 Lei Ni
Lei Ni Chia-Chen Chuang
Chia-Chen Chuang Li Zuo
Li Zuo- 1Radiologic Sciences and Respiratory Therapy Division, School of Health and Rehabilitation Sciences, Davis Heart and Lung Research Institute, The Ohio State University College of Medicine, The Ohio State University Wexner Medical Center, Columbus, OH, USA
- 2Department of Pulmonary Medicine, Ruijin Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
- 3Shanghai Key Laboratory of Meteorology and Health, Pudong Meteorological Service, Shanghai, China
- 4Interdisciplinary Biophysics Program, The Ohio State University, Columbus, OH, USA
Chronic obstructive pulmonary disease (COPD) is a common airway disorder. In particular, acute exacerbations of COPD (AECOPD) can significantly reduce pulmonary function. The majority of AECOPD episodes are attributed to infections, although environmental stress also plays a role. Increasing urbanization and associated air pollution, especially in developing countries, have been shown to contribute to COPD pathogenesis. Elevated levels of particulate matter (PM) in polluted air are strongly correlated with the onset and development of various respiratory diseases. In this review, we have conducted an extensive literature search of recent studies of the role of PM2.5 (fine PM) in AECOPD. PM2.5 leads to AECOPD via inflammation, oxidative stress (OS), immune dysfunction, and altered airway epithelial structure and microbiome. Reducing PM2.5 levels is a viable approach to lower AECOPD incidence, attenuate COPD progression and decrease the associated healthcare burden.
Introduction
According to the most recent Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines, COPD is defined as “a common preventable and treatable disease” characterized by “persistent airflow limitation that is usually progressive and associated with enhanced chronic inflammatory response in the airways and the lung to noxious particles and gases” (GOLD, 2015). Conservative estimates of COPD incidence hover around 210 million cases worldwide, excluding those that are under-recognized or under-diagnosed (Soriano and Lamprecht, 2012). As reported by the World Health Organization (WHO), it is estimated that COPD is poised to become the third largest leading cause of death in the world by 2020 (GOLD, 2015). Due to its high morbidity and mortality, COPD imposes a significant economic burden, contributing to a direct cost of $29.5 billion and an indirect cost of $20.4 billion in the United States annually (National Heart, Lung, and Blood Institute, 2009).
The exacerbation phase of COPD, known as AECOPD, is associated with symptoms such as increased dyspnea, cough, sputum and/or purulent sputum production (GOLD, 2015). AECOPD reduces respiratory function and negatively affects disease progression and prognosis, leading to high healthcare costs (McGuire et al., 2001). Nevertheless, the management of AECOPD remains a clinical challenge. Currently, AECOPD pathogenesis can be attributed to infectious and non-infectious factors. Specifically, bacterial and viral infections are the most prevalent causes of AECOPD (Perotin et al., 2013). In the sputum of 40–60% of AECOPD patients, bacteria such as Haemophilus influenzae, Moraxella catarrhalis, and Streptococcus pneumoniae can be detected. In addition, airway viral infections, including rhinovirus, respiratory syncytial virus (RSV) and influenza virus may contribute to AECOPD pathogenesis (Perotin et al., 2013).
Non-infectious factors, mainly atmospheric pollution, are also implicated in AECOPD. Notably, inhalable particulate matter (PM), which is a major player in air pollution, is involved in various chronic airway inflammatory diseases. Recent studies have demonstrated that PM2.5 (aerodynamic diameter ≤ 2.5 μm) is closely correlated with AECOPD morbidity and mortality (Sarnat et al., 2001; Dominici et al., 2003; Laden et al., 2006). For instance, Li et al. reported that a 10 μg/m3 increase in atmospheric PM2.5 raised the total mortality of COPD by 2.5% in cities worldwide (Li et al., 2015). PM2.5 induces AECOPD through a variety of mechanisms, including oxidative stress (OS), airway inflammation, airway epithelial damage, and inhibition of local airway immunity (Ling and van Eeden, 2009). In this review, we performed a systematic search of PubMed and Medline from 2000 to 2015 for literature related to the role of PM2.5 in AECOPD pathogenesis. We aim to investigate PM2.5 involvement in AECOPD and the significance of managing air pollution in preventing exacerbations of COPD.
PM2.5 Structure and Composition
PM refers to the dispersed solid, liquid or solid-liquid suspensions in the air (Wilson et al., 2002). The diameter, composition, and origin of PM play an important role in toxicity and biological pathogenicity (Valavanidis et al., 2008). PM measuring greater than 10 μm in diameter can be trapped in the mucus lining within the nose and respiratory tract, and can be readily eliminated through normal breathing activities (Squadrito et al., 2001). However, PM with a diameter less than 10 μm is more difficult to remove from the respiratory tract when inhaled (Squadrito et al., 2001). In particular, PM2.5 (also known as the fine particulate) reaches distal airways and deposits in alveolar regions (Squadrito et al., 2001). The soluble components of PM2.5 may enter the blood circulation through alveolar capillaries as ultrafine PM, whereas the insoluble portions sediment in the lung, thereby leading to detrimental health outcomes including airflow obstruction (Ling and van Eeden, 2009; Brook et al., 2010). Consequently, PM2.5 is commonly associated with cardiovascular diseases and chronic airway diseases, such as COPD and atherosclerosis. A tendency for PM to accumulate in the centribular emphysematous lesion and induce persistent inflammation has suggested its association with COPD pathogenesis, characterized by emphysema (Ling and van Eeden, 2009). Accordingly, Bose et al. showed the correlations between indoor PM2.5 and systemic inflammation in COPD patients. Yet such relationship was not found with coarse fraction of particles (PM2.5−10) (Bose et al., 2015).
The extent of PM2.5 deposition in the lung is determined by the inhaled concentration, tissue structure, and the clearing ability of airway cilia. COPD patients with persistent airway obstruction and abnormal lung architecture may display higher levels of PM2.5 pulmonary deposits when compared with healthy individuals (Ling and van Eeden, 2009). The resulting damage to airway cilia and the reduced ability to perform airway clearance prevent the timely elimination of PM2.5 from the airway and lungs (Ling et al., 2011). In a polluted environment, each alveolus comes in contact with an average of 1500 particulate molecules in a 24 h period. Approximately 50% of the PM deposits occur in the alveolus, of which 96% are composed of PM2.5 (Valavanidis et al., 2008). Further, the generation of PM2.5 is primarily attributed to industrial combustion, traffic emission, and agricultural activities (Laden et al., 2000; Saliba et al., 2010). Its chemical composition varies with different environmental sources.
PM2.5 as a Carrier
The characterization of PM2.5 chemical composition reveals its toxicity, which is relevant to the adverse health effects imposed by PM2.5 (Bell et al., 2007). PM2.5 may absorb organic molecules, transition metals, reactive gases, microbial components, and minerals, serving as a carrier for these potentially harmful molecules (Kleeman et al., 2000; Sillanpaa et al., 2006). In particular, Bell et al. have found that several components, including chlorine, zinc, and bromide, showed strong seasonal correlations; that is, higher concentration of these components were detected in PM2.5 during winter (Bell et al., 2007).
Iron is a transient metal that is commonly found in PM2.5. Soluble metal ions absorbed by PM2.5 are considered a key factor in the impairment of lung cell. Redox-active metals trigger a series of catalytic reactions and free radical formation, leading to lipid peroxidation in the cell membrane and inflammation (Harrison and Yin, 2000; Meng and Zhang, 2006; Schwarze et al., 2006). PM2.5 containing higher levels of heavy metals may damage the macrophages, leading to reduced phagocytosis (Zhou and Kobzik, 2007). Zhou and Kobzik reported that the chelation of iron substantially reversed its toxicity to macrophage cells (Zhou and Kobzik, 2007). Iron attached to the surface of particulate can disturb redox balance by inducing the release of hydroxyl radicals, further damaging DNA and generating an OS marker 8-hydroxy-2-deoxyguanine (8-OHdG) (Knaapen et al., 2002). High levels of soluble metal zinc in PM have been shown to increase the levels of neutrophils, macrophages and protein in mouse bronchoalveolar lavage (BAL) (Adamson et al., 2000). Adamson et al. also detected focal fibrosis in the alveolar mesenchyme, which demonstrated that zinc in PM2.5 was related to chronic or acute pulmonary inflammation (Adamson et al., 2000). Furthermore, Dergham et al. reported that the transient metals, as well as organic chemicals, attached to PM2.5 to upregulate the gene expression of xenobiotic-metabolizing enzymes, cytochrome P4501a1 and 1b1 in human bronchial epithelial cells (Dergham et al., 2012).
