 Yanjun Lin1
Yanjun Lin1 Huili Zhang
Huili Zhang- 1Department of Cardiology, Shanghai Ninth People's Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, China
- 2Department of Oral and Maxillofacial Pathology, Shanghai Ninth People's Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, China
Hydrogen sulfide has been suggested to play an essential role in atherogenesis. There is a paucity of information about the association between H2S and angiotensin converting enzyme 2 (ACE2), a novel homolog of ACE. Therefore, the aim of the study was to explore the role of H2S in atherosclerosis with respect to ACE2 both in vitro and in vivo. Here, a murine model of acutely disturbed flow-induced atherosclerosis by left common carotid artery (LCA) partial ligation was utilized. We found that carotid partial ligation in high-fat fed apoE−/− mice significantly inhibited endogenous H2S synthesis in LCA. Application of NaHS, an H2S donor considerably attenuated the severity of atherosclerosis with upregulating carotid expression of ACE2, thus converting pro-atherosclerotic angiotensin II (Ang II) to anti-atherosclerotic angiotensin 1-7 (Ang-(1-7)). The anti-atherosclerotic effect of NaHS was dramatically abolished by treatment with MLN-4760, an ACE2 inhibitor. In contrast, blockage of H2S formation by DL-propargylglycine exacerbated the burden of atherosclerotic plaques accompanied by inhibiting carotid expression of ACE2. At the cellular level, NaHS dose-dependently promoted the expression of ACE2 and conversion from Ang II to Ang-(1-7) in unstimulated or LPS-stimulated endothelial cells, thus exerting anti-inflammatory properties. The anti-inflammatory effect of NaHS was abrogated by pretreatment with DX600, a selective ACE2 inhibitor. In conclusion, these data provide direct evidences that endogenous H2S insufficiency exists in acute flow disturbance-induced atherosclerosis and that application of H2S may protect against atherosclerosis via upregulating ACE2 expression in endothelial cells.
Introduction
Hydrogen sulfide (H2S) is recently considered to be a novel gaseous mediator, which is endogenously produced during cysteine metabolism mainly by two pyridoxal phosphate-dependent enzymes, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) (Renga, 2011). It has become clear that H2S exerts various effects in mammalian cardiovascular tissues. Endogenous H2S has been suggested to be involved in the pathogenesis of atherosclerosis (Lefer, 2007; Elsey et al., 2010). Wang et al. found for the first time the association between H2S and atherosclerosis in apoE−/−mice (Wang et al., 2009). The causative effect of H2S in atherosclerosis was further confirmed in CSE gene-deficient mice (Mani et al., 2013). Deletion of CSE gene in mice led to decreased H2S formation, elevated blood pressure and impaired endothelium-dependent vasorelaxation (Yang et al., 2008). As a result, CSE knockout mice fed a high cholesterol diet predisposed to develop early atherosclerotic lesions (Mani et al., 2013). Mice with both CSE and apoE gene knockout had more extensive atherosclerosis burden than those with either apoE or CSE knockout (Mani et al., 2013). Furthermore, various studies have investigated the precise mechanisms by which H2S hinders the development of atherosclerosis. H2S might protect vascular tissue from atherogenic damage by inhibiting vascular intimal proliferation, reducing adhesion molecules expression, suppressing oxidative stress and limiting foam cell formation (Laggner et al., 2007; Meng et al., 2007; Zhao et al., 2011). However, due to the complexity of the atherogenic process, the anti-atherogenic mechanism of H2S is still far from clear.
It is well-known that renin-angiotensin system (RAS) is a key modulator of cardiovascular function and plays essential roles in endothelial dysfunction and atherosclerosis (Dzau, 2005; Savoia and Schiffrin, 2006). Angiotensin-converting enzyme (ACE)/angiotensin II (Ang II)/angiotensin II type 1 receptor (AT1R) axis is the main pathway of RAS and contributes to the pathogenesis of atherosclerosis (Dzau, 2005; Savoia and Schiffrin, 2006). Recently, the understanding of RAS has been greatly expanded after a homolog of ACE, namely ACE2 was identified. Unlike ACE, ACE2 stimulates the degradation of Ang II into Ang-(1-7), an anti-inflammatory vasodilator and anti-trophic heptapeptide (Donoghue et al., 2000; Oudit et al., 2003; Danilczyk and Penninger, 2006), therefore exerting protective effect in atherosclerosis. Moreover, the anti-atherosclerotic effect of ACE2 has been proposed by overexpression of ACE2 in mice or rabbits, as characterized by an increase in tissue Ang-(1-7) and a decrease in tissue Ang II (Dong et al., 2008; Lovren et al., 2008). Overexpression of ACE2 promoted the stability of atherosclerotic lesions by suppressing macrophage infiltration, decreasing lipid deposition, increasing collagen content and lowering matrix metalloproteinase activity in plaques (Dong et al., 2008). However, ACE2-deficicency in LDLR−/− or apoE−/− mice exacerbated the development of atherosclerosis (Thomas et al., 2010; Thatcher et al., 2011). Thus, ACE2 may provide a therapeutic target in the treatment of atherosclerotic cardiovascular diseases.
In light of the importance of RAS in atherosclerosis, some studies explored the relationship between H2S metabolism and RAS. It was found that H2S improved endothelial function and myocardial remodeling via downregulating Ang II/AT1R pathway in renovascular hypertensive rats (Xue et al., 2015; Liu et al., 2017). Application of NaHS, an H2S donor, inhibited hyperglycemia-induced ACE-Ang II-AT1R activation in cultured renal mesangial cells and kidneys from diabetic rats (Xue et al., 2013). Previous studies have investigated the role of H2S in ACE-Ang II- AT1R axis, the classical pathway of RAS. However, there is little information about the link between H2S and ACE2 in cardiovascular system. Given this background, this study was designed to probe the possible effect of H2S on ACE2-Ang-(1-7) in vitro and in vivo. Our data indicate that deficiency of endogenous H2S formation accompanies the development and progression of disturbed flow-induced atherosclerosis. Supplement of H2S upregulated ACE2 expression and production of Ang-(1-7) in endothelial cells, resulting in attenuation of atherosclerosis.
Materials and Methods
Cell Culture
Human umbilical vein endothelial cells (HUVECs) were obtained from American Type Culture Collection (ATCC). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, USA), 100 units/ml penicillin and 100 mg/ml streptomycin. Cells were plated in 6-well plates or 100-mm tissue culture dishes 1 day before experiments. Near-confluent cultures were starved overnight in medium containing 0.5% FBS before NaHS treatment or stimulation with LPS.
Cell Treatment
All treatments were performed in serum-free culture medium with penicillin and streptomycin. Cells were washed twice with serum-free culture medium and pre-incubated with saline or NaHS (50, 100, and 200 μM) for 24 h. Some cells were then stimulated with LPS (100 ng/ml) for 24 h in the continuous presence of NaHS. In the time course experiment, cells were pre-incubated with saline or NaHS (100 μM) for 0, 6, 24, or 48 h. For experiments using inhibitors, cells were pre-treated with DX600 (1 μM, an ACE2 inhibitor, Phoenix Pharmaceuticals; Pedersen et al., 2011) for 2 h before pre-incubation with NaHS (100 μM) for 24 h and subsequent stimulation with LPS (100 ng/ml) for 24 h.
Animal Model
C57BL/6J male apoE−/− mice were purchased from the Animal Center of the Beijing University, Beijing, China. The study was carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals of the Shanghai JiaoTong University School of Medicine. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Shanghai JiaoTong University School of Medicine (Permit Number: [2015]-117). At age of 8 weeks, mice were randomly assigned to partial ligation of left carotid artery (LCA) or sham operation, followed by being fed a high-fat diet for 4 weeks that contained 10% fat from lard and was supplemented with 2% (w/w) cholesterol. LCA was partially ligated as previously described (Sullivan, 2002; Nam et al., 2009) with slight modifications. In brief, mice were intrapeitoneally anesthetized by the mixture of xylazine (10 mg/kg) and ketamine (80 mg/kg). The neck was epilated and then disinfected with iodophor. LCA was exposed by a ventral midline incision (4–5 mm) in the neck. Except that the superior thyroid artery remained intact, left external carotid, internal carotid and occipital artery were ligated with 6–0 silk (Figure 1A). The skin was sutured and mice were then kept in a warm chamber until recovery. After LCA partial ligation or sham operation, mice were randomly given saline, NaHS (1 mg/kg/day, i.p.) or DL-propargylglycine (PAG, 10 mg/kg/day, i.p.). Some NaHS-treated mice were intraperitoneally given MLN-4760, a selective ACE2 inhibitor (0.5 mg/kg, daily, Millennium Pharmaceuticals) 2 weeks after initiation of NaHS treatment (Ye et al., 2012). Four weeks postligation, all the mice were sacrificed. Samples of carotid arteries and blood were collected and stored at −80°C. At the beginning and the end of the study, systolic blood pressure (SBP) was monitored by a tail cuff system (Blood Pressure Analysis System BP- 98AW monitor, Japan).
