, N. (2008). Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J. Immunol. 180, 4218–4226. doi: 10.4049/jimmunol.180.6.4218
Ma, B., Cao, W., Li, W., Gao, C., Qi, Z., Zhao, Y., et al. (2014). Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res. 24, 912–924. doi: 10.1038/cr.2014.84
Ma, J., Sawai, H., Ochi, N., Matsuo, Y., Xu, D., Yasuda, A., et al. (2009). PTEN regulates angiogenesis through PI3K/Akt/VEGF signaling pathway in human pancreatic cancer cells. Mol. Cell. Biochem. 331, 161–171. doi: 10.1007/s11010-009-0154-x
Ma, Y., Hwang, R. F., Logsdon, C. D., and Ullrich, S. E. (2013). Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. 73, 3927–3937. doi: 10.1158/0008-5472.CAN-12-4479
Maehama, T., and Dixon, J. E. (1998). The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378. doi: 10.1074/jbc.273.22.13375
Majewski, N., Nogueira, V., Bhaskar, P., Coy, P. E., Skeen, J. E., Gottlob, K., et al. (2004). Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 16, 819–830. doi: 10.1016/j.molcel.2004.11.014
Makinoshima, H., Takita, M., Saruwatari, K., Umemura, S., Obata, Y., Ishii, G., et al. (2015). Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/mammalian Target of Rapamycin (mTOR) axis is responsible for aerobic glycolysis mediated by glucose transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. J. Biol. Chem. 290, 17495–17504. doi: 10.1074/jbc.M115.660498
Makohon-Moore, A., and Iacobuzio-Donahue, C. A. (2016). Pancreatic cancer biology and genetics from an evolutionary perspective. Nat. Rev. Cancer 16, 553–565. doi: 10.1038/nrc.2016.66
Mangerich, A., Knutson, C. G., Parry, N. M., Muthupalani, S., Ye, W., Prestwich, E., et al. (2012). Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. U.S.A. 109, E1820–E1829. doi: 10.1073/pnas.1207829109
Mao, Y., Xi, L., Li, Q., Cai, Z., Lai, Y., Zhang, X., et al. (2016). Regulation of cell apoptosis and proliferation in pancreatic cancer through PI3K/Akt pathway via Polo-like kinase 1. Oncol. Rep. 36, 49–56. doi: 10.3892/or.2016.4820
Massihnia, D., Avan, A., Funel, N., Maftouh, M., Van Krieken, A., Granchi, C., et al. (2017). Phospho-Akt overexpression is prognostic and can be used to tailor the synergistic interaction of Akt inhibitors with gemcitabine in pancreatic cancer. J. Hematol. Oncol. 10:9. doi: 10.1186/s13045-016-0371-1
Mazure, N. M., Chen, E. Y., Laderoute, K. R., and Giaccia, A. J. (1997). Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood 90, 3322–3331.