The biological components of PM2.5 are potentially involved in the activation of pulmonary immunity (Alexis et al., 2006). Specially, Alexis et al. observed an up-regulation of TNF-α mRNA expression solely in the non-heat-treated PM2.5−10 group (Alexis et al., 2006). During the lipopolysaccharide (LPS)-mediated immune reaction, the expression of mCD14 on the surface of macrophages is higher in the non-heat-treated group than in the heat-treated group (Alexis et al., 2006). It is shown that heat treatment inactivates heat-sensitive biological components on the surface of PM2.5 such as LPS (from gram-negative bacteria) and the peptidoglycans (from gram-positive bacteria) (Alexis et al., 2006).
PM2.5 and Airway Inflammation in COPD
Exacerbation of airway inflammation is a key feature in AECOPD (Figure 1). Indeed, inflammatory biomarkers such as tumor necrosis factor alpha (TNF-α), interleukin (IL)-6 and IL-8 were significantly elevated in the induced sputum of AECOPD patients, while the concentrations returned to lower values during the recovery (Bhowmik et al., 2000; Aaron et al., 2001). PM2.5 enhanced airway inflammation via any of the following mechanisms (Figure 1): (1) Release of pro-inflammatory cytokines (e.g., TNF-α and IL-1) and oxygen radicals due to active phagocytosis of PM2.5 mainly by alveolar macrophages (AMs); (2) Production of chemoattractive and pro-inflammatory mediators, such as granulocyte macrophage colony-stimulating factor (GM-CSF), by lung epithelial cells results in enhanced leukocyte recruitment; (3) Generation of oxygen free radicals and proteases from the damaged alveolar epithelial or immune cells (Ling and van Eeden, 2009; Valavanidis et al., 2013).
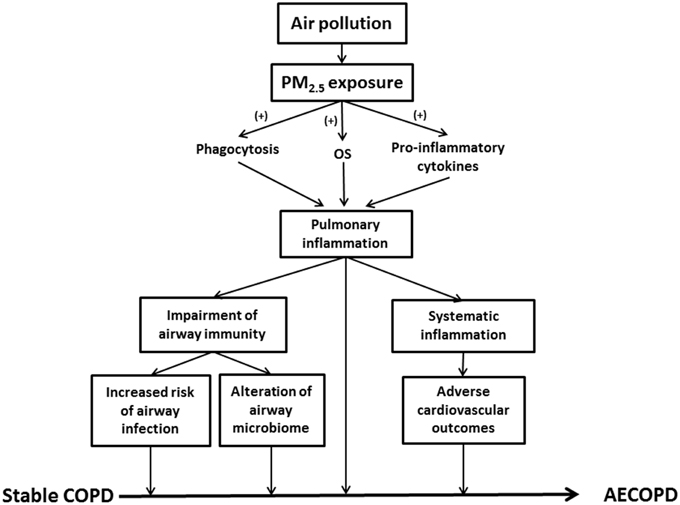
Figure 1. This schematic summarizes the role of PM2.5 in AECOPD. PM2.5, particulate matter ≤ 2.5 μm; OS, oxidative stress; COPD, chronic obstructive pulmonary disease; AECOPD, acute exacerbations of COPD.
Immune cells, such as AMs, directly interact with PM2.5, playing an important role in the elimination of inhaled particulate. As mentioned previously, PM2.5 may stimulate AM to release inflammatory mediators such as arachidonic acid (AA), TNF-α and IL-6, leading to airway inflammation (Table 1; Pozzi et al., 2003). In addition, PM2.5 has been shown to compromise the functional capacity of phagocytic cells and influence the expression of these cells (Ling and van Eeden, 2009). Exhaled nitric oxide (FeNO), an indicator of airway inflammation, is positively correlated with the number of airway inflammatory cells. Notably, the ambient air pollution-induced PM2.5 leads to increased FeNO levels in the elderly (Adamkiewicz et al., 2004). In a study with healthy, nonsmoking subjects, an increase in sputum neutrophils was observed after 4 h exposure to diesel exhaust particle (DEP) (Nightingale et al., 2000). Levels of inflammatory factors, including TNF-α, IL-1β, IL-6, and IL-8, were significantly elevated after PM2.5 exposure to human bronchial epithelial cells (Cachon et al., 2014).
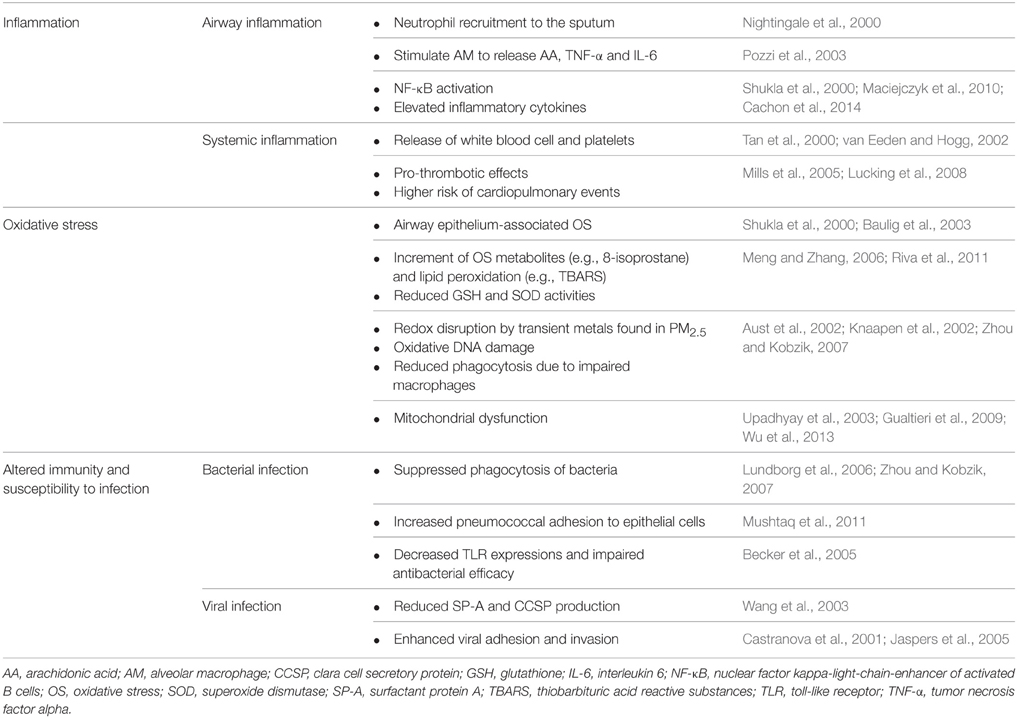
Table 1. A summary of PM2.5 involvement in AECOPD pathogenesis.
PM2.5 initiates a series of genetic mechanisms via nuclear factor kappa-light-chain-enhancement of activated B cells (NF-κB) thereby leading to a reaction cascade involving cytokine release (Dagher et al., 2007). PM2.5-induced epithelial OS facilitates the binding of NF-κB to DNA resulting in increased mRNA expression of NF-κB-related downstream inflammatory cytokines such as TNF-α, TNF-β, and IL-6 (Shukla et al., 2000). These inflammatory cytokines further increases inflammatory cell infiltration and airway mucus secretion, thereby aggravating airway stenosis and breathing difficulty (Shukla et al., 2000). Additionally, the metals in PM2.5 trigger human bronchial epithelial cells to release proinflammatory cytokines via NF-κB activation. PM2.5 also induces elevated gene expression of intercellular adhesion molecules, resulting in the adhesion of epithelial and inflammatory cells (Table 1; Maciejczyk et al., 2010).
Systemic inflammation is highly associated with the incidence and progression of COPD (Groenewegen et al., 2008). The levels of blood inflammatory markers, such as c-reactive protein (CRP), fibrinogen, and TNF-α, have been shown to elevate in stable COPD (Gan et al., 2004; Karadag et al., 2008). Whether inhaled PM2.5 can directly integrate into systemic circulation still remains a controversy (Brown et al., 2002; Kreyling et al., 2002; Nemmar et al., 2002). PM2.5 contributes to the systemic inflammatory response and the functional alteration of multiple organs (van Eeden et al., 2005). PM2.5 exacerbates COPD systemic inflammation via: (1) activation of inflammatory cells and related inflammatory mediators that infiltrate into the systemic circulation through lung tissue; (2) stimulation of hepatic synthesis of CRP and fibrinogen; (3) stimulation of bone marrow function leading to increased white blood cell and platelet counts; and (4) damaged endothelial system and increased cardiovascular morbidity and mortality (van Eeden and Hogg, 2002; van Eeden et al., 2005).
