 Thomas H. Holm1,2
Thomas H. Holm1,2 Karin Lykke-Hartmann1,2,3*
Karin Lykke-Hartmann1,2,3*- 1Department of Biomedicine, Aarhus University, Aarhus, Denmark
- 2Department of Molecular Biology and Genetics, Centre for Membrane Pumps in Cells and Disease-PUMPKIN, Danish National Research Foundation, Aarhus University, Aarhus, Denmark
- 3Aarhus Institute of Advanced Studies, Aarhus University, Aarhus, Denmark
The transmembrane Na+-/K+ ATPase is located at the plasma membrane of all mammalian cells. The Na+-/K+ ATPase utilizes energy from ATP hydrolysis to extrude three Na+ cations and import two K+ cations into the cell. The minimum constellation for an active Na+-/K+ ATPase is one alpha (α) and one beta (β) subunit. Mammals express four α isoforms (α1−4), encoded by the ATP1A1-4 genes, respectively. The α1 isoform is ubiquitously expressed in the adult central nervous system (CNS) whereas α2 primarily is expressed in astrocytes and α3 in neurons. Na+ and K+ are the principal ions involved in action potential propagation during neuronal depolarization. The α1 and α3 Na+-/K+ ATPases are therefore prime candidates for restoring neuronal membrane potential after depolarization and for maintaining neuronal excitability. The α3 isoform has approximately four-fold lower Na+ affinity compared to α1 and is specifically required for rapid restoration of large transient increases in [Na+]i. Conditions associated with α3 deficiency are therefore likely aggravated by suprathreshold neuronal activity. The α3 isoform been suggested to support re-uptake of neurotransmitters. These processes are required for normal brain activity, and in fact autosomal dominant de novo mutations in ATP1A3 encoding the α3 isoform has been found to cause the three neurological diseases Rapid Onset Dystonia Parkinsonism (RDP), Alternating Hemiplegia of Childhood (AHC), and Cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS). All three diseases cause acute onset of neurological symptoms, but the predominant neurological manifestations differ with particularly early onset of hemiplegic/dystonic episodes and mental decline in AHC, ataxic encephalopathy and impairment of vision and hearing in CAPOS syndrome and late onset of dystonia/parkinsonism in RDP. Several mouse models have been generated to study the in vivo consequences of Atp1a3 modulation. The different mice show varying degrees of hyperactivity, gait problems, and learning disability as well as stress-induced seizures. With the advent of several Atp1a3-gene or chemically modified animal models that closely phenocopy many aspects of the human disorders, we will be able to reach a much better understanding of the etiology of RDP, AHC, and CAPOS syndrome.
The Na+/K+ ATPases: Expression and Function
The Na+/K+ ATPases are transmembrane ion-pumps located at the plasma membrane of all mammalian cells. With each pump cycle, the Na+/K+ ATPases utilize the energy from hydrolysis of one adenosine triphosphate (ATP) to extrude three Na+ ions and import two K+ ions into the cell. The minimum constellation of an active pump consists of an alpha (α) and a beta (β) subunit (Kaplan, 2002; Bublitz et al., 2011; Palmgren and Nissen, 2011). The α subunit is responsible for the catalytic and pharmacological properties (Blanco et al., 1994) whereas the β and optional γ subunits may have regulatory functions (Jaisser et al., 1994; Béguin et al., 1997; Hilbers et al., 2016).
Mammals express four Na+/K+ ATPase α isoforms (α1−4) of which α1−3 are expressed in the CNS. While α1 is expressed ubiquitously and considered to maintain housekeeping cellular functions, the α2 isoform is expressed primarily in astrocytes and developing neurons and α3 isoform is restricted to neurons (McGrail et al., 1991; Bøttger et al., 2011). Thus, the cell type-specific distribution of the α isoforms suggests that each isoform has a particular function. Several suggestions have been made to elaborate on the specific role of the α3 isoform in neurons (Reviewed in Dobretsov and Stimers, 2005). Although the α3 isoform is expressed in many CNS neurons, several neuronal subsets lack the expression of this isoform (Hieber et al., 1991; McGrail et al., 1991; Bøttger et al., 2011). The ongoing hypothesis is thus that while the ubiquitously expressed α1 isoform maintains neuronal housekeeping functions, the α3 isoform serves as a reserve pump that only becomes activated when the intracellular Na+ concentration [Na+]i is high, e.g., after repeated action potentials (Brodsky and Guidotti, 1990; Jewell and Lingrel, 1991; Munzer et al., 1994; Blanco et al., 1995; Zahler et al., 1997; Monteith and Blaustein, 1998; Crambert et al., 2000), supported by the fact that the kinetics between the different isoform favors this scenario (Reviewed in Dobretsov and Stimers, 2005).
Neuronal Activity and α3
In the CNS, the Na+ and K+ gradients across the plasma membrane are essential for regulating neuronal excitability, and for multiple cellular functions, including cell volume regulation and Na+-coupled secondary transport. The distinguishing feature of α3 Na+/K+-ATPases is their several-fold lower affinity for activation by cytoplasmic Na+ compared to that of α1 Na+/K+-ATPases (Crambert et al., 2000). In rapidly firing neurons, therefore, when action potentials increase the intracellular Na+ concentration, [Na+]i, beyond saturating levels of the “housekeeping” α1 Na+/K+-ATPases, activation of α3 Na+/K+-ATPases continues to increase as [Na+]i rise. The α3 isoform thus protects neurons against catastrophic elevation of [Na+]i (Dobretsov and Stimers, 2005), and also of [Ca2+]i (because Na+/Ca2+ exchange is weakened) and general loss of the Na+ electrochemical gradient. As the α3 isoform is detected in several basal ganglia neuronal subsets (McGrail et al., 1991; Bøttger et al., 2011), reduced α3 activity may therefore interfere with reuptake of neurotransmitters such as glutamate, γ-aminobutyric acid (GABA) and dopamine (Kristensen et al., 2011), in those neurons.
The neurotransmitter transporters (NTTs) use ion gradients for the active transport, generally by co-transport of Na+ and Cl−. As an example, the glycine transporters are functionally coupled to the Na+ electrochemical gradient, which is actively generated and maintained by the Na+/K+-ATPases. The glycine transporter, GlyT2 cotransports three Na+ and one Cl− for every glycine (López-Corcuera et al., 1998), generating large rises in [Na+]i that must be efficiently reduced by the Na+/K+-ATPase to preserve ion homeostasis, which is absolutely necessary for synaptic transmission and neuronal excitability. The α3 co-localize and interacts with GlyT at the synapse in spinal cord neurons (de Juan-Sanz et al., 2013). As GlyT belongs to the solute carrier 6 (SLC6) family of highly homologous NTTs, it is possible that the transporter for dopamine, norepinephrine, serotonin, and GABA can affected by Na+/K+-ATPase activity loss (Kristensen et al., 2011). It is well-documented that dopamine increases the activity of the Na+/K+-ATPase in an organ-specific manner (Reviewed in Zhang et al., 2013). Moreover, evidence of direct interaction between Na+/K+-ATPase and dopamine receptors has also been reported (Hazelwood et al., 2008). Dopamine modulates the Na+/K+-ATPase by affecting both dopamine and other hormones. It has been shown that the D2 receptor stimulates striatal Na+/K+-ATPase activity after a short-term morphine treatment, in contrast to a long-term morphine treatment that inhibited striatal Na+/K+-ATPase activity (Wu et al., 2007).
Failure of the Na+/K+-ATPase to maintain Na+ and K+ gradients will cause a decrease in Na+ and K+ currents (through voltage-dependent channels). This will lead to a decrease in the membrane potential, action potential and most likely a loss of neuronal excitability. This will further affect sodium-coupled co- and counter-transport. Moreover, it can also affect Na+/H+ and Na+/Ca2+ exchange, essential for cellular functions, and could lead to increased intracellular Ca2+ and acidification. Not surprisingly, mutations in the ATP1A3 gene have been associated with neurological disorders.
ATP1A3-Related Diseases
Mutations in the ATP1A3 gene are associated with three related rare neurological disorders, Rapid-onset Dystonia-Parkinsonism (RDP) (de Carvalho Aguiar et al., 2004), Alternating Hemiplegia of Childhood (AHC) (Heinzen et al., 2012; Rosewich et al., 2012), and recently, Cerebellar ataxia, Areflexia, Pes cavus, Optic atrophy, and Sensorineural hearing loss (CAPOS) syndrome (Demos et al., 2014).
Presently 12 ATP1A3 missense mutations have been associated with RDP (Heinzen et al., 2014). Classical RDP patients typically develop stress-induced permanent dystonia and Parkinsonism in late adolescence or early adulthood. Other 59 different ATP1A3 de novo missense mutations are associated with AHC (Heinzen et al., 2014; Rosewich et al., 2014; Sasaki et al., 2014; Ulate-Campos et al., 2014; Yang et al., 2014; Panagiotakaki et al., 2015; Viollet et al., 2015). AHC is characterized by onset of hemiplegic/quadriplegic episodes within 18 months of birth. Other, more variable neurological abnormalities of AHC include choreathosis, ataxia, developmental delays, seizures, and high incidence of neuropsychiatric disorders (Demos et al., 2014).
Recently, the mutations causing RDP and AHC were mapped according to their amino acid position in the α3 isoform showing their location and the number of patients identified that harbors these mutations (Heinzen et al., 2014).
So far, only a single missense mutation in ATP1A3 is associated with CAPOS syndrome (Demos et al., 2014; Heimer et al., 2015). CAPOS patients show onset of symptoms at the age of 6 months to 5 years. CAPOS syndrome is characterized by episodes of ataxic encephalopathy, weakness, and loss of hearing and sight (Brashear et al., 2014).
Interestingly, a recent report identified a G316S mutation in the α3 isoform associated with Adult Rapid-onset Ataxia (Sweadner et al., 2016). The clinical examination noted most RDP symptoms except dystonia.
