 Fei Wang
Fei Wang Masayo Koide
Masayo Koide George C. Wellman
George C. Wellman
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 07 April 2017
Sec. Vascular Physiology
Volume 8 - 2017 | https://doi.org/10.3389/fphys.2017.00210
Voltage-dependent calcium channels (VDCCs) play an essential role in regulating cerebral artery diameter and it is widely appreciated that the L-type VDCC, CaV1.2, encoded by the CACNA1C gene, is a principal Ca2+ entry pathway in vascular myocytes. However, electrophysiological studies using 10 mM extracellular barium ([Ba2+]o) as a charge carrier have shown that ~20% of VDCC currents in cerebral artery myocytes are insensitive to 1,4-dihydropyridine (1,4-DHP) L-type VDDC inhibitors such as nifedipine. Here, we investigated the hypothesis that the concentration of extracellular divalent cations can influence nifedipine inhibition of VDCC currents. Whole-cell VDCC membrane currents were obtained from freshly isolated rat cerebral artery myocytes in extracellular solutions containing Ba2+ and/or Ca2+. In the absence of [Ca2+]o, both nifedipine-sensitive and -insensitive calcium currents were observed in 10 mM [Ba2+]o. However, VDCC currents were abolished by nifedipine when using a combination of 10 mM [Ba2+]o and 100 μM [Ca2+]o. VDCC currents were also completely inhibited by nifedipine in either 2 mM [Ba2+]o or 2 mM [Ca2+]o. The biophysical characteristics of all recorded VDCC currents were consistent with properties of a high-voltage activated VDCC, such as CaV1.2. Further, VDCC currents recorded in 10 mM [Ba2+]o ± 100 μM [Ca2+]o or 2 mM [Ba2+]o exhibited similar sensitivity to the benzothiazepine L-type VDCC blocker, diltiazem, with complete current inhibition at 100 μM. These data suggest that nifedipine inhibition is influenced by both Ca2+ binding to an external site(s) on these channels and surface charge effects related to extracellular divalent cations. In sum, this work demonstrates that the extracellular environment can profoundly impact VDCC current measurements.
Members of the voltage-dependent calcium channel (VDCC) family are involved in a multitude of physiological processes throughout the body. The classification of VDCC subtypes is based on the amino acid sequence of the channels' pore-forming alpha1-subunit, which principally determines gating properties such as the voltage dependence of channel activation/inactivation and sensitivity to pharmacological agents (Catterall et al., 2003; Alexander et al., 2015). It is widely appreciated that the L-type VDCC, CaV1.2, encoded by the gene CACNA1C, represents an important extracellular calcium ([Ca2+]o) entry pathway involved in the regulation of bulk cytosolic Ca2+ and contraction of vascular smooth muscle, including cerebral artery myocytes (Moosmang et al., 2003; Nystoriak et al., 2009). For example, dynamic CaV1.2 gating in response to pressure-dependent changes in smooth muscle membrane potential plays a central role in modulating cerebral artery diameter and the autoregulation of cerebral blood flow (Knot and Nelson, 1998; Davis and Hill, 1999; Nystoriak et al., 2011). Further, 1,4-dihydropyridines (1,4-DHPs), a class of compounds including nifedipine and nimodipine, that are selective inhibitors of L-type VDCCs have been used extensively in the treatment of a wide range of cardiovascular disorders, including protection against cerebral vasospasm following subarachnoid hemorrhage (Wellman, 2006; Young et al., 2015).
Radioligand binding assays used to examine interactions between 1,4-DHPs and CaV1.2 have shown the binding of these two entities to be extremely dependent on the nature and concentration of divalent cations present (Ebata et al., 1990; Yamada et al., 1990; Hille, 2001; Nakajima et al., 2002). Yamada et al., using porcine coronary artery preparations, found that Ca2+ is required for high affinity binding of 1,4-DHPs to CaV1.2, whereas Ba2+ had the opposite effect, i.e., inhibition of ligand/channel binding. Additional evidence indicates that it is a Ca2+ binding site on the extracellular surface of CaV1.2 that promotes high affinity 1,4-DHP binding (Ebata et al., 1990). However, because Ba2+ permeates several types of VDCCs more effectively than Ca2+, Ba2+ has commonly been used as a charge carrier during electrophysiological recordings to enhance the amplitude of VDCC membrane currents in cells having low channel number.
Using 10 mM extracellular barium ([Ba2+]o) as a charge carrier, several groups have shown that ~20% of VDCC currents in cerebral artery myocytes are insensitive to high concentrations (1 μM) of nifedipine (Nikitina et al., 2007; Kuo et al., 2010; Abd El-Rahman et al., 2013; Harraz and Welsh, 2013a). The identity of these currents, often referred to as nifedipine-insensitive calcium currents (NICCs), is controversial and several VDCCs other than CaV1.2 have been implicated. The goal of the present study was to examine the influence of [Ba2+]o and [Ca2+]o on the efficacy of the 1,4-DHP, nifedipine, to inhibit VDCC membrane currents in freshly isolated rat cerebral artery myocytes. Our data indicate that the use of Ba2+ and/or Ca2+ as charge carriers not only influences VDCC current density, but also impacts the ability of nifedipine to inhibit these currents.
All procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (eighth edition, 2011) and followed protocols approved by the Institutional Animal Care and Use Committee at the University of Vermont.
Male Sprague-Dawley rats (300–350 g) were euthanized by decapitation under deep anesthesia with pentobarbital (60 mg/kg). Brains were removed from the skull and placed in cold (~3°C) artificial cerebrospinal fluid (aCSF) of the following composition: 125 mM NaCl, 3 mM KCl, 18 mM NaHCO3, 1.25 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2, and 5 mM glucose, pH 7.30. Posterior cerebral arteries (PCAs) were dissected from the brain and arachnoid membrane, then kept in cold aCSF prior to cell isolation.
