 Preeya T. Shah
Preeya T. Shah Jiang Liu
Jiang Liu- Department of Pharmacology, Physiology and Toxicology, Joan C. Edwards School of Medicine, Marshall University, Huntington, WV, USA
Na/K-ATPase signaling has been implicated in different physiological and pathophysiological conditions. Accumulating evidence indicates that oxidative stress not only regulates the Na/K-ATPase enzymatic activity, but also regulates its signaling and other functions. While cardiotonic steroids (CTS)-induced increase in reactive oxygen species (ROS) generation is an intermediate step in CTS-mediated Na/K-ATPase signaling, increase in ROS alone also stimulates Na/K-ATPase signaling. Based on literature and our observations, we hypothesize that ROS have biphasic effects on Na/K-ATPase signaling, transcellular sodium transport, and urinary sodium excretion. Oxidative modulation, in particular site specific carbonylation of the Na/K-ATPase α1 subunit, is a critical step in proximal tubular Na/K-ATPase signaling and decreased transcellular sodium transport leading to increases in urinary sodium excretion. However, once this system is overstimulated, the signaling, and associated changes in sodium excretion are blunted. This review aims to evaluate ROS-mediated carbonylation of the Na/K-ATPase, and its potential role in the regulation of pump signaling and sodium reabsorption in the renal proximal tubule (RPT).
Accumulating evidence suggests that excessive dietary salt intake may play a role in the pathogenesis of hypertension, with more pronounced effects seen in salt-sensitive patients (Calhoun et al., 2008). Consequently, modest restriction of dietary salt and diuretic therapy are often recommended for treatment of resistant hypertension, particularly within the salt-sensitive sub-group (He and MacGregor, 2004; Calhoun et al., 2008). Long-term blood pressure (BP) regulation is highly associated with renal sodium handling (Guyton, 1991). Recent studies observe the CTS-activated Na/K-ATPase signaling pathway to contribute to RPT sodium handling and salt sensitivity (Buckalew, 2005; Meneton et al., 2005; Schoner and Scheiner-Bobis, 2007, 2008; Bagrov and Shapiro, 2008; Fedorova et al., 2010; Liu and Xie, 2010). Various intercellular and extracellular functions are regulated by the signaling function of the Na/K-ATPase. Discussion of Na/K-ATPase signaling and the downstream physiological and pathophysiological implications can be found in several references (Bertorello and Sznajder, 2005; Buckalew, 2005; Aperia, 2007; Liu and Shapiro, 2007; Schoner and Scheiner-Bobis, 2007, 2008; Bagrov and Shapiro, 2008; Bagrov et al., 2009; Blaustein et al., 2009; Li and Xie, 2009; Fedorova et al., 2010; Liu and Xie, 2010). Based on our recent observations, we focus on the effect of oxidative (carbonylation) modification of Na/K-ATPase and sodium handling in RPTs.
Protein Carbonylation and Cell Signaling
Biologically, electron reduction of oxygen (O2) leads to generation of ROS including superoxide (), hydrogen peroxide (H2O2), and hydroxyl radical (HO›). ROS is able to oxidize various types of biological molecules including proteins, lipids, and DNA, leading to their functional changes. Through Fenton's reaction, H2O2 (generated by or via other mechanisms) is reduced to HO› by coupling oxidation of reduced ferrous ion (Fe2+) to ferric ion (Fe3+). This metal-catalyzed oxidation (MCO) process oxidizes proteins by introducing carbonyl groups (such as aldehydes, ketones, or lactams) into the side chains of certain amino acids (such as proline, arginine, lysine and threonine; Stadtman and Berlett, 1991; Stadtman and Levine, 2000; Nyström, 2005). Unlike this direct (primary) carbonylation, indirect (secondary) carbonylation on lysine, cysteine, and histidine can occur by reactive carbonyl compounds generated from other types of oxidation, such as lipid and carbohydrate oxidation via Michael addition reactions and formation of Schiff bases. Protein carbonylation is a well-recognized marker of oxidative stress because of its stability, its effect on protein functions, and its link to various biological and pathological conditions. Oxidative stress has been implicated in the aging process, various conditions like ischemia-reperfusion and hyperoxia, and various human diseases like Alzheimer's disease, chronic lung disease, chronic renal failure, diabetes, and sepsis (Stadtman and Levine, 2000; Dalle-Donne et al., 2003, 2006a,b). Since the Fenton reaction involves the conversion of H2O2 to HO•, any specie of ROS with H2O2 as an intermediate and/or end product may stimulate the reaction. In biological systems, H2O2 is one of the most common end products of most ROS generating systems.
Oxidative modification of protein, reversible and irreversible, dynamically regulates protein structure, function, and trafficking, as well as cellular signaling and function (Go and Jones, 2013). Direct protein carbonylation is very stable and “chemically” irreversible (Stadtman and Berlett, 1991; Nyström, 2005). Recent studies from the Suzuki laboratory have demonstrated the role of the carbonylation/decarbonylation process in ROS signal transduction in which thiol groups were responsible for decarbonylation via enzymatic processes, likely through thioredoxin reductase (Wong et al., 2008, 2010, 2012, 2013).
CTS, the Na/K-ATPase, and Renal Sodium Handling
CTS, also known as endogenous digitalis-like substances, include plant-derived glycosides and vertebrate-derived aglycones (Schoner and Scheiner-Bobis, 2007, 2008). Although, the production and secretion of endogenous CTS are not completely understood, they appear to be regulated by angiotensin II and adrenocorticotropic hormone (Hamlyn et al., 1991; Laredo et al., 1997; Schoner and Scheiner-Bobis, 2007; Bagrov et al., 2009). CTS are present in measurable amounts under normal physiological conditions, and are elevated under a number of pathological states. Different species of endogenous CTS show variations in kinetics and tissue action in response to salt loading in both animal models and in human hypertensive patients (Haddy and Pamnani, 1998; Fedorova et al., 2005; Manunta et al., 2006; Schoner and Scheiner-Bobis, 2007, 2008).
The Na/K-ATPase belongs to the P-type ATPase family and consists of two non-covalently linked α and β subunits. Several α and β isoforms, expressed in a tissue-specific manner, have been identified and functionally characterized (Sweadner, 1989; Blanco and Mercer, 1998; Kaplan, 2002; Sanchez et al., 2006). The α1 subunit contains multiple structural motifs that interact with soluble, membrane and structural proteins (Jordan et al., 1995; Beggah and Geering, 1997; Feschenko et al., 1997; Zhang et al., 1998, 2006a; Yudowski et al., 2000; Lee et al., 2001; Xie and Cai, 2003; Barwe et al., 2005; Song et al., 2006; Tian et al., 2006). Binding to these proteins not only regulates the ion pumping function of the enzyme, but it also conveys signal transducing functions to the Na/K-ATPase (Xie and Cai, 2003; Kaplan, 2005; Kaunitz, 2006; Schoner and Scheiner-Bobis, 2007; Li and Xie, 2009).
It has been hypothesized for years that increases in endogenous CTS enhance natriuresis and diuresis by direct inhibition of renal tubular Na/K-ATPase, leading to reduced renal reabsorption of filtered sodium (Blaustein, 1977; Haddy et al., 1979; de Wardener and Clarkson, 1985). The first unequivocal demonstration of ouabain-like substance in human plasma was reported 25 years ago (Hamlyn et al., 1991). In vivo experiments suggest the essential role of endogenous CTS in modulating renal sodium excretion and BP with different approaches. First, administration of some (e.g., ouabain) but not all CTS induces natriuresis (Foulkes et al., 1992; Yates and McDougall, 1995). Second, in transgenic mice expressing ouabain-sensitive Na/K-ATPase α1 subunit, both acute salt load and ouabain infusion augment natriuretic responses, which may be inhibited by administration of an anti-digoxin antibody fragment (Dostanic-Larson et al., 2005; Loreaux et al., 2008). Third, immune-neutralization of endogenous CTS prevents CTS mediated natriuretic and vasoconstrictor effects (Fedorova et al., 2001, 2002; Bagrov and Shapiro, 2008; Nesher et al., 2009). Fourth, administration of the ouabain-antagonist, rostafuroxin (previously PST 2238) not only prevents ouabain induced Na/K-ATPase signaling, but also prevents ouabain-induced increase in BP (Ferrandi et al., 2004). Finally, in humans, high salt intake increases circulating endogenous CTS (Manunta et al., 2006; Anderson et al., 2008; Bagrov and Shapiro, 2008). Increased CTS excretion is directly linked to enhanced RPT-mediated fractional Na+ excretion, but inversely related to age and to age-dependent increase in salt-sensitivity (Anderson et al., 2008).
