 Sunil Jit R. J. Logantha1†
Sunil Jit R. J. Logantha1† Mathis K. Stokke2,3,4†
Mathis K. Stokke2,3,4† Andrew J. Atkinson1
Andrew J. Atkinson1 Sanjay R. Kharche1
Sanjay R. Kharche1 Sajida Parveen1
Sajida Parveen1 Yawer Saeed1Ivar Sjaastad2,3
Yawer Saeed1Ivar Sjaastad2,3 Ole M. Sejersted2,3
Ole M. Sejersted2,3 Halina Dobrzynski1*
Halina Dobrzynski1*- 1Institute of Cardiovascular Sciences, University of Manchester, Manchester, UK
- 2Institute for Experimental Medical Research, Oslo University Hospital and University of Oslo, Oslo, Norway
- 3Center for Heart Failure Research, University of Oslo, Oslo, Norway
- 4Clinic for Internal Medicine, Lovisenberg Deaconess Hospital AS, Oslo, Norway
Background: The sarcoplasmic reticulum Ca2+-ATPase (SERCA2) pump is an important component of the Ca2+-clock pacemaker mechanism that provides robustness and flexibility to sinus node pacemaking. We have developed transgenic mice with reduced cardiac SERCA2 abundance (Serca2 KO) as a model for investigating SERCA2's role in sinus node pacemaking.
Methods and Results: In Serca2 KO mice, ventricular SERCA2a protein content measured by Western blotting was 75% (P < 0.05) lower than that in control mice (Serca2 FF) tissue. Immunofluorescent labeling of SERCA2a in ventricular, atrial, sinus node periphery and center tissue sections revealed 46, 45, 55, and 34% (all P < 0.05 vs. Serca2 FF) lower labeling, respectively and a mosaic pattern of expression. With telemetric ECG surveillance, we observed no difference in basal heart rate, but the PR-interval was prolonged in Serca2 KO mice: 49 ± 1 vs. 40 ± 1 ms (P < 0.001) in Serca2 FF. During exercise, heart rate in Serca2 KO mice was elevated to 667 ± 22 bpm, considerably less than 780 ± 17 bpm (P < 0.01) in Serca2 FF. In isolated sinus node preparations, 2 mM Cs+ caused bradycardia that was equally pronounced in Serca2 KO and Serca2 FF (32 ± 4% vs. 29 ± 5%), indicating no change in the pacemaker current, If. Disabling the Ca2+-clock with 2 μM ryanodine induced bradycardia that was less pronounced in Serca2 KO preparations (9 ± 1% vs. 20 ± 3% in Serca2 FF; P < 0.05), suggesting a disrupted Ca2+-clock. Mathematical modeling was used to dissect the effects of membrane- and Ca2+-clock components on Serca2 KO mouse heart rate and sinus node action potential. Computer modeling predicted a slowing of heart rate with SERCA2 downregulation and the heart rate slowing was pronounced at >70% reduction in SERCA2 activity.
Conclusions: Serca2 KO mice show a disrupted Ca2+-clock-dependent pacemaker mechanism contributing to impaired sinus node and atrioventricular node function.
Introduction
The sinus node is the dominant pacemaker of the heart, aptly placed in the roof of the right atrium. It consists of specialized pacemaker cells that spontaneously depolarize during diastole and rhythmically generate action potentials (Keith and Flack, 1907; Silverman et al., 2006; Dobrzynski et al., 2007). The diastolic depolarization results from the synergistic interaction between an ensemble of surface membrane ion channel currents and intracellular Ca2+ release signals. Together, they regulate the rate and rhythm of the spontaneous action potentials in the sinus node (Figure 1; Dobrzynski et al., 2007; Lakatta et al., 2010). The voltage- and time-dependent gating of the surface membrane channels works in a cyclic fashion (the membrane voltage-clock) and underlies the early phase of the diastolic depolarization. This phase involves a voltage-dependent deactivation of outward currents and activation of inward currents (Dobrzynski et al., 2013). The hyperpolarization-activated cyclic nucleotide-gated (HCN) or funny current (If) is the main inward current, and thus a vital component of the voltage-clock and essential to pacemaking in the sinus node. Mice with a cardiac specific conditional knockout (KO) of the HCN4 channel have reduced If in sinus node cardiomyocytes and exhibit severe bradycardia (up to 50% reduction in heart rate; Baruscotti et al., 2011). Similarly, humans with HCN4 gene mutations exhibit sinus bradycardia (Dobrzynski et al., 2013).
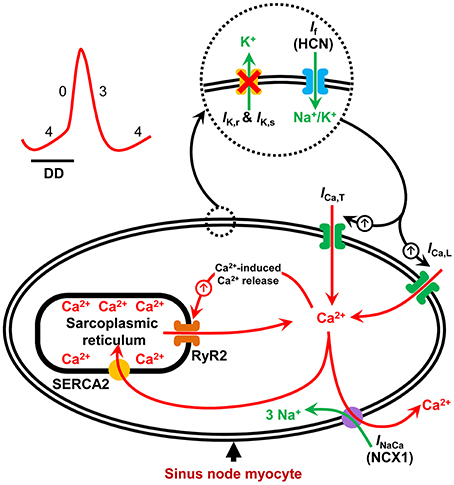
Figure 1. Schematic illustration of pacemaker mechanisms in the sinus node myocyte. Sinus node action potential is preceded by a slow diastolic depolarization (DD) which brings the membrane potential up to the threshold level for excitation. The DD is a result of synergistic interaction between the membrane voltage-clock and the subcellular Ca2+-clock. The membrane voltage-clock comprises plasma membrane bound, voltage-dependent ion channels and their corresponding ionic currents. At the beginning of the DD, there is voltage-dependent deactivation of outward K+ currents (IK, r and IK, s) and activation of inward currents: hyperpolarization-activated cyclic nucleotide-gated (HCN) or funny current (If), T-type Ca2+ current (ICa, T) and L-type Ca2+ current (ICa, L) amongst others. The Ca2+-clock contributes to sinus node DD through localized Ca2+ release from the sarcoplasmic reticulum via the ryanodine receptor (RYR2). Increased levels of cytosolic Ca2+ activates the electrogenic Na+–Ca2+ exchanger (NCX1) generating an inward current (INaCa) that imparts a steep, exponential increase to the late phase of DD. SERCA2 refills the sarcoplasmic reticulum with Ca2+ and is hence crucial to sinus node pacemaking.
In contrast to the early phase, the latter phase of the diastolic depolarization in sinus node pacemaking cells depends on localized, sarcoplasmic reticulum-generated, intracellular Ca2+ release via the ryanodine receptor, referred to as the intracellular Ca2+-clock. The released Ca2+ activates inward Na+-Ca2+ exchange, generating an inward current (INaCa) by exchanging one Ca2+ for three Na+ (Figure 1; Bogdanov et al., 2006). Acute inhibition of INaCa is reported to stop pacemaking in single rabbit sinus node myocytes (Bogdanov et al., 2001). The localized release of Ca2+ from the sarcoplasmic reticulum is dependent upon the sarcoplasmic reticulum Ca2+-load which is regulated by the sarcoplasmic reticulum Ca2+-ATPase (SERCA) pump (Vinogradova et al., 2010). Inhibition of SERCA2 with cyclopiazonic acid induces a concentration-dependent suppression of spontaneous sinus node myocyte firing rate by up to 50% (Vinogradova et al., 2010; Yaniv et al., 2014).
