 Raquel R. Bartz
Raquel R. Bartz Hagir B. Suliman
Hagir B. Suliman Claude A. Piantadosi1,2,3,4
Claude A. Piantadosi1,2,3,4- 1Department of Anesthesiology, Duke University School of Medicine, Durham, NC, USA
- 2Department of Medicine, Duke University School of Medicine, Durham, NC, USA
- 3Department of Pathology, Duke University School of Medicine, Durham, NC, USA
- 4Durham Veterans Affairs Hospital, Durham, NC, USA
Oxidative and nitrosative stress are primary contributors to the loss of myocardial tissue in insults ranging from ischemia/reperfusion injury from coronary artery disease and heart transplantation to sepsis-induced myocardial dysfunction and drug-induced myocardial damage. This cell damage caused by oxidative and nitrosative stress leads to mitochondrial protein, DNA, and lipid modifications, which inhibits energy production and contractile function, potentially leading to cell necrosis and/or apoptosis. However, cardiomyocytes have evolved an elegant set of redox-sensitive mechanisms that respond to and contain oxidative and nitrosative damage. These responses include the rapid induction of antioxidant enzymes, mitochondrial DNA repair mechanisms, selective mitochondrial autophagy (mitophagy), and mitochondrial biogenesis. Coordinated cytoplasmic to nuclear cell-signaling and mitochondrial transcriptional responses to the presence of elevated cytoplasmic oxidant production, e.g., H2O2, allows nuclear translocation of the Nfe2l2 transcription factor and up-regulation of downstream cytoprotective genes such as heme oxygenase-1 which generates physiologic signals, such as CO that up-regulates Nfe212 gene transcription. Simultaneously, a number of other DNA binding transcription factors are expressed and/or activated under redox control, such as Nuclear Respiratory Factor-1 (NRF-1), and lead to the induction of genes involved in both intracellular and mitochondria-specific repair mechanisms. The same insults, particularly those related to vascular stress and inflammation also produce elevated levels of nitric oxide, which also has mitochondrial protein thiol-protective functions and induces mitochondrial biogenesis through cyclic GMP-dependent and perhaps other pathways. This brief review provides an overview of these pathways and interconnected cardiac repair mechanisms.
Intact adult cardiomyocytes can be injured not only by ischemia, but also by various forms of oxidative and nitrosative stress following ischemia/reperfusion after myocardial infarction, sepsis-induced myocardial dysfunction and demand ischemia (Ferrari et al., 1989; Loeper et al., 1991a,b; Iqbal et al., 2002; Supinski et al., 2009). A major site of intracellular damage, particularly in the presence of pre-existing metabolic disease, such as diabetes, is the large population of mitochondria, which occupy 30% of the cardiomyocyte cytoplasmic volume (Laguens and Gómez-Dumm, 1967; Kane et al., 1975; Jennings and Ganote, 1976). However, the heart has an elegant system of anti-oxidant defenses and cell repair mechanisms that respond rapidly to and protect cardiomyocytes against oxidative and nitrosative stress, and which govern the maintenance and restoration of functional mitochondrial populations. This system involves the transcriptional relation of genes responsible for mitochondrial quality control (QC), an integrated process designed to optimize energy homeostasis. In cardiomyocytes, many of these mechanisms are redox-sensitive, and this short review concentrates on those mechanisms that are most important to the maintenance of cardiomyocyte function during periods of oxidative and nitrosative stress, and thereby serve to preserve cardiomyocyte viability and oppose apoptosis and necrosis.
Production of Reactive Oxygen and Nitrogen Species
As highly metabolic cells, cardiomyocytes maintain a high cellular store of phosphocreatine and adenosine triphosphate (ATP), which is required for continuous cardiac function. The large-scale process of generating adenosine triphosphate (ATP) from carbon substrate, which in the heart relies mostly on fatty acids, also leads to the production of reactive oxygen and reactive nitrogen species (ROS/RNS) by the mitochondrial electron transport chain (ETC). This ROS production is primarily in the form of superoxide (·) and RNS in the form of peroxinitrite. Superoxide is generated by the incomplete one-electron reduction of oxygen mainly at Complexes I and III (Cadenas et al., 1977; Turrens et al., 1985; Aon et al., 2003; Chen et al., 2003; Murphy, 2009) and is highly reactive. Under normal mitochondrial conditions, · undergoes rapid dismutation either spontaneously or by mitochondrial (Mn) superoxide dismutase (SOD2) to hydrogen peroxide (H2O2). H2O2 exits the mitochondrion to the cytoplasm, where it is relatively soluble, and in the cytoplasm undergoes further catalysis to water (H2O) and oxygen (O2) by catalase (Cat), glutathione peroxidases, glutathione, thioredoxin, and the peroxiredoxins (Balaban et al., 2005; Aon et al., 2012). Additionally, thioredoxin reductase-2 has been shown to control thioredoxin-2 and peroxiredoxin-3 and thus controlling H2O2 emission from the mitochondria independent of glutathione reduction (Stanley et al., 2011). However, under certain circumstances, H2O2 in concert with endogenous production of carbon monoxide (CO) and nitric oxide (NO) serve as important redox signals for anti-oxidant protection and for the cellular repair mechanisms discussed in this review.
Tissue specific H2O2 production and its related signaling effects appear to be dependent on factors such as age, diet, and exercise capacity. For instance, elevated mitochondrial H2O2 is found in cardiac tissues of sedentary rats, and decreases with both exercise and high-fat, high sucrose diets unlike in skeletal muscle where a high-fat, high-sucrose diet leads to greatly elevated mitochondrial H2O2. These tissue-specific differences are due mainly to different levels of thioredoxin-2 reductase expression in cardiac compared to skeletal muscle in sedentary animals (Fisher-Wellman et al., 2013). Although redox-specific signaling capacities of different muscle types were not examined in this study, other studies have shown that when cellular ROS (H2O2) production rates are properly balanced by the presence of intracellular anti-oxidant enzymes like SOD2 and Cat, there is little or no oxidative stress and the intracellular homeostasis is maintained. However, when myocardial mitochondrial ROS generation exceeds the local antioxidant capacity, such as during ischemia/reperfusion and in sepsis (Ide et al., 1999; Gauthier et al., 2013; Cortassa et al., 2014), oxidative mitochondrial damage becomes problematic, and multiple intracellular adaptive mechanisms are up-regulated. These mechanisms result in the recruitment of cell pro-survival processes that afford tissue protection and prevent the progression to apoptosis and/or necrosis.
