 Nikolay Naumenko1†
Nikolay Naumenko1† Jenni Huusko1†
Jenni Huusko1† Tomi Tuomainen1
Tomi Tuomainen1 Jussi T. Koivumäki1Mari Merentie1
Jussi T. Koivumäki1Mari Merentie1 Erika Gurzeler1Kari Alitalo2
Erika Gurzeler1Kari Alitalo2 Riikka Kivelä2
Riikka Kivelä2 Seppo Ylä-Herttuala1,3
Seppo Ylä-Herttuala1,3 Pasi Tavi1*
Pasi Tavi1*- 1A.I. Virtanen Institute for Molecular Sciences, University of Eastern Finland, Kuopio, Finland
- 2Wihuri Research Institute and Translational Cancer Biology Program, University of Helsinki, Biomedicum Helsinki, Helsinki, Finland
- 3Heart Center and Gene Therapy Unit, Kuopio University Hospital, Kuopio, Finland
Vascular endothelial growth factor B (VEGF-B) is a potent mediator of vascular, metabolic, growth, and stress responses in the heart, but the effects on cardiac muscle and cardiomyocyte function are not known. The purpose of this study was to assess the effects of VEGF-B on the energy metabolism, contractile, and electrophysiological properties of mouse cardiac muscle and cardiac muscle cells. In vivo and ex vivo analysis of cardiac-specific VEGF-B TG mice indicated that the contractile function of the TG hearts was normal. Neither the oxidative metabolism of isolated TG cardiomyocytes nor their energy substrate preference showed any difference to WT cardiomyocytes. Similarly, myocyte Ca2+ signaling showed only minor changes compared to WT myocytes. However, VEGF-B overexpression induced a distinct electrophysiological phenotype characterized by ECG changes such as an increase in QRSp time and decreases in S and R amplitudes. At the level of isolated TG cardiomyocytes, these changes were accompanied with decreased action potential upstroke velocity and increased duration (APD60–70). These changes were partly caused by downregulation of sodium current (INa) due to reduced expression of Nav1.5. Furthermore, TG myocytes had alterations in voltage-gated K+ currents, namely decreased density of transient outward current (Ito) and total K+ current (Ipeak). At the level of transcription, these were accompanied by downregulation of Kv channel-interacting protein 2 (Kcnip2), a known modulatory subunit for Kv4.2/3 channel. Cardiac VEGF-B overexpression induces a distinct electrophysiological phenotype including remodeling of cardiomyocyte ion currents, which in turn induce changes in action potential waveform and ECG.
Introduction
The Vascular Endothelial Growth Factor (VEGF) family consists of five proteins with regulative roles in blood and lymphatic vessel growth and development (Bry et al., 2014). The angiogenic potency of VEGFs makes them attractive candidates for therapy of ischemic heart (Vuorio et al., 2012). However, among the VEGFs, the effects of VEGF-B spread beyond vessel regulation as it has been implicated in the regulation of myocardial metabolism, growth, and stress-response (Karpanen et al., 2008; Huusko et al., 2012; Kivela et al., 2014). VEGF-B is expressed in a variety of tissues with highest expression in heart and oxidative skeletal muscle (Hagberg et al., 2010). Alternative splicing of the VEGF-B gene produces two isoforms, VEGF-B167 and VEGF-B186, which both mediate their effects by binding to VEGFR-1 (Olofsson et al., 1998) and NRP-1 receptors (Makinen et al., 1999). When overexpressed, VEGF-B activates Akt/mTORC1 and ERK1/2 MAPK pathways (Kivela et al., 2014). Data from genetic rodent models suggest that VEGF-B is not a prerequisite for cardiac development or function, but rather serves as a regulatory factor. Supporting this, VEGF-B deficient mice are viable and show only mild cardiac phenotype with either atrial conduction defect (Aase et al., 2001) or retarded growth accompanied with slightly impaired coronary vasculature and reduced ischemic tolerance ex vivo (Bellomo et al., 2000). Similarly, VEGF-B deficient rats have no obvious abnormalities in either contractile or vascular function (Kivela et al., 2014). Transgenic overexpression of VEGF-B in mouse (Karpanen et al., 2008) or rat heart (Bry et al., 2010; Kivela et al., 2014) induces metabolic remodeling, moderate hypertrophy, and myocardial capillary enlargement with improved ischemic tolerance. VEGF-B delivery into myocardium with adenoviruses in rodent or pig hearts consistently produces an increase in the density or diameter of the myocardial capillaries, and AAV-VEGF-B delivery appears to protect the heart against ischemia-, pacing-, or load-induced cardiomyopathy (Bry et al., 2014). So far, studies on cardiac effects of VEGF-B have been mainly conducted in the context of angiogenic therapy to enhance perfusion of the heart and metabolic effects on the myocardium. However, the effects of VEGF-B on myocardial function at the level of cardiac muscle cells are not known. The purpose of this study was to assess the effects of VEGF-B on the energy metabolism, contractile, and electrophysiological properties of the mouse heart.
Materials and Methods
Experimental Animals
The present studies employed 2–4 months old transgenic (TG) mice overexpressing human VEGF-B167 and VEGF-B186 isoforms (Supplementary Figure S1) under the cardiac-specific myosin heavy chain promoter (Bry et al., 2010). VEGF-B TG mice were bred into the C57Bl/6JOlaHsd background for over 10 generations. TG-negative littermates (WT) were used as controls in all experiments. The animals were kept in standard housing conditions in The National Laboratory Animal Center of The University of Eastern Finland. Diet and water were provided ad libitum. All animal experiments were performed according the guidelines of Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes, the procedures were also approved by the Animal Experiment Board in Finland and carried out in accordance with the guidelines of the Experimental Animal Committee of the University of Eastern Finland.
Echocardiography and Electrocardiography
Ten TG and ten WT littermate controls were used for echocardiographic and electrocardiographic measurements performed with the Vevo2100 Ultrasound System (VisualSonics Inc., Toronto, ON, Canada). A high-frequency ultrasound probe (MS400) operating at 30 MHz with a minimum of 300 frames per second was used. The animals were anesthetized with isoflurane (induction: 4.5% isoflurane, 450 mL air, maintenance: 2.0% isoflurane, 200 mL air, Baxter International Inc., Deerfield, IL, USA). Mice were placed in a supine position on a heated platform (THM100, Indus Instruments, Houston, TX, USA) to maintain the body temperature at 36–37°C. Ejection fraction (EF), left ventricle anterior wall thickness (LVAW), left ventricle volume (LV Vol), and left ventricle mass (LV Mass) were determined from parasternal short axis M-Mode measurements. EF was calculated by Vevo2100 software using the Teicholz formula. To obtain the ECG signal, the paws of the mice were connected to the electrode pads on the platform using ECG gel and fixed with a skin tape. The recorded ECG represents the standard limb lead II. The mouse heart rate and respiration were monitored during anesthesia via ECG pads. The raw data of ECG were analyzed with a Matlab-based ECG analysis program (Kubios HRV, version 2.0 beta 4, Department of Physics, University of Eastern Finland, Kuopio, Finland), which was modified specially for analyzing mouse ECG (Merentie et al., 2015). Time intervals (QRS, QRSp, and QTc time) and amplitudes of R and S wave were analyzed from the mean curve generated from a 30-s ECG recording. QTc time was calculated as QT ms/(R − R0/100 ms)1/2 (Mitchell et al., 1998).