We hypothesized that reduced Serca2 in the sinus node disrupts the Ca2+-clock component of pacemaking and contributes to pacemaker dysfunction. This hypothesis was tested in a Serca2 conditional KO mouse model in which cardiac specific Serca2 excision can be induced at any age by exposure to tamoxifen.
Methods
Homozygous, conditional cardiac specific Serca2 KO and age-matched control (Serca2 FF) mice were studied. Mice with Serca2 downregulation in the heart were generated using the Cre-lox system conditional gene KO strategy as described previously by Andersson et al. (2009). At 12 weeks of age, male Serca2 KO (Serca2flox∕flox Tg(αMHC-MerCreMer); n = 22) and control (Serca2flox∕flox; FF, n = 20) mice were injected with 1 mg of tamoxifen intraperitoneally to induce Serca2 excision in cardiomyocytes. All experiments were carried out 7 weeks after tamoxifen administration in accordance with the Norwegian National Committee for Animal Welfare Act, consistent with the NIH guidelines (NIH publication No. 85-23, revised 1996). SERCA2a abundance and expression pattern was investigated by Western blotting and immunohistochemistry (n = 10). Telemetry transmitters were implanted in one cohort of mice (n = 16) as previously described (Stokke et al., 2010). ECG was recorded under baseline conditions, during maximal exercise, and after a subsequent i.p. injection of adrenalin (0.5 mg/kg). In vitro electrophysiological investigation of the sinus node was carried out by recording extracellular potentials in isolated sinus node preparations (n = 16). Tissue preparations of Wistar rat hearts (n = 13), with intact sinus and AV nodes were used in some investigations.
Mathematical modeling of the mouse heart rate was performed using our biophysically detailed single pacemaker cell model of the mouse sinus node (Kharche et al., 2011). This robustly validated model describes the mouse sinus node electrophysiology based on a spectrum of membrane ionic currents (membrane voltage-clock), which are coupled to the intracellular Ca2+ dynamics (Ca2+-clock). The model code was implemented in C programming language, and was integrated using an implicit backward difference formula that provided a high accuracy of O(dt5), computational efficiency and unconditional numerical stability. The model code, including dependencies, is available from the authors on request. Standard library based code is available from the model repository, ModelsDB (https://senselab.med.yale.edu/modeldb/ShowModel.cshtml?model=141274). All data are expressed as mean ± s.e.m. and n refers to the number of hearts. Either an unpaired t-test or one-way ANOVA followed by Tukey's multiple comparisons post-test was conducted to determine significant differences. The difference was considered to be significant when P < 0.05. Detailed methodology is available in the Supplementary Material.
Results
The body weight of mice in the Serca2 FF group and Serca2 KO group were 32 ± 2 g (n = 12) and 32 ± 3 g (n = 14), respectively at 7 weeks after tamoxifen administration (Figure 2A).
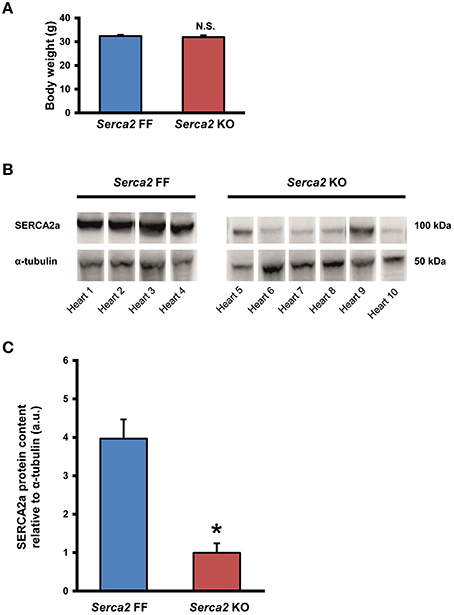
Figure 2. SERCA2a expression in ventricular myocardium. (A) Body weight of Serca2 FF and Serca2 KO mice. (B) Western blots of SERCA2a protein and α-tubulin expression in ventricular myocardium of Serca2 FF (left; n = 4) and Serca2 KO (right; n = 6) hearts. (C) Mean values of SERCA2a band density normalized to α-tubulin in Serca2 FF and Serca2 KO samples. *P < 0.05 vs. Serca2 FF.
SERCA2a Expression in the Ventricular Myocardium
SERCA2a protein expression in the ventricular myocardium was investigated by Western blot and immunohistochemistry. Western blot analysis (Figure 2B) revealed that SERCA2a protein content normalized to α-tubulin content was reduced from 4.0 ± 0.5 a.u. (n = 4) in Serca2 FF to 1.0 ± 0.3 a.u. (n = 6; P < 0.05) in Serca2 KO ventricular myocardium (Figure 2C). The distribution and expression pattern of SERCA2a protein in ventricular myocardium was assessed by immunolabelling tissue sections with a SERCA2a antibody (Supplement Table 1). In ventricular myocytes, labeling occurred adjacent to the outer cell membrane, as well as in an internal striated pattern, consistent with earlier reports of SERCA2 protein expression in ventricular myocytes (Musa et al., 2002). Figures 3A–D shows the typical distribution of SERCA2a protein in ventricular cross sections of Serca2 FF (n = 4; Figures 3A,C) and Serca2 KO (n = 6; Figures 3B,D) mouse hearts. Similar patterns of expression were observed in all Serca2 FF and Serca2 KO tissue sections. At both low (Figure 3A) and high (Figure 3C) magnification, SERCA2a expression in the Serca2 FF tissue was uniform with all cardiomyocytes expressing the SERCA2a protein. However, in the Serca2 KO tissue sections, expression appeared reduced, mosaic-like and heterogeneous, i.e., some cells expressed and some did not (Figures 3B,D). The bright red signal in cardiomyocytes in Figures 3B,D represent ventricular myocytes expressing SERCA2a protein in the Serca2 KO heart sections. The negative controls did not show fluorescence of any significance (Supplement Figure 1). Quantification of immunofluorescence measurements showed reduction in SERCA2a protein expression from 116 ± 4 a.u. (n = 4) in Serca2 FF to 62 ± 6 a.u. (n = 6; P < 0.05) in Serca2 KO sections (Figure 3G). The SERCA2a content measured by Western blot and immunohistochemistry in the individual heart samples was plotted (Supplement Figure 2). SERCA2a protein expression measured by either of the two techniques revealed a similar downregulation.
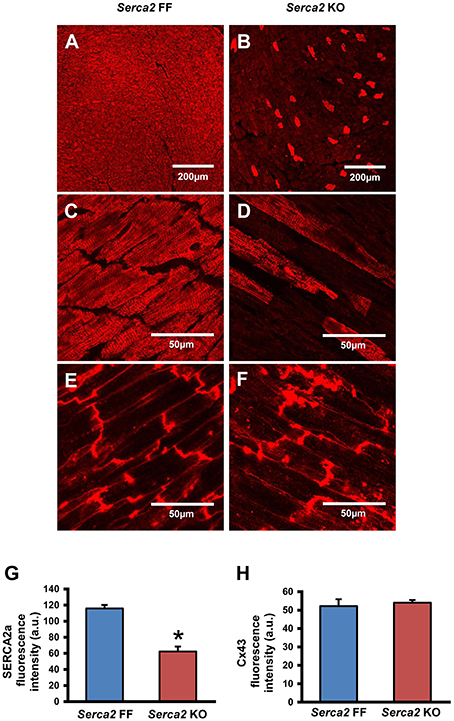
Figure 3. Immunohistochemical detection of SERCA2a and Cx43 in ventricular myocardium. (A–D) Representative immunofluorescence images of SERCA2a protein labeling (red signal) in ventricular myocardium of Serca2 FF (A,C) and Serca2 KO (B,D) hearts. (E,F), high magnification images of Cx43 expression in ventricular myocardium of Serca2 FF and Serca2 KO hearts. (G,H) bar charts showing mean SERCA2a (G) and Cx43 (H) content in the ventricular myocardium of Serca2 FF (blue bars; n = 4) and Serca2 KO (red bars; n = 6) hearts. *P < 0.05 vs. Serca2 FF.
