
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 30 June 2016
Sec. Cardiac Electrophysiology
Volume 7 - 2016 | https://doi.org/10.3389/fphys.2016.00266
This article is part of the Research Topic J-wave and fragmented QRS: differences, similarities and mutual association View all 5 articles
 Ben N. Mercer1Gordon A. Begg1
Ben N. Mercer1Gordon A. Begg1 Stephen P. Page1,2Christopher P. Bennett2
Stephen P. Page1,2Christopher P. Bennett2 Muzahir H. Tayebjee1
Muzahir H. Tayebjee1 Saagar Mahida1,2*
Saagar Mahida1,2*The early repolarization (ER) pattern on the 12-lead electrocardiogram is characterized by J point elevation in the inferior and/or lateral leads. The ER pattern is associated with an increased risk of ventricular arrhythmias and sudden cardiac death (SCD). Based on studies in animal models and genetic studies, it has been proposed that J point elevation in ER is a manifestation of augmented dispersion of repolarization which creates a substrate for ventricular arrhythmia. A competing theory regarding early repolarization syndrome (ERS) proposes that the syndrome arises as a consequence of abnormal depolarization. In recent years, multiple clinical studies have described the characteristics of ER patients with VF in more detail. The majority of these studies have provided evidence to support basic science observations. However, not all clinical observations correlate with basic science findings. This review will provide an overview of basic science and genetic research in ER and correlate basic science evidence with the clinical phenotype.
The early repolarization pattern (ER) on the 12-lead electrocardiogram (ECG) is characterized by elevation of the J point in the inferior and/or lateral leads. The J point refers to the junction between the end of the QRS complex and the start of the ST segment. Elevation of the J point typically manifests as a slurring or notching of the terminal part of the QRS complex. Over the years the description of the ER pattern has varied significantly. Based on the most recent expert consensus, in order for ER to be present, the following criteria have to be met; (1) end-QRS notch or slur on the downslope of a prominent R-wave, (2) J point is ≥0.1 mV in 2 or more contiguous leads, excluding leads V1–V3, and (3) QRS duration is < 120 ms (MacFarlane et al., 2015; Figure 1).
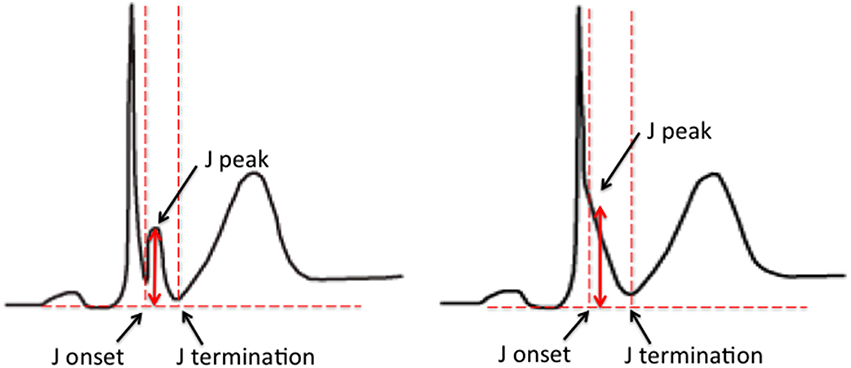
Figure 1. ER pattern on the surface ECG. The examples illustrate an end-QRS notch (left) and slur (right) on the downslope of a prominent R-wave. Current criteria for a diagnosis of ER include; (1) end-QRS notch or slur on the downslope of a prominent R-wave, (2) J point is ≥0.1 mV in 2 or more contiguous leads, excluding leads V1–V3, and (3) QRS duration is < 120 ms (Figure 1; MacFarlane et al., 2015).
The ER pattern is frequently observed in the general population, with a reported prevalence of 2–31% (Merchant et al., 2009; Maury and Rollin, 2013) but is probably closer to 5–10%. For decades, ER was regarded as a benign ECG variant in healthy populations (Goldman, 1953; Wasserburger and Alt, 1961). However, in recent years accumulating evidence has challenged this view. Multiple case-control and population-based studies have demonstrated that ER is associated with an increased risk of ventricular fibrillation (VF) and sudden cardiac death (SCD; Otto et al., 1984; Garg et al., 1998; Kalla et al., 2000; Horigome et al., 2003; Takeuchi et al., 2003; Haïssaguerre et al., 2008; Sinner et al., 2010; Derval et al., 2011; Haruta et al., 2011; Olson et al., 2011; Rollin et al., 2012; Rosso et al., 2012; Aizawa et al., 2013; Wu et al., 2013). It is important to emphasize however, that ER is common in the general population, and only a small subset of patients with the ECG pattern have an increased risk of SCD. The reported association between ER pattern and VF in these studies has led to the acknowledgment of a clinical syndrome, labeled the early repolarization syndrome (ERS).
The emergence of reports linking ER and SCD has led to significant interest into the pathophysiological basis of the ECG pattern. An in-depth understanding of the mechanistic link between ER and ventricular arrhythmogenesis is likely to have important implications for risk stratification and potentially therapy. This review will provide an overview of mechanistic and genetic research in ERS and correlate basic scientific observations with the clinical phenotype.
The first studies exploring the mechanistic basis of J point elevation on the ECG emerged in the 1950's (Osborn, 1953). Osborn reported the appearance of J point elevation secondary to severe hypothermia and acidosis in a canine model. Although it was unclear which of the two factors caused the ECG change, he speculated that the appearance of J waves (in lead II of the surface ECG) represented a “current of injury.” J point elevation was consistently associated with VF in the hypothermic dogs. Santos et al subsequently reported inferior J point elevation (also in lead II) in hypothermic dogs; see Figure 2. However, J point elevation was not associated with acidosis and the authors concluded that the pattern was induced by hypothermia (Santos and Kittle, 1958).
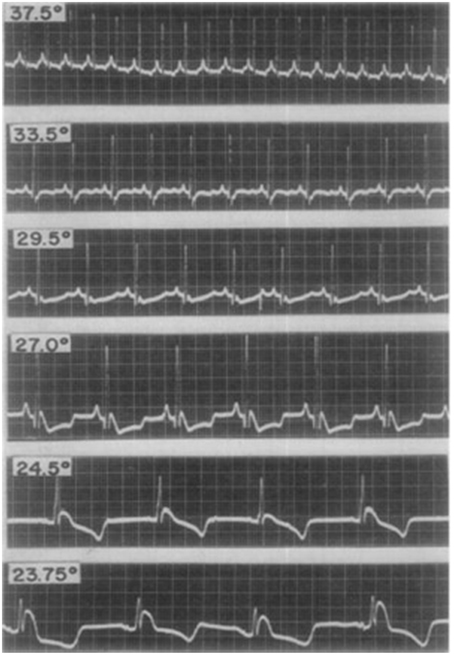
Figure 2. Electrocardiographic changes from early studies in a dog with experimental hypothermia. Progressively lower temperatures (centigrade) were observed to be associated with more pronounced J point elevation with a notch in the terminal portion of the QRS complex. Reproduced with permission (Santos and Kittle, 1958).
In order to correlate the appearance of J waves on the ECG with the ventricular action potential (AP), West et al performed a series of experiments in anesthetized open-chest dogs. They demonstrated that induced hypothermia resulted in J point elevation in ECG lead II with a progressively more pronounced phase 1 “notch” of the ventricular AP. The magnitude of J point elevation was directly proportional with the magnitude of the notch. Furthermore, the AP notch and J point elevation were rate dependent, with more pronounced changes at slower heart rates (West et al., 1959). Based on the observed response of the notch to changing heart rates, the authors argued that J point elevation was unlikely to represent a “current of injury.”
A more in-depth insight into the mechanistic basis of J point elevation came almost four decades later from an elegant series of experiments from Antzelevitch et al. (Yan and Antzelevitch, 1996). In an ex vivo model of arterially perfused canine left and right ventricular wedges, they simultaneously recorded transmural ECGs and endocardial, epicardial, and mid-myocardial APs and correlated J waves with transmural AP characteristics (Yan and Antzelevitch, 1996). Consistent with the observations from West et al. hypothermia simultaneously resulted in an increase in the J wave amplitude and a more prominent phase 1 notch in the epicardial AP but not the endocardial AP. They also demonstrated that direct pharmacological inhibition of the Ito current with 4-AP resulted in a reduction in the epicardial AP notch and corresponding reduction in the magnitude of the J wave. Furthermore, pacing the ventricular wedges with progressively more premature stimuli resulted in a progressive reduction in the amplitude of the J wave as well as the epicardial phase 1 AP notch. The observation that shortening the pacing cycle length attenuated the J wave in the transmural ECG was attributed to a progressively slower recovery of the Ito current at shorter cycle lengths. These observations suggest that a J wave is a manifestation of a transmural voltage gradient created by a more prominent epicardial AP notch relative to the endocardial AP notch. Furthermore, the Ito current is an important mediator of the transmural heterogeneity of the phase 1 notch. Overall the findings from these experiments form the basis of the repolarization theory for ERS.
