 Zhi Zhao Liu
Zhi Zhao Liu Alexander Bullen
Alexander Bullen Prabhleen Singh
Prabhleen Singh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 28 June 2017
Sec. Renal Physiology and Pathophysiology
Volume 8 - 2017 | https://doi.org/10.3389/fphys.2017.00385
This article is part of the Research Topic Hypoxia in kidney disease View all 12 articles
Chronic kidney disease (CKD) is a significant health problem associated with high morbidity and mortality. Despite significant research into various pathways involved in the pathophysiology of CKD, the therapeutic options are limited in diabetes and hypertension induced CKD to blood pressure control, hyperglycemia management (in diabetic nephropathy) and reduction of proteinuria, mainly with renin-angiotensin blockade therapy. Recently, renal oxygenation in pathophysiology of CKD progression has received a lot of interest. Several advances have been made in our understanding of the determinants and regulators of renal oxygenation in normal and diseased kidneys. The goal of this review is to discuss the alterations in renal oxygenation (delivery, consumption and tissue oxygen tension) in pre-clinical and clinical studies in diabetic and hypertensive CKD along with the underlying mechanisms and potential therapeutic options.
Chronic kidney disease (CKD) is a worldwide public health problem. In the United States, 14% of adults suffer from CKD (Coresh et al., 2007). Besides its impact on health, CKD and end-stage renal disease (ESRD) require substantial healthcare resources (Honeycutt et al., 2013). Despite the resources committed to the treatment of CKD and improvements in the quality of dialysis therapy, CKD patients continue to experience significant mortality and morbidity and a reduced quality of life. Numerous studies have elucidated the underlying mechanisms of CKD and identified new therapeutic targets, but success in clinical translation has been limited. Among various pathways, imbalance between oxygen delivery and consumption leading to tissue hypoxia has been recognized as an important contributor to CKD development and progression (Fine et al., 1998; Norman and Fine, 2006; O'Connor, 2006; Mimura and Nangaku, 2010). The aim of this review is to discuss the role of renal oxygenation (oxygen delivery, consumption and tissue oxygen tension) in CKD progression and potential therapeutic options based on recent literature.
The major function of kidney is to remove waste products and excess fluid from the body to maintain homeostasis. The kidneys filter about 180 liters of fluid daily. Nearly 99% of the of filtered sodium is reabsorbed and this is the primary energy consuming process in the kidneys powered by the Na- and K-ATPase (sodium-potassium pump) in the basolateral membranes of tubular cells in the kidney. About 60–70% of total sodium reabsorption takes place in the proximal tubule, and another 25–30% in the thick ascending limb of the loop of Henle, and less than 10% in the distal convoluted tubule and collecting duct (Palmer and Schnermann, 2015). The sodium-potassium pump transports three sodium atoms out of the cell and two potassium atoms into the cell with the cost of hydrolyzing one ATP molecule (Mandel and Balaban, 1981). To retrieve nearly 99% of filtered Na, a continuously large amount of ATP (>1.7 × 1024 molecules) is required.
Both oxidative and glycolytic pathways produce ATP. Compared to 2 molecules of ATP production in glycolysis per molecule of glucose, oxidative phosphorylation, generating 30–36 molecules of ATP, is more efficient (Soltoff, 1986). Hence, sufficient oxygen delivery to the kidney is needed to generate the required ATP via mitochondrial oxidative phosphorylation. Renal blood flow in the kidney is inhomogeneous, with cortex being well-perfused, but only 10–15% of perfusion directed to medulla to preserve osmotic gradients and enhance urinary concentration (Chou et al., 1990). Moreover, the arterial-to-venous oxygen shunting in medulla, which results from a counter-current exchange of oxygen before arterial blood reaches the renal microcirculation, decreases oxygen availability in this region (Evans et al., 2008). Lastly, the high metabolic requirements of the thick ascending limbs also contribute to hypoxia particularly in the outer medullary region, where the transport-related oxygen demand is high, while the delivery is lower than the cortex (Aukland and Krog, 1960; Mandel and Balaban, 1981; O'Connor, 2006). These particular features of tubular transport and metabolism can aggravate outer medullary hypoxia.
There is a close relation between GFR, renal sodium reabsorption, and oxygen consumption (Kiil et al., 1961; Blantz et al., 2007). In most organs, an increased demand of oxygen is met by an increased blood flow to increase oxygen delivery. However, an increase in blood flow to the kidney results in a simultaneously increased tubular sodium load due to increased GFR. Since sodium reabsorption is the major determinant of renal oxygen consumption (Lassen et al., 1961; Torelli et al., 1966), increased renal blood flow besides increasing oxygen supply also increases oxygen demand attributed to increased reabsorptive sodium load. It has been shown that renal oxygen extraction remains stable over a wide range of renal blood flow (Levy, 1960), indicating the increased oxygen delivery by renal blood flow is directly counteracted by increased oxygen consumption. So, maneuvers that increase GFR and thus the tubular sodium load also increase oxygen consumption and the effect on renal oxygenation is not always predictable.
According to the data from National Institute of Diabetes and Digestive and Kidney Diseases, as of 2014, nearly 10% of the US population has diabetes (Diabetes-NIDDK1). Diabetic nephropathy develops in nearly 40% of patients with diabetes and is the leading cause of CKD in patients with ESRD (Reutens, 2013). There has been significant advances in our understanding of the various pathways in the pathogenesis of diabetic nephropathy: hyperglycemia, glomerular hyperfiltration, activation of the polyol, renin-angiotensin and the protein kinase C pathways, advanced glycation end products, and genetic susceptibility (Brownlee, 2005). However, clinical translation to successful therapeutics for diabetic nephropathy has been limited. Since Fine et al. proposed kidney hypoxia as a mediator of progressive kidney disease (Fine et al., 1998), numerous experimental and clinical studies have provided evidence in support of chronic hypoxia being a common pathway for various progressive kidney diseases including diabetic nephropathy (Nangaku, 2006; Palm, 2006; Singh et al., 2008a, 2013; Mimura and Nangaku, 2010; Palm and Nordquist, 2011).
In experimental streptozotocin-induced diabetes, Ries et al. demonstrated that hypoxia was present in all compartments of the diabetic kidney, particularly in the outer medulla, using blood oxygen level-dependent (BOLD) MRI and diffusion-weighted imaging (Ries et al., 2003). Palm et al. showed a decrease in renal oxygen tension in the diabetic kidney, particular in medulla, due to the augmented oxygen consumption (Palm et al., 2003). Intrarenal microcirculation was not changed, but deoxyhemoglobin signals rose in diabetic kidney and oxygen consumption by tubular cells was significantly enhanced (Palm et al., 2003, 2004). Later the same group confirmed their findings using BOLD imaging (Edlund et al., 2009). Another study, demonstrated that hypoxic changes can be detected as early as 2 days in the diabetic kidney (dos Santos et al., 2007). Subsequently, Rosenberger et al. provided information on cellular localization of hypoxia in thick ascending limbs and a lesser extent in the collecting ducts using immunohistochemical staining with pimonidazole, a molecular hypoxia probe, and hypoxia inducible factor (HIF) (Rosenberger et al., 2008).