Mehla, K., and Singh, P. K. (2014). MUC1: a novel metabolic master regulator. Biochim. Biophys. Acta 1845, 126–135. doi: 10.1016/j.bbcan.2014.01.001
Melstrom, L. G., Salabat, M. R., Ding, X. Z., Milam, B. M., Strouch, M., Pelling, J. C., et al. (2008). Apigenin inhibits the GLUT-1 glucose transporter and the phosphoinositide 3-kinase/Akt pathway in human pancreatic cancer cells. Pancreas 37, 426–431. doi: 10.1097/MPA.0b013e3181735ccb
Mosdell, D. M., and Doberneck, R. C. (1991). Morbidity and mortality of ostomy closure. Am. J. Surg. 162, 633–636; discussion: 636–637. doi: 10.1016/0002-9610(91)90125-W
Motzer, R. J., Escudier, B., Oudard, S., Hutson, T. E., Porta, C., Bracarda, S., et al. (2008). Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372, 449–456. doi: 10.1016/S0140-6736(08)61039-9
Murthy, D., Attri, K. S., and Gokhale, R. S. (2013). Network, nodes and nexus: systems approach to multitarget therapeutics. Curr. Opin. Biotechnol. 24, 1129–1136. doi: 10.1016/j.copbio.2013.02.009
Nath, S., Daneshvar, K., Roy, L. D., Grover, P., Kidiyoor, A., Mosley, L., et al. (2013). MUC1 induces drug resistance in pancreatic cancer cells via upregulation of multidrug resistance genes. Oncogenesis 2:e51. doi: 10.1038/oncsis.2013.16
Notta, F., Chan-Seng-Yue, M., Lemire, M., Li, Y., Wilson, G. W., Connor, A. A., et al. (2016). A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 538, 378–382. doi: 10.1038/nature19823
Nussinov, R., Muratcioglu, S., Tsai, C. J., Jang, H., Gursoy, A., and Keskin, O. (2015). The key role of calmodulin in KRAS-driven adenocarcinomas. Mol. Cancer Res. 13, 1265–1273. doi: 10.1158/1541-7786.MCR-15-0165
Ohlund, D., Handly-Santana, A., Biffi, G., Elyada, E., Almeida, A. S., Ponz-Sarvise, M., et al. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596. doi: 10.1084/jem.20162024
Okkenhaug, K., Bilancio, A., Farjot, G., Priddle, H., Sancho, S., Peskett, E., et al. (2002). Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 297, 1031–1034. doi: 10.1126/science.1073560
Okkenhaug, K., Graupera, M., and Vanhaesebroeck, B. (2016). Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discov. 6, 1090–1105. doi: 10.1158/2159-8290.CD-16-0716
Pandol, S., Edderkaoui, M., Gukovsky, I., Lugea, A., and Gukovskaya, A. (2009). Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 7, S44–S47. doi: 10.1016/j.cgh.2009.07.039
Payne, S. N., Maher, M. E., Tran, N. H., Van De Hey, D. R., Foley, T. M., Yueh, A. E., et al. (2015). PIK3CA mutations can initiate pancreatic tumorigenesis and are targetable with PI3K inhibitors. Oncogenesis 4:e169. doi: 10.1038/oncsis.2015.28
Peng, Y. P., Xi, C. H., Zhu, Y., Yin, L. D., Wei, J. S., Zhang, J. J., et al. (2016). Altered expression of CD226 and CD96 on natural killer cells in patients with pancreatic cancer. Oncotarget 7, 66586–66594. doi: 10.18632/oncotarget.11953
Peng, Y. P., Zhang, J. J., Liang, W. B., Tu, M., Lu, Z. P., Wei, J. S., et al. (2014). Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer 14:738. doi: 10.1186/1471-2407-14-738
Peng, Y. P., Zhu, Y., Zhang, J. J., Xu, Z. K., Qian, Z. Y., Dai, C. C., et al. (2013). Comprehensive analysis of the percentage of surface receptors and cytotoxic granules positive natural killer cells in patients with pancreatic cancer, gastric cancer, and colorectal cancer. J. Transl. Med. 11:262. doi: 10.1186/1479-5876-11-262