Connexin 43 (Cx43), a major gap junction connexin in the heart, was labeled in adjacent ventricular tissue sections and used as a marker of ventricular cardiomyocytes (Figures 3E,F). The quantified immunofluorescence of Cx43 labeling was 52 ± 4 a.u. (n = 4) in Serca2 FF and 54 ± 2 a.u. (n = 6) in Serca2 KO sections. The Cx43 protein expression remained unchanged (Figure 3H).
Telemetry ECG
ECG was recorded in conscious animals with the aid of implanted telemetry electrodes (Figure 4A). The baseline heart rate measured in freely moving Serca2 FF and Serca2 KO animals was 536 ± 27 and 536 ± 31 beats per minute (bpm), respectively (both groups n = 8, Figures 4B,C). The treadmill test was performed as a gradual, stepwise increase in the running speed until exhaustion. Serca2 FF achieved a maximal heart rate of 780 ± 17 bpm, significantly higher than the 667 ± 22 bpm achieved by Serca2 KO (P < 0.01 vs. Serca2 FF, Figures 4A–C). The distance traveled till exhaustion in Serca2 FF was 117 ± 16 m and that in Serca2 KO was 56 ± 6 m (P < 0.01). The maximum running speed was significantly higher in Serca2 FF (18 ± 1 vs. 12 ± 1 m/min in Serca2 KO, P < 0.05). Subsequently, animals were injected intraperitoneally (i.p.) with adrenaline (0.5 mg/kg bw) and at 10 min after adrenaline administration, the heart rates in Serca2 FF and Serca2 KO animals was 681 ± 9 and 581 ± 23 bpm, respectively (P < 0.01, Figures 4A–C). The ECG PR-intervals at baseline, during maximal exercise, and after a subsequent i.p. injection of adrenalin in Serca2 FF was 40 ± 1, 31 ± 1, and 37 ± 2 ms and the corresponding values for Serca2 KO were 49 ± 1 (P < 0.001 vs. Serca2 FF), 37 ± 2 (P < 0.05 vs. Serca2 FF) and 51 ± 4 ms (P < 0.05 vs. Serca2 FF), respectively (Figures 4D,E).
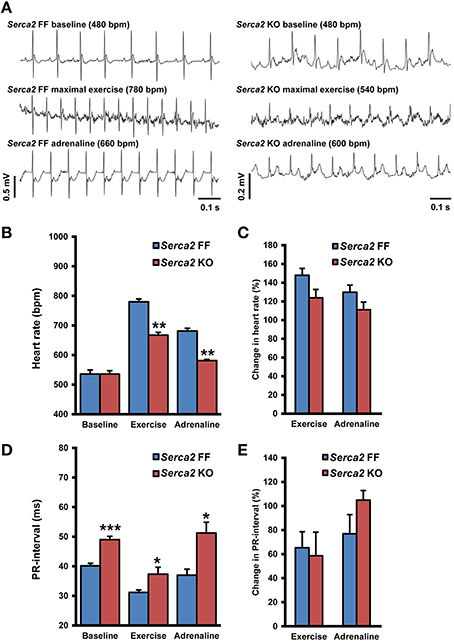
Figure 4. Telemetry ECG parameters. (A) Representative records of telemetry ECG in Serca2 FF (left) and Serca2 KO (right) mice under baseline conditions, maximal exercise, and after a subsequent i.p. injection of adrenaline (0.5 mg/kg). (B) Heart rate measurements in freely moving Serca2 FF (blue bars) and Serca2 KO (red bars) mice during baseline conditions, maximal exercise, and after a subsequent i.p. injection of adrenalin (0.5 mg/kg). (C) Change in heart rate during exercise and after adrenaline injection presented as a percentage of corresponding baseline values. (D) The PR-interval in Serca2 FF (blue bars) and Serca2 KO (red bars) mice during baseline conditions, maximal exercise, and after adrenaline administration. (E) Change in PR-interval presented as a percentage of corresponding baseline values. *, **, and *** denote P < 0.05, P < 0.01, and P < 0.0001, respectively vs. corresponding Serca2 FF.
Sarcoplasmic Reticulum Ca2+ and Atrioventricular Node
In rat right atrial preparations, the role of sarcoplasmic reticulum Ca2+ in sinus and atrioventricular (AV) node function was assessed in vitro. Ryanodine (2 μM) prolonged the sinus node intrinsic cycle length from 241.1 ± 9.63 ms (n = 13) to 369.8 ± 25.4 ms (n = 10; P < 0.001). The tissue was overdrive paced and the atrio-His (AH) interval (a measure of conduction through the AV node) and the AV node Wenckebach cycle length were measured. Ryanodine (2 μM) prolonged the AH-interval from 47 ± 6.1 ms to 59 ± 7.7 ms (P ≤ 0.01) and the AV node Wenckebach cycle length from 161.5 ± 11.7 ms to 205.1 ± 22.7 ms (P ≤ 0.05).
Sinus Node Pacemaking in Serca2 KO Mice
The intrinsic beating/heart rate measured in isolated sinus node preparations at 37°C was 415 ± 16 bpm (n = 8) in Serca2 FF and 431 ± 30 bpm (n = 8) in Serca2 KO (Figure 5A) tissues. The response to pharmacological block of the membrane- and Ca2+-clock was measured by separately treating tissues with 2 mM Cs+ (for If block) and 2 μM ryanodine (for disabling Ca2+ release via the ryanodine receptor). In the presence of Cs+, the sinus rate reduced to 294 ± 59 bpm (n = 8) in Serca2 FF and 291 ± 33 bpm (n = 8) in Serca2 KO (Figure 5A) mice. The response to ryanodine was different in Serca2 KO compared to Serca2 FF mice. The sinus rate dropped to 323 ± 26 bpm (n = 7) in Serca2 FF mice whereas in Serca2 KO mice, the drop in sinus rate was to 377 ± 26 bpm (n = 7; Figures 5A,B). In other words, the effect of ryanodine was less pronounced in Serca2 KO tissue in comparison with Serca2 FF tissue.
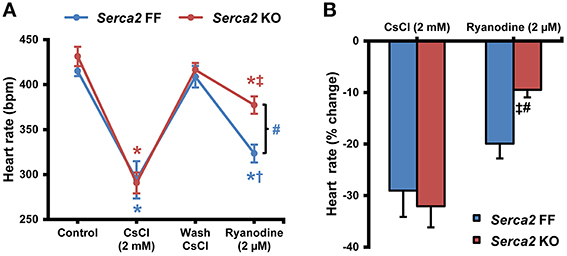
Figure 5. Sinus node pacemaking in Serca2 FF vs. Serca2 KO mice. (A) Change in heart rate of isolated sinus node preparations from Serca2 FF and Serca2 KO in response to treatment with 2 mM CsCl and 2 μM ryanodine. (B) Percentage reduction of heart rate in vitro induced by CsCl (2 mM) and ryanodine (2 μM) treatment. The CsCl (2 mM) and ryanodine (2 μM) measurements are presented as percentage of control and wash CsCl values, respectively. Serca2 FF data are shown in blue and Serca2 KO data are shown in red color. *, †, and ‡ represent P < 0.05 vs. control, wash CsCl and CsCl (2 mM) respectively. Blue symbols are used for Serca2 FF and red for Serca2 KO data. #represents P < 0.05 vs. Serca2 KO ryanodine response (Serca2 FF, n = 8 and Serca2 KO, n = 8).