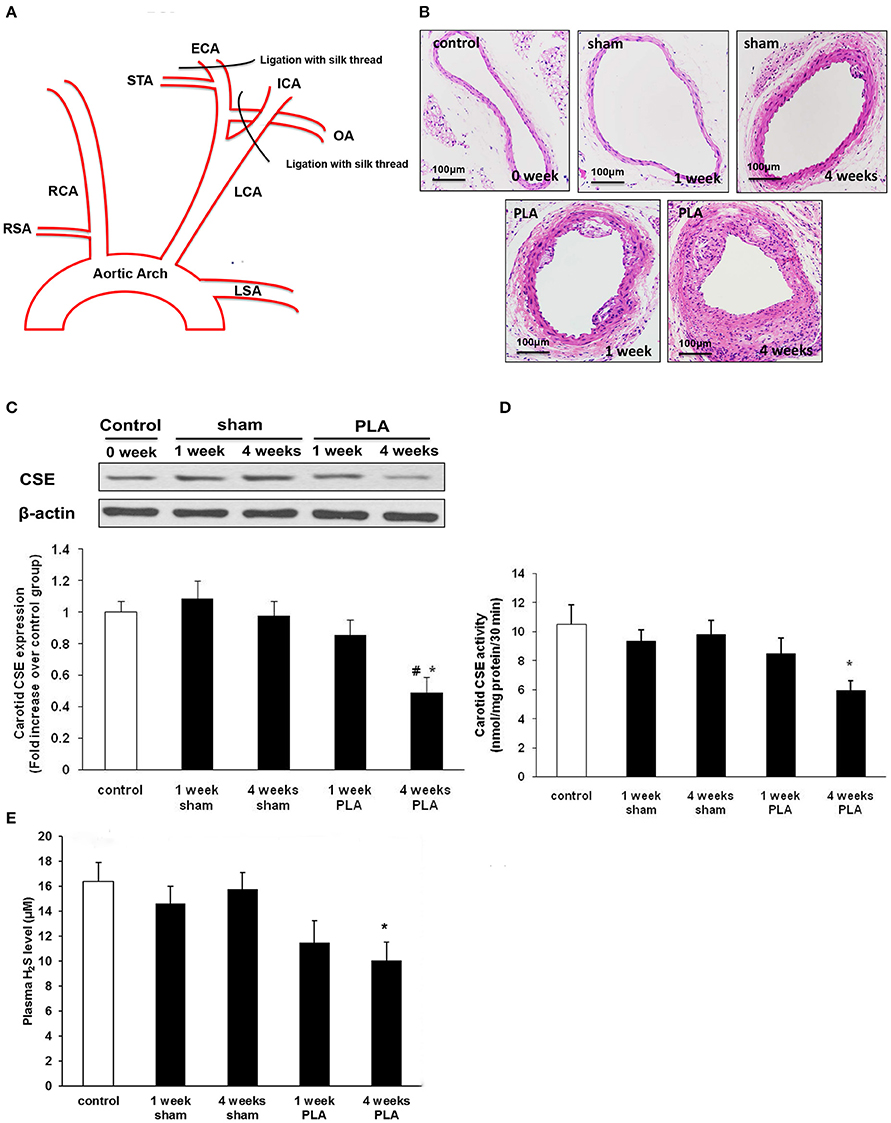
Figure 1. (A) Schematic diagram of low and oscillatory flow-induced atherosclerosis by partial ligation of LCA (PLA). Three branches of LCA, external carotid artery (ECA), internal carotid artery (ICA), and occipital artery (OA) were ligated with 6-0 silk suture, while leaving the superior thyroid artery (STA) intact. (B) Representative light microscopy images of hematoxylin and eosin-stained LCA cross sections were taken from high-fat fed apoE−/− mice 1 or 4 weeks after PLA or sham operation. Scale bar for histological images = 100 μm. (C–E) Alterations in H2S biosynthesis during PLA induced atherosclerosis in high-fat fed apoE−/− mice. CSE expression (C) and activity (D) in LCA, and plasma H2S level (E) were assayed at indicated time points (1 or 4 weeks after PLA or sham operation). Results shown are the mean ± SEM (n = 6 animals in each group). *P < 0.05 for the comparison between mice 4 weeks after sham operation and mice 4 weeks after PLA. #P < 0.05 for the comparison between mice 1 week after PLA and mice 4 weeks after PLA.
Histological Examination and Masson Staining
Anesthetized mice underwent left ventricle perfusion with buffered saline and 10% neutralized formalin at 100 mmHg. LCA was collected en bloc with the trachea and esophagus. LCA were embedded in paraffin. Fifteen serial sections (5 μm) were taken 500–1,000 μm proximal to the location of ligation. Five sections from each mouse were stained with hematoxylin and eosin. The staining were then examined by light microscopy (n = 6 mice in each group, objective lens magnification of × 20; eyepiece magnification of × 10). The lesions were quantified by Image J software (National Institutes of Health, USA) as described in the literatures (Lessner et al., 2002; Sullivan, 2002; Nam et al., 2009; Zhang et al., 2012, 2015). Measurement of intima and media of carotid arteries was acquired by tracing the border of the lumen and the internal and external elastic laminae (Figure S1). The mean area of the intima and media was measured by tracing multiple sections, and the ratio of the area of intima to media was calculated. Sections were also stained with Masson's trichrome to display collagen components in a given plaque. To calculate proportions of collagen content to neointima, we measured intimal lesion areas and Masson's trichrome positive blue areas in at least three sequential sections (Tasaki et al., 2013). Every morphological parameter was quantified by one investigator blinded for the treatment.
Immunohistochemistry of ACE2
LCA were deparaffinized in xylene and rehydrated in aqueous solutions with decreasing alcohol content, followed by a wash in PBST (1 × PBS with 0.5% Tween 20, pH 7.4). Antigen retrieval was achieved by heating the slides in 10 mM sodium citrate (PH 6.0) at 95°C for 20 min. Slides were gradually cooled down at room temperature and washed in H2O and PBST. To inactivate endogenous peroxidase, sections were treated with 3% H2O2 for 15 min, followed by incubation with 10% normal goat serum for 20 min to block nonspecific staining. After incubation overnight at 4°C with primary antibody (rabbit anti-mouse ACE2 in a dilution of 1:75, Abcam, USA), slides were incubated with secondary antibody (HRP-conjugated, goat anti-rabbit IgG in a dilution of 1:200, Santa Cruz Biotechnology, USA) for 2 h at room temperature. Specific staining for ACE2 was developed by the reaction with 3, 3′-diaminobenzidine and counterstaining was applied with hematoxylin. The immunohistochemical staining was viewed by light microscopy (objective lens magnification of × 40; eyepiece magnification of × 10).
Real-Time RT-PCR
Total RNA from cells was extracted using Trizol ® reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. The concentration of isolated RNA was assayed by measuring the absorbance at 260 nm and the integrity of RNA was visualized by ethidium bromide staining of 18S and 28S on a denaturing agarose gel. One microgram RNA was reversely transcribed using iScript™ cDNA Synthesis Kit (Biorad, USA) at 25°C for 5 min, 42°C for 30 min, followed by 85°C for 5 min. The resulting cDNA was then used as a template for real time PCR amplification. The forward and reverse primers of ACE2, ACE, Mas, and β-actin gene were shown in Table 1. Real-time PCR was performed using Lightcycler 2.0 system (Roche Applied Science, USA). Relative expression of gene mRNA was analyzed using a comparative method described in the user bulletin. The data were calculated with 2−ΔΔCT method and normalized to β-actin expression.

Table 1. Real time PCR primers.
Western Immunoblot
Cells (3 × 106) were collected and lysated at 4°C using radioimmunoprecipitation assay lysis buffer. Tissues of LCA were homogenized on ice in radioimmunoprecipitation assay lysis buffer. Cell lysates or tissue homogenates were centrifuged at 14,000 g for 10 min at 4°C. Protein concentration in the soluble fraction was determined by Bradford method. Proteins (20 μg) were size-fractionated by 8% SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were blocked for 2–3 h with 5% nonfat milk and then probed overnight at 4°C with rabbit polyclonal anti-ACE2 (1:500, molecular weight 97 kDa, Abcam, USA), β-actin antibodies (1:1,000, Santa Cruz Biotechnology, USA) and mouse monoclonal anti-CSE antibody (1:1,000, molecular weight 45 kDa, Abnova, Taiwan) respectively, followed by secondary antibody for 2 hs with a 1:2,000 dilution of HRP-conjugated, goat anti-rabbit IgG or goat anti-mouse IgG (Santa Cruz Biotechnology, USA). The blots were visualized using a standard enhanced chemiluminescence system.
ELISA
MCP-1, TNF- α and IL-6 (Quantikine, R&D systems), as well as Ang-(1-7) and Ang II (Cloud-Clone Corp., USA) were assayed using ELISA kits according to the manufacturers' instructions. Results for the levels of Ang-(1-7) and Ang II in carotid arteries were expressed as pg/mg protein after correction for the protein concentration in tissue homogenates (determined using the Bradford assay).
Measurement of Plasma H2S
Ten percent Trichloroacetic acid (120 μl), 1% zinc acetate (60 μl), 20 μM N, N-dimethyl-p-phenylenediamine sulfate (40 μl) in 7.2 M hydrochloride acid and 30 μM FeCl3 (40 μl) in 1.2 M hydrochloride acid were mixed with plasma (120 μl) and distilled water (100 μl). After 10 min, the absorbance of the mixture was assayed by spectrophotometry at 670 nm (Tecan Systems Inc.). Plasma level of H2S was calculated using a standard curve of NaHS with a concentration from 3.125 to 100 μM.
H2S Synthesizing Activity Assay
H2S synthesizing activity in LCA was measured as described previously (Zhang et al., 2006). In brief, 4.5% w/v tissue homogenate (430 μl) in 100 mM potassium phosphate buffer (pH 7.4) was mixed with 20 mM L-cysteine (20 μl), 2 mM pyridoxyal 5′-phosphate (20 μl) and saline (30 μl). The reaction started in tightly sealed tubes after tubes were transferred from ice to water bath at 37°C. After incubation for half an hour, 1% zinc acetate (250 μl) was added and trapped the evolved H2S, followed by 10% trichloroacetic acid (250 μl) to cease the reaction. Afterward, 20 μM N, N-dimethyl-p-phenylenediamine sulfate (133 μl) in 7.2 M hydrochloride acid was added, immediately followed by 30 μM FeCl3 (133 μl) in 1.2 M hydrochloride acid. The absorbance of the resulting mixture was assayed by spectrophotometry at 670 nm (Tecan Systems Inc). H2S concentration was calculated using a standard curve of NaHS with a range from 3.125 to 100 μM. Results were expressed as nmoles H2S produced per mg protein in tissue homogenates (determined using the Bradford assay).