Mitochondrial Redox Signaling of Mitochondrial Biogenesis
One of the most important adaptive mechanisms in response to oxidative stress is genetic and involves the anti-oxidant response element (ARE) transcriptional pathway which responds to the presence of chemical electrophiles and to elevated cytoplasmic H2O2 content (Itoh et al., 1997). When cytoplasmic electrophilic or oxidative ([H2O2]) stress increases, the cytoplasmic protein and binding partner of Nuclear factor erythroid-derived-like 2 (Nfe2l2 or Nrf-2), Kelch-like ECH-associated protein 1 (Keap1), releases Nfe2l2, which translocates to the nucleus (Itoh et al., 1999b). In the nucleus, Nfe2l2 binds to ARE promoter regions of genes that carry an RTGACnnnGC motif including phase II detoxifying enzymes, certain anti-oxidant enzymes such as SOD2, cytoprotective enzymes such as heme oxygenase-1 (HO-1), and genes for signaling proteins required for mitochondrial biogenesis such as Nuclear Respiratory Factor-1 (NRF-1), and for mitochondrial DNA (mtDNA) repair such as 8-oxoguanine glycosylase (Ogg1), and several proteins discovered more recently that are required for mitophagy (Rushmore et al., 1991; Favreau and Pickett, 1995; Prestera et al., 1995; Alam et al., 1999; Itoh et al., 1999a,b; Jaloszynski et al., 2007; Cherry et al., 2014; Chang et al., 2015). Each of these proteins and related pathways function to protect cells from oxidative stress and to prevent apoptosis/cell death, especially via maintenance of mitochondrial biogenesis and mitophagy, which together comprise an integrated mitochondrial quality control (QC) system (Figure 1).
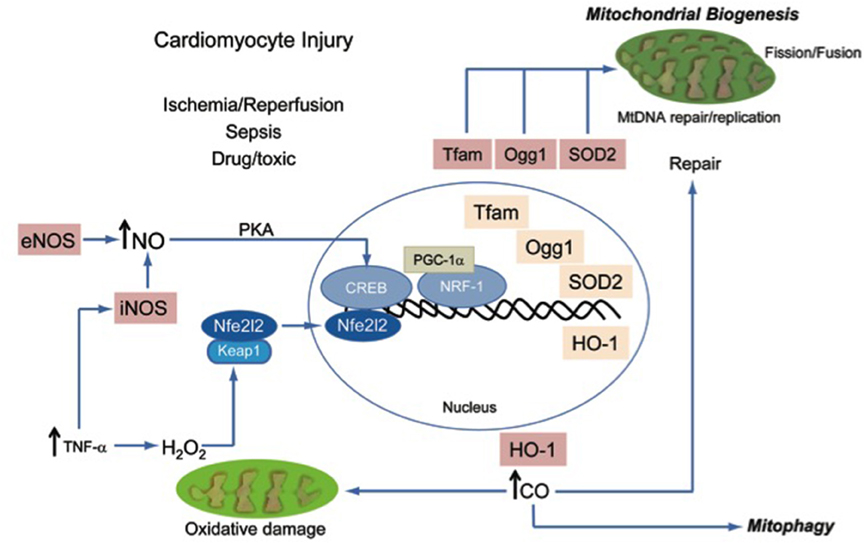
Figure 1. A schematic diagram of known processes that are recruited to maintain mitochondrial quality control and to prevent energy failure during oxidative and nitrosative injury.
A metabolically active tissue, such as myocardium, may be particularly susceptible to ROS. For instance, the inflammatory cascade triggered by local TNF-α production as a result of myocardial infarction leads to mtDNA damage, to lipid peroxidation, and to protein oxidation (carbonylation). All of these oxidized products are potential stimuli for induction of cellular apoptosis (Irwin et al., 1999). In order to maintain adequate function in the face of this damage, the critical constituents of the mitochondria, such as mtDNA, proteins, and lipids must be repaired or replaced. However, massive damage cannot be repaired and the organelle must be removed by mitophagy. For instance, hyper-stimulation of the innate immune system, such as Toll-like receptor 4 by endotoxin and other microbial products released into the circulation leads to oxidative and nitrosative stress-induced cardiomyocyte dysfunction and subsequent mtDNA damage in the form of oxidized mtDNA lesions which must ultimately be removed or repaired in order for mtDNA replication and mitochondrial biogenesis to occur (Suliman et al., 2004). On the other hand, activation of mitophagy, a form of selective macroautophagy by which cells remove dysfunctional and ROS-producing mitochondria, isolates and contains these damage-causing organelles, and hence essentially functions as a macro antioxidant system. The proper ordering of these molecular repair mechanisms is necessary in order for mitochondrial biogenesis to ensue smoothly.
As mentioned, mitochondrial biogenesis requires the coordination of both the mitochondrial and the nuclear genomes and the synthesis, activation and nuclear translocation of several transcription factors and their nuclear co-activators including NRF-1, NRF-2 (mouse ortholog GABP-α), and PGC-1α, PGC-1β, and PRC (Evans and Scarpulla, 1990; Chau et al., 1992; Scarpulla, 1997, 2002; Wu et al., 1999; Andersson and Scarpulla, 2001; Lin et al., 2002; Kelly and Scarpulla, 2004; Gleyzer et al., 2005; Vercauteren et al., 2008). These nuclear transcription factors and their co-activators are responsible for the regulation of genes whose products regulate the hundreds of nuclear-encoded mitochondrial proteins (NEMPs) involved in the many functions of cardiac mitochondria including oxidative phosphorylation and calcium homeostasis. NEMPs also regulate the function of the mitochondrial genome (mtDNA). After synthesis, these proteins are imported into mitochondria by specialized outer and inner membrane protein complexes (Mokranjac and Neupert, 2009), also under the control of NRF-1.