Pinho, V., Russo, R. C., Amaral, F. A., De Sousa, L. P., Barsante, M. M., De Souza, D. G., et al. (2007). Tissue- and stimulus-dependent role of phosphatidylinositol 3-kinase isoforms for neutrophil recruitment induced by chemoattractants in vivo. J. Immunol. 179, 7891–7898. doi: 10.4049/jimmunol.179.11.7891
Pirhonen, J. P., Erkkola, R. U., Ekblad, U. U., and Nyman, L. (1990). Single dose of nifedipine in normotensive pregnancy: nifedipine concentrations, hemodynamic responses, and uterine and fetal flow velocity waveforms. Obstet. Gynecol. 76, 807–811. doi: 10.1097/00006250-199011000-00016
Polo, M. L., Riggio, M., May, M., Rodriguez, M. J., Perrone, M. C., Stallings-Mann, M., et al. (2015). Activation of PI3K/Akt/mTOR signaling in the tumor stroma drives endocrine therapy-dependent breast tumor regression. Oncotarget 6, 22081–22097. doi: 10.18632/oncotarget.4203
Posor, Y., Eichhorn-Gruenig, M., Puchkov, D., Schoneberg, J., Ullrich, A., Lampe, A., et al. (2013). Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 499, 233–237. doi: 10.1038/nature12360
Pour, P. M., Egami, H., and Takiyama, Y. (1991). Patterns of growth and metastases of induced pancreatic cancer in relation to the prognosis and its clinical implications. Gastroenterology 100, 529–536. doi: 10.1016/0016-5085(91)90226-B
Putz, E. M., Prchal-Murphy, M., Simma, O. A., Forster, F., Koenig, X., Stockinger, H., et al. (2012). PI3Kdelta is essential for tumor clearance mediated by cytotoxic T lymphocytes. PLoS ONE 7:e40852. doi: 10.1371/journal.pone.0040852
Pylayeva-Gupta, Y., Das, S., Handler, J. S., Hajdu, C. H., Coffre, M., Koralov, S. B., et al. (2016). IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Discov. 6, 247–255. doi: 10.1158/2159-8290.CD-15-0843
Raina, D., Kharbanda, S., and Kufe, D. (2004). The MUC1 oncoprotein activates the anti-apoptotic phosphoinositide 3-kinase/Akt and Bcl-xL pathways in rat 3Y1 fibroblasts. J. Biol. Chem. 279, 20607–20612. doi: 10.1074/jbc.M310538200
Reid, M. D., Basturk, O., Thirabanjasak, D., Hruban, R. H., Klimstra, D. S., Bagci, P., et al. (2011). Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod. Pathol. 24, 1612–1619. doi: 10.1038/modpathol.2011.113
Roghanian, A., Fraser, C., Kleyman, M., and Chen, J. (2016). B cells promote pancreatic tumorigenesis. Cancer Discov. 6, 230–232. doi: 10.1158/2159-8290.CD-16-0100
Rucki, A. A., Foley, K., Zhang, P., Xiao, Q., Kleponis, J., Wu, A. A., et al. (2017). Heterogeneous stromal signaling within the tumor microenvironment controls the metastasis of pancreatic cancer. Cancer Res. 77, 41–52. doi: 10.1158/0008-5472.CAN-16-1383
Ruggeri, B. A., Huang, L., Wood, M., Cheng, J. Q., and Testa, J. R. (1998). Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol. Carcinog. 21, 81–86. doi: 10.1002/(SICI)1098-2744(199802)21:2<81::AID-MC1>3.0.CO;2-R
Ryschich, E., Notzel, T., Hinz, U., Autschbach, F., Ferguson, J., Simon, I., et al. (2005). Control of T-cell-mediated immune response by HLA class I in human pancreatic carcinoma. Clin. Cancer Res. 11, 498–504.
