 Christine C. Helms
Christine C. Helms Mark T. Gladwin2,3
Mark T. Gladwin2,3
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 22 February 2018
Sec. Vascular Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.00125
This article is part of the Research Topic Regulation of Vascular Function by Circulating Blood View all 12 articles
Erythrocytes regulate vascular function through the modulation of oxygen delivery and the scavenging and generation of nitric oxide (NO). First, hemoglobin inside the red blood cell binds oxygen in the lungs and delivers it to tissues throughout the body in an allosterically regulated process, modulated by oxygen, carbon dioxide and proton concentrations. The vasculature responds to low oxygen tensions through vasodilation, further recruiting blood flow and oxygen carrying erythrocytes. Research has shown multiple mechanisms are at play in this classical hypoxic vasodilatory response, with a potential role of red cell derived vasodilatory molecules, such as nitrite derived nitric oxide and red blood cell ATP, considered in the last 20 years. According to these hypotheses, red blood cells release vasodilatory molecules under low oxygen pressures. Candidate molecules released by erythrocytes and responsible for hypoxic vasodilation are nitric oxide, adenosine triphosphate and S-nitrosothiols. Our research group has characterized the biochemistry and physiological effects of the electron and proton transfer reactions from hemoglobin and other ferrous heme globins with nitrite to form NO. In addition to NO generation from nitrite during deoxygenation, hemoglobin has a high affinity for NO. Scavenging of NO by hemoglobin can cause vasoconstriction, which is greatly enhanced by cell free hemoglobin outside of the red cell. Therefore, compartmentalization of hemoglobin inside red blood cells and localization of red blood cells in the blood stream are important for healthy vascular function. Conditions where erythrocyte lysis leads to cell free hemoglobin or where erythrocytes adhere to the endothelium can result in hypertension and vaso constriction. These studies support a model where hemoglobin serves as an oxido-reductase, inhibiting NO and promoting higher vessel tone when oxygenated and reducing nitrite to form NO and vasodilate when deoxygenated.
How erythrocytes modulate vascular tone has been widely studied over the last two decades. The vasodilation of the vasculature under hypoxic conditions has inspired much research ranging from the effect of oxygen partial pressure on smooth muscle cell contractility and endothelial nitric oxide synthase (eNOS) activity to nitrite reduction by hemoglobin (Hb) inside erythrocytes and subsequent production of nitric oxide (Figure 1). Here we review how red blood cells (RBCs) and hemoglobin regulate vascular function and blood flow.
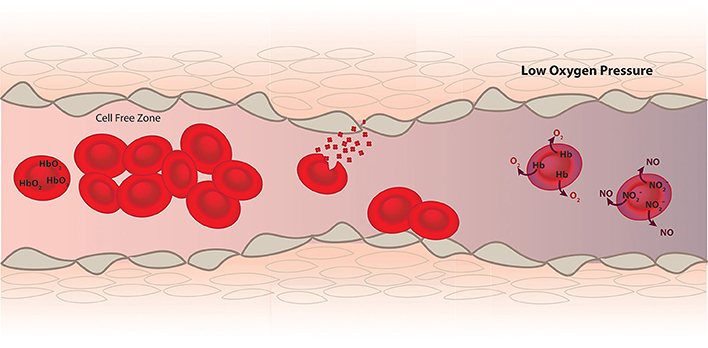
Figure 1. RBCs function as a transporter of oxygen from the lungs to the tissue and help establish hemostasis and vascular function. Since Hb inside RBCs is a very effective scavenger of NO, a vasodilator produced by the endothelium (brown), mechanism such as a cell free zone created by fluid dynamics, RBC membrane and internal diffusion minimize NO scavenging. In diseases where hemolysis and RBC adhesion occur these mechanisms to minimize NO scavenging are compromised and vasoconstriction occurs. However, in addition to scavenging NO data supports a role of deoxygenated RBCs in the production of NO leading to vasodilation under hypoxic conditions.
Many biochemistry texts provide thorough descriptions of the basic mechanisms of oxygen delivery by hemoglobin. Therefore, we will limit our discussion to a brief introduction. The hemoglobin molecule is a heterotetramer of hemoglobin alpha-beta dimers. Under high oxygen tension, each monomer can bind an O2 molecule. As hemoglobin binds oxygen, conformational changes occur to the protein and the binding affinity of the protein for molecular oxygen increases. Thus, binding is cooperative so that binding subsequent molecules of oxygen becomes easier after initial binding. Cooperative binding can be understood by a two-state model: The high affinity R-state and the low affinity T-state (Monod et al., 1965; Eaton et al., 1999). When hemoglobin has one or less oxygen molecules bound it is virtually always in the tense T-state. When it has 3–4 oxygen molecules bound it is virtually always in the R-state. Once 2 or more oxygen bind to a hemoglobin molecule that has no oxygen it will undergo a conformational change to the R-state where its oxygen affinity is now higher and it binds more oxygen. This homotropic regulation leads to a sigmoidal curve of hemoglobin fractional saturation vs. partial pressure of oxygen and allows hemoglobin to function as an efficient transporter of oxygen from the lungs to the tissues.
The complexity of oxygen delivery goes beyond the homotropic regulation. The pH of the blood also effects oxygen binding. The Bohr Effect, reported in the early 1900's, describes how increased acidity which occurs at low oxygen tension leads to protonation and stabilization of the T-state of hemoglobin promoting oxygen release. Additionally, carbon dioxide and 2,3-bisphosphate also act as heterotropic regulators, both decreasing the affinity of Hb for oxygen.
Therefore, RBCs contribute to vascular function through the delivery of oxygen by hemoglobin.
One mechanism through which oxygen tension and therefore RBCs effect vascular function is the contractile force of smooth muscle cells. Chang and Detar reported that a reduction in oxygen pressure led to a reduced contractile tension of helical strips cut from aorta, femoral arteries and small arteries from skeletal muscle (Chang and Detar, 1980). Additionally Taggart et al saw a decrease in smooth muscle force in hypoxia indicating smooth muscle response to oxygen may be independent of factors from the endothelium (Taggart and Wray, 1998). Potential mechanics of smooth muscle response to oxygen tension are increased ATP-sensitive K+ efflux or decreased voltage-sensitive Ca+2 influx (Franco-Obregón et al., 1995; Taggart and Wray, 1998). These effects have been recently reviewed (Jackson, 2016).
Alternatively, low oxygen tension effects the endothelial production of nitric oxide (NO) by eNOS, which requires oxygen as a substrate along with L-arginine (Palmer and Moncada, 1989; Kwon et al., 1990). Many researchers have shown diminished relaxation by endothelial cells under low oxygen tension (Furchgott and Zawadzki, 1980; De Mey and Vanhoutte, 1983; Lieberthal et al., 1989). Specifically, the apparent Km of eNOS for O2 is 4 μM and NOS activity begins to slow down once oxygen tensions starts falling below 1% (7.6 torr or 10 μM) (Abu-Soud et al., 2000; van Faassen et al., 2009). In summary, through the management of oxygen delivery RBCs effect eNOS function and smooth muscle contractility.
