 Anita Tripathi
Anita Tripathi Kavita Goswami
Kavita Goswami Neeti Sanan-Mishra
Neeti Sanan-Mishra- Plant Molecular Biology Group, International Centre for Genetic Engineering and Biotechnology, New Delhi, India
microRNAs (miRs) are a class of 21–24 nucleotide long non-coding RNAs responsible for regulating the expression of associated genes mainly by cleavage or translational inhibition of the target transcripts. With this characteristic of silencing, miRs act as an important component in regulation of plant responses in various stress conditions. In recent years, with drastic change in environmental and soil conditions different type of stresses have emerged as a major challenge for plants growth and productivity. The identification and profiling of miRs has itself been a challenge for research workers given their small size and large number of many probable sequences in the genome. Application of computational approaches has expedited the process of identification of miRs and their expression profiling in different conditions. The development of High-Throughput Sequencing (HTS) techniques has facilitated to gain access to the global profiles of the miRs for understanding their mode of action in plants. Introduction of various bioinformatics databases and tools have revolutionized the study of miRs and other small RNAs. This review focuses the role of bioinformatics approaches in the identification and study of the regulatory roles of plant miRs in the adaptive response to stresses.
Abiotic Stresses and their Impact on Yield
Plants are exposed to a wide array of environmental fluctuations that lead to various physiological and metabolic changes, which in turn adversely affect the growth and productivity. Abiotic stresses are the principal cause of decrement in crop production globally and are responsible for lowering the average yield of major crops by more than 50% (Mahajan and Tuteja, 2005; Rodríguez et al., 2005). The World Meteorological Organization has reported that the years from 2001 to 2010 were considered to be the warmest period after 1850 (Oosterhuis, 2013). The climate change models have predicted that in coming time the occurrence and severity of such stresses will increase, leading to a decrease in agricultural production by about 70% (Cramer et al., 2011; Hasanuzzaman et al., 2013c; Ghosh and Xu, 2014).
The different abiotic stress conditions may be segregated into 35 different types that can be sorted as 11 groups, viz. cold, heat, drought, flooding, radiations (UV and light), wind, salinity, heavy metal toxicity, nutrient deprivation in soil, and oxidative stress (Mahajan and Tuteja, 2005). These stresses act by affecting plant growth at the molecular, biological, and physiological levels (Figure 1). The most studied abiotic stress conditions are cold, high temperature, salt, and drought stress. Plants cannot escape from these stresses because of their sessile nature but, they have developed sophisticated systems to cope up with them (Nakashima et al., 2009; Pfalz et al., 2012; Upadhyaya and Panda, 2013). The response to abiotic stresses is usually multigenic and involves altering the expression of nucleic acids, proteins and other macromolecules (Figure 1). Several excellent reviews are available that discuss the impact of these stresses on plants in details (Cramer et al., 2011; Shanker and Venkateswarlu, 2011; Duque et al., 2013; Hasanuzzaman et al., 2013a; Rejeb et al., 2014; Petrov et al., 2015; Sodha and Karan, 2015).
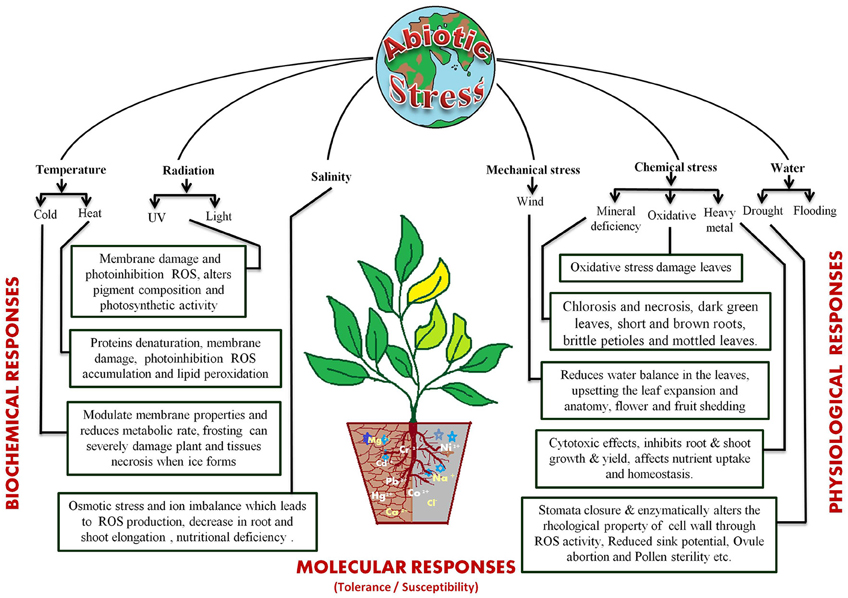
Figure 1. Various abiotic stresses and their physiological effects on plants.