The Na/K-ATPase Signaling and Salt Sensitivity
Although historical focus has largely been on the direct inhibition of the Na/K-ATPase ion-exchange activity and sodium reabsorption in RPTs by CTS, this does not appear to be the predominant mechanism for several reasons. In contrast, the newly appreciated signaling function of Na/K-ATPase has been widely confirmed and provides a realistic, mechanistic framework that we will discuss further. We have observed that the renal Na/K-ATPase and its signaling play a key role in regulating renal sodium handling (Liu et al., 2002, 2004, 2005, 2011; Periyasamy et al., 2005; Oweis et al., 2006; Cai et al., 2008; Yan et al., 2013).
Decreases in basolateral Na/K-ATPase activity alone do not appear sufficient to decrease net sodium reabsorption across the renal tubular epithelium. In porcine RPT LLC-PK1 cells, ouabain activates the Na/K-ATPase signaling pathways and consequently redistributes the basolateral Na/K-ATPase and the apical sodium/hydrogen exchanger isoform 3 (NHE3) in a coordinated manner; this leads to symmetrical reduction of cell surface Na/K-ATPase and NHE3 expression, and ultimately decreases net transcellular sodium transport (Liu et al., 2002, 2004, 2005; Oweis et al., 2006; Cai et al., 2008; Figure 1). In this experimental model, the concentrations of ouabain used in vitro were chosen to mimic the concentrations of CTS seen in vivo with salt loading. No significant acute change in intracellular Na+ concentration was observed (Cai et al., 2008), further suggesting the coordination of the downregulation of both apical and basolateral sodium transporters. This Na/K-ATPase signaling mediated regulation of renal tubular epithelial ion transporters was additionally confirmed in in vivo studies (Periyasamy et al., 2005; Liu et al., 2011).
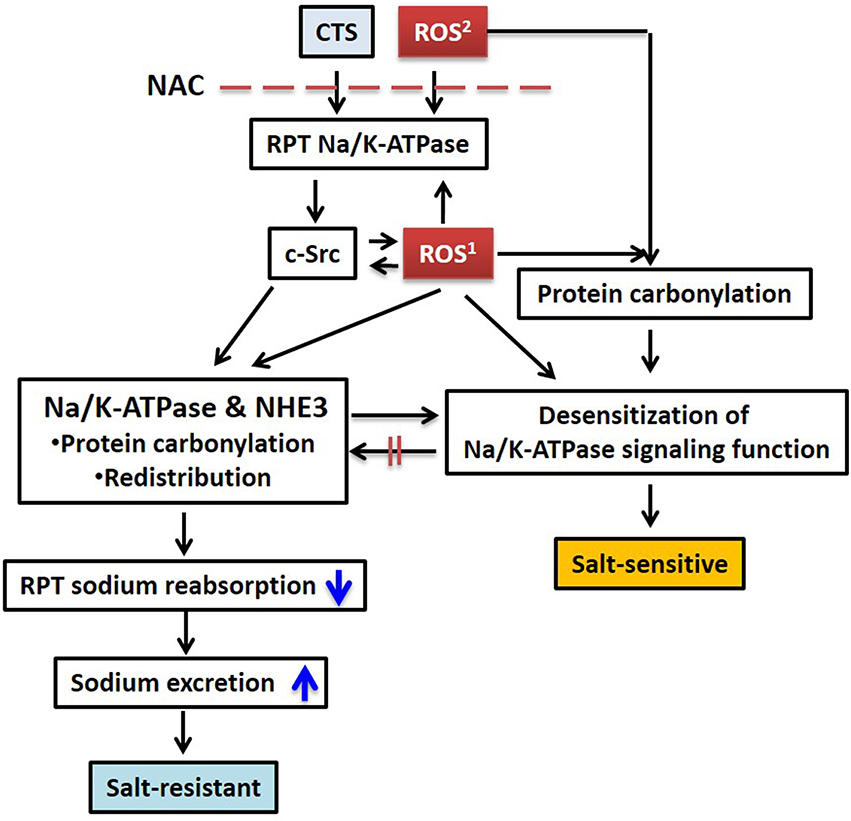
Figure 1. Schematic illustration of CTS and ROS mediated sodium handling in RPT and its relation to salt sensitivity. CTS induced intracellular ROS generation and extracellularly generated ROS stimulates Na/K-ATPase/c-Src signaling, protein carbonylation, and transporter redistribution. Pretreatment with NAC blocks this process. ROS1, CTS induced intracellular ROS generation through Na/K-ATPase signaling; ROS2, extracellular ROS generated by glucose oxidase or other stimuli. Please also see Figure 2. RPT, renal proximal tubule; NAC, N-acetyl-L-cysteine.
The Dahl salt-resistant (R) and salt-sensitive (S) strains were developed from Sprague Dawley rat strain by selective breeding, depending on the resistance or susceptibility to the hypertensive effects of high dietary sodium (Dahl et al., 1962). In these strains, sodium handling within the RPT is an essential determinant of their different BP responses (Dahl et al., 1974; Rapp, 1982; Rapp and Dene, 1985; Mokry and Cuppen, 2008). At the cost of elevated systolic BP, Dahl S rats rid excess sodium primarily via pressure-natriuresis. In contrast, Dahl R rats counterbalance salt loading via significant reduction of renal sodium reabsorption without increasing BP. In vivo studies indicate that impaired RPT Na/K-ATPase signaling appears to be causative of experimental Dahl salt-sensitivity (Liu et al., 2011). Specifically, in Dahl R rats (Jr strain), a high salt diet (2% NaCl for 7 days) and exposure to ouabain activates RPT Na/K-ATPase signaling and stimulates coordinated redistribution of Na/K-ATPase and NHE3, resulting with increases in renal sodium excretion. However, this does not occur in age- and gender-matched Dahl S rats (Jr strain; Liu et al., 2011). At present, we do not have a simple explanation for this occurrence. First, the α1 subunit is essentially the only α isoform expressed in RPTs (Blanco and Mercer, 1998; Summa et al., 2004) and genes coding α1 subunit and NHE3 (in rat chromosomes 1 and 2, respectively) are not located in identified and proposed BP quantitative trait loci (Joe, 2006). Second, there is no difference in α1 gene (Atp1a1) coding (Mokry and Cuppen, 2008), α1 ouabain-sensitivity (Nishi et al., 1993), and α1 expression (Liu et al., 2011) between these two strains. Third, acute salt-loading increases circulating CTS (ouabain and MBG) in both S and R rats (Fedorova et al., 2000). These observations suggest that there must be resistance to CTS signaling in the Dahl S rat, a phenomenon that we only partially understand and will discuss further below.