In addition to oxygen delivery, hemoglobin affects vascular function as a strong scavenger of NO. Furchgott and coworkers discovered that there is an agent produced in the endothelium that relaxes blood vessels, the endothelium relaxation factor (EDRF) (Furchgott and Zawadzki, 1980). The EDRF was then identified as NO (Ignarro et al., 1987; Palmer et al., 1987) that acts via activation of soluble guanylyl cyclase (Arnold et al., 1977). Endothelial nitric oxide synthase located on endothelial cell membranes produces NO that binds to soluble guanylyl cyclase in smooth muscles cells triggering a cell-signaling cascade that results in vasodilation. Chu et al. demonstrated the physiological regulation of blood flow and vasomotion by endogenously produced NO in awake dogs (Chu et al., 1991) and Quyyumi et al. showed atherosclerosis risk factors altered the response of coronary arteries to diminished NO production (Quyyumi et al., 1995). Additionally, NO has the anti-thrombotic effect of diminishing platelet activation (Schafer et al., 1980) and the anti-inflammatory effect of inhibiting leukocyte adhesion to the endothelium (Kubes et al., 1991). NO also leads to indirect vasodilation by inhibiting sympathetic vasoconstriction (Zanzinger et al., 1994). This is only a very small selection of a strong body of work that displays the role of NO in vascular function, which has been recently reviewed (Lundberg et al., 2015).
Since hemoglobin is present in the blood at a concentration of 10 mM in heme and ferrous heme has a strong affinity for NO with a dissociation constant of KD 10−10 to 10−11 M (Cooper, 1999), and the association rate constant is 107-108 M−1s−1 (Cassoly and Gibson, 1975), you might expect there to be no free NO at all.
Moreover, oxygenated hemoglobin reacts with NO to form nitrate (dioxygenation of NO) extremely fast (6–8 × 108 M−1s−1) (Doyle and Hoekstra, 1981; Eich et al., 1996; Herold et al., 2001) inactivating the NO, so that calculations predict that NO could not act as the EDRF (Lancaster, 1994; Vaughn et al., 1998).
However, as demonstrated above in the work by Chu et al. endothelial NO does play a physiological role in vascular function (Chu et al., 1991).
One can explain this paradox of NO bioavailability and Hb concentration in the blood by considering Hb encapsulation by RBCs. Experiments where RBCs are mixed with NO in suspension or in a stopped flow apparatus show that RBCs scavenge NO 1,000 times slower than cell-free Hb (Liu et al., 1998; Vaughn et al., 2000). Researchers have attributed this change in the rate of scavenging to a few mechanisms. One mechanism is the diffusion of NO to the RBC, which contributes to an unstirred layer around the cell (Coin and Olson, 1979; Liu et al., 1998, 2002; Azarov et al., 2005). A second mechanism is the membrane of the RBC acting as a barrier to diffusion (Vaughn et al., 2000; Huang et al., 2001, 2007). Third is diffusion of NO inside the RBC (Vaughn et al., 2000; Sakai et al., 2008). Our work suggests that rate limitations due to diffusion to the RBC are predominant (Azarov et al., 2005, 2011).
In addition to these changes under static conditions, a cell free zone created by the fluid dynamics of blood flow further reduces NO scavenging. The velocity of blood near the vessel wall is slower than in the center of the vessel due to friction. This creates a pressure gradient that pushes RBCs toward the center of the vessel away from the NO producing endothelium. Bugliarello and Sevilla, Cokelet and Liao et al. experimentally showed the presence of a cell free zone and computational modeling by Butler et al. supported its ability to reduce NO scavenging (Bugliarello and Sevilla, 1970; Cokelet, 1972; Butler et al., 1998; Liao et al., 1999). Subsequently, Liao et al experimentally demonstrated its ability to reduce NO scavenging using arteriole myography (Liao et al., 1999). Therefore, the compartmentalization of Hb inside the RBC and the localization of RBCs to the center of vessels have a strong influence on vascular function by minimizing NO scavenging.
In disease, RBC membrane abnormalities promote thrombosis (Ataga, 2009). Setty et al. showed exposure of phosphatidylserine on RBC membranes correlates with plasma indicators of thrombosis (Setty et al., 2001). In addition, changes to the RBC membrane lead to RBC adhesion to the endothelium. Diseases such as malaria, beta-thalassemia, diabetes mellitus, and sickle cell disease show increased RBC adhesion to endothelial cells (Kaul et al., 1989; Hovav et al., 1999; Cooke et al., 2004). Adhesion disrupts the cell free zone, which brings RBCs and Hb closer to the endothelium. Hoover et al. showed increased adhesion of RBCs from sickle cell patients to endothelial cells (Hoover et al., 1979) and Hebbel et al further demonstrated sickle RBC adhesion and suggested the adhesion may be a pathogenic factor in microvascular occlusion (Hebbel et al., 1980b). Hebbel also demonstrated a correlation between erythrocyte adhesion and sickle cell disease severity (Hebbel et al., 1980a). Kaul et al. developed a two-stage model of RBC adhesion initiating vaso-occlusion (Kaul et al., 1989). Thus, a role of RBC adhesion in initiating vaso-occlusive crisis is a widely supported hypothesis (Manwani and Frenette, 2013).
In addition to adhesion, polymerization of hemoglobin in sickle cell disease leads to rigid, inflexible red blood cells, which effect blood rheology. Moreover, oxygenated sickle RBC also displays changes in cell mechanics and therefore blood rheology, attributed to altered hemoglobin concentration in the cell and changes to the cell membrane (Nash et al., 1984). Changes in blood rheology affect tissue perfusion and may affect NO production through changes in shear stress on vessel walls. In addition to sickle cell disease, many other diseases display altered RBC deformability. In hypertension RBC deformability decreases, blood viscosity increases and RBCs form more stable aggregates (Cicco and Pirrelli, 1999). These changes exacerbate hypertension and effect oxygen transport (Cicco and Pirrelli, 1999). Diabetes and hypercholesterolemia, both risk factors for cardiovascular disease, show abnormal RBC membrane architecture and impaired deformability, which alter blood rheology (Shin et al., 2005; Tomaiuolo, 2014). It is difficult to determine if altered RBC deformability and therefore blood rheology are concurrent or contributing factors to the changes in vascular function in these diseases.
Work by Bor-Kucukatay et al. suggest vascular function, specifically nitric oxide production could play a role in RBC deformability and rheological alterations to RBCs during hypertension (Bor-Kucukatay et al., 2000, 2003). They suggest NO improves RBC deformability (Bor-Kucukatay et al., 2003). However, other labs have found that NO does not have a direct effect on RBC deformability (Barodka et al., 2014; Belanger et al., 2015). On the other hand, select NO donors, sodium nitroprusside and nitrite, protect against calcium-influx induced RBC dehydration and loss of deformability (Barodka et al., 2014; Belanger et al., 2015; Wajih et al., 2017). Use of sodium nitroprusside and nitrite to improve rheology in diseases affected by poor RBC deformability would thus be worth exploring.
As reviewed above, encapsulation of Hb inside RBCs greatly reduces its ability to scavenge NO produced by the endothelium. However, in multiple diseases RBC hemolysis occurs introducing cell-free Hb into the vasculature (Reiter et al., 2002; Sobolewski et al., 2005). The pathology associated with hemoglobin-based blood substitutes demonstrated the hypertensive effects of cell-free Hb (Doherty et al., 1998). Additionally, there is support for the implication of hemolysis in vaso-occlusive crisis (VOC) as hemolytic transfusion reactions can precipitate VOC (Jang et al., 2011; Manwani and Frenette, 2013).