Langendorff Heart Perfusion Model
For ex vivo heart studies, conventional Langendorff perfusion was used (Bell et al., 2011). Isolated hearts were perfused with 37°C Krebs-Henzeleit buffer solution and paced by surface electrodes at 8 Hz in all experiments. To record left ventricular pressure (LVP), a water-filled PVC balloon was inserted into the left ventricle (LV). Heart rate and LVP were monitored with an HSE ISOTECH Pressure Transducer and recorded with a data acquisition board (PCI-6052E, National Instruments, Austin, TX, USA). Collected data were analyzed with WinEDR software (University of Strathclyde, Glasgow, UK). To assess the substrate specificity (glucose vs. palmitate) of the isolated hearts, the buffer solution was supplemented with 1.2 mM sodium palmitate (Sigma-Aldrich, St. Louis, MO, USA) conjugated to fatty acid-free bovine serum albumin (BSA, Sigma-Aldrich, St. Louis, MO, USA) (Belke et al., 1999) or with 11 mM glucose and unconjugated BSA.
Single Cell Isolation
Adult mouse ventricular myocytes were obtained by enzymatic dissociation as described before (AfCS Procedure Protocol PP00000125). Briefly, isolated hearts were placed in a Langendorff apparatus for perfusion (37°C, 3 mL/min) with a trypsin (Sigma) and liberase (Roche Applied Science) solution. After that, ventricles were cut into small pieces and gently minced with a Pasteur pipette. The concentration of Ca2+ in solution was increased slowly up to 1 mM. The suspension of cells was placed on laminin-coated coverslips and stored in an incubator (5% CO2 at 37°C).
Size Measurement of the Isolated Cardiomyocytes
Adult cardiomyocyte size was assessed with a Coulter Counter Z2 (Beckman Coulter) equipped with a 200 μm aperture and data analyzed with Accucomp software (Beckman Coulter). 20% of the isolated cells from one heart were suspended in 2 mL medium and 10 mL Beckman Isoton II diluent (Beckman Coulter) was added. The metered volume was 1 mL, main gain 32 and aperture current 0.250. From each analysis mean and median cell volumes of the cell population above the size of 20,000 fL were recorded. The cell population below 20,000 fL (equal to a spherical particle with 17 μm diameter) was contaminated with smaller cells and cell debris, and therefore discarded from the analysis (Supplementary Figure S2).
Western Blot
Frozen ventricular tissue was lysed with a TissueLyser II (Qiagen) and protein concentration of the lysate was measured with the Bio-Rad Protein Assay. Ten to fifty micrograms of protein was loaded onto an 8–12% SDS polyacrylamide gel. After gel electrophoresis, proteins were transferred to a nitrocellulose membrane (0.2 μm, Bio-Rad Laboratories), which was blocked with 5% BSA. For blotting of the large (240 kDa) Nav1.5 protein, a gradient gel (4–15%, Bio-Rad Laboratories, #456–1,083) was used and protein samples transferred to a 0.4 μm membrane. Membranes were incubated with primary antibody, after which incubation with Cy5 (GE Healthcare, PA45012) or Cy3 (PA43010)-linked secondary antibody was performed and blots visualized with the ECL Plus Detection Kit (GE Healthcare) using a Typhoon 9400 scanner (GE Healthcare). The antibodies used were: Anti-Nav1.5 (Alomone labs, ASC-005), Anti-VEGF-B (R&D Systems, Af751), and β-actin (Cell Signaling Technology, 4967). Ponceau S staining solution (Sigma-Aldrich, St. Louis, MO, USA) was used as per manufacturer's instructions to visualize total membrane protein as a loading control for the blotted Nav1.5 protein.
Metabolic Analysis of Isolated Cardiomyocytes
To assess mitochondrial respiration, a Seahorse XF24 Analyzer (Seahorse Bioscience, North Billerica, MA, USA) was used as described in the Online Data Supplement. To assess the substrate specificity (glucose vs. palmitate) of the isolated cardiomyocytes, the assay buffer was supplemented with 200 μM sodium palmitate (Sigma-Aldrich, St. Louis, MO, USA) conjugated to fatty acid-free bovine serum albumin (BSA, Sigma-Aldrich, St. Louis, MO, USA; Belke et al., 1999) or with 25 mM glucose and unconjugated BSA.
Gene Expression
Primers and probes designed to detect mouse mRNA transcripts in Taqman-based RT-qPCR are listed in Supplementary Table S1.
Patch-Clamp Experiments
For individual currents or action potential measurements, an Axopatch-200B amplifier and Digidata 1440A A/D-D/A and Clampex 10 software (Axon Instruments, Sunnyvale, CA, USA) were used as previously (Karppinen et al., 2016). Previously described solutions and protocols were used for L-type Ca2+ current (Xu et al., 2011), K+ currents (Rivard et al., 2009), Na+-currents (Sato et al., 2009; Sossalla et al., 2010), and for action potential measurements (Yang et al., 2005). Late INa was estimated as integration of the current from 50 to 250 ms of the beginning of the depolarizing pulse (250 ms) without and with ranolazine (10 μM) in the bath solution. Current-voltage relations for steady-state activation and inactivation for calcium and sodium currents were determined by fitting a Boltzmann function (I/Imax = [1 + exp ((V − V1/2)/k)]−1), yielding the membrane potential of the half-maximal activation (V1/2-activation) and inactivation (V1/2-inactivation) and slope factor (k). The current recovery from inactivation curves were fitted using a single exponential function. Data analysis was made using Clampfit10 software (Molecular Devices Inc., USA).
Ca2+ Measurements
Adult cardiomyocytes were loaded with Fluo-4-acetoxymethyl (AM)-ester (10 μM; 0.02% pluronic acid, Invitrogen, Carlsbad, CA, USA), calcium signals were recorded with a confocal microscope, and data were analyzed as previously (Tavi et al., 2005; Korhonen et al., 2010).
Computer Modeling
Some of the in vitro experiments were replicated in silico to evaluate the contribution of individual VEGF-B related changes on AP morphology, using mathematical model of mouse ventricular myocyte (Koivumaki et al., 2009). The VEGF-B related modifications were implemented to mutant model variants based on experimental data, details are available in the Supplementary Methods.
Statistical Testing
Data and statistical analyses were made using Origin9 software (OriginLab Corp., Northampton, MA, USA). For statistical analysis, one-way ANOVA with Fisher's post-hoc comparison was performed for all ion currents and calcium transient frequency dependence, as well as Student's t-tests where appropriate, were applied at a level of significance of P < 0.05. Data are given as mean values ± SEM.