Thus, it appears that the ATP1A3-related disorders arise from autosomal dominant mutations with variable penetrance (Demos et al., 2014). Affected patients typically present in the context of an acute onset of paroxysmal, episodic neurological symptoms that may include hemiplegia, dystonia, ataxia, or seizures. Some symptoms may persist after resolution, such as neurodevelopmental delays, attention deficits, hyperactivity, trunk instability, dystonia, or ataxia (Mikati et al., 2000; Shafer et al., 2005; Panagiotakaki et al., 2010; Heinzen et al., 2012, 2014; Sweney et al., 2015). Although the ATP1A3-related neurological disorders are considered clinically distinct, the phenotypic spectrum of each disease continues to expand (Heinzen et al., 2014; Rosewich et al., 2014; Sweney et al., 2015). In support, there have recently been identified patients with intermediate, non-classical symptoms (Sasaki et al., 2013; Termsarasab et al., 2015).
Genotype-Phenotype—Affect Protein Function—Cause Symptoms
A recent case study of 35 AHC patients showed relatively mild symptoms in patients carrying the D801N mutation, whereas the E815K mutation was associated with far more severe symptoms (Sasaki et al., 2014). In fact, the same ATP1A3 mutation may result in quite different time of onset and disease progression (Dobyns et al., 1993; Oblak et al., 2014), emphasizing that other factors, such as genetic background and epigenetic regulation play a large role in disease penetrance.
Recent functional studies suggest that most RDP mutations do not reach the cell surface (Heinzen et al., 2012). In contrast, the majority of AHC mutant proteins exert dominant negative effects on the wild type protein at the cell surface (Clapcote et al., 2009; Li et al., 2015), thus explaining the more severe phenotypes associated with AHC. The majority of AHC mutations (>70%) are located within the transmembrane helices of the Na+/K+-ATPase protein (Heinzen et al., 2012). Homology modeling of the two most common mutations, D801N and E815K suggested different mutational consequences: In the D801N mutation, a positive dipole was formed, which through electrostatic repulsion directly affected passage of K+ ions (Kirshenbaum et al., 2013). In contrast, the E815K mutation prevented inward H+ currents through the Na+/K+-ATPase. Intracellular H+ currents through the Na+/K+-ATPase were recently identified (Vedovato and Gadsby, 2014) and are known regulators of neuronal excitability (Takahashi and Copenhagen, 1996), thus suggesting a correlation between severity and loss of H+ transport. The significance of this discovery however remains to be determined in vivo.
ATP1A3-Modified Mouse Models
Four genetically modified mouse models targeting the Atp1a3 gene has been reported, and have been extensively used to study in vivo functions of the α3 isoform (Table 1). A detailed comparison of mouse models looking at clinical features in AHC and Atp1a3 mouse models, as well as behavioral testing has been reported (Hunanyan et al., 2015).
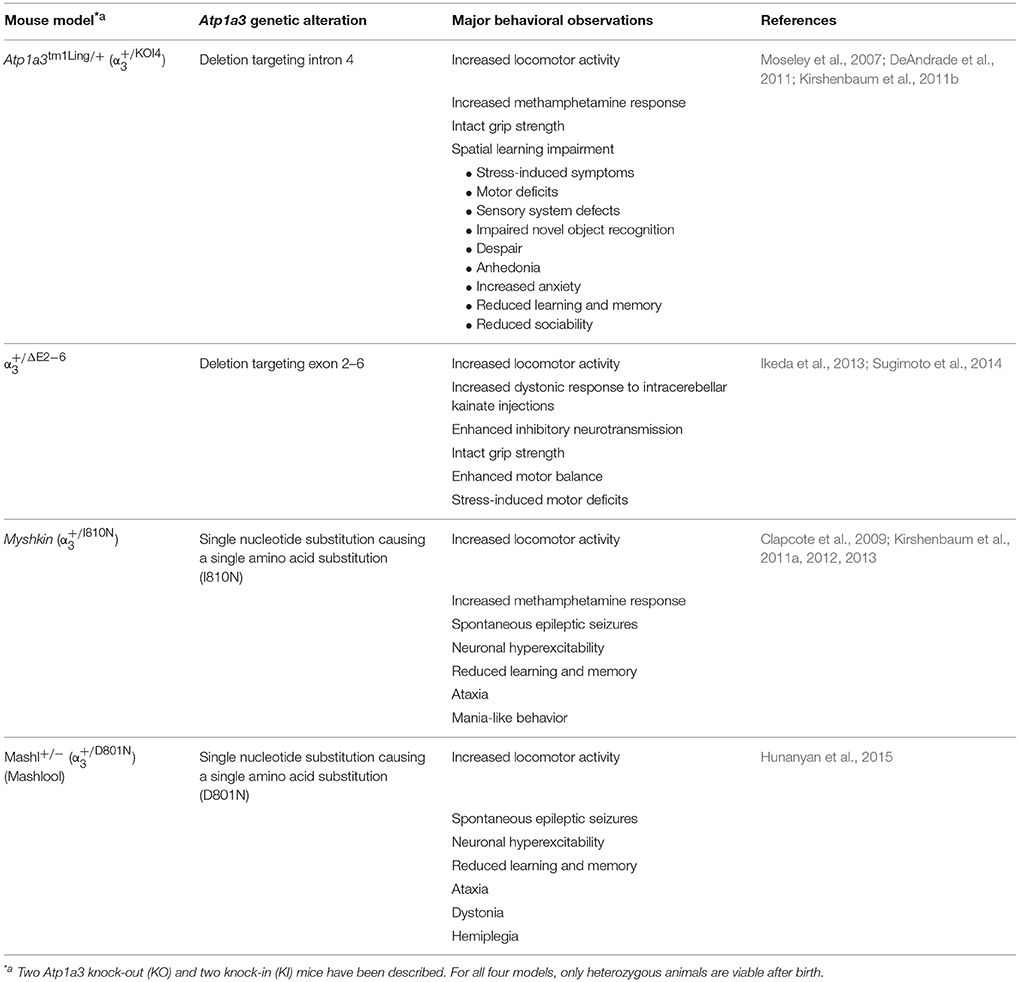
Table 1. Atp1a3 gene modified animals.
The Atp1a3tm1Ling∕+ was produced by introducing a single base pair mutation in intron 4 (), causing an aberrant splicing of Atp1a3, effectively knocking out the allele (Moseley et al., 2007). The was generated by an eGFP-Atp1a3 gene replacement strategy, knocking out one Atp1a3 allele () (Ikeda et al., 2013). The Myshkin mouse () (Clapcote et al., 2009) expressed the I810N AHC mutation (Panagiotakaki et al., 2015). The Mashlool mouse (, Mashl+∕−) (Hunanyan et al., 2015) was generated to study the effects of the most common mutation in AHC, D801N (Heinzen et al., 2012).
The cardiotonic steroids (CTS) constitute a group of organic compounds that show high binding affinity toward Na+/K+-ATPases and inhibit the catalytic activity by stabilizing the enzyme's phosphorylated E2-P state (Yatime et al., 2011; Laursen et al., 2013). CTS such as ouabain have been used for more than 50 years to study the function of Na+/K+-ATPases in vitro and in acute animal models. An ouabain-perfused mouse model presumably targeting the α3 isoform in cerebellum and basal ganglia (Calderon et al., 2011) supplements the findings summarized in Table 1.
Basic Characterization
Hippocampal lysates showed a significant reduction in α3 protein expression in the mice and a compensatory upregulation of α1 expression but not of α2 (Moseley et al., 2007). In contrast, whole brain lysates from the Myshkin mice (Myk/+ and Myk/Myk) showed no changes in protein expression levels of the α1, α2, and α3 isoforms relative to wild type, suggesting that the Myshkin mutation is a functional null allele of the Atp1a3 gene, which encodes a normally expressed, but inactive enzyme, as revealed by reduced total ATPase activity (Clapcote et al., 2009). In support of this, the authors observed normal plasma membrane localization of the Myskhin α3 isoform expressed in COS cells with only small amounts retained in the ER (Clapcote et al., 2009).
AHC patients tend to be smaller and weigh less—presumably due to eating difficulties (Neville and Ninan, 2007; Panagiotakaki et al., 2010). Although abilities to chew and swallow have not been addressed for any of the mouse models, it is interesting that the Myshkin and male Mashl+∕− mice were reported to have smaller body sizes (Clapcote et al., 2009; Hunanyan et al., 2015). Myshkin mice crossed with mice expressing a bacterial artificial chromosome expressing wild type Atp1a3 (Atp1a3 BAC) regained approximately 80% of wild type total Na+/K+-ATPase activity and showed normal body size, thus supporting the theory that Na+/K+-ATPase activity loss correlates with symptoms (Clapcote et al., 2009).
Effects on Motor Function, Balance, and Coordination
The motor dysfunction in RDP and AHC i.e., ataxia, dystonia, unsteady gait, and tip-toing are reflected in motor function, balance problems, and gait disturbances (Brashear et al., 1997; Linazasoro et al., 2002; Svetel et al., 2010).
Combined, the Atp1a3tm1Ling∕+ and mouse models displayed mild motor symptoms, whereas the Myshkin and the Mashl+∕− mice recapitulated a much broader spectrum of ATP1A3-disease related symptoms. Motor tests showed these defects were caused by dyscoordination rather than lack of muscle strength as grip strength was intact (Kirshenbaum et al., 2013; Hunanyan et al., 2015), and thus corresponds to reports of AHC patients.
Adult Atp1a3tm1Ling∕+ and mice displayed normal motor function when tested on the balance beam and accelerated rotarod (DeAndrade et al., 2011; Sugimoto et al., 2014). However, restraint caused a stress-induced deterioration of motor performance of mice on the balance beam and rotarod and a significant drop in ATPase activity from 85 to 67% (DeAndrade et al., 2011).
Similarly, restraint stress significantly reduced the hanging times of the mice (Sugimoto et al., 2014). In contrast to Atp1a3tm1Ling∕+ and mice, distinct motor symptoms were apparent in naïve Myshkin and the Mashl+∕− mice: Both strains showed ataxia and tremor on the balance beam but only the Myshkin mice performed poorly on the accelerated rotarod (Kirshenbaum et al., 2013; Hunanyan et al., 2015).