ROS and the Na/K-ATPase Signaling
It is well established that an increase in oxidative stress occurs in many forms of experimental hypertension (Kitiyakara et al., 2003; Touyz, 2004; Wilcox, 2005; Vaziri and Rodriguez-Iturbe, 2006; Welch, 2006). We and others have observed that a high salt diet stimulates endogenous CTS release and ROS generation within the RPT (Moe et al., 1991; Yang et al., 2008; Panico et al., 2009; McDonough, 2010; Banday and Lokhandwala, 2011; Liu et al., 2011). The increases in ROS (Meng et al., 2002; Kitiyakara et al., 2003; Taylor et al., 2006) regulate physiological processes including renal tubular ion transport, fluid reabsorption, and sodium excretion (Moe et al., 1991; Zhang et al., 2002; Garvin and Ortiz, 2003; Han et al., 2005; Yang et al., 2008; Panico et al., 2009; Wang et al., 2009; Banday and Lokhandwala, 2011; Liu et al., 2011; Schreck and O'Connor, 2011). In particular, increases in ROS regulate the activity and cellular distribution of the basolateral Na/K-ATPase as well as the apical NHE3 and sodium/glucose cotransporter, at least under normal circumstances (Moe et al., 1991; Fisher et al., 2001; Silva and Soares-da-Silva, 2007; Yang et al., 2007, 2008; Panico et al., 2009; Crajoinas et al., 2010; Johns et al., 2010; Liu et al., 2011). In our in vitro studies with LLC-PK1 cells, we have observed that ouabain stimulates generation of ROS which is critical in CTS-mediated Na/K-ATPase signaling, transporter trafficking, and 22Na+ flux (Yan et al., 2013). Pre-treatment with higher doses, but not a low dose, of anti-oxidant N-acetyl-L-cysteine (NAC) attenuated the effect of ouabain on c-Src activation and transcellular 22Na+ flux, suggesting a role of basal physiological redox status in the initiation of ouabain induced Na/K-ATPase signaling. This is analogous to the observation that the Na/K-ATPase activity is redox-sensitive with an “optimal redox potential range” (Petrushanko et al., 2006). While CTS stimulates ROS generation and Na/K-ATPase signaling in different in vitro and in vivo models (Xie et al., 1999; Liu et al., 2000, 2006; Tian et al., 2003; Kennedy et al., 2006a,b; Elkareh et al., 2007), glucose oxidase-induced H2O2 alone also stimulates Na/K-ATPase signaling, promotes Na/K-ATPase endocytosis, and inhibits active transcellular 22Na+ transport (Liu et al., 2006; Yan et al., 2013). The phenomenon of redox-sensitivity of the Na/K-ATPase has been demonstrated within many animal species, tissues, and cell types. Oxidative modification can affect Na/K-ATPase activity through different mechanisms. For example, S-glutathionylation is the formation of a mixed disulphide (cysteine-S-S-glutathione) between cysteine-SH with glutathione-SH or thiol-disulfide exchange. S-glutathionylation of cysteine residue(s) of the Na/K-ATPase α subunit can block the intracellular ATP-binding site, leading to inhibition of its enzymatic activity (Petrushanko et al., 2012). Ouabain-induced S-glutathionylation of cysteine of the Na/K-ATPase β1 subunit, a process affected by Na/K-ATPase conformational poise (Liu et al., 2012), reduces α1/β1 association and enzymatic activity by stabilizing the enzyme in an E2-prone conformation (Figtree et al., 2009). Oxidants and oxidative modification of the Na/K-ATPase can lead to functional changes (Kim and Akera, 1987; Xie et al., 1990; Huang et al., 1992; Mense et al., 1997; Thevenod and Friedmann, 1999; Zhang et al., 2002; Ellis et al., 2003; Bogdanova et al., 2006; Liu et al., 2006; Reifenberger et al., 2008; Blaustein et al., 2009; Figtree et al., 2009; White et al., 2009; Bibert et al., 2011; Figtree et al., 2012; Petrushanko et al., 2012; Soares-da-Silva, 2012) and formation of Na/K-ATPase oligomeric structure (Dobrota et al., 1999). As partner of Na/K-ATPase signaling, tyrosine kinase c-Src and lipid rafts (including caveolae structural component caveolins) are also redox-sensitive and critical in redox signaling platform formation (Seshiah et al., 2002; Zuo et al., 2005; Touyz, 2006; Zhang et al., 2006b; Han et al., 2008). This suggests a redox-sensitive Na/K-ATPase signaling and its possible role in ROS regulation.
Both ouabain and glucose oxidase-induced H2O2 stimulate Na/K-ATPase signaling and neutralization of the increase in ROS attenuated ouabain-induced effects (Xie et al., 1999; Liu et al., 2000, 2006; Tian et al., 2003; Kennedy et al., 2006a; Elkareh et al., 2007; Yan et al., 2013; Wang et al., 2014). We further observed that both ouabain and glucose oxidase-induced H2O2 stimulate direct protein carbonylation of Pro222 and Thr224 residues of the Na/K-ATPase α1 subunit (α1 carbonylation) in LLC-PK1 cells (Yan et al., 2013). The Pro222 and Thr224 are located in peptide 211VDNSSLTGESEPQTR225 [UniProtKB/Swiss-Prot No P05024 (AT1A1_PIG)]. While the α1 subunit is highly conserved amongst human, pig, rat, and mouse (the homology is over 98.5%), the identified peptide is 100% identical amongst these four species (Table 1). This peptide is located in the actuator (A) domain of α1 subunit, and Pro222/Thr224 are highly exposed and facing the nucleotide binding (N) domain of the α1 subunit. Upon ouabain binding, Na/K-ATPase undergoes conformational changes, in which the A domain is rotated to the N domain. Structure-function analysis indicates that these conformational changes may affect binding of the α1 subunit to signaling molecules such as c-Src and PI3K (Yatime et al., 2011). In addition, the peptide also contains the TGES motif that is the anchor of A domain rotation (Yatime et al., 2011). In immunoprecipitated α1 subunit, both ouabain and glucose oxidase do not induce formation of advanced glycation end products (AGEs) adducts. Like ouabain, glucose oxidase is able to activate Na/K-ATPase signaling, leading to reduction of transcellular22Na+ transport.

Table 1. Partial alignment of α1 subunit of human, pig, rat, and mouse.
Recent studies suggest that, in biological systems, protein carbonylation is reversible (decarbonylation) and may function as regulatory mechanism of cell signaling (Wong et al., 2008, 2010, 2012, 2013). We also observed a decarbonylation mechanism, which apparently reverses the carbonylation of the Na/K-ATPase α1 subunit induced by CTS (Yan et al., 2013). Removal of ouabain from the culture medium clearly reverses ouabain-mediated carbonylation; inhibition of de novo protein synthesis as well as degradation pathways through lysosome and proteasome does not affect this decarbonylation, which is still poorly understood. It is possible that carbonylation modification might stabilize the Na/K-ATPase in a certain conformational status favoring ouabain binding to the Na/K-ATPase α1 subunit and ouabain-Na/K-ATPase signaling, as seen in S-glutathionylation of cysteine residue(s) of the Na/K-ATPase (Figtree et al., 2009; Petrushanko et al., 2012). Nevertheless, the underlying mechanism might be physiologically significant since the carbonylation/decarbonylation process could be an important regulator of the RPT Na/K-ATPase signaling and sodium handling.