Statistics
The data were expressed as mean ± SEM. The significance of differences among groups was evaluated by analysis of variance (ANOVA) with post-hoc Tukey's test when comparing three or more groups. The significance of differences between two groups was evaluated by T-test. A P < 0.05 was regarded as statistically significant.
Results
Alterations of Endogenous H2S Synthesis during Disturbed Flow-Induced Atherosclerosis
Atherosclerosis is known to be closely associated with disturbed flow characterized by low and oscillatory shear stress (Sullivan, 2002; Pedersen et al., 2011). However, studies directly linking H2S to disturbed flow condition in atherogenesis are lacking. Here, we investigated the alterations of H2S metabolism in a murine model of acutely disturbed flow-induced atherosclerosis by partial carotid ligation (Figure 1A). ApoE−/− mice underwent either LCA partial ligation or sham operation and then were fed a high-fat diet for 1 or 4 weeks. By the first week, LCA showed slight evidences of atherosclerotic lesions as determined by H&E staining (Figure 1B). By 4 weeks, LCA developed accelerated atherosclerosis (Figure 1B). However, only minor or no lesions were observed in carotid arteries isolated from mice fed a high-fat diet for 1 or 4 weeks after sham operation (Figure 1B).
Then, we examined the time-dependent alterations of carotid H2S bio-synthesis in disturbed flow-induced atherosclerosis. As shown in Figures 1C,D, H2S synthesizing activity and CSE expression in LCA decreased in a time-dependent fashion, with a significant reduction 4 weeks after ligation. Furthermore, plasma H2S concentration gradually declined in a time-dependent manner (Figure 1E). There was a statistically significant reduction in plasma H2S level by 4 weeks after ligation. These data suggest both local and systemic H2S insufficiency in LCA partial ligation induced atherosclerosis. In another word, acute flow disturbance induced by partial ligation inhibited carotid CSE expression and CSE activity, thus resulting in an evident decline in production of endogenous H2S.
Alterations of ACE2 in Carotid Arteries during Disturbed Flow-Induced Atherosclerosis
Next, we explored the alterations of carotid ACE2 over time in disturbed flow-induced atherosclerosis (Figure 2A). Immunostaining showed that ACE2 was finely expressed in vascular endothelial cells from normal mice. By 1 week postligation, expression of ACE2 in LCA endothelim was significantly upregulated. However, by 4 weeks, the endothelial expression of ACE2 dramatically decreased with the progression of atherosclerosis (Figures 2A,B). Four weeks after ligation, LCA endothelium exhibited less intense staining with ACE2 than 1 week after ligation. As a result, the level of Ang-(1-7) in LCA was high at the beginning of ligation and then gradually reduced in a time dependent manner (Figure 2C). The level of Ang II in LCA changed oppositely (Figure 2D). These data indicate that disturbed flow in carotid arteries initially induced the endothelial expression of ACE2 in an attempt to inhibit the initiation of atherosclerosis. However, with the development and progression of atherosclerosis, endothelial ACE2 expression was inhibited, therefore shifting a balance from anti-atherosclerotic Ang-(1-7) to pro-atherosclerotic Ang II.
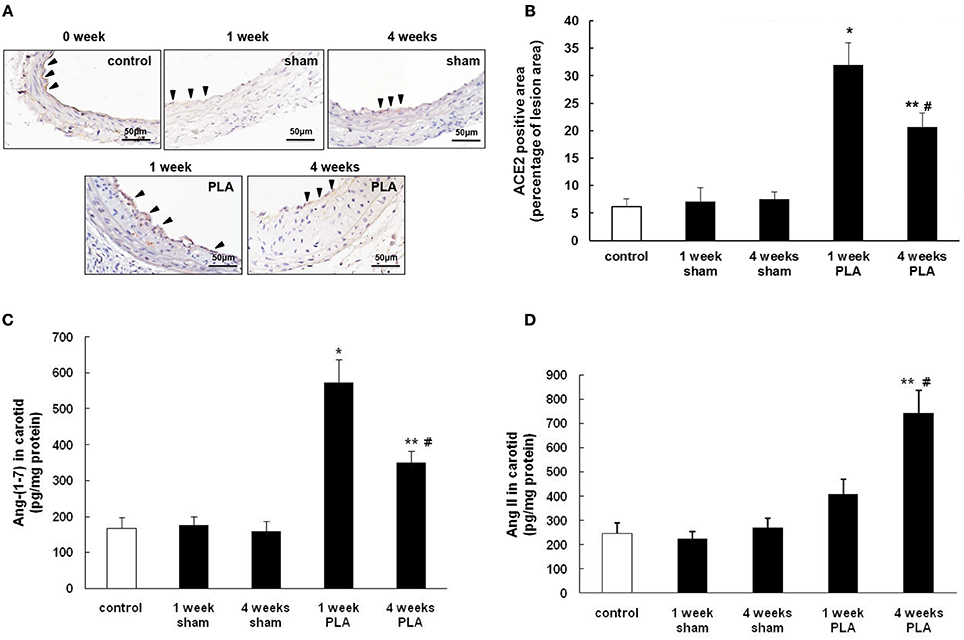
Figure 2. Alterations in ACE2 expression during PLA induced atherosclerosis in high-fat fed apoE−/− mice. (A) Representative immunohistochemical images of LCA cross sections were taken from high-fat fed apoE−/− mice 1 or 4 weeks after PLA or sham operation. Scale bar for histological images = 50 μm. (B) LCA sections from mice 1 or 4 weeks after PLA or sham operation were quantified immunohistochemically for ACE2 positive staining. Levels of Ang-(1-7) (C) and Ang II (D) in LCA were assayed by ELISA at indicated time points (1 or 4 weeks after PLA or sham operation). Results shown are the mean ± SEM (n = 6 animals in each group). Arrowheads represented positive staining for ACE2 in endothelial cells. *P < 0.05 for the comparison between mice 1 week after sham operation and mice 1 week after PLA. **P < 0.05 for the comparison between mice 4 weeks after sham operation and mice 4 weeks after PLA. #P < 0.05 for the comparison between mice 1 week after PLA and mice 4 weeks after PLA.
Effect of H2S on Disturbed Flow-Induced Atherosclerosis
In the time course study, we found that the biosynthesis of H2S significantly decreased 4 weeks after PLA whereas the carotid ACE2 expression started rising 1 week after PLA (Figures 1, 2). This finding suggested that H2S seems to play a part in regulating ACE2 expression in the advanced stage of atherosclerosis (4 weeks after PLA) but not in the early phase of atherosclerosis (1 week after PLA). In addition to H2S, there are some other factors contributing to ACE2 expression in different stages of atherosclerosis. Here, we investigated the effect of H2S on carotid ACE2 expression 4 weeks after PLA.
NaHS (1 mg/kg/day, i.p.), DL-propargylglycine (PAG, 10 mg/kg/day, i.p.) or saline was randomly given to mice after partial ligation. After 4 weeks, the extent of atherosclerotic lesions was assessed by histological analysis (Figure 3A). Histological analysis showed that NaHS treatment significantly impeded the plaque development, as characterized by alleviated neointimal hyperplasia and less atherosclerotic lesions in LCA (Figures 3B,C). On the other hand, inhibition of H2S formation by PAG induced advanced atherosclerosis, as evidenced by more severe neointimal hyperplasia and more obvious narrowness in LCA (Figures 3B,C). Masson's staining revealed that NaHS reduced the amount of collagen deposits whereas PAG enhanced hyperplasia of collagen fibers in neointimal lesions (Figures 3D,E). In addition, NaHS or PAG had negligible effect on body weight, plasma lipid profiles, and blood pressure (Table 2).