Both Myshkin and the Mashl+∕− mouse strains showed abnormal stride length (Kirshenbaum et al., 2013; Hunanyan et al., 2015). A similar phenotype was induced by restraint stress in the mice (Sugimoto et al., 2014).
The Cerebellum-Basal Ganglia Circuitry
Dystonia, Ataxia, and Parkinsonism
Dystonia, ataxia, and Parkinsonism are core symptoms of RDP. A recent study showed that dystonia and ataxia could be reproduced by partially blocking the Na+/K+-ATPase in the cerebellum of mice using ouabain whereas disruption of the basal ganglia circuit caused Parkinsonism i.e., rigidity, akinesia, tremor, and hunched posture (Calderon et al., 2011). The symptoms induced by ouabain perfusion were shown to develop over several days and to be highly dependent on the ouabain concentration used. Interestingly, mice perfused with low concentrations of ouabain into the cerebellum and basal ganglia developed symptoms corresponding to high concentration of ouabain when subjected to stress. Thus, reflecting the stress-sensitive nature of ATP1A3-related diseases.
Cerebellar dysfunction has been observed in several rodent dystonia strains (Lorden et al., 1992; LeDoux and Lorden, 1998; Richter et al., 1998; Campbell et al., 1999; Fremont et al., 2014) and in patients suffering from AHC (Saito et al., 1998; Yamashita et al., 2005; Sasaki et al., 2009) and RDP (Oblak et al., 2014; Liu et al., 2015).
The mice showed prolonged dystonic periods after intracerebellar kainate injections (Ikeda et al., 2013). Despite reduced motor performance of both naïve Myshkin and the Mashl+∕− mice, only the Mash+∕− mice were reported to develop stress-induced generalized dystonia (Hunanyan et al., 2015). Mild dystonia is common among AHC patients carrying the Mashl+∕− equivalent D801N mutation (Panagiotakaki et al., 2015), whereas only one of two AHC patients known to carry the Myskhin, I810N mutation, showed dystonia (Yang et al., 2014; Panagiotakaki et al., 2015). Presently, it is therefore not possible to determine if the differences observed can be attributed specifically to the ATP1A3 mutation or to genetic background.
The deep cerebellar nuclei (DCN) integrate inhibitory signals from GABAergic Purkinje neurons and excitatory glutamatergic mossy fibers and climbing fiber pathways and constitute the majority output fibers from the cerebellum. The DCN connects to premotor and motor nuclei in the basal ganglia (Faull, 1978; Faull and Carman, 1978). Abnormal output from the DCN will have profound effects on motor function. The Purkinje neurons are the main output to the DCN and play a central role in DCN signal integration and overall cerebellar function.
Purkinje neurons are characterized by a high Na+ channel density and short spike duration during which large amounts of Na+ ions enter the neurons, emphasizing the requirement for functional a Na+/K+-ATPase (Llinás and Sugimori, 1980; Raman and Bean, 1997; Carter and Bean, 2009). Cerebellar Purkinje neurons only express the Na+/K+-ATPase α3 isoform (Peng et al., 1997) and are therefore particularly sensitive to ATP1A3 mutations. Furthermore, in developing mice, reduced Na+/K+-ATPase α3 expression caused a build-up of [Na+]i and [Ca2+]i in the terminals of molecular-layer interneurons resulting in enhanced inhibition of Purkinje neurons (Ikeda et al., 2013). This observation is potentially very interesting, as it offers a possible mechanism to explain how Na+/K+-ATPase perturbations may affect neuronal plasticity and motor learning.
Although further experiments are required to determine the extent of this observation, it is interesting, that cortical neurons isolated from the Myshkin mice showed increased [Ca2+]i as well (Kirshenbaum et al., 2011a). This observation could imply that other neuronal populations may be affected. Indeed, Na+/K+-ATPase α3 showed co-localization with markers for several populations of GABAergic neurons in the rodent and human brain (Bøttger et al., 2011; Ikeda et al., 2013; Paciorkowski et al., 2015), suggesting these populations to be sensitive to ATP1A3 perturbations as well. Supporting GABA dysfunction, hippocampal neurons from the Myshkin and Mashl+∕− mice showed increased excitability in response to high frequency stimulation (Clapcote et al., 2009; Hunanyan et al., 2015).
Hemiplegia
Hemiplegia is specific to AHC (Sweney et al., 2015). The mechanism driving this symptom is poorly understood. Single-photon emission computed tomography (SPECT) scans of AHC patients showed malperfusion leading up to or during events in several cases (Kanazawa et al., 1991; Zupanc et al., 1991; Aminian et al., 1993; Wong et al., 1993). However, there have also been examples, where this phenotype was absent (Sweney et al., 2009; Sasaki et al., 2011). Supporting a vascular phenotype, the vasodilator, flunarizine is currently among the most widely used prophylaxis of hemiplegia in AHC (Casaer and Azou, 1984; Bourgeois et al., 1993; Mikati et al., 2000). So far, there has been no investigation of this mechanism in the current animal models. Of the four mouse models, only the Mashl+∕− mice were reported to develop stress-induced episodes of hemiplegia and quadriplegia (Hunanyan et al., 2015).
Neuropsychiatric Symptoms—Mania
Previous studies have shown intracerebroventricular (ICV) injections of ouabain in high concentrations to be CNS stimulatory and convulsive (Doggett and Spencer, 1971; Davidson et al., 1978; Corazzi et al., 1985; Haglund and Schwartzkroin, 1990; Lees et al., 1990; Yu, 2003). In rats, this approach was used to induce mania (El-Mallakh et al., 2003; Riegel et al., 2009). Episodes mimicking manic periods of bipolar mood disorder are common among AHC patients. Especially the manic episodes are particularly detrimental, as the children are at high risk of injury (Personal communications, see Acknowledgement).
There is a strong link in literature between Na+/K+-ATPase dysfunction and bipolar mood disorder (el-Mallakh and Wyatt, 1995). Bipolar patients showed reduced Na+/K+-ATPase ATP1A2 gene expression in isolated erythrocytes (Hokin-Neaverson and Jefferson, 1989a,b; Looney and el-Mallakh, 1997) and in the temporal cortex (Rose et al., 1998) and reduced Na+/K+-ATPase ATP1A3 expression in prefrontal cortex (Tochigi et al., 2008).
A prominent feature of manic episodes is hyperactivity. Although direct comparison of hyperactivity levels is impossible, all Atp1a3 mouse models showed open field hyperlocomotion (Kirshenbaum et al., 2011a; Ikeda et al., 2013; Hunanyan et al., 2015).
Exploration-based anxiety tests revealed all Atp1a3 mouse strains to be less anxious, to have increased impulsivity and risk-taking and a reduced habituation, all of which are symptoms of mania (Moseley et al., 2007; Kirshenbaum et al., 2011a; Ikeda et al., 2013; Hunanyan et al., 2015; Termsarasab et al., 2015).
In further support of mania-like behavior, the Myshkin mice showed changes to circadian rhythm (Kirshenbaum et al., 2011a). These symptoms, along with hyperactivity were reversed by treating the Myshkin mice with the mood stabilizers, lithium and valproate (Kirshenbaum et al., 2011a).
ICV injection of ouabain in rats caused phosphorylation of ERK and AKT in the hippocampus (Ruktanonchai et al., 1998; Kim et al., 2008; Yu et al., 2010). Similar increases in phosphorylated ERK and AKT were observed in the Myshkin mice. Both signaling pathways have been implicated in the control of behavioral excitement in rodents (Prickaerts et al., 2006; Creson et al., 2009; Engel et al., 2009; Ackermann et al., 2010), making them potential targets for future mood stabilizers. Correspondingly, open field hyperactivity and open arm visits were reduced after administration of the ERK inhibitor, SL327 (Kirshenbaum et al., 2011a). The Atp1a3-BAC transgenic Myshkin showed a Na+/K+-ATPase activity increase from 58 to 74% relative to wild type levels and a partial normalization of AKT phosphorylation (Clapcote et al., 2009; Kirshenbaum et al., 2011a). Correspondingly, treating the mice with the ouabain inhibitor, rostafuroxin, had a normalizing effect on hyperlocomotion (Kirshenbaum et al., 2011a).
Although the manic phase seems most prevalent in all mouse models, there have been examples of the Atp1a3tm1Ling∕+ mice being able to recapitulate the depression-like symptoms of bipolar disorder. When subjected to a chronic variable stress protocol, the mice showed prominent symptoms of anxiety, including reduced exploration of open areas and attention deficits during novel object and sociability tests. At this point, the mice showed a Na+/K+-ATPase activity of 67% relative to wild type levels (Kirshenbaum et al., 2011b). Interestingly, these symptoms did not occur in wild type mice, suggesting that ATP1A3 mutations may increase vulnerability to stress.
Epilepsy
Epilepsy and bipolar disorder share a common pathophysiology (Mazza et al., 2007) and is often comorbid in human patients (Mula et al., 2010). According to a recent study, approximately half of all AHC patients experience at least one epileptic seizure (Panagiotakaki et al., 2010).
Reduced Na+/K+-ATPase activity has been reported in genetic animal models of epilepsy and in hippocampal tissue from epileptic patients (Brines et al., 1995; Fernandes et al., 1996; Vaillend et al., 2002) and have been proposed as a causal factor in myoclonus epilepsy and ragged red fibers disease, a rare form of inherited epilepsy (McNamara, 1994). Also, Na+/K+-ATPase activity was decreased in the brain of rodents after chemical induction of seizures using the convulsant, pentylenetetrazol (Schneider Oliveira et al., 2004; Marquezan et al., 2013). Recently, two children with catastrophic early life epilepsy were shown to carry novel ATP1A3 mutations (Paciorkowski et al., 2015).
As described, ouabain is convulsive when administered to rodents intraventricularly in sufficiently high concentrations (Doggett and Spencer, 1971; Davidson et al., 1978; Corazzi et al., 1985; Haglund and Schwartzkroin, 1990; Lees et al., 1990; Yu, 2003; Alonso et al., 2013).