Oxidative (Carbonylation) Modification and Salt Sensitivity, a Hypothesis
Based on our data and literatures, we propose that carbonylation modification of RPT Na/K-ATPase α1 subunit has biphasic effects. (1) Physiological and controllable α1 carbonylation stimulates Na/K-ATPase signaling and sodium excretion, rendering salt resistance (Figure 2A) whereas (2) prolonged exposure to oxidant stress leads to overstimulated α1 carbonylation and desensitized Na/K-ATPase signaling, effecting salt sensitivity (Figure 2B). First, Dahl S rats show considerably higher basal levels of oxidative stress than R rats, and high salt diets increase renal oxidative stresses that contribute to salt-sensitive hypertension (Meng et al., 2002; Kitiyakara et al., 2003; Taylor et al., 2006). Second, while high salt diets increase circulating CTS, we have observed that a high salt diet (HS, 2% NaCl for 7 days) stimulates the Na/K-ATPase signaling in isolated RPTs from Dahl R but not S rats (i.e., impaired Na/K-ATPase signaling in S rats; Liu et al., 2011). Third, in RPT LLC-PK1 cells, CTS- and H2O2-mediated redox-sensitive Na/K-ATPase signaling and α1 carbonylation is involved in this signaling process, in a feed-forwarding mechanism (Yan et al., 2013). Fourth, high but not low concentration of NAC is able to prevent α1 carbonylation and Na/K-ATPase signaling (Yan et al., 2013). Even though it is still not clear of the carbonylation/decarbonylation process, it is reasonable to postulate that prolonged excessive α1 carbonylation (by CTS and/or other factors) might overcome the decarbonylation capacity, leading to desensitization or termination of the Na/K-ATPase signaling function. This is reminiscent of the observations in clinical trials using antioxidant supplements. The beneficial effect of antioxidant supplements is controversial and not seen in most clinical trials with administration of antioxidant supplements (reviewed in Touyz, 2004; Munzel et al., 2010). Low doses of antioxidant supplementation may be ineffective, but high doses may be even dangerous since excess antioxidants might become pro-oxidants if they cannot promptly be reduced in the anti-oxidant chain (Huang et al., 2006). It appears that the balance of the redox status, within a physiological range, may be critical in order to maintain beneficial ROS signaling.
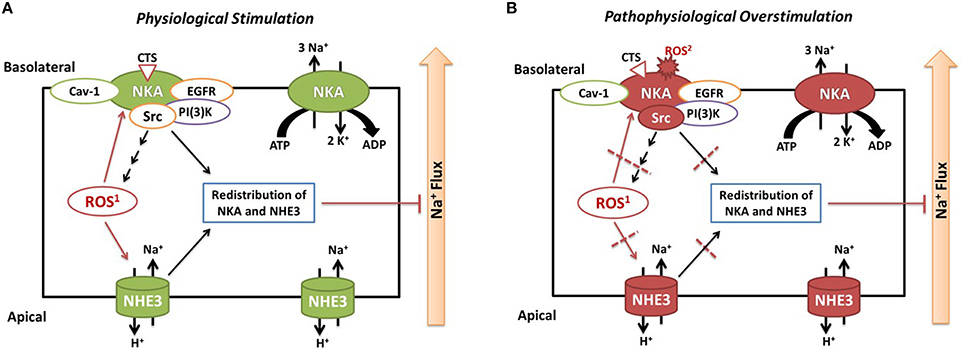
Figure 2. Illustrated hypothesis of α1 carbonylation on Na/K-ATPase signaling and sodium handling. Panel (A) shows events under physiological stimulation and Panel (B) shows events under pathophysiological overstimulation. ROS1, CTS induced intracellular ROS generation through Na/K-ATPase signaling; ROS2, extracellular ROS generated by glucose oxidase or other stimuli.
Perspective
The Na/K-ATPase has recently emerged as a therapeutic target (Aperia, 2007; Yatime et al., 2009). A clearer understanding of the mechanisms whereby a CTS-ROS-Na/K-ATPase signaling axis counterbalancing salt retention would have major pathophysiological and therapeutic implications, and further explain the progressive impairment of renal sodium handling under excessive oxidative stresses such as hypertension, aging, obesity, and diabetes. Impairment of coordinated regulation of the basolateral Na/K-ATPase and the apical NHE3 antiporter is implicated in salt-sensitive BP changes. Furthermore, recent evidence suggests both the Na/K-ATPase and, its adjacent signaling counterpart, c-Src to be redox-sensitive. Although carbonylation modification of the Na/K-ATPase is involved in the Na/K-ATPase signaling, a more thorough mechanistic understanding is necessary. Some pertinent questions remain to be resolved, such as the possible effect of carbonylation on CTS binding affinity, Na/K-ATPase conformational change, mechanisms of carbonylation/decarbonylation, and the destiny of the carbonylated Na/K-ATPase.
Author Contributions
PS, RM, YY, JS, and JL discussed the topic and wrote the manuscript. YY, JS, and JL reviewed and commended on the manuscript. PS and RM did the final edit.
Funding
This work was partially supported by NIH grants HL109015 and HL071556 to JS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anderson, D. E., Fedorova, O. V., Morrell, C. H., Longo, D. L., Kashkin, V. A., Metzler, J. D., et al. (2008). Endogenous sodium pump inhibitors and age-associated increases in salt sensitivity of blood pressure in normotensives. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1248–R1254. doi: 10.1152/ajpregu.00782.2007
Aperia, A. (2007). New roles for an old enzyme: Na,K-ATPase emerges as an interesting drug target. J. Intern. Med. 261, 44–52. doi: 10.1111/j.1365-2796.2006.01745.x
Bagrov, A. Y., and Shapiro, J. I. (2008). Endogenous digitalis: pathophysiologic roles and therapeutic applications. Nat. Clin. Pract. Nephrol. 4, 378–392. doi: 10.1038/ncpneph0848
Bagrov, A. Y., Shapiro, J. I., and Fedorova, O. V. (2009). Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 61, 9–38. doi: 10.1124/pr.108.000711
Banday, A. A., and Lokhandwala, M. F. (2011). Angiotensin II-mediated biphasic regulation of proximal tubular Na+/H+ exchanger 3 is impaired during oxidative stress. Am. J. Physiol. Renal Physiol. 301, F364–F370. doi: 10.1152/ajprenal.00121.2011
Barwe, S. P., Anilkumar, G., Moon, S. Y., Zheng, Y., Whitelegge, J. P., Rajasekaran, S. A., et al. (2005). Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol. Biol. Cell 16, 1082–1094. doi: 10.1091/mbc.e04-05-0427
Beggah, A. T., and Geering, K. (1997). Alpha and beta subunits of Na,K-ATPase interact with BiP and calnexin. Ann. N.Y. Acad. Sci. 834, 537–539. doi: 10.1111/j.1749-6632.1997.tb52311.x
Bertorello, A. M., and Sznajder, J. I. (2005). The dopamine paradox in lung and kidney epithelia: sharing the same target but operating different signaling networks. Am. J. Respir. Cell Mol. Biol. 33, 432–437. doi: 10.1165/rcmb.2005-0297tr
Bibert, S., Liu, C. C., Figtree, G. A., Garcia, A., Hamilton, E. J., Marassi, F. M., et al. (2011). FXYD proteins reverse inhibition of the Na+-K+ pump mediated by glutathionylation of its β1 subunit. J. Biol. Chem. 286, 18562–18572. doi: 10.1074/jbc.M110.184101
Blanco, G., and Mercer, R. W. (1998). Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am. J. Physiol. 275, F633–F650.
Blaustein, M. P. (1977). Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am. J. Physiol. Cell Physiol. 232, C165–C173.