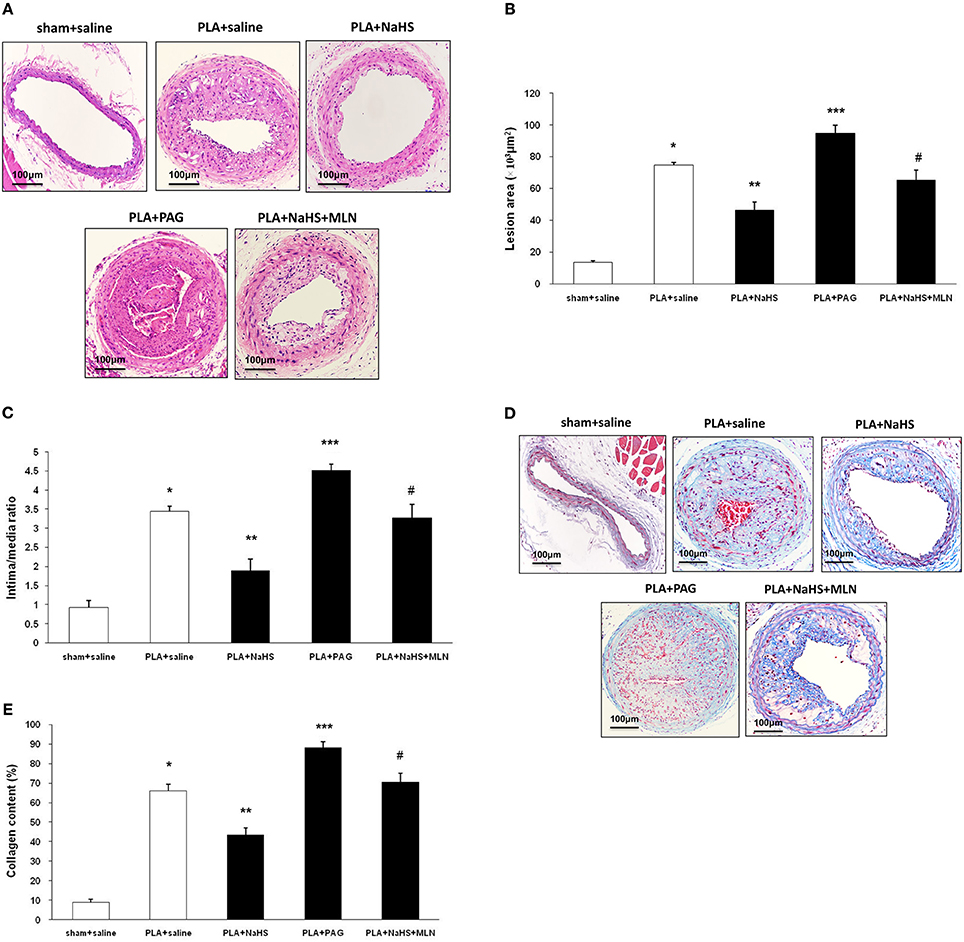
Figure 3. Effect of NaHS or PAG on PLA-induced atherosclerosis in LCA. Representative light microscopy images of hematoxylin and eosin-stained (A) or Masson trichrome-stained (D) LCA cross sections were taken from sham-operated mice with saline, PLA mice with NaHS, PLA mice with PAG, or PLA mice with NaHS and MLN (intervention with MLN-4760 for 14 days). Scale bar for histological images = 100 μm. Lesion area of intima (B), intima/media ratio (C) and collagen deposits (E) in the neointimal hyperplasia in LCA were measured as described in Materials and Methods. Results shown are the mean ± SEM (n = 6 animals in each group). *P < 0.05 for the comparison between sham+saline and PLA+saline. **P < 0.05 for the comparison between PLA+saline and PLA+NaHS. ***P < 0.05 for the comparison between PLA+saline and PLA+PAG. #P < 0.05 for the comparison between PLA+NaHS and PLA+NaHS+MLN.
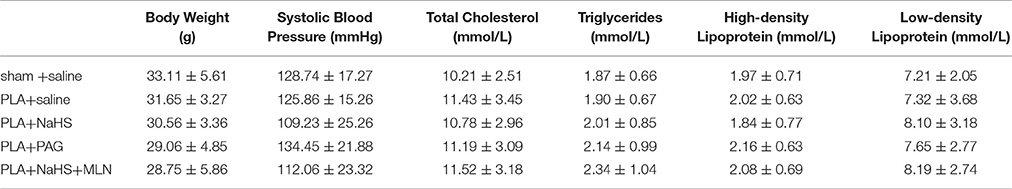
Table 2. Effects of treatment with NaHS on blood pressure and plasma lipids.
Effect of H2S on ACE2-Ang-(1-7) Expression in Atherosclerosis
Immunohistochemistry revealed that supplement with NaHS enhanced the expression of ACE2 in endothelial cells whereas PAG inhibited it in atherosclerotic lesions (Figures 4A,B). Then, we used Western blot to quantify the ACE2 expression level in LCA. Consistent with the findings obtained by immunostaining, NaHS significantly upregulated carotid ACE2 expression while PAG downregulated it (Figure 4C). As a result, NaHS notably promoted the local production of Ang (1-7) in LCA but PAG inhibited it (Figure 4D). The carotid level of Ang II was blunted by NaHS while it was raised by PAG (Figure 4E).
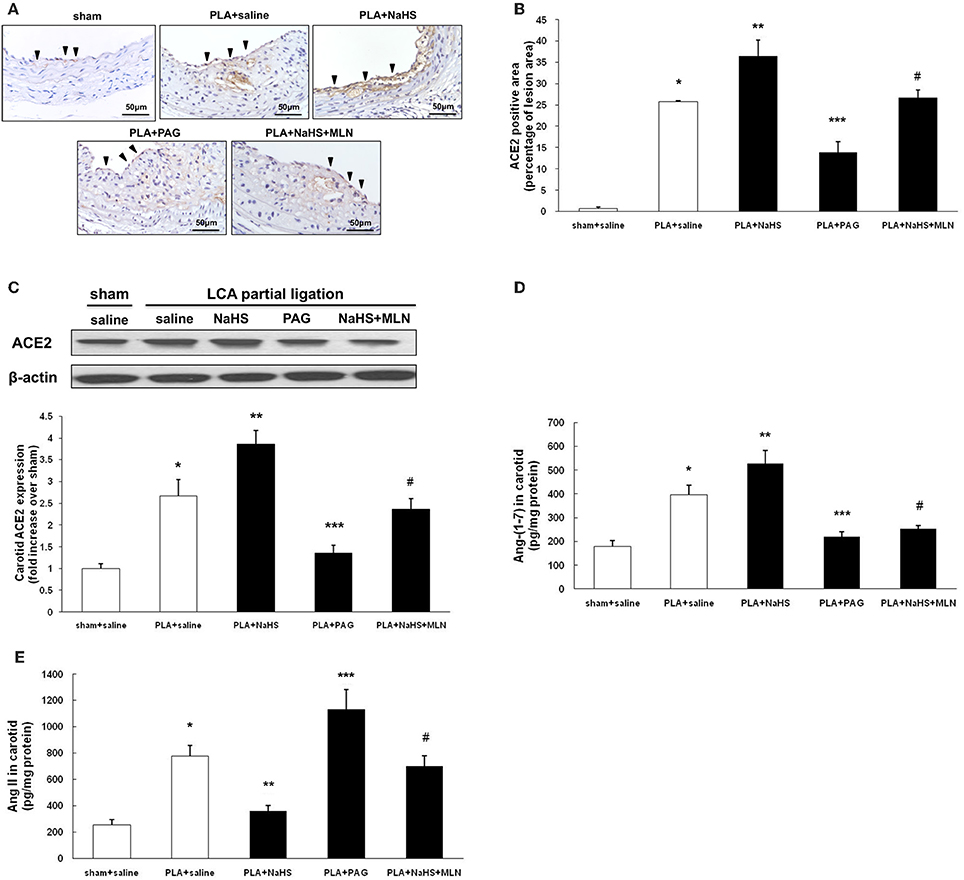
Figure 4. Effect of NaHS or PAG on ACE2 expression in LCA from mice with PLA-induced atherosclerosis. (A) Representative immunohistochemical images of LCA cross sections were taken from sham-operated mice with saline, PLA mice with NaHS, PLA mice with PAG, or PLA mice with NaHS and MLN (intervention with MLN-4760 for 14 days). Scale bar for histological images = 50 μm. (B) LCA sections were quantified immunohistochemically for ACE2 positive staining. (C) ACE2 expression in LCA was measured by Western blot as described in Materials and Methods. Levels of Ang (1-7) (D) and Ang II (E) in LCA were assayed by ELISA. Results shown are the mean ± SEM (n = 6 animals in each group). Arrowheads represented positive staining for ACE2 in endothelial cells. *P < 0.05 for the comparison between sham+saline and PLA+saline. **P < 0.05 for the comparison between PLA+saline and PLA+NaHS. ***P < 0.05 for the comparison between PLA+saline and PLA+PAG. #P < 0.05 for the comparison between PLA+NaHS and PLA+NaHS+MLN.
In order to reinforce the influence of H2S on ACE2-Ang-(1-7), MLN-4760, a selective and potent inhibitor against mouse ACE2 was applied (Ye et al., 2012). As shown in Figures 4D,E, blockage of ACE2 activity by MLN-4760 significantly reversed the elevation of carotid Ang-(1-7) level and the decline of carotid Ang II level induced by NaHS. The anti-atherosclerotic property of NaHS was also significantly abolished by treatment with MLN-4760 (Figures 3A,D).
Effect of H2S on ACE2 in Endothelial Cells
Since ACE2 is highly expressed in carotid endothelium, we sought to examine the effect of H2S on ACE2-Ang-(1-7) in HUVECs. We found that NaHS time-dependently (Figures 5A–C) and dose-dependently (Figures 5D–F) increased the mRNA and protein expression of ACE2 in HUVECs. Subsequently, NaHS enhanced the production of Ang-(1-7) (Figures 5G,I) but decreased the level of Ang II (Figures 5H,J) in a time-dependent and dose-dependent manner. Moreover, similar effect of NaHS was observed in LPS-stimulated HUVECs (Figure 6). LPS at a concentration of 100 ng/ml significantly downregulated the expression of ACE2 and Ang-(1-7) but increased the level of Ang II in HUVECs. The inhibition of endothelial ACE2-Ang-(1-7) expression induced by LPS was reversed by NaHS in a dose dependent manner (Figure 6). NaHS also dose-dependently reduced the level of Ang II in LPS-stimulated HUVECs (Figure 6E). As Mas receptor is a functional receptor for Ang-(1-7) (Santos et al., 2003), we evaluated the effect of NaHS on the mRNA expression of Mas in HUVECs. NaHS did not alter the mRNA expression of Mas in unstimulated or LPS-stimulated HUVECs (Figure S2A). In addition, we examined the effect of H2S on ACE, the classical component of RAS in HUVECs. After treatment with NaHS, the mRNA expression level of ACE remained unchangeable in unstimulated or LPS-stimulated HUVECs (Figure S2B).