PM2.5, Airway Immunity, and Infection
The relationship between PM2.5 and airway immune function is complex. PM2.5 activates innate immunity and amplifies inflammatory reactions. For example, PM2.5 stimulates airway epithelial cells to secrete the chemokine MIP-3α (CCL20), thereby recruiting dendritic cells (DC) to airways (Reibman et al., 2003). In addition, DEP up-regulates the co-stimulatory molecules (CD40 and CD86) and facilitates T helper 2 (Th2)-mediated immune response (van Zijverden et al., 2000). The expression of MHC class II molecule was also up-regulated by DEP exposure (Inoue et al., 2009). Interestingly, DEP exposure prior to viral infection increases the expression of the toll-like receptor 3 (TLR 3) and pro-inflammatory cytokines in human pulmonary epithelial cells, thus enhancing the innate immunity (Ciencewicki et al., 2006). PM2.5 exposure alters the distal airway microbiome of COPD patients, and diminishes its ability to resist microbial infection by suppressing the function of immune cells (Figure 1). An elevation of PM2.5 level leads to enhanced hospitalization for pneumonia (Tsai and Yang, 2014). According to the study of Matthews et al. PM2.5 in urban areas may inhibit the generation of primary T helper type 1 (Th1) cell and reduce the levels of IFN-γ, IL-5, and IL-13, which compromises IFN-γ-mediated antibacterial and antiviral immunity in the lungs (Matthews et al., 2014).
As the first line of host defense in the lungs, AMs are primarily responsible for eliminating harmful microorganisms and inhaled foreign particles, playing an important role in innate immunity (Nicod, 2005). Zhou et al. studied the impact of concentrated ambient particles (CAPs) on murine macrophage cell lines. The results indicated that CAPs suppress the internalization of S. pneumoniae by macrophages (Zhou and Kobzik, 2007). In addition, several studies showed that exposure to PM2.5 impairs the phagocytic capacity of human AM (Lundborg et al., 2006; Xu et al., 2008). Becker et al. found that PM2.5 exposure decreased the expression of phagocytosis-related receptor, CD11b, on the surface of AM, although to a lesser extent compared with PM10 (Becker et al., 2003). These studies demonstrated that PM2.5 reduced the functional airway defenses when combating viruses, bacteria and other microorganisms by inhibiting the phagocytic function of AM.
Further, PM2.5 also affects the function of circulating immune cells. Acute PM exposure stimulates polymorphonuclear leukocytes (PMN)-associated respiratory burst, and superoxide dismutase (SOD) supplementation may diminish such an effect (Marchini et al., 2014). Oxidative DNA damage of the lymphocytes has been reported to significantly associate with transition metal-rich PM2.5 (Sørensen et al., 2005). Fujimaki et al. studied the adjuvant impact of DEP on systemic IgE generation in an ovalbumin (OA)-treated mouse model. The experimental group of mice was administered DEP and OA, an antigen that induces allergic pulmonary responses (Fujimaki et al., 2001). After immunization, levels of serum anti-OA IgE antibody were markedly higher in the experimental group than those in the control group treated with OA only. The result indicated that exposure to DEP was responsible for the increased systemic IgE response to OA (Fujimaki et al., 2001). PM2.5 is related to airway hyperresponsiveness and systemic IgE response (Ogino et al., 2014). Asthma-COPD overlap syndrome (ACOS), one of the clinical phenotypes of COPD, shows evidence of both airway hyperresponsiveness and fixed airflow obstruction (Zeki et al., 2011). Thus, PM2.5 exposure and high levels of serum IgE are risk factors for acute exacerbation of ACOS.
In healthy populations, lower portions of the airways remain aseptic. However, bacterial colonization is commonly observed in the lower airway of stable COPD as well as other chronic lung diseases. Indeed, a higher prevalence of pathogenic infection and altered lung defense have been implicated in exacerbated COPD (Patel et al., 2002; Sethi et al., 2002). Potentially pathogenic bacteria (PPB) present in the BAL fluid include: S. pneumoniae, Moraxella catarrhalis, Haemophilus influenza, and Pseudomonas aeruginosa. The colonization by PPB is closely related to chronic airway inflammation in COPD (Sethi et al., 2006). PM2.5 increases the risk of bacterial infections in COPD patients via several mechanisms. Researchers found that PM2.5 particles carry bacteria and bacteria-derived components (Menetrez et al., 2001; Alghamdi et al., 2014). Therefore, they may alter the microbiome in the distal airway of COPD patients and subsequently lead to AECOPD (Figure 1). Another study demonstrated that PM2.5 exposure promoted pneumococcal adhesion to human epithelial cells, thereby enhancing pneumococcal pneumonia susceptibility (Table 1; Mushtaq et al., 2011). In addition, suppression of pulmonary immune function is one of the primary factors leading to bacterial infections. The carbonaceous core of DEP absorbs multiple organic materials such as PAH and transient metals. DEP is more likely to be inhaled into lower airways and alveolar tissues (Castranova et al., 2001; Yanagisawa et al., 2003). DEP diminishes pulmonary antimicrobial ability, increasing the susceptibility to infection. PM2.5 reduces bacterial elimination from the lungs via suppressed production of LPS-stimulated proinflammatory cytokines, such as IL-1 and TNF-α, and weakened phagocytosis (Castranova et al., 2001). PM2.5 decreases the expression of toll-like receptors and suppresses TNF and IL-8 release in AM (Table 1), impairing the antibacterial efficacy of the respiratory system (Becker et al., 2005).
Moreover, Listeria monocytogenes (L. monocytogenes) is a common gram-positive, facultative anaerobic bacterium. AMs and neutrophils are effector cells that can eliminate L. monocytogenes (Yang et al., 2001). Yang et al. observed that DEP reduces the responsiveness of AM to L. monocytogenes and its ability to secret antimicrobial oxidants, such as nitric oxide, thus impairing the airway cleaning competence (Yang et al., 2001). Moreover, IL-12 plays an important role in Th1 cell anti-L. monocytogenes infection's immune response. Researchers found that DEP decreases cell-mediated immune responses upon L. monocytogenes infection via AM-secreted IL-12 suppression (Yin et al., 2002). S. pneumoniae infection and sepsis are one of the most common factors for increased hospitalization in the elderly above the age of 70 (Marrie, 2000). S. pneumoniae is also a common pathogenic bacterium of AECOPD (Pérez-Trallero et al., 2011). Epidemiological studies have shown that air pollution increases the vulnerability to bacterial pneumonia (Medina-Ramón et al., 2006; Neupane et al., 2010). Zelikoff et al. established rat models through intratracheal instillation of S. pneumoniae and observed the impact of PM2.5 on the pulmonary antibacterial immune function. The results demonstrated that rats exposed to PM2.5 had decreased levels of PMN in BAL fluid (Zelikoff et al., 2003). In addition, the expression of inflammatory mediators such as TNF-α and IL-1 associated with mobilization and activation of PMN, was all decreased (Zelikoff et al., 2003). The mortality of PM2.5-exposed group was significantly higher than air-exposed control rats. The study revealed that PM2.5 exposure may increase the severity of bacterial infections by inhibiting the host anti-microorganism immunity (Zelikoff et al., 2003).
PM2.5 exposure can increase the risk of respiratory viral infections (Wang et al., 2003). Surfactant protein A (SP-A) and clara cell secretory protein (CCSP) are both important components of innate immune defense mediators for antiviral respiratory infection (Wang et al., 2003; Derscheid and Ackermann, 2013). Harrod et al. found that DEP-exposed mice have significantly higher gene expression of RSV in lung tissues than control mice (Wang et al., 2003). In addition, the number of inflammatory cells in BAL fluid of both RSV-infected and uninfected mice that were exposed to high-level DEP, was increased (Wang et al., 2003). Lung histopathology examination revealed airway epithelial remodeling with the loss of the normal appearance of bronchial epithelium, and mucous cell metaplasia. Levels of airway epithelium CCSP production and the SP-A secretion in the DEP group were both reduced (Table 1; Wang et al., 2003). These results suggest that DEP exposure can decrease lung immune defenses against viruses. Likewise, Lambert et al. observed a reduced expression of IFN-γ, a cytokine essential for microbial defenses, in RSV-infected mice preexposed to ultrafine particles, suggesting increased body's susceptibility to RSV upon PM2.5 exposure (Lambert et al., 2003). Additionally, DEP exposures can increase the number of infected A549 cells by enhancing OS, the ability of adhesion and invasion of the influenza virus (Jaspers et al., 2005). DEP promotes the replication of the influenza virus in the lungs through the inhibition of antiviral interferon production (Table 1; Castranova et al., 2001). These results support that PM2.5 can increase the susceptibility to the influenza virus in airway epithelial cells.