SERCA2 Downregulation in Sinus Node
Histological staining with Masson's trichrome was performed in tissue sections that were cut perpendicular to the crista terminalis of Serca2 FF and Serca2 KO right atrial tissue. The morphology and architecture of the sinus node was not affected in Serca2 KO mice (data not shown). The histology images were used to locate the sinus node and identify suitable sections for labeling with immunofluorescent markers. HCN4 was used as the positive marker to delineate sinus node myocytes and Cx43 was used to label atrial muscle (Liu et al., 2007). Typical low magnification images of HCN4 and Cx43 are shown in Figure 6 (top and middle row). Adjacent sections labeled for SERCA2a are shown in Figure 6 (bottom row). Representative high magnification images of the sinus node center, sinus node periphery and atrial muscle are shown in Figure 7A. The SERCA2a expression pattern in these sections was similar to that seen in sections from the ventricular myocardium: in the Serca2 FF sinus node center, sinus node periphery and atrial muscle, SERCA2a protein was ubiquitously expressed in all myocytes (Figure 7A, top row). In contrast, in the Serca2 KO sections, SERCA2a expression was reduced and mosaic-like in all three regions (Figure 7A, bottom row). The quantified fluorescence measurements in Serca2 FF sections were 147 ± 14, 139 ± 10, and 110 ± 9 a.u. (n = 4) in the sinus node center, sinus node periphery and atrial muscle, respectively (Figure 7B). In Serca2 KO sections, fluorescence measurements were reduced to 97 ± 12, 62 ± 9, and 60 ± 4 (n = 6; all P < 0.05 vs. corresponding Serca2 FF values). HCN4 and Cx43 expression patterns were similar in Serca2 FF and Serca2 KO sections (Supplement Figures 3, 4).
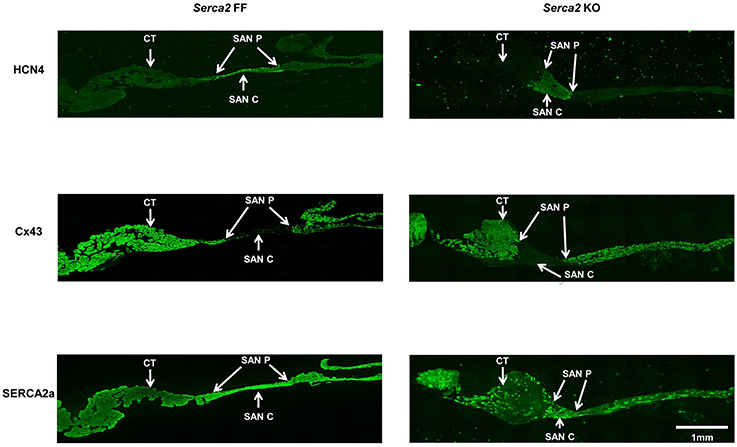
Figure 6. Immunolabelling of HCN4, Cx43, and SERCA2 in right atrial sections from Serca2 FF and Serca2 KO hearts. Top row, right atrial sections from Serca2 FF (left) and Serca2 KO (right) hearts showing HCN4 labeling (green signal) in the sinus node periphery (SAN P) and center (SAN C). Middle row, connexin 43 (Cx43) labeling (green signal) in the right atrium. Bottom row, right atrial sections showing SERCA2a labeling (green signal) throughout the tissue section in Serca2 FF (left) and a mosaic pattern of SERCA2a labeling in Serca2 KO (right). Abbreviations: CT, crista terminalis; SAN P, sinus node periphery; SAN C, sinus node center.
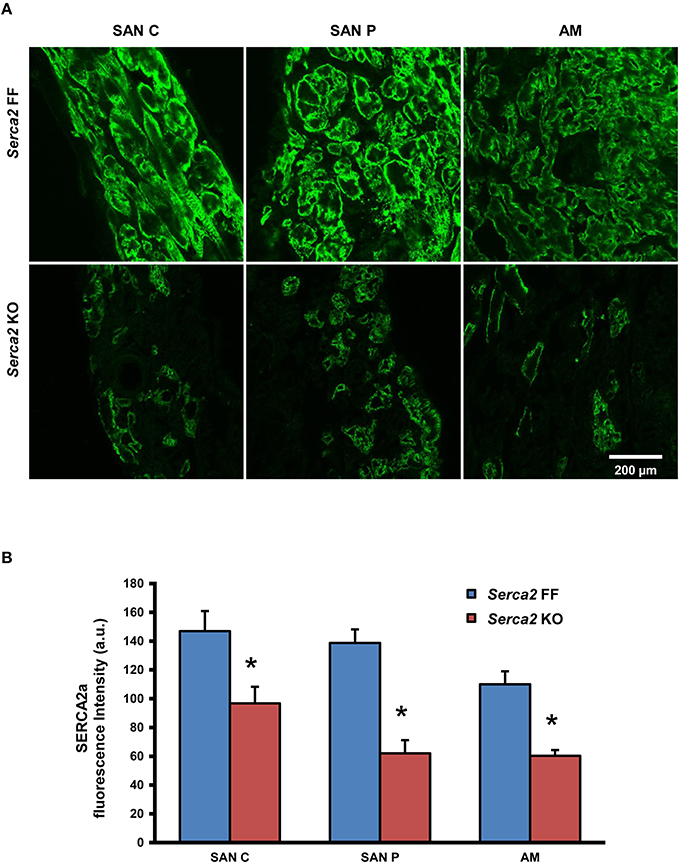
Figure 7. SERCA2a protein expression in the right atrium and sinus node. (A) Representative high magnification images showing SERCA2 labeling (green signal) in the sinus node center (SAN C), sinus node periphery (SAN P) and atrial muscle (AM) of Serca2 FF (top row) and Serca2 KO (bottom row) right atrial tissue sections. (B) SERCA2a fluorescence intensity in the sinus node center (SAN C), sinus node periphery (SAN P) and atrial muscle (AM) of Serca2 FF (blue bars) and Serca2 KO (red bars) tissue sections. * denotes P < 0.05 vs. corresponding Serca2 FF tissue values.
Mathematical Modeling of Mouse Sinus Node Action Potential
Mathematical modeling was used to dissect the role of SERCA2 in sinus node pacemaking and to gain mechanistic insights. The results of the simulations are presented in Figure 8. The relationships between SERCA2 downregulation (0–99%) and heart rate under various conditions is shown in Figure 8A. Pacemaking in the sinus node, measured as heart rate, was significantly more erratic at low levels of SERCA2 (99% downregulation) than at physiological levels of SERCA2 (0% downregulation) (red symbols, Figure 8A). At physiological values of SERCA2, heart rate of 283 bpm was observed. Downregulation of RyR2 channel (proportional to SERCA2 downregulation) did not affect the heart rate at 0% SERCA2 downregulation; however, it increased the arrhythmic region at >90% SERCA2 downregulation (yellow symbols, Figure 8A). The increase of L-type Ca2+ current (ICa, L) caused an overall increase of heart rate at all levels of SERCA2 (blue symbols, Figure 8A). The heart rate at 0% SERCA2 downregulation was 287 bpm. On the other hand, increasing the Na+-Ca2+ exchanger current (INaCa) resulted in lowering of heart rate: 272 bpm at 0% SERCA2 downregulation (green symbols, Figure 8A). When all the above alterations were implemented simultaneously to emulate the experimental observations, i.e. enhanced Ca2+ flux through the L-type Ca2+ channel and the Na+-Ca2+ exchanger in Serca2 KO mouse (Andersson et al., 2009), the resultant heart rate (black symbols, Figure 8A), was lower than in the case without remodeling (red symbols, Figure 8A). The slope of the remodeled heart rate curve is shown in Figure 8B. It was observed that the slope was small for low levels of SERCA2 downregulation and numerically large at >70% downregulation.