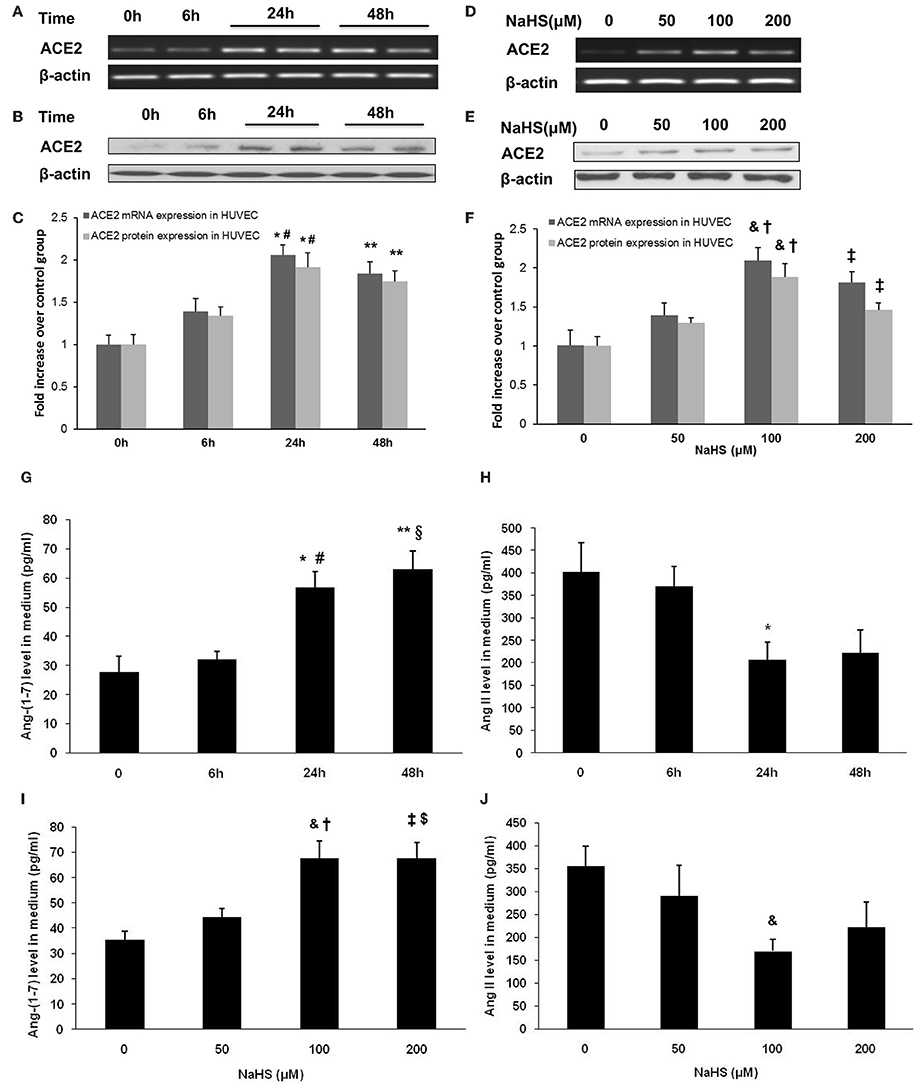
Figure 5. Effect of NaHS on ACE2 and Ang-(1-7) expression in HUVECs. In the time course study, cells were pre-incubated with saline or NaHS (100 μM) for 0, 6, 24 or 48 h (A–C,G,H). In the dose ranging study, cells were pre-incubated with saline or NaHS (50, 100, and 200 μM) for 24 h (D–F,I,J). ACE2 mRNA (A,C,D,F) and protein expression (B,C,E,F) were analyzed by real time PCR and Western blot respectively. Levels of Ang (1-7) (G,I) and Ang II (H,J) in culture medium were assayed by ELISA. The data are means ± SEM of at least three independent experiments. *P < 0.05 for the comparison between control at baseline and HUVECs treated with NaHS for 24 h. **P < 0.05 for the comparison between control at baseline and HUVECs treated with NaHS for 48 h. #P < 0.05 for the comparison between HUVECs treated with NaHS for 6 h and HUVECs treated with NaHS for 24 h. §P < 0.05 for the comparison between HUVECs treated with NaHS for 6 h and HUVECs treated with NaHS for 48 h. &P < 0.05 for the comparison between control at baseline and HUVECs treated with NaHS at a concentration of 100 μM. †P < 0.05 for the comparison between HUVECs treated with NaHS at a concentration of 50 μM and HUVECs treated with NaHS at a concentration of 100 μM. ‡P < 0.05 for the comparison between control at baseline and HUVECs treated with NaHS at a concentration of 200 μM. $P < 0.05 for the comparison between HUVECs treated with NaHS at a concentration of 50 μM and HUVECs treated with NaHS at a concentration of 200 μM.
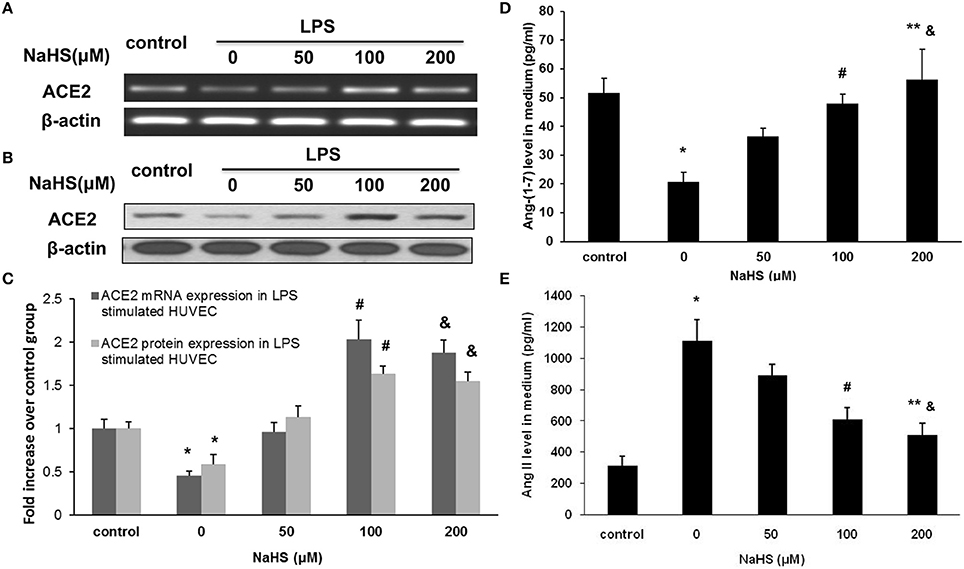
Figure 6. Effect of NaHS on ACE2 and Ang-(1-7) expression in HUVECs stimulated with LPS (100 ng/ml). HUVECs were pre-incubated with saline or NaHS (50, 100, and 200 μM) for 24 h and subsequently stimulated with saline or LPS (100 ng/ml) for 24 h. ACE2 mRNA (A,C) and protein (B,C) expression were analyzed by real time PCR and Western blot respectively. Levels of Ang (1-7) (D) and Ang II (E) in culture medium were assayed by ELISA. The data are means ± SEM of at least three independent experiments. *P < 0.05 for the comparison between control at baseline and LPS-stimulated HUVECs treated with saline. #P < 0.05 for the comparison between LPS-stimulated HUVECs treated with saline and LPS-stimulated HUVECs treated with NaHS at a concentration of 100 μM. &P < 0.05 for the comparison between LPS-stimulated HUVECs treated with saline and LPS-stimulated HUVECs treated with NaHS at a concentration of 200 μM. **P < 0.05 for the comparison between LPS-stimulated HUVECs treated with NaHS at a concentration of 50 μM and LPS-stimulated HUVECs treated with NaHS at a concentration of 200 μM.
H2S Inhibits the Production of Cytokines and Chemokine in Endothelial Cells by an ACE2 Dependent Mechanism
As shown in Figure 7, the production of TNF-α, IL-6, and MCP-1 was dose-dependently repressed by NaHS in either unstimulated or LPS-stimulated HUVECs. The inhibitory effect of NaHS was significantly blunted by DX600, a selective ACE2 inhibitor (Nam et al., 2009), suggesting that H2S may inhibit endothelial activation through an ACE2-dependent mechanism (Figure 8).