PM2.5, Oxidative Stress, and AECOPD
The chronic inflammatory reaction and OS in COPD are not limited to the lungs. Instead, systemic oxidant-antioxidant imbalance and low-grade systemic inflammation are observed in COPD (Donaldson et al., 2005; van Eeden et al., 2005). During normal metabolic processes, ROS are formed constantly in the cells (Zuo et al., 2013). Particularly in COPD development, ROS are released excessively by leukocytes, such as macrophages, in the airway due to the enhanced phagocytosis of exogenous molecules. Impaired alveolar epithelial cells also contribute to the elevated level of ROS. Subsequently, the endogenous antioxidants are unable to neutralize overproduced ROS thereby leading to OS (Zuo et al., 2014).
AECOPD patients exhale higher levels of 8-isoprostane in the lungs, indicating pulmonary OS (Biernacki et al., 2003). Several studies have suggested that some components of PM2.5, such as transient metals, polycyclic aromatic hydrocarbon (PAH) and carbonaceous material stimulate ROS release from pulmonary epithelial cells. PM2.5-induced ROS generation, such as superoxide and hydrogen peroxide, further damage cilia and contribute to mucus hypersecretion, airway stenosis and dyspnea (Dagher et al., 2007; Gualtieri et al., 2010; Seriani et al., 2015). Riva et al. reported that a small dose of PM2.5 causes functional and histological lung damage in mouse models. Mice treated with intranasal instillation of PM2.5 suspension showed an increased resistance in lung ventilation and a decrease in tissue elasticity (Riva et al., 2011). A pathological examination revealed alveolar collapse and lung biochemical analysis indicated increased neutrophil activity (Riva et al., 2011). Increased OS metabolites, such as 8-isoprostane and thiobarbituric acid, accompanied by elevated catalase activity and a reduction in GSH/GSSG, were also observed (Riva et al., 2011). Accordingly, a dose-dependent decrease of GSH levels and SOD activities was seen in rat lungs exposed to fine particles in a dust storm. The level of thiobarbituric acid reactive substances (TBARS) also increased along with fine particle exposure, indicating the occurrence of lipid peroxidation (Table 1; Meng and Zhang, 2006). Cytokines such as IL-6 and IL-1β induced by ROS/inflammation enter the blood circulation and stimulate liver cells to produce acute phase proteins (e.g., CRP and fibrinogen). As a result, additional systemic inflammation and cardiovascular events are prompted (van Eeden et al., 2005). Meng and Zhang studied the impact of fine particles from a dust storm on systemic OS in a rat model. After 24 h of PM2.5 exposure, the antioxidants SOD and GSH were significantly decreased within the lung and liver tissues. TBARS, the biomarkers of endogenous lipid peroxidation, increased in the lungs, heart, and liver demonstrating that PM2.5 not only affected the lungs, but also caused systemic OS (Meng and Zhang, 2006).
As the first line of defense, airway epithelial cells are most susceptible to the toxicity of PM2.5. Specifically, the damaged epithelium is known to release ROS and inflammatory mediators (Baulig et al., 2003). In an in vitro study, which examined the impact of PM2.5 on epithelial lung A549 cells, Deng et al. found that these cells release lactate dehydrogenase (LDH), produce ROS, and reduce intracellular levels of SOD and catalase. PM2.5-exposed A549 cells also showed an up-regulation of autophagy-related protein markers, such as microtubule-associated protein light chain-3 (LC3), Atg5, and Beclin in both time- and concentration-dependent manner (Deng et al., 2013). These results suggest that PM2.5 may damage the lung tissue and impair lung function through epithelial cells autophagy induced by OS. PM2.5 significantly induced the mRNA expression of pro-apoptotic genes such as p53 and Bax, while decreasing the mRNA expression of anti-apoptotic genes, such as Bcl-2 in human epithelial lung L132 cells. As a result, PM2.5 caused apoptosis of L132 via multiple pathways related to OS (Dagher et al., 2006).
PM2.5 may generate a toxic effect in lung epithelium via oxidative damage in the mitochondria (Table 1). Upadhyay et al. reported that airborne PM causes mitochondrial dysfunction in A549 cells via iron-derived free radicals. Reduction in mitochondrial membrane potential and caspase-9 activation was detected in the study (Upadhyay et al., 2003). Alterations in membrane integrity, permeability, and mitochondrial structure result in epithelial cell damage (Gualtieri et al., 2009; Wu et al., 2013). In human epithelial lung cells (L132), PM2.5 has been attributed to the elevation of OS markers such as malondialdehyde (MDA) (Garçon et al., 2006). PM2.5 also promotes the secretion of TNF-α, inducible nitric oxide synthase (iNOS), and nitric oxide (NO) in a dose- and time-dependent manner (Garçon et al., 2006). Further, PM2.5 causes endothelial damage by inducing inflammatory response (Montiel-Davalos et al., 2010). Moreover, Nurkiewicz et al. observed increased LDH activities and albumin protein levels in the experimental rat groups exposing to PM2.5 (residual oil fly ash; mean diameter 2.2 μm) (Nurkiewicz et al., 2004).
COPD patients often develop other systemic diseases, including heart disease, degeneration of skeletal muscle function, malnutrition, osteoporosis, diabetes, and depression (Chatila et al., 2008). Cardiovascular diseases such as atherothrombotic disease, pulmonary hypertension, and chronic congestive heart failure, are commonly found in COPD subjects (Sin and Man, 2003; van Eeden et al., 2005). Inflammatory mediators, such as TNF-α, IL-6, IL-1β, and GM-CSF, impair endothelium and elevate white blood cell counts (van Eeden et al., 2005). There were increased levels of PMN as well as leukocyte rolling and adhesion in the microvascular walls (Nurkiewicz et al., 2004). PM2.5 exposure may aggravate cardiac afterload by increasing peripheral arterial pressure (Vincent et al., 2001; Nurkiewicz et al., 2004). Epidemiological studies showed that PM2.5 is tightly related to cardiovascular incidence and hospitalization (Le Tertre et al., 2002; Dominici et al., 2006). For example, the increased hospital admissions for heart failure is considered to be associated with short-term exposure of PM2.5 (Dominici et al., 2006). A multi-country epidemiological study demonstrated that an increase of 10 μg/m3 in PM2.5−10 size, was associated with a 0.76% increase in cardiovascular deaths and 0.58% in respiratory deaths (Analitis et al., 2006). A study using an animal model showed that short-term exposure to PM2.5 causes OS damage in both cardiac and pulmonary tissues (Gurgueira et al., 2002).
Studies have also indicated that systemic inflammation is induced by atmospheric pollution and inhaled PM may stimulate bone marrow to release white blood cells and blood platelets. This can increase blood viscosity, which promotes inflammatory responses and impairments in both pulmonary and cardiovascular tissues (Tan et al., 2000; van Eeden and Hogg, 2002). Since cardiac and pulmonary functions are interrelated, cardiovascular issues may contribute to AECOPD complications. Zuo et al. demonstrated that DEP induces NADPH oxidase-dependent ROS production in a diabetic model of rat cardiomyocyte, thereby exacerbating contractile dysfunction. Such adverse effect can be attenuated by the co-culture with antioxidants (Zuo et al., 2011a,b). Thrombotic events, especially deep vein thrombosis and pulmonary embolisms, are one of the frequent inducing factors and complications of AECOPD (Erelel et al., 2002). There are several ways that PM2.5 can alter hemostasis. By utilizing hamster models, Nemmar et al. reported that inhaled DEP can trigger platelet activation and aggregation, promoting thrombus formation (Nemmar et al., 2004). Likewise, the study on human subjects revealed that PM2.5 demonstrates pro-thrombotic effects through suppressing tissue plasminogen activator release and impairing endogenous fibrinolysis (Table 1; Mills et al., 2005; Lucking et al., 2008). Therefore, exposure to PM2.5 can trigger acute myocardial infarction and atherothrombosis. From the data analysis of Third National Health and Nutrition Examination Survey (NHANES III), researchers found that air pollution is responsible for increased white cell counts, platelet counts, and fibrinogens. These increased inflammatory cells and mediators may potentially lead to higher blood viscosity, further contributing to adverse cardiovascular outcomes (Schwartz, 2001).