Primarily fluctuations in available water, temperature and soil salt content are recognized as the basic environmental stress factors. The scarcity of water because of less rainfall, paucity of soil water and excessive evaporation, is probably the most common factor, limiting the crop's growth (de Oliveira et al., 2013). Water deficit negatively affects plant growth and development by modulating nutrient uptake, photosynthesis, hormonal levels, water potential etc. This often results in tissue dehydration leading to senescence (Kaiser, 1987; Aroca et al., 2001, 2012; Kacperska, 2004; Wahid and Close, 2007). Under low water conditions plants activate their protective machinery to enhance water uptake and reduce water loss. However, deficiency of sufficient water supply or drought limits the root hydraulic conductivity (Nobel and Cui, 1992; North and Nobel, 1997; Aroca et al., 2012) thereby affecting water uptake and resulting in physiological drought condition for the plant (Bréda et al., 1995; Duursma et al., 2008; Aroca et al., 2012). Similarly, when the water level goes above the optimal levels it results in flooding which causes hypoxic conditions, stimulate the reactive oxygen species (ROS) and induces ethylene production that restricts aerobic respiration (Bailey-Serres and Voesenek, 2008; Perata et al., 2011).
Fluctuations in atmospheric temperature due to climate change are also exerting an adverse affect at physical and cellular levels. High temperatures change the cellular state, lipid composition, membrane fluidity, and organelle properties. They induce oxidative stress and reduce the water content of the soil, causing physiological drought in plants (Wahid and Close, 2007; Giri, 2011; Hasanuzzaman et al., 2013b; Goswami et al., 2014). They also affect flowering by decreasing the number of flowers, reducing pollen viability and flower fertility (Matsui et al., 2000; Prasad et al., 2000, 2006; Suzuki et al., 2001) and cause embryo damage during the early stages of seed germination (Grass, 1994; Hasanuzzaman et al., 2013b). Low temperatures also confer osmotic and oxidative stress on plants (Chinnusamy et al., 2007; Aroca et al., 2012). They reduce metabolic rate, increase rigidification of the cellular membrane, cause flower abortion, fertilization breakdown and negatively impact seed filling (Thakur et al., 2010; Zinn et al., 2010; Hedhly, 2011).
The temperature increases along with poor irrigation practices increase soil salinity. This has emerged as an important stress which inhibits plant's growth at every stage by inducing osmotic stress and ion toxicity (Diédhiou and Golldack, 2006; Joseph and Mohanan, 2013; Roychoudhury and Chakraborty, 2013). Salinity majorly affects roots by decreasing water use efficiency and ion exclusion, which adversely affects the root elongation, spike development and plant height (Choi et al., 2003; Alam et al., 2004; Diédhiou and Golldack, 2006; Mahmood et al., 2009; Aroca et al., 2012; Hakim, 2013; Pierik and Testerink, 2014).
The various environmental stresses result in osmotic and oxidative stresses, which inhibit metabolic reactions (Chinnusamy et al., 2007). Oxidative damage is one of the main reasons for loss of productivity and is triggered by increase in reactive oxygen species (ROS) that includes superoxide radicals (2O−), hydroxyl radicals (OH), and hydrogen peroxide (H2O2) (Mittler, 2002; Apel and Hirt, 2004; Bartels and Sunkar, 2005; Foyer and Noctor, 2005; Addo-Quaye et al., 2009). The ROS are responsible for nucleic acid damage, protein oxidation, and lipid peroxidation (Foyer et al., 1994). Plants have developed intrinsic mechanisms to avoid the oxidative stresses that includes recruitment of enzymatic scavengers, like superoxide dismutase (SOD), ascorbate peroxidase, glutathione peroxidase, glutathione S-transferase, catalase, and non-enzymatic low molecular mass molecules, such as ascorbate, tocopherol, carotenoids, and glutathione (Mittler, 2002; Mittler et al., 2004).
Basics of microRNA
The discovery of regulatory small RNAs (sRNAs) that block specific messenger RNAs (mRNAs) at the post-transcriptional levels (PTGS or post-transcriptional gene silencing) by cleavage or translational repression (Sunkar et al., 2006; Shi et al., 2012) or interfere with transcription (TGS or transcriptional gene silencing) by directing DNA methylation of genes (Wu and Zhang, 2010) have unlocked a new avenue in gene expression regulation. The sRNAs constitute a large family represented by many species of RNA molecules distinguished from each other by their size, biogenesis, mode of action, regulatory role etc. (Axtell and Bowman, 2008; Sanan-Mishra et al., 2009; Lima et al., 2011; Meng et al., 2011a; Zheng et al., 2012).