Cerebral artery myocytes were enzymatically dissociated from PCAs as previously described (Koide et al., 2007). Briefly, arteries were first incubated in isolation solution containing 0.3 mg/ml papain and 0.7 mg/ml dithioerythritol at 37°C for 17 min. PCA segments were then transferred to isolation solution containing 0.7 mg/ml type F collagenase and 0.3 mg/ml type H collagenase and incubated at 37°C for an additional 15 min. After the second incubation period, arteries were washed three times with cold isolation solution and triturated gently using a fire-polished Pasteur pipette to disperse individual myocytes. Cells were kept in isolation solution on ice and used for study within 6 h. The isolation solution contained: 55 mM NaCl, 80 mM sodium glutamate, 5.6 mM KCl, 10 mM HEPES, 2 mM MgCl2, 100 μM CaCl2, and 10 mM glucose (pH 7.30).
The conventional whole cell configuration of the patch clamp technique was utilized to measure whole cell VDCC membrane currents of cerebral artery myocytes using an Axopatch 200B amplifier (Molecular Devices Corp., Sunnyvale, CA). The pipette solution contained 130 mM CsCl, 10 mM EGTA, 10 mM HEPES, 2 mM ATP, 0.5 mM GTP, 5 mM phosphocreatine, and 10 mM glucose with pH adjusted to 7.20 with 1 M CsOH. Patch pipettes had resistances between 3 and 5 MΩ when filled with pipette solution. The extracellular solution contained 125 mM NaCl, 10 mM HEPES, 10 mM glucose, 1 mM MgCl2, 5 mM KCl and charge carrier (10 mM Ba2+ or 10 mM Ba2+ plus 100 μM Ca2+ or 2 mM Ba2+ or 2 mM Ca2+) with pH adjusted to 7.40 with 1 M NaOH. Osmolarity of intracellular and extracellular solutions were measured using a VAPRO vapor pressure osmometer (Wescor Inc.) and were 295 ± 5 mOsm. Pipette offset, whole-cell capacitance, and series resistance were compensated manually. Cell capacitance of myocytes were 13.78 ± 0.31, 14.14 ± 0.29, 13.21 ± 0.27, and 13.68 ± 0.34 pF in 10 mM [Ba2+]o, 10 mM [Ba2+]o plus 100 μM [Ca2+]o, 2 mM [Ba2+]o, and 2 mM [Ca2+]o,respectively, and not significantly different between groups. Series resistance measurements were 11.97 ± 1.31, 12.86 ± 1.98, 10.19 ± 1.59, and 11.77 ± 0.94 MΩ in 10 mM [Ba2+]o, 10 mM [Ba2+]o plus 100 μM [Ca2+]o, 2 mM [Ba2+]o, and 2 mM [Ca2+]o, respectively, and were also not significantly different between groups.
Myocytes were voltage clamped at a holding potential of −80 mV for ~5 min prior to the start of an experimental series. During this initial period, single depolarizing steps to +10 mV were applied to cells to ensure current amplitudes were stable before starting experimental protocols. Voltage protocols were driven and data were acquired using pCLAMP 9.2 software (Molecular Devices Corp., Sunnyvale, CA). Current traces were filtered at 1 kHz with a low-pass filter, subjected to P/4 leak subtraction and digitized at 5 kHz. Current-voltage relationships were obtained in response to a series of successive 10 mV depolarizing voltage steps (800 ms in duration) from the holding potential of −80 mV to +50 mV at 5 s intervals. Steady-state activation curves were also obtained using this protocol by measuring currents at the end of the 800 ms voltage step and dividing these measurements by peak currents obtained for each cell to obtain I/Imax. These values were then plotted against test voltage to calculate the half-activation voltage (V0.5act). In some cells, a voltage ramp protocol was used that applied a continuous depolarization from −80 to +50 mV over the course of 300 ms. Concentration-response relationships for the inhibition of VDCC currents by nifedipine and diltiazem were obtained in separate cells where currents were evoked by a single 200 ms duration voltage step from a holding potential of −80 to +10 mV. Solution changes were made via perfusion of the recording chamber (chamber volume: ~500 μl) for 10 min at a flow rate of ~1.9 ml/min. Steady-state inactivation curves were obtained using a double-step voltage protocol that applied a 5 s conditioning pulse step from −80 to +40 mV (10 mV increments) followed by a return to −80 mV for 15 ms and then 150 ms test voltage step to 10 mV. All current recordings were done at room temperature (20–22°C).
All chemicals and reagents including nifedipine and diltiazem were purchased from Sigma-Aldrich (St. Louis, MO) with the exception of papain, which was purchased from Worthington Biochemical Corp. (Lakewood, NJ).
Data were analyzed using Clampfit 9.2 software (Molecular Devices Corp., Sunnyvale, CA) as current density by dividing current amplitude by cell capacitance and are expressed as mean ± standard error of the mean (SEM). The number of cells in each dataset is denoted as “n” followed by the number of rats from which cells were obtained. The paired t-test was used for paired-sample analysis. For multiple group comparisons, data were initially evaluated by one-way ANOVA, with a p < 0.05 considered statistically significant followed by paired comparisons between two groups using Tukey's multiple comparisons post-test. The voltages for half-maximal activation (V0.5act) and half-maximal inactivation (V0.5inact) were obtained from Boltzman fit of steady-state activation and inactivation curves. Concentration-inhibition curves for nifedipine or diltiazem were calculated after normalization to currents in the absence of drug and fitted to the following equation: y = Imax/{1 + ([drug]/Ki)nH}+r, where Imax, [drug], Ki, nH, and r represent the maximum currents density, concentration of drug, binding affinity of drug, Hill coefficient, and drug resistant currents, respectively (Ebata et al., 1990). The IC50 of a drug was determined from concentration-inhibition curves.
