 Eduardo D. Costa
Eduardo D. Costa Bruno A. Rezende
Bruno A. Rezende Steyner F. Cortes
Steyner F. Cortes Virginia S. Lemos
Virginia S. Lemos
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 02 June 2016
Sec. Integrative Physiology
Volume 7 - 2016 | https://doi.org/10.3389/fphys.2016.00206
This article is part of the Research TopicCelebrating Twenty Years of the Brazilian Symposium on Cardiovascular PhysiologyView all 23 articles
The family of nitric oxide synthases (NOS) has significant importance in various physiological mechanisms and is also involved in many pathological processes. Three NOS isoforms have been identified: neuronal NOS (nNOS or NOS 1), endothelial NOS (eNOS or NOS 3), and an inducible NOS (iNOS or NOS 2). Both nNOS and eNOS are constitutively expressed. Classically, eNOS is considered the main isoform involved in the control of the vascular function. However, more recent studies have shown that nNOS is present in the vascular endothelium and importantly contributes to the maintenance of the homeostasis of the cardiovascular system. In physiological conditions, besides nitric oxide (NO), nNOS also produces hydrogen peroxide (H2O2) and superoxide () considered as key mediators in non-neuronal cells signaling. This mini-review highlights recent scientific releases on the role of nNOS in vascular homeostasis and cardiovascular disorders such as hypertension and atherosclerosis.
Since the early 80s, nitric oxide (NO) is considered an essential endothelium-derived molecule, crucial to the maintenance of cardiovascular homeostasis (Furchgott and Zawadzki, 1980). Later on, it became evident that a decrease in the bioavailability of NO participated in several cardiovascular disorders such as atherosclerosis (Napoli et al., 2006) and hypertension (Hermann et al., 2006).
NO is biologically generated by a family of three nitric oxide synthase enzymes (NOS) isoforms: neuronal nitric oxide synthase (nNOS or NOS1), inducible nitric oxide synthase (iNOS or NOS2), and endothelial nitric oxide synthase (eNOS or NOS3). Although nNOS is abundantly expressed in neurons, and associated with the control of neuronal functions (Bredt et al., 1990; Bredt and Snyder, 1992) it is known that this isoform is also expressed in many non-neuronal cells such as in the endothelium and smooth muscle cells of several types of vessels in animals (Boulanger et al., 1998; Loesch et al., 1998; Schwarz et al., 1999) and human (Buchwalow et al., 2002). Recent studies show consistent evidence that this isoform exhibits relevant physiological role in the control of vascular homeostasis (Kurihara et al., 1998; Fleming, 2003; Hagioka et al., 2005; Seddon et al., 2008, 2009).
Besides NO, nNOS also produces H2O2 in physiological conditions that contributes to endothelium-dependent vascular relaxation (Capettini et al., 2008, 2010). Impairment in endothelial nNOS-derived H2O2 production has been implicated in the endothelial dysfunction in atherosclerosis (Rabelo et al., 2003; Capettini et al., 2011) and hypertension (Silva et al., 2016). Given the importance of nNOS in health and disease, this mini-review highlights recent scientific releases on the role of nNOS in vascular homeostasis and vascular mal functioning linked to hypertension and atherosclerosis.
nNOS gene is positioned on chromosome 12 (12q24.2) and distributed over a region greater than 200 kb in human genomic DNA (Hall et al., 1994). It consists of 4299 nucleotides encoding 1434 amino acids (Boissel et al., 1998). nNOS exists as a monomer/dimer mixture, being the dimer the active form. Each monomer consists of two domains: N-terminal (catalytic or oxygenase) and C-terminal (reductase). The N-terminal domain binds to the thiolate-linked heme group, tetrahydrobiopterin (BH4), a redox co-factor; L-arginine the substrate, and the zinc ion. The C-terminal domain has binding sites for flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), and nicotinamide adenine dinucleotide phosphate (NADPH; Masters et al., 1996; Sagami et al., 2001; Feng et al., 2014).
A sequence of 40–50 amino acids inserted in the FMN domain is related to nNOS auto-inhibition by destabilizing calmodulin (CaM) binding to the enzyme and inhibiting intra- and inter-module electron transferring. This interaction occurs in low intracellular Ca2+ concentration ([Ca2+]i), taking part in the modulation of nNOS activity (Salerno et al., 1997; Daff et al., 1999; Garcin et al., 2004). Similarly, nNOS has a tail sequence of 21–42 amino acids at the C-terminal, related to the enzyme inhibition. Removal of this extension results in increased transference rates of electron flow in the reductase domain (Roman et al., 2000). Deletion of the auto-inhibitory domain and C-terminal tail results in CaM-independent electron transferring through the reductase domain, despite CaM is still required to promote electron transference from the FMN domain to the heme for NO production (Roman and Masters, 2006).
The dimerization maintained by the N-terminal domain is crucial for the catalytic activity of nNOS. Otherwise, the transport of electrons and formation of nNOS products do not exist (Stuehr, 1997). Dimer formation has the participation of residues from the oxygenase domain that form a “hook” which reaches across to the oxygenase domain of the other subunit to coordinate dimer formation (Crane et al., 1998). Zinc binding has a contribution in dimer stabilization (Hemmens et al., 2000). The disulfide bonds formed by cysteine residues along the nNOS molecule and BH4 binding are also important to stabilize nNOS dimeric form (Hemmens et al., 1998; Kamada et al., 2005).
Phosphorylation of nNOS has been shown to be the critical stage in the activation/inactivation of this isoform. Several phosphatases and kinases including protein kinase A, CaM-kinases (CaM-KI and CaMKII), protein kinase C, and phosphatase 1 may regulate the activity of nNOS. For instance, CaM-KI and CaM-KII phosphorylate Ser741 and Ser852, respectively, resulting in reduced activity of the enzyme through inhibition of CaM binding (Song et al., 2004). Phosphorylation on Ser1412 (in rat) or Ser1212 (in human) residue is associated with increased activity of nNOS (Chen et al., 2000; Adak et al., 2001).
The deficiency of L-arginine or BH4 may produce nNOS uncoupling and the enzyme synthesize superoxide instead of NO. Recently, it has been reported impaired NO signaling due to nNOS uncoupling in brain arteries of obese rats and consequent oxidative stress and vasoconstriction (Katakam et al., 2012). Moreover, nNOS uncoupling is associated with penile arteries constriction with erectile dysfunction in a model of metabolic syndrome (Sanchez et al., 2012).