Conclusion
Although the etiology and pathogenesis of AECOPD have not been fully elucidated, studies have shown that air pollution and PM2.5 exposure are closely related to AECOPD. PM2.5 induces both pulmonary and systemic OS and inflammation, affecting the patient's respiratory and immune function, respiratory microbiome, as well as cardiovascular system. Understanding the mechanisms of PM2.5-induced AECOPD contributes to patient risk management, which may reduce the likelihood of acute exacerbations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by Shanghai Key Laboratory of meteorological and health Fund QXJK201303 (China) and OSUCOM-HRS Fund 013000 (USA). We appreciate the assistance from Zan Xu, Benjamin Pannell, Andrew Graef, Alexander Ziegler, and Sundus Mohammad during the manuscript preparation.
Abbreviations
COPD, chronic obstructive pulmonary disease; AECOPD, acute exacerbation of COPD; PM, particulate matter; OS, oxidative stress; ROS, reactive oxygen species; AM, alveolar macrophage; DEP, diesel exhaust particle.
References
Aaron, S. D., Angel, J. B., Lunau, M., Wright, K., Fex, C., Le Saux, N., et al. (2001). Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 163, 349–355. doi: 10.1164/ajrccm.163.2.2003122
Adamkiewicz, G., Ebelt, S., Syring, M., Slater, J., Speizer, F. E., Schwartz, J., et al. (2004). Association between air pollution exposure and exhaled nitric oxide in an elderly population. Thorax 59, 204–209. doi: 10.1136/thorax.2003.006445
Adamson, I. Y. R., Prieditis, H., Hedgecock, C., and Vincent, R. (2000). Zinc is the toxic factor in the lung response to an atmospheric particulate sample. Toxicol. Appl. Pharmacol. 166, 111–119. doi: 10.1006/taap.2000.8955
Alexis, N. E., Lay, J. C., Zeman, K., Bennett, W. E., Peden, D. B., Soukup, J. M., et al. (2006). Biological material on inhaled coarse fraction particulate matter activates airway phagocytes in vivo in healthy volunteers. J. Allergy Clin. Immunol. 117, 1396–1403. doi: 10.1016/j.jaci.2006.02.030
Alghamdi, M. A., Shamy, M., Redal, M. A., Khoder, M., Awad, A. H., and Elserougy, S. (2014). Microorganisms associated particulate matter: a preliminary study. Sci. Total Environ. 479–480, 109–116. doi: 10.1016/j.scitotenv.2014.02.006
Analitis, A., Katsouyanni, K., Dimakopoulou, K., Samoli, E., Nikoloulopoulos, A. K., Petasakis, Y., et al. (2006). Short-term effects of ambient particles on cardiovascular and respiratory mortality. Epidemiology 17, 230–233. doi: 10.1097/01.ede.0000199439.57655.6b
Aust, A. E., Ball, J. C., Hu, A. A., Lighty, J. S., Smith, K. R., Straccia, A. M., et al. (2002). Particle characteristics responsible for effects on human lung epithelial cells. Res. Rep. Health Eff. Inst. 110, 1–65. discussion: 67–76.
Baulig, A., Garlatti, M., Bonvallot, V., Marchand, A., Barouki, R., Marano, F., et al. (2003). Involvement of reactive oxygen species in the metabolic pathways triggered by diesel exhaust particles in human airway epithelial cells. Am. J. Physiol. Lung. Cell. Mol. Physiol. 285, L671–L679. doi: 10.1152/ajplung.00419.2002
Becker, S., Mundandhara, S., Devlin, R. B., and Madden, M. (2005). Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: further mechanistic studies. Toxicol. Appl. Pharmacol. 207, 269–275. doi: 10.1016/j.taap.2005.01.023
Becker, S., Soukup, J. M., Sioutas, C., and Cassee, F. R. (2003). Response of human alveolar macrophages to ultrafine, fine, and coarse urban air pollution particles. Exp. Lung Res. 29, 29–44. doi: 10.1080/01902140303762
Bell, M. L., Dominici, F., Ebisu, K., Zeger, S. L., and Samet, J. M. (2007). Spatial and temporal variation in PM2.5 chemical composition in the United States for health effects studies. Environ. Health Perspect. 115, 989–995. doi: 10.1289/ehp.9621
Bhowmik, A., Seemungal, T. A., Sapsford, R. J., and Wedzicha, J. A. (2000). Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax 55, 114–120. doi: 10.1136/thorax.55.2.114
Biernacki, W. A., Kharitonov, S. A., and Barnes, P. J. (2003). Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax 58, 294–298. doi: 10.1136/thorax.58.4.294
Bose, S., Hansel, N., Tonorezos, E., Williams, D., Bilderback, A., Breysse, P., et al. (2015). Indoor particulate matter associated with systemic inflammation in COPD. J. Environ. Prot. 6, 566–572. doi: 10.4236/jep.2015.65051
Brook, R. D., Rajagopalan, S., Pope, C. A. III., Brook, J. R., Bhatnagar, A., Diez-Roux, A. V., et al. (2010). Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation 121, 2331–2378. doi: 10.1161/CIR.0b013e3181dbece1
Brown, J. S., Zeman, K. L., and Bennett, W. D. (2002). Ultrafine particle deposition and clearance in the healthy and obstructed lung. Am. J. Respir. Crit. Care Med. 166, 1240–1247. doi: 10.1164/rccm.200205-399OC
Cachon, B. F., Firmin, S., Verdin, A., Ayi-Fanou, L., Billet, S., Cazier, F., et al. (2014). Proinflammatory effects and oxidative stress within human bronchial epithelial cells exposed to atmospheric particulate matter (PM2.5 and PM > 2.5) collected from Cotonou, Benin. Environ. Pollut. 185, 340–351. doi: 10.1016/j.envpol.2013.10.026
Castranova, V., Ma, J. Y., Yang, H. M., Antonini, J. M., Butterworth, L., Barger, M. W., et al. (2001). Effect of exposure to diesel exhaust particles on the susceptibility of the lung to infection. Environ. Health Perspect. 109(Suppl. 4), 609–612. doi: 10.1289/ehp.01109s4609
Chatila, W. M., Thomashow, B. M., Minai, O. A., Criner, G. J., and Make, B. J. (2008). Comorbidities in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 5, 549–555. doi: 10.1513/pats.200709-148ET
Ciencewicki, J., Brighton, L., Wu, W. D., Madden, M., and Jaspers, I. (2006). Diesel exhaust enhances virus- and poly(I:C)-induced Toll-like receptor 3 expression and signaling in respiratory epithelial cells. Am. J. Physiol. Lung. Cell. Mol. Physiol. 290, L1154–L1163. doi: 10.1152/ajplung.00318.2005
Dagher, Z., Garçon, G., Billet, S., Gosset, P., Ledoux, F., Courcot, D., et al. (2006). Activation of different pathways of apoptosis by air pollution particulate matter (PM2.5) in human epithelial lung cells (L132) in culture. Toxicology 225, 12–24. doi: 10.1016/j.tox.2006.04.038
Dagher, Z., Garçon, G., Billet, S., Verdin, A., Ledoux, F., Courcot, D., et al. (2007). Role of nuclear factor-kappa B activation in the adverse effects induced by air pollution particulate matter (PM2.5) in human epithelial lung cells (L132) in culture. J. Appl. Toxicol. 27, 284–290. doi: 10.1002/jat.1211
