 Julian A. G. Glassford
Julian A. G. Glassford
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Physiol. , 17 February 2017
Sec. Integrative Physiology
Volume 8 - 2017 | https://doi.org/10.3389/fphys.2017.00088
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is a debilitating multi-systemic chronic illness of unknown etiology, classified as a neurological disorder by the World Health Organization (WHO). The symptomatology of the condition appears to emanate from a variety of sources of chronic neurological disturbance and associated distortions, and chronicity, in noxious sensory signaling and neuroimmune activation. This article incorporates a summary review and discussion of biomedical research considered relevant to this essential conception perspective. It is intended to provide stakeholders with a concise, integrated outline disease model in order to help demystify this major public health problem. The primary etiopathological factors presented are: (A) Postural/biomechanical pain signaling, affecting adverse neuroexcitation, in the context of compression, constriction, strain, or damage of vertebral-regional bone and neuromuscular tissues; (B) Immune mediated inflammatory sequelae, in the context of prolonged immunotropic neurotrophic infection—with lymphotropic/gliotropic/glio-toxic varieties implicated in particular; (C) A combination of factors A and B. Sustained glial activation under such conditions is associated with oxidative and nitrosative stress, neuroinflammation, and neural sensitivity. These processes collectively enhance the potential for multi-systemic disarray involving endocrine pathway aberration, immune and mitochondrial dysfunction, and neurodegeneration, and tend toward still more intractable synergistic neuro-glial dysfunction (gliopathy), autoimmunity, and central neuronal sensitization.
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is an idiopathic, heterogeneous condition involving distortion of homeostasis across multiple organ systems. It is chiefly characterized by myalgia, fatigue, neurocognitive dysfunction, and, critically, delayed muscle recovery (Paul et al., 1999) and the intensification of symptoms following physical exertion: “Post Exertional Malaise” (PEM), or “Post Exertional Neuroimmune Exhaustion” (PENE) (VanNess et al., 2010).
This highly intrusive disease involves varying degrees of physical disability and cognitive deficits, associated psychosocial difficulties, and significantly reduced quality of life (Winger et al., 2015) and life expectancy (Jason et al., 2006), across a patient population numbering in the millions worldwide. Complete/spontaneous recovery is extremely rare and conventional treatment strategies rarely deliver even modest direct, objective and, sustained symptomatic improvement. Thus, ME/CFS constitutes a particularly enigmatic, debilitating, and costly major public health issue, and the advancement of our understanding of its essence, hence, a pressing area of biomedical enquiry (Arroll, 2014).
The central focus of this work is the proposition that ME/CFS constitutes the symptomatic manifestation of enhanced nervous sensitivity attributable to a neuroinflammatory etiopathology associated with abnormal nociceptive and neuroimmune activity. The article reviews, highlights, and interconnects numerous relevant disease features, processes, and concepts from the biomedical literature, the ultimate aim of which is to provide stakeholders with an instructive pathophysiological conceptual framework.
Postural and biomechanically induced neuroexcitatory neuroinflammation has been posited as a partial explanation for PEM/PENE (Rowe et al., 2013), and sustained muscle activity and associated cortical excitability have been observed in ME/CFS (Brouwer and Packer, 1994).
A number of clinical and physical therapy researchers have, over the course of recent decades, variously formed associations between ME/CFS, orthostatic intolerance, and Ehlers-Danlos syndrome (Rowe et al., 1999), ME/CFS and joint hypermobility (Barron et al., 2002), and ME/CFS and innate (flat thoracic spine), osteochondritis, or trauma-linked vertebral defects (Perrin, 2007). These issues, along with other connective tissue disorders, fractures, and other factors that reduce range of motion (ROM), a physical impediment common among ME/CFS patients (Rowe et al., 2014), plus lengthy periods of reduced movement of neuromuscular tissues, may be associated with diminished neural motility and increased neuromuscular tension/strain. Both during and 24 h following strain-inducing experimental physical maneuvers, ME/CFS patients report significantly greater symptom intensity elevation than do sham exposed patient and healthy control cohorts (Rowe et al., 2016). Along with neuropathies and peri-neural adhesions, these issues represent potential ongoing sources of nociceptive input and glial activation, resulting in enhanced peripheral and central nervous sensitivity (Rowe et al., 2013). Peri-neural adhesions are fibrous bonds proximal to nerve tissues, often formed during post-operative/injury healing.
Any residual central sensitivity may, in turn, effect heightened sensitivity of relevant portions of the peripheral nervous system (PNS) to further input (Rowe et al., 2014). Furthermore, neurons may continue to fire after the initiating stimulation has ceased (Löscher and Ebert, 1996): a phenomenon observed in relation to persistent, relatively low threshold stimulation, colloquially referred to as “kindling” (Jason et al., 2011). Thus, the potential exists for the emergence of an insidious peripheral-central neurogenic sensitization loop, which would conceivably have the power to modulate the impact of symptom worsening events in ME/CFS. Hypothesized pathological products of these processes include increased resting muscle tone, affected by enhanced tension in/stimulation from proximal peri-neural fibers subject to sensitization, and aggravation of vascular and autonomic tone (Rowe et al., 2013).
Nociceptive afferent input excites post-synaptic neurons and may also be read by glia, triggering cellular responses e.g., via the stimulated neuronal release of chemical mediators that bind to glial receptors (Ren and Dubner, 2008). Calcium ion (Ca(2+)) influx into astrocytes following stimulation causes central terminals of the nociceptor to release a host of neuroactive signal molecules. These include the primary neuroexcitatory neurotransmitter, glutamate, nitric oxide (NO), and pro-inflammatory cytokines: Tumor necrosis factor alpha (TNF-α) and interleukin-1 beta (IL-1β) (Ricci et al., 2009).
Activated microglia behave similarly in responding to immune challenge/inflammation (Renno et al., 1995), also inducing superoxide production. Superoxide and NO are free radical substrates of the potent, toxic oxidant peroxynitrite (ONOO–), and hence sources of oxidative and nitrosative (O+NS) damage, both individually and, particularly, when combined (Barger et al., 2007). Along with the abovementioned stimuli, glia may also be primed to respond more harshly by exposure to toxins and electromagnetic fields (EMF), including non-ionizing, radio frequency (RFR) electromagnetic radiation (EMR) (Hao et al., 2010), autoimmune processes (Colton, 2009), and the effects of aging (Norden and Godbout, 2012).
Glia propagate inflammatory signals and may cause chronic pain e.g., involving allodynia and hyperalgesia (Yasui et al., 2014), via the impact of bidirectional neuro-glial signaling; glia may be activated by way of neuronal stimulation (as described above) and the inflammatory cytokines that they release may, in turn, couple to neuronal glutamate receptors, thereby enhancing neuroexcitation (Ren and Dubner, 2008). Synergistic interactions may result, prompting the dysregulation of glial functions (gliopathy) and associated maladaptive hyper-activation (microgliosis) (Ji et al., 2013). Magnetic resonance imaging (MRI) reveals changes in the structure of the brain in ME/CFS that may indeed be attributable to astrocyte dysfunction (Barnden et al., 2011).