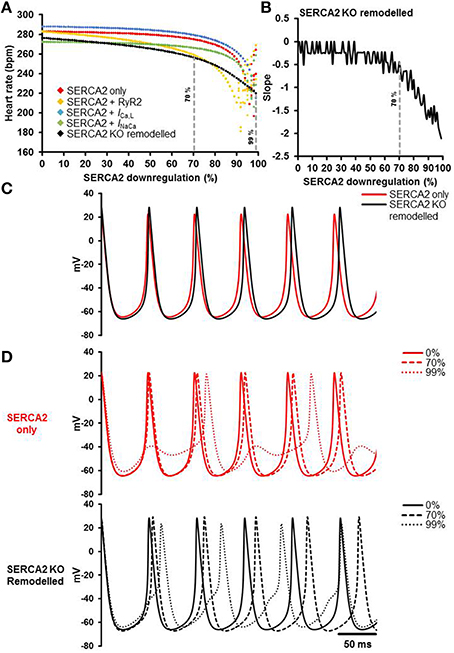
Figure 8. Simulation of the effects of SERCA2 downregulation on the sinus node action potential. (A) Relationship between SERCA2 downregulation (0–99% reduction) and heart rate (bpm) under various conditions. Red symbols represent heart rates with changes to SERCA2 only (i.e., with all other parameters unchanged). Yellow, blue and green symbols represent introduction of changes to RyR2 (proportional to SERCA2), ICa, L (1.3 fold increase), and INaCa (2 fold increase), respectively. The combined effect of all changes is represented in black (SERCA2 KO remodeled). (B) Slope of the SERCA2 KO remodeled curve. (C) Action potential profiles of SERCA2 only (red) and SERCA2 KO remodeled (black) at 0% SERCA2 downregulation are shown. (D) Typical action potential profiles under SERCA2 only in red (top) and SERCA2 KO remodeled in black (bottom) are shown. Solid, dashed and dotted lines represent 0, 70, and 99% SERCA2 downregulation respectively. Modeling data generated by SK.
Action potential profiles of SERCA2 only (red) and SERCA2 KO remodeled (black) at 0% SERCA2 downregulation is shown in Figure 8C. Corresponding action potential profiles at 0, 70, and 99% SERCA2 downregulation are shown in Figure 8D. Without any remodeling, beating/heart rates measured at 0 and 70% SERCA2 downregulation were 283 bpm (red solid line, Figure 8D) and 275 bpm (red dashed line, Figure 8D) respectively; however, at 99% downregulation the beating/heart rate was substantially reduced to 220 bpm (red dotted line, Figure 8D) and the region with >95% SERCA2 downregulation was arrhythmic. Under remodeled conditions, the corresponding heart rates were 276 bpm (black solid line, Figure 8D), 256 bpm (black dashed line, Figure 8D) and 220 bpm (black dotted line, Figure 8D) respectively. In the remodeled case at 99% SERCA2 downregulation, the maximum diastolic potential was less negative (by ~3 mV), the action potential upstroke was biphasic and the heart rate was substantially bradycardic (black dotted line, Figure 8D).
Discussion
We have investigated the consequence of substantial Serca2 downregulation on the sinus node of the adult mouse heart. Cardiomyocyte-specific excision of Serca2 in adult mice resulted in reduced SERCA2a protein abundance in the sinus node, atrial muscle and ventricular muscle. In vivo, baseline heart rate was not affected; however, during exercise and in the presence of adrenaline, heart rate was lower than Serca2 FF. In vitro, the intrinsic sinus node beating rate remained unaltered in Serca2 KO. Separate assessment of the voltage- and Ca2+-clock components of sinus node pacemaking revealed an unaltered voltage-clock and a diminished Ca2+-clock. Our Serca2 KO mouse model provides novel information on the importance of SERCA2a, and thus the sarcoplasmic reticulum Ca2+ clock, in the sinus node pacemaking mechanism.
In our study, we have demonstrated that SERCA2a downregulation in the sinus node of the adult mouse heart can be achieved by relying on the α-MHC promoter to drive the expression of the Cre-recombinase enzyme to cause cardiac specific Serca2 deletion. The reduction in SERCA2a protein content occurs in the sinus node, as well as atrial and ventricular tissues, suggesting Cre expression in all of these tissues. α-MHC is a contractile protein and mRNA corresponding to α-MHC has been reported in the ventricular myocardium, as well as in the atrium and sinus node (Nakao et al., 1997; Tellez et al., 2011). Thus, Cre-recombinase would be abundantly expressed in these tissues resulting in Serca2 deletion. Our observations are consistent with a recent study of a cardiac specific HCN4 gene KO mouse model wherein the α-MHC promoter linked Cre-Lox system of gene deletion induced substantial downregulation of HCN4 protein in the sinus node (Baruscotti et al., 2011).
In the ventricular myocardium, we found that SERCA2a protein expression was reduced by ~75% at 7 weeks after tamoxifen administration. The mosaic pattern of SERCA2a expression observed in Serca2 KO heart tissue was an interesting, yet unexpected finding of this study. It should be borne in mind that, conditional gene KO techniques based on excision of lox-P-flanked DNA segments by Cre-recombinase have some inherent pitfalls (Schmidt-Supprian and Rajewsky, 2007). LoxP-flanked target genes differ in their sensitivity to Cre-facilitated recombination and Cre can damage genomic DNA. Also, the efficiency of gene KO using Cre-loxP technology is tamoxifen dose-dependent (Schmidt-Supprian and Rajewsky, 2007; Bersell et al., 2013). Tamoxifen administration to mice expressing MerCreMer protein is known to induce severe, transient dilated cardiomyopathy that is accompanied by transient reduction in SERCA2 and phospholamban mRNA (Koitabashi et al., 2009). Thus, data obtained immediately after tamoxifen induced gene inactivation, is often unreliable. In the present study, hearts were harvested at 7 weeks post tamoxifen administration, giving sufficient time for the transient cardiomyopathy to resolve (Koitabashi et al., 2009). Western blot data showed substantial reduction in SERCA2a protein content in the left ventricular myocardium, in agreement with an earlier study of this animal model (Andersson et al., 2009). Several other studies have administered tamoxifen as 4–5 injections on successive days at a safe dose (1–2 mg/day) (Danielian et al., 1998; Andersson et al., 2009; Hougen et al., 2010; Baruscotti et al., 2011). Our results indicate that a single dose of tamoxifen (1 mg/day) is sufficient to induce efficient Cre-recombination and substantial Serca2 downregulation. However, the downregulation is heterogeneous, unlike the homogeneous Serca2 downregulation previously observed by us in ventricular tissue for the single dose tamoxifen injection (Hougen et al., 2010). The single tamoxifen dose (1 mg/day) may be on the threshold limit for efficient MerCreMer-activation (as opposed to four injections). Thus, some cells might not be exposed to an adequate concentration, giving rise to a mosaic pattern of downregulation as seen elsewhere (Hayashi and McMahon, 2002).