Protein-protein interaction is one of the key events in controlling the enzymatic activity of NOS. There are numerous proteins that may have physical interaction with nNOS in a variety of roles including activation, inhibition, and trafficking within the cell.
The increase in [Ca2+]i and its subsequent binding to CaM is the main modulatory event of nNOS activation (Bredt and Snyder, 1990). The first step of nNOS activation consists of binding Ca2+ in CaM C-terminal domain. In sequence, the CaM C-terminal domain binds to nNOS. Then, in a similar way, Ca2+ binds to the CaM N-terminal domain, which also binds to nNOS and causes the activation of nNOS by the displacement of the auto-inhibitory domain of the enzyme. When the [Ca2+]i decrease, CaM dissociates from nNOS, and it becomes inactive again (Weissman et al., 2002).
Caveolins are scaffolding proteins situated at the caveolae, the flask-shaped non-clathrin-coated invaginations of the plasma membrane (Sowa, 2012). In skeletal muscle, nNOS directly interacts with caveolin-3, involving two distinct and physically separated caveolin scaffolding domains. This interaction inhibits nNOS activity (Venema et al., 1997). In a rat model of myocardial infarction, nNOS upregulation is associated with an increased binding with caveolin-3 (Bendall et al., 2004). Moreover, caveolin-1 interacts with the oxygenase and reductase nNOS domains inhibiting electron transfers (Sato et al., 2004).
The NH2-terminus of nNOS has a binding site for the protein PIN (Jaffrey and Snyder, 1996). This endogenous protein inhibits nNOS by destabilizing the dimer isoform. Curiously, some studies have shown that PIN plays a physiological role in the control of insulin secretion (Lajoix et al., 2006). Moreover, neurogenic erectile dysfunction (NED) may be caused by impairment of nNOS regulation by PIN (Gonzalez-Cadavid and Rajfer, 2004).
The nNOS PDZ domain has 80–120 amino acid residues located in the NH2-terminus. The PDZ domain participates in the formation of active nNOS dimers and interacts with other proteins in different regions of the cell (Roman et al., 2002). A study to assess potential ligands for PDZ domain of nNOS was conducted by screening 13 billion different peptides and had found that this motif binds to peptides ending with Asp-X-Val.
NO formation through L-arginine is catalyzed by nNOS in two steps: the hydroxylation of L-arginine to the intermediate Nω-hydroxy-L-arginine (NOHA), which is then oxidized to L-citrulline and NO (Papale et al., 2012). In the first step, NADPH transfers electrons to FAD and FMN, which have the capacity to reduce molecular oxygen to superoxide () (Figure 1). At the same time, an electron from flavin-mononucleotide (FMNH) reduces the heme group (Fe3+ to Fe2+). The reduction of Fe3+ enables O2 linking resulting in an Fe2+ complex. The electron from the complex alternates between Fe2+ and O2, resulting in the complex Fe3+. In the deficiency of L-arginine or NOHA, Fe3+ transfers an electron to O2 liberating superoxide (). Studies have revealed that the heme group of nNOS oxidase domain is responsible for 90% of formation by this enzyme (Yoneyama et al., 2001). Alternatively, the intermediate Fe3+ can receive an electron, forming Fe3+ that interacts with H+ and releases H2O2 and Fe3+.
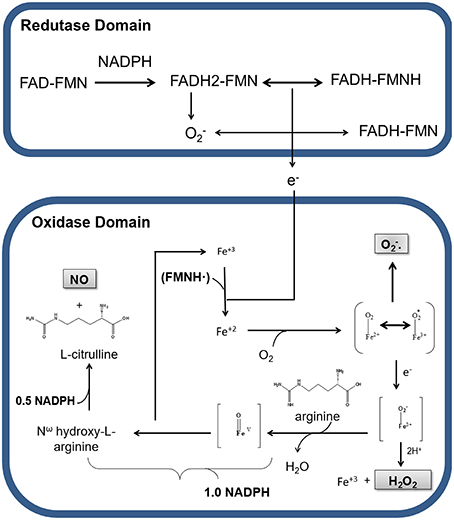
Figure 1. Production of nitric oxide (NO), hydrogen peroxide (H2O2), and superoxide anion () by nNOS in physiological conditions. Representation of electrons transport between the reductase and oxidase domains of nNOS. Nicotinamide adenine dinucleotide phosphate (NADPH) transfers electrons to the flavin adenine dinucleotide (FAD) and flavin adenine mononucleotide (FMN) in the reductase domain. In this process, oxygen receives electrons, being converted in superoxide (). In the presence of Ca2+/CaM electrons from the reductase domain, enable nNOS Fe3+ to bind O2 and to form O2-Fe2+, in the oxidase domain. During electronic switching in the heme group, H2O2 is produced, with consequent release of Fe3+. In the presence of BH4 and NADPH, a nitrogen group is inserted into L-arginine, generating the intermediate Nω-hydroxy-L-arginine, which is there after transformed in NO and L-citrulline.
In order to make the catalysis of L-arginine possible, BH4 cofactor must be binding to Fe3+ present in heme group. Electrons from BH4 cofactor are responsible for the formation of peroxy complexes (Fe3+-OOH−) with consequent hydroxylation of L-arginine, resulting in the formation of NOHA and regeneration of Fe3+ from heme group. In the next step, NOHA participates in another oxidation-reduction cycle by binding to Fe3+, which will receive more electrons from the reductase group, resulting in the cleavage of NOHA and release of water, L-citrulline and NO (Abu-Soud et al., 1994, 2000; Rosen et al., 2002).
Therefore, during the enzymatic formation of NO cycle, nNOS also generates H2O2 and (Figure 1). The production of these reactive oxygen species (ROS) by nNOS can occur even at saturating concentrations of L-arginine or NOHA in steps before the formation of NO (Rosen et al., 2002; Tsai et al., 2005; Weaver et al., 2005). At the expense of , the production of H2O2 is strongly increased by BH4 (Rosen et al., 2002).
Emerging evidence shows that nNOS has a physiologically relevant role in the control of the cardiovascular system. Here, we outline the recent advances on the role of nNOS in the vascular function.