Cell-free hemoglobin reactions in the vasculature and surrounding tissues through extravasation may add to disease conditions (Schaer et al., 2013). In vitro findings suggest cell-free hemoglobin has the potential to release hemin and take part in oxidative reactions resulting in oxidative stress and inflammation (Miller et al., 1997; Jia et al., 2007; Manwani and Frenette, 2013). Patients with elevated plasma Hb show a reduction in NO-dependent blood flow response, consistent with a decrease in vasodilatory response to NO donor sodium nitroprusside by patients with SCD (Rother et al., 2005).
In addition to releasing Hb, hemolysis also releases arginase, an enzyme that converts L-arginine to ornithine, into the blood stream. L-arginine is the substrate for nitric oxide synthesis by eNOS and therefore, the release of arginase further diminishes NO production and vascular function (Morris et al., 2003; Schnog et al., 2004). Hemolysis and changes to RBC membrane proteins in disease effect vascular function through promoting thrombosis, initiating vascular occlusion, scavenging NO, and oxidative stress.
Red blood cell breakdown not only decreases NO bioavailability through NO scavenging by cell-free Hb, but also by production of red blood cell microparticles (Donadee et al., 2011; Liu et al., 2013). Like cell-free Hb, red cell microparticles (on the order of 50-100 nm in diameter) scavenge NO hundreds of times faster than Hb encapsulated in the RBC, but not quite as fast as cell-free Hb (Donadee et al., 2011). In addition, these particles enter the cell-free zone (Liu et al., 2013). Red cell hemolysis and micropartcile formation have been proposed to contribute to poor outcomes associated with transfusion of older stored blood due to NO scavenging (Gladwin and Kim-Shapiro, 2009; Donadee et al., 2011). Substantial evidence suggests that extravasation plays a major role in NO dysregulation, so that proper compartmentalization of Hb is key (Kim-Shapiro and Patel, 2016; Schaer et al., 2016).
RBC hemolysis contributes to the vascular pathology of diseases and disorders such as thalassemia, hereditary spherocytosis, Glucose-6-phosphate dehydrogenase deficiency, paroxysmal nocturnal, hemoglobinuria, and autoimmune hemolytic anemia (Johnson et al., 1979; Rother et al., 2005). Additionally, these diseases and disorders also lead to the increased formation of RBC microparticles (Piccin et al., 2007; Westerman and Porter, 2016). Moreover, hemolysis is also associated with blood transfusion, hemodialysis and cardiac bypass surgery (Meyer et al., 2010).
In contrast to the role played by RBCs in diminishing NO. Research shows deoxy-RBCs promote vasodilation in the presence of nitrite (Cosby et al., 2003; Jensen and Agnisola, 2005; Crawford et al., 2006). Other mechanisms of vasodilatory action by RBCs have been proposed. Researchers continue to debate the mechanism or mechanisms of hypoxic vasodilation which include: (1) ATP release by RBCs due to deoxygenation, (2) SNO-Hb formation and S-nitrosothiol release and delivery during oxy/deoxy hemoglobin cycling, and (3) nitrite reduction by hemoglobin to NO.
ATP activates purinoceptors on endothelial cells leading to the production of NO and alteration of vascular tone (Ralevic and Burnstock, 1991). In line with the mechanism of ATP release by RBC under hypoxia, Ellsworth et al. showed that RBCs release more ATP under low PO2 and low pH than RBCs under normoxia and normal pH (Ellsworth et al., 1995). Additionally, they demonstrated intraluminal ATP increased vessel diameter and flow rate (Ellsworth et al., 1995). Following this work, Dietrich et al. showed perfusion of RBCs at low PO2 caused vessel dilation and an increased concentration of ATP in the effluent (Dietrich et al., 2000).
Addition of nitrite to deoxyRBCs showed an enhanced effect on vasodilation when infused (Cosby et al., 2003; Jensen and Agnisola, 2005; Crawford et al., 2006). In support of ATP release being responsible for this enhanced dilation in the presence of nitrite, Cao et al. measured an increased synthesis and hypoxic release of RBC ATP in the presence of nitrite (Cao et al., 2009). However, research showed a vasodilatory response in the presence of nitrite and RBCs when NOS inhibitors L-NAME and L-NMMA were used, suggesting a different mechanism of dilation is responsible for the enhancement by nitrite (Crawford et al., 2006; Liu et al., 2015).
The second proposed mechanism for the effect deoxyRBC and nitrite on vascular function is the formation of SNO-Hb in the presence of nitrite and subsequent release of NO from deoxygenated SNO-Hb. Research has demonstrated the hypoxic release of NO from SNO-Hb and the ability of the released NO to relax vessels (Stamler et al., 1997; McMahon et al., 2002). Diesen et al. showed a decrease in RBC SNO-Hb concentration correlated with abolished vessel hypoxic dilation (Diesen et al., 2008). The proposed mechanism of release is a reduction in the stability of SNO-Hb when Hb transitions from the R-state to the T-state and work by Doctor et al. further supports the proposal coupling SNO-Hb concentration to oxygen saturation (Doctor et al., 2005). However, for this mechanism to be relevant in vivo SNO-Hb must form during the oxygenation/deoxygenation cycle of RBCs. This leads to the question of how SNO-Hb forms. Huang et al. and Xu et al. ruled out allosterically-controlled transfer of NO from HbNO to the beta-93 cysteine of Hb (Xu et al., 2003; Huang et al., 2006). In addition, the role of nitrite in the formation of a semi-stable metHb-NO intermediate capable of SNO-Hb formation was ruled out by Basu et al. (Angelo et al., 2006; Nagababu et al., 2006; Basu et al., 2007). Lastly, Isbell et al. demonstrated nitrite associated NO bioactivity in beta-93 knockout mice, demonstrating SNO-Hb is not responsible for nitrite associated hypoxic vasodilation (Isbell et al., 2007). While data supports the release of NO from SNO-Hb under deoxygenated conditions, research has yet to elucidate a physiological mechanism of SNO-Hb formation therefore bringing to question the physiological relevance of SNO-Hb in vascular function.
The last proposed mechanism of RBC vasodilation in the presence of nitrite is the reduction of nitrite to NO by Hb. Brooks first studied the nitrite Hb interaction in 1937 and this work was later extended by Doyle in 1981 (Brooks, 1937; Doyle et al., 1981). Nitrite reacts with deoxygenated Hb to form metHb and NO (Brooks, 1937; Doyle et al., 1981). NO then reacts rapidly with deoxygenated Hb, at a rate dependent on the conformation state of the Hb (Huang et al., 2005a,b), to form HbNO. In vitro studies have rejected a significant role in the reduction of nitrite by other RBC molecules such as xanthine oxidoreductase and carbonic anhydrase supporting the hypothesis that hemoglobin plays a dominant role (Liu et al., 2015). Due to the rapid scavenging of NO by Hb the question of how NO bioactivity escapes the RBC still remains. However, data continues to grow in support of NO bioactivity escaping the deoxy-RBC in the presence of nitrite. For example, platelets activation and aggregation by ADP is diminished in the presence of deoxyRBCs and nitrite (Srihirun et al., 2012; Park et al., 2014; Liu et al., 2015; Wajih et al., 2016). Platelets stimulated by ADP in the presence of nitrite alone do not show reduced activation (Srihirun et al., 2012). The addition of a NO scavenger to platelets stimulated by ADP in the presence of deoxy-RBCs and nitrite abrogates the diminished activation (Wajih et al., 2016). These data strongly support a role of RBCs in bioactivating nitrite.