Gliopathy can come about following exposure to “glio-toxins”: Immunosuppressive mycotoxins produced by certain species of fungi, including house mold (Aikawa and Suzuki, 1985). In one study, 93% of ME/CFS patients tested positive for mycotoxins (Brewer et al., 2013), compared with 0% of controls at similar detection thresholds (Hooper et al., 2009).* Gliopathies are also believed to be initiated, more broadly, by concurrent increases in extracellular glutamate and cytokine production (Hulsebosch, 2008).
Enhanced glial activation results in neuroexcitation, neuroinflammation and neurodegeneration, indicated by significantly reduced white and gray matter in ME/CFS (Puri et al., 2012), and hence (additional) glial multiplication subject to neuronal damage; at the symptomatic level it is associated with cognitive deficits, common in ME/CFS (Shanks et al., 2013), and depressive-like behavior (Norden and Godbout, 2012), also fairly common in the condition (Maes and Twisk, 2010). Widespread low-level neuroinflammation has been detected in the brains of ME/CFS patients, with neuropsychological symptom severity associated with the level of neuroinflammation in one study (Nakatomi et al., 2014), and with self-reported chronic pain in another (Ickmans et al., 2015). Furthermore, animal testing appears to indicate that microglial induction of IL-1β may be a prerequisite for immunologically induced fatigue (Ifuku et al., 2014),** and that gender-specific relationships may exist concerning suppression of T cell contact-mediated glial activation (Brahmachari and Pahan, 2010) and relative prolongation of glial responses following neurotoxin exposure (Ciesielska et al., 2009). These findings accord with numerous human studies highlighting relatively high prevalence of ME/CFS among females (Reyes et al., 2003).
* Elevated levels of pro-inflammatory cytokines paired with O+NS stress are suspected as being behind mitochondrial dysfunction in the disease (Morris and Maes, 2012). Associated depletion of antioxidants may account for some of the damage to fatty acids, proteins, and mitochondrial DNA and membranes associated with ME/CFS, via the activity of reactive oxygen and nitrogen species (ROS/RNS) (Maes and Twisk, 2010). The anomalous presence of mycotoxins may play a role here too (Brewer et al., 2013), and EMR exposure represents a complicating factor within this thematic e.g., given its capacity to influence fungal activity/biotoxicity (Velizarov et al., 1999) and lipid peroxidation (Mailankot et al., 2009).
** IL-1β mediates substances that may induce excitotoxicity (Mohebiany and Schneider, 2013), and inflammatory cytokine activity could play an important role in the aberration of endocrine (Jason et al., 2011) and mitochondrial dysfunction (Myhill et al., 2009).
Peripheral pain can contribute to “central sensitization,” or “wind-up”: The enhancement of nociceptive pathways in the central nervous system (CNS), a process arguably manifest in hyperalgesia in ME/CFS (Nijs et al., 2012) and sister condition Fibromyalgia Syndrome (FMS) (Woolf, 2011), and one which has been used to theoretically elucidate aspects of ME/CFS symptomatology (Meeus and Nijs, 2007). Wind-up takes place under long-term potentiation (LTP), in the context of temporal summation effected by repetitive nociceptive stimulation (which may be low/subclinical in intensity) (Woolf, 1991).
ME/CFS patients have demonstrably low nociceptive thresholds of the muscle tissues (Vecchiet et al., 1996), and many have significant cerebral (brain) hypoperfusion (Hamre, 1995), including in the parahippocampus (Gay et al., 2016) and brain stem (Costa et al., 1995), which may contribute to abnormal function of the locus ceruleus, involved in the control of descending inhibitory nociceptive pathways (Meeus and Nijs, 2007). Along with NO and pro-inflammatory cytokine activity, excitotoxicity may play a role in migraine pathogenesis (Longoni and Ferrarese, 2006). Migraines have been detected in 82% of ME/CFS cases and factors implicated in migraine pathology likely contribute to the type of headaches and cognitive impairments seen in the disease. Consistent with the above, the cortical spreading depression (CSD) model of migraine describes dysfunction in brain energetics, the brainstem, and thalamocortical tracts associated with changes in cerebrovascular dynamics and ischemia affecting loss of descending inhibitory nociceptive processes (Rayhan et al., 2013).
Pro-inflammatory cytokines can alter the metabolism of serotonin (5-HT) and dopamine (Felger and Miller, 2012),* effecting dysregulation of associated neurotransmitters, including glutamate, norepinephrine (NE, noradrenalin), and corticosteroids. 5-HT and NE are the major neurotransmitters involved in descending nociceptive modulation, and NE increases blood flow/pressure by increasing vascular tone, triggers glucose release, and can suppress neuroinflammation. Glucocorticoid deficiencies** are associated with interrupted nociceptive modulation, for example via the disruption of neuroendocrine-mediated glial activation mechanisms (Schwartzman, 2012).
* Serotonin status fluctuation (Badawy et al., 2005) and dopaminergic modulation abnormalities (Georgiades et al., 2003) have been observed in ME/CFS.
** Both increased resistance to glucocorticoid-immune signaling (Kavelaars et al., 2000) and increased glucocorticoid receptor sensitivity have been implicated in ME/CFS (Visser et al., 2001).
Adenosine triphosphate (ATP) activity influences glutamate and gamma-Aminobutyric acid (GABA) release, and hence the glutamate:GABA ratio. GABA is the primary neuroinhibitory neurotransmitter in the CNS. ATP depletion is observed in ME/CFS (Morris and Maes, 2013), and is associated with N-methyl-D-aspartate (NMDA) stimulation and hypersensitivity (Pall, 2003). Furthermore, low neuronal energy production markedly increases sensitivity to glutamate excitotoxicity (Blaylock and Maroon, 2012). NMDA receptor activation results in increased levels of ONOO– (Pall, 2003) and triggers loss of mitochondrial membrane potential and apoptosis (Hardingham et al., 2002).
ME/CFS patients tend to have low cardiac output and the vast majority experience orthostatic intolerance (OI) (Miwa, 2015), a manifestation of dysautonomia relating to inadequate blood circulation and attributable to hypovolemia/blood pooling in the extremities. Various regulatory neurotransmitter/(neuro)endocrine abnormalities (Pall, 2007), as well as (related) sympathetic nervous system (SNS) dominance (Freeman and Komaroff, 1997), and diminished cardiac mass (Hollingsworth et al., 2012), may account for such features. Additionally, cerebral vascular control appears closely related to skeletal muscle pH, found to be elevated in ME/CFS at rest (Jones et al., 2010), and it has been hypothesized that compromised skeletal muscle cellular membrane function may lead to a degree of acidity equalization between the skeletal muscle intracellular environment (raised pH) and the blood (lowered pH). Consistent with these relations, abnormally prolonged cerebral vasoconstriction following orthostatic challenge is routinely observed in ME/CFS (He et al., 2013); this has ramifications for neural health, as per the links outlined in the previous section, plus other deleterious effects including cerebral hypoxia and neurocognitive deficits (Ocon, 2013).
