 Luis Sobrevia1,2,3*
Luis Sobrevia1,2,3* Rocío Salsoso1,3
Rocío Salsoso1,3 Bárbara Fuenzalida1
Bárbara Fuenzalida1 Eric Barros1
Eric Barros1 Lilian Toledo1
Lilian Toledo1 Luis Silva1
Luis Silva1 Carolina Pizarro1
Carolina Pizarro1 Mario Subiabre1
Mario Subiabre1 Roberto Villalobos1
Roberto Villalobos1 Joaquín Araos1
Joaquín Araos1 Fernando Toledo4
Fernando Toledo4 Marcelo González5,6
Marcelo González5,6 Jaime Gutiérrez1,7Marcelo Farías1
Jaime Gutiérrez1,7Marcelo Farías1 Delia I. Chiarello1
Delia I. Chiarello1 Fabián Pardo1
Fabián Pardo1 Andrea Leiva1
Andrea Leiva1- 1Cellular and Molecular Physiology Laboratory, Division of Obstetrics and Gynecology, Faculty of Medicine, School of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2Faculty of Medicine and Biomedical Sciences, University of Queensland Centre for Clinical Research, University of Queensland, Herston, QLD, Australia
- 3Department of Physiology, Faculty of Pharmacy, Universidad de Sevilla, Seville, Spain
- 4Department of Basic Sciences, Faculty of Sciences, Universidad del Bío-Bío, Chillán, Chile
- 5Vascular Physiology Laboratory, Department of Physiology, Faculty of Biological Sciences, Universidad de Concepción, Concepción, Chile
- 6Group of Research and Innovation in Vascular Health (GRIVAS-Health), Chillán, Chile
- 7Cellular Signaling and Differentiation Laboratory, Health Sciences Faculty, Universidad San Sebastián, Santiago, Chile
Gestational diabetes mellitus (GDM) is a disease of the mother that associates with altered fetoplacental vascular function. GDM-associated maternal hyperglycaemia result in fetal hyperglycaemia, a condition that leads to fetal hyperinsulinemia and altered L-arginine transport and synthesis of nitric oxide, i.e., endothelial dysfunction. These alterations in the fetoplacental endothelial function are present in women with GDM that were under diet or insulin therapy. Since these women and their newborn show normal glycaemia at term, other factors or conditions could be altered and/or not resolved by restoring normal level of circulating D-glucose. GDM associates with metabolic disturbances, such as abnormal handling of the locally released vasodilator adenosine, and biosynthesis and metabolism of cholesterol lipoproteins, or metabolic diseases resulting in endoplasmic reticulum stress and altered angiogenesis. Insulin acts as a potent modulator of all these phenomena under normal conditions as reported in primary cultures of cells obtained from the human placenta; however, GDM and the role of insulin regarding these alterations in this disease are poorly understood. This review focuses on the potential link between insulin and endoplasmic reticulum stress, hypercholesterolemia, and angiogenesis in GDM in the human fetoplacental vasculature. Based in reports in primary culture placental endothelium we propose that insulin is a factor restoring endothelial function in GDM by reversing ERS, hypercholesterolaemia and angiogenesis to a physiological state involving insulin activation of insulin receptor isoforms and adenosine receptors and metabolism in the human placenta from GDM pregnancies.
Introduction
A large number of pregnant women are diagnosed with gestational diabetes mellitus (GDM), a disease that appears in pregnancy, courses with maternal hyperglycaemia and leads to fetal hyperglycaemia and hyperinsulinemia [American Diabetes Association (ADA), 2015]. These women are subjected to a calories-controlled diet with the final goal of reducing glycaemia to values as those in normal pregnancies. Interestingly, even when the mother and newborn from GDM under diet protocol show normal glycaemia at birth, metabolic alterations reducing the reactivity of the placenta and umbilical cord vessels (i.e., fetoplacental vasculature) are still seen (Sobrevia et al., 2015). GDM results in reduced fetoplacental vascular dilation in response to insulin or adenosine (an endogenous vasodilator nucleoside) via mechanisms including altered expression of adenosine receptors (ARs) and/or insulin receptors forms A (IR-A) and B (IR-B), L-arginine and adenosine membrane transport and transporters expression in the human fetoplacental macrovascular and microvascular endothelium. GDM results in higher synthesis of nitric oxide (NO) and expression of the endothelial NO synthase (eNOS), and L-arginine transport (the substrate for eNOS), a phenomenon attributed to ARs activation by adenosine in primary cultures of human umbilical vein endothelial cells (HUVECs) and human placental microvascular endothelial cells (hPMECs) (Westermeier et al., 2011, 2015a; Salomón et al., 2012). Insulin reverses this disease's associated alterations, requiring ARs activation (Guzmán-Gutiérrez et al., 2016) via selective activation of IR-A and/or IR-B, and ARs activation leading to IR-A-associated mitogenic or IR-B-associated metabolic phenotype in these cell types (Westermeier et al., 2015a,b).
GDM triggers a variety of stressor signals leading to abnormal function of intracellular structures, including the endoplasmic reticulum (ER). GDM increases the activity of molecules associated with ER stress (ERS) (Marciniak and Ron, 2006). Insulin signal is altered in ERS (Ron and Walter, 2007; Sáez et al., 2014) and GDM (Sáez et al., 2014), a condition ending in insulin resistance. Interestingly, ERS and GDM course with altered activity of several transcription factors, such as the pro-apoptotic transcription factor growth arrest and DNA damage 153 (GADD153) (also referred as C/EBP homologous protein 10 or CHOP). Even when is known that human CHOP (hCHOP) is activated by NO leading to reduced expression of SLC291A gene [for human equilibrative nucleoside transporter 1 (hENT1)] (Farías et al., 2010), and that hCHOP activity is modulated by insulin (Sáez et al., 2014), nothing is clear regarding a potential involvement of ARs and/or IRs in this phenomenon. On the other hand, pregnant women coursing with supraphysiological hypercholesterolemia show altered fetoplacental NO-dependent and L-arginine transport-dependent vascular reactivity when plasma level of total cholesterol (TCh) is >280 mg/dL (Leiva et al., 2015). However, the vascular effect of maternal dyslipidaemia, or whether ERS and changes in cell signaling and/or expression of ARs or IRs in these alterations is not yet reported. Since ERS and maternal dyslipidaemia modulate angiogenesis (Gutiérrez et al., 2016), and because GDM associates with placental endothelium and trophoblast release of pro-angiogenic factors, dysfunction of these cell types in ERS or maternal dyslipidaemia could result in accelerated angiogenesis.
Thus, in this review, we have emphasized the possibility that an abnormal metabolic state in pregnancy, as seen in GDM, leads to fetoplacental disturbances resulting in ERS, uncontrolled angiogenesis, or lipid metabolism. The involvement of insulin modulation of human fetoplacental vasculature function and its consequences in these phenomena are discussed.
Gestational Diabetes Mellitus
GDM is a disease that first appears or is identified during pregnancy [American Diabetes Association (ADA), 2015], associates with abnormal vascular function of the placenta (Colomiere et al., 2009; Haas, 2014), and leads to deleterious consequences to the fetus development and growth as well as to the health of the mother (König et al., 2014; Lappas, 2014). The incidence of this disease of pregnancy is ~7% worldwide [Ferrara et al., 2004; Dabelea et al., 2005; American Diabetes Association (ADA), 2015]. With the goal of reaching maternal glycaemia in a normal range, so to avoid deleterious consequences of hyperglycaemia in the growing fetus, patients diagnosed with GDM are subjected to controlled diet (plus a suggested routine of exercise) or treated with insulin [i.e., insulin therapy; Verier-Mine, 2010; American Diabetes Association (ADA), 2015; Sobrevia et al., 2015].
GDM causes an abnormal supply of nutrients (e.g., D-glucose, amino acids) to the fetus [Leach, 2011; American Diabetes Association (ADA), 2015; Sobrevia et al., 2015], a phenomenon that depends on the fetoplacental vascular tone and blood flow. Since the distal segment of the umbilical cord and the placenta lack of innervation (Fox and Khong, 1990; Marzioni et al., 2004), local regulation of the vascular tone results from the synthesis, release, and bioactivity of endothelium-derived vasodilators and vasoconstrictors (Pearson and Gordon, 1985; Olsson and Pearson, 1990). The endothelium of the human fetoplacental vasculature is a monolayer directly facing fetal blood and corresponds to the epithelium underlying the syncytiotrophoblast layer (Burton and Jauniaux, 2015). Thus, the endothelium is the first target for a variety of circulating molecules in the fetal blood. Equally, it is exposed to maternal blood molecules and/or their metabolites that cross or are released from the syncytiotrophoblast. Interestingly, the level of the endogenous nucleoside adenosine, a potent vasodilator, is increased in human umbilical whole blood (Maguire et al., 1998; Westermeier et al., 2011), or umbilical vein blood (Westermeier et al., 2015a), but not in umbilical arteries blood (Salomón et al., 2012), in GDM pregnancies where the mother was under diet compared with normal pregnancies. These findings were paralleled by reduced uptake of adenosine in HUVECs and hPMECs. Thus, an altered adenosine handling (e.g., uptake, release, metabolism) by the microvascular and macrovascular endothelium of the fetoplacental unit in GDM was proposed (Sobrevia et al., 2015; Westermeier et al., 2015b).
GDM also associates with increased expression of SLC7A1 gene coding for hCAT isoform 1 (hCAT-1) and higher L-arginine/NO signaling pathway activity in HUVECs (Guzmán-Gutiérrez et al., 2016). These alterations result from activation of the L-arginine transport and NO synthesis as a result of activation of ARs (i.e., ALANO signaling pathway) in this cell type from GDM. The extracellular level of adenosine is mainly, if not only, maintained by the activity of the Na+-independent hENT1 and hENT2 in the fetoplacental endothelium (Sobrevia et al., 2015). Altered expression and/or activity of these membrane transporters result in changes in extracellular adenosine concentration, thus altering its normal and broad modulatory actions on cell function (Fredholm et al., 2011; Fredholm, 2014; Verkhratsky and Burnstock, 2014; Burnstock, 2016).
Insulin and Adenosine Receptors in GDM
The insulin receptor splice variants IR-A and IR-B result from the absence or presence of a 12-amino acid segment (encoded by exon 11) at the C-terminal of the extracellular α-subunit (Westermeier et al., 2015b). IR-A and IR-B are differentially expressed, including the fetoplacental tissue, and signal via preferential mechanisms depending on their binding affinities for insulin, receptors internalization, receptors recycling time, and intracellular signaling (Westermeier et al., 2015b). It is reported that insulin via activation of IR-A and/or IR-B modulates the expression and activity of hCAT-1, eNOS, hENT1, and hENT2 in HUVECs and hPMECs (Table 1). These findings show that insulin modulates L-arginine and adenosine transport reversing the GDM-associated alterations in these mechanisms to values in human fetoplacental endothelium from normal pregnancies.