The nuclear transcription factor NRF-1 and its co-activator, PGC-1α, indirectly regulate the mitochondrial genome by the up-regulation of mitochondrial transcription factor A (Tfam) and mitochondrial transcription factor B which enable transcription of the mitochondrial genome (Carter and Avadhani, 1994; Virbasius and Scarpulla, 1994; Dairaghi et al., 1995; Carrodeguas et al., 1996; Shadel and Clayton, 1996; Larsson et al., 1998; Falkenberg et al., 2002; Ekstrand et al., 2004; Gleyzer et al., 2005). Oxidative stress occurring in the myocardium as a result of ischemia/reperfusion injury or other cytotoxic insults, for example, in the form of cancer chemotherapeutics or in response to sepsis-induced oxidative stress leads to nuclear translocation of NRF-1 and up-regulation of its co-activator, PGC-1α (Suliman et al., 2004; Hickson-Bick et al., 2008). Transcription factor binding by one of the PGC-1α family members such as PGC-1β or PRC is required for mitochondrial biogenesis. In conjunction, adaptive cytoprotective mechanisms regulated by the endogenous gaseous signaling molecules are able to stimulate mitochondrial biogenesis.
Physiologic Carbon Monoxide (CO) and Mitochondrial Biogenesis
A major example of such a cytoprotective pathway induced by oxidative and nitrosative stress involves the up-regulation of HO-1 (Hmox1), which catalyzes the breakdown of free heme, a cellular toxin. During periods of oxidative stress, HO-1 levels increase in the heart, and the enzyme produces physiologic carbon monoxide (CO) by breakage of the heme ring to form biliverdin (Maines, 1988; Maulik et al., 1996; Sharma et al., 1996). Although often known for its damaging properties to the cell, CO produced endogenously by HO-1 has been shown to offer multifaceted cytoprotection. For instance, CO, through the functional consequences of reduced (Fe2+) heme protein binding, induces targeted ROS signaling that initiates cellular repair mechanisms and mitochondrial biogenesis. The latter occurs in part through promotion of the cytoplasmic release and translocation of Nfe2l2 (Nrf-2) into the nucleus. As mentioned Nfe2l2 nuclear translocation leads not only to the induction of specific anti-oxidant enzymes, but also activation of NRF-1-dependent mitochondrial biogenesis (Suliman et al., 2007b; Piantadosi et al., 2008). Moreover, this redox-pathway is closely coupled to the mitophagy process that sequesters and eliminates damaged mitochondria from the cell (Strohecker and White, 2014; Chang et al., 2015). Current technological limits make cellular CO measurements difficult, particularly with respect to the spatial localization of CO production. Even though the local concentration at which CO is toxic is unknown, when circulating carboxyhemoglobin concentrations exceeds 15%, the decrease in hemoglobin oxygen carrying capacity is reduced enough to produce symptoms of CO poisoning.
The Nfe2l2 pathway for HO-1 induction and subsequent mitochondrial biogenesis are important for cytoprotection. It has been found using siRNA techniques that reduction of Nfe2l2 (Nrf-2) activity in Hep9C2 cardiomyoblasts results in reduced cell survival in response to hypoxia with or without reoxygenation (Kolamunne et al., 2013). In experimental models of congestive heart failure (CHF), decreased levels of certain cytochrome oxidase subunits, PGC-1α and the mitochondrial transcription factor, Tfam have been reported (Garnier et al., 2003). A cardiomyopathy induced in humans by a widely used chemotherapeutic agent, doxorubicin has been shown to directly damage mtDNA (Palmeira et al., 1997). Protection against doxorubicin-induced cardiomyopathy by HO-1 activation in the heart leads to the induction of mitochondrial biogenesis from the generation of physiological CO (Suliman et al., 2007a). Subsequent investigations have also shown that CO generated from HO-1 overexpression stimulates mitochondrial SOD-2 up-regulation and increases H2O2 production, which activates AKT/PKB, deactivates GSK3β and induces NRF-1 leading to the stimulation of mitochondrial biogenesis (Piantadosi et al., 2008). It has also been shown that the administration of a CO-releasing molecule leads to increased HO-1 expression levels and protects mice through restoration of cardiac mitochondrial biogenesis in response to sepsis. This suggests a mechanism by which the physiological stimulation of CO-dependent pro-survival mechanisms might have some future therapeutic potential (Lancel et al., 2009). In addition, transgenic mice overexpressing HO-1 is also protective against diabetic cardiomyopathy in mice, and this set of circumstances is associated with restored expression of Amp-associated kinase (AMPK) and AKT/PKB activation compared with mice with HO-1 deletion (Zhao et al., 2013). Such observations provide evidence of molecular linkage between the redox salvage pathways outlined here and the strictly energy-and/or calcium-dependent pathways of mitochondrial QC in the heart covered elsewhere (Calì et al., 2013; Dorn, 2015; Song et al., 2015).
A part of the mitochondrial QC program requires mitophagy—the uptake of dysfunctional mitochondria into lysozomes and their cellular degradation. An important role for Nfe2l2 (Nrf-2) and the HO-1/CO system has been shown in autophagic and presumably mitophagic pathways (Unuma et al., 2013a,b; Liu et al., 2014). Identification of damaged mitochondria and their packaging into autophagosomes requires the up-regulation and mitochondrial targeting of certain redox-sensitive proteins, most visibly p62 or Sequestosome-1 (Hamacher-Brady et al., 2006; Fujita et al., 2011; Huang et al., 2011; Stepkowski and Kruszewski, 2011; Darvekar et al., 2014; Haga et al., 2014; Chang et al., 2015). The p62 protein is an autophagy receptor that interacts directly with both the cargo to be degraded and the autophagy modifier protein, LC3. It is required for the formation and autophagic degradation of polyubiquitin-containing bodies, called ALIS (aggresome-like induced structures), serving to link ALIS to autophagosome formation (Fujita et al., 2011). This process maintains recycling of dysfunctional mitochondria within the cell.