Recent studies have demonstrated the presence of tissue hypoxia before the presence of markers of kidney injury in diabetes. Friederich-Persson et al. reported kidney hypoxia resulting from increased mitochondrial oxygen consumption, independent of hyperglycemia and oxidative stress, was associated with tubulointerstitial damage, proteinuria and infiltration of inflammatory cells (Friederich-Persson et al., 2013). They concluded that kidney tissue hypoxia, per se, may be sufficient to initiate the development of nephropathy. Others have demonstrated the presence of tissue hypoxia in the kidney before the onset of albuminuria in diabetic mice (Franzén et al., 2016). Functional MRI imaging techniques have also been used to assess intrarenal oxygenation in humans, specifically in diabetic patients. Inoue et al. demonstrated parenchymal hypoxia and fibrosis in the renal cortex of CKD patients with or without diabetes using BOLD MRI and diffusion-weighted MRI, respectively (Inoue et al., 2011). Yin et al. reported reduced cortical and medullary oxygenation in patients with type 2 diabetes with nephropathy (Yin et al., 2012). Medullary hypoxia was more prominent and present at earlier stages, with cortical hypoxia becoming more apparent with worsening renal function in diabetic patients. Pruijm studied the role renin-angiotensin system (RAS) in renal oxygenation in early stage type 2 diabetic patients with evidence of nephropathy (albuminuria and/or hypertension) (Pruijm et al., 2013). Short-term RAS blockade with ace-inhibitor or angiotension receptor blocker did not increase renal tissue oxygenation. Although not specifically examined in diabetes, animal studies have demonstrated an increase renal oxygenation in both normal and remnant rat kidney with RAS inhibition (Norman et al., 2003; Deng et al., 2009). Short duration of RAS blockade or modest changes in oxygenation not detected by BOLD MRI may explain the discrepancy.
Other studies have examined renal oxygenation in diabetes before any evidence of nephropathy and found similar baseline renal oxygenation in cortical and medullary oxygenation in diabetes compared to controls with BOLD MRI (Epstein et al., 2002; Economides et al., 2004). However, remarkably in both studies, patients with diabetes and even those at risk of diabetes did not show any improvement in medullary oxygenation with water diuresis, which was observed in healthy controls. This suggests an early impairment of adaptive vasodilation in the renal medulla in diabetes. Conversely, in a study in patients with moderate to severe diabetic nephropathy, no differences in cortical oxygenation but higher medullary oxygenation compared to controls was observed (Wang et al., 2011). The authors speculate that hypoxia noted in other studies in early stages of diabetes may be due to high oxygen consumption to support increased tubular reabsorption during the stage of glomerular hyperfiltration. In their study, reduced GFR and the associated decrease in tubular sodium reabsorption may result in lower oxygen consumption and the apparent increase in medullary oxygenation. The other possibility may be related to the technical aspects of BOLD MRI measurements. BOLD MRI measures tissue oxygenation indirectly by the changes in the deoxyhemoglobin concentration in the capillaries supplying the tissue. In advanced nephropathy, tissue fibrosis, reduction in peritubular capillaries, and changes in tissue oxygen extraction may lead to discrepancies between capillary deoxyhemoglobin concentrations and tissue oxygenation. Technical aspects of BOLD MRI and standard protocols for these measurements have been extensively discussed in recent papers (Simon-Zoula et al., 2006; Pruijm et al., 2016; Hirakawa et al., 2017). Characteristics of the studies discussed is summarized in Table 1.
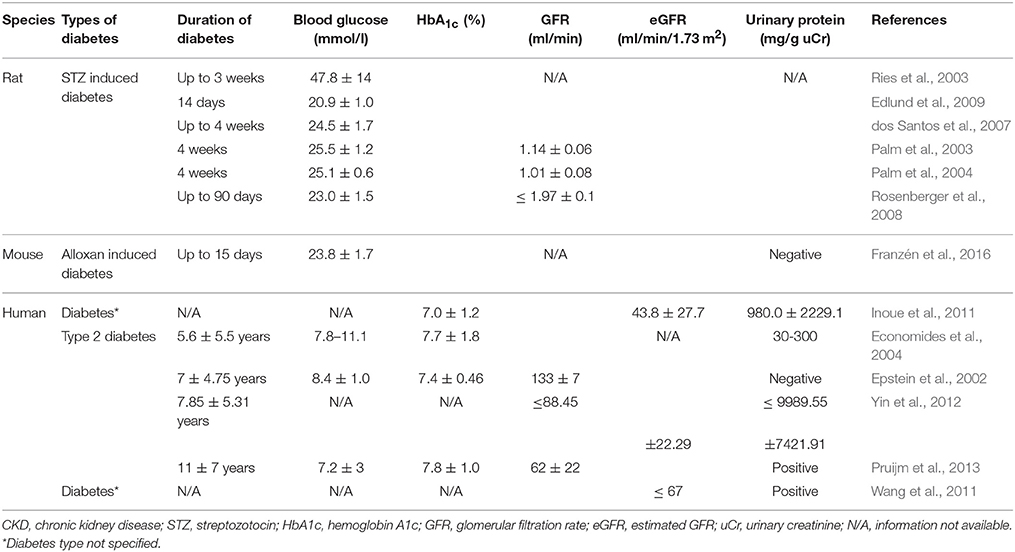
Table 1. Characteristics of studies in diabetes/diabetic CKD.
Nearly 30% of adults in the United States hypertension according to the National Health and Nutrition Examination Survey 2011–2012 (Ostchega et al., 2008). Hypertension can affect each renal compartment: vessels, glomeruli and tubulointerstitium (Meyrier, 2015). Hypertensive nephropathy is the second only to diabetic nephropathy as the most common cause of ESRD in the US (Udani et al., 2011). Franz Volhard and Theodor Fahr first introduced the term hypertensive nephrosclerosis in 1918 (Meyrier, 2015) and several studies investigating the pathophysiology and potential treatments have been published. Over the last two decades, the role of renal oxygenation in hypertensive nephrosclerosis has been investigated.