General statistical surveys are suggestive of a heritable aspect to ME/CFS, with the relative risk of disease development among first degree relatives almost 3 times the norm (Albright et al., 2011). It is worth noting that inflammatory reactivity is of course a function of genetic variation in cytokine genotypes, which underpin the severity of “sickness behavior” in ME/CFS (Vollmer-Conna et al., 2008) e.g., in the context of chronic neurotropic infection (VanElzakker, 2013).
Polymorphism in the TNF-α promoter gene TNF-857 and significant decrease of interferon-gamma (IFNγ) low producers have been observed in ME/CFS (Carlo-Stella et al., 2006), implying an inherent pro-inflammatory immunomodulatory disposition. Patients have also been found to have polymorphisms of transient receptor potential channels (TRPs): TRPM1 and TRPM3 (Marshall-Gradisnik et al., 2015), affecting mechanoreceptor nociceptive and mood-linked neurological status, and appear to have peculiar genes and gene activity relating more specifically to aspects of central nociception and stress mediation (CDC, 2006). Abnormally high expression of P2X purinoceptor 7 protein encoding P2RX7, a gene that regulates nociception (and inflammatory pain in particular), has been noted (Light et al., 2013) and there is also evidence of an array of serotonergic genetic abnormalities; these include differences in genes responsible for 5-HT production (Narita et al., 2003) as well as distinct genotypic and allelic frequencies of the 5-HT receptor 2A (5-HT2A) encoding HTR2A gene (Bozzinia et al., 2012). 5-HT2A receptors are believed to mediate neuronal excitation and anxiety, their expression appears to be up-regulated post-infection (Couch et al., 2015), and their antagonists have been shown to mitigate chronic pain (Bardin, 2011).
The term “encumbrance” is used in this article to denote the constriction, strain, or damage of neuromuscular tissues, as well as associated compression of proximal osseous tissues.
Core intensive/high neuro-dynamic intensity physical activity increases the risk of encumbrance-linked health problems, particularly during spinal development. Related risk factors also include acute neuromuscular strains, neuropathies,* and peri-neural adhesions, as well as relatively indirect processes such as the evolution of connective tissue, hypermobility, and inflammatory disorders. Such issues may be underpinned by genetic vulnerability concerning/the epigenetic impact of psychological trauma (Heim et al., 2009) and psychosocial stress (Prins et al., 2008), together with a broader array of neurotoxic (Giordano and Costa, 2012)/immunotoxic stressors (Dietert, 2014), on the development and functioning of (neuro)anatomic, (neuro)endocrine, and (neuro)immune systems.
* Animal testing reveals that peripheral nerve injury may alter blood-spinal cord barrier (BSCB) integrity (Echeverry et al., 2011), and viruses, inflammatory cytokines, RFR EMR, ONOO–, O+NS stress, and psychological stress may similarly compromise the blood-brain barrier (BBB) (Bested et al., 2001). This renders the central nervous system (CNS) relatively vulnerable to the permeation of pollutants from the blood.
Common postural risk factors associated with constriction/biomechanical strain of neuromuscular tissues subject to abnormal tension and related sensitivity may include: (1) Flexion of the hip or ankle beyond 70 degrees; (2) Flexion of the neck/arching of the back associated with tucking of the chin and forward/stooped head positions; (3) Lower back slumping/jamming/immobilization e.g., in the absence of sufficient ischial support (Rowe et al., 2016). Factor 3 is a potential source of decreased lumbar lordosis, and constriction/compression of proximal dorsal root ganglia (DRG) and osseous tissues, and hence of (enhanced) nociceptive stimulation (Mörl and Bradl, 2013).
Relative inactivity may also have ramifications for the health of neuromuscular tissues, for example via heightened nociceptive/neuroinflammatory responses in the context of enhanced neuromuscular tension/strain resulting from diminished neurodynamic and neuromuscular motility/flexibility (Rowe et al., 2013).
Chronic sleep deficiencies in ME/CFS influence insulin-like growth factor 1 (IGF-1) synthesis, neural sensitivity (Schuh-Hofer et al., 2013), and neurotoxin clearance (Xie et al., 2013). IGF-1 is a hormone stimulated by growth hormone, particularly during deeper “slow-wave” sleep; ME/CFS patients typically experience little sleep at stages III and IV (Fischler et al., 1997) and have lower serum levels of IGF-1 (Berwaerts et al., 1998), with quality of sleep correlated with circulating levels of pro-inflammatory cytokines, and severity and frequency of symptoms, in the disease (Milrad et al., 2017). Additionally, pro-inflammatory cytokines appear to directly dampen IGF-1 pathways (Puche and Castilla-Cortázar, 2012). IGF-1 plays an important role in many biological processes, notably including: Myelination (Liang et al., 2007) and early recovery from demyelination (Mason et al., 2000), and mitochondrial nutrient and inhibitory neurotransmitter synthesis.
The picture that emerges from the literature indicates that disease pathogenesis is a function of the following primary etiopathologies: (A) Chronic peripheral nociception/neuroinflammation associated with encumbrance of neuromuscular/osseous tissues (Figure 1); (B) Chronic immune activation associated with marked antigenic activity; (C) A combination of factors A and B.
A) Under postural/biomechanical challenge, sensitive neuromuscular tissues subject to encumbrance trigger noxious nociceptive input and glial activity. Responding glial cells propagate inflammatory signals, releasing pro-inflammatory cytokines and effecting central nociceptor terminal release of neuroactive molecules, including glutamate and NO.
B) Persistent neuroimmune stimulation, above systemic tolerance thresholds, associated with inadequate immunological responses to neurotrophic infection, often involving lymphotropic/gliotropic microorganisms (Hickie et al., 2006), may lead to immune suppression/exhaustion. In the context of immunological functional impairment tending toward autoimmunity (Bradley et al., 2013), this may involve somewhat circular processes of initial infection, inflammation, paired with enhanced serotonin receptor expression (Couch et al., 2015), and hence raised (inflammatory) pain, immunodeficiency, and progression/reactivation of opportunistic intercurrent (Smith and Thomas, 2015) and latent infections (Broderick et al., 2010); well represented among these pathogens (Nicolson et al., 2003) are those known to target sites of autoimmune inflammation (Posnett and Yarilin, 2005). Consistent with this picture, recurrent viral infections and concordant chronic systemic inflammation appear to be a hallmark of the disease (Raison et al., 2009).
C) Neuromuscular tissue inflaming anatomical stimuli modulate local immune responses. Equally, immune stimulation affecting altered serotonergic receptor activity, glial activation, and inflammation of neuromuscular tissues proximal to infection sites also enhances sensitivity of said tissues to postural/biomechanical stimulation. Neural excitation threshold reduction affected by select pathogenic strains may also play a role here (Oldstone, 1989).
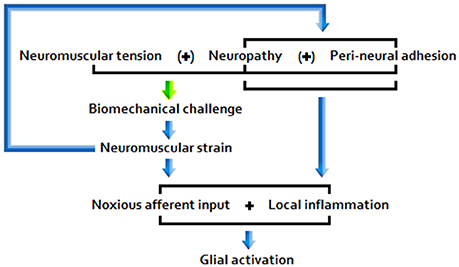
Figure 1. Encumbrance pathway “A”.