Sagona, A. P., Nezis, I. P., Pedersen, N. M., Liestol, K., Poulton, J., Rusten, T. E., et al. (2010). PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat. Cell Biol. 12, 362–371. doi: 10.1038/ncb2036
Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. doi: 10.1126/science.1106148
Scapini, P., Lapinet-Vera, J. A., Gasperini, S., Calzetti, F., Bazzoni, F., and Cassatella, M. A. (2000). The neutrophil as a cellular source of chemokines. Immunol. Rev. 177, 195–203. doi: 10.1034/j.1600-065X.2000.17706.x
Scheid, M. P., and Woodgett, J. R. (2001). PKB/AKT: functional insights from genetic models. Nat. Rev. Mol. Cell Biol. 2, 760–768. doi: 10.1038/35096067
Schild, C., Wirth, M., Reichert, M., Schmid, R. M., Saur, D., and Schneider, G. (2009). PI3K signaling maintains c-myc expression to regulate transcription of E2F1 in pancreatic cancer cells. Mol. Carcinog. 48, 1149–1158. doi: 10.1002/mc.20569
Schlieman, M. G., Fahy, B. N., Ramsamooj, R., Beckett, L., and Bold, R. J. (2003). Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br. J. Cancer 89, 2110–2115. doi: 10.1038/sj.bjc.6601396
Schmid-Kotsas, A., Gross, H. J., Menke, A., Weidenbach, H., Adler, G., Siech, M., et al. (1999). Lipopolysaccharide-activated macrophages stimulate the synthesis of collagen type I and C-fibronectin in cultured pancreatic stellate cells. Am. J. Pathol. 155, 1749–1758. doi: 10.1016/S0002-9440(10)65490-9
Schonleben, F., Qiu, W., Ciau, N. T., Ho, D. J., Li, X., Allendorf, J. D., et al. (2006). PIK3CA mutations in intraductal papillary mucinous neoplasm/carcinoma of the pancreas. Clin. Cancer Res. 12, 3851–3855. doi: 10.1158/1078-0432.CCR-06-0292
Shan, T., Chen, S., Chen, X., Lin, W. R., Li, W., Ma, J., et al. (2017). Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol. Rep. 37, 1971–1979. doi: 10.3892/or.2017.5479
Shi, C., Washington, M. K., Chaturvedi, R., Drosos, Y., Revetta, F. L., Weaver, C. J., et al. (2014). Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab. Invest. 94, 409–421. doi: 10.1038/labinvest.2014.10
Shi, P., Yin, T., Zhou, F., Cui, P., Gou, S., and Wang, C. (2014). Valproic acid sensitizes pancreatic cancer cells to natural killer cell-mediated lysis by upregulating MICA and MICB via the PI3K/Akt signaling pathway. BMC Cancer 14:370. doi: 10.1186/1471-2407-14-370
Shibata, D., Almoguera, C., Forrester, K., Dunitz, J., Martin, S. E., Cosgrove, M. M., et al. (1990). Detection of c-K-ras mutations in fine needle aspirates from human pancreatic adenocarcinomas. Cancer Res. 50, 1279–1283.
Shibuya, K. C., Goel, V. K., Xiong, W., Sham, J. G., Pollack, S. M., Leahy, A. M., et al. (2014). Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PLoS ONE 9:e96565. doi: 10.1371/journal.pone.0096565
Shiga, K., Hara, M., Nagasaki, T., Sato, T., Takahashi, H., and Takeyama, H. (2015). Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers 7, 2443–2458. doi: 10.3390/cancers7040902
Shukla, S. K., Gunda, V., Abrego, J., Haridas, D., Mishra, A., Souchek, J., et al. (2015). MUC16-mediated activation of mTOR and c-Myc reprograms pancreatic cancer metabolism. Oncotarget 6, 19118–19131. doi: 10.18632/oncotarget.4078
Shukla, S. K., Purohit, V., Mehla, K., Gunda, V., Chaika, N. V., Vernucci, E., et al. (2017). MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 32, 71–87.e77. doi: 10.1016/j.ccell.2017.06.004