There are several reports implicating the participation of nNOS in cerebral blood flow (CBF; Pelligrino et al., 1993; Santizo et al., 2000; Chi et al., 2003). Intraperitoneal injections of the selective nNOS inhibitor 7-nitroindazole (7-NI) depressed baseline CBF in rats (Montécot et al., 1997; Gotoh et al., 2001). Moreover, 7-NI decreased cerebral capillary flow in rats (Hudetz et al., 1998) and global CBF in cats (Hayashi et al., 2002). In rats, during hyperbaric conditions, it was found that the increase in CBF in the cortex prior to the appearance of electrical discharges was completely inhibited by 7-NI (Hagioka et al., 2005).
Aside from cerebral flow, it has been suggested that nNOS-derived NO regulates renal circulation. In the presence of, S-methyl-L-thiocitrulline (SMTC) a nNOS inhibitor, the vasoconstrictor response to angiotensin II is increased in the efferent arteriole (Ichihara et al., 1998). Additional evidence was obtained from nNOS−∕− mice, where genetic deletion of nNOS decreases medullary blood flow in response to angiotensin II (Mattson and Meister, 2005). In nNOS−∕− mice Vallon et al. (2001) also found that the feedback control of glomerular vascular tone is attenuated.
Similarly, studies in isolated vessels demonstrate the participation of nNOS in the control of vascular function. In pial arterioles of eNOS−∕− mice acetylcholine induced an nNOS-cGMP-dependent vasodilation (Meng et al., 1996, 1998). Another work confirmed the presence of nNOS in the endothelium of coronary arteries of eNOS−∕− mice and showed that shear stress activated endothelial nNOS-derived NO release, compensating the absence of eNOS-derived NO (Huang et al., 2002). In aorta of nNOS−∕− mice the vasodilator response induced by acetylcholine is reduced (Nangle et al., 2004). In small mesenteric arteries of female rats the inhibition of endothelial nNOS contributes to the decrease in the relaxation induced by estrogen. Furthermore, the same study showed that estrogen rapidly increased the nNOS activity and nNOS-mediated NO production in human umbilical vein endothelial cells (Lekontseva et al., 2011). A year later, the same group demonstrated that nNOS contributed to the estrogen-mediated vascular relaxation of mesenteric artery in young, but not in ovariectomized and aging female rats. In the ovariectomized and aging group nNOS functionally became a source of (Lekontseva et al., 2012).
Corroborating the above findings, NO release from nNOS also seems to be important in the control of vascular tone in humans. Expression of nNOS was found in human aorta, carotid, radial and mammary artery (Buchwalow et al., 2002), saphenous vein (Webb et al., 2006), and lung capillary endothelial cells (Lührs et al., 2002).
The first evidence that nNOS had a function in vascular regulation in humans was obtained from children suffering from Duchene muscular dystrophy (DMD). It was shown that nNOS-derived NO present in skeletal muscle acts in the blood flow and oxygen transport. nNOS expression is reduced in children with DMD resulting in increased vasoconstrictor response (Sander et al., 2000).
Later on, Seddon et al. (2008) showed the relationship between nNOS and the regulation of blood flow in human. Selective in vivo inhibition of nNOS with SMTC in healthy men promoted a reduction in the brachial artery baseline flow. This effect was eliminated in the presence of L-arginine. A similar reduction was observed with the non-selective inhibitor of NOS (L-NMMA) but required a 20-fold higher dose. This study suggested that nNOS-derived NO has a significant role in the physiological regulation of microvascular tone in vivo (Seddon et al., 2008). In another work, the same group investigated the in vivo effects of SMTC in human coronary dilatation. The infusion of SMTC in healthy patients reduced baseline coronary blood flow and coronary artery diameter measured by angiography. They concluded that local nNOS-derived NO is a key physiological regulator of human coronary vascular tone in vivo (Seddon et al., 2009).
All the above works suggesting NO as the mediator of nNOS function in the regulation of vascular tone were based on the assumption that NO was the only physiological vasodilator product of nNOS activation. Our group was the first to show the importance of nNOS-derived H2O2 in the endothelium-dependent vascular relaxation. We showed that nNOS was constitutively expressed in the endothelium of the mouse aorta and mesenteric resistance artery. Stimulation of those vessels with acetylcholine promoted increase in H2O2 production. Pharmacological selective nNOS inhibition and nNOS knockdown decreased endothelium-dependent vascular relaxation and H2O2 production. Finally, incubation of the vessels with catalase, an enzyme that degrades H2O2 into O2 and H2O, decreased vascular relaxation (Capettini et al., 2008, 2010; Silva et al., 2016). The participation of nNOS in vascular homeostasis in physiological and pathological conditions is summarized in Table 1.