Sufficiently acute/sustained exposure to glio-toxins (B, C) and/or extracellular glutamate and pro-inflammatory cytokine elevation stemming from other neuroinflammatory stimuli (A, B, C) is associated with elevated, and potentially increasingly primed, glial activity and sickness behavior, as well as neuroinflammtory (excitotoxic) neurotoxicity and O+NS stress, and raised peripheral nervous sensitivity. Signal molecules released in relation to these effects act as to lower the threshold and opening characteristics of neuronal receptor channels, hence enhancing central neuronal sensitivity. See Figure 2 for a basic model of adverse products of glial activation and their downstream effects.
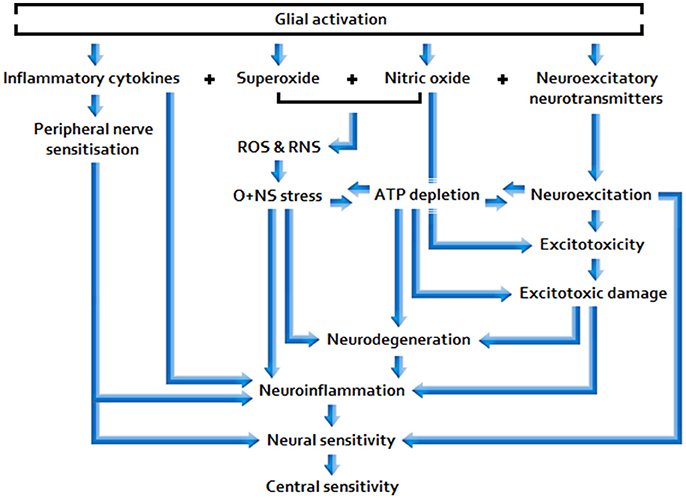
Figure 2. Effects of sustained glial activation.
Sustained glial activation enhances the potential for longer lasting, more intractable multi-systemic disarray, including space for: Synergistic neuro-glial distortions (i.e., gliopathy/microgliosis), neuroendocrine pathway aberration, mitochondrial dysfunction, and immunodeficiency; hence, glial-simulative neurological activity may have the capacity to fundamentally determine disease severity and prolongation. Given that low-dose Naltrexone (LDN) inhibits glial activation, the reported efficacy of this drug in ME/CFS and FMS would appear consistent with this proposition (Younger et al., 2013).
Optimal management/treatment of the encumbrance-linked abnormalities outlined in this article and the supporting literature warrants further study (Rowe et al., 2014). At a minimum, avoiding the noted “common postural risk factors” (flexion/slumping) and intense/repetitive neuro-dynamic movement of sensitive neuromuscular tissues is advisable. The use of special furniture/accessories may aid relevant behavior change e.g., rocking kneeling chairs/wobble cushions and vertebral support belts/braces.
Manual physical therapy focused on mild functional exercises and gentle stretching, intended to gradually increase ROM and reduce neuromuscular tension, may prove beneficial. Physical therapists should be aware, however, that the DRG may be sensitive/inflamed and that manipulation of this region, as well as direct nerve mobilization (particularly in the direction of strain), has the capacity to exacerbate symptoms considerably (Rowe et al., 2013).
Overcoming physical activity avoidance and facilitating physical rehabilitation are considered important recovery goals (Nijs et al., 2013). If progress here is to be optimally sustained, then related endeavors will, thus, remain within dynamic, individualized parameters, reflecting: Levels of strength and stamina (Eyskens et al., 2015), peripheral sensitivity (Staud et al., 2015), limited energy envelopes (Jason et al., 2009), and delayed mitochondrial energy (Lengert and Drossel, 2015) and muscular recovery (Paul et al., 1999).
Complementary dietary supplementation regimens might include: Omega-3 PUFAs (Maes and Twisk, 2010), CoQ10 (Chang et al., 2012), Withania Somnifera (Ashwagandha, Indian Ginseng) (Sankar et al., 2007), N-acetylcysteine (NAC) (Dean et al., 2011),* vitamin B12 (Zoccolella et al., 2009), curcumin (contained in turmeric) (Blaylock and Maroon, 2012), zinc (Hambidge and Krebs, 2007), magnesium (Cox et al., 1991), 2-aminoethanesulfonic acid (L-Taurine) (Leon et al., 2009), and carnitine (L-Carnitine) (Maes and Twisk, 2010). These substances reportedly aid healthy modulation of glutamatergic, neurotropic, and other relevant inflammatory pathways, protection against/the reversal of excitotoxicity, the rebalancing of glutamate:GABA, and mitochondrial and brain function.
A precautionary approach may be considered sensible with respect to additional factors that could conceivably complicate pathophysiological processes outlined in this article. These may include: Exposure to toxicants e.g., chemical pesticides (Dietert, 2014)/heavy metals (Giordano and Costa, 2012), mold (Aikawa and Suzuki, 1985), and both low frequency (ELF) (Rowland et al., 1998) and high frequency RFR (Pall, 2016) EMFs/EMR, as well as psychological stress (Prins et al., 2008), dehydration (Rowe et al., 1999), and exhaustion (Blaylock and Maroon, 2012).
The author confirms being the sole contributor of this work and approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Special thanks to Megan Arroll, Mark Livingstone, Raymond Perrin, and Peter Rowe for their feedback and encouragement.
Aikawa, H., and Suzuki, K. (1985). Enteric gliopathy in niacin-deficiency induced by CNS glio-toxin. Brain Res. 334, 354–356. doi: 10.1016/0006-8993(85)90231-8
Albright, F., Light, K., Light, A., Bateman, L., and Cannon-Albright, L. (2011). Evidence for a heritable predisposition to Chronic Fatigue Syndrome. BMC Neurol. 11:62. doi: 10.1186/1471-2377-11-62
Arroll, A. (2014). Chronic Fatigue Syndrome What You Need to Know About CFS/ME. London: Sheldon Press.