Welch et al. were the first to show that a lower tissue oxygenation in both the cortex and medulla in angiotensin II (Ang II)-infused or spontaneously hypertensive rats (Welch et al., 2001, 2003). They further demonstrated that oxygenation was restored with the administration of superoxide dismutase mimetic, tempol, or an angiotensin receptor blocker, candesartan. In spontaneously hypertensive rats, they also observed a reduction in the ratio of tubular sodium transport by oxygen consumption (TNa/QO2). In an acute hypertension model with intravenous infusion of Ang II, reduced the oxygenation in the cortex and isolated proximal tubules and reduced TNa/QO2 was observed (Welch et al., 2005). All the effects were reversed by tempol, demonstrating the role of oxidative stress in this model. Recently, Emans et al. studied the effects of Ang II on cortical pO2 by telemetry in conscious rats (Emans et al., 2016). Using exogenous Ang II intravenously or activated RAS in transgenic Cyp1a1Ren2 rats, they found reduced renal cortical PO2 preceding the development of tissue injury. Zhu et al. showed that chronic infusion of Ang II in uninephrectomized rats increased tissue hypoxia as measured by immunostaining with pimonidazole, as well as increased urinary albumin excretion and collagen accumulation (Zhu et al., 2011). The albuminuria and collagen accumulation was attenuated by gene silencing of HIF-1α, linking upregulation of HIF-1α with fibrosis in this model. In another model of hypertension, renal neurogenic hypertension, induced by intrarenal injection of phenol, was shown to renal increase HIF-1α expression (Koeners et al., 2014). Interestingly, pretreatment with cobalt chloride, known HIF-1α activator, attenuated the development of hypertension and renal vasoconstriction.
Subtotal nephrectomy (STN), by uninephrectomy and partial renal infarction in the contralateral kidney, is a well-established, representative model of hypertensive CKD. Increased oxygen consumption factored for sodium reabsorption or nephron number has been observed in this model. Harris et al. observed a threefold increase in oxygen consumption per nephron in STN kidney at 4 weeks (Harris et al., 1988). Similar results were observed by Nath et al. at 3 weeks in the STN kidney (Nath et al., 1990). We have extensively studied the early hemodynamic, transport and metabolic adaptations in the 1-week STN kidney (Deng et al., 2009, 2010; Singh et al., 2009, 2012; Singh and Thomson, 2014). A major adaptation to nephron loss in early STN is hyperfiltration in the remaining nephrons (Singh et al., 2009). Hyperfiltration in the remaining nephrons results in hyperreabsorption, and this is associated with increased oxygen consumption. Indeed, we have previously reported the high oxygen consumption factored for sodium reabsorption at 1 week after STN (Deng et al., 2009, 2010). We have also demonstrated improved renal oxygenation with RAS blockade and HIF-1α activation (Deng et al., 2009, 2010). Manotham et al. showed evidence of tissue hypoxia in the STN kidney as early as 4 days and more prominently at 7 days by using pimonidazole immunostaining (Manotham et al., 2004). Tissue hypoxia persisted until interstitial damage developed. Treatment with Ang II blocker, olmesartan, prevented vascular changes and ameliorated tubular hypoxia after nephron loss.
In terms of clinical evidence, Textor et al. showed that African-American, hypertensive patients had elevated renal medullary volumes and blood flow, and lower medullary oxygenation compared to white hypertensive patients (Textor et al., 2012). Pruijm et al. investigated the effect of sodium intake on renal oxygenation in normotensive and hypertensive subjects after 1 week of high sodium and 1 week of low-sodium diet using BOLD MRI (Pruijm et al., 2010). They found that low sodium intake was associated with an increased renal medullary oxygenation but no changes in renal cortical oxygenation in both normo- and hypertensive individuals. In hypertensive patients, medullary oxygenation correlated with 24-h sodium excretion but no correlation with segmental renal sodium reabsorption was seen as observed in normotensive group. They speculated that this may be due to enhanced proximal sodium reabsorption and salt sensitivity observed in hypertensive patients (Burnier et al., 2006).
Schachiger et al. studied the role of RAS activation in renal oxygenation in humans and showed an acute decrease in cortical oxygenation by BOLD MRI after Ang II bolus injection (Schachinger et al., 2006). Subsequently, another study demonstrated a dose-dependent decrease of renal blood flow accompanied by a minor decrease of oxygenation in the cortex, not in the medulla with continuous Ang II infusion in healthy humans (Bel et al., 2016). As discussed above, parenchymal hypoxia and fibrosis in the renal cortex of CKD patients using BOLD MRI and diffusion-weighted MRI has been shown (Inoue et al., 2011). Other recent studies have demonstrated increased renal oxygenation, particularly in the renal medulla, with RAS blockade in healthy subjects and in CKD patients (Stein et al., 2012; Siddiqi et al., 2014; Vink et al., 2015). Characteristics of the studies discussed is summarized in Table 2.
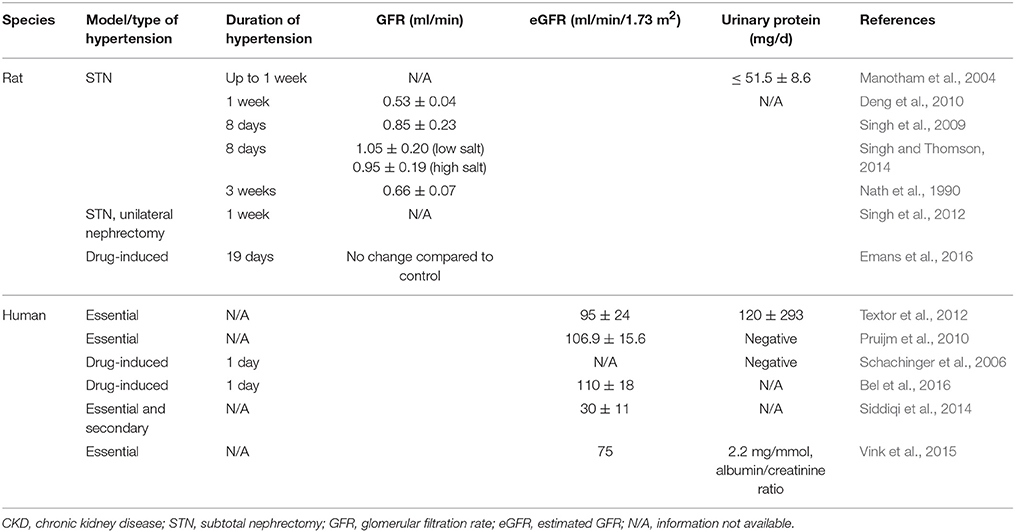
Table 2. Characteristics of studies in hypertension/hypertensive CKD.
Tubulointerstitial injury, which encompasses tubular atrophy and interstitial fibrosis, is a hallmark of progressive CKD regardless of its cause. Several studies have validated the role of tissue hypoxia in the progression of kidney disease (Kang et al., 2002; Nangaku, 2006; Heyman et al., 2008; Singh et al., 2013). Compromised oxygen delivery due to structural and functional changes impairing blood flow has been demonstrated. Tubulointerstitial hypoxia in advanced CKD can result from glomerular injury and interstitial fibrosis (Heyman et al., 2008). Glomerulosclerosis decreases the blood flow to downstream peritubular capillaries leading to insufficient oxygen delivery. Interstitial fibrosis further impairs oxygen delivery from the peritubular capillaries to the tubules. Recent studies have demonstrated peritubular capillary dysfunction as an important factor in the initiation of renal hypoxia (Ohashi et al., 2000; Kang et al., 2002; Matsumoto et al., 2004; Kawakami et al., 2014). Animal and human biopsy specimens in CKD have demonstrated that tubulointerstitial injury is preceded by rarefaction of the peritubular capillaries (Bohle et al., 1996; Choi et al., 2000; Babickova et al., 2017).