However, once NO is formed inside a RBC, it must somehow escape rapid scavenging by the intraerythrocytic Hb. Proposed mechanisms of escape of NO bioactivity from the RBC have included S-nitrosothiol formation and the role of the RBC membrane. Research by Basu et al and Roche et al suggested S-nitrosothiol formation could occur through a metHb-nitrite mediated N2O3 formation (Basu et al., 2007; Roche et al., 2013). Pawloski proposed a mechanism where low molecular weight S-nitrosothiols could transnitrosylate AE1, an abundant RBC ion transporter, and eventually escape the RBC (Pawloski et al., 2001). Another proposed mechanism, is an increased affinity of metHb-NO or Hb-NO+ for the RBC membrane, which in turn could also lead to NO escape through nitrosylation of AE1 (Salgado et al., 2015). Lastly, Wajih et al have published evidence supporting a role of RBC membrane nitrosation in the escape of NO bioactivity (Wajih et al., 2016). While this is a start in determining the mechanism of NO bioactivity, much work remains.
Other heme-globin nitrite reducers may play a role in modulating vascular function. Deoxymyoglobin reduces nitrite faster than Hb (Shiva et al., 2007). Nitrite reduction by vascular smooth muscle myoglobin has been shown to contribute significantly to vasodilation in a murine model (Ormerod et al., 2010). Neuroglobin and cytoglobin also have nitrite reducing activity that is modulated by cysteine oxidation state (Tiso et al., 2011; Li et al., 2012; Tejero et al., 2014; Amdahl et al., 2017). Globin X from fish red blood cells reduces nitrite about 200 times faster than Hb and may represent a primordial function of heme-globins in nitrite reduction (Corti et al., 2016).
The production of NO by deoxyRBCs in the presence of nitrite and other NO donors provide a potential therapeutic role for plasma nitrite in reducing vasoconstriction, RBC adhesion, and potentially improving RBC deformability (Space et al., 2000; Bor-Kucukatay et al., 2003; Lundberg et al., 2008; Horn et al., 2011). NO administration can neutralize the NO scavenging ability of cell-free Hb by preferentially reacting, converting oxygenated hemoglobin to non-scavenging methemoglobin (Reiter et al., 2002). Antioxidants aim to improve blood rheology and vascular function through reduction of oxidative stress, shown by researchers to exacerbate hemolysis and endothelial dysfunction (Fibach and Rachmilewitz, 2008; den Hartog et al., 2010). Hemin and hemoglobin scavenger proteins also provide a potential therapeutic against damage caused by cell free hemoglobin and hemin (Schaer et al., 2013). Lastly, adhesion molecule inhibitors may act as a therapeutic against RBC adhesion to the endothelium; for example, Hebbel et al. showed adhesion molecule inhibitor HDAC reduced endothelial activation (Hebbel et al., 2009; Vinchi and Tolosano, 2013). A recent trial of a compound that interferes with adhesion of neutrophils and other circulating blood cells to endothelial cells successfully decreased the frequency of painful crises in patients with sickle cell disease (Ataga et al., 2017).
Red blood cells play an important role in vascular function. Through the compartmentalization of Hb, RBCs deliver oxygen, minimize NO scavenging, sense oxygen tension, and deliver NO bioactivity in hypoxia. In healthy conditions, RBCs promote hemostasis through the well-regulated delivery of oxygen and the balance of NO scavenging and production. As data has shown, the RBC, once only viewed as a sink for NO, has the ability to produce NO bioactivity under hypoxia. The important role of the RBC in vascular function becomes more evident in diseases that alter or compromise the RBC membrane leading to conditions of oxidative stress, hypertension, thrombosis, and vaso-occlusion.
CH, MG, and DK-S together conceptualized the article and its content. CH wrote the initial draft of the manuscript and further edited the manuscript. MG and DK-S heavily edited the manuscript.
This work was supported by the National Institutes of Health grant number HL058091 and HL098032.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abu-Soud, H. M., Ichimori, K., Presta, A., and Stuehr, D. J. (2000). Electron transfer, oxygen binding, and nitric oxide feedback inhibition in endothelial nitric-oxide synthase. J. Biol. Chem. 275, 17349–17357. doi: 10.1074/jbc.M000050200
Amdahl, M. B., Sparacino-Watkins, C., Corti, P., Gladwin, M. T., and Tejero, J. (2017). Efficient reduction of vertebrate cytoglobins by the cytochrome b5/cytochrome b5 reductase/NADH System. Biochemistry 56, 3993–4004. doi: 10.1021/acs.biochem.7b00224
Angelo, M., Singel, D. J., and Stamler, J. S. (2006). An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc. Natl. Acad. Sci.U.S.A. 103, 8366–8371. doi: 10.1073/pnas.0600942103
Arnold, W. P., Mittal, C. K., Katsuki, S., and Murad, F. (1977). Nitric oxide activates guanylate cyclase and increases guanosine 3':5'-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. U.S.A. 74, 3203–3207. doi: 10.1073/pnas.74.8.3203
Ataga, K. (2009). Hypercoagulability and thrombotic complications in hemolytic anemias. Haematologica 94, 1481–1484. doi: 10.3324/haematol.2009.013672
Ataga, K. I., Kutlar, A., Kanter, J., Liles, D., Cancado, R., Friedrisch, J., et al. (2017). Crizanlizumab for the prevention of pain crises in sickle cell disease. N. Engl. J. Med. 376, 429–439. doi: 10.1056/NEJMoa1611770
Azarov, I., Huang, K., Basu, S., Gladwin, M., Hogg, N., and Kim-Shapiro, D. (2005). Nitric oxide scavenging by red blood cells as a function of hematocrit and oxygenation. J. Biol. Chem. 280:39024. doi: 10.1074/jbc.M509045200
Azarov, I., Liu, C., Reynolds, H., Tsekouras, Z., Lee, J., Gladwin, M., et al. (2011). Mechanisms of slower nitric oxide uptake by red blood cells and other hemoglobin-containing vesicles. J. Biol. Chem. 286:33567. doi: 10.1074/jbc.M111.228650
Barodka, V., Mohanty, J. G., Mustafa, A. K., Santhanam, L., Nyhan, A., Bhunia, A. K., et al. (2014). Nitroprusside inhibits calcium-induced impairment of red blood cell deformability. Transfusion 54, 434–444. doi: 10.1111/trf.12291
Basu, S., Grubina, R., Huang, J., Conradie, J., Huang, Z., Jeffers, A., et al. (2007). Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat. Chem. Biol. 3, 785–794. doi: 10.1038/nchembio.2007.46
Belanger, A. M., Keggi, C., Kanias, T., Gladwin, M. T., and Kim-Shapiro, D. B. (2015). Effects of nitric oxide and its congeners on sickle red blood cell deformability. Transfusion 55, 2464–2472. doi: 10.1111/trf.13134
Bor-Kucukatay, M., Wenby, R. B., Meiselman, H. J., and Baskurt, O. K. (2003). Effects of nitric oxide on red blood cell deformability. Am. J. Physiol. Heart Circul. Physiol. 284, H1577–H1584. doi: 10.1152/ajpheart.00665.2002
Bor-Kucukatay, M., Yalcin, O., Gokalp, O., Kipmen-Korgun, D., Yesilkaya, A., Baykal, A., et al. (2000). Red blood cell rheological alterations in hypertension induced by chronic inhibition of nitric oxide synthasis in rats. Clin. Hemorheol. Microcircul. 22, 267–275.