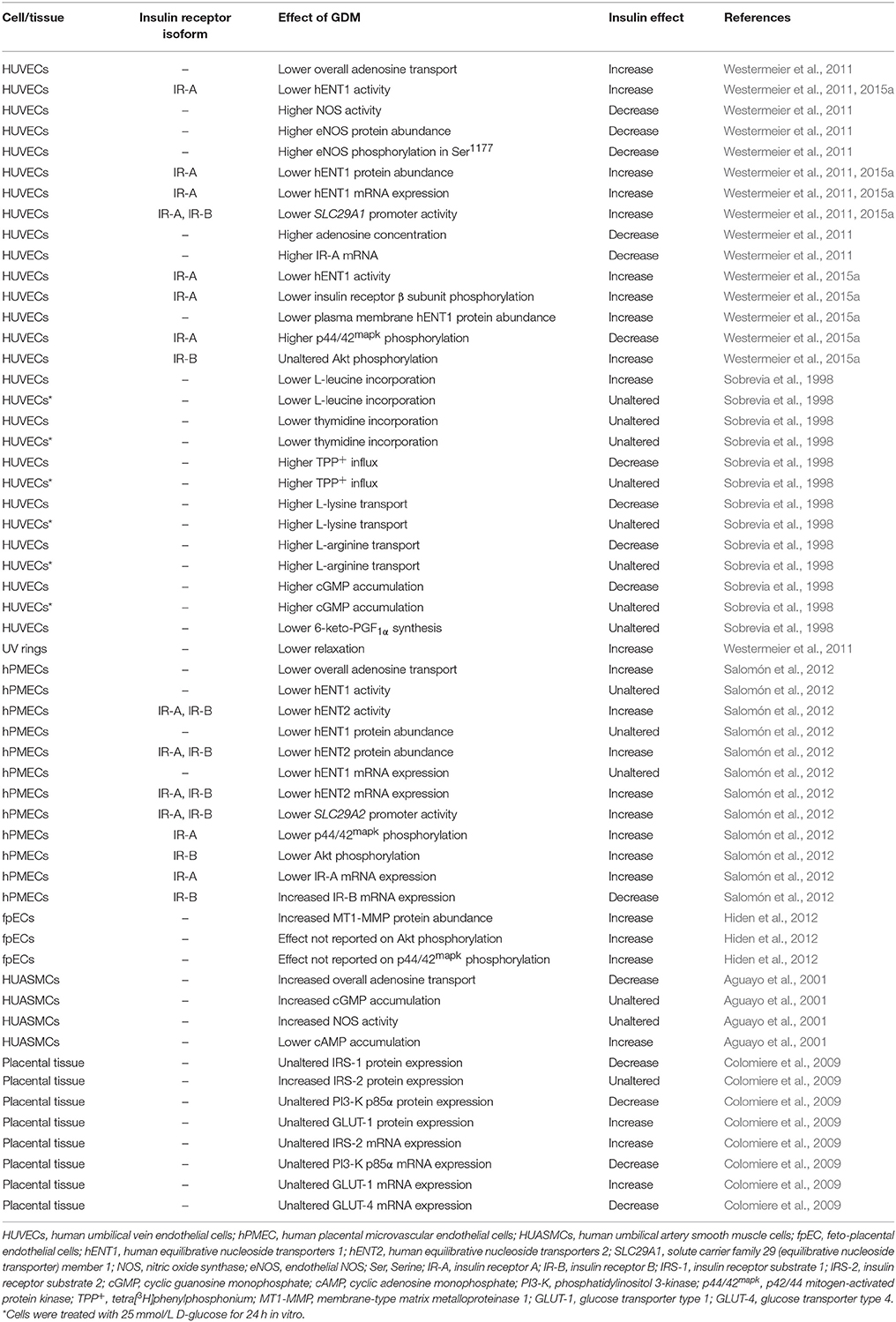
Table 1. Effect of insulin on human fetoplacental vasculature in GDM.
Adenosine causes relaxation of human umbilical vein rings (in vitro) from normal pregnancies requiring activation of endothelial ARs with a major contribution of A2A adenosine receptors (A2AAR) compared with A1 (A1AR), A2B (A2BAR), or A3 (A3AR) isoforms (Westermeier et al., 2011; Guzmán-Gutiérrez et al., 2016). ARs are expressed in HUVECs (Wyatt et al., 2002) and hPMECs (Escudero et al., 2008), of which A2AAR predominates. Despite the increase in ALANO signaling pathway in response to A2AAR activation by adenosine in HUVECs from normal or GDM pregnancies, characterization of ARs-associated cell signaling and a role for other than A2AAR in this phenomenon are still unknown (Fredholm, 2014; Verkhratsky and Burnstock, 2014; Sobrevia et al., 2015; Burnstock, 2016). We recently showed that A1AR expression and activation are required for insulin reversal of GDM-increased hCAT-1-mediated L-arginine transport and NO synthesis in HUVECs (Guzmán-Gutiérrez et al., 2016). However, nothing is reported addressing the possibility of a potential differential expression and/or cell signaling of ARs accounting for these effects in the fetoplacental endothelium from normal pregnancies, or in mothers with GDM [Verier-Mine, 2010; American Diabetes Association (ADA), 2015; Sobrevia et al., 2015].
Interestingly, an increase in the extracellular level of adenosine correlates with cardiovascular pathophysiological factors, including shear stress. Since one cellular mechanism explaining the increased level of adenosine and reduced hENT1-mediated transport in HUVECs from GDM is a higher activity of hCHOP transcription factor which is one key component of ERS response in this disease (Farías et al., 2010). Even more, GDM-associated increase in hCHOP activity is attenuated by insulin in HUVECs. Thus, ERS is an abnormal metabolic condition associated with GDM, but the role of insulin in this phenomenon is unclear.
Endoplasmic Reticulum Stress
The normal function of ER is essential for the synthesis and processing of secretory and membrane proteins, lipid biosynthesis, and calcium storage (Marciniak and Ron, 2006). The ER is highly sensitive to alterations in cellular environmental changes and acts as a quality control station allowing the transit of correctly folded proteins and retaining unfolded or misfolded proteins (Hetz et al., 2015). Thus, ER plays a key role in the general cellular response to nutrient overload or deprivation, the abnormal increase in the synthesis of secretory proteins, expression of mutant, or misfolded proteins and microbial infections, among others (Ron and Walter, 2007). These “stressor signals” disrupts ER homeostasis and accumulates unfolded proteins in the ER lumen, a phenomenon referred as ERS. In order to adapt ER function to this stress condition, the unfolded protein response (UPR) or ERS are activated (Marciniak and Ron, 2006; Zhang and Kaufman, 2006; Ron and Walter, 2007; Hetz et al., 2015).
An integrated ERS response involves transcriptional activation of multiple genes mediated by inositol-requiring enzyme 1 α (IRE1α) and activating transcription factor 6 (ATF6). It leads to a general decrease in protein translation and selective expression of specific mRNAs mediated by double-stranded RNA-dependent protein kinase (PKR)-like ER-associated kinase (PERK) (Marciniak and Ron, 2006). Thus, IRE1α, ATF6, and PERK are referred as ERS sensors. Interestingly, ERS response is also associated with activation of multiple transcription factors, including X-box binding protein-1 (XBP1) and activating transcription factor 4 (ATF4), regulating the expression of genes involved in the final adaptive effects of UPR. Under normal conditions, the UPR pathway functions as a physiological adaptive mechanism (Hetz et al., 2015). However, when a primary stimulus is too persistent or severe, the ERS response could lead to irreversible cell damage and programed cell death through hCHOP stimulation (Marciniak and Ron, 2006; Zhang and Kaufman, 2006; Ron and Walter, 2007). ERS response is thus critical for a normal cellular homeostasis, and plays relevant roles in the pathogenesis of multiple diseases such as GDM, DMT1, DMT2, obesity, inflammation, cardiovascular disorders, viral infections, neurodegeneration, and cancer (Marciniak and Ron, 2006; Zhang and Kaufman, 2006; Ron and Walter, 2007; Díaz-Villanueva et al., 2015; Salvadó et al., 2015).
ERS in GDM
Since the UPR is a general homeostatic mechanism for cellular defense, GDM-associated alterations could be different in maternal and fetal tissues. The multiple functional alterations described in human fetal endothelial cells from pregnancies with GDM include reduced expression and activity of hENT1 in HUVECs (Farías et al., 2010), likely due to ARs activation by adenosine (Burnstock, 2002, 2016; Fredholm, 2014). Interestingly, hCHOP, a key component of ERS response, act as an NO-dependent transcriptional repressive factor of SLC29A1 (for hENT1) expression in HUVECs from GDM pregnancy. An increase in the expression and activity (i.e., DNA binding) of hCHOP has been implicated in the apoptotic branch of UPR, especially when the stressor stimuli overcome the compensatory capacity of the ER in this phenomenon (Eizirik et al., 2008; Hotamisligil, 2010). Thus, GDM-associated alterations in adenosine transport in human fetal endothelial cells could be partially explained by activation of the ER-related hCHOP transcription factor. Indeed, increased expression of hCHOP in HUVECs from GDM pregnancies could be considered as an index of cellular stress. These findings are consistent with the detection of hCHOP induction in cells exposed to an elevated extracellular level of homocysteine (Outinen et al., 1999), suggesting its involvement in endothelial dysfunction caused by hyperhomocysteinemia in patients with diabetes mellitus (Austin et al., 2004; Ndrepepa et al., 2006; Sharma et al., 2006). Thus, expression of the ERS marker hCHOP in HUVECs from GDM pregnancies rise the possibility that UPR is active in this cell type and eventually in other maternal and fetal tissues in this pathology. To date, a key component of the ERS pathway referred as apoptosis signal-regulating kinase 1 (ASK1) is activated in mothers with diabetes mellitus, and plays a causal role in a defective neural tube formation (Wang et al., 2015). In addition, ERS response may also be involved in maternal diabetes-associated cardiac malformations, affecting the embryonic cardiogenesis period (Zhao, 2012).
Insulin and ERS in GDM
The ERS response associates with a development of insulin resistance in the context of obesity or DMT2 (Ozcan et al., 2004, 2006; Sáez et al., 2014). The c-Jun N-terminal kinase (JNK) is activated through an IRE-1α-dependent phosphorylation in response to ERS in endothelial cells from humans with diabetes mellitus and in animal models of diabetes (Eizirik et al., 2008; Hotamisligil, 2010; Figure 1). Activation of JNK leads to phosphorylation of serine307 on insulin receptor substrate 1 (IRS-1), thus inhibiting insulin signaling pathway, a condition that turns into a stage of insulin resistance due to defective downstream signaling, including reduced protein kinase B/Akt (Akt) activation and NO synthesis (Taniguchi et al., 2006; Hotamisligil, 2010). Additionally, ERS correlates with activation of an inflammatory response inducing interleukin 1α (IL-1α) and interleukin 1β (IL-1β) secretion in adipose tissue of pregnant women that are obese or with GDM (Liong and Lappas, 2015). Since IL-1β is a major contributor to the pathophysiology of obesity in pregnancy and GDM (Colomiere et al., 2010; Liong and Lappas, 2015), inhibition of ERS-induced IL-1β synthesis may be a potential therapeutic approach to improve pregnancy complications associated with maternal obesity and GDM, including altered insulin resistance. Thus, a growing body of evidence addresses that ERS may be activated in maternal and fetal tissues in GDM pregnancy. Since insulin signaling could be under modulation by key components of ERS pathway, it is possible that both, maternal and fetal insulin response may be reduced under conditions of ERS. Looking for eventual ERS alleviating interventions in pregnancy may contribute to the prevention of GDM-associated, ERS-related alterations of insulin biological effects.