Several mechanisms can explain why neurons are vulnerable to ATP1A3 insults. Seizures are often associated with a loss in metabolic energy (Araujo et al., 2014). Na+/K+-ATPases are highly sensitive to such perturbations, as they require approximately 50% of the energy available to the brain under normal circumstances (Attwell and Laughlin, 2001). Reduced Na+/K+-ATPase activity may cause hyperexcitability due to increased [K+]o and membrane depolarization (Haglund and Schwartzkroin, 1990; Somjen, 2002). Further effects arise from the post-tetanic buildup of [Na+]i (Azarias et al., 2013) and the following inhibition of the Ca2+/Na+ exchanger causing accumulation of [Ca2+]i and subsequent effects on gene transcription (Lyons and West, 2011), neurotransmitter release (Neher and Sakaba, 2008), and synaptic plasticity (Zucker, 1999).
The Myshkin mice showed epileptic seizures and neuronal hyperexcitability (Clapcote et al., 2009). Supporting the correlation between Na+/K+-ATPase α3 activity and seizure resistance, the epileptic seizures did not occur in Atp1a3-BAC transgenic Myshkin mice (Clapcote et al., 2009).
Furthermore, the link between ATP1A3-disease mutations and epilepsy was observed in a Chinese 12-year old boy with the I810N mutation, who was reported to have AHC with developmental delay and epilepsy (Yang et al., 2014).
The Mashl+∕− mice showed flurothyl-indiced seizures, focal epileptogenesis (via kindling) and demonstrated spontaneous recurrent seizures and neuronal excitability (Hunanyan et al., 2015). Similar predisposition to epileptogenesis has been observed in humans affected by AHC.
There have been no reports of epilepsy in either of the Atp1a3tm1Ling∕+ and mouse models (naïve or stressed), although the mice showed increased sensitivity to cerebellar kainate injections (Ikeda et al., 2013).
Epilepsy is associated with cognitive decline in human patients (Bergen, 2006). Correspondingly, signs of hippocampal necrosis and glial activation were initially reported for the Myshkin mice maintained on the 129S1/SvImJ strain background. Hippocampal pathology disappeared once the mice were crossed into the C57BL/6NCr strain (Kirshenbaum et al., 2011a). Also, once maintained in the C57BL/6NCr strain for 20 generations, the Myshkin mouse strain no longer developed spontaneous seizures (Kirshenbaum et al., 2011a). This observation parallels previous publications identifying the 129S1/SvImJ and C57BL/6 strains as relatively resistant to kainate-induced seizures whereas only the C57BL/6 strain was resistant to kainate-induced cell death (Schauwecker, 2002; McLin and Steward, 2006).
Memory
In a comprehensive report of 157 AHC patients, mental retardation was recorded in at least 92% of the cases (Panagiotakaki et al., 2010). Likewise, cognitive decline has been described in RDP patients (Cook et al., 2014).
Overall, naïve Myshkin and Mashl+∕− mice displayed poor memory performance, whereas the performance of the Atp1a3tm1Ling∕+ and mice to some degree was dependent on stress. The Atp1a3tm1Ling∕ mice thus showed no learning in locating a hidden platform in the Morris water maze (Moseley et al., 2007). In contrast the Atp1a3tm1Ling∕+ mice performed normally in a novel object recognition test, but showed significantly worse performance after subjection to a CVS protocol (Kirshenbaum et al., 2011b).
The Myshkin mice performed significantly worse in contextual- and cued-dependent fear conditioning tests. In contrast, Mashl+∕− mice showed impaired novel object recognition, but intact cued-dependent fear memory (Hunanyan et al., 2015).
The dorsal part of the hippocampus plays a central role in learning and spatial memory, whereas the ventral hippocampus primarily regulates emotional and motivated behaviors through interaction with the amygdala (Fanselow and Dong, 2010). Accordingly, ouabain injection into these brain regions caused impairments in spatial learning (Zhan et al., 2004) and fear-dependent memory, respectively (Mizumori et al., 1987).
This could suggest a difference in the performance of the ventral hippocampus between the two Myshkin and Masl+∕− strains. However, the strong hyperactivity of the both strains may have interfered with the readout of the conditioning tests, as the tests rely on the ability to suppress movement. Furthermore, the Myshkin mice showed reduced learning in a conditioned taste aversion test, suggesting also hippocampus-independent memory functions were affected (Reilly et al., 1993; Purves et al., 1995).
Due to a high voltage dependency and a high permeability for Ca2+, the N-methyl-D-aspartate receptors (NMDR) are important for triggering several different forms of synaptic plasticity, including long term potentiation and long-term depression (Cull-Candy et al., 2001). The Atp1a3tm1Ling∕+ mice showed reduced hippocampal expression of the NMDA NR1 subunit (Moseley et al., 2007), suggesting increased neuronal activity (Kvajo et al., 2004). Interestingly, NR1 expression was unaffected in the Myshkin mice (Clapcote et al., 2009). This observation is unexpected, as the Myshkin mice developed spontaneous seizures. However, direct comparison is not possible due to the FVB background of the Myshkin mice.
Effects on Neurotransmitter Homeostasis and Circuitry
As previously exemplified, decrease in Na+/K+-ATPase activity is associated with neuronal hyperexcitability and the release of neurotransmitters. Particularly dopamine, serotonin, and norepinephrine are involved in regulating movement and behavior (Perona et al., 2008).
In support of a dopamine phenotype, all naïve Atp1a3 mouse models displayed hyperlocomotion in the open field test, which was further induced by amphetamine (Moseley et al., 2007; Kirshenbaum et al., 2011a). High Performance Liquid Chromatography (HPLC) analysis showed no changes in striatal levels of dopamine, serotonin, or their metabolites in the Atp1a3tm1Ling∕+ mice (DeAndrade et al., 2011). In cerebrospinal fluid (CSF) samples obtained from two RDP patients, low levels of the dopamine metabolite, homovanilic acid, was reported (Brashear et al., 1998). However, this observation remains to be confirmed as a diagnostic criterion for RDP. A recent study reported normal CSF neurotransmitter levels in AHC patients (Fons et al., 2012).
Additional Neurological Symptoms
Some ATP1A3-disease mutation related patients show additional neurological symptoms that range from mild limb cramping sometimes decades before developing RDP (Brashear et al., 2007) to dysfunction of the autonomic nervous system with cardiac repolarization problems (Novy et al., 2014; Jaffer et al., 2015) excessive or lack of perspiration, skin discoloration, gastrointestinal problems and changes in body temperature leading up to or during attacks in AHC patients (Mikati et al., 2000; Fons et al., 2012).
The Myshkin mice showed increased systolic and diastolic blood pressure, but normal heart rate (Kirshenbaum et al., 2013). Most likely, this effect is related to ATP1A3 expression in cardiomyocytes (Zahler et al., 1993). The Mashl+∕− mice and stressed female Atp1a3tm1Ling∕+ mice showed delayed temperature response (DeAndrade et al., 2011; Hunanyan et al., 2015), suggesting impaired thermoception. The α3 Na+/K+-ATPase expression was recently reported in dorsal root ganglion γ motor neurons located in the spinal cord of mice (Edwards et al., 2013). It is therefore very likely to have implications for motor control also. Somatosensory evoked potentials (SEP) during the interictal period showed abnormal recovery cycle in a recent case report of seven AHC patients, suggesting multilevel somatosensory system hyperexcitability (Vollono et al., 2014). Sensory abnormalities have been proposed to play a role in the pathophysiology of dystonia as the basal ganglia and other motor areas are heavily connected to the somatosensory system (Tinazzi et al., 2003). Future studies may elucidate if a similar interaction affects the pathophysiology of ATP1A3-related diseases.
Summary
The current Atp1a3 mouse models recapitulate to a large part the symptoms of RDP and AHC.
Through the collaborative efforts of the ATP1A3-disease research community, it has recently been possible to carry out several studies on relatively large patient groups. Such studies continue to be invaluable not only in the search for common denominators but also for establishing the animal models for the ATP1A3-diseases.
Previous studies using CTS to study Na+/K+-ATPase function suggest a strong correlation between reduced Na+/K+-ATPase activity and severity of symptoms. The present Atp1a3 mouse models seem to support this as the Atp1a3tm1Ling∕+ and mice (with mild Na+/K+-ATPase reduction) showed relatively mild symptoms whereas the Myshkin and Mashl+∕− models carrying AHC mutations (with larger Na+/K+-ATPase reduction), recapitulated most of the key phenotypes. As a proof of concept, most symptoms of the Myshkin mouse were rescued by increasing the Na+/K+-ATPase activity. Future experiments will be required to establish if similar approaches can be translated into a possible treatment.
Animal models represent valuable tools to study the pathology of human diseases. The biological questions can be assessed by different model system, depending on the nature of the study. Different advantages and disadvantages for designing and generating mouse models. Knock-out mouse models represent loss-of-function studies of a gene, while knock-in allows to explore the consequences of a single amino acid mutation inroduced into the genome. Both knock-out and knock-in models are can be designed as conditional models, allowing to knock-out, or introduce a mutation (knock-in) in single cell populations or organs, using the Cre LoxP system (Branda and Dymecki, 2004). Blocking Na+/K+-ATPase activity by infusion of ouabain (Fremont et al., 2014) or RNAi tools (Fremont et al., 2015) into specific brain areas represent a another model to investigate the function in specific brain areas in rodent models.
The highly variable nature of ATP1A3-disease related symptoms are becoming increasingly apparent. Despite recent advances in elucidating the etiology of individual ATP1A3 mutations, large variations are reported even for patients with the same mutation. It is very likely that genetic background, epigenetic as well as environmental factors play a central role in disease penetrance.
Author Contributions
TH and KL discussed, outlined and co-wrote the review.