The microRNA (miR) represents a major sub-family of endogenously transcribed sequences, ranging in length from 21 to 24 nt (Carrington and Ambros, 2003; Eldem et al., 2013). They have been established as a major regulatory class that inhibits gene expression in a sequence-dependent manner. The lin-4 and let-7 regulatory RNAs are accepted as the naissance member of the miR family (Lee et al., 1993; Reinhart et al., 2002), which is conserved across animal and plant species. Though there is no conservation between the animal and plant sequences, but high conservation is observed among plant miRs (Reinhart et al., 2002). An exception is provided by Ath-miR854 and Ath-miR855, which regulate levels of transcript encoding the oligouridylate binding protein 1b (UBP1b) (Arteaga-Vázquez et al., 2006). The target transcript of miR854 performs similar functions in plants as well as in animals (Arteaga-Vázquez et al., 2006).
microRNA Biogenesis
Each miR arises in the nucleus from an independent transcription unit, comprising of its own promoter, transcribing region and terminator, by utilizing the basic machinery for DNA-dependent RNA polymerase II mediated transcription (Kurihara and Watanabe, 2004; Lee et al., 2004; Xie et al., 2005a; Kim et al., 2011). Plant miR genes are present throughout the genome, although majority of the loci in plants are generally found in genomic (intergenic) regions that are not protein coding (Jones-Rhoades et al., 2006; Wahid et al., 2010). Comparatively lesser number of plant miRs are present in the introns (Lagos-Quintana et al., 2001; Lau et al., 2001; Chen, 2008; Nozawa et al., 2010; Wahid et al., 2010) and are rarely found in the exons (Olena and Patton, 2010; Li et al., 2011). Two miRs, miR436, and miR444, were mapped to the exonic regions of the protein-coding genes J023035E19 (AK120922) and J033125N22 (AK103332), respectively (Sunkar et al., 2005). It is hypothesized that the miRs control the host gene expression via a negative feedback loop mechanism that affects alternative splicing and cytoplasmic movement of transcripts (Slezak-Prochazka et al., 2013). Recently, CDC5 was identified as a MYB-related DNA binding protein that positively regulates miR production (Zhang et al., 2013a) by binding to their promoters and through interaction with the RNase III enzyme DCL1 (Dicer-Like 1). The large pri-miRs (primary transcripts) contain a 5′-cap and 3′-polyA tail and are stabilized in the nucleus by DDL (Dawdle) which is a RNA binding protein (Yu et al., 2008).
The pri-miRs are further processed into hairpin loop structured pre-miRs (precursor miRs) in the D bodies (Dicing bodies) or SmD3-bodies (small nuclear RNA binding protein D3 bodies) (Kurihara et al., 2006; Fang and Spector, 2007; Fujioka et al., 2007) by a protein complex containing the DCL1 (Schauer et al., 2002) and the CBC (Cap-Binding protein Complex) (Kim et al., 2008). The accuracy of DCL1 mediated pri-miR processing is promoted by both HYL1 (Hyponastic Leaves 1), and the C2H2-zinc finger protein, SE (Serrate) (Kurihara et al., 2006; Dong et al., 2008; Manavella et al., 2012a). This activity is also aided by DRB (Double strand RNA-Binding) protein (Han et al., 2004; Kurihara et al., 2006; Vazquez, 2006). Recently the G-patch domain protein TGH (Tough) was identified as another active player which is responsible for enhancing the DCL1 activity (Ren et al., 2012). It has been shown that HYL1 binds double stranded (ds) region on the pri-miR (Hiraguri et al., 2005; Rasia et al., 2010; Yang et al., 2010), TGH binds the single-stranded (ss) RNA region (Ren et al., 2012) and SE possibly binds at ssRNA/dsRNA junctions (Machida et al., 2011). It was also observed that HYL1 is a phospho-protein that directly interacts with CPL1 (C-terminal domain Phosphatase-Like 1) protein, to maintain its hypo-phosphorylated state (Manavella et al., 2012a). Thus, CPL1 also plays a critical role in accurate miR processing though it is not directly required for DCL1 activity (Manavella et al., 2012a). It was observed that CPL1 directly interacts with SE and a mutation in SE can affect phosphorylation status of HYL1 by preventing recruitment of CPL1 (Manavella et al., 2012a). Thus, the proposed model for the pri-miR processing indicates association of multiple RNA binding proteins with definite regions to maintain the structural determinants for recruiting and directing DCL1 activity. The DCL1, HYL1, SE, and TGH seem to interact directly (Kurihara et al., 2006; Lobbes et al., 2006; Yang et al., 2006; Qin et al., 2010; Machida et al., 2011; Ren et al., 2012) and are colocalized in the D bodies as shown by bimolecular fluorescence complementation. However, it has not been demonstrated whether they represent a stable plant microprocessor complex (Fang and Spector, 2007; Fujioka et al., 2007; Song et al., 2007; Manavella et al., 2012b; Ren et al., 2012).