Antzelevitch and colleagues have also provided valuable insights into the potential mechanisms of ventricular arrhythmia in ERS. Using the aforementioned canine model, with ventricular wedges from the inferior and lateral wall, they pharmacologically modeled ERS using a combination of Ito and IK-ATP agonists and ICa and INa antagonists. These ionic currents were targeted based on evidence from the above studies as well genetic studies in ERS (discussed in subsequent sections). The authors demonstrated marked epicardial dispersion of repolarization in the ERS model, with a loss of the phase 2 AP dome and AP shortening in some epicardial regions but not others. Heterogeneous repolarization allowed propagation of the AP dome, resulting in local re-excitation, which manifested as closely coupled extrasystolic activity (phase 2 re-entry). The interaction between triggering extrasystoles and a ventricular substrate with marked repolarization heterogeneity resulted in VF (Koncz et al., 2014). See Figure 3 for a schema demonstrating phase 2 re-entry. The study also investigated the effects of vagal modulation and pharmacological therapy on arrhythmia susceptibility and described differences in arrhythmia susceptibility in the inferior and lateral left ventricle (discussed in detail in subsequent sections).
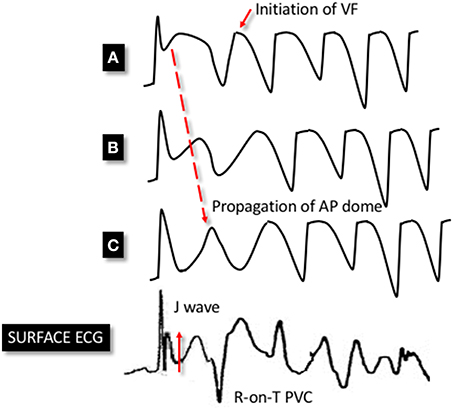
Figure 3. Schema demonstrating phase 2 reentry. Accentuation of the phase 1 notch of the AP is seen in a heterogenous manner across epicardial sites. This leads to an exaggerated phase 1 notch and phase 2 dome at point (A), whereas the phase 1 notch is accentuated to such an extent that the area is repolarised at point (C), resulting in the loss of the phase 2 dome. This area is no longer refractory and therefore allows the phase 2 dome at point (B) to be conducted to epicardial site (C). This results in extra systolic activity that manifests on the ECG as an R-on-T premature ventricular complex (PVC). This closely-coupled PVC initiates circus re-entry and PMVT/VF. The dispersion of the phase 1 notch manifests as J point elevation on the surface ECG.
Based on the above studies in animal models, ERS has widely been regarded as a repolarization abnormality, with J point elevation attributed to regional differences in the phase 1 AP notch. It is important to emphasize however that there remains controversy regarding the mechanistic basis of ERS. A competing theory proposes that ERS is a manifestation of a depolarization abnormality. The depolarization theory for ERS is largely based on parallels drawn with Brugada syndrome (BrS). BrS is characterized by J point elevation in the right precordial leads. Based on the clinical overlap between BrS and ERS, it has been proposed that BrS is a right ventricular variant of ERS (Benito et al., 2008; Haïssaguerre et al., 2008; Belhassen et al., 2009; Haïssaguerre et al., 2009b).
The mechanistic basis of BrS is also a subject of debate and the question regarding whether it is a repolarization abnormality or a depolarization abnormality remains unresolved (Meregalli et al., 2005). Indeed, uncertainty remains as to whether BrS is a primary channelopathy, a structural cardiac disease, or a combination of the two (Hoogendijk et al., 2010). The argument supporting BrS as a channelopathy is based on the identification of sodium channel mutations in BrS patients. However, it has been noted that such mutations are only present in ~20% of patients with BrS and the cause and effect of such mutations is not clear (Crotti et al., 2012). Some authors have therefore suggested that classifying BrS as a channelopathy is premature (Coronel et al., 2006). The argument supporting a structural cardiac disease is based on the identification right ventricular fibrosis and fatty infiltration, and wall motion abnormalities in BrS patients (Takagi et al., 2001; Frustaci et al., 2005; Zumhagen et al., 2009).
The depolarization theory for BrS proposes that a delay in activation in the right ventricular outflow tract (RVOT) results in a “current-to-load” mismatch, which manifests as J point and ST segment elevation (Hoogendijk et al., 2013). Evidence to support this theory is predominantly based on identification of fractionated electrograms and conduction slowing in the RVOT of BrS patients (Postema et al., 2008; Lambiase et al., 2009; Sacher et al., 2014). Studies in animal models have also demonstrated that sodium channel blockade results in excitation failure, manifesting as a BrS-type ST segment elevation (Hoogendijk et al., 2011). Based on computer modeling, it has been proposed that the Ito and ICaL currents play important roles in modulating excitation failure.
There is a relative paucity of direct evidence to support the depolarization theory in ERS. In contrast to BrS patients, structural abnormalities (fibrosis and fatty infiltration), and conduction slowing (fractionated electrograms) have not been reported ERS patients. Based on responses to drugs such as quinidine and isoproterenol and genetic studies (discussed in the next section), the Ito, ICaL, and INa currents have been implicated in ERS. Sequential activation of the INa, Ito, and ICaL play important roles in depolarization and propagation of the AP(Nerbonne and Kass, 2005). Perturbations in these currents could therefore result in regional conduction delay and/or excitation failure in the inferior and lateral walls, resulting in an ER pattern. It is important to note however, that in contrast to BrS patients, ERS patients do not display significant ST segment elevation, which would argue against excitation failure in ERS. There is some evidence to suggest that conduction slowing without excitation failure may still lead to J-point elevation (Coronel et al., 2005). However, conduction slowing has not been demonstrated in patients with ERS thus far.
Multiple ion channel mutations have been associated with ERS (Figure 4). The most frequently reported association has been between the KCNJ8 gene and ERS. KCNJ8 encodes the Kir6.1 subunit of the KATP channel. Haissaguerre et al. reported a KCNJ8 mutation in a case of ERS and VF storm (Haïssaguerre et al., 2009a). The same KCNJ8 mutation (S422L) was observed by Medeiros-Domingo et al. in 2 out of a series of 101 patients with ERS and BrS. Both the affected cases had ERS. Functional characterization of the mutation revealed a gain-of-function in the KATP channel (Medeiros-Domingo et al., 2010). Barajas-Martinez et al. also identified the KCNJ8 S422L mutation in 4 out of 204 ERS and BrS patients. They reported that the gain-of-function effect in KATP seen with this mutation is a consequence of a reduced sensitivity of the channel to intracellular ATP (Barajas-Martínez et al., 2012). More recently, mutations in ABCC9, which encodes the ATP-binding cassette transporter of IK−ATP have been identified in ERS patients. After screening a cohort of 150 patients with ERS and BrS, Hu et al identified four ERS patients with ABCC9 mutations (Hu et al., 2014). Functional analysis revealed that the mutation indirectly augments the IK−ATP current.
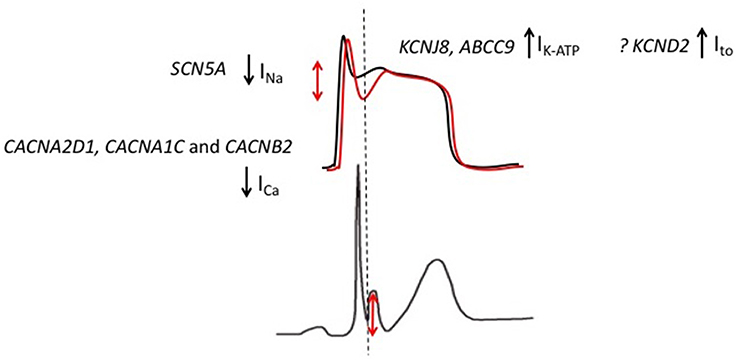
Figure 4. Genetic mutations implicated in patients with ERS. Gain-of-function mutations in KCNJ8 (ERS) and KCND2 (in an atypical J wave syndrome) result in augmented KATP and Ito currents, respectively. Loss-of-function mutations in CACNA2D1, CACNA1C, and CACNB2 result in an attenuated ICa while SCN5A mutations are predicted to result in an attenuation of INa.The overall effect is an outward shift in the balance of currents, which is predicted to preferentially accentuate the epicardial AP notch (red).