Results
VEGF-B Mice Display Normal Heart Function and Cardiomyocyte Hypertrophy without Metabolic Changes
VEGF-B has been shown to induce multiple adaptations in the heart, including angiogenesis and muscle growth (Bry et al., 2014). According to echocardiographic analysis, the VEGF-B mice had normal systolic and diastolic heart function and mild signs of cardiac hypertrophy (Figure 1). However, there was a significant increase in the volume and cell capacitance of isolated VEGF-B cardiomyocytes (Figure 1B, Supplementary Figure S2). This was accompanied with increased expression of the hypertrophy markers Nppb and Acta1 (Figure 1C), indicating an induction of hypertrophic pathways. Cardiac VEGF-B overexpression in rats has been shown to induce metabolic changes with decreased expression of fatty acid oxidation genes and increased concentration of intermediates of glucose metabolism (Kivela et al., 2014). We therefore assessed the oxidative and anaerobic metabolism and substrate preference of isolated cardiomyocytes with a Seahorse extracellular flux analyser. Extracellular acidification (ECAR), which reflects glycolysis rate, did not differ between the TG and WT cardiomyocytes and neither was there a difference in oxygen consumption (OCR) between the groups (Figure 2A). Moreover, we did not find changes in the energy substrate preferences; basal and maximal ECAR and OCR were similar in WT and TG animals when the energy source was either glucose or fatty acid (palmitate, Figure 2A). To assess the effect of energy substrate on the contractile function of the heart, we fed Langendorff-perfused hearts either glucose or palmitate and measured LV function. We saw no difference between TG and WT LV function with either glucose or palmitate (Figure 2B). In line with these findings, expression of genes related to cellular and mitochondrial energy metabolism was similar in TG and WT hearts (Figure 2C). Collectively, our data suggest that in isolated TG cardiomyocytes, changes in energy metabolism are not evident. Furthermore, the VEGF-B induced metabolic changes do not involve changes in substrate uptake, as suggested earlier (Kivela et al., 2014).
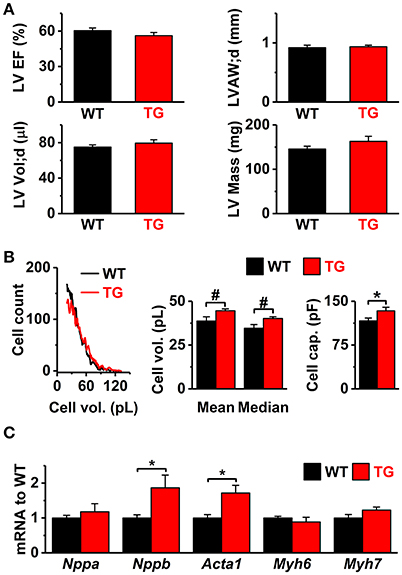
Figure 1. In vivo cardiac function, cell size, and gene expression of hypertrophy markers in VEGF-B mice. (A) In vivo echocardiography data of left ventricle (LV): ejection fraction (LV EF%, upper left), diastolic anterior wall thickness (LVAW;d, upper right), diastolic volume (LV Vol;d, lower left), and mass (LV Mass, lower right, n = 10 for both groups). (B) Size of the isolated cardiomyocytes. Left panel: representative Coulter Counter histograms. Middle panel: average of means and medians from populations of cells isolated from hearts (WT; six hearts, 4152 ± 1770 cells/analysis TG; seven hearts, 6,040 ± 1,533 cells/analysis). Right panel: means of cell capacitances measured in whole-cell experiments (WT n = 8/51 and TG n = 10/58; animals/cells). (C) Gene markers of cardiac hypertrophy [mRNA to WT ratio: natriuretic peptide A (Nppa), natriuretic peptide B (Nppb), skeletal actin (Acta1), myosin heavy polypeptide 6 (Myh6), and myosin heavy polypeptide 7 (Myh7); WT n = 7 and TG n = 6]. *P < 0.05, #P < 0.001.
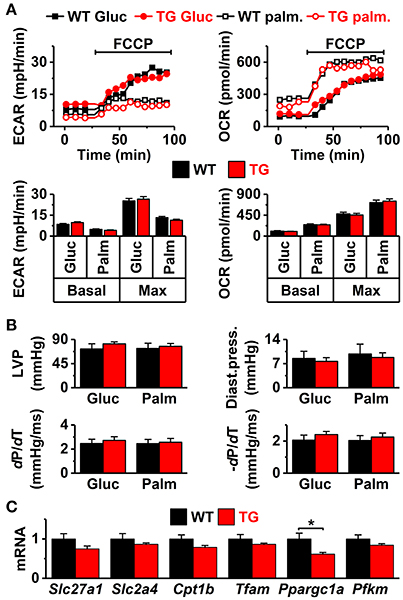
Figure 2. Cell energy metabolism and ex vivo cardiac function. (A) Extracellular acidification and oxygen consumption in isolated cardiomyocytes supplemented with either glucose (Gluc) or palmitate (Palm). Top panels: representative plots of extracellular acidification rate (ECAR, left) and oxygen consumption rate (OCR, right). Bottom panels: basal and FCCP-induced maximal ECAR (left) and OCR [right, n = 35 wells with ~2,000 cells each, from seven (7 WT and 7 TG mice) independent paired assays]. (B) Ex vivo systolic heart function in glucose- (Gluc) and palmitate-based (Palm) solutions. Systolic pressure (LVP, upper left), diastolic pressure (Diast.press., upper right), systole rise slope (dP/dT, lower left), and systole decay slope (−dP/dT, lower right, n = 6 in both groups). (C) Ventricular expression of the genes (normalized to WT) related to energy metabolism: fatty acid transporter (Slc27a1), glucose transporter (Slc2a4), carnitine palmitoyltransferase 1B (Cpt1b), mitochondrial transcription factor A (Tfam), muscle phosphofructokinase (Pfkm), and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Ppargc1a) (WT n = 7 and TG n = 6) *P < 0.05.
VEGF-B Overexpression Augments Ca2+ Signals in Ventricular Myocytes
Next, we studied if VEGF-B overexpression affects the [Ca2+]i signaling of the myocytes. To challenge the cells, we first stimulated Fluo-4-loaded isolated cardiomyocytes at different frequencies (0.5–10 Hz, Figure 3A). The amplitude of [Ca2+]i transients was higher in TG cardiomyocytes at low stimulus frequencies (0.5–2 Hz, Figure 3A). The time constant of [Ca2+]i transient decay, which indicates calcium extrusion rate, was shorter but only at the lowest pacing frequency (0.5 Hz, Figure 3A). SR Ca2+ loading, estimated by rapid application of caffeine (10 mM; Figure 3B) was higher in TG myocytes (Figure 3B). While VEGF-B TG myocytes showed increased [Ca2+]i transient amplitude and SR-load, the fractional release, assessed by the ratio of stimulus- and caffeine-induced [Ca2+]i transients, was not altered in TG vs. WT myocytes. These changes were accompanied with downregulation of Atp2a2 (Serca2a) expression, while Atp2a2/phospholamban ratio was not altered (Supplementary Figure S3B). Moreover, caffeine induced [Ca2+]i transient was shorter in TG animals (Figure 3B) which could reflect increased INCX. Taken together, although VEGF-B TG myocytes show augmented calcium transients (CaTs) and increased SR load, these changes are not due to extensive remodeling and are likely to have a relatively small impact on cardiomyocyte function at physiological heart rates in vivo.