Brooks, J. (1937). The action of nitrite on haemoglobin in the absence of oxygen. Proc. R. Soc. London B. Biol. Sci. 123, 368–382. doi: 10.1098/rspb.1937.0057
Bugliarello, G., and Sevilla, J. (1970). Velocity distribution and other characteristics of steady and pulsatile blood flow in fine glass tubes. Biorheology 7, 85–107. doi: 10.3233/BIR-1970-7202
Butler, A., Megson, I., and Wright, P. (1998). Diffusion of nitric oxide and scavenging by blood in the vasculature. Biochim. Biophys. Acta 1425:168. doi: 10.1016/S0304-4165(98)00065-8
Cao, Z., Bell, J. B., Mohanty, J. G., Nagababu, E., and Rifkind, J. M. (2009). Nitrite enhances RBC hypoxic ATP synthesis and the release of ATP into the vasculature: a new mechanism for nitrite-induced vasodilation. Am. J. Physiol. Heart Circul. Physiol. 297, H1494–H1503. doi: 10.1152/ajpheart.01233.2008
Cassoly, R., and Gibson, Q. H. (1975). Conformation, co-operativity and ligand binding in human hemoglobin. J. Mol. Biol. 91, 301–313. doi: 10.1016/0022-2836(75)90382-4
Chang, A. E., and Detar, R. (1980). Oxygen and vascular smooth muscle contraction revisited. Am. J. Physiol. Heart Circ. Physiol. 5:H716. doi: 10.1152/ajpheart.1980.238.5.H716
Chu, A., Chambers, D. E., Lin, C. C., Kuehl, W. D., Palmer, R. M., Moncada, S., et al. (1991). Effects of inhibition of nitric oxide formation on basal vasomotion and endothelium-dependent responses of the coronary arteries in awake dogs. J. Clin. Invest. 87, 1964–1968. doi: 10.1172/JCI115223
Cicco, G., and Pirrelli, A. (1999). Red blood cell (RBC) deformability, RBC aggregability and tissue oxygenation in hypertension. Clin. Hemorheol. Microcircul. 21, 169–177.
Coin, J. T., and Olson, J. S. (1979). The rate of oxygen uptake by human red blood cells. J. Biol. Chem. 254, 1178–1190.
Cokelet, G. R. (1972). “The Rheology of Human Blood” in Biomechanics: Its Foundation and Objectives, eds Y. C. Fung, N. Perrone, and M. Anliker (Englewood Cliffs, NJ: Prentice Hall), 63–103.
Cooke, B. M., Mohandas, N., and Coppel, R. L. (2004). Malaria and the red blood cell membrane. Semin. Hematol. 41, 173–188. doi: 10.1053/j.seminhematol.2004.01.004
Corti, P., Xue, J., Tejero, J., Wajih, N., Sun, M., Stolz, D. B., et al. (2016). Globin X is a six-coordinate globin that reduces nitrite to nitric oxide in fish red blood cells. Proc. Natl. Acad. Sci.U.S.A. 113, 8538–8543. doi: 10.1073/pnas.1522670113
Cosby, K., Partovi, K., Crawford, J., Patel, R., Reiter, C., Martyr, S., et al. (2003). Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 9:1498. doi: 10.1038/nm954
Crawford, J. H., Isbell, T. S., Huang, Z., Shiva, S., Chacko, B. K., Schechter, A. N., et al. (2006). Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood 107, 566–574. doi: 10.1182/blood-2005-07-2668
De Mey, J. G., and Vanhoutte, P. M. (1983). Anoxia and endothelium-dependent reactivity of the canine femoral artery. J. Physiol. 335, 65–74.
den Hartog, G. J. M., Boots, A. W., Adam-Perrot, A., Brouns, F., Verkooijen, I. W. C. M., Weseler, A. R., et al. (2010). Erythritol is a sweet antioxidant. Nutrition 26, 449–458. doi: 10.1016/j.nut.2009.05.004
Diesen, D. L., Hess, D. T., and Stamler, J. S. (2008). Hypoxic vasodilation by red blood cells: evidence for an S-nitrosothiol based signal. Circ.Res. 103, 545–553. doi: 10.1161/CIRCRESAHA.108.176867
Dietrich, H. H., Ellsworth, M. L., Sprague, R. S., and Dacey, R. G. (2000). Red blood cell regulation of microvascular tone through adenosine triphosphate. Am. J. Physiol. Heart Circul. Physiol. 278, H1294–H1298. doi: 10.1152/ajpheart.2000.278.4.H1294
Doctor, A., Platt, R., Sheram, M. L., Eischeid, A., McMahon, T., Maxey, T., et al. (2005). Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc. Natl. Acad. Sci. U.S.A. 102, 5709–5714. doi: 10.1073/pnas.0407490102
Doherty, D. H., Dolye, M. P., Curry, S. R., Vali, R. J., Fattor, T. J., Olson, J. S., et al. (1998). Rate of reaction with nitric oxide determines the hypertensive effect of cell-free hemoglobin. Nat. Biotechnol. 16, 672–676. doi: 10.1038/nbt0798-672
Donadee, C., Raat, N. J. H., Kanias, T., Tejero, J., Lee, J. S., Kelley, E. E., et al. (2011). Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion clinical perspective. Circulation 124, 465–476. doi: 10.1161/CIRCULATIONAHA.110.008698
Doyle, M. P., and Hoekstra, J. W. (1981). Oxidation of nitrogen oxides by bound dioxygen in hemoproteins. J. Inorg. Biochem. 14, 351–358. doi: 10.1016/S0162-0134(00)80291-3
Doyle, M. P., Pickering, R. A., DeWeert, T. M., Hoekstra, J. W., and Pater, D. (1981). Kinetics and mechanism of the oxidation of human deoxyhemoglobin by nitrites. J. Biol. Chem. 256, 12393–12398.
Eaton, W. A., Henry, E. R., Hofrichter, J., and Mozzarelli, A. (1999). Is cooperative oxygen binding by hemoglobin really understood? Nat. Struct. Mol. Biol. 6, 351–358. doi: 10.1038/7586
Eich, R. F., Li, T., Lemon, D. D., Doherty, D. H., Curry, S. R., Aitken, J. F., et al. (1996). Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry 35, 6976–6983.
Ellsworth, M. L., Forrester, T., Ellis, C. G., and Dietrich, H. H. (1995). The erythrocyte as a regulator of vascular tone. Am. J. Physiol. Heart Circ. Physiol. 269:H2155.
Fibach, E., and Rachmilewitz, E. (2008). The role of oxidative stress in hemolytic anemia. Curr. Mol. Med. 8, 609–619. doi: 10.2174/156652408786241384
Franco-Obregón, A., Ureña, J., and López-Barneo, J. (1995). Oxygen-sensitive calcium channels in vascular smooth muscle and their possible role in hypoxic arterial relaxation. Proc. Natl. Acad. Sci. U.S.A. 92, 4715–4719.