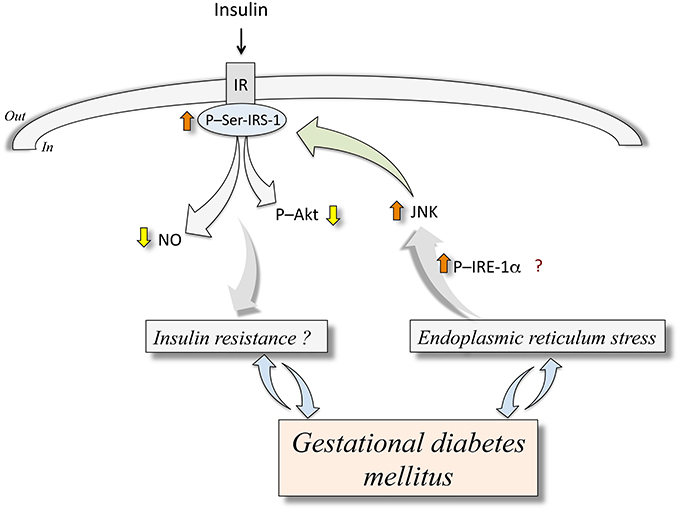
Figure 1. Endoplasmic reticulum stress and abnormal insulin signaling in human fetoplacental endothelium from gestational diabetes mellitus. Gestational diabetes mellitus is a disease that associates with endoplasmic reticulum stress (ERS). The latter is an abnormal metabolic condition that could (?) lead to increased (⇧) phosphorylation of inositol-requiring enzyme 1α (P-IRE-1α) resulting in higher c-Jun N-terminal kinase (JNK) activity. This phenomenon causes phosphorylation of insulin receptor substrate 1 at serine307 (P-Ser-IRS-1) ending in lower insulin receptor (IR)-associated cell signaling in response to insulin. This altered response to insulin results in reduced (⇩) synthesis of nitric oxide (NO) and phosphorylation of protein kinase B/Akt (P-Akt). These mechanisms are proposed to be potentially involved in insulin resistance (Insulin resistance?) in the human fetoplacental endothelium. At present, it is unclear whether GDM causes insulin resistance or ERS, or are these abnormal metabolic conditions that result in GDM clinical manifestations. Composed from information reported by Ozcan et al. (2004, 2006), Taniguchi et al. (2006), Eizirik et al. (2008), Hotamisligil (2010), Sáez et al. (2014), Westermeier et al. (2011, 2015a).
Placental Angiogenesis
One of the main steps in the placenta formation is the development of its highly structured and specialized net of blood vessels. Formation of placental blood vessels occurs with (i) vasculogenesis, which begins at the end of the third week of gestation and corresponds to the formation of the first vascular plexus from pluripotent progenitor cells which then differentiate into endothelial cells, and (ii) angiogenesis, which begins at the end of the fourth week of gestation and where the first vascular plexus are expanded and remodeled (Charnock-Jones et al., 2004; Gutiérrez et al., 2016). This process is finely tuned and regulated by different angiogenic factors including the vascular endothelial growth factor (VEGF), placental growth factor (PlGF), angiopoietins (ANG), fibroblast growth factor 2 (FGF2), and the insulin/insulin-like growth factors (INS/IGF) system (Burton et al., 2010; Burton and Jauniaux, 2015). Expression of these factors is highly regulated throughout gestation and is mainly attributed to trophoblast cells, Hofbauer cells, and smooth muscle cells, (Chen and Zheng, 2014). Since expression of angiogenic factors as well as the angiogenic process itself are under regulation by glycaemia, insulin, and hypoxia (Hadjipanayi and Schilling, 2013; Brocato et al., 2014; Chen and Zheng, 2014; Cvitic et al., 2014), vasculogenesis and angiogenesis processes at the fetoplacental vasculature are susceptible to alterations by a diabetic environment, such as in GDM or pregestational diabetes mellitus. Besides activation of the angiogenic factors, activation of ERS and UPR pathways (Paridaens et al., 2014) and dyslipidaemia (Oh et al., 2016), are also involved in physiological and pathological angiogenesis involving these molecules in the human placenta.
Angiogenesis in GDM
Placenta hypervascularization in women with DMT1, DMT2, or GDM, is reported (Cvitic et al., 2014; Huynh et al., 2015; Jarmuzek et al., 2015). DMT1 and DMT2 affect the entire process (vasculogenesis and angiogenesis; Jirkovská et al., 2002; Nelson et al., 2009; Jarmuzek et al., 2015) while GDM seems to impact the microvascular remodeling at angiogenesis (Jarmuzek et al., 2015). At 3rd trimester of pregnancy, the effect of DMT1, DMT2, and GDM on these phenomena is similar resulting in increased branching and surface area of villous capillaries (Teasdale, 1981; Jirkovská et al., 2002). Since GDM associates with developing longer umbilical cords compared with normal pregnancies (Georgiadis et al., 2014), it is suggested that placental hypervascularization in diabetes mellitus is mainly attributed to increased angiogenesis (Jirkovská et al., 2002; Leach, 2011; Figure 2). The later is partially explained by a placental hypoxia condition resulting from the fetal hyperglycaemia in diabetes mellitus. Fetal hyperglycaemia triggers fetal hyperinsulinemia, over-activating fetal metabolism leading to increased oxygen demand (Hytinantti et al., 2000; Taricco et al., 2009; Jarmuzek et al., 2015). Thus, it is likely that fetal hypoxia promotes the expression of angiogenic factors during the physiological placental angiogenesis at the 1st trimester of pregnancy when the oxygen level is reduced (Jauniaux et al., 2000). Since the level of FGF-2 is also regulated by hypoxia (Wang et al., 2009; Seo et al., 2013) and is increased in both the placenta and umbilical cord blood in diabetic pregnancies (Arany and Hill, 1998; Grissa et al., 2010), this growth factor emerges as a candidate to explain the hypervascularization in placentas from diabetes mellitus. Knowing that insulin is an angiogenic factor in endothelial cells (Liu et al., 2009), fetal hyperinsulinemia would have profound effects on placental and fetal vascular changes associated with maternal diabetes mellitus in pregnancy (Lassance et al., 2013). Interestingly, in another set of studies VEGF expression was shown to be lower in human placentas likely due to increased expression of the ERS-maker GRP78 (Aditiawarman, 2014). Thus, responses of ERS to a stressor will also result in an altered synthesis and/or release of proangiogenic factors in the human placenta vascular bed. This information is complemented by the proposed role of IRE1α, ATF6, and PERK as sensors of ERS (Marciniak and Ron, 2006; Zhang and Kaufman, 2006; Ron and Walter, 2007) or UPR (Ghosh et al., 2010; Hetz et al., 2015) constituting potential novel upstream regulatory pathways of angiogenesis via modulation of VEGF transcription in the human placenta (Iwawaki et al., 2009). Whether insulin modulates angiogenesis in the human placenta via changes in expression and/or function of ERS markers is unknown.
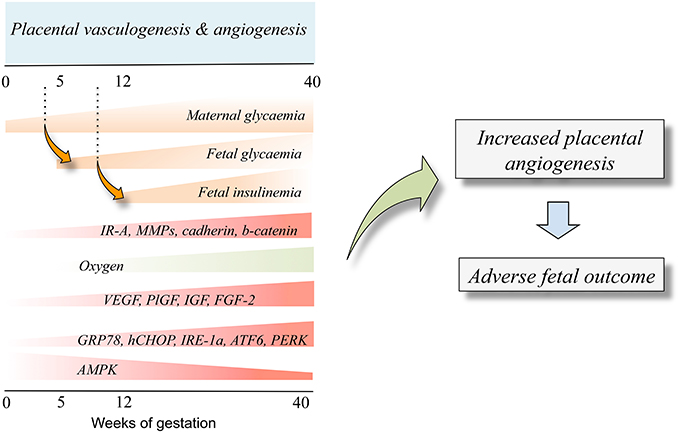
Figure 2. Fetal insulinemia and altered angiogenesis in fetoplacental endothelium from gestational diabetes mellitus. With the progression of pregnancy up to the 40th weeks of gestation, the maternal glycaemia increases, and could reach supraphysiological levels in pregnancies where the mother is diagnosed with gestational diabetes mellitus. The maternal hyperglycaemia results in increased fetal glycaemia from about the 5th week of gestation (dotted line), a condition resulting in a supraphysiological increase of fetal insulinemia from the 12th week of gestation. Increased fetal insulinemia results in altered placental vascular development and growth leading to angiogenesis alterations (Placental vasculogenesis and angiogenesis). Thus, an adverse fetal outcome is seen as a result of abnormal angiogenesis. Cell signaling mechanisms involved in this phenomenon include altered expression and/or activity of several molecules that are responsive to insulin (IR-A, MMPs, cadherin, b-catenin). Equally, a low oxygen level at the beginning of pregnancy increases the expression of proangiogenic growth factors (VEGF, PlGF, IGF, FGF-2) and increased (GRP78, CHOP, IRE-1α, ATF6, PERK) or reduced (AMPK) expression and/or activity. Composed from information reported by Babawale et al. (2000), Jirkovská et al. (2002), Easwaran et al. (2003), Baumüller et al. (2015), Westermeier et al. (2015b).
Insulin, Angiogenesis, and ERS in GDM
Insulin stimulates the formation of new blood vessels in vivo (Martínez-Jiménez et al., 2013), stimulates the formation of longer and branched blood vessels (Liu et al., 2009), and promotes microvascular endothelial cells migration (Liu et al., 2009). Thus, fetal hyperinsulinemia in GDM pregnancies could result in enhanced branching angiogenesis (Jirkovská et al., 2002) stimulating endothelial cells proliferation. This phenomenon is likely mediated by activation of IRs isoforms, which are expressed at villus branching spots in this cell type in the process of angiogenesis. At 1st trimester of pregnancy the IRs are mainly expressed by the syncytiotrophoblast, and in a lesser extent by cytotrophoblasts; however at term, IRs are mainly expressed in the fetoplacental vasculature (Desoye et al., 1994, 1997; Hiden et al., 2006). Thus, it is feasible that insulin regulates fetoplacental angiogenesis, but not, or in a minor degree, placental vasculogenesis at late pregnancy (Figure 2). Interestingly, mothers with GDM that were under insulin therapy show increased metalloproteases activity, probably mediated by activation of IR-A isoform. This is potentially due to higher levels of insulin-like growth factor 2 (IGF-2) as reported in placental endothelial cells from normal pregnancies (Hiden et al., 2012). Thus, IR-A is likely involved in proangiogenic pathways in GDM. On the other hand, placental histology in women with GDM that were under insulin therapy and show normal glycaemia at 3rd trimester of pregnancy, a decrease in the proangiogenic factors cadherin and b-catenin was reported (Babawale et al., 2000; Easwaran et al., 2003). Thus, GDM associates could course with impaired placental barrier leading to angiogenesis. However, the latter is contrary to the reported increase in expression of these molecules in these patients (Baumüller et al., 2015), highlighting a potential stimulatory effect of insulin involving these adhesion molecules to lead angiogenesis in the placenta (Table 2).
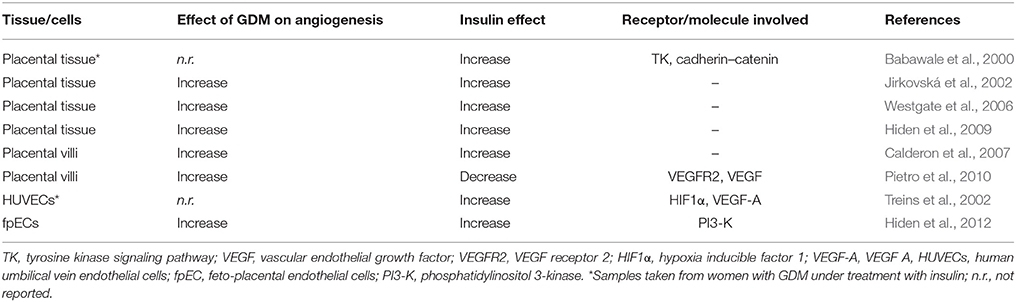
Table 2. Effect of insulin on angiogenesis in the human fetoplacental vasculature in GDM.