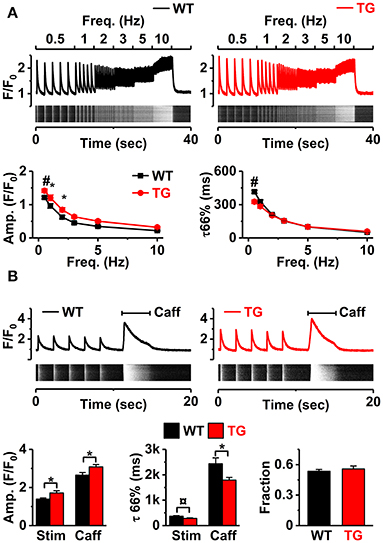
Figure 3. Ca2+ handling in VEGF-B TG ventricular myocytes. (A) Representative [Ca2+]i transients (line-scans) and their profiles in Fluo4-loaded cardiomyocytes upon pacing at different stimulus frequencies in WT and TG mice (upper), frequency dependence of amplitudes (lower left), and time constants (lower right) of the [Ca2+]i transients (n = 16 myocytes for WT and n = 22 myocytes for TG, at least three animals were used for both groups). (B) Representative line-scans and profiles of stimulus- and caffeine-induced [Ca2+]i pulses in WT (upper left) and TG mice (upper right). Amplitudes (lower left) and time constant of decays (lower middle) of stimulus- (Stim) and caffeine-induced (Caff) transients, and fractional release (lower right, WT n = 23 myocytes and TG n = 26 myocytes, at least three animals were used for both groups). *P < 0.05, ¤P < 0.01 and #P < 0.001.
VEGF-B Induces Alterations of Electrophysiological Properties of the Mouse Heart
VEGF-B deficiency has been shown to interfere with atrial conduction in mice (Aase et al., 2001), indicating that VEGF-B regulates mechanisms maintaining electrophysiological properties of the heart. Thus, VEGF-B overexpression might result in remodeling of electrical properties of myocardium. Comparison of the ECGs of WT and VEGF-B TG mice indicated drastic changes in the ECG waveform of TG hearts (Figures 4A,B). Specifically, TG hearts had decreased amplitudes of R- and S-waves accompanied by an increase in the QRSp time which reveals early repolarization in the heart (Figure 4B), whereas QRS width was not changed. ECG changes, such as those seen in VEGF-B TG hearts, can be induced by changes in the electrical properties of myocytes or passive properties of the heart tissue (Dhein et al., 2014). VEGF-B TG hearts had no signs of fibrosis based on LV relaxation (Figure 2B) and histology (data not shown) or changes in the expression patterns of connexin isoforms (Supplementary Figure S3A), but isolated ventricular myocytes had significant changes in action potential (AP) waveform. However, TG myocytes had no significant changes in APD90 they had increased duration of AP at 60 and 70% of repolarization (~40 and ~45% in APD60 and APD70, respectively; Figures 4C,D). Furthermore, TG myocytes had an increase in the rise time and slowing of maximum rise slope of the AP upstroke (Figure 4C). These results suggest that ECG changes are due to remodeling of the electrical properties of the myocytes involving both depolarizing and repolarizing currents.
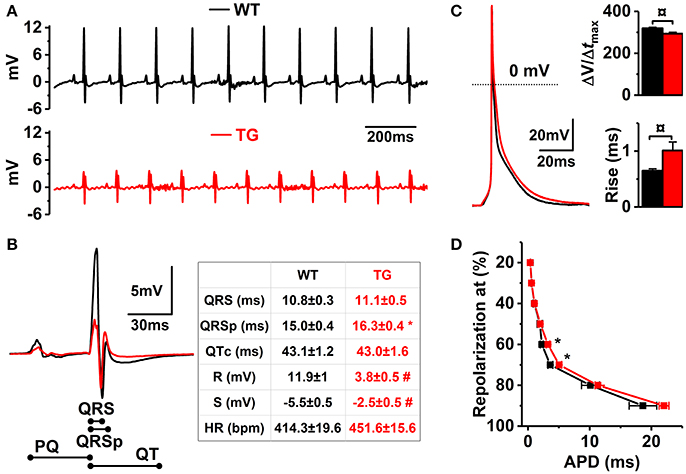
Figure 4. Electrophysiological properties of VEGF-B TG hearts and isolated cardiomyocytes. (A) Representative ECG recordings from WT (black line) and VEGF-B TG (red line) mice. (B) Enlarged superimposed ECG complexes (left panel) with summary of the ECG parameters (right panel, n = 10 for both groups). (C) Representative superimposed AP traces from isolated myocytes (left) and means of the AP maximum rise slope (upper right) and rise time (lower right). (D) AP durations at different percentages of repolarization (WT n = 17 and TG n = 34; 4 animals for both group). *P < 0.05, ¤P < 0.01 and #P < 0.001.
VEGF-B TG Myocytes Have Depressed INa
To specify the ion current changes causing ECG and AP changes in VEGF-B TG hearts, we first measured L-type Ca2+ channel current (ICaL) from cardiomyocytes isolated from TG and WT hearts. We saw no differences in the I–V-dependence, density, activation/inactivation, or expression of the Cacna1c gene encoding the pore-forming subunit of the L-type channel (Figure 5A, Supplementary Table S2). In contrast to ICaL the other major depolarizing current, INa was significantly decreased in VEGF-B TG myocytes (Figure 5B). The decrease of the INa density was accompanied with downregulation of the Scn5a mRNA and Nav1.5 protein (Figure 5C). Moreover, INa from VEGF-B TG myocytes showed a rightward shift in both voltage-dependent activation and inactivation (Figure 5D, Supplementary Table S3) without a change in recovery time after inactivation (Figure 5D, Supplementary Table S3). Another property of Na-channels subject to modifications is the slowly inactivating component (Gintant et al., 1984), also known as the late Na-current. Since the late INa has been linked to ECG changes such as those seen in VEGF-B mice (Moreno and Clancy, 2012), we assessed the INa-L in VEGF-B TG myocytes. However, we did not find differences in late INa between WT and TG myocytes either with INa-L blocker ranolazine or by a voltage clamp protocol specific for late Na+-current even in condition when physiological [Na+]out concentration was used (Supplementary Figure S4). Altogether, decreased INa gives a reasonable explanation for the ECG anomalies (Figures 4A,B) and decrease in the AP upstroke velocity (Figure 4C).