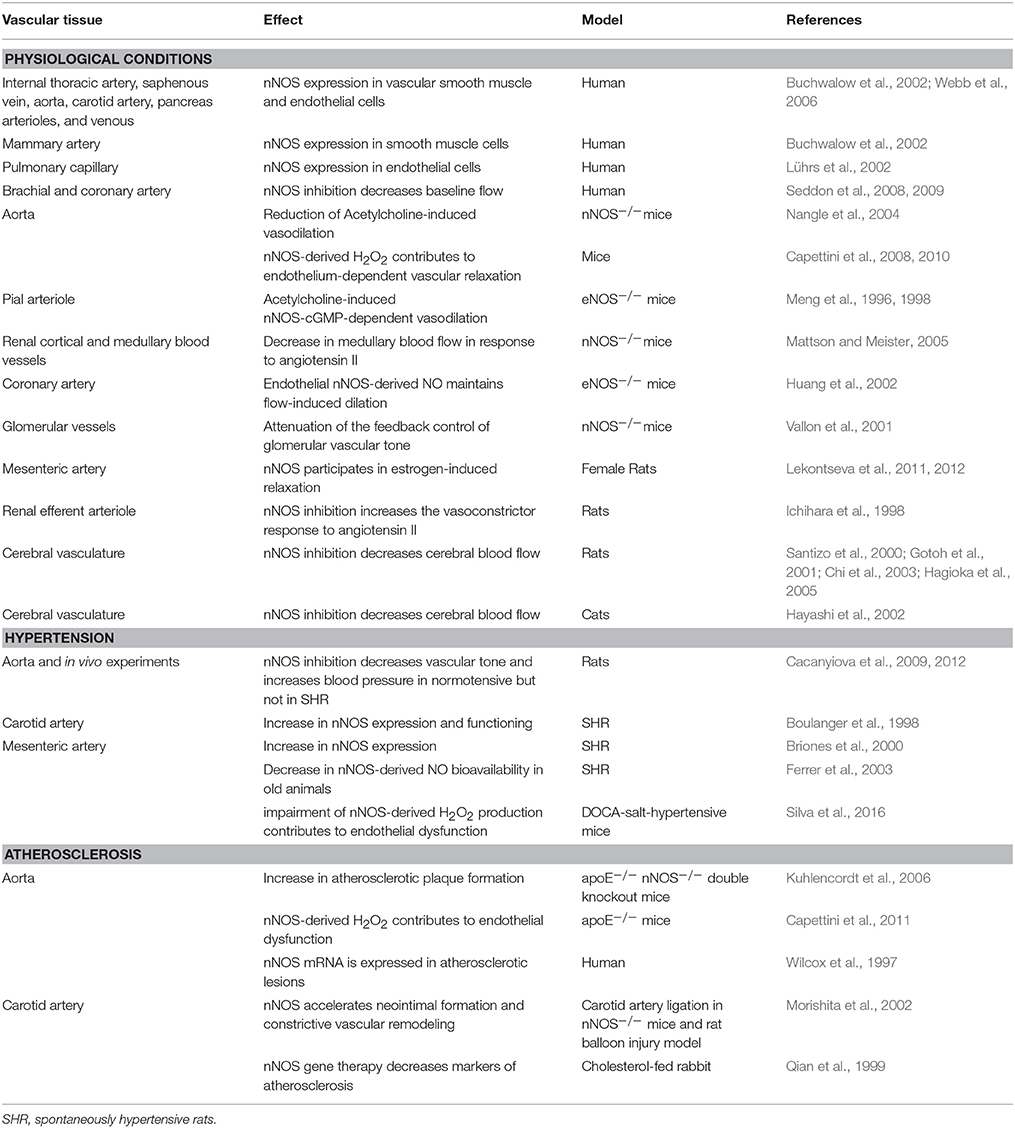
Table 1. Participation of nNOS in the control of vascular function in physiological conditions and during hypertension and atherosclerosis.
Several studies have indicated that the imbalance in nNOS expression and/or activity is involved in the mechanism of pathogenesis of hypertension. In mesenteric arteries from spontaneously hypertensive rats (SHR), nNOS expression was ~2 times higher than in vessels from control animals (Briones et al., 2000). A similar result showing increased expression of nNOS in vascular smooth muscle cells was found in carotid arteries from SHR. It was shown that activation of nNOS on stimulation by Angiotensin II occurs in hypertensive but not in normotensive animals (Boulanger et al., 1998). Interestingly, in SHR rats the expression and activity of nNOS are decreased in the adrenal gland. Chronic treatment of SHR with antihypertensive drugs, increased the expression and activity of nNOS in the adrenal gland, suggesting that normalization of blood pressure (BP) may be in part related to an increase in nNOS (Qadri et al., 2001).
BP and vascular function were evaluated in normotensive rats chronically treated (6 weeks) with the selective nNOS inhibitor 7-NI. A significant increase in systolic BP was observed in the first 2 weeks of treatment. Corroborating the in vivo study, isolated vessels showed an attenuated relaxant response to acetylcholine in the aorta. These results show that nNOS participates in the regulation of BP and vascular tone (Cacanyiova et al., 2009). In contrast, in SHR, treatment with 7-NI had no effect in blood pressure or acetylcholine-induced vasodilatation in the aorta (Cacanyiova et al., 2009, 2012), suggesting that nNOS function was lost in hypertension.
A recent study revealed that impairment of nNOS-derived H2O2 pathway participates in the endothelial dysfunction and increase in blood pressure in DOCA-salt-hypertensive mice (Silva et al., 2016). This study showed that 1-(2-trifluoromethylphenyl) imidazole, a selective nNOS inhibitor, and catalase, exhibited a more pronounced reduction of acetylcholine-induced decrease in blood pressure in normotensive than in hypertensive mice. Moreover, selective nNOS inhibition and catalase had a greater inhibitory effect in acetylcholine-induced vasodilatation in control compared to DOCA-salt mice. Also, acetylcholine-induced H2O2 production and the expression and functioning of nNOS were considerably diminished in the resistance mesenteric arteries of DOCA-salt mice.
The first evidence that nNOS plays a vasculoprotective role in atherosclerosis came from a work by Wilcox et al. (1997) that showed a correlation between the progression of plaque formation and nNOS mRNA. In 1999, Qian et al. performed experiments with recombinant adenoviruses expressing nNOS transferred to carotid of hypercholesterolemic rabbits and showed a marked reduction in expression of adhesion molecules and infiltration of inflammatory cells. Additionally, a reduction in lipid deposition was observed after gene transfer. In another work, nNOS−∕− mice exhibited accelerated neointimal formation and constrictive vascular remodeling caused by blood flow disruption in a model of carotid artery ligation. It was also observed that selective inhibition of nNOS decreased cGMP production, inducing an increase in vasoconstrictor response and accelerating neointimal formation in a rat balloon injury model (Morishita et al., 2002). Using a double knockout mouse (nNOS DKO) that combined genetic deletion of nNOS (nNOS−∕−) with a model of atherosclerosis (apoE−∕−), Kuhlencordt et al. (2006) showed that the absence of nNOS accelerated the atherosclerotic plaque lesion. After 14 weeks following a “Western-type” atherogenic diet, nNOS DKO animals showed 66% increase of lesion area, compared to apoE−∕− control mice.
nNOS-derived H2O2 also seems to participate in endothelial dysfunction in atherosclerosis. Capettini et al. (2011) showed that selective pharmacological inhibition of nNOS, nNOS knockdown and catalase reduced the vasodilator effect of acetylcholine, diminished NO and abolished endothelial-dependent H2O2 production in wild-type mice, but had no effect in ApoE−∕− animals. In addition, nNOS functioning was decreased in ApoE−∕− mice compared to controls.
This mini-review summarizes puzzling information on the role of nNOS in the control of vascular homeostasis under physiological and diseases conditions. Recent data indicates that nNOS is constitutively expressed in the endothelial cells of different types of vessels in animals and human. More importantly, nNOS-derived products such as NO and H2O2 play an important role in the control of vascular function and blood pressure. Finally, nNOS participates in the physiopathology of hypertension and atherosclerosis.