Using 10 mM [Ba2+]o as a charge carrier, VDCC currents were elicited in rat cerebral artery myocytes by a series of 10 mV depolarizing steps from a holding potential −80 mV. In nominally Ca2+ free 10 mM Ba2+ bath solution, a maximum VDCC current density of −7.30 ± 0.35 pA/pF (n = 8 cells from four animals) was observed at +10 mV (Figures 1A,C). In the presence of nifedipine (1 μM), VDCC currents were reduced, but not abolished (Figures 1B,C). At +10 mV, the nifedipine-insensitive current density (−1.62 ± 0.48 pA/pF) was ~22% of total VDCC current density measured in these cells. To examine the impact of [Ca2+]o on VDCC currents when 10 mM [Ba2+]o is used as the primary charge carrier, the above voltage-step protocol was repeated using myocytes bathed in a combination of 10 mM [Ba2+]o and 100 μM [Ca2+]o (Figures 1D–F). The VDCC current density recorded at +10 mV (−7.28 ± 0.54 pA/pF; n = 11 cells, seven animals) was not significantly different compared to VDCC current density measured in 10 mM [Ba2+]o, alone (Figure 1C). However, the addition of 100 μM [Ca2+]o dramatically influenced the efficacy of nifedipine to inhibit VDCC currents. As illustrated in Figure 1F, inward VDCC currents obtained in the combination of 10 mM [Ba2+]o and 100 μM [Ca2+]o were completely abolished by 1 μM nifedipine. To further investigate the impact of [Ca2+]o on VDCC currents, single depolarizing voltage steps from a holding potential of −80 to +10 mV were used to elicit VDCC currents in the presence of increasing concentrations of nifedipine (10−10–10−6 M) followed by drug washout (Figures 2A–E and Table 2). In 10 mM [Ba2+]o, nifedipine caused a concentration-dependent decrease in VDCC currents with an IC50 of 6.02 ± 0.36 nM (n = 5 cells from five animals) with ~20% of the current insensitive to 1 μM nifedipine. Addition of 100 μM [Ca2+]o to the 10 mM [Ba2+]o bath solution again facilitated the complete inhibition of VDCC currents by 1 μM nifedipine, but did not change VDCC sensitivity to nifedipine (IC50 value 5.12 ± 0.41 nM; n = 4 cell from three animals). Upon washout of 1 μM nifedipine, current density was not significantly different when compared to current density obtained prior to the start of the nifedipine concentration-response curve (Table 2). Current-voltage (I-V) plots shown in Figures 1C,F are consistent with the activation of high-voltage activated VDCCs, i.e., maximum current density was observed at ~+10 mV. To obtain higher resolution with respect to the voltage-dependent activation of VDCC currents, voltage ramp protocols were used that applied a continuous depolarization from a holding potential of −80 to +50 mV over the course of 300 ms (Figures 2D–F). This approach also enabled the study of the same cell using 10 mM [Ba2+]o as the charge carrier in the presence and absence of 100 μM [Ca2+]o (Figures 2D–F). No difference was detected in the membrane potential that evoked maximum VDCC currents in 10 mM [Ba2+]o in the absence or presence of 100 μM [Ca2+]o (9.99 ± 0.08 vs. 10.01 ± 0.08 mV; n = 5 cells from three animals; p > 0.05). Maximum VDCC current density in cells bathed in 10 mM [Ba2+]o was also not significantly different in the absence or presence of 100 μM [Ca2+]o (Figures 2G–I). Voltages for half-maximal inactivation (V0.5inact) were ~−13.9, −32.5, and −19.9 mV in 10 mM [Ba2+]o, 10 mM [Ba2+]o + nifedipine (1 μM) and 10 mM [Ba2+]o + 100 μM [Ca2+]o, respectively (Figures 3A–D and Table 1) Voltages for half-maximal activation (V0.5act) were similar in 10 mM [Ba2+]o ± 100 μM [Ca2+]o, but were shifted modestly (~+8 mV) in 10 mM [Ba2+]o plus 1 μM nifedipine (Figure 3E and Table 1). In addition to 1,4-DHPs (e.g., nifedipine), L-type VDCCs are inhibited by benzothiazepines such as diltiazem. Figure 4 illustrates the concentration dependent inhibition by diltiazem of VDCC currents in 10 mM [Ba2+]o ± 100 μM [Ca2+]o. In contrast to the actions of nifedipine, diltiazem (100 μM) caused complete inhibition of 10 mM [Ba2+]o VDCC currents irrespective of the presence or absence of 100 μM [Ca2+]o. Upon washout of 100 μM diltiazam, current density was not significantly different when compared to a current density obtained prior to the start of the diltiazem concentration-response curve (Table 2).
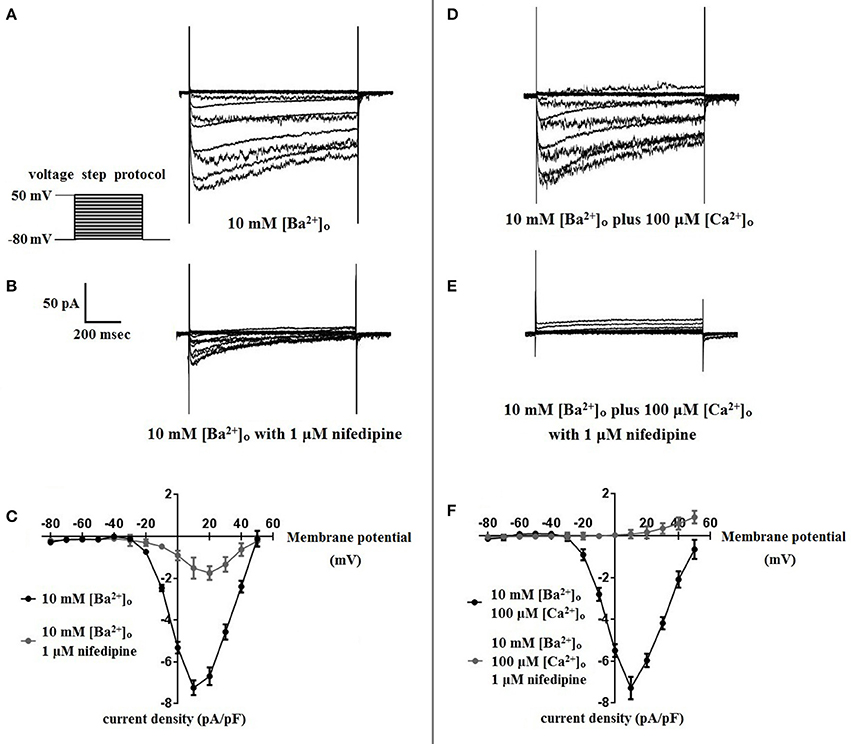
Figure 1. Nifedipine inhibition of VDCC currents obtained in 10 mM [Ba2+]o ± 100 μM [Ca2+]o. VDCC currents were elicited in rat cerebral artery myocytes in response to successive 10 mV depolarizing voltage steps from a holding potential of −80 mV. (A,B) VDCC currents obtained using 10 mM [Ba2+]o as the charge carrier in nominally Ca2+-free extracellular solution in the absence (A) and presence (B) of nifedipine (1 μM). (C) Summary data demonstrating that ~20 % of VDCC currents using 10 mM [Ba2+]o were not inhibited by 1 μM nifedipine. (D,E) VDCC currents obtained using 10 mM [Ba2+]o plus 100 μM [Ca2+]o in the absence (D) and presence (E) of nifedipine (1 μM). (F) Summary data demonstrating that VDCC currents were completely inhibited by 1 μM nifedipine in 10 mM [Ba2+]o plus 100 μM [Ca2+]o.