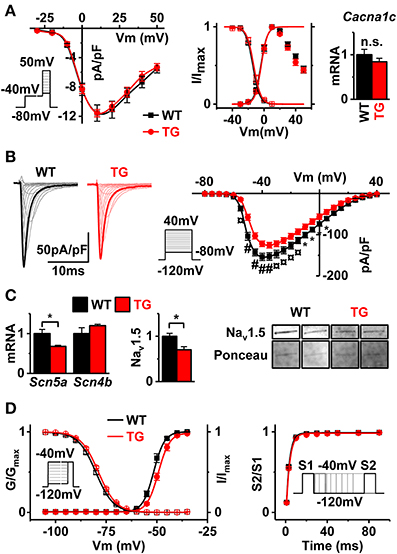
Figure 5. Depolarizing currents in VEGF-B TG myocytes. (A) I–V curves of ICaL densities (left panel, WT n = 9 and TG n = 11, myocytes from three animals in both groups), voltage-dependent activation/inactivation of ICaL (middle panel, Boltzmann fitting) and gene expression of α-subunit calcium channel (Cacna1c, right panel). (B) Typical sodium channel current traces (left panel) and I–V curves (right panel) (WT black line, n = 19 and TG red line n = 14 myocytes from 3 mice were used for both groups). (C) Gene expression of sodium channel subunits Scn5a and Scn4b (left, WT n = 7 and TG n = 6) and Western blot of Nav1.5 from membrane protein fraction. Nav1.5 western blot signal normalized to Ponceau stain (middle, WT n = 3, TG n = 4). Representative SCN5a bands and Ponceau stain from the same region of the membrane (right). (D) Voltage-dependent activation and inactivation of sodium channels (left panel) and recovery time from inactivation (right panel) (see Table S3 for fitting results, WT n = 19 myocytes and TG n = 15 myocytes from three animals in both groups). Refer to insets for voltage-step protocols. *P < 0.05, ¤P < 0.01 and #P < 0.001.
VEGF-B Affects the Expression and Density of Potassium Currents
In general, the AP repolarization is determined by the potassium currents, of which transient outward and delayed rectifier currents are the most prominent ones in mouse ventricular myocytes (Nerbonne, 2004). While there was relatively small change in the total K+ current (~18%; Figure 6A), VEGF-B overexpression shown drastic decreasing of transient outward current (Ito, Figure 6B). Moreover, decreasing of Ito in VEGF-B animals was accompanied with significant acceleration of inactivation of this current (by ~1.7-fold). The ultra-rapid component of delayed rectifier current as well as IK1 and IKss were unaffected (IKur, IK1, and IKss Figures 6C,D). Channels responsible for fast and slow Ito are assembled by α-subunits Kv4.2 and Kv1.4, encoded by genes Kcna4 and Kcnd2, respectively (Nerbonne, 2004). However, expression of these genes was not different between WT and TG myocytes; instead, we saw downregulation of Kcnip2 Kv channel-interacting protein 2 (ß-subunit), a known modulatory subunit of the Kv4.2/3 channel (Figure 6E). This indicates that VEGF-B regulates AP repolarization partly by affecting the expression of potassium channel subunits and accessory proteins.
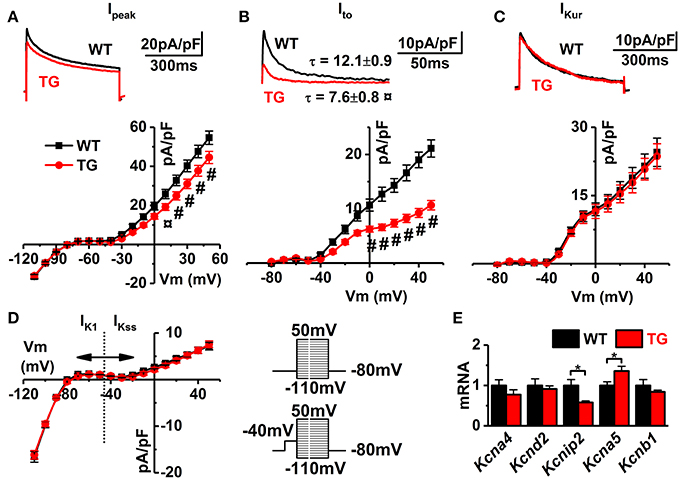
Figure 6. Voltage-gated potassium currents. Representative superimposed original traces (upper, obtained at + 50 mV) and I–V curves (lower) of (A) total potassium (Ipeak), (B) transient outward (Ito), and (C) ultra-rapid (IKur) currents. (D) I–V curves of K1 and steady state currents (IK1/IKss, WT black and TG red lines, WT n = 8 and TG n = 17; from four animals). Refer to insets for voltage-step protocols (upper without and lower with inactivation prepulse). (E) Gene expressions of potassium channel subunits (WT n = 7 and TG n = 6). *P < 0.05, ¤P < 0.01 and #P < 0.001.
Effects of VEGF-B-Related Alterations on Electrophysiology
To elucidate the isolated contribution of VEGF-B-related modifications of INa, Ito, and INCX on electrophysiology (Figures 5, 6), we replicated the AP experiments (Figure 4) in silico, together with the effect of EGTA Ca2+ buffer used in experiments (Figure 7A), using a computational model of mouse ventricular myocyte (Koivumaki et al., 2009). Model simulations recapitulated well the emergent changes in the AP morphology (Figure 7B). As the comparison of simulated APs shows (Figure 7B), the sodium and potassium channel modifications partially cancel out each other, and thus the sum effect on repolarization is smaller in the virtual VEGF-B myocyte. The reduced INa (Figure 7C) lead to a 12.0% decrease in maximum upstroke velocity of the AP. The indirect effect of slower AP repolarization is that the inactivation of IKss, ICaL, and IKur is decelerated (Figure 7D).
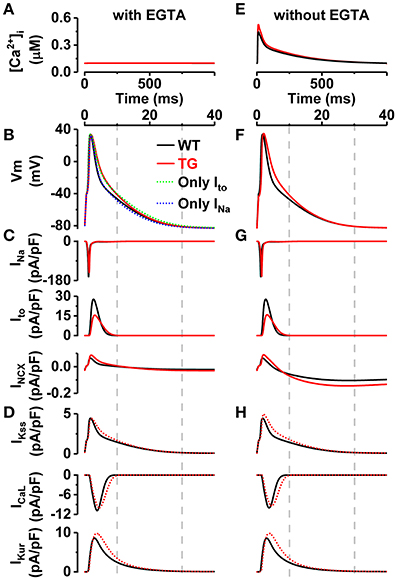
Figure 7. Effect of VEGF-B-related electrophysiological alterations explored in silico. From top to the bottom: (A,E) [Ca2+]i transients, (B,F) action potentials, (C,G) currents introduced into model INa, Ito, INCX (solid red lines), and (D,H) resulting currents IKss, ICaL, IKur (dotted red lines) in simulated pacing experiments, in the presence (A–D) and absence (E–H) of EGTA. Note, the different time scale in [Ca2+]i transients panels. The VEGF-B TG model variant (red lines) accounts for the (1) decreased INa (−18%), (2) reduced Ito (conductance −49%, and inactivation rate +59%), and (3) increased NCX current (+36%) as found in experiments.