The hairpin looped pre-miRs thus formed are further processed by DCL1 to produce miR/miR* duplex (Xie et al., 2005b; Sanan-Mishra et al., 2009; Naqvi et al., 2012). Recently a proline-rich protein, SIC (Sickle), was identified to co-localize with HYL1 foci (Zhan et al., 2012) and it was found to play an important role in the accumulation of mature miR duplex (Zhan et al., 2012). The strands of the duplex are protected from uridylation and degradation by the activity of a methyltransferase protein known as HEN1 (Hua Enhancer 1) which covalently attaches a methyl residue at the 3′ ribose of last nucleotide from each strand (Li et al., 2005a; Yu et al., 2005). The miR duplexes are transported to the cytoplasm by HST (Hasty), the ortholog of Exportin-5 (Park et al., 2005), where the miR strand guides the AGO1 (Argonaute 1) containing RNA-induced silencing complex (RISC) complex to the target transcript (Baumberger and Baulcombe, 2005; Qi et al., 2005).
microRNA Function
Plant miRs generally control the expression of their targets transcripts by cleavage and translational repression (Chen, 2009). Brodersen et al. concluded that central matches in miR:target-mRNA duplex tend to cleave target mRNA, regardless of a few mismatches in other regions, while central mismatches in miR:target mRNA duplex lead to translational repression (Brodersen et al., 2008). It was hypothesized that the rapid fine-tuning of the target transcripts by translation repression is required for the reversible modulation of the negative regulators of stress responses whereas the on-off switching of target gene expression by cleavage was important in regulating developmental processes, which require permanent determination of cell fates (Baumberger and Baulcombe, 2005).
In plants, miRs regulate various biological processes such as, growth and development, pattern formation, organ polarity, signal transduction, and hormone homeostasis etc. (Palatnik et al., 2003; Dugas and Bartel, 2004; Jones-Rhoades et al., 2006; Mallory and Vaucheret, 2006; Mishra and Mukherjee, 2007; Cai et al., 2009; Voinnet, 2009; Sanan-Mishra et al., 2013). In past few years the role of miRs in response to diseases and environmental stresses has been highlighted (Fujii et al., 2005; Sunkar et al., 2007; Zhou et al., 2010; Lima et al., 2011; Meng et al., 2011a; Zheng et al., 2012; Mittal et al., 2013; Sharma et al., 2015). These are supported by reports on mutants of the miR biogenesis or pathways exhibiting defective phenotypes (Laufs et al., 2004; Zhong and Ye, 2004; Millar and Gubler, 2005; Ori et al., 2007; Chen et al., 2010; Rubio-Somoza and Weigel, 2011). The stress regulated miRs may be engaged in many biological pathways that re-program intricate procedures of physiology and metabolism (Khraiwesh et al., 2012) as suggested by their differential expression patterns in tissues in presence or absence of stress (Covarrubias and Reyes, 2010).
Identification of Stress-associated microRNAs
The identification of plant miR families began in the year 2000, with direct cloning and sequencing (Llave et al., 2002; Park et al., 2002; Reinhart et al., 2002). However, this was an uphill task owing to their small size, methylation status and multiple occurrences in genome. The numbers however increased rapidly with the advancement in cloning techniques and computational algorithms. In the past few years high throughput sequencing and screening protocols has caused an exponential increase in number of miRs, identified and functionally annotated from various plant species (Rajagopalan et al., 2006; Fahlgren et al., 2007; Jagadeeswaran et al., 2010; Rosewick et al., 2013). This is best exemplified by the establishment of miRBase, a biological database that acts as an archive of miR sequences and annotations (Griffiths-Jones, 2004; Griffiths-Jones et al., 2008; Kozomara and Griffiths-Jones, 2014). The first release of miRBase in the year 2002 included total 5 miRs from only 1 plant species, Arabidopsis thaliana. This was followed by the inclusion of Oryza sativa, in miRBase in the year 2003. Thereafter miRs reported from Medicago truncatula, Glycine Max, and Populus trichocarpa were included in the year 2005. The current version (release 21) includes 48,496 mature plant miRs derived from 6992 hairpin precursors reported in 73 plant species (Figure 2).