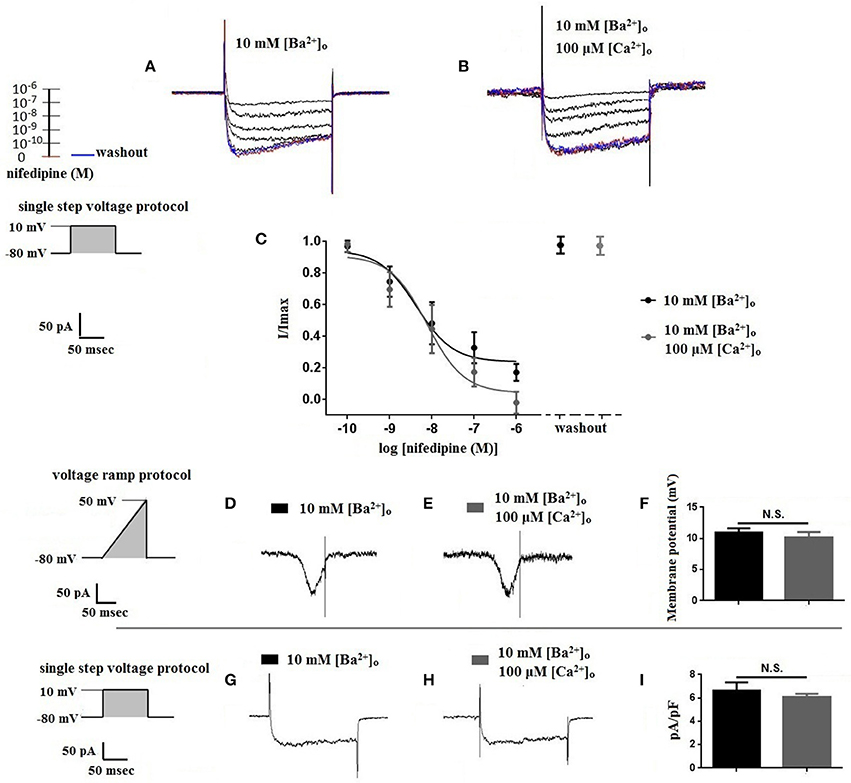
Figure 2. Properties of VDCC currents obtained using 10 mM [Ba2+]o ± 100 μM [Ca2+]o. (A–C) VDCC currents elicited by depolarizing voltage steps to +10 mV from a holding potential of −80 mV. (A) Effect of increasing concentrations of nifedipine in 10 mM [Ba2+]o. (C) Effect of increasing concentrations of nifedipine in 10 mM [Ba2+]o plus 100 μM [Ca2+]o. Traces in red represent currents obtained in cells prior to the addition of nifedipine. Traces in blue represent current traces obtained in cells following washout of 1 μM nifedipine. (C) Concentration-inhibition curves of nifedipine on VDCC currents obtained in bath solution containing 10 mM [Ba2+]o ± 100 μM [Ca2+]o. (D–F) VDCC currents evoked using a voltage ramp protocol that applied a continuous membrane potential depolarization from −80 to +50 mV over the course of 300 ms. (D) VDCC currents from a cerebral artery myocyte in bath solution containing 10 mM [Ba2+]o. (E) VDCC currents from the same cell depicted in (D) in bath solution containing 10 mM [Ba2+]o plus 100 μM [Ca2+]o. (F) Summary data demonstrating that the membrane potential evoking maximum VDCC current density was not different in bath solution containing 10 mM [Ba2+]o ± 100 μM [Ca2+]o (paired t-test, p > 0.05). (G–I) VDCC currents evoked by voltage steps to +10 mV in cells in bath solution containing 10 mM [Ba2+]o ± 100 μM [Ca2+]o. Maximum current density at +10 mV was not different between groups (paired t-test, p > 0.05).