Nitric Oxide (NO) Induced Signaling of Mitochondrial Biogenesis
Another major gaseous physiological mediator produced both constitutively and during ischemia/reperfusion and other types of cardiac damage is nitric oxide (NO). Although release of NO from ischemia may interact directly with ROS such as · to produce peroxinitrite (McBride et al., 1999; Paolocci et al., 2001), other evidence is beginning to emerge that NO plays a direct role in mitochondrial biogenesis as well as in mitochondrial QC in the heart. Using HL-1 cardiomyocytes, NO donors have been demonstrated to activate mitochondrial biogenesis (Vettor et al., 2014). Intracellular NO is produced by endothelial NO synthase (eNOS/NOS1), inducible NO synthase (iNOS/NOS2), or neuronal NO synthase (nNOS/NOS3). NO generated through NOS1 activity leads to the induction of mitochondrial biogenesis through guanylate cyclase activation and enhanced cyclic GMP and protein kinase A (PKA) activity (Nisoli et al., 2003, 2004; Signorile et al., 2014). Additionally, exercise-induced myocardial mitochondrial biogenesis is inhibited in mice with eNOS/NOS1 deletion (Vettor et al., 2014). And it has been shown that the renal hematopoietic hormone erythropoietin (EPO) induces mitochondrial biogenesis through EPO receptors in the mouse heart, and that this response is absent in mice lacking eNOS/NOS1 (Carraway et al., 2010).
The inducible NOS (NOS2 or iNOS) is also an important responder to oxidative stress and inflammation. NOS2 is induced by events that stimulate NF-κb nuclear translocation. Although not as well-studied within the heart as eNOS, in other organ systems the up-regulation of this enzyme isoform also promotes mitochondrial biogenesis (Suliman et al., 2010). Additionally, transgenic mice overexpressing NOS2 targeted to cardiomyocytes exhibit smaller infarct sizes in an ischemia/reperfusion model, and NOS2/iNOS overexpression has been found to inhibit the mitochondrial permeability transition, and to prevent ROS formation. However, it was not actually determined whether the mitochondrial biogenesis genetic program was up-regulated during the process (West et al., 2008).
Although beyond the scope of this review, it is important to note that controversy exists as to the presence of NOS within the mitochondria given NOS activity within the mitochondria has been previously reported (Giulivi et al., 1998; Ghafourifar et al., 1999). Most authors report an NOS1 (nNOS) variant (Holmqvist and Ekstrom, 1997; Kanai et al., 2001). Additionally, other authors have suggested localization of NOS2 to the mitochondrial by immunolabeling in addition to other organelles (Buchwalow et al., 1997) The role of so-called mtNOS is not clear and more work is needed to understand whether it has any role in mitochondrial biogenesis or mitochondrial QC in the heart.
Important to note, NO bioactivity plays a rather complex set of protective roles in the myocardium beyond those involved in mitochondrial QC. These include the ability of NO to form protein-thiol or SNO adducts in proteins in the form of protein nitrosylation (Stamler et al., 2001; Hogg et al., 2007; Broniowska and Hogg, 2012). These events in their early stages, such as the formation of SNO-proteins in mitochondria, are usually reversible, but rapidly can become chemically irreversible if allowed to persist or operate out of regulation (Anand and Stamler, 2012). The protective aspects appear to be related to the temporary inhibition of electron transport and to the regulation of ROS production by the organelles (Piantadosi, 2012). The extent to which this type of mechanism operates in the ischemic or failing heart and their impact on mitochondrial QC programming has not yet been fully explored, although recent descriptions have provided proof of principle of their biological importance (Ozawa et al., 2013; Sun et al., 2015).
In summary, the cytoprotective responses that have evolved to allow heart cells to respond, survive, and adapt to oxidative and nitrosative stress involve several novel, redox-based, signaling mechanisms. These are adaptive processes that occur generally before overt energy failure and result in heme oxygenase-1 up-regulation, nuclear translocation of Nfe2l2, induction of NRF-1 and its co-activator PGC-1α, the transcription of Tfam and its importation into mitochondria, the up-regulation of mtDNA repair enzymes and ultimately to mtDNA replication and to mitochondrial biogenesis. Both mitochondrial biogenesis and other organelle repair mechanisms occur in response to intracellular oxidative stress, but mitochondrial H2O2, production as well as endogenous enzymatic CO and NO production are able to induce the transcriptional program for mitochondrial biogenesis. This process along with the complementary and closely interrelated process of mitophagy protects and preserves a well-functioning population of mitochondria and allows the cell to reduce the ongoing contribution of damaged mitochondria to overall cellular oxidative stress. Together, these mechanisms are highly cardio-protective and preserve cardiomyocyte viability by limiting the activation of intrinsic apoptosis and by preventing energy failure and necrosis. A more thorough molecular understanding of the pathways that regulate and enhance mitochondrial QC has the potential to lead to novel therapeutic targets in a wide variety of heart diseases including ischemic, metabolic, and toxic cardiomyopathies.
Author Contributions
RB, HS, CP contributed equally to this design, draft, editing of the article.
Funding
Grant support (RB: K08-GM087429 and CP: R01-AI095424, P01-HL08801) was provided by the National Institutes of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
eNOS, endothelial nitric oxide synthase; iNOS, inducible nitric oxide synthase; NO, nitric oxide; TNF-α, tumor necrosis factor-α; H2O2, hydrogen peroxide; PKA, protein kinase A; Nfe2l2, nuclear factor (erythroid-derived 2)-like 2; Keap1, kelch-like ECH-associated protein 1; creb, cyclicAMP responsive element binding protein; PGC-1α, peroxisome proliferator-activated receptor gamma, coactivator 1α; NRF-1, nuclear respiratory factor-1; Tfam, transcription factor A, mitochondrial; OGG-1, 8-oxoguanine DNA glycosylase; SOD2, superoxide dismutase 2; HO-1, heme-oxygenase 1; CO, carbon monoxide; mtDNA, mitochondrial DNA.