It is interesting to notice that the metformin, an insulin sensitizer currently used to treat DMT2, is also an activator of the AMP-activated protein kinase (AMPK), which was shown to inhibit ERS restoring endothelial cell dysfunction in high fat diet-induced obese mice (Cheang et al., 2014). Since AMPK is a molecule whose activation may result in reducing ERS-markers activation (particularly PERK), and its expression is low in a mice model of GDM (Yao et al., 2015), it is feasible that it is involved in the GDM-associated increase in placental angiogenesis. Interestingly, AMPK activity was also reduced in skeletal muscle in obese pregnant women with GDM (Boyle et al., 2014), complementing the observations regarding AMPK as a potential ERS-marker in mice GDM. Other studies have recently shown that atherogenesis is also a process that involves ERS-factors activation (GRP78 and CHOP) and endothelial dysfunction in rabbits (Kruzliak et al., 2015). In addition, treatment of HUVECs with low-density lipoprotein (LDL) activated UPR and interleukins expression (Gora et al., 2010). Certainly, further research is necessary in order to understand whether insulin treatment during pregnancy in women with GDM or coursing without or with supraphysiological hypercholesterolaemia (Leiva et al., 2015) leads to a beneficial or detrimental result on overall angiogenic mechanisms involving or not ERS and/or UPS pathways in the human fetoplacental vasculature.
Maternal Dyslipidaemia
Dyslipidaemia is defined as the elevated blood level of triglycerides (hypertriglyceridemia) and TCh (hypercholesterolaemia) including increased LDL and reduced high-density lipoprotein (HDL) levels [National Cholesterol Education Program (NCEP), 2002]. This pathological condition is recognized as the main risk factor for the development of cardiovascular disease [National Cholesterol Education Program (NCEP), 2002; Arsenault et al., 2011]. GDM also courses with maternal dyslipidaemia affecting fetal development and growth (Desoye and Hauguel-de Mouzon, 2007; Sanchez-Vera et al., 2007; Marseille-Tremblay et al., 2008; Schaefer-Graf et al., 2008). Indeed, hypercholesterolaemia was shown to contribute to endothelial dysfunction in the fetal vasculature in this disease (Reece, 2010; Sreckovic et al., 2014). Interestingly, GDM-associated increase in the maternal plasma lipids may result in abnormal transport of these molecules across the placenta into the developing fetus, a phenomenon likely regulated by insulin (Herrera and Desoye, 2015). Thus, alterations of a maternal lipids profile in GDM could lead to alterations in the fetal circulating level of lipids or to a defective composition of lipid macromolecules making HDL or other lipids less functional in the fetus (Leiva et al., 2015).
Dyslipidaemia in GDM
GDM courses with increased maternal TCh and triglycerides altering the expression and function of proteins involved in triglycerides and cholesterol homeostasis (Marseille-Tremblay et al., 2008; Radaelli et al., 2009; Herrera and Ortega-Senovilla, 2010; Herrera and Desoye, 2015). These changes include increased expression of genes related to lipid transport and metabolism such as the fatty acyl-CoA ligases (FACLs), which catalyse conversion of fatty acids into fatty acyl-CoA esters required for the synthesis of triglycerides, increased cholesterol and membrane phospholipids, higher expression and activity of placental fatty acid binding proteins (FABPs), endothelial and lipoprotein lipases that favor the breakdown of maternal triglycerides into fatty acids (Radaelli et al., 2009; Figure 3). Increased FABPs found in GDM pregnancies leads to binding fatty acids from the maternal circulation to export these to the fetal circulation. FACLs could favor the synthesis of triglycerides in the fetal circulation, and endothelial lipases and lipoprotein lipases could increase the breakdown of maternal triglycerides favoring the uptake of the fatty acids by the trophoblast. Additionally, placental expression of fatty acid synthase (FAS) is increased in placentas from GDM (Marseille-Tremblay et al., 2008), suggesting that lipid metabolism in the placenta is altered by this pathological condition. Interestingly, in placental tissue and trophoblast from GDM a higher level of lipid droplets has been reported, suggesting that lipid content is higher in this pathological condition compared with normal pregnancies (Elchalal et al., 2005; Scifres et al., 2011).
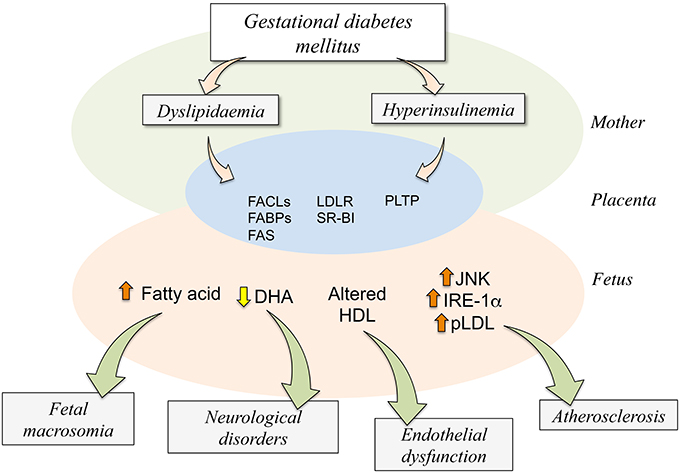
Figure 3. Potential consequences of dyslipidaemia and hyperinsulinemia on the human fetoplacental unit from gestational diabetes mellitus. Gestational diabetes mellitus (GDM) results in maternal (Mother) metabolic alterations leading to dyslipidaemia and hyperinsulinemia. These two abnormal metabolic conditions are associated with higher expression and activity of several molecules involved in the placental transport (FABPs,) and metabolism (FACLs, FAS, PLTP) of lipids or its receptors (LDLR, SR-BI) in the human trophoblast (Placenta). These changes result in altered transplacental transport of several signaling molecules via the trophoblast barrier, ending in increased (⇧) fatty acid, reduced (⇩) docosahexaenoic acid (DHA), or altered composition or function of high-density lipoprotein (HDL) in the fetoplacental circulation (Fetus). Since these changes in the capacity of transport by the placenta increased levels of endoplasmic reticulum stress (JNK, IRE-1α) and atherosclerotic [phospholipolyzed LDL (pLDL)] markers are detected in the fetal endothelium. Adverse fetal outcome results from alterations in the fetal circulating or tissue levels of these molecules in GDM compared with normal pregnancies. Increased level of fatty acids regards with a higher incidence of fetal macrosomia while a decrease in the fetal plasma level of DHA associates with increased number of neurological disorders. Additionally, less functional HDL could potentially result in endothelial dysfunction in the newborn, and atherosclerosis could result from increased ERS markers. Composed of information reported by Ethier-Chiasson et al. (2007), Marseille-Tremblay et al. (2008), Herrera and Ortega-Senovilla (2010), Scifres et al. (2011), Olmos et al. (2012), Pagán et al. (2013), Araújo et al. (2013), Sreckovic et al. (2013), Herrera and Desoye (2015).
GDM effect on lipoprotein receptors expression has also been reported. Whereas, in normal pregnancies maternal hypercholesterolaemia associates with lower expression of LDL receptor (LDLR) in homogenized placenta (Ethier-Chiasson et al., 2007; Desoye et al., 2011), expression of the HDL scavenger receptor class B type I (SR-BI) and LDLR is increased in GDM compared with normal pregnancies (Dubé et al., 2013). Thus, GDM affects maternal and neonatal lipid profiles perhaps predisposing the fetus to future metabolic diseases (Dubé et al., 2013). Another protein involved in the lipoprotein metabolism that is also modified by GDM in placental cells is the phospholipid transfer protein (PLTP), which is involved in the metabolism of fetal HDL and directly related with the HDL remodeling leading to a larger HDL molecule (Tzotzas et al., 2009). PLTP is expressed in endothelial cells of the placental vasculature (Marceau et al., 2005; Scholler et al., 2012a). GDM associates with upregulation of PLTP in the placental endothelium (Scholler et al., 2012b), a phenomenon due to the hyperinsulinemia and hyperglycaemia. PLTP increased expression could also be a phenomenon associated with the increased concentration of HDL described in newborns from GDM pregnancies (Merzouk et al., 2000; Scholler et al., 2012a,b; Sreckovic et al., 2014).
Insulin, Dyslipidaemia, and ERS in GDM
In normal and GDM pregnancies transfer of lipids from the maternal to the fetal blood across the placenta is a process highly regulated by the level of insulin (Table 3). Interestingly, incubation of trophoblast cells from normal pregnancies with insulin and fatty acids (i.e., concomitant conditions in GDM) increase lipid droplets formation, suggesting that trophoblast is involved in packaging lipids. The mechanisms involved in this phenomenon include insulin-stimulated overexpression of adipophilin (a protein involved in fatty acid uptake and storage in adipocytes; Elchalal et al., 2005). However, human trophoblast from GDM pregnancies shows higher expression of FABPs isoform 4 (FABP4), which was suggested as likely responsible for the associated increase in placenta lipid droplets (Scifres et al., 2011). However, increased FABP4 expression and lipid droplets formation are not regulated by insulin in these cells. Thus, insulin-modulated mechanisms involved in lipid droplets formation are likely to be different in normal compared with GDM pregnancies. Other findings show that GDM-associated reduction in the mother-to-placenta transfer of the fatty acid docosahexaenoic acid (DHA) is worsened in mothers with GDM under insulin therapy (Pagán et al., 2013; Larqué et al., 2014; Sobrevia et al., 2015). Interestingly, DHA uptake is increased by insulin in human trophoblast from normal pregnancies (Araújo et al., 2013). Thus, a potential increase in DHA uptake is unlikely in trophoblast from GDM pregnancies since plasma insulin in the fetoplacental circulation is higher in GDM compared with normal pregnancies (~75 vs. ~40 pmol/L; Westermeier et al., 2011, 2015a; Salomón et al., 2012; Guzmán-Gutiérrez et al., 2016). HDL metabolism at the fetal circulation is also altered in GDM pregnancies mainly due to upregulation of PLTP expression and activity (Scholler et al., 2012a; Sreckovic et al., 2014). Since insulin increases this protein expression in human placental endothelium, it is feasible that this hormone contributes to the synthesis of a larger HDL molecule as seen in GDM as well as with the increase in maternal-to-fetal cholesterol transfer (Scholler et al., 2012a,b). Worryingly, increased fatty acids or decreased DHA in the fetal circulation under maternal dyslipidaemia and hyperinsulinemia associates with fetal macrosomia (Herrera and Desoye, 2015) and neurological disorders (Araújo et al., 2013; Larqué et al., 2014; Figure 3).