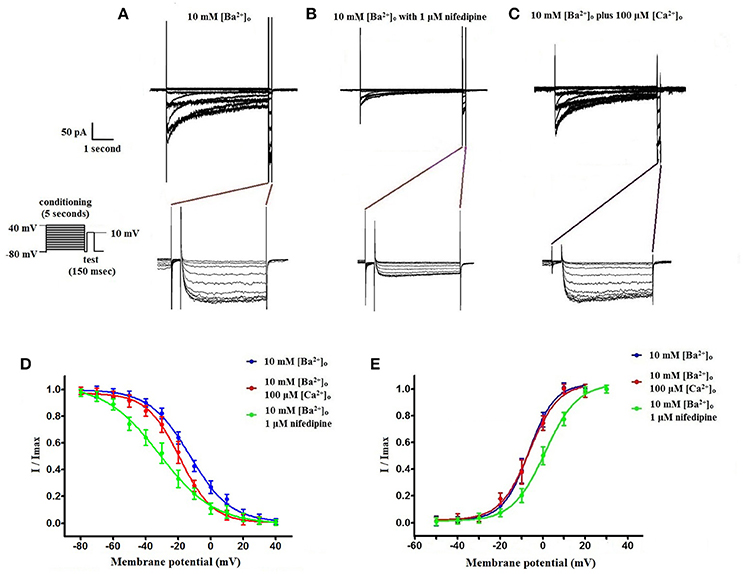
Figure 3. Voltage-dependent inactivation and activation characteristics of VDCC currents recorded in 10 mM [Ba2+]o ± 100 μM [Ca2+]o. (A–C) Current recordings obtained from cells using a double-step voltage protocol that applied a 5 s conditioning pulse step to examine steady-state VDCC inactivation. Recordings obtained in extracellular solution containing 10 mM [Ba2+]o (A), 10 mM [Ba2+]o with 1 μM nifedipine (B), and 10 mM [Ba2+]o plus 100 μM [Ca2+]o (C). (D) Steady-state inactivation curves obtained using the voltage protocol illustrated in (A–C). (E) VDCC activation curves derived from currents elicited by successive 10 mV depolarizing voltage steps from a holding potential of −80 mV.

Table 1. VDCC current properties.
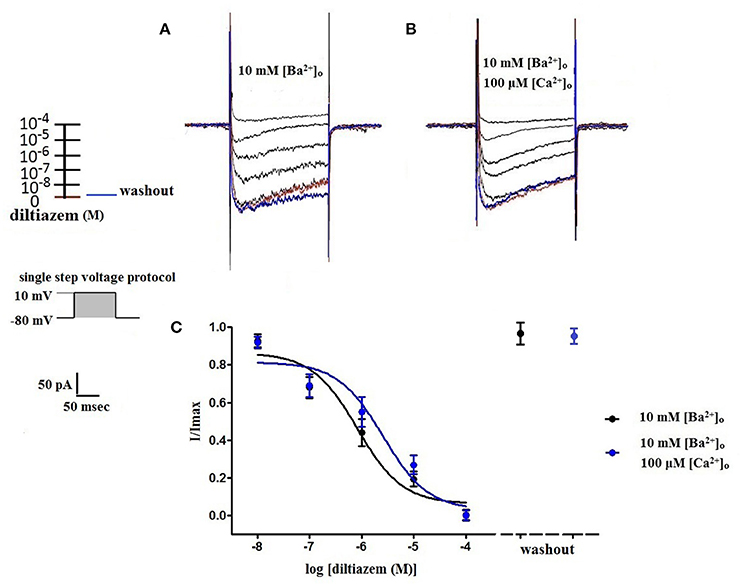
Figure 4. Diltiazem abolishes VDCC currents in 10 mM [Ba2+]o ± 100 μM [Ca2+]o. VDCC currents elicited by depolarizing voltage steps to +10 mV from a holding potential of −80 mV in bath solutions containing 10 mM [Ba2+]o (A) or 10 mM [Ba2+]o plus 100 μM [Ca2+]o (B). Traces in red represent currents obtained in cells prior to the addition of diltiazem. Traces in blue represent current traces obtained in cells following washout of 100 μM diltiazem. (C) Concentration-inhibition curves of diltiazem on VDCC currents obtained in extracellular solution containing 10 mM [Ba2+]o ± 100 μM [Ca2+]o.
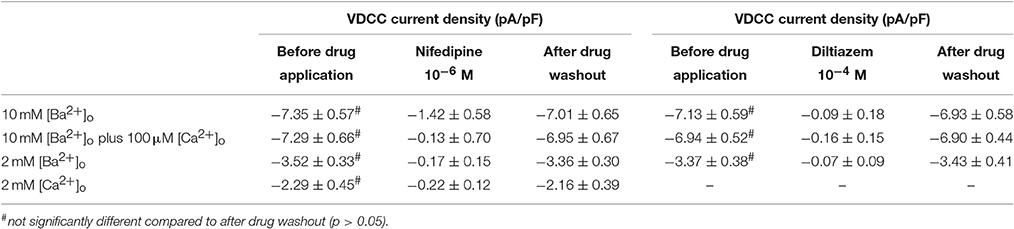
Table 2. VDCC current density ± nifedipine or diltiazem.
Surface charges, influenced by the concentration of extracellular divalent cations can impact VDCC properties, including drug/ligand binding (Kass and Krafte, 1987). To examine the impact of divalent cation concentration on nifedipine inhibition, VDCC currents were recorded in cerebral artery myocytes in Ca2+-free extracellular solution containing 2 mM [Ba2+]o as a charge carrier (Figures 5A–C). As expected, maximum current density in 2 mM [Ba2+]o (−3.77 ± 0.37 pA/pF at +10 mV; n = 13 cells from eight animals) was decreased compared to VDCC current density obtained in 10 mM [Ba2+]o. However, in contrast to currents obtained in 10 mM [Ba2+]o, VDCC currents were completely inhibited by 1 μM nifedipine in 2 mM [Ba2+]o. Similarly, VDCC currents were abolished by 1 μM nifedipine when 2 mM [Ca2+]o was used in place of 2 mM [Ba2+]o as a charge carrier (Figures 5D–F). Nifedipine IC50 values in 2 mM [Ba2+]o were 1.85 ± 0.29 nM, n = 5 cells from two animals) and were 3.35 ± 0.34 nM, (n = 4 cells from four animals, Figure 6) in 2 mM [Ca2+]o. Diltiazem IC50 value of VDCC currents recorded in 2 mM [Ba2+]o was 1.88 ± 0.25 μM, n = 4 from three animals. Upon washout of nifedipine and diltiazem, current density was not significantly different when compared to a current density obtained prior to drug application (Figure 6 and Table 2). Values obtained for V0.5act and V0.5inact were similar between cells bathed in solution containing 2 mM [Ba2+]o and 2 mM [Ca2+]o (Table 1).