Furthermore, we studied the possible interdependence of AP changes and the apparent changes in CaT amplitude with simulations of more physiological conditions (without EGTA; Figure 7E). While the enhanced NCX activity (Figure 7G) should result in increased Ca2+ extrusion and thus reduced CaT amplitude, the simulations demonstrate that due the reduced Ito the initial repolarization phase is much slower (Figure 7F), which decelerates the inactivation of ICaL (Figure 7H) and thus increases the integral of Ca2+ influx by +39%. As a result, the CaT amplitude is increased by 24% (Figure 7E).
Discussion
Here, we studied the effect of VEGF-B overexpression in mouse heart at the level of cardiac myocytes. We report that cardiac-specific overexpression of VEGF-B induces mild changes in cardiomyocyte calcium signaling and growth without pathological metabolic remodeling. Interestingly, our results show that VEGF-B induces marked changes in the electrophysiological properties of cardiomyocytes, including changes in densities of both depolarizing and repolarizing currents, ECG, and action potential parameters.
Particular changes in mouse ECG complex correlate with certain cardiac conditions such as aging, infarction, and LV hypertrophy (LVH) (Merentie et al., 2015); these changes can be used to estimate disease progression (Sysa-Shah et al., 2015). VEGF-B TG hearts show two distinct ECG changes. First is increase in the width of the QRSp which together with unaffected QRS represents prolongation of early repolarization (Liu et al., 2004; Boukens et al., 2013, 2014). That prolongation fits very good to lengthening of APD60–70 which we observed in TG cardiomyocytes. That change of QRSp is common to LVH (Merentie et al., 2015) and some forms of sodium channelopathies (Remme et al., 2006; Watanabe et al., 2011). And second is decreased amplitude of the R and S waves, which has been shown to associate with acute infarction or ischemia in mouse heart (Gehrmann et al., 2001; Merentie et al., 2015) and e.g., sodium channel blocker Ajmaline (Boukens et al., 2013). VEGF-B TG hearts show only modest signs of organ level hypertrophy and no infarctions, so that the ECG anomalies cannot be considered as signs of cardiac disease, but they rather reflect more specific changes in cardiomyocytes electrophysiology. Interpretation of mouse ECG is not straightforward (Boukens et al., 2014), but ECG changes of the VEGF-B mice shown here suggest changes in both depolarizing and repolarizing currents, which predispose the heart to arrhythmias upon stress. Cell level electrophysiological changes such as AP prolongation predispose cardiomyocytes to afterdepolarization and triggered arrhythmias (Janse, 2004; Aiba and Tomaselli, 2010). Consequently, APD changes are common findings in heart diseases, such as heart failure and hypertrophy (Wickenden et al., 1998), and in a variety genetic mouse models of human long QT syndrome (Salama and London, 2007). Due to short APD in mouse ventricular cardiomyocytes, the depolarizing currents such as L-type Ca2+ current contribute less to APD than in species with long AP, such as humans. Instead, mouse AP repolarization is dominated by potassium currents, especially transient outward currents (Ito,f, Ito,s) (Nerbonne, 2004). VEGF-B TG myocytes have an increase in APD due to decreased Ito. At the level of K+ channel transcripts, TG myocytes have suppressed expression of Kcnip2. Although translation of ion channel transcripts to functional channels is subject to various post-translational modifications and multiple regulative steps in channels assembly, the protein resulting from Kcnip2 transcript has been shown to be a regulator of Ito (Kuo et al., 2001) which is in line with our findings and corresponds well with the previous findings from Kcnip2−/− mice (Thomsen et al., 2009).
Some of the ECG and action potential findings from VEGF-B TG hearts cannot be solely explained by the changes in potassium currents. These include a decrease in the amplitude of the ECG complex (R and S waves), increased duration of the QRSp, and the absence of QT lengthening usually associated with an increase in APD. These changes suggested that the depolarization phase of the ventricles of VEGF-B mice is also affected and the ECG changes reflect combinatory effects of both depolarizing and repolarizing currents of the cardiomyocytes. In line with this, action potentials of VEGF-B mouse ventricular myocytes not only had increased duration, but also a decrease in maximum upstroke velocity as well as an increase in the rise time of the initial depolarization. The upstroke phase of the AP is dependent on the Na+-current density and therefore it was not a surprise that it was accompanied by a reduction in INa density and a slight rightward shift in the activation-inactivation curve of the INa in TG cardiomyocytes. Unlike the potassium channel remodeling, INa changes are not commonly associated with cardiac hypertrophy or cardiac failure progression (Hill, 2003). Interestingly, according to previous data from mouse models where the INa downregulation occur as a secondary effect (Gavillet et al., 2006; Hesse et al., 2007; Cerrone et al., 2012; Rizzo et al., 2012; Han et al., 2015), or as a result of expression of mutated Nav1.5 (Remme et al., 2006; Abriel, 2007; Watanabe et al., 2011), the electrophysiological findings in mouse heart are similar to those in VEGF-B TG mice. Prototypically these include decreases in the QRS complex amplitude, reduced upstroke velocity of the AP, and reduced conduction velocity. Therefore, it appears that the electrophysiological phenotype of the VEGF-B TG hearts is greatly impacted by the downregulation of the Na+-current. According to the simulations with a mathematical model, the measured changes in Ito and INa densities together with the changes in their activation/inactivation properties explain well the measured changes in the AP shape. Interestingly, the modeling also suggests that the modest changes in calcium transient amplitude at low pacing frequencies actually results from the AP lengthening. That is, slower initial AP repolarization decelerate the inactivation of the ICaL and consequently increases to total Ca2+ influx via ICaL during AP in VEGF-B TG myocytes.
The effects of VEGF-B on the structure and function of the heart have been studied with a wide variety of animal models and approaches (Bry et al., 2014). It has become evident that some of the VEGF-B effects show species or isoform specificity, whereas others depend on methods used for overexpression or knockdown. The TG mice analyzed in this study showed a relatively mild tendency to hypertrophy, indicating that the effect of VEGF-B on cardiac phenotype is graded, producing milder changes with less powerful transgene expression. Collectively the existing data show that VEGF-B induces a spectrum of changes in the heart ranging from capillary density and structure and growth of the myocardium to more diffuse changes in energy substrate metabolism and signaling. Bearing this in mind it is a surprise that the cardiomyocyte-level phenotype of the VEGF-B TG mice show minimal alterations in Ca2+ signaling, contraction, or structure. Instead, TG hearts have a very distinct and specific electrophysiological phenotype originating from precise changes in ion current densities. This suggests that VEGF-B triggers pathways mediating cardiomyocyte-specific alterations in electrophysiology without directly affecting the contractile function of the whole heart.