As discussed in the previous section, the Ito current has been implicated as an important contributor to J wave syndromes in animal studies. On the basis of these reports, Perrin et al. performed genetic screening in a cohort of 31 ERS patients and 20 Brugada syndrome patients for mutations in genes encoding Ito channel subunits (KCNA4, KCND2, and KCND3). They also screened the KCNJ8 gene. They did not identify mutations in any patients with infero-lateral ERS. A single mutation in KCND2 (D612N) was identified in a patent with an unusual J wave syndrome with evidence of QRS notching in the anterior leads. Functional characterization revealed a gain-of-function effect with an increased Ito current density.
Mutations in ion channels that encode inward currents have also been implicated in ERS. Burashnikov and colleagues reported multiple mutations in L-type calcium channel subunit genes (CACNA2D1, CACNA1C, and CACNB2) after screening 24 ERS patients (Burashnikov et al., 2010). Preliminary functional analysis revealed that the mutations are associated with loss-of-function type modulation (Napolitano and Antzelevitch, 2011). Watanabe et al. identified mutations in the SCN5A gene in three ER patients after screening a cohort of 26 patients. Functional studies demonstrated a loss-of-function effect, with all three mutations failing to generate an INa current (Watanabe et al., 2011).
The findings from genetic studies are generally consistent with the repolarization theory for ERS. Although, it should be noted that only one mutation has been identified in the Ito channel. The Ito and IK−ATP currents play important roles during the early phases of the AP and are more abundantly expressed in the epicardium relative to the endocardium. Therefore, gain-of-function mutations in Ito and IK−ATP are predicted to increase the transmural voltage gradient by preferentially augmenting the epicardial phase 1 notch (Antzelevitch and Barajas-Martinez, 2010; Benito et al., 2010). Loss-of-function ICa and INa channel mutations on the other hand are predicted to reduce depolarizing forces. As a result of higher epicardial expression of the aforementioned Ito and IK−ATP currents, the epicardium is more susceptible to the reduction in depolarizing forces (Meregalli et al., 2005). Therefore, loss-of-function ICa and INa channel mutations are also predicted to augment transmural voltage gradients during the early phases of the AP. Overall, gain-of-function IK−ATP and Ito channel mutations and loss-of-function ICa and INa channel mutations are predicted to result in an overall outward shift in the balance of currents, which in turn is predicted to accentuate the epicardial AP notch Figure 4; (Koncz et al., 2014; Patocskai et al., 2016). Of note, based on results from genetic studies, pharmacological augmentation of IK−ATP and Ito and attenuation of ICa and INa have been used to recapitulate both the ECG pattern and arrhythmic features of ERS in a canine ventricular wedge model (Koncz et al., 2014).
The reported genetic mutations may also support the depolarization theory for ERS. Loss-of-function INa channel mutations are predicted to directly reduce conduction velocity by decreasing the AP upslope. Ito and ICa channel mutations also have the potential to influence conduction velocity by altering the early phase of the AP (Yan and Antzelevitch, 1999; Hoogendijk et al., 2011, 2013). Evidence to support this theory has come from a recent study from Hoogendijk and colleagues. In porcine ex vivo model, they demonstrated that sodium channel blockade results in excitation failure and ST elevation on a pseudo ECG. Furthermore, using computer modeling, they demonstrated that manipulation of the Ito and ICa channels also influences current-to-load matching (Hoogendijk et al., 2011). Specifically, Ito reduced the current available for conduction while, ICaL had the opposite effect. On the basis of these observations, it is plausible that the reported mutations influence the ERS phenotype through altered depolarization.
As discussed in previous sections, the repolarization theory regarding the mechanistic basis of ERS proposes that J point elevation is a manifestation of augmented heterogeneity of the AP notch, which predisposes to phase 2 re-entry and ventricular arrhythmia. The depolarization theory on the other hand proposes that J point elevation is a manifestation of delayed activation of specific regions of the myocardium, a “current-to-load” mismatch. To date, the evidence in support of the repolarization theory has been more compelling.
Since the reports of an association between ER and SCD, multiple clinical studies have described the characteristics of ER patients who have had VF. The majority of clinical observations have been correlated with the repolarization theory (Figure 5). There are however some aspects of the clinical syndrome that may support the depolarization theory. There also remain some clinical features of ERS that are unexplained. The correlation between mechanistic theories and the clinical syndrome is discussed below.
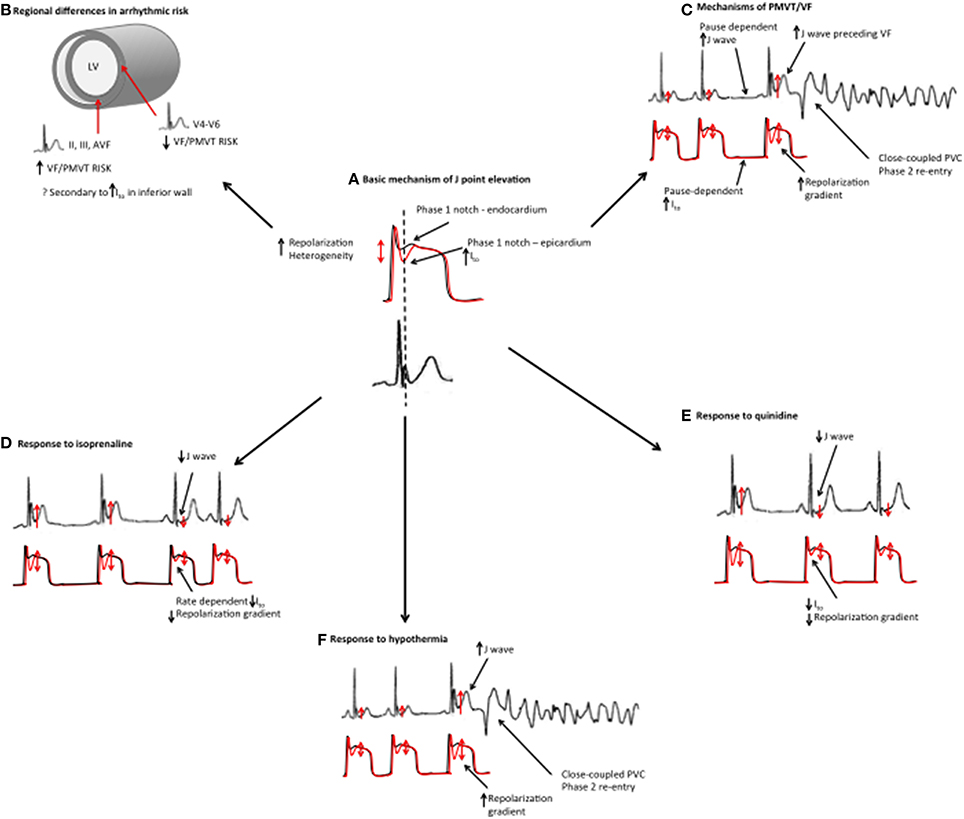
Figure 5. Mechanistic links between basic science studies and clinical features in ERS. (A) Basic science studies have demonstrated that J point elevation is a manifestation of increased transmural repolarization heterogeneity. (B) Patients with an inferior ER pattern have a higher risk of ventricular arrhythmias, which could potentially be related to increased Ito in the inferior wall. (C) ERS patients demonstrate more pronounced J point elevation immediately preceding VF episodes. Higher J waves may reflect augmented repolarization heterogeneity. J wave augmentation is pause-dependent, which may, be a reflection of Ito channel kinetics (slow temporal recovery from activation). The proposed mechanism of VF/PMVT is phase 2 reentry. (D) Isoproterenol suppresses VF/PMVT and reduces J point elevation in ERS, an effect that is thought to be mediated by elevated heart rates and reduced Ito channel recovery. (E) Quinidine also suppresses VF/PMVT in ERS by blocking the Ito current. (F) Hypothermia results in an augmented transmural repolarization gradient and an increased susceptibility to VF/PMVT.