Isolated ventricular myocytes of Serca2 KO mice show significantly lower sarcoplasmic reticulum Ca2+ content (Swift et al., 2008, 2012; Andersson et al., 2009; Stokke et al., 2010). Given that we used an identical mouse model (albeit with one tamoxifen injection instead of four), and that the SERCA2 protein is substantially downregulated, it is reasonable to expect a significant degree of reduction in sarcoplasmic reticulum Ca2+ content in sinus node myocytes. Reduced sarcoplasmic reticulum Ca2+ content decreases the open probability of the ryanodine receptor and the occurrence of localized Ca2+ release events that govern the late phase of the diastolic depolarization in the sinus node (Bogdanov et al., 2006). Also, the kinetics of the sarcoplasmic reticulum Ca2+ pumping by SERCA2 is known to regulate sinus node beating rates (Vinogradova et al., 2010). In vitro, pharmacological blockade of either the ryanodine receptor with ryanodine or the SERCA2 pump with cyclopiazonic acid, inhibits the sinus node beating rate (Rigg and Terrar, 1996; Vinogradova et al., 2010; Yaniv et al., 2014). One might then ask why the Serca2 KO mice baseline heart rates measured in vivo (536 ± 31 vs. 536 ± 27 bpm in Serca2 FF) and sinus node beating rates measured in vitro (431 ± 30 vs. 415 ± 16 bpm in Serca2 FF tissues) were not different from those in Serca2 FF? In the present study, although SERCA2a is significantly downregulated (by 70–75%) in Serca2 KO hearts, some degree of residual SERCA2a activity will exist and is likely to contribute to the maintenance of baseline heart/sinus rates. Previously, we have observed baseline bradycardia in Serca2 KO with >95% SERCA2a downregulation and have demonstrated that very low SERCA levels are capable of partially refilling the sarcoplasmic reticulum (Andersson et al., 2009; Louch et al., 2010). The residual SERCA2a activity is however, below optimal during exercise and in the presence of adrenaline resulting in smaller heart rate increases. Additionally, it is possible that compensatory mechanisms may have come into play during the 7 week period to allow for the Serca2 downregulation. Blocking If slowed pacemaking that was equally pronounced in Serca2 KO and control mice (Figure 4) and hence it is unlikely that If compensates for the loss of the Ca2+-clock. It is possible that some other compensatory mechanism exists. Pacemaking in the sinus node is also dependent on the voltage-dependent deactivation of outward currents: the rapid and slow delayed rectifier K+ currents (IK, r and IK, s) and activation of inward currents such as the L- and T-type Ca2+ currents (ICa, L and ICa, T), INaCa, tetrodotoxin-sensitive Na+ current (INa) and sustained inward current (Ist), amongst others (Dobrzynski et al., 2013; Logantha et al., 2014; Morris and Kalman, 2014).
Studies on isolated ventricular myocytes of Serca2 KO mice have shown larger ICa, L and INaCa density, indicating a greater Ca2+-influx and extrusion across the plasma membrane (Andersson et al., 2009). Using mathematical modeling, we evaluated the consequences of increased ICa, L and INaCa on the heart rate of Serca2 KO. Downregulation of SERCA2 alone caused a reduction in heart rate that was pronounced at >70% SERCA2 downregulation. At >95% SERCA2 downregulation, pacemaking was arrhythmic, i.e., non-periodic. When SERCA2 downregulation was coupled with a proportional reduction of Ca2+ flux through the RyR2, the region of arrhythmic pacemaking was increased. This is in agreement with our previous work, as well as work by others (Kharche et al., 2011; Maltsev and Lakatta, 2013). As the membrane- and Ca2+-clocks in our model are based on extensive experimental data, it was possible to simulate the phenomenon of how SERCA2 alterations affect heart rate. In the sinus node model, ICa, L regulates the upstroke of the action potential and the heart rate (Kharche et al., 2011). On the other hand, INaCa mainly provides calcium homeostasis. Therefore, in the current study, increasing ICa, L increased the heart rate and INaCa augmentation resulted in lower heart rate measurements due to a lower level of cytosolic Ca2+. Simultaneously altering multiple parameters (conductance of SERCA2, RyR2, ICa, L, and INaCa) does not result in a straightforward overlay of the outcomes (i.e., heart rates) because the cell model is complex and non-linear (Kharche et al., 2009). Implementing the remodeling as a simultaneous change of function in RyR2, ICa, L, and INaCa resulted in reduced heart rates. The heart rate changes minimally upto 70% SERCA2 downregulation; however, at >70% downregulation the reduction in heart rate is much pronounced. This is consistent with our experimental observations and it is likely that similar compensatory remodeling occurs in the sinus node which helps maintain baseline heart rate in vivo and in vitro. Although, mathematical models cannot encompass all the physiological processes in a sinus node cell, our model is capable of dissecting the important mechanisms that regulate pacemaking (See Model Limitations in Supplementary Material).
In vivo, with the sympathetic innervation intact, the resting heart rates of both Serca2 KO and Serca2 FF mice are higher than that observed in vitro in isolated right atrial preparations. In our study, Serca2 KO mice with 75% reduction in left ventricular SERCA2a protein content show a smaller increase in heart rate vs. Serca2 FF in response to exercise and adrenaline injection. This is consistent with the smaller increase in heart rate in response to isoproterenol observed in Serca2 KO mice with >95% SERCA2a downregulation (Boardman et al., 2014). Catecholamine mediated acceleration of sinus node pacemaking is crucially dependent on the Ca2+-clock (Gao et al., 2010; Lakatta et al., 2010; Liu et al., 2011).
The ECG PR-interval, a measure of AV node conduction, was significantly prolonged in Serca2 KO mice. In the present study, we also show that ryanodine, by disrupting sarcoplasmic reticulum Ca2+ releases, prolongs the AH-interval and the Wenckebach cycle length in isolated rat AV node preparations. This is consistent with previous work from our laboratory that showed that ryanodine increases the spontaneous cycle length of intact AV node preparations (Nikmaram et al., 2008). Additionally, both ryanodine and SERCA inhibitors (thapsigargin and cyclopiazonic acid) prolong the spontaneous cycle length in isolated AV node myocytes and the beating rate is therefore dependent on sarcoplasmic reticulum function (Ridley et al., 2008; Cheng et al., 2011). The prolonged PR-interval in Serca2 KO mice is indicative of first degree AV-block and points at the important role for sarcoplasmic reticulum Ca2+ in AV node function.
Clinical Perspectives and Future Applications
SERCA2 is an important Ca2+ handling protein in the sinus node. Serca2 expression as well as protein abundance is significantly altered in the diseased or aged sinus node (Tellez et al., 2011; Benoist et al., 2014). It has been long established that SERCA2a expression and activity is decreased in human heart failure of most etiologies and SERCA2a is a promising target for gene therapy in heart failure (Unverferth et al., 1988; Mercadier et al., 1990; Hasenfuss et al., 1994; Bers et al., 2003; Hayward et al., 2014). However, it has not been clear whether Serca2 loss is a basis for or a result of sinus node disease and heart failure. Here we show for the first time that Serca2 downregulation can cause pacemaker dysfunction, which might contribute to further deterioration in an already failing heart. The Serca2 KO mouse developed for this study is a valuable tool in the investigation of the role of the sarcoplasmic reticulum in sinus node and AV node function. Previously, such investigations were limited by the use of pharmacological tools to block the SERCA2 pump or ryanodine receptor. The Serca2 KO mouse offers a suitable platform for mechanistic studies at the level of the single cell, tissue and the whole animal.