Badawy, A. A., Morgan, C. J., Llewelyn, M. B., Albuquerque, S. R., and Farmer, A. (2005). Heterogeneity of serum tryptophan concentration and availability to the brain in patients with the chronic fatigue syndrome. J. Psychopharmacol. 19, 385–391. doi: 10.1177/0269881105053293
Bardin, L. (2011). The complex role of serotonin and 5-HT receptors in chronic pain. Behav. Pharmacol. 22, 390–404. doi: 10.1097/FBP.0b013e328349aae4
Barger, S. W., Goodwin, M. E., Porter, M. M., and Beggs, M. L. (2007). Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J. Neurochem. 101, 1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x
Barnden, L., Crouch, B., Kwiatek, R., Burnet, R., Mernone, A., Chryssidis, S., et al. (2011). A brain MRI study of chronic fatigue syndrome: evidence of brainstem dysfunction and altered homeostasis. NMR Biomed. 24, 302–1312. doi: 10.1002/nbm.1692
Barron, D. F., Cohen, B. A., Geraghty, M. T., Violand, R., and Rowe, P. C. (2002). Joint hypermobility is more common in children with chronic fatigue syndrome than in healthy controls. J. Pediatr. 141, 421–425. doi: 10.1067/mpd.2002.127496
Berwaerts, J., Moorkens, G., and Abs, R. (1998). Secretion of growth hormone in patients with chronic fatigue syndrome. Growth Horm. IGF Res. 8(Suppl. 2), 127–129. doi: 10.1016/S1096-6374(98)80036-1
Bested, A. C., Saunders, P. R., and Logan, A. (2001). Chronic fatigue syndrome: neurological findings may be related to blood–brain barrier permeability. Med. Hypotheses 57, 231–237. doi: 10.1054/mehy.2001.1306
Blaylock, R. L., and Maroon, J. (2012). Natural plant products and extracts that reduce immunoexcitotoxicity-associated neurodegeneration and promote repair within the central nervous system. Surg. Neurol. Int. 3:19. doi: 10.4103/2152-7806.92935
Bozzinia, S., Boiocchib, C., Carlo-Stellaa, N., Ricevutic, G., and Cucciaa, M. (2012). The possible underworld of Chronic Fatigue Syndrome from neurotransmitters polymorphisms to disease. J. Neurol. Res. 2, 16–24. doi: 10.4021/jnr86w
Bradley, A. S., Ford, B., and Bansal, A. (2013). Altered functional B cell subset populations in patients with chronic fatigue syndrome compared to healthy controls. Clin. Exp. Immunol. 172, 73–80. doi: 10.1111/cei.12043
Brahmachari, S., and Pahan, K. (2010). Gender-specific expression of beta1 integrin of VLA-4 in myelin basic protein-primed T cells: implications for gender bias in multiple sclerosis. J. Immunol. 184, 6103–6113. doi: 10.4049/jimmunol.0804356
Brewer, J. H., Thrasher, J. D., Straus, D. C., Madison, R. A., and Hooper, D. (2013). Detection of Mycotoxins in patients with chronic fatigue syndrome. Toxins 5, 605–617. doi: 10.3390/toxins5040605
Broderick, G., Fuite, J., Kreitz, A., Vernon, S., Klimas, N., and Fletcher, M. (2010). A formal analysis of cytokine networks in Chronic Fatigue Syndrome. Brain Behav. Immun. 24, 1209–1217. doi: 10.1016/j.bbi.2010.04.012
Brouwer, B., and Packer, T. (1994). Corticospinal excitability in patients diagnosed with chronic fatigue syndrome. Muscle Nerve 17, 1210–1212. doi: 10.1002/mus.880171012
Carlo-Stella, N., Badulli, C., De Silvestri, A., Bazzichi, L., Martinetti, M., Lorusso, L., et al. (2006). A first study of cytokine genomic polymorphisms in CFS: positive association of TNF-857 and IFNgamma 874 rare alleles. Clin. Exp. Rheumatol. 24, 179–182.
CDC (2006). Press Briefing Transcripts: Chronic Fatigue Syndrome. Available online at: http://www.cdc.gov/media/transcripts/t060420.htm (Accessed September 25, 2016).
Chang, Y., Huang, S., and Wang, S. (2012). Coenzyme Q10 inhibits the release of glutamate in rat cerebrocortical nerve terminals by suppression of voltage-dependent calcium influx and mitogen-activated protein kinase signaling pathway. J. Agric. Food Chem. 60, 11909–11918. doi: 10.1021/jf302875k
Ciesielska, A., Joniec, I., Kurkowska-Jastrzebska, I., Cudna, A., Przybyłkowski, A., Członkowska, A., et al. (2009). The impact of age and gender on the striatal astrocytes activation in murine model of Parkinson's disease. Inflamm. Res. 58, 747–753. doi: 10.1007/s00011-009-0026-6
Colton, C. (2009). Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 4, 399–418. doi: 10.1007/s11481-009-9164-4
Costa, D., Tannock, C., and Brostoff, J. (1995). Brainstem perfusion is impaired in chronic fatigue syndrome. QJM 88, 767–773.
Couch, Y., Xie, Q., Lundberg, L., Sharp, T., and Anthony, D. (2015). A model of Post-Infection Fatigue is associated with increased TNF and 5-HT2A receptor expression in mice. PLoS ONE 10:e0130643. doi: 10.1371/journal.pone.0130643. eCollection 2015
Cox, I. M., Campbell, M. J., and Dowson, D. (1991). Red blood cell magnesium and chronic fatigue syndrome. Lancet 337, 757–760. doi: 10.1016/0140-6736(91)91371-Z
Dean, O., Giorlando, F., and Berk, M. (2011). N-acetylcysteine in psychiatry: current therapeutic evidence and potential mechanisms of action. J. Psychiatry Neurosci. 36, 78–86. doi: 10.1503/jpn.100057
Dietert, R. R. (2014). Developmental immunotoxicity, perinatal programming, and noncommunicable diseases: focus on human studies. Adv Med. 2014:867805. doi: 10.1155/2014/867805
Echeverry, S., Shi, X., Rivest, S., and Zhang, J. (2011). Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. J. Neurosci. 31, 10819–10828. doi: 10.1523/JNEUROSCI.1642-11.2011
Eyskens, J. B., Nijs, J., D'Août, K., Sand, A., Wouters, K., and Moorkens, G. (2015). Timed loaded standing in female chronic fatigue syndrome compared with other populations. J. Rehabil. Res. Dev. 52, 21–29. doi: 10.1682/JRRD.2014.03.0086
Felger, J. C., and Miller, A. (2012). Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front. Neuroendocrinol. 33, 315–327. doi: 10.1016/j.yfrne.2012.09.003
Fischler, B., Le Bon, O., Hoffmann, G., Cluydts, R., Kaufman, L., and De Meirleir, K. (1997). Sleep anomalies in the chronic fatigue syndrome. a comorbidity study. Neuropsychobiology 35, 115–122. doi: 10.1159/000119331
Freeman, R., and Komaroff, A. (1997). Does the chronic fatigue syndrome involve the autonomic nervous system? Am. J. Med. 102, 357–364. doi: 10.1016/S0002-9343(97)00087-9
Gay, C., Robinson, M., Lai, S., O'Shea, A., Craggs, J., Price, D., et al. (2016). Abnormal resting-state functional connectivity in patients with chronic fatigue syndrome: results of seed and data-driven analyses. Brain Connect. 6, 48–56. doi: 10.1089/brain.2015.0366
Georgiades, E., Behan, W., Kilduff, L., Hadjicharalambous, M., Mackie, E., Wilson, J., et al. (2003). Chronic fatigue syndrome: new evidence for a central fatigue disorder. Clin. Sci. 105, 213–218. doi: 10.1042/CS20020354
Giordano, G., and Costa, L. (2012). Developmental neurotoxicity: some old and new issues. ISRN Toxicol. 2012:814795. doi: 10.5402/2012/814795
Hambidge, K. M., and Krebs, N. F. (2007). Zinc deficiency: a special challenge. J. Nutr. 137, 1101–1105.
Hamre, H. (1995). [Chronic fatigue syndrome–a review of the literature]. [Article in Norwegian]. Tidsskr Nor Laegeforen. 115, 3042–3045.