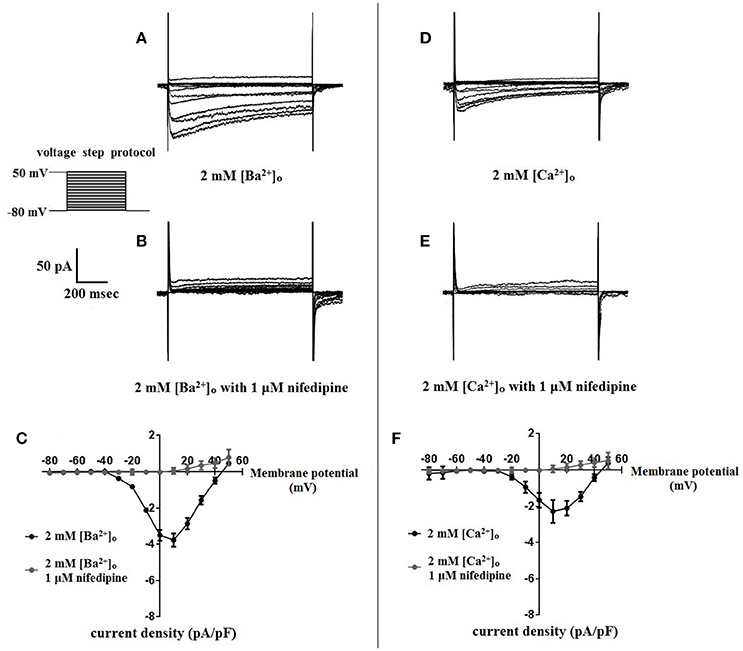
Figure 5. Nifedipine inhibition of VDCC currents obtained in 2 mM [Ba2+]o or 2 mM [Ca2+]o. VDCC currents were elicited in rat cerebral artery myocytes in response to successive 10 mV depolarizing voltage steps from a holding potential of −80 mV. (A,B) VDCC currents obtained using 2 mM [Ba2+]o as the charge carrier in nominally Ca2+-free extracellular solution in the absence (A) and presence (B) of nifedipine (1 μM). (C) Summary data demonstrating that VDCCs currents were completely inhibited by 1 μM nifedipine in bath solution containing 2 mM [Ba2+]o. (D,E) VDCC currents obtained using 2 mM [Ca2+]o in the absence (D) and presence (E) of nifedipine (1 μM). (F) Summary data demonstrating that VDCC currents were completely inhibited by 1 μM nifedipine in bath solution containing 2 mM [Ca2+]o.
Figure 6. Concentration-dependent inhibition of VDCC currents by nifedipine in 2 mM [Ba2+]o or 2 mM [Ca2+]o. VDCC currents elicited by depolarizing voltage steps to +10 mV from a holding potential of −80 mV. (A) Effect of increasing concentrations of nifedipine in bath solution containing 2 mM [Ba2+]o. (B) Effect of increasing concentrations of nifedipine in bath solution containing 2 mM [Ca2+]o. Traces in red represent currents obtained in cells prior to the addition of nifedipine. Traces in blue represent current traces obtained in cells following washout of 1 μM nifedipine. (C) Concentration-inhibition curves of nifedipine on VDCC currents obtained in bath solution containing either 2 mM [Ba2+]o or 2 mM [Ca2+]o.
This study demonstrates that the composition of extracellular divalent cations can profoundly alter the inhibition of whole-cell VDCC currents by 1,4-DHPs such as nifedipine. Our data indicate that nifedipine inhibition of VDCC currents in rat cerebral artery myocytes can be influenced by two factors: (1) the presence or absence of 100 μM [Ca2+]o when using 10 mM [Ba2+]o as a charge carrier— an effect likely mediated by the interaction of Ca2+ with an extracellular VDCC binding site and (2) the concentration of the extracellular divalent cation used as a charge carrier— an effect likely mediated by a change in surface charge. Further, these findings support the functional presence of a single population of high-voltage activated VDCCs in rat cerebral artery myocytes and demonstrate that the inhibition of these channels by nifedipine can be influence by the extracellular environment.
As depicted in Figure 1, nifedipine-resistant VDCC currents are absent when 100 μM Ca2+ is included in the extracellular solution during conventional whole-cell current recordings. This finding is consistent with a body of previous work indicating that the extracellular VDCC binding site for Ca2+ is coupled to a 1,4-DHP binding site on the channel and that [Ca2+]o is required for high affinity 1,4-DHP binding (Ebata et al., 1990; Knaus et al., 1992; Nakajima et al., 2002). Further, a K0.5 of 300 nM for [Ca2+]o to stabilize the high affinity binding domain for 1,4-DHPs was reported in L-type VDCC purified from rabbit skeletal muscle (Staudinger et al., 1991). In our study, the addition of 100 μM [Ca2+]o enabled the complete inhibition of VDCC currents by 1 μM nifedipine, but did not change VDCC sensitivity to nifedipine, with IC50 values similar ± [Ca2+]o (Figure 2). This observation is consistent with the work of Yamada et al. that reported a reduction in the maximum number of high affinity 1,4-DHP binding sites either in the presence of [Ba2+]o or the absence of [Ca2+]o in porcine coronary artery preparations (Yamada et al., 1990). In addition to 1,4-DHPs, L-type VDCCs are selectively inhibited by two other classes of compounds, benzothiazepines, and phenylalkylamines, with each class of inhibitors having distinct binding sites to the VDCC alpha1 subunit (Hille, 2001). We found the efficacy of the benzothiazepine, diltiazem, to be independent of [Ca2+]o. In effect, diltiazem (100 μM) completely abolished 10 mM [Ba2+]o VDCC currents irrespective of the presence or absence of [Ca2+]o (Figure 4), consistent with the existence of independent binding sites for 1,4-DHPs and benzothiazepines. This observation supports the work of other groups that have shown that L-type VDCC mutations that prevent 1,4-DHP binding do not alter channel inhibition by diltiazem (Yamaguchi et al., 2000; Walsh et al., 2007).