References
Alam, J., Stewart, D., Touchard, C., Boinapally, S., Choi, A. M., and Cook, J. L. (1999). Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 274, 26071–26078. doi: 10.1074/jbc.274.37.26071
Anand, P., and Stamler, J. S. (2012). Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J. Mol. Med. 90, 233–244. doi: 10.1007/s00109-012-0878-z
Andersson, U., and Scarpulla, R. C. (2001). Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell. Biol. 21, 3738–3749. doi: 10.1128/MCB.21.11.3738-3749.2001
Aon, M. A., Cortassa, S., Marbán, E., and O'Rourke, B. (2003). Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 278, 44735–44744. doi: 10.1074/jbc.M302673200
Aon, M. A., Stanley, B. A., Sivakumaran, V., Kembro, J. M., O'Rourke, B., Paolocci, N., et al. (2012). Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J. Gen. Physiol. 139, 479–491. doi: 10.1085/jgp.201210772
Balaban, R. S., Nemoto, S., and Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495. doi: 10.1016/j.cell.2005.02.001
Broniowska, K. A., and Hogg, N. (2012). The chemical biology of S-nitrosothiols. Antioxid. Redox Signal. 17, 969–980. doi: 10.1089/ars.2012.4590
Buchwalow, I. B., Schulze, W., Kostic, M. M., Wallukat, G., and Morwinski, R. (1997). Intracellular localization of inducible nitric oxide synthase in neonatal rat cardiomyocytes in culture. Acta Histochem. 99, 231–240. doi: 10.1016/S0065-1281(97)80046-3
Cadenas, E., Boveris, A., Ragan, C. I., and Stoppani, A. O. (1977). Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 180, 248–257. doi: 10.1016/0003-9861(77)90035-2
Calì, T., Ottolini, D., Negro, A., and Brini, M. (2013). Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 1832, 495–508. doi: 10.1016/j.bbadis.2013.01.004
Carraway, M. S., Suliman, H. B., Jones, W. S., Chen, C. W., Babiker, A., and Piantadosi, C. A. (2010). Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ. Res. 106, 1722–1730. doi: 10.1161/CIRCRESAHA.109.214353
Carrodeguas, J. A., Yun, S., Shadel, G. S., Clayton, D. A., and Bogenhagen, D. F. (1996). Functional conservation of yeast mtTFB despite extensive sequence divergence. Gene Expr. 6, 219–230.
Carter, R. S., and Avadhani, N. G. (1994). Cooperative binding of GA-binding protein transcription factors to duplicated transcription initiation region repeats of the cytochrome c oxidase subunit IV gene. J. Biol. Chem. 269, 4381–4387.
Chang, A. L., Ulrich, A., Suliman, H. B., and Piantadosi, C. A. (2015). Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic. Biol. Med. 78, 179–189. doi: 10.1016/j.freeradbiomed.2014.10.582
Chau, C. M., Evans, M. J., and Scarpulla, R. C. (1992). Nuclear respiratory factor 1 activation sites in genes encoding the gamma-subunit of ATP synthase, eukaryotic initiation factor 2 alpha, and tyrosine aminotransferase. Specific interaction of purified NRF-1 with multiple target genes. J. Biol. Chem. 267, 6999–7006.
Chen, Q., Vazquez, E. J., Moghaddas, S., Hoppel, C. L., and Lesnefsky, E. J. (2003). Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem. 278, 36027–36031. doi: 10.1074/jbc.M304854200
Cherry, A. D., Suliman, H. B., Bartz, R. R., and Piantadosi, C. A. (2014). Peroxisome proliferator-activated receptor gamma co-activator 1-alpha as a critical co-activator of the murine hepatic oxidative stress response and mitochondrial biogenesis in Staphylococcus aureus sepsis. J. Biol. Chem. 289, 41–52. doi: 10.1074/jbc.M113.512483
Cortassa, S., O'Rourke, B., and Aon, M. A. (2014). Redox-optimized ROS balance and the relationship between mitochondrial respiration and ROS. Biochim. Biophys. Acta 1837, 287–295. doi: 10.1016/j.bbabio.2013.11.007
Dairaghi, D. J., Shadel, G. S., and Clayton, D. A. (1995). Human mitochondrial transcription factor A and promoter spacing integrity are required for transcription initiation. Biochim. Biophys. Acta 1271, 127–134. doi: 10.1016/0925-4439(95)00019-Z
Darvekar, S. R., Elvenes, J., Brenne, H. B., Johansen, T., and Sjøttem, E. (2014). SPBP is a sulforaphane induced transcriptional coactivator of NRF2 regulating expression of the autophagy receptor p62/SQSTM1. PLoS ONE 9:e85262. doi: 10.1371/journal.pone.0085262
Dorn, G. W. II. (2015). Mitochondrial dynamism and heart disease: changing shape and shaping change. EMBO Mol. Med. 7, 865–877. doi: 10.15252/emmm.201404575
Ekstrand, M. I., Falkenberg, M., Rantanen, A., Park, C. B., Gaspari, M., Hultenby, K., et al. (2004). Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 13, 935–944. doi: 10.1093/hmg/ddh109
Evans, M. J., and Scarpulla, R. C. (1990). NRF-1: a trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 4, 1023–1034. doi: 10.1101/gad.4.6.1023
Falkenberg, M., Gaspari, M., Rantanen, A., Trifunovic, A., Larsson, N. G., and Gustafsson, C. M. (2002). Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat. Genet. 31, 289–294. doi: 10.1038/ng909