In ERS patients with electrical storm, Nam et al. reported a marked increase in the J wave amplitude prior to the onset of VF. They also identified closely coupled ventricular premature beats (VPBs) triggering VF episodes. The authors recorded the Tpeak-Tend, a marker of repolarization in these patients and found significant differences in the Tpeak-Tend between ECG leads around the time of VF (Nam et al., 2010). These clinical observations are consistent with increased dispersion of repolarization and phase 2 re-entry preceding VF episodes, which correlates with an accentuation of the phase 1 AP notch preceding phase 2 re-entry in animal models (Patocskai et al., 2016).
Aizawa and colleagues also reported that patients with ERS display higher J wave amplitudes preceding VF episodes. Moreover, they reported pause-dependent accentuation of J point elevation preceding VF episodes (Aizawa et al., 2012). These observations are consistent with cellular electrophysiology studies demonstrating slow temporal recovery of the Ito channel from inactivation, which results in an augmented Ito current at slower heart rates (Yan and Antzelevitch, 1996). In keeping with the Ito channel kinetics, administration of acetylcholine has been reported to increase the phase 1 AP notch which in turn increases dispersion of repolarization and susceptibility to VF (Koncz et al., 2014). These data may also explain why VF is more frequently observed in patients with ERS during sleep or low levels of activity (Antzelevitch and Yan, 2015).
Invasive mapping studies in patients with ERS using monophasic action potential (MAP) catheters have demonstrated more prominent phase 1 AP notching in the inferior and lateral left ventricular epicardium (Mahida et al., 2015). These findings provide direct support for basic science reports demonstrating that a prominent epicardial phase 1 AP notch underlies J point elevation (Koncz et al., 2014). Invasive studies have also reported that amongst patients with an inferior ER pattern, short-coupled VPBs that potentially trigger VF originate from the inferior wall of the left ventricle (Haïssaguerre et al., 2008). These observations are consistent with studies in ex vivo models of ERS demonstrating that local re-excitation (phase 2 re-entry) and closely coupled extrasystolic activity, occurs in regions with augmented dispersion of repolarization.
The distribution of J point elevation has been proposed to influence risk of VF in ERS. Multiple clinical studies have demonstrated that J point abnormalities in the lateral ECG leads are associated with the lowest risk of VF whilst J point abnormalities in the inferior leads and global J point abnormalities are associated with progressively increasing risk of VF (Antzelevitch and Yan, 2010). These findings are consistent with animal studies demonstrating higher levels of Ito in the inferior wall of canine myocardium and in experimental conditions that mimic ERS, this region of myocardium is more vulnerable to VT/VF (Koncz et al., 2014). Further evidence has come from studies demonstrating a higher expression of Ito channel subunits in the inferior wall of the canine LV (Szentadrassy et al., 2005). It should be noted, however, that although current data offers explanation as to why an inferior pattern confers a higher risk of VF, they do not explain why an ER pattern in the lateral leads does not increase risk despite J point elevation.
A horizontal or downsloping morphology of the ST segment has been reported to be a marker of increased arrhythmic risk in cohorts of patients with the ER pattern (Tikkanen et al., 2011). To our knowledge there is no mechanistic data to explain this phenomenon. There are, however, some parallels that can be drawn from the BrS. Provocative testing in BrS patients with class 1c drugs can result in a type 1 ECG, with downsloping ST segment elevation being characteristic. It could be speculated that the horizontal/downsloping ST segments observed in ERS are analogous to those seen in BrS and are therefore representative of similar cellular and ionic abnormalities. It is important to note however that this proposed link is speculative; while downsloping ST segments are occasionally observed in ERS the T wave inversion observed in BrS are not seen in ERS.
A relatively high incidence if late potentials (LP) on signal-averaged ECG (SAECG) has been reported in ERS patients (Abe et al., 2010). The LP have been reported to demonstrate dynamic circadian changes, with more pronounced LP parameters at night time. LP are thought to represent areas of delayed ventricular conduction. Therefore, the identification of late potentials in ERS may support the depolarization theory of ERS. Further evidence to support this notion comes from studies in BrS patients. Invasive studies in BrS patients have demonstrated that LP on the SAECG are correlated with delayed local electrograms recorded during electrophysiology studies (Nagase et al., 2002). As discussed previously however, extrapolating results from BrS to ERS should be performed with caution.
Quinidine has been demonstrated to suppress VF storm in patients with ERS (Haïssaguerre et al., 2009a,b). One of the first reports on the efficacy of quinidine in ERS came from Haïssaguerre et al. (2009a). In a young patient with VF storm that was refractory to multiple antiarrhythmic drugs, only quinidine was effective at preventing VF. In a subsequent multicentre study, the same group demonstrated that quinidine effectively suppressed arrhythmia in 9 out of 9 ERS patients with VF storm (Haïssaguerre et al., 2009b). Their clinical observations are in keeping with basic science studies. Quinidine has consistently been demonstrated to abrogate the ER pattern, and the associated exaggerated phase 1 notch in in vitro models of ERS (Koncz et al., 2014; Szél and Antzelevitch, 2014). Quinidine is predicted to exert its antiarrhythmic effect in ERS patients by suppressing the Ito current (Antzelevitch and Yan, 2015).
Isoproterenol has been shown to effectively suppresses electrical storm in ERS patients in multiple studies (Haïssaguerre et al., 2009a). Furthermore, isoproterenol consistently normalizes J waves in patients with lateral ERS. In patients with inferior ERS, the response to beta adrenergic stimulation was more heterogenous (Roten et al., 2012a). Consistent with these clinical observations, studies in canine models have demonstrated that isoproterenol normalizes J point elevation (Yan and Antzelevitch, 1999). The effect of isoproterenol is likely to be mediated by a combination of heart rate acceleration, which is predicted to reduce Ito, and an increase in ICa, that restores the phase 2 dome and hence transmural AP homogeneity (Koncz et al., 2014; Patocskai et al., 2016). The distinctive and heterogenous clinical response of inferior ER to isoproterenol remains unclear. As discussed previously, Ito expression has been reported to be higher in the inferior wall (Koncz et al., 2014). Therefore, isoproterenol would be expected to have a homogenous effect in inferior ERS patients. A potential explanation may relate to the fact that the study included a heterogenous cohort of patients which included patients with VF, patients with syncope and asymptomatic patients. Therefore, not all patients in the study had the malignant form of ERS.
In addition to the reported efficacy of quinidine and isoproterenol in the aforementioned studies, a number of antiarrhythmic drugs including beta-blockers, lidocaine, mexiletine, and verapamil were demonstrated to be ineffective (Haïssaguerre et al., 2009b). Supporting evidence for these observations has come from both basic science and genetic studies. Beta-blockers and verapamil slow the heart rate, which is predicted to increase Ito channel recovery from inactivation (Yan and Antzelevitch, 1996). In turn, enhanced Ito channel recovery increases the phase 1 AP notch and dispersion of repolarization. In the specific case of verapamil, based on studies in canine models, calcium channel blockade leads to a loss of the phase 2 dome of the AP, an effect that is also predicted to enhance dispersion of repolarization (Fish and Antzelevitch, 2004). The failure of sodium channel blockers such as lidocaine and mexiletine to suppress arrhythmia in ERS is consistent with the reports of loss-of-function SCN5A mutations underlying ERS and animal studies demonstrating that INa blockade with pilsicainide increases susceptibility to VF (Watanabe et al., 2011; Koncz et al., 2014). Overall, the observations from basic science studies predict that beta-blockers, sodium channel blockers, and calcium channel blockers could actually have a proarrhythmic effect in ERS.
As discussed previously, ERS demonstrates significant overlap with BrS, which is typically associated with an accentuated J wave following administration of the sodium channel blocker ajmaline (Antzelevitch and Yan, 2015). Based on genetic studies reporting loss-of-function SCN5A mutations in ERS and basic science studies demonstrating more prominent AP notching and J waves following INa blockade, ajmaline would be predicted to accentuate J waves in ERS patients. Paradoxically however, a number of studies have reported that ajmaline attenuates J waves (Kawata et al., 2012; Roten et al., 2012b). Of note, this response to ajmaline has not been consistent in all studies. A recent case report involving epicardial left ventricular mapping in an ERS patient demonstrated that administration of pilsicainide, an INa blocker, resulted in augmentation of epicardial J waves in the lateral left ventricle. This change was however not observed on the surface ECG (Nakagawa et al., 2014). The mechanisms underlying these discrepant observations remain to be elucidated.