Author Contributions
MS, IS, OS, SL, and HD conceived the project, secured funding and planned the experiments. MS performed telemetry ECG monitoring and analysis. SL carried out the in vitro electrophysiology experiments and collected tissue samples for Western blot and immunohistochemistry. AA performed Western blot. SL, SP, and HD were responsible for cryosectioning, histology staining, immunolabelling, immunofluorescence quantification and analysis. YS performed the experiments on the isolated rat atrioventricular node. AA, SP, and YS were supervised by HD. SL provided the experimental data input to SK for the mathematical modeling. SK designed and performed mathematical modeling and interpreted the simulation results. SL generated the figures and prepared the first draft of the manuscript. All authors contributed to the revision of manuscript and have approved the manuscript.
Funding
SL was awarded a travel grant by the Physiological Society, London and financial support by the University of Oslo for a visit to Oslo to carry out electrophysiological investigations and collect tissue samples. He also received salary support from the British Heart Foundation (Programme Grant RG/11/18/29257).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Professor Mark R. Boyett, University of Manchester, U.K. for his efforts in initiating the collaboration between the Universities of Manchester (UK) and Oslo (Norway). We are also thankful for his advice on the electrophysiological investigations and comments on the manuscript. MS, IS, and OS are members of KG Jebsen Cardiac Research Center in Oslo, Norway.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphys.2016.00197
References
Andersson, K. B., Birkeland, J. A., Finsen, A. V., Louch, W. E., Sjaastad, I., Wang, Y., et al. (2009). Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J. Mol. Cell. Cardiol. 47, 180–187. doi: 10.1016/j.yjmcc.2009.03.013
Baruscotti, M., Bucchi, A., Viscomi, C., Mandelli, G., Consalez, G., Gnecchi-Rusconi, T., et al. (2011). Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc. Natl. Acad. Sci. U.S.A. 108, 1705–1710. doi: 10.1073/pnas.1010122108
Benoist, D., Stones, R., Benson, A. P., Fowler, E. D., Drinkhill, M. J., Hardy, M. E., et al. (2014). Systems approach to the study of stretch and arrhythmias in right ventricular failure induced in rats by monocrotaline. Prog. Biophys. Mol. Biol. 115, 162–172. doi: 10.1016/j.pbiomolbio.2014.06.008
Bers, D. M., Eisner, D. A., and Valdivia, H. H. (2003). Sarcoplasmic reticulum Ca2+ and heart failure: roles of diastolic leak and Ca2+ transport. Circ. Res. 93, 487–490. doi: 10.1161/01.RES.0000091871.54907.6B
Bersell, K., Choudhury, S., Mollova, M., Polizzotti, B. D., Ganapathy, B., Walsh, S., et al. (2013). Moderate and high amounts of tamoxifen in αMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Dis. Model. Mech. 6, 1459–1469. doi: 10.1242/dmm.010447
Boardman, N. T., Aronsen, J. M., Louch, W. E., Sjaastad, I., Willoch, F., Christensen, G., et al. (2014). Impaired left ventricular mechanical and energetic function in mice after cardiomyocyte-specific excision of Serca2. Am. J. Physiol. Heart Circ. Physiol. 306, H1018–H1024. doi: 10.1152/ajpheart.00741.2013
Bogdanov, K. Y., Maltsev, V. A., Vinogradova, T. M., Lyashkov, A. E., Spurgeon, H. A., Stern, M. D., et al. (2006). Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ. Res. 99, 979–987. doi: 10.1161/01.RES.0000247933.66532.0b
Bogdanov, K. Y., Vinogradova, T. M., and Lakatta, E. G. (2001). Sinoatrial nodal cell ryanodine receptor and Na+-Ca2+ exchanger: molecular partners in pacemaker regulation. Circ. Res. 88, 1254–1258. doi: 10.1161/hh1201.092095
Cheng, H., Smith, G. L., Hancox, J. C., and Orchard, C. H. (2011). Inhibition of spontaneous activity of rabbit atrioventricular node cells by KB-R7943 and inhibitors of sarcoplasmic reticulum Ca2+ ATPase. Cell Calcium 49, 56–65. doi: 10.1016/j.ceca.2010.11.008
Danielian, P. S., Muccino, D., Rowitch, D. H., Michael, S. K., and McMahon, A. P. (1998). Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 8, 1323–1326. doi: 10.1016/S0960-9822(07)00562-3
Dobrzynski, H., Anderson, R. H., Atkinson, A., Borbas, Z., D'Souza, A., Fraser, J. F., et al. (2013). Structure, function and clinical relevance of the cardiac conduction system, including the atrioventricular ring and outflow tract tissues. Pharmacol. Ther. 139, 260–288. doi: 10.1016/j.pharmthera.2013.04.010
Dobrzynski, H., Boyett, M. R., and Anderson, R. H. (2007). New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation 115, 1921–1932. doi: 10.1161/CIRCULATIONAHA.106.616011
Gao, Z., Chen, B., Joiner, M. L., Wu, Y., Guan, X., Koval, O. M., et al. (2010). If and SR Ca2+ release both contribute to pacemaker activity in canine sinoatrial node cells. J. Mol. Cell. Cardiol. 49, 33–40. doi: 10.1016/j.yjmcc.2010.03.019
Hasenfuss, G., Reinecke, H., Studer, R., Meyer, M., Pieske, B., Holtz, J., et al. (1994). Relation between myocardial function and expression of sarcoplasmic reticulum Ca2+-ATPase in failing and nonfailing human myocardium. Circ. Res. 75, 434–442. doi: 10.1161/01.RES.75.3.434
Hayashi, S., and McMahon, A. P. (2002). Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244, 305–318. doi: 10.1006/dbio.2002.0597
Hayward, C., Patel, H., and Lyon, A. (2014). Gene therapy in heart failure. SERCA2a as a therapeutic target. Circ. J. 78, 2577–2587. doi: 10.1253/circj.CJ-14-1053
Hougen, K., Aronsen, J. M., Stokke, M. K., Enger, U., Nygard, S., Andersson, K. B., et al. (2010). Cre-loxP DNA recombination is possible with only minimal unspecific transcriptional changes and without cardiomyopathy in Tg(alphaMHC-MerCreMer) mice. Am. J. Physiol. Heart Circ. Physiol. 299, H1671–H1678. doi: 10.1152/ajpheart.01155.2009
Keith, A., and Flack, M. (1907). The form and nature of the muscular connections between the primary divisions of the vertebrate heart. J. Anat. Physiol. 41, 172–189.