Furchgott, R. F., and Zawadzki, J. V. (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288, 373–376. doi: 10.1038/288373a0
Gladwin, M. T., and Kim-Shapiro, D. B. (2009). Storage lesion in banked blood due to hemolysis-dependent disruption of nitric oxide homeostasis. Curr. Opin. Hematol. 16, 515–523. doi: 10.1097/MOH.0b013e32833157f4
Hebbel, R. P., Boogaerts, M. A. B., Eaton, J. W., and Steinberg, M. H. (1980a). Erythrocyte adherence to endothelium in sickle-cell anemia. N. Engl. J. Med. 302, 992–995. doi: 10.1056/NEJM198005013021803
Hebbel, R. P., Vercellotti, G. M., Pace, B. S., Solovey, A. N., Kollander, R., Abanonu, C. F., et al. (2009). The HDAC inhibitors trichostatin A and suberoylanilide hydroxamic acid exhibit multiple modalities of benefit for the vascular pathobiology of sickle transgenic mice. Blood 115, 2483–2490. doi: 10.1182/blood-2009-02-204990
Hebbel, R. P., Yamada, O., Moldow, C. F., Jacob, H. S., White, J. G., and Eaton, J. W. (1980b). Abnormal adherence of sickle erythrocytes to cultured vascular endothelium: possible mechanism for microvascular occlusion in sickle cell disease. J. Clin. Invest. 65, 154–160. doi: 10.1172/JCI109646
Herold, S., Exner, M., and Nauser, T. (2001). Kinetic and mechanistic studies of the NO center dot-mediated oxidation of oxymyoglobin and oxyhemoglobin. Biochemistry 40, 3385–3395. doi: 10.1021/bi002407m
Hoover, R., Rubin, R., Wise, G., and Warren, R. (1979). Adhesion of normal and sickle erythrocytes to endothelial monolayer cultures. Blood 54:872.
Horn, P., Cortese-Krott, M., Keymel, S., Kumara, I., Burghoff, S., Schrader, J., et al. (2011). Nitric oxide influences red blood cell velocity independently of changes in the vascular tone. Free Radic. Res. 45, 653–661. doi: 10.3109/10715762.2011.574288
Hovav, T., Goldfarb, A., Artmann, G., Yedgar, S., and Barshtein, G. (1999). Enhanced adherence of β-thalassaemic erythrocytes to endothelial cells. Br. J. Haematol. 106, 178–181. doi: 10.1046/j.1365-2141.1999.01489.x
Huang, K., Han, T. H., Hyduke, D. R., Vaughn, M. W., Van Herle, H., Hein, T. W., et al. (2001). Modulation of nitric oxide bioavailability by erythrocytes. Proc. Natl. Acad. Sci. U.S.A. 98, 11771–11776. doi: 10.1073/pnas.201276698
Huang, K. T., Azarov, I., Basu, S., Huang, J., and Kim-Shapiro, D. (2006). Lack of allosterically controlled intramolecular transfer of nitric oxide from the heme to cysteine in the β-93 subunit of hemoglobin. Blood 107, 2602–2604. doi: 10.1182/blood-2005-10-4104
Huang, K. T., Huang, Z., and Kim-Shapiro, D. B. (2007). Nitric oxide red blood cell membrane permeability at high and low oxygen tension. Nitric Oxide 16, 209–216. doi: 10.1016/j.niox.2006.11.002
Huang, K. T., Keszler, A., Patel, N., Patel, R. P., Gladwin, M. T., Kim-Shapiro, D. B., et al. (2005a). The reaction between nitrite and deoxyhemoglobin: reassessment of reaction kinetics and stoichiometry. J. Biol. Chem. 280, 31126–31131. doi: 10.1074/jbc.M501496200
Huang, Z., Shiva, S., Kim-Shapiro, D., Patel, R. P., Ringwood, L. A., Irby, C. E., et al. (2005b). Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J. Clin. Invest. 115, 2099–2107. doi: 10.1172/JCI24650
Ignarro, L. J., Byrns, R. E., Buga, G. M., and Wood, K. S. (1987). Endothelium-derived relaxing factor from pulmonary-artery and vein possesses pharmacological and chemical-properties identical to those of nitric-oxide radical. Circ. Res. 61, 866–879. doi: 10.1161/01.RES.61.6.866
Isbell, T. S., Gladwin, M. T., and Patel, R. P. (2007). Hemoglobin oxygen fractional saturation regulates nitrite-dependent vasodilation of aortic ring bioassays. Am. J. Physiol. 293, H2565–H2572. doi: 10.1152/ajpheart.00759.2007
Jackson, W. F. (2016). Arteriolar oxygen reactivity: where is the sensor and what is the mechanism of action? J. Physiol. 594, 5055–5077. doi: 10.1113/JP270192
Jang, J., Hod, E. A., Spitalnik, S. L., and Frenette, P. S. (2011). CXCL1 and its receptor, CXCR2, mediate murine sickle cell vaso-occlusion during hemolytic transfusion reactions. J. Clin. Invest. 121, 1397–1401. doi: 10.1172/JCI45336
Jensen, F. B., and Agnisola, C. (2005). Perfusion of the isolated trout heart coronary circulation with red blood cells: effects of oxygen supply and nitrite on coronary flow and myocardial oxygen consumption. J. Exp. Biol. 208, 3665–3674. doi: 10.1242/jeb.01815
Jia, Y., Buehler, P. W., Boykins, R. A., Venable, R. M., and Alayash, A. I. (2007). Structural basis of peroxide-mediated changes in human hemoglobin: a novel oxidative pathway. J. Biol. Chem. 282, 4894–4907. doi: 10.1074/jbc.M609955200
Johnson, G. J., Allen, D. W., Cadman, S., Fairbanks, V. F., White, J. G., Lampkin, B. C., et al. (1979). Red-cell-membrane polypeptide aggregates in glucose-6-phosphate dehydrogenase mutants with chronic hemolytic disease. A clue to the mechanism of hemolysis. N. Engl. J. Med. 301, 522–527. doi: 10.1056/NEJM197909063011004
Kaul, D. K., Fabry, M. E., and Nagel, R. L. (1989). Microvascular sites and characteristics of sickle cell adhesion to vascular endothelium in shear flow conditions: pathophysiological implications. Proc. Natl. Acad. Sci. U.S.A. 86, 3356–3360. doi: 10.1073/pnas.86.9.3356
Kim-Shapiro, D., and Patel, R. P. (2016). Compartmentalization is key in limiting nitric oxide scavenging by cell-free hemoglobin. Am. J. Respir. Crit. Care Med. 193, 1072–1074. doi: 10.1164/rccm.201512-2481ED
Kubes, P., Suzuki, M., and Granger, D. N. (1991). Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. U.S.A. 88, 4651–4655. doi: 10.1073/pnas.88.11.4651
Kwon, N. S., Nathan, C. F., Gilker, C., Griffith, O. W., Matthews, D. E., and Stuehr, D. J. (1990). L-citrulline production from L-arginine by macrophage nitric oxide synthase. The ureido oxygen derives from dioxygen. J. Biol. Chem. 265, 13442–13445.