Author Contributions
PT, JH, and NN designed the research. SY, KA, and RK provided the material. NN, JH, TT, MM, JK, and EG conducted the experiments, collected, and analyzed data. PT and NN wrote the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by the Academy of Finland (PT: #267637), Sigrid Juselius Foundation, and Finnish Foundation for Cardiovascular Research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Anne Karppinen for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphys.2017.00373/full#supplementary-material
References
Aase, K., von Euler, G., Li, X., Ponten, A., Thoren, P., Cao, R., et al. (2001). Vascular endothelial growth factor-B-deficient mice display an atrial conduction defect. Circulation 104, 358–364. doi: 10.1161/01.CIR.104.3.358
Abriel, H. (2007). Roles and regulation of the cardiac sodium channel Na v1.5: recent insights from experimental studies. Cardiovasc. Res. 76, 381–389. doi: 10.1016/j.cardiores.2007.07.019
Aiba, T., and Tomaselli, G. F. (2010). Electrical remodeling in the failing heart. Curr. Opin. Cardiol. 25, 29–36. doi: 10.1097/HCO.0b013e328333d3d6
Belke, D. D., Larsen, T. S., Lopaschuk, G. D., and Severson, D. L. (1999). Glucose and fatty acid metabolism in the isolated working mouse heart. Am. J. Physiol. 277(4 Pt 2), R1210–R1217.
Bell, R. M., Mocanu, M. M., and Yellon, D. M. (2011). Retrograde heart perfusion: the Langendorff technique of isolated heart perfusion. J. Mol. Cell. Cardiol. 50, 940–950. doi: 10.1016/j.yjmcc.2011.02.018
Bellomo, D., Headrick, J. P., Silins, G. U., Paterson, C. A., Thomas, P. S., Gartside, M., et al. (2000). Mice lacking the vascular endothelial growth factor-B gene (Vegfb) have smaller hearts, dysfunctional coronary vasculature, and impaired recovery from cardiac ischemia. Circ. Res. 86, E29–E35. doi: 10.1161/01.RES.86.2.e29
Boukens, B. J., Hoogendijk, M. G., Verkerk, A. O., Linnenbank, A., van Dam, P., Remme, C. A., et al. (2013). Early repolarization in mice causes overestimation of ventricular activation time by the QRS duration. Cardiovasc. Res. 97, 182–191. doi: 10.1093/cvr/cvs299
Boukens, B. J., Rivaud, M. R., Rentschler, S., and Coronel, R. (2014). Misinterpretation of the mouse ECG: ‘musing the waves of Mus musculus.’ J. Physiol. 592, 4613–4626. doi: 10.1113/jphysiol.2014.279380
Bry, M., Kivela, R., Holopainen, T., Anisimov, A., Tammela, T., Soronen, J., et al. (2010). Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation 122, 1725–1733. doi: 10.1161/CIRCULATIONAHA.110.957332
Bry, M., Kivela, R., Leppanen, V. M., and Alitalo, K. (2014). Vascular endothelial growth factor-B in physiology and disease. Physiol. Rev. 94, 779–794. doi: 10.1152/physrev.00028.2013
Cerrone, M., Noorman, M., Lin, X., Chkourko, H., Liang, F. X., van der Nagel, R., et al. (2012). Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc. Res. 95, 460–468. doi: 10.1093/cvr/cvs218
Dhein, S., Seidel, T., Salameh, A., Jozwiak, J., Hagen, A., Kostelka, M., et al. (2014). Remodeling of cardiac passive electrical properties and susceptibility to ventricular and atrial arrhythmias. Front. Physiol. 5:424. doi: 10.3389/fphys.2014.00424
Gavillet, B., Rougier, J. S., Domenighetti, A. A., Behar, R., Boixel, C., Ruchat, P., et al. (2006). Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ. Res. 99, 407–414. doi: 10.1161/01.RES.0000237466.13252.5e
Gehrmann, J., Frantz, S., Maguire, C. T., Vargas, M., Ducharme, A., Wakimoto, H., et al. (2001). Electrophysiological characterization of murine myocardial ischemia and infarction. Basic Res. Cardiol. 96, 237–250. doi: 10.1007/s003950170054
Gintant, G. A., Datyner, N. B., and Cohen, I. S. (1984). Slow inactivation of a tetrodotoxin-sensitive current in canine cardiac Purkinje fibers. Biophys. J. 45, 509–512. doi: 10.1016/S0006-3495(84)84187-9
Hagberg, C. E., Falkevall, A., Wang, X., Larsson, E., Huusko, J., Nilsson, I., et al. (2010). Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 464, 917–921. doi: 10.1038/nature08945
Han, Z., Jiang, Y., Yang, Y., Li, X., Yang, Z., Cao, K., et al. (2015). Deletion of PDK1 causes cardiac sodium current reduction in mice. PLoS ONE 10:e0122436. doi: 10.1371/journal.pone.0122436
Hesse, M., Kondo, C. S., Clark, R. B., Su, L., Allen, F. L., Geary-Joo, C. T., et al. (2007). Dilated cardiomyopathy is associated with reduced expression of the cardiac sodium channel Scn5a. Cardiovasc. Res. 75, 498–509. doi: 10.1016/j.cardiores.2007.04.009
Hill, J. A. (2003). Electrical remodeling in cardiac hypertrophy. Trends Cardiovasc. Med. 13, 316–322. doi: 10.1016/j.tcm.2003.08.002
Huusko, J., Lottonen, L., Merentie, M., Gurzeler, E., Anisimov, A., Miyanohara, A., et al. (2012). AAV9-mediated VEGF-B gene transfer improves systolic function in progressive left ventricular hypertrophy. Mol. Ther. 20, 2212–2221. doi: 10.1038/mt.2012.145
Janse, M. J. (2004). Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc. Res. 61, 208–217. doi: 10.1016/j.cardiores.2003.11.018
Karpanen, T., Bry, M., Ollila, H. M., Seppanen-Laakso, T., Liimatta, E., Leskinen, H., et al. (2008). Overexpression of vascular endothelial growth factor-B in mouse heart alters cardiac lipid metabolism and induces myocardial hypertrophy. Circ. Res. 103, 1018–1026. doi: 10.1161/CIRCRESAHA.108.178459
Karppinen, S., Rapila, R., Naumenko, N., Tuomainen, T., Koivumäki, J. T., Hänninen, S. L., et al. (2016). Ca2+-activated K+ current is essential for maintaining excitability and gene transcription in early embryonic cardiomyocytes. Acta Physiol. 216, 101–111. doi: 10.1111/apha.12540
Kivela, R., Bry, M., Robciuc, M. R., Rasanen, M., Taavitsainen, M., Silvola, J. M., et al. (2014). VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol. Med. 6, 307–321. doi: 10.1002/emmm.201303147
Koivumaki, J. T., Korhonen, T., Takalo, J., Weckstrom, M., and Tavi, P. (2009). Regulation of excitation-contraction coupling in mouse cardiac myocytes: integrative analysis with mathematical modelling. BMC Physiol. 9:16. doi: 10.1186/1472-6793-9-16
Korhonen, T., Rapila, R., Ronkainen, V. P., Koivumaki, J. T., and Tavi, P. (2010). Local Ca2+ releases enable rapid heart rates in developing cardiomyocytes. J. Physiol. 588(Pt 9), 1407–1417. doi: 10.1113/jphysiol.2009.185173
Kuo, H. C., Cheng, C. F., Clark, R. B., Lin, J. J., Lin, J. L., Hoshijima, M., et al. (2001). A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell 107, 801–813. doi: 10.1016/S0092-8674(01)00588-8
Liu, G., Iden, J. B., Kovithavongs, K., Gulamhusein, R., Duff, H. J., and Kavanagh, K. M. (2004). In vivo temporal and spatial distribution of depolarization and repolarization and the illusive murine T wave. J. Physiol. 555(Pt 1), 267–279. doi: 10.1113/jphysiol.2003.054064
Makinen, T., Olofsson, B., Karpanen, T., Hellman, U., Soker, S., Klagsbrun, M., et al. (1999). Differential binding of vascular endothelial growth factor B splice and proteolytic isoforms to neuropilin-1. J. Biol. Chem. 274, 21217–21222. doi: 10.1074/jbc.274.30.21217
Merentie, M., Lipponen, J. A., Hedman, M., Hedman, A., Hartikainen, J., Huusko, J., et al. (2015). Mouse ECG findings in aging, with conduction system affecting drugs and in cardiac pathologies: development and validation of ECG analysis algorithm in mice. Physiol. Rep. 3:e12639. doi: 10.14814/phy2.12639
Mitchell, G. F., Jeron, A., and Koren, G. (1998). Measurement of heart rate and Q-T interval in the conscious mouse. Am. J. Physiol. 274(3 Pt 2), H747–H751.