Siegel, R. L., Miller, K. D., and Jemal, A. (2016). Cancer statistics, 2016. CA Cancer J. Clin. 66, 7–30. doi: 10.3322/caac.21332
Singh, P. K., Behrens, M. E., Eggers, J. P., Cerny, R. L., Bailey, J. M., Shanmugam, K., et al. (2008). Phosphorylation of MUC1 by Met modulates interaction with p53 and MMP1 expression. J. Biol. Chem. 283, 26985–26995. doi: 10.1074/jbc.M805036200
Singh, P. K., and Hollingsworth, M. A. (2006). Cell surface-associated mucins in signal transduction. Trends Cell Biol. 16, 467–476. doi: 10.1016/j.tcb.2006.07.006
Singh, P. K., Wen, Y., Swanson, B. J., Shanmugam, K., Kazlauskas, A., Cerny, R. L., et al. (2007). Platelet-derived growth factor receptor beta-mediated phosphorylation of MUC1 enhances invasiveness in pancreatic adenocarcinoma cells. Cancer Res. 67, 5201–5210. doi: 10.1158/0008-5472.CAN-06-4647
Smith, J. P., Cooper, T. K., Mcgovern, C. O., Gilius, E. L., Zhong, Q., Liao, J., et al. (2014). Cholecystokinin receptor antagonist halts progression of pancreatic cancer precursor lesions and fibrosis in mice. Pancreas 43, 1050–1059. doi: 10.1097/MPA.0000000000000194
Son, J., Lyssiotis, C. A., Ying, H., Wang, X., Hua, S., Ligorio, M., et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105. doi: 10.1038/nature12040
Stahelin, R. V., Karathanassis, D., Bruzik, K. S., Waterfield, M. D., Bravo, J., Williams, R. L., et al. (2006). Structural and membrane binding analysis of the Phox homology domain of phosphoinositide 3-kinase-C2alpha. J. Biol. Chem. 281, 39396–39406. doi: 10.1074/jbc.M607079200
Stanger, B. Z., Stiles, B., Lauwers, G. Y., Bardeesy, N., Mendoza, M., Wang, Y., et al. (2005). Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell 8, 185–195. doi: 10.1016/j.ccr.2005.07.015
Storz, P. (2017). KRas, ROS and the initiation of pancreatic cancer. Small GTPases 8, 38–42. doi: 10.1080/21541248.2016.1192714
Strouch, M. J., Cheon, E. C., Salabat, M. R., Krantz, S. B., Gounaris, E., Melstrom, L. G., et al. (2010). Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin. Cancer Res. 16, 2257–2265. doi: 10.1158/1078-0432.CCR-09-1230
Sulli, G., Di Micco, R., and D'adda Di Fagagna, F. (2012). Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 12, 709–720. doi: 10.1038/nrc3344
Tan, M. C., Goedegebuure, P. S., Belt, B. A., Flaherty, B., Sankpal, N., Gillanders, W. E., et al. (2009). Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J. Immunol. 182, 1746–1755. doi: 10.4049/jimmunol.182.3.1746
Tape, C. J., Ling, S., Dimitriadi, M., Mcmahon, K. M., Worboys, J. D., Leong, H. S., et al. (2016). Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell 165, 910–920. doi: 10.1016/j.cell.2016.03.029
Tassi, E., Braga, M., Longhi, R., Gavazzi, F., Parmiani, G., Di Carlo, V., et al. (2009). Non-redundant role for IL-12 and IL-27 in modulating Th2 polarization of carcinoembryonic antigen specific CD4 T cells from pancreatic cancer patients. PLoS ONE 4:e7234. doi: 10.1371/journal.pone.0007234
Tassi, E., Gavazzi, F., Albarello, L., Senyukov, V., Longhi, R., Dellabona, P., et al. (2008). Carcinoembryonic antigen-specific but not antiviral CD4+ T cell immunity is impaired in pancreatic carcinoma patients. J. Immunol. 181, 6595–6603. doi: 10.4049/jimmunol.181.9.6595
Thomas, D. A., and Massague, J. (2005). TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380. doi: 10.1016/j.ccr.2005.10.012