Favreau, L. V., and Pickett, C. B. (1995). The rat quinone reductase antioxidant response element. Identification of the nucleotide sequence required for basal and inducible activity and detection of antioxidant response element-binding proteins in hepatoma and non-hepatoma cell lines. J. Biol. Chem. 270, 24468–24474. doi: 10.1074/jbc.270.41.24468
Ferrari, R., Cargnoni, A., Curello, S., Boffa, G. M., and Ceconi, C. (1989). Effects of iloprost (ZK 36374) on glutathione status during ischaemia and reperfusion of rabbit isolated hearts. Br. J. Pharmacol. 98, 678–684. doi: 10.1111/j.1476-5381.1989.tb12643.x
Fisher-Wellman, K. H., Mattox, T. A., Thayne, K., Katunga, L. A., La Favor, J. D., Neufer, P. D., et al. (2013). Novel role for thioredoxin reductase-2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J. Physiol. (Lond). 591, 3471–3486. doi: 10.1113/jphysiol.2013.254193
Fujita, K., Maeda, D., Xiao, Q., and Srinivasula, S. M. (2011). Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc. Natl. Acad. Sci. U.S.A. 108, 1427–1432. doi: 10.1073/pnas.1014156108
Garnier, A., Fortin, D., Deloménie, C., Momken, I., Veksler, V., and Ventura-Clapier, R. (2003). Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. (Lond). 551, 491–501. doi: 10.1113/jphysiol.2003.045104
Gauthier, L. D., Greenstein, J. L., Cortassa, S., O'Rourke, B., and Winslow, R. L. (2013). A computational model of reactive oxygen species and redox balance in cardiac mitochondria. Biophys. J. 105, 1045–1056. doi: 10.1016/j.bpj.2013.07.006
Ghafourifar, P., Schenk, U., Klein, S. D., and Richter, C. (1999). Mitochondrial nitric-oxide synthase stimulation causes cytochrome c release from isolated mitochondria. Evidence for intramitochondrial peroxynitrite formation. J. Biol. Chem. 274, 31185–31188. doi: 10.1074/jbc.274.44.31185
Giulivi, C., Poderoso, J. J., and Boveris, A. (1998). Production of nitric oxide by mitochondria. J. Biol. Chem. 273, 11038–11043. doi: 10.1074/jbc.273.18.11038
Gleyzer, N., Vercauteren, K., and Scarpulla, R. C. (2005). Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 25, 1354–1366. doi: 10.1128/MCB.25.4.1354-1366.2005
Haga, S., Ozawa, T., Yamada, Y., Morita, N., Nagashima, I., Inoue, H., et al. (2014). p62/SQSTM1 plays a protective role in oxidative injury of steatotic liver in a mouse hepatectomy model. Antioxid. Redox Signal. 21, 2515–2530. doi: 10.1089/ars.2013.5391
Hamacher-Brady, A., Brady, N. R., and Gottlieb, R. A. (2006). Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 281, 29776–29787. doi: 10.1074/jbc.M603783200
Hickson-Bick, D. L., Jones, C., and Buja, L. M. (2008). Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J. Mol. Cell. Cardiol. 44, 411–418. doi: 10.1016/j.yjmcc.2007.10.013
Hogg, N., Broniowska, K. A., Novalija, J., Kettenhofen, N. J., and Novalija, E. (2007). Role of S-nitrosothiol transport in the cardioprotective effects of S-nitrosocysteine in rat hearts. Free Radic. Biol. Med. 43, 1086–1094. doi: 10.1016/j.freeradbiomed.2007.06.016
Holmqvist, B., and Ekström, P. (1997). Subcellular localization of neuronal nitric oxide synthase in the brain of a teleost; an immunoelectron and confocal microscopical study. Brain Res. 745, 67–82. doi: 10.1016/S0006-8993(96)01128-6
Huang, C., Andres, A. M., Ratliff, E. P., Hernandez, G., Lee, P., and Gottlieb, R. A. (2011). Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE 6:e20975. doi: 10.1371/journal.pone.0020975
Ide, T., Tsutsui, H., Kinugawa, S., Utsumi, H., Kang, D., Hattori, N., et al. (1999). Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res. 85, 357–363. doi: 10.1161/01.RES.85.4.357
Iqbal, M., Cohen, R. I., Marzouk, K., and Liu, S. F. (2002). Time course of nitric oxide, peroxynitrite, and antioxidants in the endotoxemic heart. Crit. Care Med. 30, 1291–1296. doi: 10.1097/00003246-200206000-00021
Irwin, M. W., Mak, S., Mann, D. L., Qu, R., Penninger, J. M., Yan, A., et al. (1999). Tissue expression and immunolocalization of tumor necrosis factor-alpha in postinfarction dysfunctional myocardium. Circulation 99, 1492–1498. doi: 10.1161/01.CIR.99.11.1492
Itoh, K., Chiba, T., Takahashi, S., Ishii, T., Igarashi, K., Katoh, Y., et al. (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236, 313–322. doi: 10.1006/bbrc.1997.6943
Itoh, K., Ishii, T., Wakabayashi, N., and Yamamoto, M. (1999a). Regulatory mechanisms of cellular response to oxidative stress. Free Radic. Res. 31, 319–324. doi: 10.1080/10715769900300881
Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, T., Igarashi, K., Engel, J. D., et al. (1999b). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86. doi: 10.1101/gad.13.1.76
Jaloszynski, P., Murata, S., Shinkai, Y., Takahashi, S., Kumagai, Y., Nishimura, S., et al. (2007). Dysfunction of Nrf2 decreases KBrO3-induced oxidative DNA damage in Ogg1-null mice. Biochem. Biophys. Res. Commun. 364, 966–971. doi: 10.1016/j.bbrc.2007.10.123
Jennings, R. B., and Ganote, C. E. (1976). Mitochondrial structure and function in acute myocardial ischemic injury. Circ. Res. 38, I80–I91.