VL defined the research topics and co-wrote the manuscript. EC, BR, and SC co-wrote the manuscript.
This work was supported by FAPEMIG (Fundação de Apoio à Pesquisa do Estado de Minas Gerais) grant APQ-00683-13 and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) grants 467147/2014-0, 305693/2014-0, and 470860/2012-0.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abu-Soud, H. M., Ichimori, K., Presta, A., and Stuehr, D. J. (2000). Electron transfer, oxygen binding, and nitric oxide feedback inhibition in endothelial nitric-oxide synthase. J. Biol. Chem. 275, 17349–17357. doi: 10.1074/jbc.M000050200
Abu-Soud, H. M., Yoho, L. L., and Stuehr, D. J. (1994). Calmodulin controls neuronal nitric-oxide synthase by a dual mechanism. Activation of intra- and interdomain electron transfer. J. Biol. Chem. 269, 32047–32050.
Adak, S., Santolini, J., Tikunova, S., Wang, Q., Johnson, J. D., and Stuehr, D. J. (2001). Neuronal nitric-oxide synthase mutant (Ser-1412 –> Asp) demonstrates surprising connections between heme reduction, NO complex formation, and catalysis. J. Biol. Chem. 276, 1244–1252. doi: 10.1074/jbc.M006857200
Bendall, J. K., Damy, T., Ratajczak, P., Loyer, X., Monceau, V., Marty, I., et al. (2004). Role of myocardial neuronal nitric oxide synthase-derived nitric oxide in β-adrenergic hyporesponsiveness after myocardial infarction-induced heart failure in rat. Circulation 110, 2368–2375. doi: 10.1161/01.CIR.0000145160.04084.AC
Boissel, J. P., Schwarz, P. M., and Förstermann, U. (1998). Neuronal-type NO synthase: transcript diversity and expressional regulation. Nitric Oxide 2, 337–349. doi: 10.1006/niox.1998.0189
Boulanger, C. M., Heymes, C., Benessiano, J., Geske, R. S., Lévy, B. I., and Vanhoutte, P. M. (1998). Neuronal nitric oxide synthase is expressed in rat vascular smooth muscle cells: activation by angiotensin II in hypertension. Circ. Res. 83, 1271–1278. doi: 10.1161/01.RES.83.12.1271
Bredt, D. S., Hwang, P. M., and Snyder, S. H. (1990). Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 347, 768–770. doi: 10.1038/347768a0
Bredt, D. S., and Snyder, S. H. (1990). Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. U.S.A. 87, 682–685. doi: 10.1073/pnas.87.2.682
Bredt, D. S., and Snyder, S. H. (1992). Nitric oxide, a novel neuronal messenger. Neuron 8, 3–11. doi: 10.1016/0896-6273(92)90104-L
Briones, A. M., Alonso, M. J., Marín, J., Balfagón, G., and Salaices, M. (2000). Influence of hypertension on nitric oxide synthase expression and vascular effects of lipopolysaccharide in rat mesenteric arteries. Br. J. Pharmacol. 131, 185–194. doi: 10.1038/sj.bjp.0703552
Buchwalow, I. B., Podzuweit, T., Bocker, W., Samoilova, V. E., Thomas, S., Wellner, M., et al. (2002). Vascular smooth muscle and nitric oxide synthase. FASEB J. 16, 500–508. doi: 10.1096/fj.01-0842com
Cacanyiova, S., Kristek, F., Gerova, M., Krenek, P., and Klimas, J. (2009). Effect of chronic nNOS inhibition on blood pressure, vasoactivity, and arterial wall structure in Wistar rats. Nitric Oxide 20, 304–310. doi: 10.1016/j.niox.2009.03.002
Cacanyiova, S., Kristek, F., Malekova, M., and Ondrias, K. (2012). Effects of chronic neuronal nitric oxide-synthase inhibition on arterial function and structure in spontaneously hypertensive rats. J. Physiol. Pharmacol. 63, 23–28.
Capettini, L. S., Cortes, S. F., Gomes, M. A., Silva, G. A., Pesquero, J. L., Lopes, M. J., et al. (2008). Neuronal nitric oxide synthase-derived hydrogen peroxide is a major endothelium-dependent relaxing factor. Am. J. Physiol. Heart Circ. Physiol. 295, H2503–H2511. doi: 10.1152/ajpheart.00731.2008
Capettini, L. S., Cortes, S. F., and Lemos, V. S. (2010). Relative contribution of eNOS and nNOS to endothelium-dependent vasodilation in the mouse aorta. Eur. J. Pharmacol. 643, 260–266. doi: 10.1016/j.ejphar.2010.06.066
Capettini, L. S., Cortes, S. F., Silva, J. F., Alvarez-Leite, J. I., and Lemos, V. S. (2011). Decreased production of neuronal NOS-derived hydrogen peroxide contributes to endothelial dysfunction in atherosclerosis. Br. J. Pharmacol. 164, 1738–1748. doi: 10.1111/j.1476-5381.2011.01500.x
Chen, Z. P., Mcconell, G. K., Michell, B. J., Snow, R. J., Canny, B. J., and Kemp, B. E. (2000). AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. Am. J. Physiol. Endocrinol. Metab. 279, E1202–E1206.