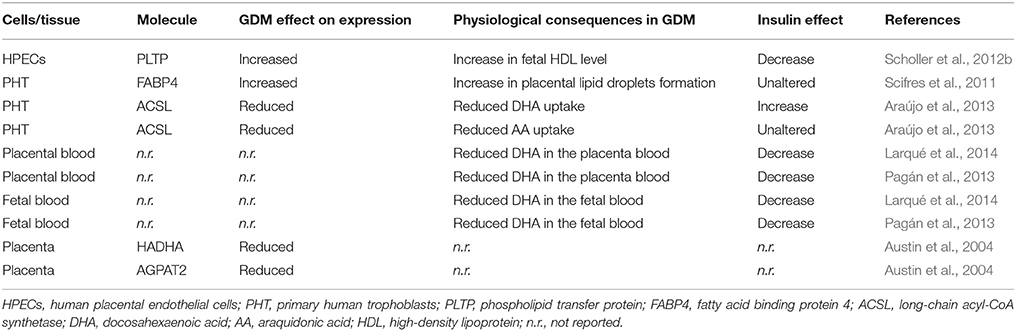
Table 3. Effect of insulin on the expression of molecules involved in lipids metabolism in GDM.
Modified lipoproteins profile has also been associated with ERS pathway as previously reviewed (Lenna et al., 2014). In brief, it has been suggested that oxidized LDL (oxLDL) may cause atherosclerosis requiring JNK and IRE-1α activation, thus involving ERS and UPR pathways in this phenomenon (Sanson et al., 2009). In addition, the pro-inflammatory phospholipolyzed low-density lipoprotein, which is increased in atherosclerotic lesions, activated UPR and interleukins expression in the treatment of HUVECs (Gora et al., 2010). Thus, fetoplacental endothelial cells are prone to activate ERS, and perhaps URP pathways in maternal dyslipidaemia. However, nothing is reported on the potential effects of insulin and the involvement of IRs isoforms in this phenomenon.
Concluding Comment
Altered vascular function in GDM pregnancies is a critical condition leading to severe dysfunction of the human placenta and altered delivery of nutrients and signaling molecules from mother-to-fetus and vice-versa (Desoye et al., 2011; Leach, 2011; Herrera and Desoye, 2015; Sobrevia et al., 2015). Several mechanisms leading to abnormal function of the human placenta regards with metabolic alterations of this organ, including ERS (Lenna et al., 2014) and metabolism of lipids (Leiva et al., 2015), as well as metabolic-derived structural modulation of the placental vascular bed, such as angiogenesis and vasculogenesis (Gutiérrez et al., 2016; Figure 4). Interestingly, GDM leads to a state where cell signaling mechanisms associated with insulin biological effects in cells from the fetoplacental vasculature, and perhaps in other vascular beds, is altered leading to a potential state of insulin resistance (Colomiere et al., 2009; Westermeier et al., 2015b). One of these metabolic conditions is ERS where key molecules (e.g., ISR-1, JNK, IRE-1α, and potentially AMPK) are apparently involved. Abnormal insulin signaling in the human fetoplacental endothelium results in a lower NO bioavailability and Akt activation, thus reducing fetoplacental vascular reactivity in vitro. These phenomena could explain the abnormal or even lack of regulation of a vectorial mother-to-fetus transplacental transport of nutrients, which could result in altered fetal growth and development, with subsequent consequences at birth and/or adulthood.
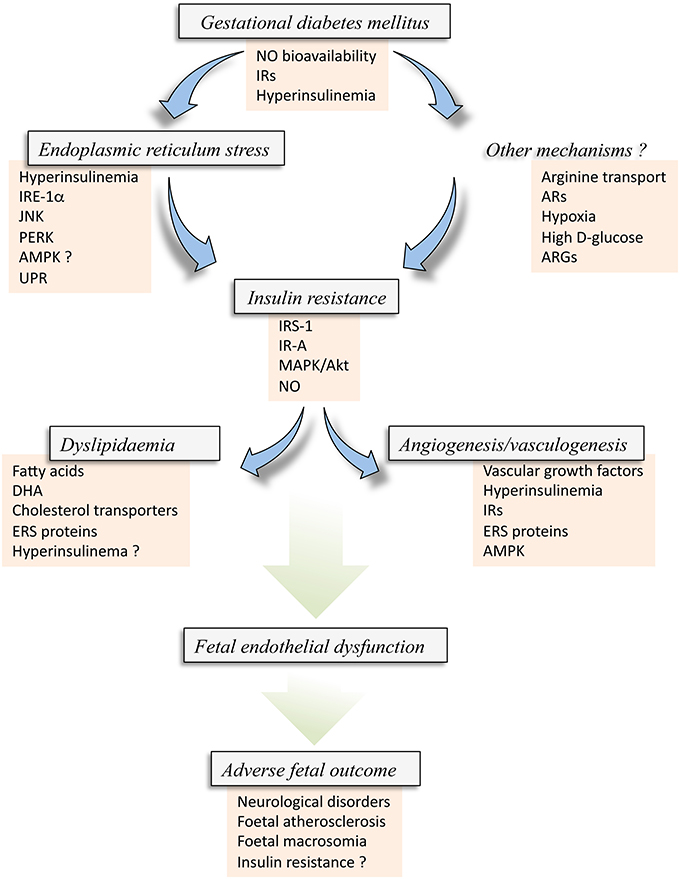
Figure 4. Insulin resistance in the fetus from gestational diabetes mellitus. Gestational diabetes mellitus (GDM) is a disease coursing with fetal hyperinsulinemia and associated with metabolic alterations that result in reduced bioavailability of nitric oxide (NO) and altered expression of insulin receptors (IRs). These alterations, and perhaps hyperinsulinemia itself, may result in abnormal expression and activity of several molecules associated with endoplasmic reticulum stress (ERS) [IRE-1α, JNK, PERK, and likely AMPK (AMPK?)], which could also lead to the activation of the unfolded protein response (UPR) pathway. ERS via this set of alterations could result in Insulin resistance in the fetus/newborn. Insulin resistance could alternatively be caused by Other mechanisms, including membrane transport of L-arginine, the substrate for NO synthesis, adenosine receptors (ARs) expression and/or activation, hypoxia or high extracellular concentration of D-glucose, and arginases (ARGs) activity, a metabolic pathway that consume L-arginine in endothelial cells. All these factors could result in increased inhibitor phosphorylation of IRS-1, perhaps involving the subtype A of IRs (IR-A) with a deficient signaling pathway mediated by p44/42mapk (MAPK) and protein kinase B/Akt. These phenomena lead to reduced synthesis and/or bioavailability of NO in the endothelial cells from the human placenta. Since insulin signaling is crucial maintaining a normal metabolism of lipids and angiogenesis and vasculogenesis in the human placenta from normal pregnancies, GDM-associated fetal insulin resistance could result in altered mother-to-fetus transplacental transfer due to altered expression and activity of cholesterol transporters, and metabolism of lipids leading to accumulation of fatty acids, docosahexaenoic acid (DHA), or cholesterol. A generalized metabolic disturbance referred as Dyslipidaemia. The latter is also associated with increased expression of ERS markers, and could also be due to hyperinsulinemia (Hyperinsulinemia ?). Additionally, insulin resistance results in a lack of modulation of physiological Angiogenesis and vasculogenesis in pregnancy where and increase in vascular growth factors, hyperinsulinemia, IRs and ERS molecules, including AMPK activity, are potentially involved. All these phenomena, i.e., GDM, ERS, angiogenesis and dyslipidaemia, develop with altered expression and activity of common molecules due to the state of insulin resistance in the fetoplacental vasculature. The final result is an abnormal function of the fetal endothelium (Fetal endothelial dysfunction) that ends with Adverse fetal outcome characterized by increased risk of fetal/newborn atherosclerosis, macrosomia, neurological disorders, and insulin resistance.
Reduced NO synthesis could also be a condition leading to abnormal angiogenesis (Gutiérrez et al., 2016). Interestingly, fetal hyperinsulinemia in GDM pregnancies results to be a key factor leading to an abnormal fetal outcome, including macrosomia (Olmos et al., 2012; Leiva et al., 2015) and endothelial dysfunction (Sobrevia et al., 2015). The possibility of a reduced vascular reactivity to insulin in GDM pregnancies is likely and it is a phenomenon that could explain the diminished response of the fetoplacental vascular endothelium to this hormone. Even when women coursing with GDM pregnancies that do not reach normal glycaemia by diet/exercise are passed into insulin therapy [Verier-Mine, 2010; American Diabetes Association (ADA), 2015; Sobrevia et al., 2015], it is unknown whether treatment with insulin in this group of women, which in fact normalizes their glycaemia, will result in normalization of the microvascular and macrovascular fetoplacental endothelial function.
In summary, we propose insulin as a key factor playing a modulatory role in GDM-associated altered angiogenesis, ERS, and metabolism of lipids in the human fetoplacental vascular bed. Since GDM courses with fetoplacental insulin resistance state at birth, the potential beneficial effect of this hormone on these phenomena is restricted. The possibility that clinical management of insulin sensitivity is considered a therapeutic target in the treatment of mothers with this disease could result in reversing along with insulin resistance, the GDM-associated alterations in ERS, angiogenesis and lipids metabolism. However, we emphasize to take with caution the broad spectrum of results reported in primary cell cultures, cell lines, or experimental models, as summarized in this review, so not to extrapolate the findings to what is happening in the mother and their newborn in GDM. Certainly, a better understanding of the mechanisms behind these alterations caused by GDM, including pregnant women with this disease but treated with diet/exercise or under insulin therapy, and the potential effect of insulin in this phenomenon, is crucial in the aim of preventing adverse fetal outcome from this disease of pregnancy.
Author Contributions
Conception and designed of the manuscript: LS, AL, MF, JG, IC, FP. Acquisition of data/information: RS, BF, EB, CP, LT, FT, MG, RV, MS, JA, LSi, IC. Analysis of data/information: LS, AL, MF, JG, FP, FT, RS, EB, BF. Interpretation of data/information: LS, AL, MF, JG, FP, LT, RS, MG, BF, EB. Compilation of tables: RS, BF, LT, CP, LS, AL, FP. The design of figures: LS, AL, JG, FP.
Funding
This work was supported by Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT 1150377, 1150344, 1121145, 11150083), VRID-Asociativo project (213.A84.014-1.0), Universidad de Concepción, and Dirección de Investigación, Universidad San Sebastián, Chile. RS, LS, RV, and MS hold a Comisión Nacional de Investigación en Ciencia y Tecnología (CONICYT) Chile–Ph.D. fellowship. RS, BF, LS, and EB hold Faculty of Medicine, PUC–Ph.D. fellowships. RV and MS hold Vicerectorate of Research, PUC-Ph.D. fellowships.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors thank Mrs Amparo Pacheco and Mrs Ninoska Muñoz from CMPL, Pontificia Universidad Católica de Chile (PUC), for excellent technical and secretarial assistance, respectively.