Deng, X., Zhang, F., Rui, W., Long, F., Wang, L., Feng, Z., et al. (2013). PM2.5-induced oxidative stress triggers autophagy in human lung epithelial A549 cells. Toxicol. In Vitro 27, 1762–1770. doi: 10.1016/j.tiv.2013.05.004
Dergham, M., Lepers, C., Verdin, A., Billet, S., Cazier, F., Courcot, D., et al. (2012). Prooxidant and proinflammatory potency of air pollution particulate matter (PM2.5-0.3) produced in rural, urban, or industrial surroundings in human bronchial epithelial cells (BEAS-2B). Chem. Res. Toxicol. 25, 904–919. doi: 10.1021/tx200529v
Derscheid, R. J., and Ackermann, M. R. (2013). The innate immune system of the perinatal lung and responses to respiratory syncytial virus infection. Vet. Pathol. 50, 827–841. doi: 10.1177/0300985813480216
Dominici, F., McDermott, A., Zeger, S. L., and Samet, J. M. (2003). National maps of the effects of particulate matter on mortality: exploring geographical variation. Environ. Health Perspect. 111, 39–43. doi: 10.1289/ehp.5181
Dominici, F., Peng, R. D., Bell, M. L., Pham, L., McDermott, A., Zeger, S. L., et al. (2006). Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. JAMA 295, 1127–1134. doi: 10.1001/jama.295.10.1127
Donaldson, G. C., Seemungal, T. A., Patel, I. S., Bhowmik, A., Wilkinson, T. M., Hurst, J. R., et al. (2005). Airway and systemic inflammation and decline in lung function in patients with COPD. Chest 128, 1995–2004. doi: 10.1378/chest.128.4.1995
Erelel, M., Cuhadaroglu, C., Ece, T., and Arseven, O. (2002). The frequency of deep venous thrombosis and pulmonary embolus in acute exacerbation of chronic obstructive pulmonary disease. Respir. Med. 96, 515–518. doi: 10.1053/rmed.2002.1313
Fujimaki, H., Ui, N., Ushio, H., Nohara, K., and Endo, T. (2001). Roles of CD4+ and CD8+ T cells in adjuvant activity of diesel exhaust particles in mice. Int. Arch. Allergy Immunol. 124, 485–496. doi: 10.1159/000053784
Gan, W. Q., Man, S. F. P., Senthilselvan, A., and Sin, D. D. (2004). Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax 59, 574–580. doi: 10.1136/thx.2003.019588
Garçon, G., Dagher, Z., Zerimech, F., Ledoux, F., Courcot, D., Aboukais, A., et al. (2006). Dunkerque city air pollution particulate matter-induced cytotoxicity, oxidative stress and inflammation in human epithelial lung cells (L132) in culture. Toxicol. In Vitro 20, 519–528. doi: 10.1016/j.tiv.2005.09.012
GOLD (2015). From the Global Strategy for the Diagnosis, Management and Prevention of COPD. Global Initiative for Chronic Obstructive Lung Disease. Available online at: http://www.goldcopd.org/
Groenewegen, K. H., Postma, D. S., Hop, W. C. J., Wielders, P. L. M. L., Schlösser, N. J. J., Wouters, E. F. M., et al. (2008). Increased systemic inflammation is a risk factor for COPD exacerbations. Chest 133, 350–357. doi: 10.1378/chest.07-1342
Gualtieri, M., Mantecca, P., Corvaja, V., Longhin, E., Perrone, M. G., Bolzacchini, E., et al. (2009). Winter fine particulate matter from Milan induces morphological and functional alterations in human pulmonary epithelial cells (A549). Toxicol. Lett. 188, 52–62. doi: 10.1016/j.toxlet.2009.03.003
Gualtieri, M., Øvrevik, J., Holme, J. A., Perrone, M. G., Bolzacchini, E., Schwarze, P. E., et al. (2010). Differences in cytotoxicity versus pro-inflammatory potency of different PM fractions in human epithelial lung cells. Toxicol. In Vitro 24, 29–39. doi: 10.1016/j.tiv.2009.09.013
Gurgueira, S. A., Lawrence, J., Coull, B., Murthy, G. G., and González-Flecha, B. (2002). Rapid increases in the steady-state concentration of reactive oxygen species in the lungs and heart after particulate air pollution inhalation. Environ. Health Perspect. 110, 749–755. doi: 10.1289/ehp.02110749
Harrison, R. M., and Yin, J. X. (2000). Particulate matter in the atmosphere: which particle properties are important for its effects on health? Sci. Total Environ. 249, 85–101. doi: 10.1016/S0048-9697(99)00513-6
Inoue, K., Koike, E., Takano, H., Yanagisawa, R., Ichinose, T., and Yoshikawa, T. (2009). Effects of diesel exhaust particles on antigen-presenting cells and antigen-specific Th immunity in mice. Exp. Biol. Med. (Maywood). 234, 200–209. doi: 10.3181/0809-RM-285
Jaspers, I., Ciencewicki, J. M., Zhang, W. L., Brighton, L. E., Carson, J. L., Beck, M. A., et al. (2005). Diesel exhaust enhances influenza virus infections in respiratory epithelial cells. Toxicol. Sci. 85, 990–1002. doi: 10.1093/toxsci/kfi141
Karadag, F., Kirdar, S., Karul, A. B., and Ceylan, E. (2008). The value of C-reactive protein as a marker of systemic inflammation in stable chronic obstructive pulmonary disease. Eur. J. Intern. Med. 19, 104–108. doi: 10.1016/j.ejim.2007.04.026
Kleeman, M. J., Schauer, J. J., and Cass, G. R. (2000). Size and composition distribution of fine particulate matter emitted from motor vehicles. Environ. Sci. Technol. 34, 1132–1142. doi: 10.1021/es981276y
Knaapen, A. M., Shi, T. M., Borm, P. J. A., and Schins, R. P. F. (2002). Soluble metals as well as the insoluble particle fraction are involved in cellular DNA damage induced by particulate matter. Mol. Cell. Biochem. 234, 317–326. doi: 10.1023/A:1015970023889
Kreyling, W. G., Semmler, M., Erbe, F., Mayer, P., Takenaka, S., Schulz, H., et al. (2002). Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J. Toxicol. Environ. Health A 65, 1513–1530. doi: 10.1080/00984100290071649
Laden, F., Neas, L. M., Dockery, D. W., and Schwartz, J. (2000). Association of fine particulate matter from different sources with daily mortality in six US cities. Environ. Health Perspect. 108, 941–947. doi: 10.1289/ehp.00108941
Laden, F., Schwartz, J., Speizer, F. E., and Dockery, D. W. (2006). Reduction in fine particulate air pollution and mortality - extended follow-up of the Harvard six cities study. Am. J. Respir. Crit. Care Med. 173, 667–672. doi: 10.1164/rccm.200503-443OC
Lambert, A. L., Trasti, F. S., Mangum, J. B., and Everitt, J. I. (2003). Effect of preexposure to ultrafine carbon black on respiratory syncytial virus infection in mice. Toxicol. Sci. 72, 331–338. doi: 10.1093/toxsci/kfg031
Le Tertre, A., Medina, S., Samoli, E., Forsberg, B., Michelozzi, P., Boumghar, A., et al. (2002). Short-term effects of particulate air pollution on cardiovascular diseases in eight European cities. J. Epidemiol. Community Health 56, 773–779. doi: 10.1136/jech.56.10.773
Li, M. H., Fan, L. C., Mao, B., Yang, J. W., Choi, A. M., Cao, W. J., et al. (2015). Short term exposure to ambient fine particulate matter (PM2.5) increases hospitalizations and mortality of chronic obstructive pulmonary disease: a systematic review and meta-analysis. Chest. doi: 10.1378/chest.15-0513. [Epub ahead of print].