Blaustein, M. P., Zhang, J., Chen, L., Song, H., Raina, H., Kinsey, S. P., et al. (2009). The pump, the exchanger, and endogenous ouabain: signaling mechanisms that link salt retention to hypertension. Hypertension 53, 291–298. doi: 10.1161/HYPERTENSIONAHA.108.119974
Bogdanova, I. P. A., Boldyrev, A., and Gassmann, M. (2006). Oxygen- and redox-induced regulation of the Na/K ATPase. Curr. Enzym. Inhib. 2, 37–59. doi: 10.2174/157340806775473490
Buckalew, V. M. (2005). Endogenous digitalis-like factors. An historical overview. Front. Biosci. 10, 2325–2334. doi: 10.2741/1701
Cai, H., Wu, L., Qu, W., Malhotra, D., Xie, Z., Shapiro, J. I., et al. (2008). Regulation of apical NHE3 trafficking by ouabain-induced activation of the basolateral Na+-K+-ATPase receptor complex. Am. J. Physiol. Cell Physiol. 294, C555–C563. doi: 10.1152/ajpcell.00475.2007
Calhoun, D. A., Jones, D., Textor, S., Goff, D. C., Murphy, T. P., Toto, R. D., et al. (2008). Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the american heart association professional education committee of the council for high blood pressure research. Hypertension 51, 1403–1419. doi: 10.1161/HYPERTENSIONAHA.108.189141
Crajoinas, R. O., Lessa, L. M., Carraro-Lacroix, L. R., Davel, A. P., Pacheco, B. P., Rossoni, L. V., et al. (2010). Posttranslational mechanisms associated with reduced NHE3 activity in adult vs. young prehypertensive SHR. Am. J. Physiol. Renal Physiol. 299, F872–F881. doi: 10.1152/ajprenal.00654.2009
Dahl, L. K., Heine, M., and Tassinari, L. (1962). Role of genetic factors in susceptibility to experimental hypertension due to chronic excess salt ingestion. Nature 194, 480–482. doi: 10.1038/194480b0
Dahl, L. K., Heine, M., and Thompson, K. (1974). Genetic influence of the kidneys on blood pressure. Evidence from chronic renal homografts in rats with opposite predispositions to hypertension. Circ. Res. 40, 94–101. doi: 10.1161/01.RES.40.4.94
Dalle-Donne, I., Aldini, G., Carini, M., Colombo, R., Rossi, R., and Milzani, A. (2006b). Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 10, 389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x
Dalle-Donne, I., Giustarini, D., Colombo, R., Rossi, R., and Milzani, A. (2003). Protein carbonylation in human diseases. Trends Mol. Med. 9, 169–176. doi: 10.1016/S1471-4914(03)00031-5
Dalle-Donne, I., Rossi, R., Colombo, R., Giustarini, D., and Milzani, A. (2006a). Biomarkers of oxidative damage in human disease. Clin. Chem. 52, 601–623. doi: 10.1373/clinchem.2005.061408
de Wardener, H. E., and Clarkson, E. M. (1985). Concept of natriuretic hormone. Physiol. Rev. 65, 658–759.
Dobrota, D., Matejovicova, M., Kurella, E. G., and Boldyrev, A. A. (1999). Na/K-ATPase under oxidative stress: molecular mechanisms of injury. Cell. Mol. Neurobiol. 19, 141–149. doi: 10.1023/A:1006909927287
Dostanic-Larson, I., Van Huysse, J. W., Lorenz, J. N., and Lingrel, J. B. (2005). The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc. Natl. Acad. Sci. U.S.A. 102, 15845–15850. doi: 10.1073/pnas.0507358102
Ellis, D. Z., Rabe, J., and Sweadner, K. J. (2003). Global loss of Na,K-ATPase and its nitric oxide-mediated regulation in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 23, 43–51.
Elkareh, J., Kennedy, D. J., Yashaswi, B., Vetteth, S., Shidyak, A., Kim, E. G., et al. (2007). Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 49, 215–224. doi: 10.1161/01.HYP.0000252409.36927.05
Fedorova, O. V., Agalakova, N. I., Talan, M. I., Lakatta, E. G., and Bagrov, A. Y. (2005). Brain ouabain stimulates peripheral marinobufagenin via angiotensin II signalling in NaCl-loaded Dahl-S rats. J. Hypertens. 23, 1515–1523. doi: 10.1097/01.hjh.0000174969.79836.8b
Fedorova, O. V., Kolodkin, N. I., Agalakova, N. I., Lakatta, E. G., and Bagrov, A. Y. (2001). Marinobufagenin, an endogenous alpha-1 sodium pump ligand, in hypertensive Dahl salt-sensitive rats. Hypertension 37, 462–466. doi: 10.1161/01.HYP.37.2.462
Fedorova, O. V., Lakatta, E. G., and Bagrov, A. Y. (2000). Endogenous Na,K pump ligands are differentially regulated during acute NaCl loading of Dahl Rats. Circulation 102, 3009–3014. doi: 10.1161/01.cir.102.24.3009
Fedorova, O. V., Shapiro, J. I., and Bagrov, A. Y. (2010). Endogenous cardiotonic steroids and salt-sensitive hypertension. Biochim. Biophys. Acta 1802, 1230–1236. doi: 10.1016/j.bbadis.2010.03.011
Fedorova, O. V., Talan, M. I., Agalakova, N. I., Lakatta, E. G., and Bagrov, A. Y. (2002). Endogenous ligand of alpha(1) sodium pump, marinobufagenin, is a novel mediator of sodium chloride–dependent hypertension. Circulation 105, 1122–1127. doi: 10.1161/hc0902.104710
Ferrandi, M., Molinari, I., Barassi, P., Minotti, E., Bianchi, G., and Ferrari, P. (2004). Organ hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and antagonized by PST 2238. J. Biol. Chem. 279, 33306–33314. doi: 10.1074/jbc.m402187200
Feschenko, M. S., Wetzel, R. K., and Sweadner, K. J. (1997). Phosphorylation of Na,K-ATPase by protein kinases. Sites, susceptibility, and consequences. Ann. N.Y. Acad. Sci. 834, 479–488. doi: 10.1111/j.1749-6632.1997.tb52306.x
Figtree, G. A., Keyvan Karimi, G., Liu, C. C., and Rasmussen, H. H. (2012). Oxidative regulation of the Na(+)-K(+) pump in the cardiovascular system. Free Radic. Biol. Med. 53, 2263–2268. doi: 10.1016/j.freeradbiomed.2012.10.539
Figtree, G. A., Liu, C. C., Bibert, S., Hamilton, E. J., Garcia, A., White, C. N., et al. (2009). Reversible oxidative modification: a key mechanism of Na+-K+ pump regulation. Circ. Res. 105, 185–193. doi: 10.1161/CIRCRESAHA.109.199547
Fisher, K. A., Lee, S. H., Walker, J., Dileto-Fang, C., Ginsberg, L., and Stapleton, S. R. (2001). Regulation of proximal tubule sodium/hydrogen antiporter with chronic volume contraction. Am. J. Physiol. Renal Physiol. 280, F922–F926.
Foulkes, R., Ferrario, R. G., Salvati, P., and Bianchi, G. (1992). Differences in ouabain-induced natriuresis between isolated kidneys of Milan hypertensive and normotensive rats. Clin. Sci. 82, 185–190. doi: 10.1042/cs0820185
Garvin, J. L., and Ortiz, P. A. (2003). The role of reactive oxygen species in the regulation of tubular function. Acta Physiol. Scand. 179, 225–232. doi: 10.1046/j.0001-6772.2003.01203.x
Go, Y. M., and Jones, D. P. (2013). The redox proteome. J. Biol. Chem. 288, 26512–26520. doi: 10.1074/jbc.r113.464131
Guyton, A. C. (1991). Blood pressure control–special role of the kidneys and body fluids. Science 252, 1813–1816. doi: 10.1126/science.2063193
Haddy, F. J., and Pamnani, M. B. (1998). Role of ouabain-like factors and Na-K-ATPase inhibitors in hypertension–some old and recent findings. Clin. Exp. Hypertens. 20, 499–508. doi: 10.3109/10641969809053228
Haddy, F. J., Pamnani, M. B., and Clough, D. L. (1979). Humoral factors and the sodium-potassium pump in volume expanded hypertension. Life Sci. 24, 2105–2117. doi: 10.1016/0024-3205(79)90108-5
Hamlyn, J. M., Blaustein, M. P., Bova, S., DuCharme, D. W., Harris, D. W., Mandel, F., et al. (1991). Identification and characterization of a ouabain-like compound from human plasma. Proc. Natl. Acad. Sci. U.S.A. 88, 6259–6263. doi: 10.1073/pnas.88.14.6259
Han, H. J., Lee, Y. J., Park, S. H., Lee, J. H., and Taub, M. (2005). High glucose-induced oxidative stress inhibits Na+/glucose cotransporter activity in renal proximal tubule cells. Am. J. Physiol. Renal Physiol. 288, F988–F996. doi: 10.1152/ajprenal.00327.2004
Han, W., Li, H. V., Villar, A. M., Pascua, A. M., Dajani, M. I., Wang, X., et al. (2008). Lipid rafts keep NADPH oxidase in the inactive state in human renal proximal tubule cells. Hypertension 51, 481–487. doi: 10.1161/HYPERTENSIONAHA.107.103275
He, F. J., and MacGregor, G. A. (2004). Effect of longer-term modest salt reduction on blood pressure. Cochrane Database Syst. Rev. 3:CD004937. doi: 10.1002/14651858.CD004937
Huang, H. Y., Caballero, B., Chang, S., Alberg, A. J., Semba, R. D., Schneyer, C. R., et al. (2006). The efficacy and safety of multivitamin and mineral supplement use to prevent cancer and chronic disease in adults: a systematic review for a national institutes of health state-of-the-science conference. Ann. Intern. Med. 145, 372–385. doi: 10.7326/0003-4819-145-5-200609050-00135
Huang, W. H., Wang, Y., and Askari, A. (1992). (Na+ + K+)-ATPase: inactivation and degradation induced by oxygen radicals. Int. J. Biochem. 24, 621–626. doi: 10.1016/0020-711X(92)90337-Z
Joe, G. M. B. (2006). Genetic Analysis of Inherited Hypertension in the Rat. Amsterdam: Elsevier Science.