Chi, O. Z., Liu, X., and Weiss, H. R. (2003). Effects of inhibition of neuronal nitric oxide synthase on NMDA-induced changes in cerebral blood flow and oxygen consumption. Exp. Brain Res. 148, 256–260. doi: 10.1007/s00221-002-1310-7
Crane, B. R., Arvai, A. S., Ghosh, D. K., Wu, C., Getzoff, E. D., Stuehr, D. J., et al. (1998). Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science 279, 2121–2126. doi: 10.1126/science.279.5359.2121
Daff, S., Sagami, I., and Shimizu, T. (1999). The 42-amino acid insert in the FMN domain of neuronal nitric-oxide synthase exerts control over Ca(2+)/calmodulin-dependent electron transfer. J. Biol. Chem. 274, 30589–30595. doi: 10.1074/jbc.274.43.30589
Feng, C., Chen, L., Li, W., Elmore, B. O., Fan, W., and Sun, X. (2014). Dissecting regulation mechanism of the FMN to heme interdomain electron transfer in nitric oxide synthases. J. Inorg. Biochem. 130, 130–140. doi: 10.1016/j.jinorgbio.2013.09.005
Ferrer, M., Sánchez, M., Minoves, N., Salaices, M., and Balfagón, G. (2003). Aging increases neuronal nitric oxide release and superoxide anion generation in mesenteric arteries from spontaneously hypertensive rats. J. Vasc. Res. 40, 509–519. doi: 10.1159/000075183
Fleming, I. (2003). Brain in the brawn: the neuronal nitric oxide synthase as a regulator of myogenic tone. Circ. Res. 93, 586–588. doi: 10.1161/01.RES.0000095380.06622.D8
Furchgott, R. F., and Zawadzki, J. V. (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288, 373–376. doi: 10.1038/288373a0
Garcin, E. D., Bruns, C. M., Lloyd, S. J., Hosfield, D. J., Tiso, M., Gachhui, R., et al. (2004). Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J. Biol. Chem. 279, 37918–37927. doi: 10.1074/jbc.M406204200
Gonzalez-Cadavid, N. F., and Rajfer, J. (2004). Molecular pathophysiology and gene therapy of aging-related erectile dysfunction. Exp. Gerontol. 39, 1705–1712. doi: 10.1016/j.exger.2004.06.022
Gotoh, J., Kuang, T. Y., Nakao, Y., Cohen, D. M., Melzer, P., Itoh, Y., et al. (2001). Regional differences in mechanisms of cerebral circulatory response to neuronal activation. Am. J. Physiol. Heart Circ. Physiol. 280, H821–H829.
Hagioka, S., Takeda, Y., Zhang, S., Sato, T., and Morita, K. (2005). Effects of 7-nitroindazole and N-nitro-l-arginine methyl ester on changes in cerebral blood flow and nitric oxide production preceding development of hyperbaric oxygen-induced seizures in rats. Neurosci. Lett. 382, 206–210. doi: 10.1016/j.neulet.2005.01.006
Hall, A. V., Antoniou, H., Wang, Y., Cheung, A. H., Arbus, A. M., Olson, S. L., et al. (1994). Structural organization of the human neuronal nitric oxide synthase gene (NOS1). J. Biol. Chem. 269, 33082–33090.
Hayashi, T., Katsumi, Y., Mukai, T., Inoue, M., Nagahama, Y., Oyanagi, C., et al. (2002). Neuronal nitric oxide has a role as a perfusion regulator and a synaptic modulator in cerebellum but not in neocortex during somatosensory stimulation–an animal PET study. Neurosci. Res. 44, 155–165. doi: 10.1016/S0168-0102(02)00122-0
Hemmens, B., Goessler, W., Schmidt, K., and Mayer, B. (2000). Role of bound zinc in dimer stabilization but not enzyme activity of neuronal nitric-oxide synthase. J. Biol. Chem. 275, 35786–35791. doi: 10.1074/jbc.M005976200
Hemmens, B., Gorren, A. C., Schmidt, K., Werner, E. R., and Mayer, B. (1998). Haem insertion, dimerization and reactivation of haem-free rat neuronal nitric oxide synthase. Biochem. J. 332, 337–342. doi: 10.1042/bj3320337
Hermann, M., Flammer, A., and Lüscher, T. F. (2006). Nitric oxide in hypertension. J. Clin. Hypertens. (Greenwich) 8, 17–29. doi: 10.1111/j.1524-6175.2006.06032.x
Huang, A., Sun, D., Shesely, E. G., Levee, E. M., Koller, A., and Kaley, G. (2002). Neuronal NOS-dependent dilation to flow in coronary arteries of male eNOS-KO mice. Am. J. Physiol. Heart Circ. Physiol. 282, H429–H436. doi: 10.1152/ajpheart.00501.2001
Hudetz, A. G., Shen, H., and Kampine, J. P. (1998). Nitric oxide from neuronal NOS plays critical role in cerebral capillary flow response to hypoxia. Am. J. Physiol. 274, H982–H989.
Ichihara, A., Inscho, E. W., Imig, J. D., and Navar, L. G. (1998). Neuronal nitric oxide synthase modulates rat renal microvascular function. Am. J. Physiol. 274, F516–F524.
Jaffrey, S. R., and Snyder, S. H. (1996). PIN: an associated protein inhibitor of neuronal nitric oxide synthase. Science 274, 774–777. doi: 10.1126/science.274.5288.774
Kamada, Y., Jenkins, G. J., Lau, M., Dunbar, A. Y., Lowe, E. R., and Osawa, Y. (2005). Tetrahydrobiopterin depletion and ubiquitylation of neuronal nitric oxide synthase. Brain Res. Mol. Brain Res. 142, 19–27. doi: 10.1016/j.molbrainres.2005.09.003
Katakam, P. V., Snipes, J. A., Steed, M. M., and Busija, D. W. (2012). Insulin-induced generation of reactive oxygen species and uncoupling of nitric oxide synthase underlie the cerebrovascular insulin resistance in obese rats. J. Cereb. Blood Flow Metab. 32, 792–804. doi: 10.1038/jcbfm.2011.181
Kuhlencordt, P. J., Hötten, S., Schödel, J., Rützel, S., Hu, K., Widder, J., et al. (2006). Atheroprotective effects of neuronal nitric oxide synthase in apolipoprotein e knockout mice. Arterioscler. Thromb. Vasc. Biol. 26, 1539–1544. doi: 10.1161/01.ATV.0000223143.88128.19
Kurihara, N., Alfie, M. E., Sigmon, D. H., Rhaleb, N. E., Shesely, E. G., and Carretero, O. A. (1998). Role of nNOS in blood pressure regulation in eNOS null mutant mice. Hypertension 32, 856–861. doi: 10.1161/01.HYP.32.5.856
Lajoix, A. D., Badiou, S., Péraldi-Roux, S., Chardés, T., Aknin, C., Tribillac, F., et al. (2006). Protein inhibitor of neuronal nitric oxide synthase (PIN) is a new regulator of glucose-induced insulin secretion. Diabetes 55, 3279–3288. doi: 10.2337/db06-0257
Lekontseva, O., Chakrabarti, S., Jiang, Y., Cheung, C. C., and Davidge, S. T. (2011). Role of neuronal nitric-oxide synthase in estrogen-induced relaxation in rat resistance arteries. J. Pharmacol. Exp. Ther. 339, 367–375. doi: 10.1124/jpet.111.183798
Lekontseva, O., Jiang, Y., Schleppe, C., and Davidge, S. T. (2012). Altered neuronal nitric oxide synthase in the aging vascular system: implications for estrogens therapy. Endocrinology 153, 3940–3948. doi: 10.1210/en.2012-1071
Loesch, A., Milner, P., and Burnstock, G. (1998). Endothelin in perivascular nerves. An electron-immunocytochemical study of rat basilar artery. Neuroreport 9, 3903–3906. doi: 10.1097/00001756-199812010-00025
Lührs, H., Papadopoulos, T., Schmidt, H. H., and Menzel, T. (2002). Type I nitric oxide synthase in the human lung is predominantly expressed in capillary endothelial cells. Respir. Physiol. 129, 367–374. doi: 10.1016/S0034-5687(01)00323-1
Masters, B. S., Mcmillan, K., Sheta, E. A., Nishimura, J. S., Roman, L. J., and Martasek, P. (1996). Neuronal nitric oxide synthase, a modular enzyme formed by convergent evolution: structure studies of a cysteine thiolate-liganded heme protein that hydroxylates L-arginine to produce NO. as a cellular signal. FASEB J. 10, 552–558.