Ling, S. H., McDonough, J. E., Gosselink, J. V., Elliott, W. M., Hayashi, S., Hogg, J. C., et al. (2011). Patterns of retention of particulate matter in lung tissues of patients with COPD: potential role in disease progression. Chest 140, 1540–1549. doi: 10.1378/chest.10-2281
Ling, S. H., and van Eeden, S. F. (2009). Particulate matter air pollution exposure: role in the development and exacerbation of chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 4, 233–243. doi: 10.2147/COPD.S5098
Lucking, A. J., Lundback, M., Mills, N. L., Faratian, D., Barath, S. L., Pourazar, J., et al. (2008). Diesel exhaust inhalation increases thrombus formation in man. Eur. Heart. J. 29, 3043–3051. doi: 10.1093/eurheartj/ehn464
Lundborg, M., Dahlén, S. E., Johard, U., Gerde, P., Jarstrand, C., Camner, P., et al. (2006). Aggregates of ultrafine particles impair phagocytosis of microorganisms by human alveolar macrophages. Environ. Res. 100, 197–204. doi: 10.1016/j.envres.2005.08.007
Maciejczyk, P., Zhong, M., Lippmann, M., and Chen, L. C. (2010). Oxidant generation capacity of source-apportioned PM2.5. Inhal. Toxicol. 22(Suppl. 2), 29–36. doi: 10.3109/08958378.2010.509368
Marchini, T., Magnani, N. D., Paz, M. L., Vanasco, V., Tasat, D., González Maglio, D. H., et al. (2014). Time course of systemic oxidative stress and inflammatory response induced by an acute exposure to Residual Oil Fly Ash. Toxicol. Appl. Pharmacol. 274, 274–282. doi: 10.1016/j.taap.2013.11.013
Marrie, T. J. (2000). Community-acquired pneumonia in the elderly. Clin. Infect. Dis. 31, 1066–1078. doi: 10.1086/318124
Matthews, N. C., Faith, A., Pfeffer, P., Lu, H., Kelly, F. J., Hawrylowicz, C. M., et al. (2014). Urban particulate matter suppresses priming of T helper type 1 cells by granulocyte/macrophage colony-stimulating factor-activated human dendritic cells. Am. J. Respir. Cell. Mol. Biol. 50, 281–291. doi: 10.1165/rcmb.2012-0465OC
McGuire, A., Irwin, D. E., Fenn, P., Gray, A., Anderson, P., Lovering, A., et al. (2001). The excess cost of acute exacerbations of chronic bronchitis in patients aged 45 and older in England and Wales. Value Health 4, 370–375. doi: 10.1046/j.1524-4733.2001.45049.x
Medina-Ramón, M., Zanobetti, A., and Schwartz, J. (2006). The effect of ozone and PM10 on hospital admissions for pneumonia and chronic obstructive pulmonary disease: a national multicity study. Am. J. Epidemiol. 163, 579–588. doi: 10.1093/aje/kwj078
Menetrez, M. Y., Foarde, K. K., and Ensor, D. S. (2001). An analytical method for the measurement of nonviable bioaerosols. J. Air. Waste Manag. Assoc. 51, 1436–1442. doi: 10.1080/10473289.2001.10464365
Meng, Z. Q., and Zhang, Q. X. (2006). Oxidative damage of dust storm fine particles instillation on lungs, hearts and livers of rats. Environ. Toxicol. Pharmacol. 22, 277–282. doi: 10.1016/j.etap.2006.04.005
Mills, N. L., Törnqvist, H., Robinson, S. D., Gonzalez, M., Darnley, K., Macnee, W., et al. (2005). Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation 112, 3930–3936. doi: 10.1161/CIRCULATIONAHA.105.588962
Montiel-Dávalos, A., Ibarra-Sánchez Mde, J., Ventura-Gallegos, J. L., Alfaro-Moreno, E., and López-Marure, R. (2010). Oxidative stress and apoptosis are induced in human endothelial cells exposed to urban particulate matter. Toxicol. In Vitro 24, 135–141. doi: 10.1016/j.tiv.2009.08.004
Mushtaq, N., Ezzati, M., Hall, L., Dickson, I., Kirwan, M., Png, K. M., et al. (2011). Adhesion of Streptococcus pneumoniae to human airway epithelial cells exposed to urban particulate matter. J. Allergy Clin. Immunol. 127, 1236–1242.e1232. doi: 10.1016/j.jaci.2010.11.039
National Heart, Lung, Blood Institute (NHLBI). (2009). Morbidity and Mortality: 2009 Chart Book Cardiovascular, Lung, and Blood Diseases. Bethesda, MD: National Institutes of Health (NIH).
Nemmar, A., Hoet, P. H., Vanquickenborne, B., Dinsdale, D., Thomeer, M., Hoylaerts, M. F., et al. (2002). Passage of inhaled particles into the blood circulation in humans. Circulation 105, 411–414. doi: 10.1161/hc0402.104118
Nemmar, A., Hoylaerts, M. F., Hoet, P. H., and Nemery, B. (2004). Possible mechanisms of the cardiovascular effects of inhaled particles: systemic translocation and prothrombotic effects. Toxicol. Lett. 149, 243–253. doi: 10.1016/j.toxlet.2003.12.061
Neupane, B., Jerrett, M., Burnett, R. T., Marrie, T., Arain, A., and Loeb, M. (2010). Long-term exposure to ambient air pollution and risk of hospitalization with community-acquired pneumonia in older adults. Am. J. Respir. Crit. Care Med. 181, 47–53. doi: 10.1164/rccm.200901-0160OC
Nicod, L. P. (2005). Lung defences: an overview. Eur. Respir Rev. 14, 45–50. doi: 10.1183/09059180.05.00009501
Nightingale, J. A., Maggs, R., Cullinan, P., Donnelly, L. E., Rogers, D. F., Kinnersley, R., et al. (2000). Airway inflammation after controlled exposure to diesel exhaust particulates. Am. J. Respir. Crit. Care Med. 162, 161–166. doi: 10.1164/ajrccm.162.1.9908092
Nurkiewicz, T. R., Porter, D. W., Barger, M., Castranova, V., and Boegehold, M. A. (2004). Particulate matter exposure impairs systemic microvascular endothelium-dependent dilation. Environ. Health Perspect. 112, 1299–1306. doi: 10.1289/ehp.7001
Ogino, K., Zhang, R., Takahashi, H., Takemoto, K., Kubo, M., Murakami, I., et al. (2014). Allergic airway inflammation by nasal inoculation of particulate matter (PM2.5) in NC/Nga mice. PLoS ONE 9:e92710. doi: 10.1371/journal.pone.0092710
Patel, I. S., Seemungal, T. A., Wilks, M., Lloyd-Owen, S. J., Donaldson, G. C., and Wedzicha, J. A. (2002). Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax 57, 759–764. doi: 10.1136/thorax.57.9.759
Pérez-Trallero, E., Marimón, J. M., Larruskain, J., Alonso, M., and Ercibengoa, M. (2011). Antimicrobial susceptibilities and serotypes of Streptococcus pneumoniae isolates from elderly patients with pneumonia and acute exacerbation of chronic obstructive pulmonary disease. Antimicrob. Agents Chemother. 55, 2729–2734. doi: 10.1128/AAC.01546-10
Perotin, J. M., Dury, S., Renois, F., Deslee, G., Wolak, A., Duval, V., et al. (2013). Detection of multiple viral and bacterial infections in acute exacerbation of chronic obstructive pulmonary disease: a pilot prospective study. J. Med. Virol. 85, 866–873. doi: 10.1002/jmv.23495
Pozzi, R., De Berardis, B., Paoletti, L., and Guastadisegni, C. (2003). Inflammatory mediators induced by coarse (PM2.5-10) and fine (PM2.5) urban air particles in RAW 264.7 cells. Toxicology 183, 243–254. doi: 10.1016/S0300-483X(02)00545-0
Reibman, J., Hsu, Y., Chen, L. C., Bleck, B., and Gordon, T. (2003). Airway epithelial cells release MIP-3alpha/CCL20 in response to cytokines and ambient particulate matter. Am. J. Respir. Cell. Mol. Biol. 28, 648–654. doi: 10.1165/rcmb.2002-0095OC
Riva, D. R., Magalhães, C. B., Lopes, A. A., Lancas, T., Mauad, T., Malm, O., et al. (2011). Low dose of fine particulate matter (PM2.5) can induce acute oxidative stress, inflammation and pulmonary impairment in healthy mice. Inhalation Toxicol. 23, 257–267. doi: 10.3109/08958378.2011.566290
Saliba, N. A., El Jam, F., El Tayar, G., Obeid, W., and Roumie, M. (2010). Origin and variability of particulate matter (PM10 and PM2.5) mass concentrations over an Eastern Mediterranean city. Atmos. Res. 97, 106–114. doi: 10.1016/j.atmosres.2010.03.011
Sarnat, J. A., Schwartz, J., and Suh, H. H. (2001). Fine particulate air pollution and mortality in 20 US cities. New Engl. J. Med. 344, 1253–1254. doi: 10.1056/NEJM200104193441614
Schwartz, J. (2001). Air pollution and blood markers of cardiovascular risk. Environ. Health Perspect. 109(Suppl. 3), 405–409. doi: 10.1289/ehp.01109s3405
Schwarze, P. E., Ovrevik, J., Låg, M., Refsnes, M., Nafstad, P., Hetland, R. B., et al. (2006). Particulate matter properties and health effects: consistency of epidemiological and toxicological studies. Hum. Exp. Toxicol. 25, 559–579. doi: 10.1177/096032706072520