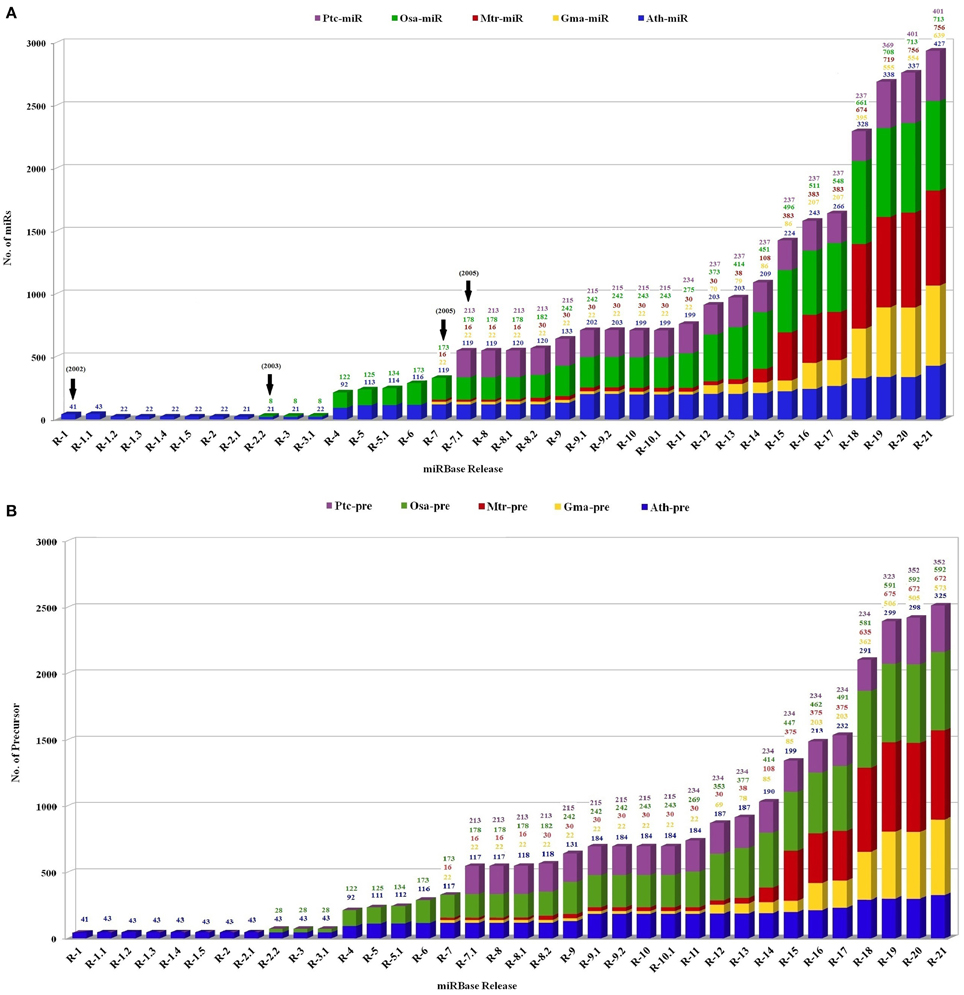
Figure 2. The status of the number of (A) mature miRs and (B) precursor miR sequences in the miRBase registry in five plant species. The plot contains the number of miRs in Medicago truncatula (red), Oryza sativa (green), Glycine max (yellow), Populus trichocarpa (violet), and Arabidopsis thaliana (blue) calculated with respect to total number reported miRs. X axis represents number of respective sequences and Y axis denotes the released versions of miRBase.
The association of plant miRs with stress was first reported in 2004 (Sunkar and Zhu, 2004). Now there are a number of reports supporting the hypothesis for the function for miRs in the adaptive response to abiotic stress including drought (Liu et al., 2008b; Zhou et al., 2010), cold (Zhou et al., 2008), salinity (Liu et al., 2008a; Sunkar et al., 2008) and nutrient deficiency (Fujii et al., 2005). 1062 miRs have been reported to be differentially expressed in 35 different abiotic stress types in 41 plant species (Zhang et al., 2013b). The detailed list of these miRs is available as Supplementary Table 1. The comparative picture of stress-induced dis-regulations of Arabidopsis and rice miRs is compiled as Figure 3.
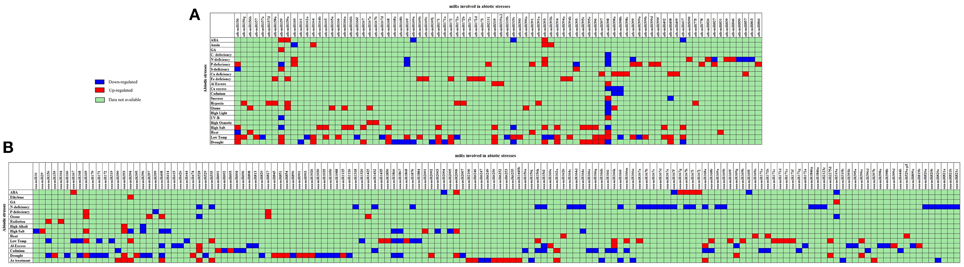
Figure 3. Status of abiotic stress regulation for conserved miRs as reported from (A) Arabidopsis and (B) rice. The X axis contains the list of abiotic stress regulated miRs, and Y axis lists the different abiotic stress studied. ABA, Abscisic Acid; GA, Gibberellic Acid; C, carbon; N, nitrogen; P, Phosphorus; S, Sulfur; UV-B, Ultra violet; Al, aluminum; Cu, cupper; Fe, Iron. The miRs down-regulated in stress are shown in Blue and miRs up-regulated in stress are shown in Red. The miRs for which the stress induced status is not available are represented by green.