Lancaster, J. R. (1994). Simulation of the diffusion and reaction of endogenously produced nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 91, 8137–8141. doi: 10.1073/pnas.91.17.8137
Li, H., Hemann, C., Abdelghany, T. M., El-Mahdy, M. A., and Zweier, J. L. (2012). Characterization of the mechanism and magnitude of cytoglobin-mediated nitrite reduction and nitric oxide generation under anaerobic conditions. J. Biol. Chem. 287, 36623–36633. doi: 10.1074/jbc.M112.342378
Liao, J. C., Hein, W. T., Vaughn, M. W., Huang, K., and Kuo, L. (1999). Intravascular flow decreases erythrocyte consumption of nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 96, 8757–8761. doi: 10.1073/pnas.96.15.8757
Lieberthal, W., Wolf, E. F., Rennke, H. G., Valeri, C. R., and Levinsky, N. G. (1989). Renal ischemia and reperfusion impair endothelium-dependent vascular relaxation. Am. J. Physiol. Renal Physiol. 256:F894.
Liu, C., Wajih, N., Liu, X., Basu, S., Janes, J., Marvel, M., et al. (2015). Mechanisms of human erythrocytic bioactivation of nitrite. J. Biol. Chem. 290, 1281–1294. doi: 10.1074/jbc.M114.609222
Liu, C., Zhao, W., Christ, G. J., Gladwin, M. T., and Kim-Shapiro, D. B. (2013). Nitric oxide scavenging by red cell microparticles. Free Radic. Biol. Med. 65, 1164–1173. doi: 10.1016/j.freeradbiomed.2013.09.002
Liu, X., Miller, M. J. S., Joshi, M. S., Sadowska-Krowicka, H., Clark, D. A., and Lancaster, J. R. (1998). Diffusion-limited reaction of free nitric oxide with erythrocytes. J. Biol. Chem. 273, 18709–18713. doi: 10.1074/jbc.273.30.18709
Liu, X. P., Samouilov, A., Lancaster, J. R. Jr., and Zweier, J. L. (2002). Nitric oxide uptake by erythrocytes is primarily limited by extracellular diffusion not membrane resistance. J. Biol. Chem. 277, 26194–26199. doi: 10.1074/jbc.M201939200
Lundberg, J. O., Gladwin, M. T., and Weitzberg, E. (2015). Strategies to increase nitric oxide signalling in cardiovascular disease. Nat. Rev. Drug Discov. 14, 623–641. doi: 10.1038/nrd4623
Lundberg, J., Weitzberg, E., and Gladwin, M. (2008). The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Disc. 7:156. doi: 10.1038/nrd2466
Manwani, D., and Frenette, P. S. (2013). Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood 122, 3892–3898. doi: 10.1182/blood-2013-05-498311
McMahon, T. J., Moon, R. E., Luschinger, B. P., Carraway, M. S., Stone, A. E., Stolp, B. W., et al. (2002). Nitric oxide in the human respiratory cycle. Nat. Med. 8, 711–717. doi: 10.1038/nm718
Meyer, C., Heiss, C., Drexhage, C., Kehmeier, E. S., Balzer, J., Mühlfeld, A., et al. (2010). Hemodialysis-induced release of hemoglobin limits nitric oxide bioavailability and impairs vascular function. J. Am. Coll. Cardiol. 55, 454–459. doi: 10.1016/j.jacc.2009.07.068
Miller, Y. I., Altamentova, S. M., and Shaklai, N. (1997). Oxidation of low-density lipoprotein by hemoglobin stems from a heme-initiated globin radical: antioxidant role of haptoglobin. Biochemistry 36, 12189–12198. doi: 10.1021/bi970258a
Monod, J., Wyman, J., and Changeux, J. P. (1965). On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118. doi: 10.1016/S0022-2836(65)80285-6
Morris, C. R., Morris, S. M. J., Hagar, W., van Warmerdam, J., Claster, S., Kepka-Lenhart, D., et al. (2003). Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease? Am. J. Respir. Crit. Care 168, 63–69. doi: 10.1164/rccm.200208-967OC
Nagababu, E., Ramasamy, S., and Rifkind, J. M. (2006). S-nitrosohemoglobin: a mechanism for its formation in conjunction with nitrite reduction by deoxyhemoglobin. Nitric Oxide 15, 20–29. doi: 10.1016/j.niox.2006.01.012
Nash, G. B., Johnson, C. S., and Meiselman, H. J. (1984). Mechanical properties of oxygenated red blood cells in sickle cell (HbSS) disease. Blood 63:73.
Ormerod, J. O. M., Ashrafian, H., Maher, A. R., Arif, S., Steeples, V., Born, G. V. R., et al. (2010). The role of vascular myoglobin in nitrite-mediated blood vessel relaxation. Cardiovasc. Res. 89, 560–565. doi: 10.1093/cvr/cvq299
Palmer, R. M. J., Ferrige, A. G., and Moncada, S. (1987). Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327, 524–526. doi: 10.1038/327524a0
Palmer, R. M. J., and Moncada, S. A. (1989). Novel citrulline-forming enzyme implicated in the formation of nitric oxide by vascular endothelial cells. Biochem. Biophys. Res. Commun. 158, 348–352. doi: 10.1016/S0006-291X(89)80219-0
Park, J. W., Piknova, B., Nghiem, K., Lozier, J. N., and Schechter, A. N. (2014). Inhibitory effect of nitrite on coagulation processes demonstrated by thrombelastography. Nitric Oxide 40, 45–51. doi: 10.1016/j.niox.2014.05.006
Pawloski, J. R., Hess, D. T., and Stamler, J. S. (2001). Export by red blood cells of nitric oxide bioactivity. Nature 409, 622–626. doi: 10.1038/35054560
Piccin, A., Murphy, W. G., and Smith, O. P. (2007). Circulating microparticles: pathophysiology and clinical implications. Blood Rev. 21, 157–171. doi: 10.1016/j.blre.2006.09.001
Quyyumi, A. A., Dakak, N., Andrews, N. P., Husain, S., Arora, S., Gilligan, D. M., et al. (1995). 3. Nitric oxide activity in the human coronary circulation. Impact of risk factors for coronary atherosclerosis. J. Clin. Invest. 95, 1747–1755. doi: 10.1172/JCI117852
Ralevic, V., and Burnstock, G. (1991). Roles of P2-purinoceptors in the cardiovascular system. Circulation 84:1.