Kharche, S., Ludtke, N., Panzeri, S., and Zhang, H. G. (2009). A global sensitivity index for biophysically detailed cardiac cell models: a computational approach. LNCS, 5528, 366–375. doi: 10.1007/978-3-642-01932-6_40
Kharche, S., Yu, J., Lei, M., and Zhang, H. (2011). A mathematical model of action potentials of mouse sinoatrial node cells with molecular bases. Am. J. Physiol. Heart Circ. Physiol. 301, H945–H963. doi: 10.1152/ajpheart.00143.2010
Koitabashi, N., Bedja, D., Zaiman, A. L., Pinto, Y. M., Zhang, M., Gabrielson, K. L., et al. (2009). Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circ Res. 105, 12–15. doi: 10.1161/CIRCRESAHA.109.198416
Lakatta, E. G., Maltsev, V. A., and Vinogradova, T. M. (2010). A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ Res. 106, 659–673. doi: 10.1161/CIRCRESAHA.109.206078
Liu, J., Dobrzynski, H., Yanni, J., Boyett, M. R., and Lei, M. (2007). Organisation of the mouse sinoatrial node: structure and expression of HCN channels. Cardiovasc. Res. 73, 729–738. doi: 10.1016/j.cardiores.2006.11.016
Liu, J., Sirenko, S., Juhaszova, M., Ziman, B., Shetty, V., Rain, S., et al. (2011). A full range of mouse sinoatrial node AP firing rates requires protein kinase A-dependent calcium signaling. J. Mol. Cell. Cardiol. 51, 730–739. doi: 10.1016/j.yjmcc.2011.07.028
Logantha, S. J. R. J., Atkinson, A. J., Boyett, M. R., and Dobrzynski, H. (2014). “Molecular basis of arrhythmias associated with the cardiac conduction system,” in Cardiac Arrhythmias. from Basic Mechanism to State-of-the-Art Management, eds A. S. Kibos, B. P. Knight, V. Essebag, S. B. Fishberger, M. Slevin, I. C. Tintoiu (London: Springer), 19–34.
Louch, W. E., Hougen, K., Mørk, H. K., Swift, F., Aronsen, J. M., Sjaastad, I., et al. (2010). Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J. Physiol. 588, 465–478. doi: 10.1113/jphysiol.2009.183517
Maltsev, V. A., and Lakatta, E. G. (2013). Numerical models based on a minimal set of sarcolemmal electrogenic proteins and an intracellular Ca2+ clock generate robust, flexible, and energy efficient cardiac pacemaking. J. Mol. Cell. Cardiol. 59, 181–195. doi: 10.1016/j.yjmcc.2013.03.004
Mercadier, J. J., Lompré, A. M., Duc, P., Boheler, K. R., Fraysse, J. B., Wisnewsky, C., et al. (1990). Altered sarcoplasmic reticulum Ca2+-ATPase gene expression in the human ventricle during end-stage heart failure. J. Clin. Invest. 85, 305–309. doi: 10.1172/JCI114429
Morris, G. M., and Kalman, J. M. (2014). Fibrosis, electrics and genetics. Perspectives in sinoatrial node disease. Circ. J. 78, 1272–1282. doi: 10.1253/circj.CJ-14-0419
Musa, H., Lei, M., Honjo, H., Jones, S. A., Dobrzynski, H., Lancaster, M. K., et al. (2002). Heterogeneous expression of Ca2+ handling proteins in rabbit sinoatrial node. J. Histochem. Cytochem. 50, 311–324. doi: 10.1177/002215540205000303
Nakao, K., Minobe, W., Roden, R., Bristow, M. R., and Leinwand, L. A. (1997). Myosin heavy chain gene expression in human heart failure. J. Clin. Invest. 100, 2362–2370. doi: 10.1172/JCI119776
Nikmaram, M. R., Liu, J., Abdelrahman, M., Dobrzynski, H., Boyett, M. R., and Lei, M. (2008). Characterization of the effects of ryanodine, TTX, E-4031 and 4-AP on the sinoatrial and atrioventricular nodes. Prog. Biophys. Mol. Biol. 96, 452–464. doi: 10.1016/j.pbiomolbio.2007.07.003
Ridley, J. M., Cheng, H., Harrison, O. J., Jones, S. K., Smith, G. L., Hancox, J. C., et al. (2008). Spontaneous frequency of rabbit atrioventricular node myocytes depends on SR function. Cell Calcium. 44, 580–591. doi: 10.1016/j.ceca.2008.04.004
Rigg, L., and Terrar, D. A. (1996). Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp. Physiol. 81, 877–880. doi: 10.1113/expphysiol.1996.sp003983
Schmidt-Supprian, M., and Rajewsky, K. (2007). Vagaries of conditional gene targeting. Nat. Immunol. 8, 665–668. doi: 10.1038/ni0707-665
Silverman, M. E., Grove, D., and Upshaw, C. B. Jr. (2006). Why does the heart beat? The discovery of the electrical system of the heart. Circulation 113, 2225–2281. doi: 10.1161/CIRCULATIONAHA.106.616771
Stokke, M. K., Hougen, K., Sjaastad, I., Louch, W. E., Briston, S. J., Enger, U. H., et al. (2010). Reduced SERCA2 abundance decreases the propensity for Ca2+ wave development in ventricular myocytes. Cardiovasc. Res. 86, 63–71. doi: 10.1093/cvr/cvp401
Swift, F., Birkeland, J. A., Tovsrud, N., Enger, U. H., Aronsen, J. M., Louch, W. E., et al. (2008). Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase alpha2-isoform in heart failure. Cardiovasc. Res. 78, 71–78. doi: 10.1093/cvr/cvn013
Swift, F., Franzini-Armstrong, C., Øyehaug, L., Enger, U. H., Andersson, K. B., Christensen, G., et al. (2012). Extreme sarcoplasmic reticulum volume loss and compensatory T-tubule remodeling after Serca2 knockout. Proc. Natl. Acad. Sci. U.S.A. 109, 3997–4001. doi: 10.1073/pnas.1120172109
Tellez, J. O., Mczewski, M., Yanni, J., Sutyagin, P., Mackiewicz, U., Atkinson, A., et al. (2011). Ageing-dependent remodelling of ion channel and Ca2+ clock genes underlying sino-atrial node pacemaking. Exp. Physiol. 96, 1163–1178. doi: 10.1113/expphysiol.2011.057752
Unverferth, D. V., Lee, S. W., and Wallick, E. T. (1988). Human myocardial adenosine triphosphatase activities in health and heart failure. Am. Heart J. 115, 139–146. doi: 10.1016/0002-8703(88)90529-7
Vinogradova, T. M., Brochet, D. X., Sirenko, S., Li, Y., Spurgeon, H., and Lakatta, E. G. (2010). Sarcoplasmic reticulum Ca2+ pumping kinetics regulates timing of local Ca2+ releases and spontaneous beating rate of rabbit sinoatrial node pacemaker cells. Circ. Res. 107, 767–775. doi: 10.1161/CIRCRESAHA.110.220517
Keywords: sinus node, atrioventricular node, Ca2+-clock, SERCA2, heart failure, telemetry, heart rate, mathematical model
Citation: Logantha SJRJ, Stokke MK, Atkinson AJ, Kharche SR, Parveen S, Saeed Y, Sjaastad I, Sejersted OM and Dobrzynski H (2016) Ca2+-Clock-Dependent Pacemaking in the Sinus Node Is Impaired in Mice with a Cardiac Specific Reduction in SERCA2 Abundance. Front. Physiol. 7:197. doi: 10.3389/fphys.2016.00197
Received: 27 January 2016; Accepted: 17 May 2016;
Published: 02 June 2016.
Edited by:
Olivier Bernus, Université de Bordeaux, FranceReviewed by:
Sandeep Pandit, University of Michigan, USAFabien Brette, Institut National de la Santé et de la Recherche Médicale, France
Copyright © 2016 Logantha, Stokke, Atkinson, Kharche, Parveen, Saeed, Sjaastad, Sejersted and Dobrzynski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Halina Dobrzynski, aGFsaW5hLmRvYnJ6eW5za2lAbWFuY2hlc3Rlci5hYy51aw==
†Joint first authors