Unnikrishnan, A., Gafken, P. R., and Tsukiyama, T. (2010). Dynamic changes in histone acetylation regulate origins of DNA replication. Nat. Struct. Mol. Biol. 17, 430–437. doi: 10.1038/nsmb.1780
Utomo, W. K., Narayanan, V., Biermann, K., Van Eijck, C. H., Bruno, M. J., Peppelenbosch, M. P., et al. (2014). mTOR is a promising therapeutical target in a subpopulation of pancreatic adenocarcinoma. Cancer Lett. 346, 309–317. doi: 10.1016/j.canlet.2014.01.014
Van Dort, M. E., Galban, S., Wang, H., Sebolt-Leopold, J., Whitehead, C., Hong, H., et al. (2015). Dual inhibition of allosteric mitogen-activated protein kinase (MEK) and phosphatidylinositol 3-kinase (PI3K) oncogenic targets with a bifunctional inhibitor. Bioorg. Med. Chem. 23, 1386–1394. doi: 10.1016/j.bmc.2015.02.053
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M., and Bilanges, B. (2010). The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11, 329–341. doi: 10.1038/nrm2882
Vasseur, R., Skrypek, N., Duchene, B., Renaud, F., Martinez-Maqueda, D., Vincent, A., et al. (2015). The mucin MUC4 is a transcriptional and post-transcriptional target of K-ras oncogene in pancreatic cancer. Implication of MAPK/AP-1, NF-kappaB and RalB signaling pathways. Biochim. Biophys. Acta 1849, 1375–1384. doi: 10.1016/j.bbagrm.2015.10.014
Venkannagari, S., Fiskus, W., Peth, K., Atadja, P., Hidalgo, M., Maitra, A., et al. (2012). Superior efficacy of co-treatment with dual PI3K/mTOR inhibitor NVP-BEZ235 and pan-histone deacetylase inhibitor against human pancreatic cancer. Oncotarget 3, 1416–1427. doi: 10.18632/oncotarget.724
Vitale, M., Cantoni, C., Pietra, G., Mingari, M. C., and Moretta, L. (2014). Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 44, 1582–1592. doi: 10.1002/eji.201344272
Von Hoff, D. D., Ervin, T., Arena, F. P., Chiorean, E. G., Infante, J., Moore, M., et al. (2013). Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703. doi: 10.1056/NEJMoa1304369
Waddell, N., Pajic, M., Patch, A. M., Chang, D. K., Kassahn, K. S., Bailey, P., et al. (2015). Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501. doi: 10.1038/nature14169
Wang, J. Y., Lian, S. T., Chen, Y. F., Yang, Y. C., Chen, L. T., Lee, K. T., et al. (2002). Unique K-ras mutational pattern in pancreatic adenocarcinoma from Taiwanese patients. Cancer Lett. 180, 153–158. doi: 10.1016/S0304-3835(01)00818-7
Wang, P., Sun, Y. C., Lu, W. H., Huang, P., and Hu, Y. (2015). Selective killing of K-ras-transformed pancreatic cancer cells by targeting NAD(P)H oxidase. Chin. J. Cancer 34, 166–176. doi: 10.1186/s40880-015-0012-z
Wei, W. T., Chen, H., Wang, Z. H., Ni, Z. L., Liu, H. B., Tong, H. F., et al. (2012). Enhanced antitumor efficacy of gemcitabine by evodiamine on pancreatic cancer via regulating PI3K/Akt pathway. Int. J. Biol. Sci. 8, 1–14. doi: 10.7150/ijbs.8.1
Whatcott, C. J., Han, H., and Von Hoff, D. D. (2015). Orchestrating the tumor microenvironment to improve survival for patients with pancreatic cancer: normalization, not destruction. Cancer J. 21, 299–306. doi: 10.1097/PPO.0000000000000140
Xie, S., Chen, M., Yan, B., He, X., Chen, X., and Li, D. (2014). Identification of a role for the PI3K/AKT/mTOR signaling pathway in innate immune cells. PLoS ONE 9:e94496. doi: 10.1371/journal.pone.0094496
Xing, F., Saidou, J., and Watabe, K. (2010). Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 15, 166–179. doi: 10.2741/3613