Funding
Danish National Research Foundation (DNRF) (PUMPKIN DNRF85).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the ATP1A3 working community for continues discussions regarding ATP1A3-related disorders. Moreover, many thanks to Jeff Wuchich, Co-founder and President of Cure AHC, USA, Dr. Tsveta Schyns, Board Secretary of EGAN, Brussels, Belgium and Chair and Project Coordinator ENRAH, Vienna, Austria, Lynn Egan, president of AHCF, Southfield, MI, USA, and Dr. Hendrick Rosewich, MD Dept. of Pediatrics and Pediatric Neurology, Georg August University Faculty of Medicine, Germany for scientific discussions regarding AHC and RDP pathology.
References
Ackermann, T. F., Kempe, D. S., Lang, F., and Lang, U. E. (2010). Hyperactivity and enhanced curiosity of mice expressing PKB/SGK-resistant glycogen synthase kinase-3 (GSK-3). Cell. Physiol. Biochem. 25, 775–786. doi: 10.1159/000315097
Alonso, E., Cano-Abad, M. F., Moreno-Ortega, A. J., Novalbos, J., Milla, J., García, A. G., et al. (2013). Nanomolar ouabain elicits apoptosis through a direct action on HeLa cell mitochondria. Steroids 78, 1110–1118. doi: 10.1016/j.steroids.2013.07.010
Aminian, A., Strashun, A., and Rose, A. (1993). Alternating hemiplegia of childhood: studies of regional cerebral blood flow using 99mTc-hexamethylpropylene amine oxime single-photon emission computed tomography. Ann. Neurol. 33, 43–47. doi: 10.1002/ana.410330108
Araujo, B., Torres, L., Stein, M., Cabral, F. R., Herai, R., Okamoto, O., et al. (2014). Decreased expression of proteins involved in energy metabolism in the hippocampal granular layer of rats submitted to the pilocarpine epilepsy model. Neurosci. Lett. 561, 46–51. doi: 10.1016/j.neulet.2013.12.040
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Azarias, G., Kruusmägi, M., Connor, S., Akkuratov, E. E., Liu, X. L., Lyons, D., et al. (2013). A specific and essential role for Na,K-ATPase alpha3 in neurons co-expressing alpha1 and alpha3. J. Biol. Chem. 288, 2734–2743. doi: 10.1074/jbc.M112.425785
Béguin, P., Wang, X., Firsov, D., Puoti, A., Claeys, D., Horisberger, J. D., et al. (1997). The gamma subunit is a specific component of the Na,K-ATPase and modulates its transport function. EMBO J. 16, 4250–4260.
Bergen, D. C. (2006). Do seizures harm the brain? Epilepsy Curr. 6, 117–118. doi: 10.1111/j.1535-7511.2006.00116.x
Blanco, G., DeTomaso, A. W., Koster, J., Xie, Z. J., and Mercer, R. W. (1994). The alpha-subunit of the Na,K-ATPase has catalytic activity independent of the beta-subunit. J. Biol. Chem. 269, 23420–23425.
Blanco, G., Sánchez, G., and Mercer, R. W. (1995). Comparison of the enzymatic properties of the Na,K-ATPase alpha 3 beta 1 and alpha 3 beta 2 isozymes. Biochemistry 34, 9897–9903.
Bøttger, P., Tracz, Z., Heuck, A., Nissen, P., Romero-Ramos, M., and Lykke-Hartmann, K. (2011). Distribution of Na/K-ATPase alpha 3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J. Comp. Neurol. 519, 376–404. doi: 10.1002/cne.22524
Bourgeois, M., Aicardi, J., and Goutières, F. (1993). Alternating hemiplegia of childhood. J. Pediatr. 122, 673–679. doi: 10.1016/s0022-3476(06)80003-x
Branda, C. S., and Dymecki, S. M. (2004). Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev. Cell 6, 7–28. doi: 10.1016/S1534-5807(03)00399-X
Brashear, A., Butler, I. J., Hyland, K., Farlow, M. R., and Dobyns, W. B. (1998). Cerebrospinal fluid homovanillic acid levels in rapid-onset dystonia-parkinsonism. Ann. Neurol. 43, 521–526. doi: 10.1002/ana.410430417
Brashear, A., DeLeon, D., Bressman, S. B., Thyagarajan, D., Farlow, M. R., and Dobyns, W. B. (1997). Rapid-onset dystonia-parkinsonism in a second family. Neurology 48, 1066–1069.
Brashear, A., Dobyns, W. B., Carvalho Aguiar, P., Borg, M., Frijns, C. J., and Gollamudi, S. (2007). The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain 130, 610–622. doi: 10.1093/brain/awl340
Brashear, A., Sweadner, K. J., Cook, J. F., Swoboda, K. J., and Ozelius, L. (2014). “ATP1A3-related neurologic disorders,” in GeneReviews®, eds R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E. Wallace, A. Amemiya, L. J. H. Bean et al. (Seattle, WA: University of Washington), 1993–2016.
Brines, M. L., Dare, A. O., and de Lanerolle, N. C. (1995). The cardiac glycoside ouabain potentiates excitotoxic injury of adult neurons in rat hippocampus. Neurosci. Lett. 191, 145–148.
Brodsky, J. L., and Guidotti, G. (1990). Sodium affinity of brain Na(+)-K(+)-ATPase is dependent on isozyme and environment of the pump. Am. J. Physiol. 258(5 Pt 1), C803–C811.
Bublitz, M., Morth, J. P., and Nissen, P. (2011). P-type ATPases at a glance. J. Cell Sci. 124(Pt 15), 2515–2519. doi: 10.1242/jcs.088716
Calderon, D. P., Fremont, R., Kraenzlin, F., and Khodakhah, K. (2011). The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat. Neurosci. 14, 357–365. doi: 10.1038/nn.2753
Campbell, D. B., North, J. B., and Hess, E. J. (1999). Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp. Neurol. 160, 268–278. doi: 10.1006/exnr.1999.7171
Carter, B. C., and Bean, B. P. (2009). Sodium entry during action potentials of mammalian neurons: incomplete inactivation and reduced metabolic efficiency in fast-spiking neurons. Neuron 64, 898–909. doi: 10.1016/j.neuron.2009.12.011
Casaer, P., and Azou, M. (1984). Flunarizine in alternating hemiplegia in childhood. Lancet 324, 579. doi: 10.1016/S0140-6736(84)90794-3
Clapcote, S. J., Duffy, S., Xie, G., Kirshenbaum, G., Bechard, A. R., Rodacker Schack, V., et al. (2009). Mutation I810N in the alpha3 isoform of Na+,K+-ATPase causes impairments in the sodium pump and hyperexcitability in the CNS. Proc. Natl. Acad. Sci. U.S.A. 106, 14085–14090. doi: 10.1073/pnas.0904817106
Cook, J. F., Hill, D. F., Snively, B. M., Boggs, N., Suerken, C. K., Haq, I., et al. (2014). Cognitive impairment in rapid-onset dystonia-parkinsonism. Mov. Disord. 29, 344–350. doi: 10.1002/mds.25790
Corazzi, L., Piccinin, G. L., Roberti, R., Marku, N., Binaglia, L., Porcellati, G., et al. (1985). Effect of various drugs producing convulsive seizures on rat brain glycerolipid metabolism. Neurochem. Res. 10, 879–885.
Crambert, G., Hasler, U., Beggah, A. T., Yu, C., Modyanov, N. N., Horisberger, J. D., et al. (2000). Transport and pharmacological properties of nine different human Na, K-ATPase isozymes. J. Biol. Chem. 275, 1976–1986. doi: 10.1074/jbc.275.3.1976
Creson, T. K., Hao, Y., Engel, S., Shen, Y., Hamidi, A., Zhuo, M., et al. (2009). The anterior cingulate ERK pathway contributes to regulation of behavioral excitement and hedonic activity. Bipolar Disord. 11, 339–350. doi: 10.1111/j.1399-5618.2009.00697.x
Cull-Candy, S., Brickley, S., and Farrant, M. (2001). NMDA receptor subunits: diversity, development and disease. Curr. Opin. Neurobiol. 11, 327–335. doi: 10.1016/s0959-4388(00)00215-4
Davidson, D. L., Tsukada, Y., and Barbeau, A. (1978). Ouabain induced seizures: site of production and response to anticonvulsants. Can. J. Neurol. Sci. 5, 405–411.
de Carvalho Aguiar, P., Sweadner, K. J., Penniston, J. T., Zaremba, J., Liu, L., Caton, M., et al. (2004). Mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 43, 169–175. doi: 10.1016/j.neuron.2004.06.028
de Juan-Sanz, J., Nunez, E., Villarejo-Lopez, L., Perez-Hernandez, D., Rodriguez-Fraticelli, A. E., Lopez-Corcuera, B., et al. (2013). Na+/K+-ATPase is a new interacting partner for the neuronal glycine transporter GlyT2 that downregulates its expression in vitro and in vivo. J. Neurosci. 33, 14269–14281. doi: 10.1523/JNEUROSCI.1532-13.2013
DeAndrade, M. P., Yokoi, F., van Groen, T., Lingrel, J. B., and Li, Y. (2011). Characterization of Atp1a3 mutant mice as a model of rapid-onset dystonia with parkinsonism. Behav. Brain Res. 216, 659–665. doi: 10.1016/j.bbr.2010.09.009
Demos, M. K., van Karnebeek, C. D., Ross, C. J., Adam, S., Shen, Y., Zhan, S. H., et al. (2014). A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet J. Rare Dis. 9:15. doi: 10.1186/1750-1172-9-15
Dobretsov, M., and Stimers, J. R. (2005). Neuronal function and alpha3 isoform of the Na/K-ATPase. Front. Biosci. 10, 2373–2396. doi: 10.2741/1704
Dobyns, W. B., Ozelius, L. J., Kramer, P. L., Brashear, A., Farlow, M. R., Perry, T. R., et al. (1993). Rapid-onset dystonia-parkinsonism. Neurology 43, 2596–2602.
Doggett, N. S., and Spencer, P. S. (1971). Pharmacological properties of centrally administered ouabain and their modification by other drugs. Br. J. Pharmacol. 42, 242–253.