Hao, Y., Yang, X., Chen, C., Yuan-Wang, Wang, X., Li, M., et al. (2010). STAT3 signalling pathway is involved in the activation of microglia induced by 2.45 GHz electromagnetic fields. Int. J. Radiat. Biol. 86, 27–36. doi: 10.3109/09553000903264507
Hardingham, G. E., Fukunaga, Y., and Bading, H. (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5, 405–414. doi: 10.1038/nn835
He, J., Hollingsworth, K., Newton, J., and Blamire, A. (2013). Cerebral vascular control is associated with skeletal muscle pH in chronic fatigue syndrome patients both at rest and during dynamic stimulation. Neuroimage Clin. 2, 168–173. doi: 10.1016/j.nicl.2012.12.006
Heim, C., Nater, U., Maloney, E., Boneva, R., Jones, J., and Reeves, W. (2009). Childhood trauma and risk for chronic fatigue syndrome: association with neuroendocrine dysfunction. Arch. Gen. Psychiatry 66, 72–80. doi: 10.1001/archgenpsychiatry.2008.508
Hickie, I., Davenport, T., Wakefield, D., Vollmer-Conna, U., Cameron, B., Vernon, S., et al. (2006). Post-infective and chronic fatigue syndromes precipitated by viral and non-viral pathogens: prospective cohort study. BMJ 333, 575–577. doi: 10.1136/bmj.38933.585764.AE
Hollingsworth, K., Hodgson, T., Macgowan, G., Blamire, A., and Newton, J. (2012). Impaired cardiac function in chronic fatigue syndrome measured using magnetic resonance cardiac tagging. J. Intern. Med. 271, 264–270. doi: 10.1111/j.1365-2796.2011.02429.x
Hooper, D. G., Bolton, V. E., Guilford, F., and Straus, D. (2009). Mycotoxin detection in human samples from patients exposed to environmental molds. Int. J. Mol. Sci. 10, 1465–1475. doi: 10.3390/ijms10041465
Hulsebosch, C. (2008). Gliopathy ensures persistent inflammation and chronic pain after spinal cord injury. Exp. Neurol. 214, 6–9. doi: 10.1016/j.expneurol.2008.07.016
Ickmans, K., Meeus, M., De Kooning, M., Lambrecht, L., Pattyn, N., and Nijs, J. (2015). Associations between cognitive performance and pain in chronic fatigue syndrome: comorbidity with Fibromyalgia does matter. Pain Phys. 18, E841–E852. doi: 10.1016/j.physio.2015.03.3465
Ifuku, M., Hossain, S., Noda, M., and Katafuchi, T. (2014). Induction of interleukin-1β by activated microglia is a prerequisite for immunologically induced fatigue. Eur. J. Neurosci. 40, 3253–3263. doi: 10.1111/ejn.12668
Jason, L., Benton, M., Torres-Harding, S., and Muldowney, K. (2009). The impact of energy modulation on physical functioning and fatigue severity among patients with ME/CFS. Patient Educ. Couns. 77, 237–241. doi: 10.1016/j.pec.2009.02.015
Jason, L. A., Corradi, K., Gress, S., Williams, S., and Torres-Harding, S. (2006). Causes of death among patients with chronic fatigue syndrome. Health Care Women Int. 27, 615–626. doi: 10.1080/07399330600803766
Jason, L., Sorenson, M., Porter, N., and Belkairous, N. (2011). An etiological model for myalgic encephalomyelitis/chronic fatigue syndrome. Neurosci. Med. 2, 14–27. doi: 10.4236/nm.2011.21003
Ji, R. R., Berta, T., and Nedergaard, M. (2013). Glia and pain: is chronic pain a gliopathy? Pain 154 (Suppl. 1), S10–S28. doi: 10.1016/j.pain.2013.06.022
Jones, D., Hollingsworth, K., Taylor, R., Blamire, A., and Newton, J. (2010). Abnormalities in pH handling by peripheral muscle and potential regulation by the autonomic nervous system in chronic fatigue syndrome. J. Intern. Med. 267, 394–401. doi: 10.1111/j.1365-2796.2009.02160.x
Kavelaars, A., Kuis, W., Knook, L., Sinnema, G., and Heijnen, C. (2000). Disturbed neuroendocrine-immune interactions in chronic fatigue syndrome. J. Clin. Endocrinol. Metab. 85, 692–696. doi: 10.1210/jcem.85.2.6379
Lengert, N., and Drossel, B. (2015). In silico analysis of exercise intolerance in myalgic encephalomyelitis/chronic fatigue syndrome. Biophys. Chem. 202, 21–31. doi: 10.1016/j.bpc.2015.03.009
Leon, R., Wu, H., Jin, Y., Wei, J., Buddhala, C., Prentice, H., et al. (2009). Protective function of taurine in glutamate-induced apoptosis in cultured neurons. J. Neurosci. Res. 87, 1185–1194. doi: 10.1002/jnr.21926
Liang, G., Cline, G., and Macica, C. (2007). IGF-1 stimulates de novo fatty acid biosynthesis by Schwann cells during myelination. Glia 55, 632–641. doi: 10.1002/glia.20496
Light, K., Agarwal, N., Iacob, E., White, A., Kinney, A., VanHaitsma, T., et al. (2013). Differing leukocyte gene expression profiles associated with fatigue in patients with prostate cancer versus Chronic Fatigue Syndrome. Psychoneuroendocrinology 38, 2983–2995. doi: 10.1016/j.psyneuen.2013.08.008
Longoni, M., and Ferrarese, C. (2006). Inflammation and excitotoxicity: role in migraine pathogenesis. Neurol. Sci. 27(Suppl. 2), S107–S110. doi: 10.1007/s10072-006-0582-2
Löscher, W., and Ebert, U. (1996). The role of the piriform cortex in kindling. Prog. Neurobiol. 50, 427–481. doi: 10.1016/S0301-0082(96)00036-6
Maes, M., and Twisk, F. (2010). Chronic fatigue syndrome: Harvey and Wessely's (bio)psychosocial model versus a bio(psychosocial) model based on inflammatory and oxidative and nitrosative stress pathways. BMC Med. 8:35. doi: 10.1186/1741-7015-8-35
Mailankot, M., Kunnath, A. P., Jayalekshmi, H., Koduru, B., and Valsalan, R. (2009). Radio frequency electromagnetic radiation (RF-EMR) from GSM (0.9/1.8GHz) mobile phones induces oxidative stress and reduces sperm motility in rats. Clinics 64, 561–565. doi: 10.1590/S1807-59322009000600011
Marshall-Gradisnik, S., Smith, P., Brenu, E., Nilius, B., Ramos, S., and Staines, D. (2015). Examination of Single Nucleotide Polymorphisms (SNPs) in Transient Receptor Potential (TRP) ion channels in Chronic Fatigue Syndrome patients. Immunol. Immunogenet. Insights 7, 1–6. doi: 10.4137/III.S25147
Mason, J., Ye, P., Suzuki, K., D'Ercole, A., and Matsushima, G. (2000). Insulin-like growth factor-1 inhibits mature oligodendrocyte apoptosis during primary demyelination. J Neurosci. 20, 5703–5708.