Johns, E. J., O'shaughnessy, B., O'Neill, S., Lane, B., and Healy, V. (2010). Impact of elevated dietary sodium intake on NAD(P)H oxidase and SOD in the cortex and medulla of the rat kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R234–R240. doi: 10.1152/ajpregu.00541.2009
Jordan, C., Püschel, B., Koob, R., and Drenckhahn, D. (1995). Identification of a binding motif for ankyrin on the alpha-subunit of Na+,K(+)-ATPase. J. Biol. Chem. 270, 29971–29975. doi: 10.1074/jbc.270.50.29971
Kaplan, J. H. (2002). Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 71, 511–535. doi: 10.1146/annurev.biochem.71.102201.141218
Kaplan, J. H. (2005). A moving new role for the sodium pump in epithelial cells and carcinomas. Sci. STKE 2005:pe31. doi: 10.1126/stke.2892005pe31
Kaunitz, J. D. (2006). Membrane transport proteins: not just for transport anymore. Am. J. Physiol. Renal Physiol. 290, F995–F996. doi: 10.1152/ajprenal.00515.2005
Kennedy, D. J., Vetteth, S., Periyasamy, S. M., Kanj, M., Fedorova, L., Khouri, S., et al. (2006a). Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 47, 488–495. doi: 10.1161/01.HYP.0000202594.82271.92
Kennedy, D. J., Vetteth, S., Xie, M., Periyasamy, S. M., Xie, Z., Han, C., et al. (2006b). Ouabain decreases sarco(endo)plasmic reticulum calcium ATPase activity in rat hearts by a process involving protein oxidation. Am. J. Physiol. Heart Circ. Physiol. 291, H3003–H3011. doi: 10.1152/ajpheart.00603.2006
Kim, M. S., and Akera, T. (1987). O2 free radicals: cause of ischemia-reperfusion injury to cardiac Na+-K+-ATPase. Am. J. Physiol. Heart Circ. Physiol. 252, H252–H257.
Kitiyakara, C., Chabrashvili, T., Chen, Y., Blau, J., Karber, A., Aslam, S., et al. (2003). Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J. Am. Soc. Nephrol. 14, 2775–2782. doi: 10.1097/01.asn.0000092145.90389.65
Laredo, J., Shah, J. R., Lu, Z. R., Hamilton, B. P., and Hamlyn, J. M. (1997). Angiotensin II stimulates secretion of endogenous ouabain from bovine adrenocortical cells via angiotensin type 2 receptors. Hypertension 29, 401–407. doi: 10.1161/01.HYP.29.1.401
Lee, K., Jung, J., Kim, M., and Guidotti, G. (2001). Interaction of the alpha subunit of Na,K-ATPase with cofilin. Biochem. J. 353, 377–385. doi: 10.1042/bj3530377
Liu, C. C., Garcia, A., Mahmmoud, Y. A., Hamilton, E. J., Galougahi, K. K. N., Fry, G. A., et al. (2012). Susceptibility of β1 Na+-K+ pump subunit to glutathionylation and oxidative inhibition depends on conformational state of pump. J. Biol. Chem. 287, 12353–12364. doi: 10.1074/jbc.M112.340893
Liu, J., Kesiry, R., Periyasamy, S. M., Malhotra, D., Xie, Z., and Shapiro, J. I. (2004). Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney Int. 66, 227–241. doi: 10.1111/j.1523-1755.2004.00723.x
Liu, J., Liang, M., Liu, L., Malhotra, D., Xie, Z., and Shapiro, J. I. (2005). Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int. 67, 1844–1854. doi: 10.1111/j.1523-1755.2005.00283.x
Liu, J., Periyasamy, S. M., Gunning, W., Fedorova, O. V., Bagrov, A. Y., Malhotra, D., et al. (2002). Effects of cardiac glycosides on sodium pump expression and function in LLC-PK1 and MDCK cells. Kidney Int. 62, 2118–2125. doi: 10.1046/j.1523-1755.2002.00672.x
Liu, J., and Shapiro, J. I. (2007). Regulation of sodium pump endocytosis by cardiotonic steroids: Molecular mechanisms and physiological implications. Pathophysiology 14, 171–181. doi: 10.1016/j.pathophys.2007.09.008
Liu, J., Tian, J., Haas, M., Shapiro, J. I., Askari, A., and Xie, Z. (2000). Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 275, 27838–27844. doi: 10.1074/jbc.M002950200
Liu, J., and Xie, Z. J. (2010). The sodium pump and cardiotonic steroids-induced signal transduction protein kinases and calcium-signaling microdomain in regulation of transporter trafficking. Biochim. Biophys. Acta 1802, 1237–1245. doi: 10.1016/j.bbadis.2010.01.013
Liu, J., Yan, Y., Liu, L., Xie, Z., Malhotra, D., Joe, B., et al. (2011). Impairment of Na/K-ATPase signaling in renal proximal tubule contributes to Dahl salt-sensitive hypertension. J. Biol. Chem. 286, 22806–22813. doi: 10.1074/jbc.m111.246249
Li, Z., and Xie, Z. (2009). The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflugers Arch. 457, 635–644. doi: 10.1007/s00424-008-0470-0
Liu, L., Li, J., Liu, J., Yuan, Z., Pierre, S. V., Qu, W., et al. (2006). Involvement of Na+/K+-ATPase in hydrogen peroxide-induced hypertrophy in cardiac myocytes. Free Radic. Biol. Med. 41, 1548–1556. doi: 10.1016/j.freeradbiomed.2006.08.018
Loreaux, E. L., Kaul, B., Lorenz, J. N., and Lingrel, J. B. (2008). Ouabain-sensitive alpha1 Na,K-ATPase enhances natriuretic response to saline load. J. Am. Soc. Nephrol. 19, 1947–1954. doi: 10.1681/ASN.2008020174
Manunta, P., Hamilton, B. P., and Hamlyn, J. M. (2006). Salt intake and depletion increase circulating levels of endogenous ouabain in normal men. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R553–R559. doi: 10.1152/ajpregu.00648.2005
McDonough, A. A. (2010). Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R851–R861. doi: 10.1152/ajpregu.00002.2010
Meneton, P., Jeunemaitre, X., de Wardener, H. E., and MacGregor, G. A. (2005). Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol. Rev. 85, 679–715. doi: 10.1152/physrev.00056.2003
Meng, S., Roberts, L. J., Cason, G. W., Curry, T. S., and Manning, R. D. (2002). Superoxide dismutase and oxidative stress in Dahl salt-sensitive and -resistant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 283, R732–R738. doi: 10.1152/ajpregu.00346.2001
Mense, M., Stark, G., and Apell, H. J. (1997). Effects of free radicals on partial reactions of the Na,K-ATPase. J. Membr. Biol. 156, 63–71. doi: 10.1007/s002329900188
Moe, O. W., Tejedor, A., Levi, M., Seldin, D. W., Preisig, P. A., and Alpern, R. J. (1991). Dietary NaCl modulates Na(+)-H+ antiporter activity in renal cortical apical membrane vesicles. Am. J. Physiol. Renal Physiol. 260, F130–F137.