Kanai, A. J., Pearce, L. L., Clemens, P. R., Birder, L. A., VanBibber, M. M., Choi, S. Y., et al. (2001). Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc. Natl. Acad. Sci. U.S.A. 98, 14126–14131. doi: 10.1073/pnas.241380298
Kane, J. J., Murphy, M. L., Bissett, J. K., deSoyza, N., Doherty, J. E., and Straub, K. D. (1975). Mitochondrial function, oxygen extraction, epicardial S-T segment changes and tritiated digoxin distribution after reperfusion of ischemic myocardium. Am. J. Cardiol. 36, 218–224. doi: 10.1016/0002-9149(75)90530-5
Kelly, D. P., and Scarpulla, R. C. (2004). Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 18, 357–368. doi: 10.1101/gad.1177604
Kolamunne, R. T., Dias, I. H., Vernallis, A. B., Grant, M. M., and Griffiths, H. R. (2013). Nrf2 activation supports cell survival during hypoxia and hypoxia/reoxygenation in cardiomyoblasts; the roles of reactive oxygen and nitrogen species. Redox Biol. 1, 418–426. doi: 10.1016/j.redox.2013.08.002
Laguens, R. P., and Gómez-Dumm, C. L. (1967). Fine structure of myocardial mitochondria in rats after exercise for one-half to two hours. Circ. Res. 21, 271–279. doi: 10.1161/01.RES.21.3.271
Lancel, S., Hassoun, S. M., Favory, R., Decoster, B., Motterlini, R., and Neviere, R. (2009). Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 329, 641–648. doi: 10.1124/jpet.108.148049
Larsson, N. G., Wang, J., Wilhelmsson, H., Oldfors, A., Rustin, P., Lewandoski, M., et al. (1998). Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 18, 231–236. doi: 10.1038/ng0398-231
Lin, J., Puigserver, P., Donovan, J., Tarr, P., and Spiegelman, B. M. (2002). Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J. Biol. Chem. 277, 1645–1648. doi: 10.1074/jbc.C100631200
Liu, A., Fang, H., Wei, W., Dirsch, O., and Dahmen, U. (2014). Ischemic preconditioning protects against liver ischemia/reperfusion injury via heme oxygenase-1-mediated autophagy. Crit. Care Med. 42, e762–e771. doi: 10.1097/CCM.0000000000000659
Loeper, J., Goy, J., Klein, J. M., Dufour, M., Bedu, O., Loeper, S., et al. (1991a). The evolution of oxidative stress indicators in the course of myocardial ischemia. Free Radic. Res. Commun. 12–13(Pt 2), 675–680. doi: 10.3109/10715769109145846
Loeper, J., Goy, J., Rozensztajn, L., Bedu, O., and Moisson, P. (1991b). Lipid peroxidation and protective enzymes during myocardial infarction. Clin. Chim. Acta 196, 119–125. doi: 10.1016/0009-8981(91)90064-J
Maines, M. D. (1988). Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 2, 2557–2568.
Maulik, N., Sharma, H. S., and Das, D. K. (1996). Induction of the haem oxygenase gene expression during the reperfusion of ischemic rat myocardium. J. Mol. Cell. Cardiol. 28, 1261–1270. doi: 10.1006/jmcc.1996.0116
McBride, A. G., Borutaité, V., and Brown, G. C. (1999). Superoxide dismutase and hydrogen peroxide cause rapid nitric oxide breakdown, peroxynitrite production and subsequent cell death. Biochim. Biophys. Acta 1454, 275–288. doi: 10.1016/S0925-4439(99)00046-0
Mokranjac, D., and Neupert, W. (2009). Thirty years of protein translocation into mitochondria: unexpectedly complex and still puzzling. Biochim. Biophys. Acta 1793, 33–41. doi: 10.1016/j.bbamcr.2008.06.021
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Nisoli, E., Clementi, E., Paolucci, C., Cozzi, V., Tonello, C., Sciorati, C., et al. (2003). Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299, 896–899. doi: 10.1126/science.1079368
Nisoli, E., Falcone, S., Tonello, C., Cozzi, V., Palomba, L., Fiorani, M., et al. (2004). Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. U.S.A. 101, 16507–16512. doi: 10.1073/pnas.0405432101
Ozawa, K., Komatsubara, A. T., Nishimura, Y., Sawada, T., Kawafune, H., Tsumoto, H., et al. (2013). S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci. Rep. 3:2202. doi: 10.1038/srep02202
Palmeira, C. M., Serrano, J., Kuehl, D. W., and Wallace, K. B. (1997). Preferential oxidation of cardiac mitochondrial DNA following acute intoxication with doxorubicin. Biochim. Biophys. Acta 1321, 101–106. doi: 10.1016/S0005-2728(97)00055-8
Paolocci, N., Biondi, R., Bettini, M., Lee, C. I., Berlowitz, C. O., Rossi, R., et al. (2001). Oxygen radical-mediated reduction in basal and agonist-evoked NO release in isolated rat heart. J. Mol. Cell. Cardiol. 33, 671–679. doi: 10.1006/jmcc.2000.1334
Piantadosi, C. A. (2012). Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 1820, 712–721. doi: 10.1016/j.bbagen.2011.03.008
Piantadosi, C. A., Carraway, M. S., Babiker, A., and Suliman, H. B. (2008). Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 103, 1232–1240. doi: 10.1161/01.RES.0000338597.71702.ad
Prestera, T., Talalay, P., Alam, J., Ahn, Y. I., Lee, P. J., and Choi, A. M. (1995). Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: regulation by upstream antioxidant-responsive elements (ARE). Mol. Med. 1, 827–837.
Rushmore, T. H., Morton, M. R., and Pickett, C. B. (1991). The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 266, 11632–11639.