Our data indicate that surface charge, influenced by the concentration of extracellular divalent cations, can also affect inhibition of L-type VDCCs by nifedipine. As shown in Figures 5, 6, decreasing the extracellular concentration of Ba2+ from 10 to 2 mM dramatically increased VDCC inhibition by nifedipine. In this case, the change in nifedipine efficacy was independent of [Ca2+]o, as the experimental series was done in nominally Ca2+-free extracellular solution. These findings are remarkably similar to those reported by Kass and Krafte examining nisoldipine block of L-type VDCC in cardiac myocytes (Kass and Krafte, 1987). These investigators provided evidence that a depolarizing shift in surface charge and the resulting impact on channel gating was largely responsible for the apparent antagonism between divalent cations and 1,4-DHPs. In our present study, V0.5act was shifted positively from −22.0 to −13.5 mV when 2 mM [Ba2+]o was replaced with 10 mM [Ba2+]o, indicative of a depolarizing shift in surface charge. However, additional mechanisms independent of the effects of surface charge on VDCC gating such as direct interaction of divalent cations with the 1,4-DHP binding site may also contribute to nifedipine-resistant currents observed in the presence of higher concentrations of divalent cations.
Our data are consistent with the presence of high-voltage activated L-type VDCCs in cerebral artery myocytes (Figures 1, 2, 5). Under physiological conditions, i.e., 2 mM [Ca2+]o, 1,4-DHPs would be predicted to inhibit these channels, as has been shown in a number of functional studies (Knot and Nelson, 1998; Gokina and Osol, 2002; Wellman et al., 2002; Ishiguro et al., 2005; Syyong et al., 2009). Interestingly, other studies using a combination of ex vivo diameter measurements, molecular biology, western blotting, and electrophysiology indicate the co-existence of nifedipine-insensitive low-voltage activated T-type VDCCs (CaV3.x family members) along with L-type VDCCs in cerebral artery myocytes (Kuo et al., 2010; Abd El-Rahman et al., 2013; Harraz and Welsh, 2013b; Harraz et al., 2015). In the current study, the current- voltage relationships that were obtained are consistent with those of high-voltage activated VDCCs, rather than with properties characteristic of those reported for T-type VDCCs (Lambert et al., 2014). Further, the modest depolarizing shift in the current-voltage relationship of nifedipine-resistant currents obtained in 10 mM Ba2+ is also not consistent with the presence of T-type channels. Presently, the reason why T-type VDCC currents were detected in the above-mentioned studies, but not in our study is unclear.
In conclusion, this study demonstrates that the composition of extracellular divalent cations can profoundly impact the binding of 1,4-DHPs to L-type VDCCs. These findings suggest that measurements of VDCC currents should be preferentially carried out in physiological concentrations of divalent cations.
FW contributed to data acquisition, data analysis, and manuscript preparation. MK contributed to data interpretation and manuscript revision. GW contributed to study design, data analysis/interpretation, and manuscript preparation.
This study was supported by the Totman Medical Research Trust, Grants from National Institutes of Health (No. P01-HL-2095488, P20-RR-16435, and R01-HL-078983), the American Heart Association (14SDG20150027), the Peter Martin Fund, the National Natural Science Foundation of China (No. 81260182, 81560206) and the Natural Science Foundation of Yunnan Province (No. FB2016121).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abd El-Rahman, R. R., Harraz, O. F., Brett, S. E., Anfinogenova, Y., Mufti, R. E., Goldman, D., et al. (2013). Identification of L- and T-type Ca2+ channels in rat cerebral arteries: role in myogenic tone development. Am. J. Physiol. Heart Circ. Physiol. 304, H58–H71. doi: 10.1152/ajpheart.00476.2012
Alexander, S. P. H., Catterall, W. A., Kelly, E., Marrion, N., Peters, J. A., Benson, H. E., et al. (2015). The concise guide to PHARMACOLOGY 2015/16: voltage-gated ion channels. Br. J. Pharmacol. 172, 5904–5941. doi: 10.1111/bph.13349
Catterall, W. A., Striessnig, J., Snutch, T. P., Perez-Reyes, E., and International Union of Pharmacology. (2003). International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol. Rev. 55, 579–581. doi: 10.1124/pr.55.4.8
Davis, M. J., and Hill, M. A. (1999). Signaling mechanisms underlying the vascular myogenic response. Physiol. Rev. 79, 387–423.
Ebata, H., Mills, J. S., Nemcek, K., and Johnson, J. D. (1990). Calcium binding to extracellular sites of skeletal muscle calcium channels regulates dihydropyridine binding. J. Biol. Chem. 265, 177–182.
Gokina, N. I., and Osol, G. (2002). Actin cytoskeletal modulation of pressure-induced depolarization and Ca2+ influx in cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 282, H1410–H1420. doi: 10.1152/ajpheart.00441.2001
Harraz, O. F., Visser, F., Brett, S. E., Goldman, D., Zechariah, A., Hashad, A. M., et al. (2015). CaV1.2/CaV3.x channels mediate divergent vasomotor responses in human cerebral arteries. J. Gen. Physiol. 145, 405–418. doi: 10.1085/jgp.201511361
Harraz, O. F., and Welsh, D. G. (2013a). Protein kinase A regulation of T-type Ca2+ channels in rat cerebral arterial smooth muscle. J. Cell. Sci. 126, 2944–2954. doi: 10.1242/jcs.128363
Harraz, O. F., and Welsh, D. G. (2013b). T-type Ca2+ channels in cerebral arteries: approaches, hypotheses, and speculation. Microcirculation 20, 299–306. doi: 10.1111/micc.12038
Hille, B. (2001). Ion Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates, Inc. Publishers, 100–108.
Ishiguro, M., Wellman, T. L., Honda, A., Russell, S. R., Tranmer, B. I., and Wellman, G. C. (2005). Emergence of a R-type Ca2+ channel (CaV2.3) contributes to cerebral artery constriction after subarachnoid hemorrhage. Circ. Res. 96, 419–426. doi: 10.1161/01.RES.0000157670.49936.da
Kass, R. S., and Krafte, D. S. (1987). Negative surface charge density near heart calcium channels. Relevance to block by dihydropyridines. J. Gen. Physiol. 89, 629–644. doi: 10.1085/jgp.89.4.629
Knaus, H. G., Moshammer, T., Kang, H. C., Haugland, R. P., and Glossmann, H. (1992). A unique fluorescent phenylalkylamine probe for L-type Ca2+ channels. Coupling of phenylalkylamine receptors to Ca2+ and dihydropyridine binding sites. J. Biol. Chem. 267, 2179–2189.