Mokry, M., and Cuppen, E. (2008). The Atp1a1 gene from inbred Dahl salt sensitive rats does not contain the A1079T missense transversion. Hypertension 51, 922–927. doi: 10.1161/HYPERTENSIONAHA.107.108415
Munzel, T., Gori, T., Bruno, R. M., and Taddei, S. (2010). Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 31, 2741–2748. doi: 10.1093/eurheartj/ehq396
Nesher, M., Dvela, M., Igbokwe, V. U., Rosen, H., and Lichtstein, D. (2009). Physiological roles of endogenous ouabain in normal rats. Am. J. Physiol. Heart Circ. Physiol. 297, H2026–H2034. doi: 10.1152/ajpheart.00734.2009
Nishi, A., Bertorello, A. M., and Aperia, A. (1993). Renal Na+,K(+)-ATPase in Dahl salt-sensitive rats: K+ dependence, effect of cell environment and protein kinases. Acta Physiol. Scand. 149, 377–384. doi: 10.1111/j.1748-1716.1993.tb09633.x
Nyström, T. (2005). Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 24, 1311–1317. doi: 10.1038/sj.emboj.7600599
Oweis, S., Wu, L., Kiela, P. R., Zhao, H., Malhotra, D., Ghishan, F. K., et al. (2006). Cardiac glycoside downregulates NHE3 activity and expression in LLC-PK1 cells. Am. J. Physiol. Renal Physiol. 290, F997–F1008. doi: 10.1152/ajprenal.00322.2005
Panico, C., Luo, Z., Damiano, S., Artigiano, F., Gill, P., and Welch, W. J. (2009). Renal proximal tubular reabsorption is reduced in adult spontaneously hypertensive rats: roles of superoxide and Na+/H+ exchanger 3. Hypertension 54, 1291–1297. doi: 10.1161/hypertensionaha.109.134783
Periyasamy, S. M., Liu, J., Tanta, F., Kabak, B., Wakefield, B., Malhotra, D., et al. (2005). Salt loading induces redistribution of the plasmalemmal Na/K-ATPase in proximal tubule cells. Kidney Int. 67, 1868–1877. doi: 10.1111/j.1523-1755.2005.00285.x
Petrushanko, I., Bogdanov, N., Bulygina, E., Grenacher, B., Leinsoo, T., Boldyrev, A., et al. (2006). Na-K-ATPase in rat cerebellar granule cells is redox sensitive. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R916–R925. doi: 10.1152/ajpregu.00038.2005
Petrushanko, I. Y., Yakushev, S., Mitkevich, V. A., Kamanina, Y. V., Ziganshin, R. H., Meng, X., et al. (2012). S-glutathionylation of the Na,K-ATPase catalytic alpha subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 287, 32195–32205. doi: 10.1074/jbc.M112.391094
Rapp, J. P. (1982). Dahl salt-susceptible and salt-resistant rats. A review. Hypertension 4, 753–763. doi: 10.1161/01.hyp.4.6.753
Rapp, J. P., and Dene, H. (1985). Development and characteristics of inbred strains of Dahl salt-sensitive and salt-resistant rats. Hypertension 7, 340–349.
Reifenberger, M. S., Arnett, K. L., Gatto, C., and Milanick, M. A. (2008). The reactive nitrogen species peroxynitrite is a potent inhibitor of renal Na-K-ATPase activity. Am. J. Physiol. Renal Physiol. 295, F1191–F1198. doi: 10.1152/ajprenal.90296.2008
Sanchez, G., Nguyen, A. N., Timmerberg, B., Tash, J. S., and Blanco, G. (2006). The Na,K-ATPase alpha4 isoform from humans has distinct enzymatic properties and is important for sperm motility. Mol. Hum. Reprod. 12, 565–576. doi: 10.1093/molehr/gal062
Schoner, W., and Scheiner-Bobis, G. (2007). Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am. J. Physiol. Cell Physiol. 293, C509–C536. doi: 10.1152/ajpcell.00098.2007
Schoner, W., and Scheiner-Bobis, G. (2008). Role of endogenous cardiotonic steroids in sodium homeostasis. Nephrol. Dial. Transpl. 23, 2723–2729. doi: 10.1093/ndt/gfn325
Schreck, C., and O'Connor, P. M. (2011). NAD(P)H oxidase and renal epithelial ion transport. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R1023–R1029. doi: 10.1152/ajpregu.00618.2010
Seshiah, P. N., Weber, D. S., Rocic, P., Valppu, L., Taniyama, Y., and Griendling, K. K. (2002). Angiotensin II stimulation of NAD(P)H oxidase activity. Circ. Res. 91, 406–413. doi: 10.1161/01.RES.0000033523.08033.16
Silva, E., and Soares-da-Silva, P. (2007). Reactive oxygen species and the regulation of renal Na+-K+-ATPase in opossum kidney cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1764–R1770. doi: 10.1152/ajpregu.00425.2007
Soares-da-Silva, E. S. A. P. (2012). “Renal redox balance and Na+,K+-ATPase regulation: role in physiology and pathophysiology,” in Oxidative Stress-Molecular Mechanisms and Biological Effects, ed D. V. Lushchak (Rijeka: InTech), 157–172.
Song, H., Lee, M. Y., Kinsey, S. P., Weber, D. J., and Blaustein, M. P. (2006). An N-terminal sequence targets and tethers Na+ pump alpha2 subunits to specialized plasma membrane microdomains. J. Biol. Chem. 281, 12929–12940. doi: 10.1074/jbc.M507450200
Stadtman, E. R., and Berlett, B. S. (1991). Fenton chemistry. Amino acid oxidation. J. Biol. Chem. 266, 17201–17211.
Stadtman, E. R., and Levine, R. L. (2000). Protein oxidation. Ann. N.Y. Acad. Sci. 899, 191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x
Summa, V., Camargo, S. M., Bauch, C., Zecevic, M., and Verrey, F. (2004). Isoform specificity of human Na(+), K(+)-ATPase localization and aldosterone regulation in mouse kidney cells. J. Physiol. 555, 355–364. doi: 10.1113/jphysiol.2003.054270
Sweadner, K. J. (1989). Isozymes of the Na+/K+-ATPase. Biochim. Biophys. Acta 988, 185–220. doi: 10.1016/0304-4157(89)90019-1
Taylor, N. E., Glocka, P., Liang, M., and Cowley, A. W. (2006). NADPH oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in Dahl S Rats. Hypertension 47, 692–698. doi: 10.1161/01.HYP.0000203161.02046.8d
Thevenod, F., and Friedmann, J. M. (1999). Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+/K(+)-ATPase through proteasomal and endo-/lysosomal proteolytic pathways. FASEB J. 13, 1751–1761.