Meeus, M., and Nijs, J. (2007). Central sensitization: a biopsychosocial explanation for chronic widespread pain in patients with fibromyalgia and chronic fatigue syndrome. Clin. Rheumatol. 26, 465–473. doi: 10.1007/s10067-006-0433-9
Milrad, S., Hall, D., Jutagir, D., Lattie, E., Ironson, G., Wohlgemuth, W., et al. (2017). Poor sleep quality is associated with greater circulating pro-inflammatory cytokines and severity and frequency of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME) symptoms in women. J. Neuroimmunol. 303, 43–50. doi: 10.1016/j.jneuroim.2016.12.008
Miwa, K. (2015). Cardiac dysfunction and orthostatic intolerance in patients with myalgic encephalomyelitis and a small left ventricle. Heart Vessels 30, 484–489. doi: 10.1007/s00380-014-0510-y
Mohebiany, A. N., and Schneider, R. (2013). Glutamate Excitotoxicity in the cerebellum mediated by IL-1β. J. Neurosci. 33, 18353–18355. doi: 10.1523/jneurosci.3946-13.2013
Mörl, F., and Bradl, I. (2013). Lumbar posture and muscular activity while sitting during office work. J. Electromyogr. Kinesiol. 23, 362–368. doi: 10.1016/j.jelekin.2012.10.002
Morris, G., and Maes, M. (2012). A neuro-immune model of Myalgic Encephalomyelitis/Chronic fatigue syndrome. Metab. Brain Dis. 28, 523–540. doi: 10.1007/s11011-012-9324-8
Morris, G., and Maes, M. (2013). Myalgic encephalomyelitis/chronic fatigue syndrome and encephalomyelitis disseminata/multiple sclerosis show remarkable levels of similarity in phenomenology and neuroimmune characteristics. BMC Med. 11:205. doi: 10.1186/1741-7015-11-205
Myhill, S., Booth, N., and Mclaren-Howard, J. (2009). Chronic fatigue syndrome and mitochondrial dysfunction. Int. J. Clin. Exp. Med. 2, 1–16.
Nakatomi, Y., Mizuno, K., Ishii, A., Wada, Y., Tanaka, M., Tazawa, S., et al. (2014). Neuroinflammation in patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: an 11C-(R)-PK11195 PET study. J. Nucl. Med. 55, 945–950. doi: 10.2967/jnumed.113.131045
Narita, M., Nishigami, N., Narita, N., Yamaguti, K., Okado, N., Watanabe, Y., et al. (2003). Association between serotonin transporter gene polymorphism and chronic fatigue syndrome. Biochem. Biophys. Res. Commun. 311, 264–266. doi: 10.1016/j.bbrc.2003.09.207
Nicolson, G., Gan, R., and Haier, J. (2003). Multiple co-infections (Mycoplasma, Chlamydia, human herpes virus-6) in blood of chronic fatigue syndrome patients: association with signs and symptoms. APMIS 111, 557–566. doi: 10.1034/j.1600-0463.2003.1110504.x
Nijs, J., Meeus, M., Van Oosterwijck, J., Ickmans, K., Moorkens, G., Hans, G., et al. (2013). Fear of movement and avoidance behaviour toward physical activity in chronic-fatigue syndrome and fibromyalgia: state of the art and implications for clinical practice. Clin. Rheumatol. 32, 1121–1129. doi: 10.1007/s10067-013-2277-4
Nijs, J., Roussel, N., Van Oosterwijck, J., De Kooning, M., Ickmans, K., Struyf, F., et al. (2012). In the mind or in the brain? Scientific evidence for central sensitisation in chronic fatigue syndrome. Eur. J. Clin Invest. 42, 203–212. doi: 10.1111/j.1365-2362.2011.02575.x
Norden, D., and Godbout, J. (2012). Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 39, 19–34. doi: 10.1111/j.1365-2990.2012.01306.x
Ocon, A. (2013). Caught in the thickness of brain fog: exploring the cognitive symptoms of Chronic Fatigue Syndrome. Front. Physiol. 4:63. doi: 10.3389/fphys.2013.00063
Pall, M. (2003). Elevated nitric oxide/peroxynitrite theory of multiple chemical sensitivity: central role of N-methyl-D-aspartate receptors in the sensitivity mechanism. Environ. Health Perspect. 111, 1461–1464. doi: 10.1289/ehp.5935
Pall, M. (2007). Explaining Unexplained Illnesses: Disease Paradigm for Chronic Fatigue Syndrome, Multiple Chemical Sensitivity, Fibromyalgia, Post-Traumatic Stress Disorder, Gulf War Syndrome, and Others. New York, NY: Harrington Park Press.
Pall, M. (2016). Microwave frequency electromagnetic fields (EMFs) produce widespread neuropsychiatric effects including depression. J. Chem. Neuroanat. 75, 43–51. doi: 10.1016/j.jchemneu.2015.08.001
Paul, L., Wood, L., Behan, W., and Maclaren, W. (1999). Demonstration of delayed recovery from fatiguing exercise in chronic fatigue syndrome. Eur. J. Neurol. 6, 63–69. doi: 10.1046/j.1468-1331.1999.610063.x
Posnett, D. N., and Yarilin, D. (2005). Amplification of autoimmune disease by infection. Arthritis Res. Ther. 7, 74–84. doi: 10.1186/ar1691
Prins, Y., Crous, L., and Louw, Q. (2008). A systematic review of posture and psychosocial factors as contributors to upper quadrant musculoskeletal pain in children and adolescents. Physiother. Theory Pract. 24, 221–242. doi: 10.1080/09593980701704089
Puche, J. E., and Castilla-Cortázar, I. (2012). Human conditions of insulin-like growth factor-I (IGF-I) deficiency. J. Transl. Med. 10:224. doi: 10.1186/1479-5876-10-224
Puri, B., Jakeman, P., Agour, M., Gunatilake, K., Fernando, K., Gurusinghe, A., et al. (2012). Regional grey and white matter volumetric changes in myalgic encephalomyelitis (chronic fatigue syndrome): a voxel-based morphometry 3 T MRI study. Br. J. Radiol. 85, e270–e273. doi: 10.1259/bjr/93889091
Raison, C., Lin, J., and Reeves, W. (2009). Association of peripheral inflammatory markers with chronic fatigue in a population-based sample. Brain Behav. Immun. 23, 327–337. doi: 10.1016/j.bbi.2008.11.005
Rayhan, R. U., Ravindran, M., and Baraniuk, J. (2013). Migraine in gulf war illness and chronic fatigue syndrome: prevalence, potential mechanisms, and evaluation. Front. Physiol. 4:181. doi: 10.3389/fphys.2013.00181
Ren, K., and Dubner, R. (2008). Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Curr. Opin. Anaesthesiol. 21, 570–579. doi: 10.1097/ACO.0b013e32830edbdf
Renno, T., Krakowski, M., Piccirillo, C., Lin, J., and Owens, T. (1995). TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J Immunol. 154, 944–953.