Edwards, I. J., Bruce, G., Lawrenson, C., Howe, L., Clapcote, S. J., Deuchars, S. A., et al. (2013). Na+/K+ ATPase alpha1 and alpha3 isoforms are differentially expressed in alpha- and gamma-motoneurons. J. Neurosci. 33, 9913–9919. doi: 10.1523/JNEUROSCI.5584-12.2013
El-Mallakh, R. S., El-Masri, M. A., Huff, M. O., Li, X. P., Decker, S., and Levy, R. S. (2003). Intracerebroventricular administration of ouabain as a model of mania in rats. Bipolar Disord. 5, 362–365. doi: 10.1034/j.1399-5618.2003.00053.x
el-Mallakh, R. S., and Wyatt, R. J. (1995). The Na,K-ATPase hypothesis for bipolar illness. Biol. Psychiatry 37, 235–244. doi: 10.1016/0006-3223(94)00201-D
Engel, S. R., Creson, T. K., Hao, Y., Shen, Y., Maeng, S., Nekrasova, T., et al. (2009). The extracellular signal-regulated kinase pathway contributes to the control of behavioral excitement. Mol. Psychiatry 14, 448–461. doi: 10.1038/sj.mp.4002135
Fanselow, M. S., and Dong, H.-W. (2010). Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65, 7. doi: 10.1016/j.neuron.2009.11.031
Faull, R. L. (1978). The cerebellofugal projections in the brachium conjunctivum of the rat. II. The ipsilateral and contralateral descending pathways. J. Comp. Neurol. 178, 519–535. doi: 10.1002/cne.901780307
Faull, R. L., and Carman, J. B. (1978). The cerebellofugal projections in the brachium conjunctivum of the rat I. The contralateral ascending pathway. J. Comp. Neurol. 178, 495–517.
Fernandes, M. J., Naffah-Mazzacoratti, M. G., and Cavalheiro, E. A. (1996). Na+K+ ATPase activity in the rat hippocampus: a study in the pilocarpine model of epilepsy. Neurochem. Int. 28, 497–500.
Fons, C., Campistol, J., Panagiotakaki, E., Giannotta, M., Arzimanoglou, A., Gobbi, G., et al. (2012). Alternating hemiplegia of childhood: metabolic studies in the largest European series of patients. Eur. J. Paediatr. Neurol. 16, 10–14. doi: 10.1016/j.ejpn.2011.08.006
Fremont, R., Calderon, D. P., Maleki, S., and Khodakhah, K. (2014). Abnormal high-frequency burst firing of cerebellar neurons in rapid-onset dystonia-parkinsonism. J. Neurosci. 34, 11723–11732. doi: 10.1523/JNEUROSCI.1409-14.2014
Fremont, R., Tewari, A., and Khodakhah, K. (2015). Aberrant purkinje cell activity is the cause of dystonia in a shRNA-based mouse model of rapid onset Dystonia-Parkinsonism. Neurobiol. Dis. 82, 200–212. doi: 10.1016/j.nbd.2015.06.004
Haglund, M. M., and Schwartzkroin, P. A. (1990). Role of Na-K pump potassium regulation and IPSPs in seizures and spreading depression in immature rabbit hippocampal slices. J. Neurophysiol. 63, 225–239.
Hazelwood, L. A., Free, R. B., Cabrera, D. M., Skinbjerg, M., and Sibley, D. R. (2008). Reciprocal modulation of function between the D1 and D2 dopamine receptors and the Na+,K+-ATPase. J. Biol. Chem. 283, 36441–36453. doi: 10.1074/jbc.M805520200
Heimer, G., Sadaka, Y., Israelian, L., Feiglin, A., Ruggieri, A., Marshall, C. R., et al. (2015). CAOS-episodic cerebellar ataxia, areflexia, optic atrophy, and sensorineural hearing loss: a third allelic disorder of the ATP1A3 gene. J. Child Neurol. 30, 1749–1756. doi: 10.1177/0883073815579708
Heinzen, E. L., Arzimanoglou, A., Brashear, A., Clapcote, S. J., Gurrieri, F., Goldstein, D. B., et al. (2014). Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. 13, 503–514. doi: 10.1016/S1474-4422(14)70011-0
Heinzen, E. L., Swoboda, K. J., Hitomi, Y., Gurrieri, F., Nicole, S., de Vries, B., et al. (2012). De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat. Genet. 44, 1030–1034. doi: 10.1038/ng.2358
Hieber, V., Siegel, G. J., Fink, D. J., Beaty, M. W., and Mata, M. (1991). Differential distribution of (Na, K)-ATPase alpha isoforms in the central nervous system. Cell. Mol. Neurobiol. 11, 253–262.
Hilbers, F., Kopec, W., Isaksen, T. J., Holm, T. H., Lykke-Hartmann, K., Nissen, P., et al. (2016). Tuning of the Na,K-ATPase by the beta subunit. Sci. Rep. 6:20442. doi: 10.1038/srep20442
Hokin-Neaverson, M., and Jefferson, J. W. (1989a). Deficient erythrocyte NaK-ATPase activity in different affective states in bipolar affective disorder and normalization by lithium therapy. Neuropsychobiology 22, 18–25.
Hokin-Neaverson, M., and Jefferson, J. W. (1989b). Erythrocyte sodium pump activity in bipolar affective disorder and other psychiatric disorders. Neuropsychobiology 22, 1–7.
Hunanyan, A. S., Fainberg, N. A., Linabarger, M., Arehart, E., Leonard, A. S., Adil, S. M., et al. (2015). Knock-in mouse model of alternating hemiplegia of childhood: behavioral and electrophysiologic characterization. Epilepsia 56, 82–93. doi: 10.1111/epi.12878
Ikeda, K., Satake, S., Onaka, T., Sugimoto, H., Takeda, N., Imoto, K., et al. (2013). Enhanced inhibitory neurotransmission in the cerebellar cortex of Atp1a3-deficient heterozygous mice. J. Physiol. 591(Pt 13), 3433–3449. doi: 10.1113/jphysiol.2012.247817
Jaffer, F., Avbersek, A., Vavassori, R., Fons, C., Campistol, J., Stagnaro, M., et al. (2015). Faulty cardiac repolarization reserve in alternating hemiplegia of childhood broadens the phenotype. Brain 138(Pt 10), 2859–2874. doi: 10.1093/brain/awv243
Jaisser, F., Jaunin, P., Geering, K., Rossier, B. C., and Horisberger, J. D. (1994). Modulation of the Na,K-pump function by beta subunit isoforms. J. Gen. Physiol. 103, 605–623.
Jewell, E. A., and Lingrel, J. B. (1991). Comparison of the substrate dependence properties of the rat Na,K-ATPase alpha 1, alpha 2, and alpha 3 isoforms expressed in HeLa cells. J. Biol. Chem. 266, 16925–16930.
Kanazawa, O., Shirasaka, Y., Hattori, H., Okuno, T., and Mikawa, H. (1991). Ictal 99mTc-HMPAO SPECT in alternating hemiplegia. Pediatr. Neurol. 7, 121–124.
Kaplan, J. H. (2002). Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 71, 511–535. doi: 10.1146/annurev.biochem.71.102201.141218
Kim, S. H., Yu, H. S., Park, H. G., Jeon, W. J., Song, J. Y., Kang, U. G., et al. (2008). Dose-dependent effect of intracerebroventricular injection of ouabain on the phosphorylation of the MEK1/2-ERK1/2-p90RSK pathway in the rat brain related to locomotor activity. Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 1637–1642. doi: 10.1016/j.pnpbp.2008.05.027
Kirshenbaum, G. S., Clapcote, S. J., Duffy, S., Burgess, C. R., Petersen, J., Jarowek, K. J., et al. (2011a). Mania-like behavior induced by genetic dysfunction of the neuron-specific Na+,K+-ATPase alpha3 sodium pump. Proc. Natl. Acad. Sci. U.S.A. 108, 18144–18149. doi: 10.1073/pnas.1108416108
Kirshenbaum, G. S., Clapcote, S. J., Petersen, J., Vilsen, B., Ralph, M. R., and Roder, J. C. (2012). Genetic suppression of agrin reduces mania-like behavior in Na+, K+ -ATPase alpha3 mutant mice. Genes Brain Behav. 11, 436–443. doi: 10.1111/j.1601-183X.2012.00800.x
Kirshenbaum, G. S., Dawson, N., Mullins, J. G., Johnston, T. H., Drinkhill, M. J., Edwards, I. J., et al. (2013). Alternating hemiplegia of childhood-related neural and behavioural phenotypes in Na+,K+-ATPase alpha3 missense mutant mice. PLoS ONE 8:e60141. doi: 10.1371/journal.pone.0060141
Kirshenbaum, G. S., Saltzman, K., Rose, B., Petersen, J., Vilsen, B., and Roder, J. C. (2011b). Decreased neuronal Na+, K+ -ATPase activity in Atp1a3 heterozygous mice increases susceptibility to depression-like endophenotypes by chronic variable stress. Genes Brain Behav. 10, 542–550. doi: 10.1111/j.1601-183X.2011.00691.x
Kristensen, A. S., Andersen, J., Jørgensen, T. N., Sørensen, L., Eriksen, J., Loland, C. J., et al. (2011). SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 63, 585–640. doi: 10.1124/pr.108.000869
Kvajo, M., Albrecht, H., Meins, M., Hengst, U., Troncoso, E., Lefort, S., et al. (2004). Regulation of brain proteolytic activity is necessary for the in vivo function of NMDA receptors. J. Neurosci. 24, 9734–9743. doi: 10.1523/JNEUROSCI.3306-04.2004
Laursen, M., Yatime, L., Nissen, P., and Fedosova, N. U. (2013). Crystal structure of the high-affinity Na+K+-ATPase-ouabain complex with Mg2+ bound in the cation binding site. Proc. Natl. Acad. Sci. U.S.A. 110, 10958–10963. doi: 10.1073/pnas.1222308110
LeDoux, M. S., and Lorden, J. F. (1998). Abnormal cerebellar output in the genetically dystonic rat. Adv. Neurol. 78, 63–78.
Lees, G. J., Lehmann, A., Sandberg, M., and Hamberger, A. (1990). The neurotoxicity of ouabain, a sodium-potassium ATPase inhibitor, in the rat hippocampus. Neurosci. Lett. 120, 159–162.