The significance of these structural changes aside, early hypoxia demonstrated in animal and human studies are likely due to altered hemodynamics and metabolism. As discussed above, alteration in renal oxygen consumption and tissue hypoxia has been demonstrated in both diabetic and non-diabetic CKD models prior to the development of structural changes (Ries et al., 2003; Manotham et al., 2004; dos Santos et al., 2007; Deng et al., 2009, 2010; Franzén et al., 2016). Moreover, kidney hypoxia resulting from increased mitochondrial oxygen consumption alone has been shown to induce kidney injury (Friederich-Persson et al., 2013). Hence, understanding of the determinants and regulation of renal oxygenation in early stages is valuable in preventing subsequent tissue injury.
Chronic hypoxia causes tissue damage via various cytokine and cell-signaling pathways including RAS, endothelin, plasmin activator inhibitor-1, adhesion molecules and growth factors (Heyman et al., 2008; Haase, 2013). Impaired nitric oxide availability has also been implicated in the pathophysiology of diabetic nephropathy (Palm et al., 2003; Prabhakar et al., 2007) and hypertensive nephropathy (Welch et al., 2003; Singh et al., 2008b; Deng et al., 2009). Nitric oxide can increase renal oxygenation by improving oxygen delivery via vasodilation and directly lowering oxygen consumption (Evans and Fitzgerald, 2005; Singh et al., 2008b). HIF, a key transcriptional regulator of cellular adaption to hypoxia, has also received significant attention. It induces various target genes that impact oxygen delivery via vasomotor regulation and cellular oxygen consumption via several pathways (Semenza, 2009). Downstream HIF target proteins include inducible NOS, heme oxygenase-1, vascular endothelial growth factor, glucose transproters, glycolytic enzymes as well as proteins involved in cell proliferation and survival (Nangaku and Eckardt, 2007; Gunaratnam and Bonventre, 2009). HIF activation induces a glycolytic phenotype by upregulating GLUT transporters to facilitate glucose entry and inducing glycolytic enzymes (Semenza, 2009). HIF also has significant effects on mitochondrial metabolism and improves efficiency of respiratory chain proteins in hypoxia (Wheaton and Chandel, 2011). It represses mitochondrial biogenesis and respiration (Papandreou et al., 2006). It also induces mitochondrial autophagy as an adaptive metabolic response in hypoxia (Zhang et al., 2008).
While HIF activation has been shown to be renoprotective role in acute kidney injury, its effect in CKD are conflicting (Nangaku and Eckardt, 2007; Haase, 2009). Pharmacological HIF activation improved renal oxygenation, function and morphology in streptozotocin-induced diabetic and in STN rats (Deng et al., 2010; Li et al., 2015; Nordquist et al., 2015). Other studies have also shown beneficial effects of systemic HIF activation with different pharmacological agents in experimental models characterized by hypoxia (Manotham et al., 2004; Tanaka et al., 2005; Song et al., 2010; Yu et al., 2012a). However, studies using genetic approaches have yielded conflicting results. HIF-1α gene silencing or HIF-1α knockout has been shown to attenuate albuminuria, collagen accumulation and the development of tubulointerstitial fibrosis (Welch et al., 2005; Wang et al., 2014). Proximal tubule specific overexpression of HIF-1α under normoxic conditions increased fibrosis while ablation ameliorated it (Higgins et al., 2007, 2008). Interestingly, the same group has subsequently shown that global activation of HIF-1α in all tubules ameliorated inflammation and fibrosis (Kobayashi et al., 2012). These results suggest that the outcomes of HIF activation are cell-specific, context-specific and timing-dependent in CKD (Yu et al., 2012b).
Lastly, the role AMP-activated kinase in the metabolism of diabetic and non-diabetic CKD is also receiving attention. AMPK is a ubiquitously expressed, highly conserved, key energy sensor and regulator of cellular metabolic activity (Hardie and Ashford, 2014). It is activated by cellular stressor such as nutrient deprivation, hypoxia or ischemia. AMPK facilitates metabolic adaptation by triggering ATP producing pathways, while inhibiting ATP consuming pathways (Gwinn et al., 2008). It stimulates mitochondrial biogenesis and cellular autophagy as survival mechanisms in low energy states (Steinberg and Kemp, 2009; Weinberg, 2011). Beneficial effects of AMPK activation in diabetes and high-fat diet induced kidney disease has been described (Lee et al., 2007; Hallows et al., 2010; Decleves et al., 2011). AMPK activation has also been shown to improve oxygen delivery and lower oxygen consumption and improve renal function and histology in STN (Satriano et al., 2013). We have also recently described the interactions between HIF and AMPK pathways in the regulation of cellular hypoxia adaptation in STN (Li et al., 2015). A schematic diagram incorporating some of the above discussed aspects in the pathophysiology of development of CKD is shown in Figure 1.
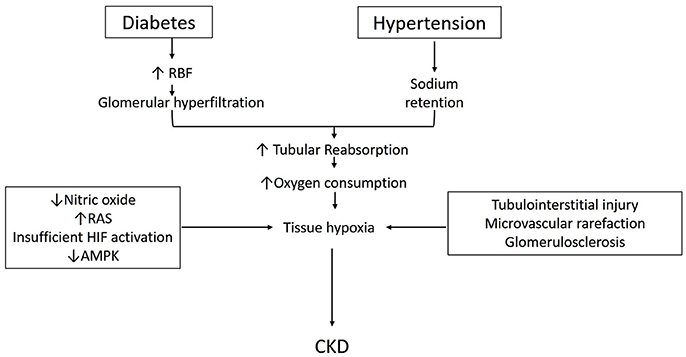
Figure 1. Schematic describing pathophysiology for the development of CKD in diabetes and hypertension (see text for details).
CKD is a major healthcare burden, associated with significant comorbidities and progression to ESRD. Available therapies for diabetic and non-diabetic CKD are inadequate in halting or slowing the progression of disease. An increasing number of experimental and clinical studies indicate that renal tissue hypoxia, resulting from mismatch between oxygen delivery and consumption is a major contributor to CKD progression. Hence, it is imperative to understand the regulation of kidney oxygenation in CKD to identify novel therapeutic targets. Interventions to restore kidney tissue oxygenation has been shown to be beneficial in animal models of CKD. Clinical studies are limited and additional investigations into therapeutic targets to improve renal oxygenation in different forms of CKD are needed to improve clinical outcomes.