Mattson, D. L., and Meister, C. J. (2005). Renal cortical and medullary blood flow responses to L-NAME and ANG II in wild-type, nNOS null mutant, and eNOS null mutant mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R991–R997. doi: 10.1152/ajpregu.00207.2005
Meng, W., Ayata, C., Waeber, C., Huang, P. L., and Moskowitz, M. A. (1998). Neuronal NOS-cGMP-dependent ACh-induced relaxation in pial arterioles of endothelial NOS knockout mice. Am. J. Physiol. 274, H411–H415.
Meng, W., Ma, J., Ayata, C., Hara, H., Huang, P. L., Fishman, M. C., et al. (1996). ACh dilates pial arterioles in endothelial and neuronal NOS knockout mice by NO-dependent mechanisms. Am. J. Physiol. 271, H1145–H1150.
Montécot, C., Borredon, J., Seylaz, J., and Pinard, E. (1997). Nitric oxide of neuronal origin is involved in cerebral blood flow increase during seizures induced by kainate. J. Cereb. Blood Flow Metab. 17, 94–99. doi: 10.1097/00004647-199701000-00012
Morishita, T., Tsutsui, M., Shimokawa, H., Horiuchi, M., Tanimoto, A., Suda, O., et al. (2002). Vasculoprotective roles of neuronal nitric oxide synthase. FASEB J. 16, 1994–1996. doi: 10.1096/fj.02-0155fje
Nangle, M. R., Cotter, M. A., and Cameron, N. E. (2004). An in vitro investigation of aorta and corpus cavernosum from eNOS and nNOS gene-deficient mice. Pflugers Arch. 448, 139–145. doi: 10.1007/s00424-003-1232-7
Napoli, C., De Nigris, F., Williams-Ignarro, S., Pignalosa, O., Sica, V., and Ignarro, L. J. (2006). Nitric oxide and atherosclerosis: an update. Nitric Oxide 15, 265–279. doi: 10.1016/j.niox.2006.03.011
Papale, D., Bruckmann, C., Gazur, B., Miles, C. S., Mowat, C. G., and Daff, S. (2012). Oxygen activation in neuronal NO synthase: resolving the consecutive mono-oxygenation steps. Biochem. J. 443, 505–514. doi: 10.1042/BJ20111644
Pelligrino, D. A., Koenig, H. M., and Albrecht, R. F. (1993). Nitric oxide synthesis and regional cerebral blood flow responses to hypercapnia and hypoxia in the rat. J. Cereb. Blood Flow Metab. 13, 80–87. doi: 10.1038/jcbfm.1993.10
Qadri, F., Arens, T., Schwartz, E. C., Häuser, W., and Dominiak, P. (2001). Angiotensin-converting enzyme inhibitors and AT1-receptor antagonist restore nitric oxide synthase (NOS) activity and neuronal NOS expression in the adrenal glands of spontaneously hypertensive rats. Jpn. J. Pharmacol. 85, 365–369. doi: 10.1254/jjp.85.365
Qian, H., Neplioueva, V., Shetty, G. A., Channon, K. M., and George, S. E. (1999). Nitric oxide synthase gene therapy rapidly reduces adhesion molecule expression and inflammatory cell infiltration in carotid arteries of cholesterol-fed rabbits. Circulation 99, 2979–2982. doi: 10.1161/01.CIR.99.23.2979
Rabelo, L. A., Cortes, S. F., Alvarez-Leite, J. I., and Lemos, V. S. (2003). Endothelium dysfunction in LDL receptor knockout mice: a role for H2O2. Br. J. Pharmacol. 138, 1215–1220. doi: 10.1038/sj.bjp.0705164
Roman, L. J., Martásek, P., and Masters, B. S. (2002). Intrinsic and extrinsic modulation of nitric oxide synthase activity. Chem. Rev. 102, 1179–1190. doi: 10.1021/cr000661e
Roman, L. J., Martásek, P., Miller, R. T., Harris, D. E., De La Garza, M. A., Shea, T. M., et al. (2000). The C termini of constitutive nitric-oxide synthases control electron flow through the flavin and heme domains and affect modulation by calmodulin. J. Biol. Chem. 275, 29225–29232. doi: 10.1074/jbc.M004766200
Roman, L. J., and Masters, B. S. (2006). Electron transfer by neuronal nitric-oxide synthase is regulated by concerted interaction of calmodulin and two intrinsic regulatory elements. J. Biol. Chem. 281, 23111–23118. doi: 10.1074/jbc.M603671200
Rosen, G. M., Tsai, P., Weaver, J., Porasuphatana, S., Roman, L. J., Starkov, A. A., et al. (2002). The role of tetrahydrobiopterin in the regulation of neuronal nitric-oxide synthase-generated superoxide. J. Biol. Chem. 277, 40275–40280. doi: 10.1074/jbc.M200853200
Sagami, I., Daff, S., and Shimizu, T. (2001). Intra-subunit and inter-subunit electron transfer in neuronal nitric-oxide synthase: effect of calmodulin on heterodimer catalysis. J. Biol. Chem. 276, 30036–30042. doi: 10.1074/jbc.M104123200
Salerno, J. C., Harris, D. E., Irizarry, K., Patel, B., Morales, A. J., Smith, S. M., et al. (1997). An autoinhibitory control element defines calcium-regulated isoforms of nitric oxide synthase. J. Biol. Chem. 272, 29769–29777. doi: 10.1074/jbc.272.47.29769
Sanchez, A., Contreras, C., Martínez, M. P., Climent, B., Benedito, S., Garcia-Sacristan, A., et al. (2012). Role of neural NO synthase (nNOS) uncoupling in the dysfunctional nitrergic vasorelaxation of penile arteries from insulin resistant obese Zucker rats. PLoS ONE 7:e36027. doi: 10.1371/journal.pone.0036027