Moreno, J. D., and Clancy, C. E. (2012). Pathophysiology of the cardiac late Na current and its potential as a drug target. J. Mol. Cell. Cardiol. 52, 608–619. doi: 10.1016/j.yjmcc.2011.12.003
Nerbonne, J. M. (2004). Studying cardiac arrhythmias in the mouse–a reasonable model for probing mechanisms? Trends Cardiovasc. Med. 14, 83–93. doi: 10.1016/j.tcm.2003.12.006
Olofsson, B., Korpelainen, E., Pepper, M. S., Mandriota, S. J., Aase, K., Kumar, V., et al. (1998). Vascular endothelial growth factor B (VEGF-B) binds to VEGF receptor-1 and regulates plasminogen activator activity in endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 95, 11709–11714. doi: 10.1073/pnas.95.20.11709
Remme, C. A., Verkerk, A. O., Nuyens, D., van Ginneken, A. C., van Brunschot, S., Belterman, C. N., et al. (2006). Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation 114, 2584–2594. doi: 10.1161/CIRCULATIONAHA.106.653949
Rivard, K., Trepanier-Boulay, V., Rindt, H., and Fiset, C. (2009). Electrical remodeling in a transgenic mouse model of alpha1B-adrenergic receptor overexpression. Am. J. Physiol. Heart Circ. Physiol. 296, H704–H718. doi: 10.1152/ajpheart.00337.2008
Rizzo, S., Lodder, E. M., Verkerk, A. O., Wolswinkel, R., Beekman, L., Pilichou, K., et al. (2012). Intercalated disc abnormalities, reduced Na+ current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc. Res. 95, 409–418. doi: 10.1093/cvr/cvs219
Salama, G., and London, B. (2007). Mouse models of long QT syndrome. J. Physiol. 578(Pt 1), 43–53. doi: 10.1113/jphysiol.2006.118745
Sato, P. Y., Musa, H., Coombs, W., Guerrero-Serna, G., Patino, G. A., Taffet, S. M., et al. (2009). Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes ina. Circ. Res. 105, 523–526. doi: 10.1161/CIRCRESAHA.109.201418
Sossalla, S., Kallmeyer, B., Wagner, S., Mazur, M., Maurer, U., Toischer, K., et al. (2010). Altered Na+ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 55, 2330–2342. doi: 10.1016/j.jacc.2009.12.055
Sysa-Shah, P., Sorensen, L. L., Abraham, M. R., and Gabrielson, K. L. (2015). Electrocardiographic characterization of cardiac hypertrophy in mice that overexpress the ErbB2 receptor tyrosine kinase. Comp. Med. 65, 295–307.
Tavi, P., Hansson, A., Zhang, S. J., Larsson, N. G., and Westerblad, H. (2005). Abnormal Ca2+ release and catecholamine-induced arrhythmias in mitochondrial cardiomyopathy. Hum. Mol. Genet. 14, 1069–1076. doi: 10.1093/hmg/ddi119
Thomsen, M. B., Sosunov, E. A., Anyukhovsky, E. P., Ozgen, N., Boyden, P. A., and Rosen, M. R. (2009). Deleting the accessory subunit KChIP2 results in loss of I(to,f) and increased I(K,slow) that maintains normal action potential configuration. Heart Rhythm 6, 370–377. doi: 10.1016/j.hrthm.2008.11.023
Vuorio, T., Jauhiainen, S., and Yla-Herttuala, S. (2012). Pro- and anti-angiogenic therapy and atherosclerosis with special emphasis on vascular endothelial growth factors. Expert Opin. Biol. Ther. 12, 79–92. doi: 10.1517/14712598.2012.641011
Watanabe, H., Yang, T., Stroud, D. M., Lowe, J. S., Harris, L., Atack, T. C., et al. (2011). Striking in vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation 124, 1001–1011. doi: 10.1161/CIRCULATIONAHA.110.987248
Wickenden, A. D., Kaprielian, R., Kassiri, Z., Tsoporis, J. N., Tsushima, R., Fishman, G. I., et al. (1998). The role of action potential prolongation and altered intracellular calcium handling in the pathogenesis of heart failure. Cardiovasc. Res. 37, 312–323. doi: 10.1016/S0008-6363(97)00256-3
Xu, L., Li, X. Y., Liu, Y., Li, H. T., Chen, J., Li, X. Y., et al. (2011). The mechanisms underlying ICa heterogeneity across murine left ventricle. Mol. Cell. Biochem. 352, 239–246. doi: 10.1007/s11010-011-0759-8
Keywords: cardiac myocytes, growth factors, EC-coupling, energy metabolism, ion channels, action potential
Citation: Naumenko N, Huusko J, Tuomainen T, Koivumäki JT, Merentie M, Gurzeler E, Alitalo K, Kivelä R, Ylä-Herttuala S and Tavi P (2017) Vascular Endothelial Growth Factor-B Induces a Distinct Electrophysiological Phenotype in Mouse Heart. Front. Physiol. 8:373. doi: 10.3389/fphys.2017.00373
Received: 20 January 2017; Accepted: 18 May 2017;
Published: 31 May 2017.
Edited by:
Antonius Baartscheer, University of Amsterdam, NetherlandsReviewed by:
Toon Van Veen, Utrecht University, NetherlandsDirk Snyders, University of Antwerp, Belgium
Marcella Rocchetti, University of Milano-Bicocca, Italy
Copyright © 2017 Naumenko, Huusko, Tuomainen, Koivumäki, Merentie, Gurzeler, Alitalo, Kivelä, Ylä-Herttuala and Tavi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pasi Tavi, pasi.tavi@uef.fi
†These authors have contributed equally to this work.