ZZL: drafted, prepared, and edited the manuscript. AB and YL: reviewed and edited the manuscript. PS: reviewed, revised and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by VA Merit BX002175 (PS), NIH-NIDDK R01 DK107852 (PS), NIH-NIDDK R03 DK101841 (PS), American Diabetes Association Grant #4-15-CKD-70 and UAB-UCSD O'Brien Center (NIH P30-DK 079337).
1. ^Diabetes-NIDDK. National Institute of Diabetes and Digestive and Kidney Diseases. https://wwwniddknihgov/health-information/diabetes (accessed 2017).
Babickova, J., Klinkhammer, B. M., Buhl, E. M., Djudjaj, S., Hoss, M., Heymann, F., et al. (2017). Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 91, 70–85. doi: 10.1016/j.kint.2016.07.038
Bel, R., Coolen, B. F., Nederveen, A. J., Potters, W. V., Verberne, H. J., Vogt, L., et al. (2016). Magnetic resonance imaging–derived renal oxygenation and perfusion during continuous, steady-state angiotensin-II infusion in healthy humans. J. Am. Heart Assoc. 5:e003185. doi: 10.1161/JAHA.115.003185
Blantz, R. C., Deng, A., Miracle, C. M., and Thomson, S. C. (2007). Regulation of kidney function and metabolism: a question of supply and demand. Trans. Am. Clin. Climatol. Assoc. 118, 23–43.
Bohle, A., Mackensen-Haen, S., and Wehrmann, M. (1996). Significance of postglomerular capillaries in the pathogenesis of chronic renal failure. Kidney Blood Press. Res. 19, 191–195. doi: 10.1159/000174072
Brownlee, M. (2005). The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54, 1615–1625. doi: 10.2337/diabetes.54.6.1615
Burnier, M., Bochud, M., and Maillard, M. (2006). Proximal tubular function and salt sensitivity. Curr. Hypertens. Rep. 8, 8–15. doi: 10.1007/s11906-006-0035-6
Choi, Y. J., Chakraborty, S., Nguyen, V., Nguyen, C., Kim, B. K., Shim, S. I., et al. (2000). Peritubular capillary loss is associated with chronic tubulointerstitial injury in human kidney: altered expression of vascular endothelial growth factor. Hum. Pathol. 31, 1491–1497. doi: 10.1053/hupa.2000.20373
Chou, S. Y., Porush, J. G., and Faubert, P. F. (1990). Renal medullary circulation: hormonal control. Kidney Int. 37, 1–13. doi: 10.1038/ki.1990.1
Coresh, J., Selvin, E., Stevens, L. A., Manzi, J., Kusek, J. W., Eggers, P., et al. (2007). Prevalence of chronic kidney disease in the United States. JAMA 298, 2038–2047. doi: 10.1001/jama.298.17.2038
Decleves, A. E., Mathew, A. V., Cunard, R., and Sharma, K. (2011). AMPK mediates the initiation of kidney disease induced by a high-fat diet. J. Am. Soc. Nephrol. 22, 1846–1855. doi: 10.1681/ASN.2011010026
Deng, A., Arndt, M. A. K., Satriano, J., Singh, P., Rieg, T., Thomson, S., et al. (2010). Renal protection in chronic kidney disease: hypoxia-inducible factor activation vs. angiotensin II blockade. Am. J. Physiol. Ren. Physiol. 299, F1365–F1373. doi: 10.1152/ajprenal.00153.2010
Deng, A., Tang, T., Singh, P., Wang, C., Satriano, J., Thomson, S. C., et al. (2009). Regulation of oxygen utilization by angiotensin II in chronic kidney disease. Kidney Int. 75, 197–204. doi: 10.1038/ki.2008.481
dos Santos, E. A., Li, L.-P., Ji, L., and Prasad, P. V. (2007). Early changes with diabetes in renal medullary hemodynamics as evaluated by fiberoptic probes and BOLD magnetic resonance imaging. Invest. Radiol. 42, 157–162. doi: 10.1097/01.rli.0000252492.96709.36
Economides, P. A., Caselli, A., Zuo, C. S., Sparks, C., Khaodhiar, L., Katsilambros, N., et al. (2004). Kidney oxygenation during water diuresis and endothelial function in patients with type 2 diabetes and subjects at risk to develop diabetes. Metab. Clin. Exp. 53, 222–227. doi: 10.1016/j.metabol.2003.09.019
Edlund, J., Hansell, P., Fasching, A., Liss, P., Weis, J., Glickson, J. D., et al. (2009). Reduced oxygenation in diabetic rat kidneys measured by T2* weighted magnetic resonance micro-imaging. Adv. Exp. Med. Biol. 645, 199–204. doi: 10.1007/978-0-387-85998-9_31
Emans, T. W., Janssen, B. J., Pinkham, M. I., Ow, C. P., Evans, R. G., Joles, J. A., et al. (2016). Exogenous and endogenous angiotensin-II decrease renal cortical oxygen tension in conscious rats by limiting renal blood flow. J. Physiol. 594, 6287–6300. doi: 10.1113/JP270731
Epstein, F. H., Veves, A., and Prasad, P. V. (2002). Effect of diabetes on renal medullary oxygenation during water diuresis. Diabetes Care 25, 575–578. doi: 10.2337/diacare.25.3.575
Evans, R. G., and Fitzgerald, S. M. (2005). Nitric oxide and superoxide in the renal medulla: a delicate balancing act. Curr. Opin. Nephrol. Hypertens. 14, 9–15. doi: 10.1097/00041552-200501000-00003
Evans, R. G., Gardiner, B. S., Smith, D. W., and O'Connor, P. M. (2008). Intrarenal oxygenation: unique challenges and the biophysical basis of homeostasis. Am. J. Physiol. Ren. Physiol. 295, F1259–F1270. doi: 10.1152/ajprenal.90230.2008
Fine, L. G., Orphanides, C., and Norman, J. T. (1998). Progressive renal disease: the chronic hypoxia hypothesis. Kidney Int. Suppl. 65, S74–S78.