The survey of literature reveals that three major approaches have been employed for the identification and expression profiling of stress induced miRs. The first approach involves the classical experimental route that included direct cloning, genetic screening, or expression profiling. The second method involved computational predictions from genomic or EST loci and the third one employed a combination of both as it was based on the prediction of miRs from High Throughput Sequencing (HTS) data. Each of these was followed by experimental validations by northern analysis, PCRs or microarrays.
Experimental Approaches
Direct cloning was the principal and conventional method for the identification of miRs (Park et al., 2002; Reinhart et al., 2002). This method was of significant consideration as it was a sequence-independent approach where a priori knowledge of miR sequence was not required. Moreover, it provided more accuracy and efficiency by giving few false positives. Several related studies led to the establishment of different protocols for sRNA isolation and adaptor mediated synthesis of a cDNA library followed by their amplification and then cloning. The clones were screened and sequenced to identify the potential miRs (Llave et al., 2002; Reinhart et al., 2002; Sunkar and Zhu, 2004). Thus, it was portrayed as a time-consuming, low throughput, laborious, and expensive approach.
However, the first report indicating the role of miRs in plant responses to environmental stresses came from the sequencing and analysis of a library of sRNAs from Arabidopsis seedlings treated with cold, dehydration, salinity, and the plant stress hormone abscisic acid (ABA). It was observed that several miRs were up-regulated or down-regulated by the abiotic stresses (Sunkar and Zhu, 2004). This strategy was used to clone miRs from the mechanical stress-treated Populus plants (Lu et al., 2005). A majority of these miRs were predicted to target developmental- and stress/defense-related genes. In our lab, 39 new miR sequences were cloned from salt-stressed basmati rice variety. This study also provided evidence for a converging functional role of miRs in managing both abiotic and biotic stresses (Sanan-Mishra et al., 2009).
The importance of miRs in abiotic stress responses was also implicated by the fact that several mutants such as hyl1, hen1, and dcl1 which are defective in miR metabolism, exhibited hypersensitivity to ABA, salt, and osmotic stresses (Lu and Fedoroff, 2000). Nonetheless, the direct evidence was provided by studies monitoring the down-regulation of miR398 expression in response to oxidative stresses, in Arabidopsis. It was later shown that miR398 targeted two Cu/Zn superoxide dismutase (CSD) transcripts, cytosolic CSD1, and chloroplastic CSD2, so stress induced reduction of miR398 was expected to improve plant tolerance. This theory was proved subsequently by analysis of transgenic lines under oxidative stress conditions (Sunkar et al., 2006).
Expression analysis by northern blot analysis revealed that miR395 and miR399 were involved in sulfate and inorganic phosphate starvation responses, respectively (Jones-Rhoades and Bartel, 2004; Fujii et al., 2005). Similarly, RNA gel blot analysis identified miRs induced by cold, ABA, dehydration, and high salinity in 2-week-old Arabidopsis seedlings (Sunkar and Zhu, 2004). The results indicated that Ath-miR393 was highly up-regulated whereas Ath-miR397b and Ath-miR402 were slightly up-regulated and Ath-miR389a.1 was down-regulated under all the stress treatments. Similarly low temperature stress condition induced the expression of Ath-miR319c but no increase in response to dehydration, NaCl or ABA (Sunkar and Zhu, 2004). These and related findings not only helped in interpreting the role of miRs during stress but unraveled the role of specific members of the miR family. A comprehensive study of Ath-miR398, revealed that the expression of miR398 precursors (with identical mature sequences) is increased under high temperature stress and that heat stress induces expression of Ath-miR398b to a much higher level than that of the Ath-miR398a,c (Guan et al., 2013). Similarly in rice, Osa-miR169g, was proven as the only drought stress induced member among the ABA responsive miR169 family (Zhao et al., 2007).