Tian, J., Cai, T., Yuan, Z., Wang, H., Liu, L., Haas, M., et al. (2006). Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 17, 317–326. doi: 10.1091/mbc.E05-08-0735
Tian, J., Liu, J., Garlid, K. D., Shapiro, J. I., and Xie, Z. (2003). Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP) channels. Mol. Cell. Biochem. 242, 181–187. doi: 10.1023/A:1021114501561
Touyz, R. M. (2004). Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension 44, 248–252. doi: 10.1161/01.HYP.0000138070.47616.9d
Touyz, R. M. (2006). Lipid rafts take center stage in endothelial cell redox signaling by death receptors. Hypertension 47, 16–18. doi: 10.1161/01.hyp.0000196730.13216.f3
Vaziri, N. D., and Rodriguez-Iturbe, B. (2006). Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2, 582–593. doi: 10.1038/ncpneph0283
Wang, X., Armando, I., Upadhyay, K., Pascua, A., and Jose, P. A. (2009). The regulation of proximal tubular salt transport in hypertension: an update. Curr. Opin. Nephrol. Hypertens. 18, 412–420. doi: 10.1097/mnh.0b013e32832f5775
Wang, Y., Ye, Q., Liu, C., Xie, J. X., Yan, Y., Lai, F., et al. (2014). Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic. Biol. Med. 71, 415–426. doi: 10.1016/j.freeradbiomed.2014.03.036
Welch, W. J. (2006). Intrarenal oxygen and hypertension. Clin. Exp. Pharmacol. Physiol. 33, 1002–1005. doi: 10.1111/j.1440-1681.2006.04478.x
White, C. N., Figtree, G. A., Liu, C. C., Garcia, A., Hamilton, E. J., Chia, K. K., et al. (2009). Angiotensin II inhibits the Na+-K+ pump via PKC-dependent activation of NADPH oxidase. Am. J. Physiol. Cell Physiol. 296, C693–C700. doi: 10.1152/ajpcell.00648.2008
Wilcox, C. S. (2005). Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R913–R935. doi: 10.1152/ajpregu.00250.2005
Wong, C. M., Bansal, G., Marcocci, L., and Suzuki, Y. J (2012). Proposed role of primary protein carbonylation in cell signaling. Redox Rep. 17, 90–94. doi: 10.1179/1351000212Y.0000000007
Wong, C. M., Cheema, A. K., Zhang, L., and Suzuki, Y. J. (2008). Protein carbonylation as a novel mechanism in redox signaling. Circ. Res. 102, 310–318. doi: 10.1161/circresaha.107.159814
Wong, C. M., Marcocci, L., Das, D., Wang, X., Luo, H., Zungu-Edmondson, M., et al. (2013). Mechanism of protein decarbonylation. Free Radic. Biol. Med. 65, 1126–1133. doi: 10.1016/j.freeradbiomed.2013.09.005
Wong, C. M., Marcocci, L., Liu, L., and Suzuki, Y. J. (2010). Cell signaling by protein carbonylation and decarbonylation. Antioxid. Redox Signal. 12, 393–404. doi: 10.1089/ars.2009.2805
Xie, Z., and Cai, T. (2003). Na+-K+–ATPase-mediated signal transduction: from protein interaction to cellular function. Mol. Interv. 3, 157–168. doi: 10.1124/mi.3.3.157
Xie, Z. J., Wang, Y. H., Askari, A., Huang, W. H., and Klaunig, J. E. (1990). Studies on the specificity of the effects of oxygen metabolites on cardiac sodium pump. J. Mol. Cell. Cardiol. 22, 911–920. doi: 10.1016/0022-2828(90)90122-i
Xie, Z., Kometiani, P., Liu, J., Li, J., Shapiro, J. I., and Askari, A. (1999). Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J. Biol. Chem. 274, 19323–19328. doi: 10.1074/jbc.274.27.19323
Yang, L. E. P., Leong, K. K., and McDonough, A. A. (2007). Reducing blood pressure in SHR with enalapril provokes redistribution of NHE3, NaPi2, and NCC and decreases NaPi2 and ACE abundance. Am. J. Physiol. Renal Physiol. 293, F1197–F1208. doi: 10.1152/ajprenal.00040.2007
Yang, L. E., Sandberg, M. B., Can, A. D., Pihakaski-Maunsbach, K., and McDonough, A. A. (2008). Effects of dietary salt on renal Na+ transporter subcellular distribution, abundance, and phosphorylation status. Am. J. Physiol. Renal Physiol. 295, F1003–F1016. doi: 10.1152/ajprenal.90235.2008
Yan, Y., Shapiro, A. P., Haller, S., Katragadda, V., Liu, L., Tian, J., et al. (2013). Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J. Biol. Chem. 288, 34249–34258. doi: 10.1074/jbc.m113.461020
Yates, N. A., and McDougall, J. G. (1995). Effect of volume expansion on the natriuretic response to ouabain infusion. Ren. Physiol. Biochem. 18, 311–320. doi: 10.1159/000173932
Yatime, L., Buch-Pedersen, M. J., Musgaard, M., Morth, J. P., Lund Winther, A. M., Pedersen, B. P., et al. (2009). P-type ATPases as drug targets: tools for medicine and science. Biochim. Biophys. Acta 1787, 207–220. doi: 10.1016/j.bbabio.2008.12.019
Yatime, L., Laursen, M., Morth, J. P., Esmann, M., Nissen, P., and Fedosova, N. U. (2011). Structural insights into the high affinity binding of cardiotonic steroids to the Na+,K+-ATPase. J. Struct. Biol. 174, 296–306. doi: 10.1016/j.jsb.2010.12.004
Yudowski, G. A., Efendiev, R., Pedemonte, C. H., Katz, A. I., Berggren, P. O., and Bertorello, A. M. (2000). Phosphoinositide-3 kinase binds to a proline-rich motif in the Na+, K+-ATPase alpha subunit and regulates its trafficking. Proc. Natl. Acad. Sci. U.S.A. 97, 6556–6561. doi: 10.1073/pnas.100128297
Zhang, A. Y., Yi, F., Zhang, G., Gulbins, E., and Li, P. L. (2006b). Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 47, 74–80. doi: 10.1161/10.1161/01.HYP.0000196727.53300.62
Zhang, C., Imam, S. Z., Ali, S. F., and Mayeux, P. R. (2002). Peroxynitrite and the regulation of Na+,K+-ATPase activity by angiotensin II in the rat proximal tubule. Nitric Oxide 7, 30–35. doi: 10.1016/S1089-8603(02)00003-4
Zhang, S., Malmersjö, S., Li, J., Ando, H., Aizman, O., Uhlen, P., et al. (2006a). Distinct role of the N-terminal tail of the Na,K-ATPase catalytic subunit as a signal transducer. J. Biol. Chem. 281, 21954–21962. doi: 10.1074/jbc.M601578200
Zhang, Z., Devarajan, P., Dorfman, A. L., and Morrow, J. S. (1998). Structure of the ankyrin-binding domain of alpha-Na,K-ATPase. J. Biol. Chem. 273, 18681–18684. doi: 10.1074/jbc.273.30.18681
Zuo, L., Ushio-Fukai, M., Ikeda, S., Hilenski, L., Patrushev, N., and Alexander, R. W. (2005). Caveolin-1 is essential for activation of Rac1 and NAD(P)H oxidase after Angiotensin II Type 1 receptor stimulation in vascular smooth muscle cells: role in redox signaling and vascular hypertrophy. Arterioscler. Thromb. Vasc. Biol. 25, 1824–1830. doi: 10.1161/01.ATV.0000175295.09607.18
Keywords: Na/K-ATPase, ROS, protein carbonylation, signaling
Citation: Shah PT, Martin R, Yan Y, Shapiro JI and Liu J (2016) Carbonylation Modification Regulates Na/K-ATPase Signaling and Salt Sensitivity: A Review and a Hypothesis. Front. Physiol. 7:256. doi: 10.3389/fphys.2016.00256
Received: 21 March 2016; Accepted: 11 June 2016;
Published: 28 June 2016.
Edited by:
Laura Andrea Dada, Northwestern University, USAReviewed by:
John D. Imig, Medical College of Wisconsin, USAAnna Bogdanova, University of Zurich, Switzerland
Copyright © 2016 Shah, Martin, Yan, Shapiro and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiang Liu, bGl1akBtYXJzaGFsbC5lZHU=