Franzén, S., Pihl, L., Khan, N., Gustafsson, H., and Palm, F. (2016). Pronounced kidney hypoxia precedes albuminuria in type 1 diabetic mice. Am. J. Physiol. Ren. Physiol. 310, F807–F809. doi: 10.1152/ajprenal.00049.2016
Friederich-Persson, M., Thörn, E., Hansell, P., Nangaku, M., Levin, M., and Palm, F. (2013). Kidney hypoxia, attributable to increased oxygen consumption, induces nephropathy independently of hyperglycemia and oxidative stress. Hypertension 62, 914–919. doi: 10.1161/HYPERTENSIONAHA.113.01425
Gunaratnam, L., and Bonventre, J. V. (2009). HIF in kidney disease and development. JASN 20, 1877–1887. doi: 10.1681/ASN.2008070804
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 30, 214–226. doi: 10.1016/j.molcel.2008.03.003
Haase, V. H. (2009). Pathophysiological consequences of HIF activation: HIF as a modulator of fibrosis. Ann. N. Y. Acad. Sci. 1177, 57–65. doi: 10.1111/j.1749-6632.2009.05030.x
Haase, V. H. (2013). Mechanisms of hypoxia responses in renal tissue. J. Am. Soc. Nephrol. 24, 537–541. doi: 10.1681/ASN.2012080855
Hallows, K. R., Mount, P. F., Pastor-Soler, N. M., and Power, D. A. (2010). Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am. J. Physiol. Ren. Physiol. 298, F1067–F1077. doi: 10.1152/ajprenal.00005.2010
Hardie, D. G., and Ashford, M. L. (2014). AMPK: regulating energy balance at the cellular and whole body levels. Physiology 29, 99–107. doi: 10.1152/physiol.00050
Harris, D. C., Chan, L., and Schrier, R. W. (1988). Remnant kidney hypermetabolism and progression of chronic renal failure. Am. J. Physiol. 254, F267–F276.
Heyman, S. N., Khamaisi, M., Rosen, S., and Rosenberger, C. (2008). Renal parenchymal hypoxia, hypoxia response and the progression of chronic kidney disease. Am. J. Nephrol. 28, 998–1006. doi: 10.1159/000146075
Higgins, D. F., Kimura, K., Bernhardt, W. M., Shrimanker, N., Akai, Y., Hohenstein, B., et al. (2007). Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest. 117, 3810–3820. doi: 10.1172/JCI30487
Higgins, D. F., Kimura, K., Iwano, M., and Haase, V. H. (2008). Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle 7, 1128–1132. doi: 10.4161/cc.7.9.5804
Hirakawa, Y., Tanaka, T., and Nangaku, M. (2017). Renal hypoxia in CKD; pathophysiology and detecting methods. Front. Physiol. 8:99. doi: 10.3389/fphys.2017.00099
Honeycutt, A. A., Segel, J. E., Zhuo, X., Hoerger, T. J., Imai, K., and Williams, D. (2013). Medical costs of CKD in the medicare population. J. Am. Soc. Nephrol. 24, 1478–1483. doi: 10.1681/ASN.2012040392
Inoue, T., Kozawa, E., Okada, H., Inukai, K., Watanabe, S., Kikuta, T., et al. (2011). Noninvasive evaluation of kidney hypoxia and fibrosis using magnetic resonance imaging. J. Am. Soc. Nephrol. 22, 1429–1434. doi: 10.1681/ASN.2010111143
Kang, D.-H., Kanellis, J., Hugo, C., Truong, L., Anderson, S., Kerjaschki, D., et al. (2002). Role of the microvascular endothelium in progressive renal disease. J. Am. Soc. Nephrol. 13, 806–816. doi: 10.1097/01.ASN.0000034910.58454.FD
Kawakami, T., Mimura, I., Shoji, K., Tanaka, T., and Nangaku, M. (2014). Hypoxia and fibrosis in chronic kidney disease: crossing at pericytes. Kidney Int. Suppl. 4, 107–112. doi: 10.1038/kisup.2014.20
Kiil, F., Aukland, K., and Refsum, H. E. (1961). Renal sodium transport and oxygen consumption. Am. J. Physiol. 201, 511–516.
Kobayashi, H., Gilbert, V., Liu, Q., Kapitsinou, P. P., Unger, T. L., Rha, J., et al. (2012). Myeloid cell-derived hypoxia-inducible factor attenuates inflammation in unilateral ureteral obstruction-induced kidney injury. J. Immunol. 188, 5106–5115. doi: 10.4049/jimmunol.1103377
Koeners, M. P., Vink, E. E., Kuijper, A., Gadellaa, N., Rosenberger, C., Mathia, S., et al. (2014). Stabilization of hypoxia inducible factor-1alpha ameliorates acute renal neurogenic hypertension. J. Hypertens. 32, 587–597. doi: 10.1097/HJH.0000000000000060
Lassen, N. A., Munck, O., and Thaysen, J. H. (1961). Oxygen consumption and sodium reabsorption in the kidney. Acta Physiol. Scand. 51, 371–384. doi: 10.1111/j.1748-1716.1961.tb02147.x
Lee, M. J., Feliers, D., Mariappan, M. M., Sataranatarajan, K., Mahimainathan, L., Musi, N., et al. (2007). A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am. J. Physiol. Ren. Physiol. 292, F617–F627. doi: 10.4049/jimmunol.1103377
Levy, M. N. (1960). Effect of variations of blood flow on renal oxygen extraction. Am. J. Physiol. 199, 13–18.
Li, H., Satriano, J., Thomas, J. L., Miyamoto, S., Sharma, K., Pastor-Soler, N. M., et al. (2015). Interactions between HIF-1alpha and AMPK in the regulation of cellular hypoxia adaptation in chronic kidney disease. Am. J. Physiol. Ren. Physiol. 309, F414–F428. doi: 10.1152/ajprenal.00463.2014
Mandel, L. J., and Balaban, R. S. (1981). Stoichiometry and coupling of active transport to oxidative metabolism in epithelial tissues. Am. J. Physiol. 240, F357–F371.
Manotham, K., Tanaka, T., Matsumoto, M., Ohse, T., Miyata, T., Inagi, R., et al. (2004). Evidence of tubular hypoxia in the early phase in the remnant kidney model. J. Am. Soc. Nephrol. 15, 1277–1288. doi: 10.1097/01.ASN.0000125614.35046.10
Matsumoto, M., Tanaka, T., Yamamoto, T., Noiri, E., Miyata, T., Inagi, R., et al. (2004). Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J. Am. Soc. Nephrol. 15, 1574–1581. doi: 10.1097/01.ASN.0000128047.13396.48
Meyrier, A. (2015). Nephrosclerosis: update on a centenarian. Nephrol. Dial. Transplant. 30, 1833–1841. doi: 10.1093/ndt/gfu366
Mimura, I., and Nangaku, M. (2010). The suffocating kidney: tubulointerstitial hypoxia in end-stage renal disease. Nat. Rev. Nephrol. 6, 667–678. doi: 10.1038/nrneph.2010.124
Nangaku, M. (2006). Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 17, 17–25. doi: 10.1681/ASN.2005070757
Nangaku, M., and Eckardt, K.-U. (2007). Hypoxia and the HIF system in kidney disease. J. Mol. Med. 85, 1325–1330. doi: 10.1007/s00109-007-0278-y
Nath, K. A., Croatt, A. J., and Hostetter, T. H. (1990). Oxygen consumption and oxidant stress in surviving nephrons. Am. J. Physiol. 258, F1354–F1362.