The variable expression patterns of the miRs in response to different stresses were captured by reverse transcription quantitative PCR (RT-PCR) in several plants including Arabidopsis (Jung and Kang, 2007; Reyes and Chua, 2007; Li et al., 2008; Liu et al., 2008a; Jia et al., 2009), rice (Liu et al., 2009), Phaseolus vulgaris, (Arenas-Huertero et al., 2009), sugarcane (Thiebaut et al., 2012), and poplar (Rossi et al., 2015). These methods captured the similarities and differences in expression profiles of conserved miRs across different plants (Zhou et al., 2010). This is exemplified by identified molecules like miR393 that is consistently up-regulated during drought stress in many plants such as Arabidopsis, Medicago, common bean, and rice (Sunkar and Zhu, 2004; Zhao et al., 2007; Arenas-Huertero et al., 2009). Whereas miR169 was found to be induced by drought and high salinity in rice (Zhao et al., 2009), but was down-regulated by drought stress treatment in Arabidopsis (Li et al., 2008). High-throughput expression profiling analysis through one-tube stem-loop RT-PCR quantified the relative expression levels of 41 rice miRs under drought, salt, cold, or ABA treatments (Ding et al., 2011).
The need for genome wide characterization of miR expression profiles established the microarray analysis as a useful tool (Garzon et al., 2006; Zhao et al., 2007). The microarray technology is a hybridization based and a relatively cost-effective assay that allows analysis of large numbers of molecules in parallel. The tiling path microarray analysis was used to identify 14 stress-inducible Arabidopsis miRs after screening 117 miRs under high-salinity, drought, and low-temperature stress conditions (Liu et al., 2008a; Zhang et al., 2008b). The results were further validated to provide evidence for cross-talk among the high-salinity, drought and low temperature stress associated signaling pathways (Liu et al., 2008a). Similar studies were performed to capture the expression patterns of miRs in response to Ultraviolet-B rays in Arabidopsis (Zhou et al., 2007), drought stress in rice (Zhao et al., 2007), cold stress in rice (Kang et al., 2010), cadmium stress in rice (Ding et al., 2011), and ABA and NaCl in Populus tremula (Jia et al., 2009).
The expression patterns also identified that tissue-specific regulation of miRs may be important for adaptation to stress. Under water deficit conditions, miR398a/b and miR408 were up-regulated in both roots and shoots of Medicago truncatula plant, but the increase was more pronounced in the shoots than in the roots. This was accompanied by the down-regulation of their corresponding targets, COX5b and plantacyanin, thereby suggesting that these miRs have a crucial role in regulation of plants responses against water deficiency (Trindade et al., 2010). In barley, miR166 was up-regulated in leaves, where as it was shown to be down-regulated in roots; and miR156a, miR171, and miR408 were induced in leaves, but unaltered in roots (Kantar et al., 2010).
The miR expression profiles were also used to compare the genotypic differences between varieties exhibiting contrasting stress sensitivities. Microarray profiles of salt-resistant and susceptible Zea mays identified 98 miRs belonging to 27 families (Ding et al., 2009). Zma-miR168 family members were induced in the salt-tolerant maize but suppressed in the salt-sensitive line. Interestingly this salt-responsive behavior of miR168 was found to be conserved in Maize and Arabidopsis (Liu et al., 2008a). miR microarray was also used to study drought-tolerant wild emmer wheat (Triticum dicoccoides) (Kantar et al., 2011), two cotton cultivars with high tolerance (SN-011) and high sensitivity (LM-6) to salinity (Yin et al., 2012) and for comparative analysis between drought-resistant and susceptible soybean (Kulcheski et al., 2011). A comparison of 12 salt-tolerant and 12 salt-susceptible genotypes in Oryza sativa, identified 12 polymorphic miR based simple sequence repeats (Mondal and Ganie, 2014). Only miR172b-SSR was different between the salinity stress tolerant and susceptible genotypes. The genotype-dependent miR profiles suggested that response of miRs to abiotic stresses varies among closely related genotypes with contrasting stress sensitivities. The result of this analysis showed that there was less diversity of miR genes in the tolerant as compared with susceptible cultivars (Mondal and Ganie, 2014).
Computational Predictions
The detection and validation of miRs by molecular cloning was supported by systematic approaches using computational techniques (Bonnet et al., 2004b). These approaches also complemented the experimental methods by identifying difficult to clone miR families such as miR395 and miR399 (Jones-Rhoades and Bartel, 2004; Adai et al., 2005) which were difficult to detect by experimental approaches due to their low expression levels. Computational predictions strategies have been quite useful in miR identification in various plant species such as Arabidopsis (Wang et al., 2004; Adai et al., 2005; Li et al., 2005b), rice (Li et al., 2005b; Zhang et al., 2005), maize (Zhang et al., 2006a, 2009b), tomato (Yin et al., 2008; Zhang et al., 2008b), foxtail millet (Khan et al., 2014), soybean (Zhang et al., 2008a), Brassica napus (Xie et al., 2007), apple (Gleave et al., 2008), grape (Carra et al., 2009), and some other plants (Zhang et al., 2005; Sunkar and Jagadeeswaran, 2008).