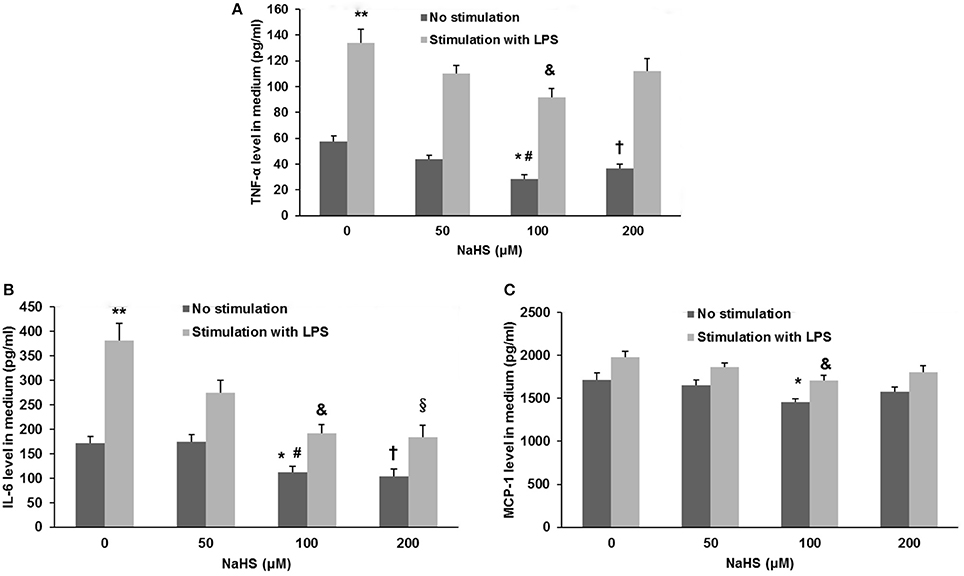
Figure 7. Effect of NaHS on cytokine and chemokine production in unstimulated or LPS-stimulated HUVECs. HUVECs were pre-incubated with saline or NaHS (50, 100, and 200 μM) for 24 h and subsequently stimulated with saline or LPS (100 ng/ml) for 24 h. Production of cytokines (TNF-α, IL-6) (A,B) and chemokine (MCP-1) (C) was assayed by ELISA. The data are means ± SEM of at least three independent experiments. *P < 0.05 for the comparison between unstimulated HUVECs treated with saline and unstimulated HUVECs treated with NaHS at a concentration of 100 μM. #P < 0.05 for the comparison between unstimulated HUVECs treated with NaHS at a concentration of 50 μM and unstimulated HUVECs treated with NaHS at a concentration of 100 μM. †P < 0.05 for the comparison between unstimulated HUVECs treated with saline and unstimulated HUVECs treated with NaHS at a concentration of 200 μM. **P < 0.05 for the comparison between unstimulated HUVECs treated with saline and LPS-stimulated HUVECs treated with saline. &P < 0.05 for the comparison between LPS-stimulated HUVECs treated with saline and LPS-stimulated HUVECs treated with NaHS at a concentration of 100 μM. §P < 0.05 for the comparison between LPS-stimulated HUVECs treated with saline and LPS-stimulated HUVECs treated with NaHS at a concentration of 200 μM.
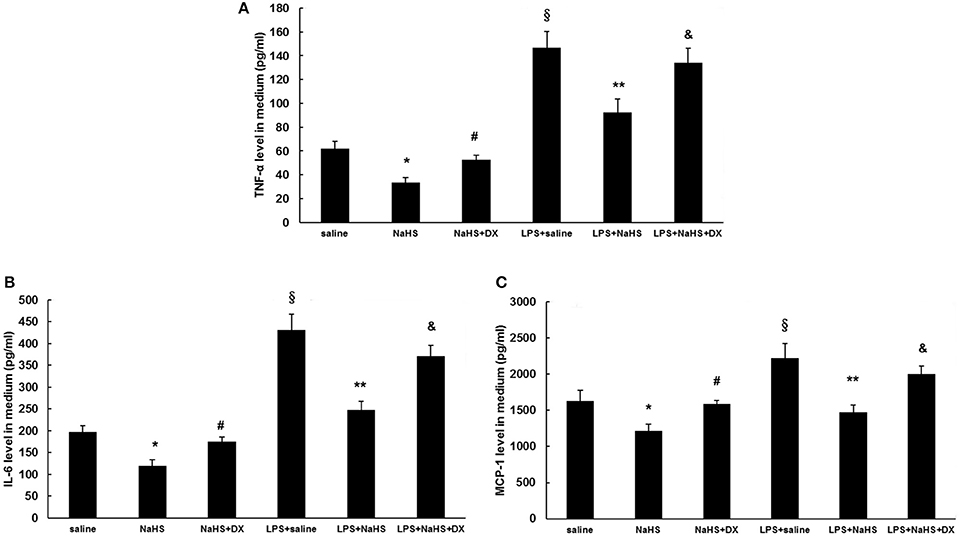
Figure 8. Effect of pretreatment with DX600, a selective ACE2 antagonist on suppressing the production of cytokines and chemokine induced by NaHS in unstimulated or LPS-stimulated HUVECs. Before pre-incubation with NaHS (100 μM) for 24 h, HUVECs were pre-treated with DX600 (1 μM) for 2 h. Then the cells were stimulated with saline or LPS (100 ng/ml) for 24 h. Production of cytokines (TNF-α, IL-6) (A,B) and chemokine (MCP-1) (C) was assayed by ELISA. The data are means ± SEM of at least three independent experiments. *P < 0.05 for the comparison between control at baseline and unstimulated HUVECs treated with NaHS. #P < 0.05 for the comparison between unstimulated HUVECs treated with NaHS and unstimulated HUVECs treated with NaHS and DX600. §P < 0.05 for the comparison between control at baseline and LPS-stimulated HUVECs treated with saline. **P < 0.05 for the comparison between LPS-stimulated HUVECs treated with saline and LPS-stimulated HUVECs treated with NaHS. &P < 0.05 for the comparison between LPS-stimulated HUVECs treated with NaHS and LPS-stimulated HUVECs treated with NaHS and DX600.
In addition to endothelial cells, positive immunostaining for ACE2 in atherosclerotic plaques was also present in foam cells and macrophages (Figure 4A). H2S has been shown to be a potent regulator of monocyte/ macrophage activation (Zhi et al., 2007; Zhang et al., 2012), which is central to the pathogenesis of atherosclerosis. Therefore, we explored whether H2S would affect the activation of macrophages via ACE2-Ang-(1-7). As shown in Figure S3, NaHS significantly inhibited the production of TNF-α and MCP-1 in unstimulated or LPS-stimulated RAW264.7 cells. The anti-inflammatory property of NaHS was not reversed by pretreatment with DX 600, suggesting that H2S likely modulates macrophage functions through other pathways but not ACE2.
Discussion
It is well established that atherosclerosis develops preferentially at particular sites in branched or curved arteries, which expose endothelial cells to disturbed flow characterized by low and oscillatory wall shear stress, even if there are various risk factors such as smoking, hyperlipidemia, diabetes and hypertension (Ku et al., 1985; VanderLaan et al., 2004). The present study utilized partial carotid ligation in high-fat fed apoE−/− mice as an animal model of acutely disturbed flow-induced atherosclerosis and then investigated the role of H2S in it. To the best of our knowledge, this is the first study to explore the alterations of H2S biosynthesis in disturbed flow-induced atherosclerosis. We found that partial carotid ligation inhibited local CSE expression and CSE activity, thus resulting in an evident decline in endogenous production of H2S. Consistent with our findings, deficit of H2S synthesis was also obtained in fat-fed apoE−/− mice or balloon injury induced neointimal hyperplasis (Meng et al., 2007; Wang et al., 2009; Zhang et al., 2012; Mani et al., 2013).
The biological importance of H2S in disturbed flow-induced atherosclerosis is further underlined by application of NaHS to manipulate endogenous H2S deficiency. NaHS treatment significantly reduced LCA atherosclerotic burden and impeded the progression of atherosclerosis. Furthermore, abolishing systemic H2S by PAG aggravated the extent of atherosclerosis in carotid arteries. These interesting findings highlight the potential role of H2S in the pathogenesis of atherosclerosis induced by low and oscillatory shear stress.
It is well documented that ACE2 plays a permissive role in reducing atherosclerosis. ACE2 is thought to counterbalance ACE by degrading pro-atherosclerotic Ang II to the putative protective peptide, Ang-(1-7) (Oudit et al., 2003; Dong et al., 2008; Lovren et al., 2008; Thomas et al., 2010; Thatcher et al., 2011). The anti-atherosclerotic properties of ACE2 were also evaluated in disturbed flow induced-atherosclerosis. We found that ACE2 was predominantly expressed in carotid endothelial cells in normal mice and that the expression of ACE2 significantly increased 1 week after partial ligation. With the progression of atherosclerosis, the expression level of ACE2 dramatically decreased. As a result, the level of Ang-(1-7) in LCA gradually reduced in a time dependent manner although it was high at the beginning of ligation. The level of Ang II in LCA changed oppositely. These data suggest that disturbed flow in carotid arteries initially upregulated the expression of ACE2 in an attempt to inhibit the initiation of atherosclerosis. However, with the development and progression of atherosclerosis, ACE2 expression decreased, thus shifting the balance from anti-atherosclerotic Ang-(1-7) to pro-atherosclerotic Ang II microenvironment. Consistent with our findings, expression of ACE2 mRNA and protein was observed in early and advanced human carotid atherosclerotic lesions (Sluimer et al., 2008). Overexpression of ACE2 by gene transfer attenuated the progression of atherosclerotic lesions in a rabbit model of atherosclerosis or mouse studies (Dong et al., 2008; Lovren et al., 2008). Knockout ACE2 gene in LDLR−/− or apoE−/− mice increased the development of atherosclerosis in aortic arch and sinus (Thomas et al., 2010; Thatcher et al., 2011). A meta-analysis involving 11,051 subjects suggests that genetic variants in ACE2 gene might have a potential effect on ACE2 activity, ACE2 level or Ang-(1-7) production and that ACE2 gene polymorphism may be a genetic risk factor for essential hypertension (Lu et al., 2012).
Furthermore, the association between H2S and ACE2 in atherosclerosis was explored in the present study. We found that application of exogenous H2S reversed partial ligation induced downregulation of ACE2 and Ang-(1-7) in LCA while blockage of H2S synthesis by PAG significantly aggravated it. The local level of Ang II was reduced by NaHS, but raised by PAG. Similar observations were obtained in cellular experiments. NaHS dose-dependently and time-dependently increased the expression of ACE2 in unstimulated or LPS-stimulated HUVECs. As a result, NaHS enhanced the production of Ang-(1-7) but decreased the level of Ang II. On the other hand, NaHS significantly inhibited the production of TNF-α, IL-6 and MCP-1 in unstimulated or LPS-stimulated HUVECs. The anti-inflammatory effect of NaHS in HUVECs was blunted by DX600. The anti-atherosclerotic benefit of NaHS was also abrogated by treatment with MLN-4760. Taken together, our findings provide solid evidences proposing that H2S plays a critical role in modulating endothelial ACE2 expression and promoting the cleavage of pro-atherosclerotic Ang II to anti-atherosclerotic Ang-(1-7), thereby impeding the development and progression of atherosclerosis. However, the precise mechanism by which H2S regulates the expression of ACE2 in endothelia cells during the initiation and progression of atherosclerosis remains elusive.