Nordquist, L., Friederich-Persson, M., Fasching, A., Liss, P., Shoji, K., Nangaku, M., et al. (2015). Activation of hypoxia-inducible factors prevents diabetic nephropathy. J. Am. Soc. Nephrol. 26, 328–338. doi: 10.1681/ASN.2013090990
Norman, J. T., and Fine, L. G. (2006). Intrarenal oxygenation in chronic renal failure. Clin. Exp. Pharmacol. Physiol. 33, 989–996. doi: 10.1111/j.1440-1681.2006.04476.x
Norman, J. T., Stidwill, R., Singer, M., and Fine, L. G. (2003). Angiotensin II blockade augments renal cortical microvascular pO2 indicating a novel, potentially renoprotective action. Nephron Physiol. 94, P39–P46. doi: 10.1159/000071289
O'Connor, P. M. (2006). Renal oxygen delivery: matching delivery to metabolic demand. Clin. Exp. Pharmacol. Physiol. 33, 961–967. doi: 10.1111/j.1440-1681.2006.04475.x
Ohashi, R., Kitamura, H., and Yamanaka, N. (2000). Peritubular capillary injury during the progression of experimental glomerulonephritis in rats. J. Am. Soc. Nephrol. 11, 47–56.
Ostchega, Y., Yoon, S. S., Hughes, J., and Louis, T. (2008). Hypertension awareness, treatment, and control–continued disparities in adults: United States, 2005–2006. NCHS Data Brief. 1–8.
Palm, F. (2006). Intrarenal oxygen in diabetes and a possible link to diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 33, 997–1001. doi: 10.1111/j.1440-1681.2006.04473.x
Palm, F., Cederberg, J., Hansell, P., Liss, P., and Carlsson, P. O. (2003). Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia 46, 1153–1160. doi: 10.1007/s00125-003-1155-z
Palm, F., Hansell, P., Ronquist, G., Waldenström, A., Liss, P., and Carlsson, P. O. (2004). Polyol-pathway-dependent disturbances in renal medullary metabolism in experimental insulin-deficient diabetes mellitus in rats. Diabetologia 47, 1223–1231. doi: 10.1007/s00125-004-1434-3
Palm, F., and Nordquist, L. (2011). Renal tubulointerstitial hypoxia: cause and consequence of kidney dysfunction. Clin. Exp. Pharmacol. Physiol. 38, 474–480. doi: 10.1111/j.1440-1681.2011.05532.x
Palmer, L. G., and Schnermann, J. (2015). Integrated control of Na transport along the nephron. Clin. J. Am. Soc. Nephrol. 10, 676–687. doi: 10.2215/CJN.12391213
Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L., and Denko, N. C. (2006). HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197. doi: 10.1016/j.cmet.2006.01.012
Prabhakar, S., Starnes, J., Shi, S., Lonis, B., and Tran, R. (2007). Diabetic nephropathy is associated with oxidative stress and decreased renal nitric oxide production. J. Am. Soc. Nephrol. 18, 2945–2952. doi: 10.1681/ASN.2006080895
Pruijm, M., Hofmann, L., Maillard, M., Tremblay, S., Glatz, N., Wuerzner, G., et al. (2010). Effect of sodium loading/depletion on renal oxygenation in young normotensive and hypertensive men. Hypertension 55, 1116–1122. doi: 10.1161/HYPERTENSIONAHA.109.149682
Pruijm, M., Hofmann, L., Zanchi, A., Maillard, M., Forni, V., Muller, M.-E., et al. (2013). Blockade of the renin-angiotensin system and renal tissue oxygenation as measured with BOLD-MRI in patients with type 2 diabetes. Diabetes Res. Clin. Pract. 99, 136–144. doi: 10.1016/j.diabres.2012.11.004
Pruijm, M., Milani, B., and Burnier, M. (2016). Blood oxygenation level-dependent MRI to assess renal oxygenation in renal diseases: progresses and challenges. Front. Physiol. 7:667. doi: 10.3389/fphys.2016.00667
Reutens, A. T. (2013). Epidemiology of diabetic kidney disease. Med. Clin. North Am. 97, 1–18. doi: 10.1016/j.mcna.2012.10.001
Ries, M., Basseau, F., Tyndal, B., Jones, R., Deminière, C., Catargi, B., et al. (2003). Renal diffusion and BOLD MRI in experimental diabetic nephropathy. Blood oxygen level-dependent. J. Magn. Reson. Imaging 17, 104–113. doi: 10.1002/jmri.10224
Rosenberger, C., Khamaisi, M., Abassi, Z., Shilo, V., Weksler-Zangen, S., Goldfarb, M., et al. (2008). Adaptation to hypoxia in the diabetic rat kidney. Kidney Internat. 73, 34–42. doi: 10.1038/sj.ki.5002567
Satriano, J., Sharma, K., Blantz, R. C., and Deng, A. (2013). Induction of AMPK activity corrects early pathophysiological alterations in the subtotal nephrectomy model of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 305, F727–F733. doi: 10.1152/ajprenal.00293.2013
Schachinger, H., Klarhöfer, M., Linder, L., Drewe, J., and Scheffler, K. (2006). Angiotensin II decreases the renal MRI blood oxygenation level-dependent signal. Hypertension 47, 1062–1066. doi: 10.1161/01.HYP.0000220109.98142.a3
Semenza, G. L. (2009). Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology 24, 97–106. doi: 10.1152/physiol.00045.2008
Siddiqi, L., Hoogduin, H., Visser, F., Leiner, T., Mali, W. P., and Blankestijn, P. J. (2014). Inhibition of the renin-angiotensin system affects kidney tissue oxygenation evaluated by magnetic resonance imaging in patients with chronic kidney disease. J Clin. Hypertens. 16, 214–218. doi: 10.1111/jch.12263
Simon-Zoula, S. C., Hofmann, L., Giger, A., Vogt, B., Vock, P., Frey, F. J., et al. (2006). Non-invasive monitoring of renal oxygenation using BOLD-MRI: a reproducibility study. NMR Biomed. 19, 84–89. doi: 10.1002/nbm.1004
Singh, D. K., Winocour, P., and Farrington, K. (2008a). Mechanisms of disease: the hypoxic tubular hypothesis of diabetic nephropathy. Nat. Clin. Pract. Nephrol. 4, 216–226. doi: 10.1038/ncpneph0757
Singh, P., Blantz, R. C., Rosenberger, C., Gabbai, F. B., Schoeb, T. R., and Thomson, S. C. (2012). Aberrant tubuloglomerular feedback and HIF-1α confer resistance to ischemia after subtotal nephrectomy. J. Am. Soc. Nephrol. 23, 483–493. doi: 10.1681/ASN.2011020130