Li, M., Jazayeri, D., Corry, B., McSweeney, K. M., Heinzen, E. L., Goldstein, D. B., et al. (2015). A functional correlate of severity in alternating hemiplegia of childhood. Neurobiol. Dis. 77, 88–93. doi: 10.1016/j.nbd.2015.02.002
Linazasoro, G., Indakoetxea, B., Ruiz, J., Van Blercom, N., and Lasa, A. (2002). Possible sporadic rapid-onset dystonia-parkinsonism. Mov. Disord. 17, 608–609. doi: 10.1002/mds.10103
Liu, Y. B., Tewari, A., Salameh, J., Arystarkhova, E., Hampton, T. G., Brashear, A., et al. (2015). A dystonia-like movement disorder with brain and spinal neuronal defects is caused by mutation of the mouse laminin β1 subunit, Lamb1. Elife 4:e11102. doi: 10.7554/eLife.11102
Llinás, R., and Sugimori, M. (1980). Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. J. Physiol. 305, 171–195.
Looney, S. W., and el-Mallakh, R. S. (1997). Meta-analysis of erythrocyte Na,K-ATPase activity in bipolar illness. Depress. Anxiety 5, 53–65.
López-Corcuera, B., Martínez-Maza, R., Núñez, E., Roux, M., Supplisson, S., and Aragón, C. (1998). Differential properties of two stably expressed brain-specific glycine transporters. J. Neurochem. 71, 2211–2219.
Lorden, J. F., Lutes, J., Michela, V. L., and Ervin, J. (1992). Abnormal cerebellar output in rats with an inherited movement disorder. Exp. Neurol. 118, 95–104.
Lyons, M. R., and West, A. E. (2011). Mechanisms of specificity in neuronal activity-regulated gene transcription. Prog. Neurobiol. 94, 259–295. doi: 10.1016/j.pneurobio.2011.05.003
Marquezan, B. P., Funck, V. R., Oliveira, C. V., Pereira, L. M., Araújo, S. M., Zarzecki, M. S., et al. (2013). Pentylenetetrazol-induced seizures are associated with Na(+),K(+)-ATPase activity decrease and alpha subunit phosphorylation state in the mice cerebral cortex. Epilepsy Res. 105, 396–400. doi: 10.1016/j.eplepsyres.2013.03.007
Mazza, M., Di Nicola, M., Della Marca, G., Janiri, L., Bria, P., and Mazza, S. (2007). Bipolar disorder and epilepsy: a bidirectional relation? Neurobiological underpinnings, current hypotheses, and future research directions. Neuroscientist 13, 392–404. doi: 10.1177/10738584070130041101
McGrail, K. M., Phillips, J. M., and Sweadner, K. J. (1991). Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na,K-ATPase. J. Neurosci. 11, 381–391.
McLin, J. P., and Steward, O. (2006). Comparison of seizure phenotype and neurodegeneration induced by systemic kainic acid in inbred, outbred, and hybrid mouse strains. Eur. J. Neurosci. 24, 2191–2202. doi: 10.1111/j.1460-9568.2006.05111.x
Mikati, M. A., Kramer, U., Zupanc, M. L., and Shanahan, R. J. (2000). Alternating hemiplegia of childhood: clinical manifestations and long-term outcome. Pediatr. Neurol. 23, 134–141. doi: 10.1016/S0887-8994(00)00157-0
Mizumori, S. J., Sakai, D. H., Rosenzweig, M. R., Bennett, E. L., and Wittreich, P. (1987). Investigations into the neuropharmacological basis of temporal stages of memory formation in mice trained in an active avoidance task. Behav. Brain Res. 23, 239–250.
Monteith, G. R., and Blaustein, M. P. (1998). Different effects of low and high dose cardiotonic steroids on cytosolic calcium in spontaneously active hippocampal neurons and in co-cultured glia. Brain Res. 795, 325–340.
Moseley, A. E., Williams, M. T., Schaefer, T. L., Bohanan, C. S., Neumann, J. C., Behbehani, M. M., et al. (2007). Deficiency in Na,K-ATPase alpha isoform genes alters spatial learning, motor activity, and anxiety in mice. J. Neurosci. 27, 616–626. doi: 10.1523/JNEUROSCI.4464-06.2007
Mula, M., Marotta, A. E., and Monaco, F. (2010). Epilepsy and bipolar disorders. Expert Rev. Neurother. 10, 13–23. doi: 10.1586/ern.09.139
Munzer, J. S., Daly, S. E., Jewell-Motz, E. A., Lingrel, J. B., and Blostein, R. (1994). Tissue- and isoform-specific kinetic behavior of the Na,K-ATPase. J. Biol. Chem. 269, 16668–16676.
Neher, E., and Sakaba, T. (2008). Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59, 861–872. doi: 10.1016/j.neuron.2008.08.019
Neville, B. G., and Ninan, M. (2007). The treatment and management of alternating hemiplegia of childhood. Dev. Med. Child Neurol. 49, 777–780. doi: 10.1111/j.1469-8749.2007.00777.x
Novy, J., McWilliams, E., and Sisodiya, S. M. (2014). Asystole in alternating hemiplegia with de novo ATP1A3 mutation. Eur. J. Med. Genet. 57, 37–39. doi: 10.1016/j.ejmg.2013.11.003
Oblak, A. L., Hagen, M. C., Sweadner, K. J., Haq, I., Whitlow, C. T., Maldjian, J. A., et al. (2014). Rapid-onset dystonia-parkinsonism associated with the I758S mutation of the ATP1A3 gene: a neuropathologic and neuroanatomical study of four siblings. Acta Neuropathol. 128, 81–98. doi: 10.1007/s00401-014-1279-x
Paciorkowski, A. R., McDaniel, S. S., Jansen, L. A., Tully, H., Tuttle, E., Ghoneim, D. H., et al. (2015). Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 56, 422–430. doi: 10.1111/epi.12914
Palmgren, M. G., and Nissen, P. (2011). P-type ATPases. Annu. Rev. Biophys. 40, 243–266. doi: 10.1146/annurev.biophys.093008.131331
Panagiotakaki, E., De Grandis, E., Stagnaro, M., Heinzen, E. L., Fons, C., Sisodiya, S., et al. (2015). Clinical profile of patients with ATP1A3 mutations in alternating hemiplegia of childhood—a study of 155 patients. Orphanet J. Rare Dis. 10, 1–13. doi: 10.1186/s13023-015-0335-5
Panagiotakaki, E., Gobbi, G., Neville, B., Ebinger, F., Campistol, J., and Nevsímalová, S. (2010). Evidence of a non-progressive course of alternating hemiplegia of childhood: study of a large cohort of children and adults. Brain 133, 3598–3610. doi: 10.1093/brain/awq295
Peng, L., Martin-Vasallo, P., and Sweadner, K. J. (1997). Isoforms of Na,K-ATPase alpha and beta subunits in the rat cerebellum and in granule cell cultures. J. Neurosci. 17, 3488–3502.
Perona, M. T., Waters, S., Hall, F. S., Sora, I., Lesch, K. P., Murphy, D. L., et al. (2008). Animal models of depression in dopamine, serotonin, and norepinephrine transporter knockout mice: prominent effects of dopamine transporter deletions. Behav. Pharmacol. 19, 566–574. doi: 10.1097/FBP.0b013e32830cd80f
Prickaerts, J., Moechars, D., Cryns, K., Lenaerts, I., van Craenendonck, H., Goris, I., et al. (2006). Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J. Neurosci. 26, 9022–9029. doi: 10.1523/JNEUROSCI.5216-05.2006
Purves, D., Bonardi, C., and Hall, G. (1995). Enhancement of latent inhibition in rats with electrolytic lesions of the hippocampus. Behav. Neurosci. 109, 366–370.
Raman, I. M., and Bean, B. P. (1997). Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 17, 4517–4526.
Reilly, S., Harley, C., and Revusky, S. (1993). Ibotenate lesions of the hippocampus enhance latent inhibition in conditioned taste aversion and increase resistance to extinction in conditioned taste preference. Behav. Neurosci. 107, 996–1004.
Richter, A., Brotchie, J. M., Crossman, A. R., and Löscher, W. (1998). [3H]-2-deoxyglucose uptake study in mutant dystonic hamsters: abnormalities in discrete brain regions of the motor system. Mov. Disord. 13, 718–725. doi: 10.1002/mds.870130419
Riegel, R. E., Valvassori, S. S., Elias, G., Réus, G. Z., Steckert, A. V., de Souza, B., et al. (2009). Animal model of mania induced by ouabain: evidence of oxidative stress in submitochondrial particles of the rat brain. Neurochem. Int. 55, 491–495. doi: 10.1016/j.neuint.2009.05.003
Rose, A. M., Mellett, B. J., Valdes, R. Jr., Kleinman, J. E., Herman, M. M., Li, R., et al. (1998). Alpha 2 isoform of the Na,K-adenosine triphosphatase is reduced in temporal cortex of bipolar individuals. Biol. Psychiatry 44, 892–897.
Rosewich, H., Ohlenbusch, A., Huppke, P., Schlotawa, L., Baethmann, M., and Carrilho, I. (2014). The expanding clinical and genetic spectrum of ATP1A3-related disorders. Neurology 82, 945–955. doi: 10.1212/wnl.0000000000000212
Rosewich, H., Thiele, H., Ohlenbusch, A., Maschke, U., Altmuller, J., Frommolt, P., et al. (2012). Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene-identification study. Lancet Neurol. 11, 764–773. doi: 10.1016/S1474-4422(12)70182-5
Ruktanonchai, D. J., El-Mallakh, R. S., Li, R., and Levy, R. S. (1998). Persistent hyperactivity following a single intracerebroventricular dose of ouabain. Physiol. Behav. 63, 403–406.
Saito, Y., Sakuragawa, N., Sasaki, M., Sugai, K., and Hashimoto, T. (1998). A case of alternating hemiplegia of childhood with cerebellar atrophy. Pediatr. Neurol. 19, 65–68.