Peroxisome proliferator-activated receptors (PPAR) are nuclear receptors and function as transcription factors. PPARα has been suggested to affect the signaling pathway of RAS (Banks and Oyekan, 2008; Ibarra-Lara et al., 2010). Stimulation of PPARα with clofibrate favored ACE2/Ang-(1–7)/ATR2 axis in aortic coarctation-induced hypertensive rats as evidenced by enhancing Ang-(1-7) and ACE2 expression in the heart (Ibarra-Lara et al., 2016). Recent studies found that H2S promoted the activation of PPARγ in macrophages (Zhang et al., 2012) and facilitated the nuclear translocation of PPARα in fat-fed apoE−/− mice, thus exerting beneficial effect on atherogenesis (Li et al., 2016). Therefore, it offers an original possibility that H2S might induce the expression of ACE2 in endothelial cells via modulating the activation of PPARα. Moreover, latest studies reported that H2S participated in regulating the expression of microRNAs (miR-129, miR-299b, and miR-369) in Ang II-induced hypertensive kidney (Weber et al., 2017). Na2S, an H2S donor alleviated ischemic and inflammatory injury in cardiomyocytes through upregulation of miR-21 (Toldo et al., 2014). Epigenetic modulation of miRNAs raises another possible way that H2S may post-transcriptionally regulate ACE2 expression through miRNAs in atherosclerosis. To further prove these assumptions and elucidate the precise mechanism for the induction of ACE2 expression by H2S, more research is warranted.
In addition to endothelial cells, macrophages are the main source of proinflammatory mediators and contribute to the initiation and development of atherosclerosis. Previous studies have implied the regulatory role of H2S in monocyte/macrophage activation (Zhi et al., 2007; Zhang et al., 2012). Here, we further investigated whether H2S affected the function of macrophages involving the pathway of ACE2-Ang-(1-7). It was found that NaHS significantly suppressed the production of TNF-α and MCP-1 in RAW264.7 cells. The anti-inflammatory property of NaHS was not reversed by pretreatment with DX 600. These findings indicate that H2S possibly inhibited the activation of macrophages through other pathways, but not ACE2 dependent pathway. Clearly, further studies are needed to elucidate the correlation between H2S and RAS in macrophages. In contrast to the anti-inflammatory effect of H2S observed in the present study, some researchers demonstrate that H2S acts as a pro-inflammatory mediator in sepsis and acute pancreatitis (Bhatia et al., 2005; Zhang et al., 2006). H2S stimulated the synthesis of pro-inflammatory cytokines in human monocyte cell line (Zhi et al., 2007). The discrepancy suggests that H2S plays various roles in different inflammatory conditions. Different cell types and animal models used in these studies may contribute to this divergence.
ACE is known to assist the conversion from Ang I to Ang II while ACE2 is a major enzyme to degrade Ang II to Ang-(1-7). Here, H2S was found to have negligible effect on the mRNA expression of ACE in endothelial cells. Thus, H2S regulated the carotid level of Ang II mainly by local ACE2. However, some studies proposed the role of H2S in regulating ACE-Ang II-AT1R axis. H2S inhibited Ang II/AT1R pathway and improved endothelial function and myocardial remodeling in renovascular hypertensive rats (Xue et al., 2015; Liu et al., 2017). NaHS also inhibited hyperglycemia-induced ACE-Ang II-AT1R activation in cultured renal mesangial cells and kidneys from diabetic rats (Xue et al., 2013). The discrepancy about the role of H2S in regulating ACE may be due to different animal models and cell lines utilized in experiments. Future work is needed to clarify this divergence and expand the understanding of the effect of H2S on RAS.
In conclusion, our findings propose deficiency of endogenous H2S formation as well as downregulation of ACE2 in carotid atherosclerosis induced by disturbed flow and high fat diet. Supplement of H2S promotes ACE2 expression and production of Ang-(1-7) in endothelial cells, resulting in attenuation of atherosclerosis.
Author Contributions
HZh and CW supervised the whole project. YL, HZe, and LG performed the major experiments. TG provided the technical support.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 81570037), the Shanghai Municipal Natural Science Foundation (Grant No. 13ZR1424200) and the Shanghai Ninth People's Hospital Integration Foundation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2017.00782/full#supplementary-material
Figure S1. Representative sections used for morphometric analysis. Sections were measured by image analysis stained with hematoxylin and eosin (H&E) and Masson's trichrome. Arrowheads indicated external elastic lamina (EEL). Small arrows indicated internal elastic lamina (IEL).
Figure S2. Effect of NaHS on Mas, a functional receptor for Ang-1-7 and ACE expression in unstimulated HUVECs or HUVECs stimulated with LPS (100 ng/ml). HUVECs were pre-incubated with saline or NaHS (100 μM) for 24 and then stimulated with saline or LPS (100 ng/ml) for 24 h. Mas (A) and ACE (B) mRNA expression were analyzed by RT-PCR.
Figure S3. Effect of pretreatment with DX600, a selective ACE2 antagonist on suppressing the production of TNF-α nd MCP-1 induced by NaHS in unstimulated or LPS-stimulated murine macrophage cell line (RAW264.7). Before pre-incubation with NaHS (100 μM) for 24 h, RAW264.7 cells were pre-treated with DX600 (1 μM) for 2 h. Then the cells were stimulated with saline or LPS (100 ng/ml) for 6 h. Production of TNF-α (A) and MCP-1 (B) was assayed by ELISA. The data are means ± SEM of at least three independent experiments. *P < 0.05 for the comparison between control at baseline and unstimulated HUVECs treated with NaHS. #P < 0.05 for the comparison between control at baseline and LPS-stimulated HUVECs treated with saline. **P < 0.05 for the comparison between LPS-stimulated HUVECs treated with saline and LPS-stimulated HUVECs treated with NaHS.
References
Banks, T., and Oyekan, A. (2008). Peroxisome proliferator-activated receptor alpha activationattenuated angiotensin type 1-mediated but enhanced angiotensin type 2-mediated hemodynamic effects to angiotensin II in the rat. J. Hypertens. 26, 468–477. doi: 10.1097/HJH.0b013e3282f2f0f3
Bhatia, M., Wong, F. L., Fu, D., Lau, H. Y., Moochhala, S. M., and Moore, P. K. (2005). Role of hydrogen sulfide in acute pancreatitis and associated lung injury. FASEB J. 19, 623–625. doi: 10.1096/fj.04-3023fje
Danilczyk, U., and Penninger, J. M. (2006). Angiotensin-converting enzyme II in the heart and the kidney. Circ. Res. 98, 463–471. doi: 10.1161/01.RES.0000205761.22353.5f
Dong, B., Zhang, C., Feng, J. B., Zhao, Y. X., Li, S. Y., Yang, Y. P., et al. (2008). Overexpression of ACE2 enhances plaque stability in a rabbit model of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 28, 1270–1276. doi: 10.1161/ATVBAHA.108.164715
Donoghue, M., Hsieh, F., Baronas, E., Godbout, K., Gosselin, M., Stagliano, N., et al. (2000). A novel angiotensin-converting enzyme–related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Cir. Res. 87, e1–e9. doi: 10.1161/01.RES.87.5.e1
Dzau, V. (2005). The cardiovscular continuum and renin-angiotensin-aldosterone system blockade. J. Hypertens. 13, S9–S17. doi: 10.1097/01.hjh.0000165623.72310.dd
Elsey, D. J., Fowkes, R. C., and Baxter, G. F. (2010). Regulation of cardiovascular cell function by hydrogen sulfide (H(2)S). Cell Biochem. Funct. 28, 95–106. doi: 10.1002/cbf.1618
Ibarra-Lara, L., Cervantes-Perez, L. G., Perez-Severiano, F., Del Valle, L., Rubio-Ruiz, E., Soria-Castro, E., et al. (2010). PPARα stimulation exerts a blood pressure lowering effect through different mechanisms in a time-dependent manner. Eur. J. Pharmacol. 627, 185–193. doi: 10.1016/j.ejphar.2009.10.039
Ibarra-Lara, L., Del Valle-Mondragón, L., Soria-Castro, E., Torres-Narváez, J. C., Pérez-Severiano, F., Sánchez-Aguilar, M., et al. (2016). Peroxisome proliferator-activated receptor-α stimulation by clofibrate favors an antioxidant and vasodilator environment in a stressed left ventricle. Pharmacol. Rep. 68, 692–702. doi: 10.1016/j.pharep.2016.03.002
Ku, D. N., Giddens, D. P., Zarins, C. K., and Glagov, S. (1985). Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arterioscler. Thromb. Vasc. Biol. 5, 293–302. doi: 10.1161/01.ATV.5.3.293