Scarpulla, R. C. (1997). Nuclear control of respiratory chain expression in mammalian cells. J. Bioenerg. Biomembr. 29, 109–119. doi: 10.1023/A:1022681828846
Scarpulla, R. C. (2002). Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim. Biophys. Acta 1576, 1–14. doi: 10.1016/S0167-4781(02)00343-3
Shadel, G. S., and Clayton, D. A. (1996). Isolation and characterization of vertebrate mitochondrial transcription factor A homologs. Meth. Enzymol. 264, 149–158. doi: 10.1016/S0076-6879(96)64016-6
Sharma, H. S., Maulik, N., Gho, B. C., Das, D. K., and Verdouw, P. D. (1996). Coordinated expression of heme oxygenase-1 and ubiquitin in the porcine heart subjected to ischemia and reperfusion. Mol. Cell. Biochem. 157, 111–116. doi: 10.1007/BF00227888
Signorile, A., Micelli, L., De Rasmo, D., Santeramo, A., Papa, F., Ficarella, R., et al. (2014). Regulation of the biogenesis of OXPHOS complexes in cell transition from replicating to quiescent state: involvement of PKA and effect of hydroxytyrosol. Biochim. Biophys. Acta 1843, 675–684. doi: 10.1016/j.bbamcr.2013.12.017
Song, M., Mihara, K., Chen, Y., Scorrano, L., and Dorn, G. W. II. (2015). Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 21, 273–285. doi: 10.1016/j.cmet.2014.12.011
Stamler, J. S., Lamas, S., and Fang, F. C. (2001). Nitrosylation. the prototypic redox-based signaling mechanism. Cell 106, 675–683. doi: 10.1016/S0092-8674(01)00495-0
Stanley, B. A., Sivakumaran, V., Shi, S., McDonald, I., Lloyd, D., Watson, W. H., et al. (2011). Thioredoxin reductase-2 is essential for keeping low levels of H(2)O(2) emission from isolated heart mitochondria. J. Biol. Chem. 286, 33669–33677. doi: 10.1074/jbc.M111.284612
Stepkowski, T. M., and Kruszewski, M. K. (2011). Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radic. Biol. Med. 50, 1186–1195. doi: 10.1016/j.freeradbiomed.2011.01.033
Strohecker, A. M., and White, E. (2014). Autophagy promotes BrafV600E-driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy 10, 384–385. doi: 10.4161/auto.27320
Suliman, H. B., Carraway, M. S., Ali, A. S., Reynolds, C. M., Welty-Wolf, K. E., and Piantadosi, C. A. (2007a). The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J. Clin. Invest. 117, 3730–3741. doi: 10.1172/jci32967
Suliman, H. B., Carraway, M. S., Tatro, L. G., and Piantadosi, C. A. (2007b). A new activating role for CO in cardiac mitochondrial biogenesis. J. Cell Sci. 120, 299–308. doi: 10.1242/jcs.03318
Suliman, H. B., Sweeney, T. E., Withers, C. M., and Piantadosi, C. A. (2010). Co-regulation of nuclear respiratory factor-1 by NFkappaB and CREB links LPS-induced inflammation to mitochondrial biogenesis. J. Cell Sci. 123, 2565–2575. doi: 10.1242/jcs.064089
Suliman, H. B., Welty-Wolf, K. E., Carraway, M., Tatro, L., and Piantadosi, C. A. (2004). Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc. Res. 64, 279–288. doi: 10.1016/j.cardiores.2004.07.005
Sun, J., Nguyen, T., Aponte, A. M., Menazza, S., Kohr, M. J., Roth, D. M., et al. (2015). Ischaemic preconditioning preferentially increases protein S-nitrosylation in subsarcolemmal mitochondria. Cardiovasc. Res. 106, 227–236. doi: 10.1093/cvr/cvv044
Supinski, G. S., Murphy, M. P., and Callahan, L. A. (2009). MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1095–R1102. doi: 10.1152/ajpregu.90902.2008
Turrens, J. F., Alexandre, A., and Lehninger, A. L. (1985). Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 237, 408–414. doi: 10.1016/0003-9861(85)90293-0
Unuma, K., Aki, T., Funakoshi, T., Yoshida, K., and Uemura, K. (2013a). Cobalt protoporphyrin accelerates TFEB activation and lysosome reformation during LPS-induced septic insults in the rat heart. PLoS ONE 8:e56526. doi: 10.1371/journal.pone.0056526
Unuma, K., Aki, T., Matsuda, S., Funakoshi, T., Yoshida, K., and Uemura, K. (2013b). Inducer of heme oxygenase-1 cobalt protoporphyrin accelerates autophagy and suppresses oxidative damages during lipopolysaccharide treatment in rat liver. Hepatol. Res. 43, 91–96. doi: 10.1111/j.1872-034X.2012.01049.x
Vercauteren, K., Gleyzer, N., and Scarpulla, R. C. (2008). PGC-1-related coactivator complexes with HCF-1 and NRF-2beta in mediating NRF-2(GABP)-dependent respiratory gene expression. J. Biol. Chem. 283, 12102–12111. doi: 10.1074/jbc.M710150200
Vettor, R., Valerio, A., Ragni, M., Trevellin, E., Granzotto, M., Olivieri, M., et al. (2014). Exercise training boosts eNOS-dependent mitochondrial biogenesis in mouse heart: role in adaptation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 306, E519–E528. doi: 10.1152/ajpendo.00617.2013
Virbasius, J. V., and Scarpulla, R. C. (1994). Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. U.S.A. 91, 1309–1313. doi: 10.1073/pnas.91.4.1309
West, M. B., Rokosh, G., Obal, D., Velayutham, M., Xuan, Y. T., Hill, B. G., et al. (2008). Cardiac myocyte-specific expression of inducible nitric oxide synthase protects against ischemia/reperfusion injury by preventing mitochondrial permeability transition. Circulation 118, 1970–1978. doi: 10.1161/CIRCULATIONAHA.108.791533
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124. doi: 10.1016/S0092-8674(00)80611-X
Keywords: reactive oxygen species (ROS), mitochondria, heart, mitochondrial biogenesis, nitric oxide synthase, oxidative stress
Citation: Bartz RR, Suliman HB and Piantadosi CA (2015) Redox mechanisms of cardiomyocyte mitochondrial protection. Front. Physiol. 6:291. doi: 10.3389/fphys.2015.00291
Received: 27 June 2015; Accepted: 02 October 2015;
Published: 26 October 2015.
Edited by:
Paul Huang, Massachusetts General Hospital, USAReviewed by:
Nazareno Paolocci, Johns Hopkins University, USACesare Indiveri, University of Calabria, Italy
Copyright © 2015 Bartz, Suliman and Piantadosi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raquel R. Bartz, cmFxdWVsLmJhcnR6QGRtLmR1a2UuZWR1