References
Aditiawarman (2014). The role of albumin and endoplasmic reticulum in pathogenesis Preeclampsia. Changes of GRP78 and placental VEGF in preeclampsia. Pregnancy Hypertens. 4, 247. doi: 10.1016/j.preghy.2014.04.022
Aguayo, C., Flores, C., Parodi, J., Rojas, R., Mann, G. E., Pearson, J. D., et al. (2001). Modulation of adenosine transport by insulin in human umbilical artery smooth muscle cells from normal or gestational diabetic pregnancies. J. Physiol. (Lond.) 534, 243–254. doi: 10.1111/j.1469-7793.2001.00243.x
American Diabetes Association (ADA) (2015). Classification and diagnosis of diabetes. Diabetes Care 38, S8–S16. doi: 10.2337/dc15-S005
Arany, E., and Hill, D. J. (1998). Fibroblast growth factor-2 and fibroblast growth factor receptor-1 mRNA expression and peptide localization in placentae from normal and diabetic pregnancies. Placenta 19, 133–142. doi: 10.1016/S0143-4004(98)90001-7
Araújo, J. R., Correia-Branco, A., Ramalho, C., Keating, E., and Martel, F. (2013). Gestational diabetes mellitus decreases placental uptake of long-chain polyunsaturated fatty acids: involvement of long-chain acyl-CoA synthetase. J. Nutr. Biochem. 24, 1741–1750. doi: 10.1016/j.jnutbio.2013.03.003
Arsenault, B. J., Barter, P., DeMicco, D. A., Bao, W., Preston, G. M., LaRosa, J. C., et al. (2011). Prediction of cardiovascular events in statin-treated stable coronary patients by lipid and nonlipid biomarkers. J. Am. Coll. Cardiol. 57, 63–69. doi: 10.1016/j.jacc.2010.06.052
Austin, R. C., Lentz, S. R., and Werstuck, G. H. (2004). Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 1, S56–S64. doi: 10.1038/sj.cdd.4401451
Babawale, M. O., Lovat, S., Mayhew, T. M., Lammiman, M. J., James, D. K., and Leach, L. (2000). Effects of gestational diabetes on junctional adhesion molecules in human term placental vasculature. Diabetologia 43, 1185–1196. doi: 10.1007/s001250051511
Baumüller, S., Lehnen, H., Schmitz, J., Fimmers, R., and Müller, A. M. (2015). The impact of insulin treatment on the expression of vascular endothelial cadherin and Beta-catenin in human fetoplacental vessels. Pediatr. Dev. Pathol. 18, 17–23. doi: 10.2350/13-11-1400-OA.1
Boyle, K. E., Hwang, H., Janssen, R. C., DeVente, J. M., Barbour, L. A., Hernandez, T. L., et al. (2014). Gestational diabetes is characterized by reduced mitochondrial protein expression and altered calcium signaling proteins in skeletal muscle. PLoS ONE 9:e106872. doi: 10.1371/journal.pone.0106872
Brocato, J., Chervona, Y., and Costa, M. (2014). Molecular responses to hypoxia-inducible factor 1 alpha and beyond. Mol. Pharmacol. 85, 651–657. doi: 10.1124/mol.113.089623
Burnstock, G. (2002). Purinergic signaling and vascular cell proliferation and death. Arterioscler. Thromb. Vasc. Biol. 22, 364–373. doi: 10.1161/hq0302.105360
Burnstock, G. (2016). Purinergic signalling and endothelium. Curr. Vasc. Pharmacol. 14, 130–145. doi: 10.2174/1570161114666151202204948
Burton, G. J., and Jauniaux, E. (2015). What is the placenta? Am. J. Obstet. Gynecol. 213, S6.e1, S6–S8. doi: 10.1016/j.ajog.2015.07.050
Burton, G. J., Jauniaux, E., and Charnock-Jones, D. S. (2010). The influence of the intrauterine environment on human placental development. Int. J. Dev. Biol. 54, 303–312. doi: 10.1387/ijdb.082764gb
Calderon, I. M., Damasceno, D. C., Amorin, R. L., Costa, R. A., Brasil, M. A., and Rudge, M. V. (2007). Morphometric study of placental villi and vessels in women with mild hyperglycemia or gestational or overt diabetes. Diabetes Res. Clin. Pract. 78, 65–71. doi: 10.1016/j.diabres.2007.01.023
Charnock-Jones, D. S., Kaufmann, P., and Mayhew, T. M. (2004). Aspects of human fetoplacental vasculogenesis and angiogenesis. I. Molecular regulation. Placenta 25, 103–113. doi: 10.1016/j.placenta.2003.10.004
Cheang, W. S., Tian, X. Y., Wong, W. T., Lau, C. W., Lee, S. S., Chen, Z. Y., et al. (2014). Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5′ adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor δ pathway. Arterioscler. Thromb. Vasc. Biol. 34, 830–836. doi: 10.1161/ATVBAHA.113.301938
Chen, D. B., and Zheng, J. (2014). Regulation of placental angiogenesis. Microcirculation 21, 15–25. doi: 10.1111/micc.12093
Colomiere, M., Permezel, M., Riley, C., Desoye, G., and Lappas, M. (2009). Defective insulin signaling in placenta from pregnancies complicated by gestational diabetes mellitus. Eur. J. Endocrinol. 160, 567–578. doi: 10.1530/EJE-09-0031
Colomiere, M., Permezel, M., and Lappas, M. (2010). Diabetes and obesity during pregnancy alter insulin signalling and glucose transporter expression in maternal skeletal muscle and subcutaneous adipose tissue. J. Mol. Endocrinol. 44, 213–223. doi: 10.1677/JME-09-0091
Cvitic, S., Desoye, G., and Hiden, U. (2014). Glucose, insulin, and oxygen interplay in placental hypervascularisation in diabetes mellitus. Biomed. Res. Int. 2014, 145846. doi: 10.1155/2014/145846
Dabelea, D., Snell-Bergeon, J. K., Hartsfield, C. L., Bischoff, K. J., Hamman, R. F., McDuffie, R. S., et al. (2005). Increasing prevalence of gestational diabetes mellitus over time and by birth cohort: Kaiser Permanente Colorado GDM Screening Program. Diabetes Care 28, 579–584. doi: 10.2337/diacare.28.3.579
Desoye, G., Gauster, M., and Wadsack, C. (2011). Placental transport in pregnancy pathologies. Am. J. Clin. Nutr. 94, S1896–S1902. doi: 10.3945/ajcn.110.000851
Desoye, G., Hartmann, M., Blaschitz, A., Dohr, G., Hahn, T., Kohnen, G., et al. (1994). Insulin receptors in syncytiotrophoblast and fetal endothelium of human placenta. Immunohistochemical evidence for developmental changes in distribution pattern. Histochemistry 101, 277–285. doi: 10.1007/BF00315915
Desoye, G., Hartmann, M., Jones, C. J., Wolf, H. J., Kohnen, G., Kosanke, G., et al. (1997). Location of insulin receptors in the placenta and its progenitor tissues. Microsc. Res. Tech. 38, 63–65.
Desoye, G., and Hauguel-de Mouzon, S. (2007). The human placenta in gestational diabetes mellitus. The insulin and cytokine network. Diabetes Care 30, S120–S126. doi: 10.2337/dc07-s203
Díaz-Villanueva, J. F., Díaz-Molina, R., and García-González, V. (2015). Protein folding and mechanisms of proteostasis. Int. J. Mol. Sci. 16, 17193–17230. doi: 10.3390/ijms160817193
Dubé, E., Ethier-Chiasson, M., and Lafond, J. (2013). Modulation of cholesterol transport by insulin-treated gestational diabetes mellitus in human full-term placenta. Biol. Reprod. 16, 1–10. doi: 10.1095/biolreprod.112.105619
Easwaran, V., Lee, S. H., Inge, L., Guo, L., Goldbeck, C., Garrett, E., et al. (2003). β-Catenin regulates vascular endothelial growth factor expression in colon cancer. Cancer Res. 63, 3145–3153.
Eizirik, D.L., Cardozo, A. K., and Cnop, M. (2008). The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 29, 42–61. doi: 10.1210/er.2007-0015
Elchalal, U., Schaiff, W. T., Smith, S. D., Rimon, E., Bildirici, I., Nelson, D. M., et al. (2005). Insulin and fatty acids regulate the expression of the fat droplet-associated protein adipophilin in primary human trophoblasts. Am. J. Obstet. Gynecol. 193, 1716–1723. doi: 10.1016/j.ajog.2005.04.012
Escudero, C., Casanello, P., and Sobrevia, L. (2008). Human equilibrative nucleoside transporters 1 and 2 may be differentially modulated by A2B adenosine receptors in placenta microvascular endothelial cells from pre-eclampsia. Placenta 29, 816–825. doi: 10.1016/j.placenta.2008.06.014
Ethier-Chiasson, M., Duchesne, A., Forest, J. C., Giguère, Y., Masse, A., Mounier, C., et al. (2007). Influence of maternal lipid profile on placental protein expression of LDLr and SR-BI. Biochem. Biophys. Res. Commun. 359, 8–14. doi: 10.1016/j.bbrc.2007.05.002
Farías, M., Puebla, C., Westermeier, F., Jo, M. J., Pastor-Anglada, M., Casanello, P., et al. (2010). Nitric oxide reduces SLC29A1 promoter activity and adenosine transport involving transcription factor complex hCHOP-C/EBPα in human umbilical vein endothelial cells from gestational diabetes. Cardiovasc. Res. 86, 45–54. doi: 10.1093/cvr/cvp410
Ferrara, A., Kahn, H. S., Quesenberry, C. P., Riley, C., and Hedderson, M. M. (2004). An increase in the incidence of gestational diabetes mellitus: Northern California, 1991-2000. Obstet. Gynecol. 103, 526–533. doi: 10.1097/01.AOG.0000113623.18286.20
Fox, S. B., and Khong, T. Y. (1990). Lack of innervation of human umbilical cord. An immunohistological and histochemical study. Placenta 11, 59–62. doi: 10.1016/S0143-4004(05)80443-6
Fredholm, B. B. (2014). Adenosine–a physiological or pathophysiological agent? J. Mol. Med. 92, 201–206. doi: 10.1007/s00109-013-1101-6
Fredholm, B. B., IJzerman, A. P., Jacobson, K. A., Linden, J., and Müller, C. E. (2011). International union of basic and clinical pharmacology. Nomenclature and classification of adenosine receptors–an update. Pharmacol. Rev. 63, 1–34. doi: 10.1124/pr.110.003285
Georgiadis, L., Keski-Nisula, L., and Harju, M. (2014). Umbilical cord length in singleton gestations: a finnish population-based retrospective register study. Placenta 35, 275–280. doi: 10.1016/j.placenta.2014.02.001
Ghosh, R., Lipson, K. L., Sargent, K. E., Mercurio, A. M., Hunt, J. S., Ron, D., et al. (2010). Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS ONE 5:e9575. doi: 10.1371/journal.pone.0009575
Gora, S., Maouche, S., Atout, R., Wanherdrick, K., Lambeau, G., Cambien, F., et al. (2010). Phospholipolyzed LDL induces an inflammatory response in endothelial cells through endoplasmic reticulum stress signaling. FASEB J. 24, 3284–3297. doi: 10.1096/fj.09-146852
Grissa, O., Yessoufou, A., Mrisak, I., Hichami, A., Amoussou-Guenou, D., Grissa, A., et al. (2010). Growth factor concentrations and their placental mRNA expression are modulated in gestational diabetes mellitus: possible interactions with macrosomia. BMC Pregnancy Childbirth 10:7. doi: 10.1186/1471-2393-10-7
Gutiérrez, J., Droppelmann, C. A., Salsoso, R., Westermeier, F., Toledo, F., Salomon, C., et al. (2016). A hypothesis for the role of RECK in angiogenesis. Curr. Vasc. Pharmacol. 14, 106–115. doi: 10.2174/1570161113666151014130746
Guzmán-Gutiérrez, E., Armella, A., Toledo, F., Pardo, F., Leiva, A., and Sobrevia, L. (2016). Insulin requires A1 adenosine receptors expression to reverse gestational diabetes-increased L-arginine transport in human umbilical vein endothelium. Purinergic Signal. 12, 175–190. doi: 10.1007/s11302-015-9491-2
Haas, T. L. (2014). Shaping and remodeling of the fetoplacental circulation: aspects of health and disease. Microcirculation 21, 1–3. doi: 10.1111/micc.12084
Hadjipanayi, E., and Schilling, A. F. (2013). Hypoxia-based strategies for angiogenic induction: the dawn of a new era for ischemia therapy and tissue regeneration. Organogenesis 9, 261–272. doi: 10.4161/org.25970
Herrera, E., and Desoye, G. (2015). Maternal and fetal lipid metabolism under normal and gestational diabetic conditions. Horm. Mol. Biol. Clin. Investig. doi: 10.1515/hmbci-2015-0025. [Epub ahead of print].