Sander, M., Chavoshan, B., Harris, S. A., Iannaccone, S. T., Stull, J. T., Thomas, G. D., et al. (2000). Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U.S.A. 97, 13818–13823. doi: 10.1073/pnas.250379497
Santizo, R., Baughman, V. L., and Pelligrino, D. A. (2000). Relative contributions from neuronal and endothelial nitric oxide synthases to regional cerebral blood flow changes during forebrain ischemia in rats. Neuroreport 11, 1549–1553. doi: 10.1097/00001756-200005150-00036
Sato, Y., Sagami, I., and Shimziu, T. (2004). Identification of caveolin-1 interacting sites in neuronal nitric-oxide synthase: molecular mechanism for inhibition of NO formation. J. Biol. Chem. 279, 8827–8836. doi: 10.1074/jbc.M310327200
Schwarz, P. M., Kleinert, H., and Förstermann, U. (1999). Potential functional significance of brain-type and muscle-type nitric oxide synthase I expressed in adventitia and media of rat aorta. Arterioscler. Thromb. Vasc. Biol. 19, 2584–2590. doi: 10.1161/01.ATV.19.11.2584
Seddon, M. D., Chowienczyk, P. J., Brett, S. E., Casadei, B., and Shah, A. M. (2008). Neuronal nitric oxide synthase regulates basal microvascular tone in humans in vivo. Circulation 117, 1991–1996. doi: 10.1161/CIRCULATIONAHA.107.744540
Seddon, M., Melikian, N., Dworakowski, R., Shabeeh, H., Jiang, B., Byrne, J., et al. (2009). Effects of neuronal nitric oxide synthase on human coronary artery diameter and blood flow in vivo. Circulation 119, 2656–2662. doi: 10.1161/CIRCULATIONAHA.108.822205
Silva, G. C., Silva, J. F., Diniz, T. F., Lemos, V. S., and Cortes, S. F. (2016). Endothelial dysfunction in DOCA-salt-hypertensive mice: role of neuronal nitric oxide synthase-derived hydrogen peroxide. Clin. Sci. (Lond). 130, 895–906. doi: 10.1042/CS20160062
Song, T., Hatano, N., Horii, M., Tokumitsu, H., Yamaguchi, F., Tokuda, M., et al. (2004). Calcium/calmodulin-dependent protein kinase I inhibits neuronal nitric-oxide synthase activity through serine 741 phosphorylation. FEBS Lett. 570, 133–137. doi: 10.1016/j.febslet.2004.05.083
Sowa, G. (2012). Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front. Physiol. 2:120. doi: 10.3389/fphys.2011.00120
Stuehr, D. J. (1997). Structure-function aspects in the nitric oxide synthases. Annu. Rev. Pharmacol. Toxicol. 37, 339–359. doi: 10.1146/annurev.pharmtox.37.1.339
Tsai, P., Weaver, J., Cao, G. L., Pou, S., Roman, L. J., Starkov, A. A., et al. (2005). L-arginine regulates neuronal nitric oxide synthase production of superoxide and hydrogen peroxide. Biochem. Pharmacol. 69, 971–979. doi: 10.1016/j.bcp.2004.12.009
Vallon, V., Traynor, T., Barajas, L., Huang, Y. G., Briggs, J. P., and Schnermann, J. (2001). Feedback control of glomerular vascular tone in neuronal nitric oxide synthase knockout mice. J. Am. Soc. Nephrol. 12, 1599–1606.
Venema, V. J., Ju, H., Zou, R., and Venema, R. C. (1997). Interaction of neuronal nitric-oxide synthase with caveolin-3 in skeletal muscle. Identification of a novel caveolin scaffolding/inhibitory domain. J. Biol. Chem. 272, 28187–28190.
Weaver, J., Porasuphatana, S., Tsai, P., Pou, S., Roman, L. J., and Rosen, G. M. (2005). A comparative study of neuronal and inducible nitric oxide synthases: generation of nitric oxide, superoxide, and hydrogen peroxide. Biochim. Biophys. Acta 1726, 302–308. doi: 10.1016/j.bbagen.2005.08.012
Webb, G. D., Lim, L. H., Oh, V. M., El Oakley, R., Lee, C. N., Wong, P. S., et al. (2006). Expression of neuronal nitric oxide synthase in the internal thoracic artery and saphenous vein. J. Thorac. Cardiovasc. Surg. 132, 1131–1136. doi: 10.1016/j.jtcvs.2006.08.001
Weissman, B. A., Jones, C. L., Liu, Q., and Gross, S. S. (2002). Activation and inactivation of neuronal nitric oxide synthase: characterization of Ca(2+)-dependent [125I]Calmodulin binding. Eur. J. Pharmacol. 435, 9–18. doi: 10.1016/S0014-2999(01)01560-6
Wilcox, J. N., Subramanian, R. R., Sundell, C. L., Tracey, W. R., Pollock, J. S., Harrison, D. G., et al. (1997). Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler. Thromb. Vasc. Biol. 17, 2479–2488. doi: 10.1161/01.ATV.17.11.2479
Keywords: neuronal nitric oxide synthase, nitric oxide, hydrogen peroxide, vascular function, hypertension, atherosclerosis
Citation: Costa ED, Rezende BA, Cortes SF and Lemos VS (2016) Neuronal Nitric Oxide Synthase in Vascular Physiology and Diseases. Front. Physiol. 7:206. doi: 10.3389/fphys.2016.00206
Received: 19 April 2016; Accepted: 20 May 2016;
Published: 02 June 2016.
Edited by:
Camille M. Balarini, Federal University of Paraíba, BrazilReviewed by:
Luciana Venturini Rossoni, University of São Paulo, BrazilCopyright © 2016 Costa, Rezende, Cortes and Lemos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Virginia S. Lemos, dnNsZW1vc0BpY2IudWZtZy5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.