Reyes, M., Nisenbaum, R., Hoaglin, D., Unger, E., Emmons, C., Randall, B., et al. (2003). Prevalence and incidence of chronic fatigue syndrome in Wichita, Kansas. Arch. Intern. Med. 163, 1530–1536. doi: 10.1001/archinte.163.13.1530
Ricci, G., Volpi, L., Pasquali, L., Petrozzi, L., and Siciliano, G. (2009). Astrocyte–neuron interactions in neurological disorders. J. Biol. Phys. 35, 317–336. doi: 10.1007/s10867-009-9157-9
Rowe, P., Barron, D., Calkins, H., Maumenee, I., Tong, P., and Geraghty, M. (1999). Orthostatic intolerance and chronic fatigue syndrome associated with Ehlers-Danlos syndrome. J. Pediatr. 135, 494–499. doi: 10.1016/S0022-3476(99)70173-3
Rowe, P., Fontaine, K., Lauver, M., Jasion, S., Marden, C., Moni, M., et al. (2016). Neuromuscular strain increases symptom intensity in chronic fatigue syndrome. PLoS ONE 11:e0159386. doi: 10.1371/journal.pone.0159386
Rowe, P., Fontaine, K., and Violand, R. (2013). Neuromuscular strain as a contributor to cognitive and other symptoms in chronic fatigue syndrome: hypothesis and conceptual model. Front. Physiol. 4:115. doi: 10.3389/fphys.2013.00115
Rowe, P., Marden, C., Flaherty, M., Jasion, S., Cranston, E., Johns, A., et al. (2014). Impaired range of motion of limbs and spine in chronic fatigue syndrome. J. Pediatr. 165, 360–366. doi: 10.1016/j.jpeds.2014.04.051
Rowland, R., Podd, J., Rapley, B., and Maisch, D. (1998). Chronic fatigue syndrome - is prolonged exposure to environmental level powerline frequency electromagnetic fields a co-factor to consider in treatment? ANCEM 17, 29–35.
Sankar, S., Manivasagam, T., Krishnamurti, A., and Ramanathan, M. (2007). The neuroprotective effect of Withania somnifera root extract in MPTP-intoxicated mice: an analysis of behavioral and biochemical variables. Cell. Mol. Biol. Lett. 12, 473–481. doi: 10.2478/s11658-007-0015-0
Schuh-Hofer, S., Wodarski, R., Pfau, D., Caspani, O., Magerl, W., Kennedy, J., et al. (2013). One night of total sleep deprivation promotes a state of generalized hyperalgesia: a surrogate pain model to study the relationship of insomnia and pain. Pain 154, 1613–1621. doi: 10.1016/j.pain.2013.04.046
Schwartzman, R. (2012). Systemic complications of complex regional pain syndrome. Neurosci. Med. 3, 225–242. doi: 10.4236/nm.2012.33027
Shanks, L., Jason, L., Evans, M., and Brown, A. (2013). Cognitive impairments associated with CFS and POTS. Front. Physiol. 4:113. doi: 10.3389/fphys.2013.00113
Smith, A., and Thomas, M. (2015). Chronic fatigue syndrome and increased susceptibility to upper respiratory tract infections and illnesses. Fatigue Biomed. Health Behav. 3, 156–163. doi: 10.1080/21641846.2015.1033271
Staud, R., Mokthech, M., Price, D., and Robinson, M. (2015). Evidence for sensitized fatigue pathways in patients with chronic fatigue syndrome. Pain 156, 750–759. doi: 10.1097/j.pain.0000000000000110
VanElzakker, M. (2013). Chronic fatigue syndrome from vagus nerve infection: a psychoneuroimmunological hypothesis. Med. Hypothese. 81, 414–423. doi: 10.1016/j.mehy.2013.05.034
VanNess, J., Stevens, S., Bateman, L., Stiles, T., and Snell, C. (2010). Postexertional malaise in women with chronic fatigue syndrome. J. Womens Health 19, 239–244. doi: 10.1089/jwh.2009.1507
Vecchiet, L., Montanari, G., Pizzigallo, E., Iezzi, S., de Bigontina, P., Dragani, L., et al. (1996). Sensory characterization of somatic parietal tissues in humans with chronic fatigue syndrome. Neurosci. Lett. 208, 117–120. doi: 10.1016/0304-3940(96)12559-3
Velizarov, S., Raskmark, P., and Kwee, S. (1999). The effects of radiofrequency fields on cell proliferation are non-thermal. Bioelectrochem. Bioenerg. 48, 177–180. doi: 10.1016/S0302-4598(98)00238-4
Visser, J., Lentjes, E., Haspels, I., Graffelman, W., Blauw, B., de Kloet, R., et al. (2001). Increased sensitivity to glucocorticoids in peripheral blood mononuclear cells of chronic fatigue syndrome patients, without evidence for altered density or affinity of glucocorticoid receptors. J. Investig. Med. 49, 195–204. doi: 10.2310/6650.2001.34047
Vollmer-Conna, U., Piraino, B., Cameron, B., Davenport, T., Hickie, I., Wakefield, D., et al. (2008). Cytokine polymorphisms have a synergistic effect on severity of the acute sickness response to infection. Clin. Infect. Dis. 47, 1418–1425. doi: 10.1086/592967
Winger, A., Kvarstein, G., Wyller, V., Ekstedt, M., Sulheim, D., Fagermoen, E., et al. (2015). Health related quality of life in adolescents with chronic fatigue syndrome: a cross-sectional study. Health Qual. Life Outcomes 13:96. doi: 10.1186/s12955-015-0288-3
Woolf, C. (2011). Central sensitization: implications for the diagnosis and treatment of pain. Pain 152(Suppl.), S2–S15. doi: 10.1016/j.pain.2010.09.030
Xie, L., Kang, H., Xu, Q., Chen, M., Liao, Y., Thiyagarajan, M., et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. doi: 10.1126/science.1241224
Yasui, M., Yoshimura, T., Takeuchi, S., Tokizane, K., Tsuda, M., Inoue, K., et al. (2014). A Chronic fatigue syndrome model demonstrates mechanical allodynia and muscular hyperalgesia via spinal microglial activation. Glia 62, 1407–1417. doi: 10.1002/glia.22687
Younger, J., Noor, N., Mccue, R., and Mackey, S. (2013). Low-dose naltrexone for the treatment of fibromyalgia: findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, crossover trial assessing daily pain levels. Arthritis Rheum. 65, 529–538. doi: 10.1002/art.37734
Keywords: chronic neurotrophic infection, glial hyper-activation, ME/CFS, neural sensitization, neuroimmune, neuroinflammation, neurotoxic neuroexcitation, noxious nociception
Citation: Glassford JAG (2017) The Neuroinflammatory Etiopathology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Front. Physiol. 8:88. doi: 10.3389/fphys.2017.00088
Received: 27 September 2016; Accepted: 01 February 2017;
Published: 17 February 2017.
Edited by:
Elisabeth Lambert, Baker IDI Heart and Diabetes Institute, AustraliaReviewed by:
Alla B. Salmina, Krasnoyarsk State Medical University named after Prof. V.F.Voino-Yasenetsky, RussiaCopyright © 2017 Glassford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julian A. G. Glassford, anVsaWFuZ2xhc3Nmb3JkQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.