Herrera, E., and Ortega-Senovilla, H. (2010). Disturbances in lipid metabolism in diabetic pregnancy — Are these the cause of the problem? Best Pract. Res. Clin. Endocrinol. Metab. 24, 515–525. doi: 10.1016/j.beem.2010.05.006
Hetz, C., Chevet, E., and Oakes, S. A. (2015). Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838. doi: 10.1038/ncb3184
Hiden, U., Glitzner, E., Hartmann, M., and Desoye, G. (2009). Insulin and the IGF system in the human placenta of normal and diabetic pregnancies. J. Anat. 215, 60–68. doi: 10.1111/j.1469-7580.2008.01035.x
Hiden, U., Lassance, L., Tabrizi, N. G., Miedl, H., Tam-Amersdorfer, C., Cetin, I., et al. (2012). Fetal insulin and IGF-II contribute to gestational diabetes mellitus (GDM)-associated up-regulation of membrane-type matrix metalloproteinase 1 (MT1-MMP) in the human feto-placental endothelium. J. Clin. Endocrinol. Metab. 97, 3613–3621. doi: 10.1210/jc.2012-1212
Hiden, U., Maier, A., Bilban, M., Ghaffari-Tabrizi, N., Wadsack, C., Lang, I., et al. (2006). Insulin control of placental gene expression shifts from mother to foetus over the course of pregnancy. Diabetologia 49, 123–131. doi: 10.1007/s00125-005-0054-x
Hotamisligil, G. (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917. doi: 10.1016/j.cell.2010.02.034
Huynh, J., Yamada, J., Beauharnais, C., Wenger, J. B., Thadhani, R. I., Wexler, D., et al. (2015). Type 1, type 2 and gestational diabetes mellitus differentially impact placental pathologic characteristics of uteroplacental malperfusion. Placenta 36, 1161–1166. doi: 10.1016/j.placenta.2015.08.004
Hytinantti, T. K., Koistinen, H. A., Teramo, K., Karonen, S. L., Koivisto, V. A., and Andersson, S. (2000). Increased fetal leptin in type I diabetes mellitus pregnancies complicated by chronic hypoxia. Diabetologia 43, 709–713. doi: 10.1007/s001250051367
Iwawaki, T., Akai, R., Yamanaka, S., and Kohno, K. (2009). Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. U.S.A. 106, 16657–16662. doi: 10.1073/pnas.0903775106
Jarmuzek, P., Wielgos, M., and Bomba-Opon, D. (2015). Placental pathologic changes in gestational diabetes mellitus. Neuro Endocrinol. Lett. 36, 101–105.
Jauniaux, E., Watson, A. L., Hempstock, J., Bao, Y. P., Skepper, J. N., and Burton, G. J. (2000). Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am. J. Pathol. 157, 2111–2122. doi: 10.1016/S0002-9440(10)64849-3
Jirkovská, M., Kubinová, L., Janácek, J., Moravcová, M., Krejcí, V., and Karen, P. (2002). Topological properties and spatial organization of villous capillaries in normal and diabetic placentas. J. Vasc. Res. 39, 268–278. doi: 10.1159/000063692
König, A. B., Junginger, S., Reusch, J., Louwen, F., and Badenhoop, K. (2014). Gestational diabetes outcome in a single center study: higher BMI in children after six months. Horm. Metab. Res. 46, 804–809. doi: 10.1055/s-0034-1375652
Kruzliak, P., Sabo, J., and Zulli, A. (2015). Endothelial endoplasmic reticulum and nitrative stress in endothelial dysfunction in the atherogenic rabbit model. Acta Histochem. 117, 762–766. doi: 10.1016/j.acthis.2015.08.003
Lappas, M. (2014). NOD1 expression is increased in the adipose tissue of women with gestational diabetes. J. Endocrinol. 222, 99–112. doi: 10.1530/JOE-14-0179
Larqué, E., Pagán, A., Prieto, M. T., Blanco, J. E., Gil-Sánchez, A., Zornoza-Moreno, M., et al. (2014). Placental fatty acid transfer: a key factor in fetal growth. Ann. Nutr. Metab. 64, 247–253. doi: 10.1159/000365028
Lassance, L., Miedl, H., Absenger, M., Diaz-Perez, F., Lang, U., Desoye, G., et al. (2013). Hyperinsulinemia stimulates angiogenesis of human fetoplacental endothelial cells: a possible role of insulin in placental hypervascularization in diabetes mellitus. J. Clin. Endocrinol. Metab. 98, E1438–E1447. doi: 10.1210/jc.2013-1210
Leach, L. (2011). Placental vascular dysfunction in diabetic pregnancies: intimations of fetal cardiovascular disease? Microcirculation 18, 263–269. doi: 10.1111/j.1549-8719.2011.00091.x
Leiva, A., Salsoso, R., Sáez, T., Sanhueza, C., Pardo, F., and Sobrevia, L. (2015). Cross-sectional and longitudinal lipid determination studies in pregnant women reveal an association between increased maternal LDL cholesterol concentrations and reduced human umbilical vein relaxation. Placenta 36, 895–902. doi: 10.1016/j.placenta.2015.05.012
Lenna, S., Han, R., and Trojanowska, M. (2014). Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life 66, 530–537. doi: 10.1002/iub.1292
Liong, S., and Lappas, M. (2015). Endoplasmic reticulum stress is increased in adipose tissue of women with gestational diabetes. PLoS ONE 10:e0122633. doi: 10.1371/journal.pone.0122633
Liu, Y., Petreaca, M., and Martins-Green, M. (2009). Cell and molecular mechanisms of insulin-induced angiogenesis. J. Cell. Mol. Med. 13, 4492–4504. doi: 10.1111/j.1582-4934.2008.00555.x
Maguire, M. H., Szabó, I., Valkó, I. E., Finley, B. E., and Bennett, T. L. (1998). Simultaneous measurement of adenosine and hypoxanthine in human umbilical cord plasma by reversed-phase high-performance liquid chromatography with photodiode-array detection and on-line validation of peak purity. J. Chromatogr. B Biomed. Sci. Appl. 707, 33–41. doi: 10.1016/S0378-4347(97)00581-1
Marceau, G., Volle, D. H., Gallot, D., Mangelsdorf, D. J., Sapin, V., and Lobaccaro, J. M. (2005). Placental expression of the nuclear receptors for oxysterols LXRalpha and LXRbeta during mouse and human development. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 283, 175–181. doi: 10.1002/ar.a.20157
Marciniak, S. J., and Ron, D. (2006). Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 86, 1133–1149. doi: 10.1152/physrev.00015.2006
Marseille-Tremblay, C., Ethier-Chiasson, M., Forest, J. C., Giguère, Y., Masse, A., Mounier, C., et al. (2008). Impact of maternal circulating cholesterol and gestational diabetes mellitus on lipid metabolism in human term placenta. Mol. Reprod. Dev. 75, 1054–1062. doi: 10.1002/mrd.20842
Martínez-Jiménez, M. A., Aguilar-García, J., Valdés-Rodríguez, R., Metlich-Medlich, M. A., Dietsch, L. J., Gaitán-Gaona, F. I., et al. (2013). Local use of insulin in wounds of diabetic patients: higher temperature, fibrosis, and angiogenesis. Plast. Reconstr. Surg. 132, 1015e–1019e. doi: 10.1097/PRS.0b013e3182a806f0
Marzioni, D., Tamagnone, L., Capparuccia, L., Marchini, C., Amici, A., Todros, T., et al. (2004). Restricted innervation of uterus and placenta during pregnancy: evidence for a role of the repelling signal Semaphorin 3A. Dev. Dyn. 231, 839–848. doi: 10.1002/dvdy.20178
Merzouk, H., Bouchenak, M., Loukidi, B., Madani, S., Prost, J., and Belleville, J. (2000). Fetal macrosomia related to maternal poorly controlled type 1 diabetes strongly impairs serum lipoprotein concentrations and composition. J. Clin. Pathol. 53, 917–923. doi: 10.1136/jcp.53.12.917
National Cholesterol Education Program (NCEP) (2002). Expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation 106, 3143–3421.
Ndrepepa, G., Kastrati, A., Braun, S., Koch, W., Kölling, K., Mehilli, J., et al. (2006). Circulating homocysteine levels in patients with type 2 diabetes mellitus. Nutr. Metab. Cardiovasc. Dis. 18, 66–73. doi: 10.1016/j.numecd.2006.03.007
Nelson, S. M., Coan, P. M., Burton, G. J., and Lindsay, R. S. (2009). Placental structure in type 1 diabetes: relation to fetal insulin, leptin, and IGF-I. Diabetes 58, 2634–2641. doi: 10.2337/db09-0739
Oh, M. J., Zhang, C., LeMaster, E., Adamos, C., Berdyshev, E., Bogachkov, Y., et al. (2016). Oxidized-LDL signals through Rho-GTPase to induce endothelial cell stiffening and promote capillary formation. J. Lipid Res. doi: 10.1194/jlr.M062539. [Epub ahead of print].
Olmos, P. R., Borzone, G. R., Olmos, R. I., Valencia, C. N., Bravo, F. A., Hodgson, M. I., et al. (2012). Gestational diabetes and pre-pregnancy overweight: possible factors involved in newborn macrosomia. J. Obstet. Gynaecol. Res. 38, 208–214. doi: 10.1111/j.1447-0756.2011.01681.x
Outinen, P. A., Sood, S. K., Pfeifer, S. I., Pamidi, S., Podor, T. J., Li, J., et al. (1999). Homocysteine-induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood 94, 959–967.