Xue, J., Sharma, V., Hsieh, M. H., Chawla, A., Murali, R., Pandol, S. J., et al. (2015). Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun 6:7158. doi: 10.1038/ncomms8158
Yamamoto, S., Tomita, Y., Hoshida, Y., Morooka, T., Nagano, H., Dono, K., et al. (2004). Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin. Cancer Res. 10, 2846–2850. doi: 10.1158/1078-0432.CCR-02-1441
Yamazaki, M., Nakamura, K., Mizukami, Y., Ii, M., Sasajima, J., Sugiyama, Y., et al. (2008). Sonic hedgehog derived from human pancreatic cancer cells augments angiogenic function of endothelial progenitor cells. Cancer Sci. 99, 1131–1138. doi: 10.1111/j.1349-7006.2008.00795.x
Yang, S., Wang, X., Contino, G., Liesa, M., Sahin, E., Ying, H., et al. (2011). Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729. doi: 10.1101/gad.2016111
Yao, J. C., Shah, M. H., Ito, T., Bohas, C. L., Wolin, E. M., Van Cutsem, E., et al. (2011). Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 364, 514–523. doi: 10.1056/NEJMoa1009290
Yap, T. A., Yan, L., Patnaik, A., Fearen, I., Olmos, D., Papadopoulos, K., et al. (2011). First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 29, 4688–4695. doi: 10.1200/JCO.2011.35.5263
Ying, H., Elpek, K. G., Vinjamoori, A., Zimmerman, S. M., Chu, G. C., Yan, H., et al. (2011). PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discov. 1, 158–169. doi: 10.1158/2159-8290.CD-11-0031
Ying, H., Kimmelman, A. C., Lyssiotis, C. A., Hua, S., Chu, G. C., Fletcher-Sananikone, E., et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. doi: 10.1016/j.cell.2012.01.058
Yip-Schneider, M. T., Wiesenauer, C. A., and Schmidt, C. M. (2003). Inhibition of the phosphatidylinositol 3'-kinase signaling pathway increases the responsiveness of pancreatic carcinoma cells to sulindac. J. Gastrointest. Surg. 7, 354–363. doi: 10.1016/S1091-255X(02)00156-7
Yoshikawa, K., Mitsunaga, S., Kinoshita, T., Konishi, M., Takahashi, S., Gotohda, N., et al. (2012). Impact of tumor-associated macrophages on invasive ductal carcinoma of the pancreas head. Cancer Sci. 103, 2012–2020. doi: 10.1111/j.1349-7006.2012.02411.x
Yuan, T. L., and Cantley, L. C. (2008). PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510. doi: 10.1038/onc.2008.245
Zahreddine, H., and Borden, K. L. (2013). Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 4:28. doi: 10.3389/fphar.2013.00028
Zhang, Y., Kwok-Shing Ng, P., Kucherlapati, M., Chen, F., Liu, Y., Tsang, Y. H., et al. (2017). A pan-cancer proteogenomic atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell 31, 820–832.e823. doi: 10.1016/j.ccell.2017.04.013
Zhong, H., Chiles, K., Feldser, D., Laughner, E., Hanrahan, C., Georgescu, M. M., et al. (2000). Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 60, 1541–1545.
Keywords: PI3K, pancreatic cancer, mucins, MUC1, cancer metabolism, tumor microenvironment, cancer therapy
Citation: Murthy D, Attri KS and Singh PK (2018) Phosphoinositide 3-Kinase Signaling Pathway in Pancreatic Ductal Adenocarcinoma Progression, Pathogenesis, and Therapeutics. Front. Physiol. 9:335. doi: 10.3389/fphys.2018.00335
Received: 30 November 2017; Accepted: 19 March 2018;
Published: 04 April 2018.
Edited by:
Andrzej Ptasznik, University of Pennsylvania, United StatesCopyright © 2018 Murthy, Attri and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pankaj K. Singh, cGFua2FqLnNpbmdoQHVubWMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.