The association between hypothermia and J point elevation has been recognized for decades. In 1938, Tomaszewski et al. described a case of a frozen man with J point elevation (Tomaszewski, 1938). Interestingly, hypothermia has been reported to increase the risk of ventricular arrhythmias in patients with ERS. Bastiaenen et al. reported a case of inferior ER who developed VF storm with more pronounced J point elevation in response to therapeutic hypothermia (Bastiaenen et al., 2010). Federman and colleagues reported a case of infero-lateral ER with hypothermia-induced VF storm (Federman et al., 2013). The association between hypothermia and J point elevation is also well established in animal models (Osborn, 1953; Santos and Kittle, 1958; Emslie-smith et al., 1959; West et al., 1959). In a canine model of ERS, hypothermia has been reported to accentuate the epicardial AP notch, which results in a heterogeneous loss of the AP dome, phase 2 re-entry and VF (Gurabi et al., 2014). Based on the clinical and basic science findings, it could be speculated that hypothermia acts as an additional “insult” in ER patients with an underlying arrhythmogenic substrate.
The ER pattern has been associated with an increased risk of arrhythmic events in patients with coronary artery disease. Multiple previous studies have demonstrated that in patients with acute myocardial infarction, the presence of ER is associated with a significantly increased of ventricular arrhythmia (Naruse et al., 2012; Rudic et al., 2012; Tikkanen et al., 2012) ER has also been associated with an increased risk of ventricular arrhythmia in patients with chronic coronary artery disease (Patel et al., 2010).
Interestingly, the mechanisms of ventricular arrhythmia in patients with acute myocardial infarction are thought to be similar to those in patients with ERS. Yan and colleagues have previously demonstrated that in an ex vivo right ventricular wedge preparation, regions of ischaemia had a loss of the Ito-mediated epicardial AP dome with phase 2 re-entry resulting in VF (Di Diego and Antzelevitch, 2011; Yan et al., 2004). As discussed previously, a similar mechanism has been reported by the same group in an ex vivo model of ERS. On the basis of these observations, it could be speculated that ER augments arrhythmic risk in acute myocardial infarction by increasing risk of phase 2 re-entry (Naruse et al., 2012).
It is important to note that in patients with coronary artery disease, particularly chronic coronary artery disease, the presence of J point elevation in the inferior could potentially be a manifestation of delayed depolarization due to “peri-infarction block” in areas of scar (Castle, 1965; Littmann, 2010; Tikkanen et al., 2012). Indeed, as discussed previously, the ER pattern could also potentially be a manifestation of a depolarization abnormality. Overall, there remains significant uncertainty regarding the mechanistic links between ER and increased risk of ventricular arrhythmia in patients with coronary artery disease.
The ER pattern is more prevalent in patients with false tendons (Nakagawa et al., 2012). False tendons are fibromuscular bands containing conduction tissue which traverse the LV cavity (Luetmer et al., 1986; Abdulla et al., 1990; Loukas et al., 2007). The mechanistic link between false tendons has yet to be elucidated. It has been proposed that false tendons may create localized stretching, which in turn results in repolarization gradients and J point elevation. An alternative hypothesis is that false tendons result in altered conduction to the inferior and lateral walls, leading to J point elevation (Nakagawa et al., 2012). It is plausible therefore that J point elevation in patients with false tendons is a manifestation of an intraventricular conduction defect or repolarization abnormality. However, at this stage, these proposed mechanisms remain speculative.
Despite significant advances in basic science studies, important gaps remain in our understanding of the mechanistic basis of ERS. Current mechanistic evidence is based primarily on studies in an ex vivo model of ERS, and debate continues with regards the fundamental mechanistic abnormality that underpins ERS, i.e., repolarization or depolarization. While these ex vivo studies have provided valuable insights, a degree of caution must be exercised when extrapolating results to a clinical situation. In the future, more refined basic science and translational studies are necessary to further our understanding of the mechanistic basis of ERS. Potential areas of future research are outlined below.
To date, classical genetic studies in ERS have identified a small number of ion channel mutations. The mechanistic link between these mutations and ERS has not been fully characterized. The recent emergence of next-generation sequencing technologies such as exome sequencing and whole-genome sequencing has significantly enhanced our ability to identify the genetic basis of inherited arrhythmia syndromes. The application of these technologies in ERS patients is predicted to identify additional causative mutations. In terms of functional characterization of the genetic variants, induced pluripotent stem cell (iPS) technology represents a promising novel technique. Patient-specific iPS cells have successfully recapitulated the cellular electrophysiology of a number of inherited arrhythmia syndromes (Moretti et al., 2010; Itzhaki et al., 2011; Yazawa et al., 2011). The application of iPS technology in genotype-positive ERS patients is likely to enhance our understanding of the mechanistic basis of the trait.
Electrocardiographic imaging ECGI is an emerging translational research tool for inherited arrhythmia syndromes (Zhang et al., 2015). The technique involves recording body surface potentials using 250 body surface electrodes and computing potentials on the epicardium using the inverse solution (Rudy and Lindsay, 2015). Information from epicardial potentials is subsequently used to derive information on ventricular activation and repolarization. Ghosh et al. recently utilized ECGI in ERS patients to demonstrate early repolarization in regions of the LV that corresponded to the ECG leads with an ER pattern (Ghosh et al., 2010). These regions also had steep activation recovery interval (ARI) gradients, which are predicted to promote re-entrant arrhythmias (Ghosh et al., 2010). In the future, studies in larger numbers of ERS patients with different ER patterns, e.g., horizontal and downsloping ST segments, may provide valuable mechanistic information. It is important to note that while ECGI has demonstrated early promise, it is associated with significant limitations. For a given set of body surface potentials, there exist multiple potential cardiac solutions when the inverse solution is applied. Therefore, assumptions have to be applied to the analysis. Furthermore, small errors in the recorded body surface potentials can result in large errors in the computed epicardial potentials (Rudy and Lindsay, 2015).
To date, the majority of mechanistic studies in ERS have focused on ion channels and their potential contribution to abnormal repolarization and/or depolarization. In addition to ion channels, proteins that mediate cell-to-cell coupling, such as connections may play important roles in the pathogenesis of ERS. Interestingly, Nademanee and colleagues demonstrated that patients with BrS have reduced RVOT expression of connection-43, which is a major determinant of electrical coupling and conduction velocity in the ventricle (Nademanee et al., 2015). In the future, research into connnexin function in patients with ERS may provide important insights into the mechanistic basis of the trait.
Our understanding of the ERS has evolved rapidly in recent years but significant questions and controversies remain. The body of basic scientific data from animal and human studies continues to grow and there is evidence that developing electrophysiological and imaging technologies are beginning to bridge the gap between the bench and the bedside. These are important developments in terms of enhancing risk stratification as well as developing effective treatment strategies.