Seriani, R., Junqueira, M. S., de Toledo, A. C., Martins, M. A., Seckler, M., Alencar, A. M., et al. (2015). Diesel exhaust particulates affect cell signaling, mucin profiles, and apoptosis in trachea explants of Balb/C mice. Environ. Toxicol. 30, 1297–1308. doi: 10.1002/tox.22000
Sethi, S., Evans, N., Grant, B. J., and Murphy, T. F. (2002). New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N. Engl. J. Med. 347, 465–471. doi: 10.1056/NEJMoa012561
Sethi, S., Maloney, J., Grove, L., Wrona, C., and Berenson, C. S. (2006). Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 173, 991–998. doi: 10.1164/rccm.200509-1525OC
Shukla, A., Timblin, C., Berube, K., Gordon, T., McKinney, W., Driscoll, K., et al. (2000). Inhaled particulate matter causes expression of nuclear factor (NF)-kappaB-related genes and oxidant-dependent NF-kappaB activation in vitro. Am. J. Respir. Cell. Mol. Biol. 23, 182–187. doi: 10.1165/ajrcmb.23.2.4035
Sillanpaa, M., Hillamo, R., Saarikoski, S., Frey, A., Pennanen, A., Makkonen, U., et al. (2006). Chemical composition and mass closure of particulate matter at six urban sites in Europe. Atmos. Environ. 40, S212–S223. doi: 10.1016/j.atmosenv.2006.01.063
Sin, D. D., and Man, S. F. (2003). Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation 107, 1514–1519. doi: 10.1161/01.CIR.0000056767.69054.B3
Sørensen, M., Schins, R. P., Hertel, O., and Loft, S. (2005). Transition metals in personal samples of PM2.5 and oxidative stress in human volunteers. Cancer Epidemiol. Biomarkers Prev. 14, 1340–1343. doi: 10.1158/1055-9965.EPI-04-0899
Soriano, J. B., and Lamprecht, B. (2012). Chronic obstructive pulmonary disease: a worldwide problem. Med. Clin. N. Am. 96, 671–680. doi: 10.1016/j.mcna.2012.02.005
Squadrito, G. L., Cueto, R., Dellinger, B., and Pryor, W. A. (2001). Quinoid redox cycling as a mechanism for sustained free radical generation by inhaled airborne particulate matter. Free Radical Biol. Med. 31, 1132–1138. doi: 10.1016/S0891-5849(01)00703-1
Tan, W. C., Qiu, D., Liam, B. L., Ng, T. P., Lee, S. H., van Eeden, S. F., et al. (2000). The human bone marrow response to acute air pollution caused by forest fires. Am. J. Respir. Crit. Care Med. 161, 1213–1217. doi: 10.1164/ajrccm.161.4.9904084
Tsai, S. S., and Yang, C. Y. (2014). Fine particulate air pollution and hospital admissions for pneumonia in a subtropical city: Taipei, Taiwan. J. Toxicol. Environ. Health A 77, 192–201. doi: 10.1080/15287394.2013.853337
Upadhyay, D., Panduri, V., Ghio, A., and Kamp, D. W. (2003). Particulate matter induces alveolar epithelial cell DNA damage and apoptosis: role of free radicals and the mitochondria. Am. J. Respir. Cell. Mol. Biol. 29, 180–187. doi: 10.1165/rcmb.2002-0269OC
Valavanidis, A., Fiotakis, K., and Vlachogianni, T. (2008). Airborne particulate matter and human health: toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J. Environ. Sci. Health C. Environ. Carcinog. Ecotoxicol. Rev. 26, 339–362. doi: 10.1080/10590500802494538
Valavanidis, A., Vlachogianni, T., Fiotakis, K., and Loridas, S. (2013). Pulmonary oxidative stress, inflammation and cancer: respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int. J. Env. Res. Public Health 10, 3886–3907. doi: 10.3390/ijerph10093886
van Eeden, S. F., and Hogg, J. C. (2002). Systemic inflammatory response induced by particulate matter air pollution: the importance of bone-marrow stimulation. J. Toxicol. Environ. Health A 65, 1597–1613. doi: 10.1080/00984100290071685
van Eeden, S. F., Yeung, A., Quinlam, K., and Hogg, J. C. (2005). Systemic response to ambient particulate matter: relevance to chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2, 61–67. doi: 10.1513/pats.200406-035MS
van Zijverden, M., van der Pijl, A., Bol, M., van Pinxteren, F. A., de Haar, C., Penninks, A. H., et al. (2000). Diesel exhaust, carbon black, and silica particles display distinct Th1/Th2 modulating activity. Toxicol. Appl. Pharmacol. 168, 131–139. doi: 10.1006/taap.2000.9013
Vincent, R., Kumarathasan, P., Goegan, P., Bjarnason, S. G., Guénette, J., Bérubé, D., et al. (2001). Inhalation toxicology of urban ambient particulate matter: acute cardiovascular effects in rats. Res. Rep. Health Eff. Inst. 104, 5–54. discussion: 55–62.
Wang, S. Z., Rosenberger, C. L., Bao, Y. X., Stark, J. M., and Harrod, K. S. (2003). Clara cell secretory protein modulates lung inflammatory and immune responses to respiratory syncytial virus infection. J. Immunol. 171, 1051–1060. doi: 10.4049/jimmunol.171.2.1051
Wilson, W. E., Chow, J. C., Claiborn, C., Fusheng, W., Engelbrecht, J., and Watson, J. G. (2002). Monitoring of particulate matter outdoors. Chemosphere 49, 1009–1043. doi: 10.1016/S0045-6535(02)00270-9
Wu, Y., McEwen, G. D., Tang, M., Yu, T., Dimmick, J. T., Zhou, A., et al. (2013). Sensing biophysical alterations of human lung epithelial cells (A549) in the context of toxicity effects of diesel exhaust particles. Cell Biochem. Biophys. 67, 1147–1156. doi: 10.1007/s12013-013-9618-4
Xu, D., Huang, N., Wang, Q., and Liu, H. (2008). Study of ambient PM2.5 on the influence of the inflammation injury and the immune function of subchronic exposure rats [Chinese]. Wei Sheng Yan Jiu 37, 423–428.
Yanagisawa, R., Takano, H., Inoue, K., Ichinose, T., Sadakane, K., Yoshino, S., et al. (2003). Enhancement of acute lung injury related to bacterial endotoxin by components of diesel exhaust particles. Thorax 58, 605–612. doi: 10.1136/thorax.58.7.605
Yang, H. M., Antonini, J. M., Barger, M. W., Butterworth, L., Roberts, B. R., Ma, J. K., et al. (2001). Diesel exhaust particles suppress macrophage function and slow the pulmonary clearance of Listeria monocytogenes in rats. Environ. Health Perspect. 109, 515–521. doi: 10.1289/ehp.01109515
Yin, X. J., Schafer, R., Ma, J. Y., Antonini, J. M., Weissman, D. D., Siegel, P. D., et al. (2002). Alteration of pulmonary immunity to Listeria monocytogenes by diesel exhaust particles (DEPs). I. Effects of DEPs on early pulmonary responses. Environ. Health Perspect. 110, 1105–1111. doi: 10.1289/ehp.021101105
Zeki, A. A., Schivo, M., Chan, A., Albertson, T. E., and Louie, S. (2011). The asthma-COPD overlap syndrome: a common clinical problem in the elderly. J. Allergy (Cairo) 2011, 861926. doi: 10.1155/2011/861926
Zelikoff, J. T., Chen, L. C., Cohen, M. D., Fang, K., Gordon, T., Li, Y., et al. (2003). Effects of inhaled ambient particulate matter on pulmonary antimicrobial immune defense. Inhal. Toxicol. 15, 131–150. doi: 10.1080/08958370304478
Zhou, H., and Kobzik, L. (2007). Effect of concentrated ambient particles on macrophage phagocytosis and killing of Streptococcus pneumoniae. Am. J. Respir. Cell. Mol. Biol. 36, 460–465. doi: 10.1165/rcmb.2006-0293OC
Zuo, L., He, F., Sergakis, G. G., Koozehchian, M. S., Stimpfl, J. N., Rong, Y., et al. (2014). Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Lung. Cell. Mol. Physiol. 307, L205–L218. doi: 10.1152/ajplung.00330.2013
Zuo, L., Nogueira, L., and Hogan, M. C. (2011a). Reactive oxygen species formation during tetanic contractions in single isolated Xenopus myofibers. J. Appl. Physiol. (1985) 111, 898–904. doi: 10.1152/japplphysiol.00398.2011
Zuo, L., Otenbaker, N. P., Rose, B. A., and Salisbury, K. S. (2013). Molecular mechanisms of reactive oxygen species-related pulmonary inflammation and asthma. Mol. Immunol. 56, 57–63. doi: 10.1016/j.molimm.2013.04.002
Keywords: AECOPD, PM2.5, oxidative stress, inflammation, alveolar macrophages
Citation: Ni L, Chuang C-C and Zuo L (2015) Fine particulate matter in acute exacerbation of COPD. Front. Physiol. 6:294. doi: 10.3389/fphys.2015.00294
Received: 27 July 2015; Accepted: 05 October 2015;
Published: 23 October 2015.
Edited by:
Yu Ru Kou, National Yang-Ming University, TaiwanCopyright © 2015 Ni, Chuang and Zuo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zuo, enVvLjRAb3N1LmVkdQ==