Sasaki, M., Ishii, A., Saito, Y., and Hirose, S. (2013). Intermediate form between alternating hemiplegia of childhood and rapid-onset dystonia-parkinsonism. Mov. Disord. 33, 390–393. doi: 10.1002/mds.25659
Sasaki, M., Ishii, A., Saito, Y., Morisada, N., Iijima, K., and Takada, S. (2014). Genotype-phenotype correlations in alternating hemiplegia of childhood. Neurology 82:e0127045. doi: 10.1212/wnl.0000000000000102
Sasaki, M., Matsufuji, H., Inui, T., and Arima, K. (2011). Absence of small-vessel abnormalities in alternating hemiplegia of childhood. Brain Dev. 33, 390–393. doi: 10.1016/j.braindev.2010.08.011
Sasaki, M., Sakuma, H., Fukushima, A., Yamada, K., Ohnishi, T., and Matsuda, H. (2009). Abnormal cerebral glucose metabolism in alternating hemiplegia of childhood. Brain Dev. 31, 20–26. doi: 10.1016/j.braindev.2008.03.008
Schauwecker, P. E. (2002). Modulation of cell death by mouse genotype: differential vulnerability to excitatory amino acid-induced lesions. Exp. Neurol. 178, 219–235. doi: 10.1006/exnr.2002.8038
Schneider Oliveira, M., Flávia Furian, A., Freire Royes, L. F., Rechia Fighera, M., de Carvalho Myskiw, J., Gindri Fiorenza, N., et al. (2004). Ascorbate modulates pentylenetetrazol-induced convulsions biphasically. Neuroscience 128, 721–728. doi: 10.1016/j.neuroscience.2004.07.012
Shafer, M. E., Mayfield, J. W., and McDonald, F. (2005). Alternating hemiplegia of childhood: a study of neuropsychological functioning. Appl. Neuropsychol. 12, 49–56. doi: 10.1207/s15324826an1201_8
Somjen, G. G. (2002). Ion regulation in the brain: implications for pathophysiology. Neuroscientist 8, 254–267. doi: 10.1177/1073858402008003011
Sugimoto, H., Ikeda, K., and Kawakami, K. (2014). Heterozygous mice deficient in Atp1a3 exhibit motor deficits by chronic restraint stress. Behav. Brain Res. 272, 100–110. doi: 10.1016/j.bbr.2014.06.048
Svetel, M., Ozelius, L. J., Buckley, A., Lohmann, K., Brajkovic, L., Klein, C., et al. (2010). Rapid-onset dystonia-parkinsonism: case report. J. Neurol. 257, 472–474. doi: 10.1007/s00415-009-5385-y
Sweadner, K. J., Toro, C., Whitlow, C. T., Snively, B. M., Cook, J. F., Ozelius, L. J., et al. (2016). ATP1A3 mutation in adult rapid-onset ataxia. PLoS ONE 11:e0151429. doi: 10.1371/journal.pone.0151429
Sweney, M. T., Newcomb, T. M., and Swoboda, K. J. (2015). The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, alternating hemiplegia of childhood, rapid-onset Dystonia-Parkinsonism, CAPOS and beyond. Pediatr. Neurol. 52, 56–64. doi: 10.1016/j.pediatrneurol.2014.09.015
Sweney, M. T., Silver, K., Gerard-Blanluet, M., Pedespan, J. M., Renault, F., Arzimanoglou, A., et al. (2009). Alternating hemiplegia of childhood: early characteristics and evolution of a neurodevelopmental syndrome. Pediatrics 123, e534–e541. doi: 10.1542/peds.2008-2027
Takahashi, K. I., and Copenhagen, D. R. (1996). Modulation of neuronal function by intracellular pH. Neurosci. Res. 24, 109–116. doi: 10.1016/0168-0102(95)00989-2
Termsarasab, P., Yang, A. C., and Frucht, S. J. (2015). Intermediate phenotypes of ATP1A3 mutations: phenotype–genotype correlations. Tremor Other Hyperkinet. Mov. 5, 336. doi: 10.7916/D8MG7NS8
Tinazzi, M., Rosso, T., and Fiaschi, A. (2003). Role of the somatosensory system in primary dystonia. Mov. Disord. 18, 605–622. doi: 10.1002/mds.10398
Tochigi, M., Iwamoto, K., Bundo, M., Sasaki, T., Kato, N., and Kato, T. (2008). Gene expression profiling of major depression and suicide in the prefrontal cortex of postmortem brains. Neurosci. Res. 60, 184–191. doi: 10.1016/j.neures.2007.10.010
Ulate-Campos, A., Fons, C., Campistol, J., Martorell, L., Cancho-Candela, R., Eiris, J., et al. (2014). [Alternating hemiplegia of childhood: ATP1A3 gene analysis in 16 patients]. Med. Clin. (Barc.) 143, 25–28. doi: 10.1016/j.medcli.2014.01.036
Vaillend, C., Mason, S. E., Cuttle, M. F., and Alger, B. E. (2002). Mechanisms of neuronal hyperexcitability caused by partial inhibition of Na+-K+-ATPases in the rat CA1 hippocampal region. J. Neurophysiol. 88, 2963–2978. doi: 10.1152/jn.00244.2002
Vedovato, N., and Gadsby, D. C. (2014). Route, mechanism, and implications of proton import during Na+/K+ exchange by native Na+/K+-ATPase pumps. J. Gen. Physiol. 143, 449–464. doi: 10.1085/jgp.201311148
Viollet, L., Glusman, G., Murphy, K. J., Newcomb, T. M., Reyna, S. P., and Sweney, M. (2015). Alternating hemiplegia of childhood: retrospective genetic study and genotype-phenotype correlations in 187 subjects from the US AHCF registry. PLoS ONE 10:e127045. doi: 10.1371/journal.pone.0127045
Vollono, C., Rinalduzzi, S., Miliucci, R., Vigevano, F., and Valeriani, M. (2014). Somatosensory system hyperexcitability in alternating hemiplegia of childhood. Eur. J. Neurol. 21, 1478-e97. doi: 10.1111/ene.12516
Wong, V., Ho, G. C., Yeung, H. W., and Ma, K. M. (1993). Alternating hemiplegia syndrome: electroencephalogram, brain mapping, and brain perfusion SPECT scan study in a Chinese girl. J. Child Neurol. 8, 221–226.
Wu, Z. Q., Chen, J., Chi, Z. Q., and Liu, J. G. (2007). Involvement of dopamine system in regulation of Na+,K+-ATPase in the striatum upon activation of opioid receptors by morphine. Mol. Pharmacol. 71, 519–530. doi: 10.1124/mol.106.029561
Yamashita, S., Hamano, S., Tanaka, M., Yoshinari, S., Minamitani, M., and Hayakawa, M. (2005). [Single photon emission computed tomography findings in a case of alternating hemiplegia of childhood in relation to migraine]. No To Hattatsu 37, 413–418.
Yang, X., Gao, H., Zhang, J., Xu, X., Liu, X., and Wu, X. (2014). ATP1A3 mutations and genotype-phenotype correlation of alternating hemiplegia of childhood in Chinese patients. PLoS ONE 9:e97274. doi: 10.1371/journal.pone.0097274
Yatime, L., Laursen, M., Morth, J. P., Esmann, M., Nissen, P., and Fedosova, N. U. (2011). Structural insights into the high affinity binding of cardiotonic steroids to the Na+,K+-ATPase. J. Struct. Biol. 174, 296–306. doi: 10.1016/j.jsb.2010.12.004
Yu, H. S., Kim, S. H., Park, H. G., Kim, Y. S., and Ahn, Y. M. (2010). Activation of Akt signaling in rat brain by intracerebroventricular injection of ouabain: a rat model for mania. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 888–894. doi: 10.1016/j.pnpbp.2010.04.010
Yu, S. P. (2003). Na(+), K(+)-ATPase: the new face of an old player in pathogenesis and apoptotic/hybrid cell death. Biochem. Pharmacol. 66, 1601–1609. doi: 10.1016/S0006-2952(03)00531-8
Zahler, R., Gilmore-Hebert, M., Baldwin, J. C., Franco, K., and Benz, E. J. Jr. (1993). Expression of alpha isoforms of the Na,K-ATPase in human heart. Biochim. Biophys. Acta. 1149, 189–194.
Zahler, R., Zhang, Z. T., Manor, M., and Boron, W. F. (1997). Sodium kinetics of Na,K-ATPase alpha isoforms in intact transfected HeLa cells. J. Gen. Physiol. 110, 201–213.
Zhan, H., Tada, T., Nakazato, F., Tanaka, Y., and Hongo, K. (2004). Spatial learning transiently disturbed by intraventricular administration of ouabain. Neurol. Res. 26, 35–40. doi: 10.1179/016164104773026507
Zhang, L. N., Li, J. X., Hao, L., Sun, Y. J., Xie, Y. H., Wu, S. M., et al. (2013). Crosstalk between dopamine receptors and the Na(+)/K(+)-ATPase (review). Mol. Med. Rep. 8, 1291–1299. doi: 10.3892/mmr.2013.1697
Zucker, R. S. (1999). Calcium- and activity-dependent synaptic plasticity. Curr. Opin. Neurobiol. 9, 305–313.
Keywords: α3 sodium ion pump, neurons, rapid onset dystonia parkinsonism (RDP), alternating hemiplegia of childhood (AHC) and cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS), mouse models
Citation: Holm TH and Lykke-Hartmann K (2016) Insights into the Pathology of the α3 Na+/K+-ATPase Ion Pump in Neurological Disorders; Lessons from Animal Models. Front. Physiol. 7:209. doi: 10.3389/fphys.2016.00209
Received: 12 March 2016; Accepted: 22 May 2016;
Published: 14 June 2016.
Edited by:
Olga Vagin, University of California, Los Angeles, USAReviewed by:
Xavier Gasull, University of Barcelona, SpainPablo Martín-Vasallo, Universidad de La Laguna, Spain
Copyright © 2016 Holm and Lykke-Hartmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karin Lykke-Hartmann, a2x5QGJpb21lZC5hdS5kaw==