BM, manuscript writing; GAB, critical revisions; SP, critical revisions; CB, critical revisions; MT, critical revisions; SM, manuscript writing and critical revisions
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdulla, A. K., Frustaci, A., Martinez, J. E., Florio, R. A., Somerville, J., and Olsen, E. G. (1990). Echocardiography and pathology of left ventricular “false Tendons”. Chest 98, 129–132. doi: 10.1378/chest.98.1.129
Abe, A., Ikeda, T., Tsukada, T., Ishiguro, H., Miwa, Y., Miyakoshi, M., et al. (2010). Circadian variation of late potentials in idiopathic ventricular fibrillation associated with J waves: insights into alternative pathophysiology and risk stratification. Heart Rhythm 7, 675–682. doi: 10.1016/j.hrthm.2010.01.023
Aizawa, Y., Chinushi, M., Hasegawa, K., Naiki, N., Horie, M., Kaneko, Y., et al. (2013). Electrical storm in idiopathic ventricular fibrillation is associated with early repolarization. J. Am. Coll. Cardiol. 62, 1015–1019. doi: 10.1016/j.jacc.2013.05.030
Aizawa, Y., Sato, A., Watanabe, H., Chinushi, M., Furushima, H., Horie, M., et al. (2012). Dynamicity of the J-wave in idiopathic ventricular fibrillation with a special reference to pause-dependent augmentation of the J-wave. J. Am. Coll. Cardiol. 59, 1948–1953. doi: 10.1016/j.jacc.2012.02.028
Antzelevitch, C., and Barajas-Martinez, H. (2010). A gain-of-function I(K-ATP) mutation and its role in sudden cardiac death associated with J-wave syndromes. Heart Rhythm 7, 1472–1474. doi: 10.1016/j.hrthm.2010.07.027
Antzelevitch, C., and Yan, G.-X. (2010). J wave syndromes. Heart Rhythm 7, 549–558. doi: 10.1016/j.hrthm.2009.12.006
Antzelevitch, C., and Yan, G.-X. (2015). J-wave syndromes: brugada and early repolarization syndromes. Heart Rhythm 12, 1852–1866. doi: 10.1016/j.hrthm.2015.04.014
Barajas-Martínez, H., Hu, D., Ferrer, T., Onetti, C. G., Wu, Y., Burashnikov, E., et al. (2012). Molecular genetic and functional association of brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm 9, 548–555. doi: 10.1016/j.hrthm.2011.10.035
Bastiaenen, R., Hedley, P. L., Christiansen, M., and Behr, E. R. (2010). Therapeutic hypothermia and ventricular fibrillation storm in early repolarization syndrome. Heart Rhythm 7, 832–834. doi: 10.1016/j.hrthm.2010.02.037
Belhassen, B., Glick, A., and Viskin, S. (2009). Excellent long-term reproducibility of the electrophysiologic efficacy of quinidine in patients with idiopathic ventricular fibrillation and brugada syndrome. Pacing Clin. Electrophysiol. 32, 294–301. doi: 10.1111/j.1540-8159.2008.02235.x
Benito, B., Guasch, E., Rivard, L., and Nattel, S. (2010). Clinical and mechanistic issues in early repolarization of normal variants and lethal arrhythmia syndromes. J. Am. Coll. Cardiol. 56, 1177–1186. doi: 10.1016/j.jacc.2010.05.037
Benito, B., Sarkozy, A., Mont, L., Henkens, S., Berruezo, A., Tamborero, D., et al. (2008). Gender differences in clinical manifestations of brugada syndrome. J. Am. Coll. Cardiol. 52, 1567–1573. doi: 10.1016/j.jacc.2008.07.052
Burashnikov, E., Pfeiffer, R., Barajas-Martinez, H., Delpón, E., Hu, D., Desai, M., et al. (2010). Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7, 1872–1882. doi: 10.1016/j.hrthm.2010.08.026
Castle, C H, and, Keane, W. M. (1965). ELECTROCARDIOGRAPHIC “PERI-INFARCTION BLOCK”: A CLINICAL AND PATHOLOGIC CORRELATION. Circulation 31, 403–408.
Coronel, R., Berecki, G., and Opthof, T. (2006). Why the brugada syndrome is not yet a disease: syndromes, diseases, and genetic causality. Cardiovasc. Res. 72, 361–363. doi: 10.1016/j.cardiores.2006.09.004
Coronel, R., Casini, S., Koopmann, T. T., Wilms-Schopman, F. J., Verkerk, A. O., de Groot, J. R., et al. (2005). Right ventricular fibrosis and conduction delay in a patient with clinical signs of brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 112, 2769–2777. doi: 10.1161/CIRCULATIONAHA.105.532614
Crotti, L., Marcou, C. A., Tester, D. J., Castelletti, S., Giudicessi, J. R., Torchio, M., et al. (2012). Spectrum and prevalence of mutations involving BrS1- through BrS12-susceptibility genes in a cohort of unrelated patients referred for brugada syndrome genetic testing: implications for genetic testing. J. Am. Coll. Cardiol. 60, 1410–1418. doi: 10.1016/j.jacc.2012.04.037
Emslie-smith, D., Sladden, G. E., and Stirling, G. R. (1959). The significance of changes in the electrocardiogram in hypothermia. Br. Heart J. 21, 343–351.
Derval, N., Simpson, C. S., Birnie, D. H., Healey, J. S., Chauhan, V., Champagne, J., et al. (2011). Prevalence and characteristics of early repolarization in the CASPER registry: cardiac arrest survivors with preserved ejection fraction registry. J. Am. Coll. Cardiol. 58, 722–728. doi: 10.1016/j.jacc.2011.04.022
Di Diego, J. M., and Antzelevitch, C. (2011). Ischemic ventricular arrhythmias: experimental models and their clinical relevance. Heart Rhythm 8, 1963–1968. doi: 10.1016/j.hrthm.2011.06.036
Federman, N. J., Mechulan, A., Klein, G. J., and Krahn, A. D. (2013). Ventricular fibrillation induced by spontaneous hypothermia in a patient with early repolarization syndrome. J. Cardiovasc. Electrophysiol. 24, 586–588. doi: 10.1111/jce.12030
Fish, J. M., and Antzelevitch, C. (2004). Role of sodium and calcium channel block in unmasking the brugada syndrome. Heart Rhythm 1, 210–217. doi: 10.1016/j.hrthm.2004.03.061
Frustaci, A., Priori, S. G., Pieroni, M., Chimenti, C., Napolitano, C., Rivolta, I., et al. (2005). Cardiac histological substrate in patients with clinical phenotype of brugada syndrome. Circulation 112, 3680–3687. doi: 10.1161/CIRCULATIONAHA.105.520999
Garg, A., Finneran, W., and Feld, G. K. (1998). Familial sudden cardiac death associated with a terminal QRS abnormality on surface 12-lead electrocardiogram in the index case. J. Cardiovasc. Electrophysiol. 9, 642–647. doi: 10.1111/j.1540-8167.1998.tb00947.x
Ghosh, S., Cooper, D. H., Vijayakumar, R., Zhang, J., Pollak, S., Haïssaguerre, M., et al. (2010). Early repolarization associated with sudden death: insights from noninvasive electrocardiographic imaging. Heart Rhythm 7, 534–537. doi: 10.1016/j.hrthm.2009.12.005
Goldman, M. J. (1953). RS-T segment elevation in mid- and left precordial leads as a normal variant. Am. Heart J. 46, 817–820. doi: 10.1016/0002-8703(53)90080-5
Gurabi, Z., Koncz, I., Patocskai, B., Nesterenko, V. V., and Antzelevitch, C. (2014). Cellular mechanism underlying hypothermia-induced ventricular tachycardia/ventricular fibrillation in the setting of early repolarization and the protective effect of quinidine, cilostazol, and milrinone. Circ. Arrhythm. Electrophysiol. 7, 134–142. doi: 10.1161/CIRCEP.113.000919
Haïssaguerre, M., Chatel, S., Sacher, F., Weerasooriya, R., Probst, V., Gildas, L., et al. (2009a). Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/K ATP channel. J. Cardiovasc. Electrophysiol. 20, 93–98. doi: 10.1111/j.1540-8167.2008.01326.x
Haïssaguerre, M., Derval, N., Sacher, F., Jesel, L., Deisenhofer, I., de Roy, L., et al. (2008). Sudden cardiac arrest associated with early repolarization. N. Engl. J. Med. 358, 2016–2023. doi: 10.1056/NEJMoa071968
Haïssaguerre, M., Sacher, F., Nogami, A., Komiya, N., Bernard, A., Probst, V., et al. (2009b). Characteristics of recurrent ventricular fibrillation associated with inferolateral early repolarization role of drug therapy. J. Am. Coll. Cardiol. 53, 612–619. doi: 10.1016/j.jacc.2008.10.044