Ozcan, U., Cao, Q., Yilmaz, E., Lee, A., Iwakoshi, N., Özdelen, E., et al. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461. doi: 10.1126/science.1103160
Ozcan, U., Yilmaz, E., Ozcan, L., Furuhashi, M., Vaillancourt, E., Smith, R. O., et al. (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140. doi: 10.1126/science.1128294
Pagán, A., Prieto-Sánchez, M. T., Blanco-Carnero, J. E., Gil-Sánchez, A., Parrilla, J. J., Demmelmair, H., et al. (2013). Materno-fetal transfer of docosahexaenoic acid is impaired by gestational diabetes mellitus. Am. J. Physiol. 305, E826–E833. doi: 10.1152/ajpendo.00291.2013
Paridaens, A., Laukens, D., Vandewynckel, Y. P., Coulon, S., Van Vlierberghe, H., Geerts, A., et al. (2014). Endoplasmic reticulum stress and angiogenesis: is there an interaction between them? Liver Int. 34, e10–e18. doi: 10.1111/liv.12457
Pearson, J. D., and Gordon, J. L. (1985). Nucleotide metabolism by endothelium. Annu. Rev. Physiol. 47, 617–627. doi: 10.1146/annurev.ph.47.030185.003153
Pietro, L., Daher, S., Rudge, M. V., Calderon, I. M., Damasceno, D. C., Sinzato, Y. K., et al. (2010). Vascular endothelial growth factor (VEGF) and VEGF-receptor expression in placenta of hyperglycemic pregnant women. Placenta 31, 770–780. doi: 10.1016/j.placenta.2010.07.003
Radaelli, T., Lepercq, J., Varastehpour, A., Basu, S., Catalano, P. M., and Hauguel-De Mouzon, S. (2009). Differential regulation of genes for fetoplacental lipid pathways in pregnancy with gestational and type 1 diabetes mellitus. Am. J. Obstet. Gynecol. 201, 209.e1–209.e10. doi: 10.1016/j.ajog.2009.04.019
Reece, E. (2010). The fetal and maternal consequences of gestational diabetes mellitus. J. Matern. Fetal Neonatal Med. 23, 199–203. doi: 10.3109/14767050903550659
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. doi: 10.1038/nrm2199
Sáez, P. J., Villalobos-Labra, R., Westermeier, F., Sobrevia, L., and Farías-Jofré, M. (2014). Modulation of endothelial cell migration by ER stress and insulin resistance: a role during maternal obesity? Front. Pharmacol. 5:189. doi: 10.3389/fphar.2014.00189
Salomón, C., Westermeier, F., Puebla, C., Arroyo, P., Guzmán-Gutiérrez, E., Pardo, F., et al. (2012). Gestational diabetes reduces adenosine transport in human placental microvascular endothelium, an effect reversed by insulin. PLoS ONE 7:e40578. doi: 10.1371/journal.pone.0040578
Salvadó, L., Palomer, X., Barroso, E., and Vázquez-Carrera, M. (2015). Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol. Metab. 26, 438–448. doi: 10.1016/j.tem.2015.05.007
Sanchez-Vera, I., Bonet, B., Viana, M., Quintanar, A., Martin, M. D., Blanco, P., et al. (2007). Changes in plasma lipids and increased low-density lipoprotein susceptibility to oxidation in pregnancies complicated by gestational diabetes: consequences of obesity. Metab. Clin. Exp. 56, 1527–1533. doi: 10.1016/j.metabol.2007.06.020
Sanson, M., Augé, N., Vindis, C., Muller, C., Bando, Y., Thiers, J. C., et al. (2009). Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 expression. Circ. Res. 104, 328–336. doi: 10.1161/CIRCRESAHA.108.183749
Schaefer-Graf, U. M., Graf, K., Kulbacka, I., Kjos, S. L., Dudenhausen, J., Vetter, K., et al. (2008). Maternal lipids as strong determinants of fetal environment and growth in pregnancies with gestational diabetes mellitus. Diabetes Care 31, 1858–1863. doi: 10.2337/dc08-0039
Scholler, M., Wadsack, C., Lang, I., Etschmaier, K., Schweinzer, C., Marsche, G., et al. (2012b). Phospholipid transfer protein in the placental endothelium is affected by gestational diabetes mellitus. J. Clin. Endocrinol. Metab. 97, 437–445. doi: 10.1210/jc.2011-1942
Scholler, M., Wadsack, C., Metso, J., Chirackal Manavalan, A. P., Sreckovic, I., Schweinzer, C., et al. (2012a). Phospholipid transfer protein is differentially expressed in human arterial and venous placental endothelial cells and enhances cholesterol efflux to fetal HDL. J. Clin. Endocrinol. Metab. 97, 2466–2474. doi: 10.1210/jc.2011-2969
Scifres, C. M., Chen, B., Nelson, D. M., and Sadovsky, Y. (2011). Fatty acid binding protein 4 regulates intracellular lipid accumulation in human trophoblasts. J. Clin. Endocrinol. Metab. 96, E1083–E1091. doi: 10.1210/jc.2010-2084
Seo, J. H., Yu, J. H., Suh, H., Kim, M. S., and Cho, S. R. (2013). Fibroblast growth factor-2 induced by enriched environment enhances angiogenesis and motor function in chronic hypoxic-ischemic brain injury. PLoS ONE 8:e74405. doi: 10.1371/journal.pone.0074405
Sharma, P., Senthilkumar, R. D., Brahmachari, V., Sundaramoorthy, E., Mahajan, A., Sharma, A., et al. (2006). Mining literature for a comprehensive pathway analysis: a case study for retrieval of homocysteine related genes for genetic and epigenetic studies. Lipids Health Dis. 5, 1. doi: 10.1186/1476-511X-5-1
Sobrevia, L., Salsoso, R., Sáez, T., Sanhueza, C., Pardo, F., and Leiva, A. (2015). Insulin therapy and fetoplacental vascular function in gestational diabetes mellitus. Exp. Physiol. 100, 231–238. doi: 10.1113/expphysiol.2014.082743
Sobrevia, L., Yudilevich, D. L., and Mann, G. E. (1998). Elevated D-glucose induces insulin insensitivity in human umbilical endothelial cells isolated from gestational diabetic pregnancies. J. Physiol. 506, 219–230. doi: 10.1111/j.1469-7793.1998.219bx.x
Sreckovic, I., Birner-Gruenberger, R., Besenboeck, C., Miljkovic, M., Stojakovic, T., Scharnagl, H., et al. (2014). Gestational diabetes mellitus modulates neonatal high-density lipoprotein composition and its functional heterogeneity. Biochim. Biophys. Acta 1841, 1619–1627. doi: 10.1016/j.bbalip.2014.07.021
Sreckovic, I., Birner-Gruenberger, R., Obrist, B., Stojakovic, T., Scharnagl, H., Holzer, M., et al. (2013). Distinct composition of human fetal HDL attenuates its anti-oxidative capacity. Biochim. Biophys. Acta 1831, 737–746. doi: 10.1016/j.bbalip.2012.12.015
Taniguchi, M., Kwak, Y. L., Jones, K. A., Warner, D. O., and Perkins, W. J. (2006). Nitric oxide sensitivity in pulmonary artery and airway smooth muscle: a possible role for cGMP responsiveness. Am. J. Physiol. 290, L1018–L1027. doi: 10.1152/ajplung.00402.2005
Taricco, E., Radaelli, T., Rossi, G., Nobile de Santis, M. S., Bulfamante, G. P., Avagliano, L., et al. (2009). Effects of gestational diabetes on fetal oxygen and glucose levels in vivo. Br. J. Obstet. Gynecol. 116, 1729–1735. doi: 10.1111/j.1471-0528.2009.02341.x
Teasdale, F. (1981). Histomorphometry of the placenta of the diabetic women: class A diabetes mellitus. Placenta 2, 241–251. doi: 10.1016/S0143-4004(81)80007-0
Treins, C., Giorgetti-Peraldi, S., Murdaca, J., Semenza, G. L., and Van Obberghen, E. (2002). Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J. Biol. Chem. 277, 27975–27981. doi: 10.1074/jbc.M204152200
Tzotzas, T., Desrumaux, C., and Lagrost, L. (2009). Plasma phospholipid transfer protein (PLTP): review of an emerging cardiometabolic risk factor. Obes. Rev. 10, 403–411. doi: 10.1111/j.1467-789X.2009.00586.x
Verier-Mine, O. (2010). Outcomes in women with a history of gestational diabetes. Screening and prevention of type 2 diabetes. Literature review. Diab. Metab. 36, 595–616. doi: 10.1016/j.diabet.2010.11.011
Verkhratsky, A., and Burnstock, G. (2014). Biology of purinergic signalling: its ancient evolutionary roots, its omnipresence and its multiple functional significance. Bioessays 36, 697–705. doi: 10.1002/bies.201400024
Wang, F., Wu, Y., Gu, H., Reece, E. A., Fang, S., Gabbay-Benziv, R., et al. (2015). Ask1 gene deletion blocks maternal diabetes–induced endoplasmic reticulum stress in the developing embryo by disrupting the unfolded protein response signalosome. Diabetes 64, 973–988. doi: 10.2337/db14-0409
Wang, K., Jiang, Y. Z., Chen, D. B., and Zheng, J. (2009). Hypoxia enhances FGF2- and VEGF-stimulated human placental artery endothelial cell proliferation: roles of MEK1/2/ERK1/2 and PI3K/AKT1 pathways. Placenta 30, 1045–1051. doi: 10.1016/j.placenta.2009.10.007
Westermeier, F., Sáez, T., Arroyo, P., Toledo, F., Gutiérrez, J., Sanhueza, C., et al. (2015b). Insulin receptor isoforms: an integrated view focused on gestational diabetes mellitus. Diab. Metab. Res. Rev. doi: 10.1002/dmrr.2729. [Epub ahead of print].
Westermeier, F., Salomón, C., Farías, M., Arroyo, P., Fuenzalida, B., Sáez, T., et al. (2015a). Insulin requires normal expression and signaling of insulin receptor A to reverse gestational diabetes-reduced adenosine transport in human umbilical vein endothelium. FASEB J. 29, 37–49. doi: 10.1096/fj.14-254219
Westermeier, F., Salomón, C., González, M., Puebla, C., Guzmán-Gutiérrez, E., Cifuentes, F., et al. (2011). Insulin restores gestational diabetes mellitus-reduced adenosine transport involving differential expression of insulin receptor isoforms in human umbilical vein endothelium. Diabetes 60, 1677–1687. doi: 10.2337/db11-0155
Westgate, J. A., Lindsay, R. S., Beattie, J., Pattison, N. S., Gamble, G., Mildenhall, L. F., et al. (2006). Hyperinsulinemia in cord blood in mothers with type 2 diabetes and gestational diabetes mellitus in New Zealand. Diabetes Care 29, 1345–1350. doi: 10.2337/dc05-1677
Wyatt, A. W., Steinert, J. R., Wheeler-Jones, C. P., Morgan, A. J., Sugden, D., Pearson, J. D., et al. (2002). Early activation of the p42/p44 MAPK pathway mediates adenosine-induced NO production in human endothelial cells: a novel calcium insensitive mechanism. FASEB J. 16, 1584–1594. doi: 10.1096/fj.01-0125com
Yao, L., Wan, J., Li, H., Ding, J., Wang, Y., Wang, X., et al. (2015). Resveratrol relieves gestational diabetes mellitus in mice through activating AMPK. Reprod. Biol. Endocrinol. 13, 118. doi: 10.1186/s12958-015-0114-0
Zhang, K., and Kaufman, R. J. (2006). Protein folding in the endoplasmic reticulum and the unfolded protein response. Handb. Exp. Pharmacol. 172, 69–91. doi: 10.1007/3-540-29717-0_3
Keywords: insulin, gestational diabetes, endoplasmic reticulum stress, angiogenesis, lipids, placenta, endothelium
Citation: Sobrevia L, Salsoso R, Fuenzalida B, Barros E, Toledo L, Silva L, Pizarro C, Subiabre M, Villalobos R, Araos J, Toledo F, González M, Gutiérrez J, Farías M, Chiarello DI, Pardo F and Leiva A (2016) Insulin Is a Key Modulator of Fetoplacental Endothelium Metabolic Disturbances in Gestational Diabetes Mellitus. Front. Physiol. 7:119. doi: 10.3389/fphys.2016.00119
Received: 21 December 2015; Accepted: 15 March 2016;
Published: 31 March 2016.
Edited by:
Agustín Guerrero-Hernández, CINVESTAV, MexicoReviewed by:
Geraldine Clough, University of Southampton, UKEric Chevet, Institut National de la Santé et de la Recherche Médicale, France
Copyright © 2016 Sobrevia, Salsoso, Fuenzalida, Barros, Toledo, Silva, Pizarro, Subiabre, Villalobos, Araos, Toledo, González, Gutiérrez, Farías, Chiarello, Pardo and Leiva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis Sobrevia, c29icmV2aWFAbWVkLnB1Yy5jbA==