Singh, P., Deng, A., Blantz, R. C., and Thomson, S. C. (2009). Unexpected effect of angiotensin AT1 receptor blockade on tubuloglomerular feedback in early subtotal nephrectomy. Am. J. Physiol. Ren. Physiol. 296, F1158–F1165. doi: 10.1152/ajprenal.90722.2008
Singh, P., Deng, A., Weir, M. R., and Blantz, R. C. (2008b). The balance of angiotensin II and nitric oxide in kidney diseases. Curr. Opin. Nephrol. Hypertens. 17, 51–56. doi: 10.1097/MNH.0b013e3282f29a8b
Singh, P., Ricksten, S.-E., Bragadottir, G., Redfors, B., and Nordquist, L. (2013). Renal oxygenation and hemodynamics in acute kidney injury and chronic kidney disease. Clin. Exp. Pharmacol. Physiol. 40, 138–147. doi: 10.1111/1440-1681.12036
Singh, P., and Thomson, S. C. (2014). Salt sensitivity of tubuloglomerular feedback in the early remnant kidney. Am. J. Physiol. Ren. Physiol. 306, F172–F180. doi: 10.1152/ajprenal.00431.2013
Soltoff, S. P. (1986). ATP and the regulation of renal cell function. Annu. Rev. Physiol. 48, 9–31. doi: 10.1146/annurev.ph.48.030186.000301
Song, Y. R., You, S. J., Lee, Y. M., Chin, H. J., Chae, D. W., Oh, Y. K., et al. (2010). Activation of hypoxia-inducible factor attenuates renal injury in rat remnant kidney. Nephrol. Dial. Transplant. 25, 77–85. doi: 10.1093/ndt/gfp454
Stein, A., Goldmeier, S., Voltolini, S., Setogutti, E., Feldman, C., Figueiredo, E., et al. (2012). Renal oxygen content is increased in healthy subjects after angiotensin-converting enzyme inhibition. Clinics (Sao Paulo). 67, 761–765. doi: 10.6061/clinics/2012(07)10
Steinberg, G. R., and Kemp, B. E. (2009). AMPK in health and disease. Physiol. Rev. 89, 1025–1078. doi: 10.1152/physrev.00011.2008
Tanaka, T., Kojima, I., Ohse, T., Ingelfinger, J. R., Adler, S., Fujita, T., et al. (2005). Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab. Invest. 85, 1292–1307. doi: 10.1038/labinvest.3700328
Textor, S. C., Gloviczki, M. L., Flessner, M. F., Calhoun, D. A., Glockner, J., Grande, J. P., et al. (2012). Association of filtered sodium load with medullary volumes and medullary hypoxia in hypertensive African Americans as compared with whites. Am. J. Kidney Dis. 59, 229–237. doi: 10.1053/j.ajkd.2011.09.023
Torelli, G., Milla, E., Faelli, A., and Costantini, S. (1966). Energy requirement for sodium reabsorption in the in vivo rabbit kidney. Am. J. Physiol. 211, 576–580.
Udani, S., Lazich, I., and Bakris, G. L. (2011). Epidemiology of hypertensive kidney disease. Nat. Rev. Nephrol. 7, 11–21. doi: 10.1038/nrneph.2010.154
Vink, E. E., de Boer, A., Hoogduin, H. J., Voskuil, M., Leiner, T., Bots, M. L., et al. (2015). Renal BOLD-MRI relates to kidney function and activity of the renin-angiotensin-aldosterone system in hypertensive patients. J. Hypertens. 33, 597–603. discussion: 603–594. doi: 10.1097/HJH.0000000000000436
Wang, Z. J., Kumar, R., Banerjee, S., and Hsu, C.-Y. (2011). Blood oxygen level-dependent (BOLD) MRI of diabetic nephropathy: preliminary experience. J. Magn. Reson. Imaging 33, 655–660. doi: 10.1002/jmri.22501
Wang, Z., Zhu, Q., Li, P.-L., Dhaduk, R., Zhang, F., Gehr, T. W., et al. (2014). Silencing of hypoxia-inducible factor-1α gene attenuates chronic ischemic renal injury in two-kidney, one-clip rats. Amer. J. Physiol. 306, F1236–F1242. doi: 10.1152/ajprenal.00673.2013
Weinberg, J. M. (2011). Mitochondrial biogenesis in kidney disease. J. Am. Soc. Nephrol. 22, 431–436. doi: 10.1681/ASN.2010060643
Welch, W. J., Baumgärtl, H., Lübbers, D., and Wilcox, C. S. (2001). Nephron pO2 and renal oxygen usage in the hypertensive rat kidney. Kidney Int. 59, 230–237. doi: 10.1046/j.1523-1755.2001.00483.x
Welch, W. J., Baumgärtl, H., Lübbers, D., and Wilcox, C. S. (2003). Renal oxygenation defects in the spontaneously hypertensive rat: role of AT1 receptors. Kidney Int. 63, 202–208. doi: 10.1046/j.1523-1755.2003.00729.x
Welch, W. J., Blau, J., Xie, H., Chabrashvili, T., and Wilcox, C. S. (2005). Angiotensin-induced defects in renal oxygenation: role of oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 288, H22–H28. doi: 10.1152/ajpheart.00626.2004
Wheaton, W. W., and Chandel, N. S. (2011). Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell. Physiol. 300, C385–C393. doi: 10.1152/ajpcell.00485.2010
Yin, W.-J., Liu, F., Li, X.-M., Yang, L., Zhao, S., Huang, Z.-X., et al. (2012). Noninvasive evaluation of renal oxygenation in diabetic nephropathy by BOLD-MRI. Eur. J. Radiol. 81, 1426–1431. doi: 10.1016/j.ejrad.2011.03.045
Yu, X., Fang, Y., Ding, X., Liu, H., Zhu, J., Zou, J., et al. (2012a). Transient hypoxia-inducible factor activation in rat renal ablation and reduced fibrosis with L-mimosine. Nephrology (Carlton). 17, 58–67. doi: 10.1111/j.1440-1797.2011.01498.x
Yu, X., Fang, Y., Liu, H., Zhu, J., Zou, J., Xu, X., et al. (2012b). The balance of beneficial and deleterious effects of hypoxia-inducible factor activation by prolyl hydroxylase inhibitor in rat remnant kidney depends on the timing of administration. Nephrol. Dial. Transplant. 27, 3110–3119. doi: 10.1093/ndt/gfr754
Zhang, H., Bosch-Marce, M., Shimoda, L. A., Tan, Y. S., Baek, J. H., Wesley, J. B., et al. (2008). Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903. doi: 10.1074/jbc.M800102200
Keywords: hypoxia, renal oxygen consumption, hypoxia inducible factor (HIF), AMPK, chronic kidney disease pathophysiology
Citation: Liu ZZ, Bullen A, Li Y and Singh P (2017) Renal Oxygenation in the Pathophysiology of Chronic Kidney Disease. Front. Physiol. 8:385. doi: 10.3389/fphys.2017.00385
Received: 15 March 2017; Accepted: 23 May 2017;
Published: 28 June 2017.
Edited by:
Fredrik Palm, Uppsala University, SwedenCopyright © 2017 Liu, Bullen, Li and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prabhleen Singh, cDFzaW5naEB1Y3NkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.