Knot, H. J., and Nelson, M. T. (1998). Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J. Physiol. 508, 199–209. doi: 10.1111/j.1469-7793.1998.199br.x
Koide, M., Penar, P. L., Tranmer, B. I., and Wellman, G. C. (2007). Heparin-binding EGF-like growth factor mediates oxyhemoglobin-induced suppression of voltage-dependent potassium channels in rabbit cerebral artery myocytes. Am. J. Physiol. Heart Circ. Physiol. 293, H1750–H1759. doi: 10.1152/ajpheart.00443.2007
Kuo, I. Y., Ellis, A., Seymour, V. A., Sandow, S. L., and Hill, C. E. (2010). Dihydropyridine-insensitive calcium currents contribute to function of small cerebral arteries. J. Cereb. Blood Flow Metab. 30, 1226–1239. doi: 10.1038/jcbfm.2010.11
Lambert, R. C., Bessaih, T., Crunelli, V., and Leresche, N. (2014). The many faces of T-type calcium channels. Pflugers Arch. 466, 415–423. doi: 10.1007/s00424-013-1353-6
Moosmang, S., Schulla, V., Welling, A., Feil, R., Feil, S., Wegener, J. W., et al. (2003). Dominant role of smooth muscle L-type calcium channel CaV1.2 for blood pressure regulation. EMBO J. 22, 6027–6034. doi: 10.1093/emboj/cdg583
Nakajima, M., Yamada, S., Uchida, S., and Kimura, R. (2002). In vivo measurement of 1,4-dihydropyridine receptors in mesenteric arteries of spontaneously hypertensive rats and effect of nifedipine and cilnidipine. Biol. Pharm. Bull. 25, 24–28. doi: 10.1248/bpb.25.24
Nikitina, E., Zhang, Z. D., Kawashima, A., Jahromi, B. S., Bouryi, V. A., Takahashi, M., et al. (2007). Voltage-dependent calcium channels of dog basilar artery. J. Physiol. 580, 523–541. doi: 10.1113/jphysiol.2006.126128
Nystoriak, M. A., Murakami, K., Penar, P. L., and Wellman, G. C. (2009). CaV1.2 splice variant with exon 9* is critical for regulation of cerebral artery diameter. Am. J. Physiol. Heart Circ. Physiol. 297, H1820–H1828. doi: 10.1152/ajpheart.00326.2009
Nystoriak, M. A., O'Connor, K. P., Sonkusare, S. K., Brayden, J. E., Nelson, M. T., and Wellman, G. C. (2011). Fundamental increase in pressure-dependent constriction of brain parenchymal arterioles from subarachnoid hemorrhage model rats due to membrane depolarization. Am. J. Physiol. Heart Circ. Physiol. 300, H803–H812. doi: 10.1152/ajpheart.00760.2010
Staudinger, R., Knaus, H. G., and Glossmann, H. (1991). Positive heterotropic allosteric regulators of dihydropyridine binding increase the Ca2+ affinity of the L-type Ca2+ channel. Stereoselective reversal by the novel Ca2+ antagonist BM 20.1140. J. Biol. Chem. 266, 10787–10795.
Syyong, H. T., Yang, H. H., Trinh, G., Cheung, C., Kuo, K. H., and van Breemen, C. (2009). Mechanism of asynchronous Ca2+ waves underlying agonist-induced contraction in the rat basilar artery. Br. J. Pharmacol. 156, 587–600. doi: 10.1111/j.1476-5381.2008.00063.x
Walsh, K. B., Zhang, J., Fuseler, J. W., Hilliard, N., and Hockerman, G. H. (2007). Adenoviral-mediated expression of dihydropyridine-insensitive L-type calcium channels in cardiac ventricular myocytes and fibroblasts. Eur. J. Pharmacol. 565, 7–16. doi: 10.1016/j.ejphar.2007.02.049
Wellman, G. C. (2006). Ion channels and calcium signaling in cerebral arteries following subarachnoid hemorrhage. Neurol. Res. 28, 690–702. doi: 10.1179/016164106X151972
Wellman, G. C., Nathan, D. J., Saundry, C. M., Perez, G., Bonev, A. D., Penar, P. L., et al. (2002). Ca2+ sparks and their function in human cerebral arteries. Stroke 33, 802−808. doi: 10.1161/hs0302.104089
Yamada, S., Kimura, R., Harada, Y., and Nakayama, K. (1990). Calcium channel receptor sites for (+)-[3H]PN 200–110 in coronary artery. J. Pharmacol. Exp. Ther. 252, 327–332.
Yamaguchi, S., Okamura, Y., Nagao, T., and Adachi-Akahane, S. (2000). Serine residue in the IIIS5–S6 linker of the L-type Ca2+ channel alpha 1C subunit is the critical determinant of the action of dihydropyridine Ca2+ channel agonists. J. Biol. Chem. 275, 41504–41511. doi: 10.1074/jbc.M007165200
Keywords: cerebral artery, vascular smooth muscle, nifedipine, patch clamp, voltage-dependent calcium channels
Citation: Wang F, Koide M and Wellman GC (2017) Nifedipine Inhibition of High-Voltage Activated Calcium Channel Currents in Cerebral Artery Myocytes Is Influenced by Extracellular Divalent Cations. Front. Physiol. 8:210. doi: 10.3389/fphys.2017.00210
Received: 05 December 2016; Accepted: 23 March 2017;
Published: 07 April 2017.
Edited by:
Maik Gollasch, Charité Universitätsmedizin Berlin, GermanyReviewed by:
Withrow Gil Wier, University of Maryland, Baltimore, USACopyright © 2017 Wang, Koide and Wellman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George C. Wellman, Z3dlbGxtYW5AdXZtLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.