Laggner, H., Muellner, M. K., Schreier, S., Sturm, B., Hermann, M., Exner, M., et al. (2007). Hydrogen sulphide: a novel physiological inhibitor of LDL atherogenic modification by HOCl. Free Radic. Res. 41, 741–747. doi: 10.1080/10715760701263265
Lefer, D. J. (2007). A new gaseous signaling molecule emerges: cardioprotective role of hydrogen sulfide. Proc. Natl. Acad. Sci. U.S.A. 104, 17907–17908. doi: 10.1073/pnas.0709010104
Lessner, S. M., Prado, H. L., Waller, E. K., and Galis, Z. S. (2002). Atherosclerotic lesions grow through recruitment and proliferation of circulating monocytes in a murine model. Am. J. Pathol. 160, 2145–2155. doi: 10.1016/S0002-9440(10)61163-7
Li, D., Xiong, Q., Peng, J., Hu, B., Li, W., Zhu, Y., et al. (2016). Hydrogen sulfide up-regulates the expression of ATP-Binding cassette transporter A1 via promoting nuclear translocation of PPARα. Int. J. Mol. Sci. 17:635. doi: 10.3390/ijms17050635
Liu, S. Y., Duan, X. C., Jin, S., Teng, X., Xiao, L., Xue, H. M., et al. (2017). Hydrogen sulfide improves myocardial remodeling via downregulated angiotensin /AT1R pathway in renovascular hypertensive rats. Am. J. Hypertens. 30, 67–74. doi: 10.1093/ajh/hpw104
Lovren, F., Pan, Y., Quan, A., Teoh, H., Wang, G., Shukla, P. C., et al. (2008). Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 295, H1377–H1384. doi: 10.1152/ajpheart.00331.2008
Lu, N., Yang, Y., Wang, Y., Liu, Y., Fu, G., Chen, D., et al. (2012). ACE2 gene polymorphism and essential hypertension: an updated meta-analysis involving 11,051 subjects. Mol. Biol. Rep. 39, 6581–6589. doi: 10.1007/s11033-012-1487-1
Mani, S., Li, H., Untereiner, A., Wu, L., Yang, G., Austin, R. C., et al. (2013). Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 127, 2523–2534. doi: 10.1161/CIRCULATIONAHA.113.002208
Meng, Q. H., Yang, G., Yang, W., Jiang, B., Wu, L., and Wang, R. (2007). Protective effect of hydrogen sulfide on balloon injury-induced neointima hyperplasia in rat carotid arteries. Am. J. Pathol. 170, 1406–1414. doi: 10.2353/ajpath.2007.060939
Nam, D., Ni, C. W., Rezvan, A., Suo, J., Budzyn, K., Llanos, A., et al. (2009). Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 297, H1535–H1543. doi: 10.1152/ajpheart.00510.2009
Oudit, G. Y., Crackower, M. A., Backx, P. H., and Penninger, J. M. (2003). The role of ACE2 in cardiovascular physiology Trends Cardiovas. Med. 13, 93–101. doi: 10.1016/S1050-1738(02)00233-5
Pedersen, K. B., Sriramula, S., Chhabra, K. H., Xia, H., and Lazartigues, E. (2011). Species-specific inhibitor sensitivity of angiotensin-converting enzyme 2 (ACE2) and its implication for ACE2 activity assays. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1293–1299. doi: 10.1152/ajpregu.00339.2011
Renga, B. (2011). Hydrogen sulfide generation in mammals: the molecular biology of cystathionine-β-synthase (CBS) and cytathionine-γ-Lyase (CSE). Inflamm. Allergy Drug Targets 10, 85–91. doi: 10.2174/187152811794776286
Santos, R. A. S., Simoes e Silva, A. C., Maric, C., Silva, D. M., Machado, R. P., de Buhr, I., et al. (2003). Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. U.S.A. 100, 8258–8263. doi: 10.1073/pnas.1432869100
Savoia, C., and Schiffrin, E. L. (2006). Inhibition of the renin angiotensin system: implications for the endothelium. Curr. Diab. Rep. 6, 274–278. doi: 10.1007/s11892-006-0060-5
Sluimer, J. C., Gasc, J. M., Hamming, I., van Goor, H., Michaud, A., van den Akker, L. H., et al. (2008). Angiotensin-converting enzyme 2 (ACE2) expression and activity in human carotid atherosclerotic lesions. J. Pathol. 215, 273–279. doi: 10.1002/path.2357
Sullivan, C. J. (2002). Flow-dependent remodeling in the carotid artery of fibroblast growth factor-2 knockout mice. Arterioscler. Thromb. Vasc. Biol. 22, 1100–1105. doi: 10.1161/01.ATV.0000023230.17493.E3
Tasaki, T., Yamada, S., Guo, X., Tanimoto, A., Wang, K. Y., Nabeshima, A., et al. (2013). Apoptosis signal-regulating kinase 1 deficiency attenuates vascular injury-induced neointimal hyperplasia by suppressing apoptosis in smooth muscle cells. Am. J. Pathol. 182, 597–609. doi: 10.1016/j.ajpath.2012.10.008
Thatcher, S. E., Zhang, X., Howatt, D. A., Lu, H., Gurley, S. B., Daugherty, A., et al. (2011). Angiotensin-converting enzyme 2 deficiency in whole body or bone marrow-derived cells increases atherosclerosis in low-density lipoprotein receptor–/– mice. Arterioscler. Thromb. Vasc. Biol. 31, 758–765. doi: 10.1161/ATVBAHA.110.221614
Thomas, M. C., Pickering, R. J., Tsorotes, D., Koitka, A., Sheehy, K., Bernardi, S., et al. (2010). Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ. Res. 107, 888–897. doi: 10.1161/CIRCRESAHA.110.219279
Toldo, S., Das, A., Mezzaroma, E., Chau, V. Q., Marchetti, C., Durrant, D., et al. (2014). Induction of microRNA-21 with exogenous hydrogen sulfide attenuates myocardial ischemic and inflammatory injury in mice. Circ. Cardiovasc. Genet. 7, 311–320. doi: 10.1161/CIRCGENETICS.113.000381
VanderLaan, P. A., Reardon, C. A., and Getz, G. S. (2004). Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler. Thromb. Vasc. Biol. 24, 12–22. doi: 10.1161/01.ATV.0000105054.43931.f0
Wang, Y., Zhao, X., Jin, H., Wei, H., Li, W., Bu, D., et al. (2009). Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 29, 173–179. doi: 10.1161/ATVBAHA.108.179333
Weber, G. J., Pushpakumar, S. B., and Sen, U. (2017). Hydrogen sulfide alleviates hypertensive kidney dysfunction through an epigenetic mechanism. Am. J. Physiol. Heart Circ. Physiol. 312, H874–H885. doi: 10.1152/ajpheart.00637.2016
Xue, H., Yuan, P., Ni, J., Li, C., Shao, D., Liu, J., et al. (2013). H2S inhibits hyperglycemia-induced intrarenal renin-angiotensin system activation via attenuation of reactive oxygen species generation. PLoS ONE 8:e74366. doi: 10.1371/journal.pone.0074366
Xue, H., Zhou, S., Xiao, L., Guo, Q., Liu, S., and Wu, Y. (2015). Hydrogen sulfide improves the endothelial dysfunction in renovascular hypertensive rats. Physiol. Res. 64, 663–672.
Yang, G., Wu, L., Jiang, B., Yang, W., Qi, J., Cao, K., et al. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322, 587–590. doi: 10.1126/science.1162667
Ye, M., Wysocki, J., Gonzalez-Pacheco, F. R., Salem, M., Evora, K., Garcia-Halpin, L., et al. (2012). Murine recombinant angiotensin-converting enzyme 2: effect on angiotensin II-dependent hypertension and distinctive angiotensin-converting enzyme 2 inhibitor characteristics on rodent and human angiotensin-converting enzyme 2. Hypertension 60, 730–740. doi: 10.1161/HYPERTENSIONAHA.112.198622
Zhang, H., Guo, C., Wu, D., Zhang, A., Gu, T., Wang, L., et al. (2012). Hydrogen sulfide inhibits the development of atherosclerosis with suppressing CX3CR1 and CX3CL1 expression. PLoS ONE 7:e41147. doi: 10.1371/journal.pone.0041147
Zhang, H., Zhi, L., Moore, P. K., and Bhatia, M. (2006). Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L1193–L1201. doi: 10.1152/ajplung.00489.2005
Zhang, X., Zhang, T., Gao, F., Li, Q., Shen, C., Li, Y., et al. (2015). Fasudil, a Rhokinase inhibitor, prevents intimamedia thickening in a partially ligated carotid artery mouse model: effects of fasudil in flowinduced vascular remodeling. Mol. Med. Rep. 12, 7317–7325. doi: 10.3892/mmr.2015.4409
Zhao, Z. Z., Wang, Z., Li, G. H., Wang, R., Tan, J. M., Cao, X., et al. (2011). Hydrogen sulfide inhibits macrophage-derived foam cell formation. Exp. Biol. Med. (Maywood) 236, 169–176. doi: 10.1258/ebm.2010.010308
Keywords: hydrogen sulfide, atherosclerosis, ACE2, Ang-(1-7), Ang II
Citation: Lin Y, Zeng H, Gao L, Gu T, Wang C and Zhang H (2017) Hydrogen Sulfide Attenuates Atherosclerosis in a Partially Ligated Carotid Artery Mouse model via Regulating Angiotensin Converting Enzyme 2 Expression. Front. Physiol. 8:782. doi: 10.3389/fphys.2017.00782
Received: 18 March 2017; Accepted: 25 September 2017;
Published: 10 October 2017.
Edited by:
Gerald A Meininger, University of Missouri, United StatesReviewed by:
Keshari Thakali, University of Arkansas for Medical Sciences, United StatesShuangtao Ma, Michigan State University, United States
Junbao Du, Peking University First Hospital, China
Copyright © 2017 Lin, Zeng, Gao, Gu, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changqian Wang, Y2hhbmdxaWFud2FuZ0Bob3RtYWlsLmNvbQ==
Huili Zhang, aHVpbGl6aGFuZzgxNUAxNjMuY29t