Reiter, C. D., Wang, X., Tanus-Santos, J. E., Hogg, N., Cannon, R. O., Schechter, A. N., et al. (2002). Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 8, 1383–1389. doi: 10.1038/nm1202-799
Roche, C. J., Cassera, M. B., Dantsker, D., Hirsch, R. E., and Friedman, J. M. (2013). Generating S-nitrosothiols from hemoglobin: mechanisms, conformational dependence, and physiological relevance. J. Biol. Chem. 288, 22408–22425. doi: 10.1074/jbc.M113.482679
Rother, R. P., Bell, L., Hillmen, P., and Gladwin, M. T. (2005). The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin. JAMA 293, 1653–1662. doi: 10.1001/jama.293.13.1653
Sakai, H., Sato, A., Masuda, K., Takeoka, S., and Tsuchida, E. (2008). Encapsulation of concentrated hemoglobin solution in phospholipid vesicles retards the reaction with no, but not, C. O, by intracellular diffusion barrier. J. Biol. Chem. 283, 1508–1517. doi: 10.1074/jbc.M707660200
Salgado, M. T., Cao, Z., Nagababu, E., Mohanty, J. G., and Rifkind, J. M. (2015). Red blood cell membrane-facilitated release of nitrite-derived nitric oxide bioactivity. Biochemistry 54, 6712–6723. doi: 10.1021/acs.biochem.5b00643
Schaer, C. A., Deuel, J. W., Schildknecht, D., Mahmoudi, L., Garcia-Rubio, I., Owczarek, C., et al. (2016). Haptoglobin preserves vascular nitric oxide signaling during hemolysis. Am. J. Respir. Crit. Care Med. 193, 1111–1122. doi: 10.1164/rccm.201510-2058OC
Schaer, D. J., Buehler, P. W., Alayash, A. I., Belcher, J. D., and Vercellotti, G. M. (2013). Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121, 1276–1284. doi: 10.1182/blood-2012-11-451229
Schafer, A. I., Alexander, R. W., and Handin, R. I. (1980). Inhibition of platelet function by organic nitrate vasodilators. Blood 55:649.
Schnog, J. B., Jager, E. H., van der Dijs, F. P., Duits, A. J., Moshage, H., Muskiet, F. D., et al. (2004). Evidence for a metabolic shift of arginine metabolism in sickle cell disease. Ann. Hematol. 83, 371–375. doi: 10.1007/s00277-004-0856-9
Setty, B. N. Y., Rao, A. K., and Stuart, M. J. (2001). Thrombophilia in sickle cell disease: the red cell connection. Blood 98, 3228–3233. doi: 10.1182/blood.V98.12.3228
Shin, S., Ku, Y., Park, M., and Suh, J. (2005). Deformability of red blood cells: a determinant of blood viscosity. J. Mech. Sci. Technol. 19:216. doi: 10.1007/BF02916121
Shiva, S., Huang, Z., Grubina, R., Sun, J., Ringwood, L. A., MacArthur, P. H., et al. (2007). Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ.Res. 100:654. doi: 10.1161/01.RES.0000260171.52224.6b
Sobolewski, P., Gramaglia, I., Frangos, J., Intaglietta, M., and van der Heyde, H. C. (2005). Nitric oxide bioavailability in malaria. Trends Parasitol. 21, 415–422. doi: 10.1016/j.pt.2005.07.002
Space, S. L., Lane, P. A., Pickett, C. K., and Weil, J. V. (2000). Nitric oxide attenuates normal and sickle red blood cell adherence to pulmonary endothelium. Am. J. Hemotol. 63, 200–204. doi: 10.1002/(SICI)1096-8652(200004)63:4<200::AID-AJH7>3.0.CO;2-Q
Srihirun, S., Sriwantana, T., Unchern, S., Kittikool, D., Noulsri, E., Pattanapanyasat, K., et al. (2012). Platelet inhibition by nitrite is dependent on erythrocytes and deoxygenation. PLoS ONE 7:e30380. doi: 10.1371/journal.pone.0030380
Stamler, J. S., Jia, L., Eu, J. P., McMahon, T. J., Demchenko, I. T., Bonaventura, J., et al. (1997). Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 276, 2034–2037. doi: 10.1126/science.276.5321.2034
Taggart, M. J., and Wray, S. (1998). Hypoxia and smooth muscle function: key regulatory events during metabolic stress. J. Physiol. 509, 315–325. doi: 10.1111/j.1469-7793.1998.315bn.x
Tejero, J., Sparacino-Watkins, C., Ragireddy, V., Frizzell, S., and Gladwin, M. T. (2014). Exploring the mechanisms of the reductase activity of neuroglobin by site-directed mutagenesis of the heme distal pocket. Biochemistry 54, 722–733. doi: 10.1021/bi501196k
Tiso, M., Tejero, J., Basu, S., Azarov, I., Wang, X., Simplaceanu, V., et al. (2011). Human neuroglobin functions as a redox-regulated nitrite reductase. J. Biol. Chem. 286, 18277–18289. doi: 10.1074/jbc.M110.159541
Tomaiuolo, G. (2014). Biomechanical properties of red blood cells in health and disease towards microfluidics. Biomicrofluidics 8:051501. doi: 10.1063/1.4895755
van Faassen, E. E., Bahrami, S., Feelisch, M., Hogg, N., Kelm, M., Kim-Shapiro, D. B., et al. (2009). Nitrite as regulator of hypoxic signaling in mammalian physiology. Med. Res. Rev. 29, 683–741. doi: 10.1002/med.20151
Vaughn, M. W., Huang, K. T., Kuo, L., and Liao, J. C. (2000). Erythrocytes possess an intrinsic barrier to nitric oxide consumption. J. Biol. Chem. 275, 2342–2348. doi: 10.1074/jbc.275.4.2342
Vaughn, M. W., Kuo, L., and Liao, J. C. (1998). Effective diffusion distance of nitric oxide in the microcirculation. Am. J. Physiol. Heart Circ. Physiol. 274:H1705.
Vinchi, F., and Tolosano, E. (2013). Therapeutic approaches to limit hemolysis-driven endothelial dysfunction: scavenging free heme to preserve vasculature homeostasis. Oxid. Med. Cell. Longev. 2013:96527. doi: 10.1155/2013/396527
Wajih, N., Basu, S., Jailwala, A., Kim, H. W., Ostrowski, D., Perlegas, A., et al. (2017). Potential therapeutic action of nitrite in sickle cell disease. Redox Biol. 12, 1026–1039. doi: 10.1016/j.redox.2017.05.006
Wajih, N., Liu, X., Shetty, P., Basu, S., Wu, H., Hogg, N., et al. (2016). The role of red blood cell S-nitrosation in nitrite bioactivation and its modulation by leucine and glucose. Redox Biol. 8, 415–421. doi: 10.1016/j.redox.2016.04.004
Westerman, M., and Porter, J. B. (2016). Red blood cell-derived microparticles: an overview. Blood Cells Molecules Dis. 59, 134–139. doi: 10.1016/j.bcmd.2016.04.003
Xu, X., Cho, M., Spencer, N. Y., Patel, N., Huang, Z., Shields, H., et al. (2003). Measurements of nitric oxide on the heme iron and ß-93 thiol of human hemoglobin during cycles of oxygenation and deoxygenation. Proc. Natl. Acad. Sci. U.S.A. 100, 11303–11308. doi: 10.1073/pnas.2033883100
Keywords: erythrocytes, hemoglobin, hemolysis, hypoxic vasodilation, nitric oxide, nitrite
Citation: Helms CC, Gladwin MT and Kim-Shapiro DB (2018) Erythrocytes and Vascular Function: Oxygen and Nitric Oxide. Front. Physiol. 9:125. doi: 10.3389/fphys.2018.00125
Received: 30 October 2017; Accepted: 07 February 2018;
Published: 22 February 2018.
Edited by:
Dan E. Berkowitz, Johns Hopkins University, United StatesReviewed by:
Suowen Xu, University of Rochester, United StatesCopyright © 2018 Helms, Gladwin and Kim-Shapiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christine C. Helms, Y2hlbG1zQHJpY2htb25kLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.