Haruta, D., Matsuo, K., Tsuneto, A., Ichimaru, S., Hida, A., Sera, N., et al. (2011). Incidence and prognostic value of early repolarization pattern in the 12-lead electrocardiogram. Circulation 123, 2931–2937. doi: 10.1161/CIRCULATIONAHA.110.006460
Hoogendijk, M. G., Opthof, T., Postema, P. G., Wilde, A. A., de Bakker, J. M., and Coronel, R. (2010). The brugada ECG pattern: a marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the brugada syndrome. Circ. Arrhythm. Electrophysiol. 3, 283–290. doi: 10.1161/CIRCEP.110.937029
Hoogendijk, M. G., Potse, M., and Coronel, R. (2013). Critical appraisal of the mechanism underlying J waves. J. Electrocardiol. 46, 390–394. doi: 10.1016/j.jelectrocard.2013.06.017
Hoogendijk, M. G., Potse, M., Vinet, A., de Bakker, J. M., and Coronel, R. (2011). ST segment elevation by current-to-load mismatch: an experimental and computational study. Heart Rhythm 8, 111–118. doi: 10.1016/j.hrthm.2010.09.066
Horigome, H., Shigeta, O., Kuga, K., Isobe, T., Sakakibara, Y., Yamaguchi, I., et al. (2003). Ventricular fibrillation during anesthesia in association with J waves in the left precordial leads in a child with coarctation of the aorta. J. Electrocardiol. 36, 339–343. doi: 10.1016/S0022-0736(03)00079-7
Hu, D., Barajas-Martínez, H., Terzic, A., Park, S., Pfeiffer, R., Burashnikov, E., et al. (2014). ABCC9 is a novel brugada and early repolarization syndrome susceptibility gene. Int. J. Cardiol. 171, 431–442. doi: 10.1016/j.ijcard.2013.12.084
Itzhaki, I., Maizels, L., Huber, I., Zwi-Dantsis, L., Caspi, O., Winterstern, A., et al. (2011). Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471, 225–229. doi: 10.1038/nature09747
Kalla, H., Yan, G.-X., and Marinchak, R. (2000). Ventricular fibrillation in a patient with prominent J (Osborn) waves and ST segment elevation in the inferior electrocardiographic leads: a brugada syndrome variant? J. Cardiovasc. Electrophysiol. 11, 95–98. doi: 10.1111/j.1540-8167.2000.tb00743.x
Kawata, H., Noda, T., Yamada, Y., Okamura, H., Satomi, K., Aiba, T., et al. (2012). Effect of sodium-channel blockade on early repolarization in inferior/lateral leads in patients with idiopathic ventricular fibrillation and brugada syndrome. Heart Rhythm 9, 77–83. doi: 10.1016/j.hrthm.2011.08.017
Koncz, I., Gurabi, Z., Patocskai, B. K., Panama, B., Szél, T., Hu, D., et al. (2014). Mechanisms underlying the development of the electrocardiographic and arrhythmic manifestations of early repolarization syndrome. J. Mol. Cell. Cardiol. 68, 20–28. doi: 10.1016/j.yjmcc.2013.12.012
Lambiase, P. D., Ahmed, A. K., Ciaccio, E. J., Brugada, R., Lizotte, E., Chaubey, S., et al. (2009). High-density substrate mapping in brugada syndrome: combined role of conduction and repolarization heterogeneities in arrhythmogenesis. Circulation 120, 106–117. doi: 10.1161/circulationaha.108.771401
Littmann, L. (2010). Early Repolarization on Electrocardiography. N. Engl. J. Med. 362, 1148. doi: 10.1056/NEJMc1000697
Loukas, M., Louis, R. G., Black, B., Pham, D., Fudalej, M., and Sharkees, M. (2007). False tendons: an endoscopic cadaveric approach. Clin. Anat. 20, 163–169. doi: 10.1002/ca.20347
Luetmer, P. H., Edwards, W. D., Seward, J. B., and Tajik, A. J. (1986). Incidence and distribution of left ventricular false tendons: an autopsy study of 483 normal human hearts. J. Am. Coll. Cardiol. 8, 179–183. doi: 10.1016/S0735-1097(86)80110-3
MacFarlane, P. W., Antzelevitch, C., Haissaguerre, M., Huikuri, H. V., Potse, M., Rosso, R., et al. (2015). The early repolarization pattern: a consensus paper. J. Am. Coll. Cardiol. 66, 470–477. doi: 10.1016/j.jacc.2015.05.033
Mahida, S., Derval, N., Sacher, F., Berte, B., Yamashita, S., Hooks, D. A., et al. (2015). History and clinical significance of early repolarization syndrome. Heart Rhythm 12, 242–249. doi: 10.1016/j.hrthm.2014.09.048
Maury, P., and Rollin, A. (2013). Prevalence of early repolarisation/j wave patterns in the normal population. J. Electrocardiol. 46, 411–416. doi: 10.1016/j.jelectrocard.2013.06.014
Medeiros-Domingo, A., Tan, B.-H., Crotti, L. J., Tester, D., Eckhardt, L., Cuoretti, A., et al. (2010). Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm 7, 1466–1471. doi: 10.1016/j.hrthm.2010.06.016
Merchant, F. M., Noseworthy, P. A., Weiner, R. B., Singh, S. M., and Ruskin, J. N, Reddy, V. Y. (2009). Ability of terminal QRS notching to distinguish benign from malignant electrocardiographic forms of early repolarization. Am. J. Cardiol. 104, 1402–1406. doi: 10.1016/j.amjcard.2009.06.062
Meregalli, P. G., Wilde, A. A. M., and Tan, H. L. (2005). Pathophysiological mechanisms of brugada syndrome: depolarization disorder, repolarization disorder, or more? Cardiovasc. Res. 67, 367–378. doi: 10.1016/j.cardiores.2005.03.005
Moretti, A., Bellin, M., Welling, A., Jung, C. B., Lam, J. T., Bott-Flügel, L., et al. (2010). Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 363, 1397–1409. doi: 10.1056/NEJMoa0908679
Nademanee, K., Raju, H., de Noronha, S. V., Papadakis, M., Robinson, L., Rothery, S., et al. (2015). Fibrosis, connexin-43, and conduction abnormalities in the brugada syndrome. J. Am. Coll. Cardiol. 66, 1976–1986. doi: 10.1016/j.jacc.2015.08.862
Nagase, S., Kusano, K. F., Morita, H., Fujimoto, Y., Kakishita, M., and Nakamura, K. (2002). Epicardial electrogram of the right ventricular outflow tract in patients with the brugada syndrome: using the epicardial lead. J. Am. Coll. Cardiol. 39, 1992–1995. doi: 10.1016/S0735-1097(02)01888-0
Nakagawa, K., Nagase, S., Morita, H., and Ito, H. (2014). Left ventricular epicardial electrogram recordings in idiopathic ventricular fibrillation with inferior and lateral early repolarization. Heart Rhythm 11, 314–317. doi: 10.1016/j.hrthm.2013.10.057
Nakagawa, M., Ezaki, K., Miyazaki, H., Wakisaka, O., Shinohara, T., Teshima, Y., et al. (2012). Electrocardiographic characteristics of patients with false tendon: possible association of false tendon with J waves. Heart Rhythm 9, 782–788. doi: 10.1016/j.hrthm.2011.12.022
Nam, G.-B., Ko, K.-H., Kim, J., Park, K.-M., Rhee, K.-S., Choi, K.-J., et al. (2010). Mode of onset of ventricular fibrillation in patients with early repolarization pattern vs. brugada syndrome. Eur. Heart J. 31, 330–339. doi: 10.1093/eurheartj/ehp423
Napolitano, C., and Antzelevitch, C. (2011). Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac voltage-dependent L-type calcium channel. Circ. Res. 108, 607–618. doi: 10.1161/CIRCRESAHA.110.224279
Naruse, Y., Tada, H., Harimura, Y., Hayashi, M., Noguchi, Y., Sato, A., et al. (2012). Early repolarization is an independent predictor of occurrences of ventricular fibrillation in the very early phase of acute myocardial infarction. Circ. Arrhythm. Electrophysiol. 5, 506–513. doi: 10.1161/CIRCEP.111.966952
Nerbonne, J. M., and Kass, R. S. (2005). Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253. doi: 10.1152/physrev.00002.2005
Olson, K. A., Viera, A. J., Soliman, E. Z., Crow, R. S., and Rosamond, W. D. (2011). Long-term prognosis associated with J-point elevation in a large middle-aged biracial cohort: the ARIC study. Eur. Heart J. 32, 3098–3106. doi: 10.1093/eurheartj/ehr264
Osborn, J. J. (1953). Experimental hypothermia; respiratory and blood pH changes in relation to cardiac function. Am. J. Physiol. 175, 389–398.
Otto, C. M., Tauxe, R. V., Cobb, L. A., Greene, H. L., Gross, B. W., Werner, J. A., et al. (1984). Ventricular fibrillation causes sudden death in southeast asian immigrants. Ann. Intern. Med. 101, 45–47. doi: 10.7326/0003-4819-101-1-45
Patel, R. B., Ng, J., Reddy, V., Chokshi, M., Parikh, K., Subacius, H., et al. (2010). Early repolarization associated with ventricular arrhythmias in patients with chronic coronary artery disease. Circ. Arrhythm.Electrophysiol. 3, 489–495. doi: 10.1161/CIRCEP.109.921130
Patocskai, B., Barajas-Martinez, H., Hu, D., Gurabi, Z., Koncz, I., and Antzelevitch, C. (2016). Cellular and ionic mechanisms underlying effects of cilostazol, milrinone and isoproterenol to suppress arrhythmogenesis in an experimental model of early repolarization syndrome. Heart Rhythm 13, 1326–1334. doi: 10.1016/j.hrthm.2016.01.024
Postema, P. G., van Dessel, P. F., de Bakker, J. M., Dekker, L. R., Linnenbank, A. C., and Linnenbank, A. C. (2008). Slow and discontinuous conduction conspire in brugada syndrome: a right ventricular mapping and stimulation study. Circ. Arrhythm. Electrophysiol. 1, 379–386. doi: 10.1161/CIRCEP.108.790543
Rollin, A., Maury, P., Bongard, V., Sacher, F., Delay, M., Duparc, A., et al. (2012). Prevalence, prognosis, and identification of the malignant form of early repolarization pattern in a population-based study. Am. J. Cardiol. 110, 1302–1308. doi: 10.1016/j.amjcard.2012.06.033
Rosso, R., Glikson, E., Belhassen, B., Katz, A., Halkin, A., Steinvil, A., et al. (2012). Distinguishing “benign” from “malignant early repolarization”: the value of the ST-segment morphology. Heart Rhythm 9, 225–229. doi: 10.1016/j.hrthm.2011.09.012
Roten, L., Derval, N., Sacher, F., Pascale, P., Scherr, D., Komatsu, Y., et al. (2012a). Heterogeneous response of J-wave syndromes to beta-adrenergic stimulation. Heart Rhythm 9, 1970–1976. doi: 10.1016/j.hrthm.2012.08.003
Roten, L., Derval, N., Sacher, F., Pascale, P., Wilton, S. B., Scherr, D., et al. (2012b). Ajmaline attenuates electrocardiogram characteristics of inferolateral early repolarization. Heart Rhythm Society 9, 232–239. doi: 10.1016/j.hrthm.2011.09.013
Rudic, B., Veltmann, C., Kuntz, E., Behnes, M., Elmas, E., Konrad, T., et al. (2012). Early repolarization pattern is associated with ventricular fibrillation in patients with acute myocardial infarction. Heart Rhythm 9, 1295–1300. doi: 10.1016/j.hrthm.2012.03.006
Rudy, Y., and Lindsay, B. D. (2015). Electrocardiographic imaging of heart rhythm disorders: from bench to bedside. Card. Electrophysiol. Clin. 7, 17–35. doi: 10.1016/j.ccep.2014.11.013
Sacher, F., Jesel, L., Jais, P., and Haïssaguerre, M. (2014). Insight into the mechanism of brugada syndrome: epicardial substrate and modification during ajmaline testing. Heart Rhythm 11, 732–734. doi: 10.1016/j.hrthm.2013.05.023
Santos, E. M., and Kittle, C. F. (1958). Electrocardiographic changes in the dog during hypothermia. Am. Heart J. 55, 415–420. doi: 10.1016/0002-8703(58)90057-7
Sinner, M. F., Reinhard, W., Müller, M., Beckmann, B.-M., Martens, E., Perz, S., et al. (2010). Association of early repolarization pattern on ecg with risk of cardiac and all-cause mortality: a population-based prospective cohort study (MONICA/KORA). PLoS Med. 7:e1000314. doi: 10.1371/journal.pmed.1000314
Szél, T., and Antzelevitch, C. (2014). Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of brugada syndrome. J. Am. Coll. Cardiol. 63, 2037–2045. doi: 10.1016/j.jacc.2014.01.067
Szentadrassy, N., Banyasz, T., Biro, T., Szabo, G., Toth, B. I., and Magyar, J. (2005). Apico-basal inhomogeneity in distribution of ion channels in canine and human ventricular myocardium. Cardiovasc. Res. 65, 851–860. doi: 10.1016/j.cardiores.2004.11.022
Takagi, M., Aihara, N., Kuribayashi, S., Taguchi, A., Shimizu, W., Kurita, T., et al. (2001). Localized right ventricular morphological abnormalities detected by electron-beam computed tomography represent arrhythmogenic substrates in patients with the brugada syndrome. Eur. Heart J. 22, 1032–1041. doi: 10.1053/euhj.2000.2424
Takeuchi, T., Sato, N., Kawamura, Y., Takahashi, F., Sato, M., Kikuchi, K., et al. (2003). A case of a short-coupled variant of torsades de pointes with electrical storm. Pacing Clin. Electrophysiol. 26(2 Pt 1), 632–636. doi: 10.1046/j.1460-9592.2003.00106.x
Tikkanen, J. T., Junttila, M. J., Anttonen, O., Aro, A. L., Luttinen, S., Kerola, T., et al. (2011). Early repolarization: electrocardiographic phenotypes associated with favorable long-term outcome. Circulation 123, 2666–2673. doi: 10.1161/CIRCULATIONAHA.110.014068
Tikkanen, J. T., Wichmann, V., Junttila, J. M., Rainio, M., Hookana, E., Lappi, O.-P., et al. (2012). Association of early repolarization and sudden cardiac death during an acute coronary event. Circ. Arrhythm. Electrophysiol. 5, 714–718. doi: 10.1161/CIRCEP.112.970863
Tomaszewski, W. (1938). Changement electrocardiographiques observes chez un homme mort de froid. Arch. Mal. Coeur Vaiss. 31, 525–528.
Wasserburger, R. H., and and, Alt, W. J. (1961). The normal RS-T segment elevation variant. Am. J. Cardiol. 8, 184–192.
Watanabe, H., Nogami, A., Ohkubo, K., Kawata, H., Hayashi, Y., Ishikawa, T., et al. (2011). Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fibrillation associated with early repolarization. Circ. Arrhythm. Electrophysiol. 4, 874–881. doi: 10.1161/CIRCEP.111.963983
West, T. C., Frederickson, E. L., and Amory, D. W. (1959). Single fiber recording of the ventricular response to induced hypothermia in the anesthetized dog: correlation with multicellular parameters. Circ. Res. 7, 880–888.
Wu, S.-H., Lin, X.-X., Cheng, Y.-J., Qiang, C.-C., and Zhang, J. (2013). Early repolarization pattern and risk for arrhythmia death: a meta-analysis. J. Am. Coll. Cardiol. 61, 645–650. doi: 10.1016/j.jacc.2012.11.023
Yan, G.-X., and Antzelevitch, C. (1996). Cellular basis for the electrocardiographic J wave. Circulation 93, 372–379. doi: 10.1161/01.CIR.93.2.372
Yan, G.-X., and Antzelevitch, C. (1999). Cellular basis for the brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation 100, 1660–1666. doi: 10.1161/01.CIR.100.15.1660
Yan, G.-X., Joshi, A., Guo, D., Hlaing, T., Martin, J., Xu, X., et al. (2004). Phase 2 reentry as a trigger to initiate ventricular fibrillation during early acute myocardial ischemia. Circulation 110, 1036–1041. doi: 10.1161/01.CIR.0000140258.09964.19
Yazawa, M., Hsueh, B., Jia, X., Pasca, A. M., Bernstein, J. A., Hallmayer, J., et al. (2011). Using induced pluripotent stem cells to investigate cardiac phenotypes in timothy syndrome. Nature 471, 230–234. doi: 10.1038/nature09855
Zhang, J., Sacher, F., Hoffmayer, K., O'Hara, T., Strom, M., Cuculich, P., et al. (2015). Cardiac electrophysiological substrate underlying the ECG phenotype and electrogram abnormalities in brugada syndrome patients. Circulation 131, 1950–1959. doi: 10.1161/CIRCULATIONAHA.114.013698
Keywords: early repolarization, ventricular fibrillation, sudden cardiac death
Citation: Mercer BN, Begg GA, Page SP, Bennett CP, Tayebjee MH and Mahida S (2016) Early Repolarization Syndrome; Mechanistic Theories and Clinical Correlates. Front. Physiol. 7:266. doi: 10.3389/fphys.2016.00266
Received: 24 March 2016; Accepted: 15 June 2016;
Published: 30 June 2016.
Edited by:
Jerzy Sacha, Opole University of Technology, PolandReviewed by:
Rob Gourdie, Virginia Tech Carilion Research Institute, USACopyright © 2016 Mercer, Begg, Page, Bennett, Tayebjee and Mahida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saagar Mahida, c2FhZ2FyN203QHlhaG9vLmNvLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.