It had been verified that a majority of known miRs are evolutionarily conserved and are expected to have homologs or orthologs in other species. So search criteria allowed up-to three sequence mismatches while looking for conserved miRs in heterologous species. Using this approach 85 conserved sequences which were showing perfect match to miRs reported in miRBase (Release 19) were predicted from Morus notabilis tissues (Jia et al., 2014). Whereas in another study 35 miR families were identified in heat stressed Brassica napus by allowing two mismatches with A. thaliana miRs (Yu et al., 2012). Thus, the conserved sequence of plant miRs and other structural features were used for developing suitable strategies and rules for identifying and annotating (Discussed in Section The Influence of Bioinformatics Approaches on microRNA Nomenclature and Annotation) new miR genes (Lagos-Quintana et al., 2001; Reinhart et al., 2002; Floyd and Bowman, 2004; Wang et al., 2004; Adai et al., 2005; Zhang et al., 2006a; Lukasik et al., 2013). One of the early comprehensive computational analysis by Jones-Rhoades and Bartel (2004) systematically identified plant miRs and their regulatory targets that are conserved between Arabidopsis and rice. Using MIRcheck algorithm they predicted that the miRs could target mRNAs like superoxide dismutases (SOD), laccases, and ATP sulfurylases that are involved in plant stress responses. Such studies lead to identification of involvement of Ath-miR398 in the ROS pathway by targeting sites on Cu/Zn-SOD (Jones-Rhoades and Bartel, 2004; Sunkar and Zhu, 2004; Lu et al., 2005; Sunkar et al., 2005) A similar approach was used in miRFinder computational pipeline, to identify 91 conserved plant miRs in rice and Arabidopsis (Bonnet et al., 2004a).
Another strategy was based on the property of miRs to bind with perfect complementarity to their target transcripts (Laufs et al., 2004). In plant species where the target sequence was available the conserved miRs could be easily predicted by using 20 mer genomic segments with not more than two mismatches as in silico probes. This target-guided strategy was adopted to identify 16 families of drought stress-associated miRs from Physcomitrella patens (Wan et al., 2011).
The computational predictions also utilized the criteria for conservation of miR sequence and key secondary structure features of pre-miRs like their characteristic fold-back structure, thermodynamic stability etc. to predict new miRs (Berezikov et al., 2006). Seventy-nine putative miRs were identified in wheat using traditional computational strategy, out of which 9 were validated by northern blot experiments (Jin et al., 2008). Subsequently bioinformatics tools like miRAlign were developed based on the requirement of structural similarity and sequence conservation between new candidates and experimentally identified miRs (Wang et al., 2005). Though numerous miR profiles were generated by the computational algorithms, this was not found to be appropriate for species with less annotated genomes (Chen and Xiong, 2012).
The non-availability of complete genome annotation was overcome by employing the Expressed Sequence Tags (EST) database. These represented the true gene expression entities so they emerged as better indicators of dynamic expressions of the miR. A detailed study by identified 123 miRs from stress-induced ESTs of 60 plant species (Zhang et al., 2005). This study confirmed that irrespective of evolutionary divergence miRs are highly conserved in plant kingdom and miR genes may exist as orthologs or homologs in different species within the same kingdom (Weber, 2005; Zhang et al., 2006b). The EST database was also used to confirm some novel miRs identified earlier by computational strategies in citrus (Song et al., 2010) and peach (Zhang et al., 2012). In a recent study ESTs of abiotic stress treated libraries of Triticum aestivum were used to identify novel miRs in drought, cold, and salt stressed cDNA libraries by searching all mature sequences deposited in the miRBase (Release 19) (Pandey et al., 2013).
High Throughput Sequencing
The recent development of HTS approaches has invoked a new era by allowing the sequencing of millions of sRNA molecules. The HTS techniques employ sequencing-by-synthesis (SBS) technology, which enable accessing the full complexity of sRNAs in plants. In addition, it provides quantitative information of the expression profiles, since the cloning frequency of each sRNA generally reflects its relative presence in the sample. The signature-based expression profiling method such as massively parallel signature sequencing (MPSS) has identified miRs that have thus far proven difficult to find by using traditional cloning or in silico predictions. Sequencing technologies are rapidly emerging as the favored alternatives to the microarray-based approaches, since direct measures of gene expression can be obtained through sequencing of random ESTs, SAGE, and MPSS. The expression patterns of the identified miR targets can then be followed in the transcriptome sequencing data to gain novel insights into plant growth and development and stress responses (Wang et al., 2010; Li et al., 2013). Though currently an expensive technique, it is expected that as the technology grows, it will become more affordable.
Complex computational algorithms are used to rapidly and rigorously sift through the HTS data for identification of putative miRs (Figure 5). These datasets have been very successful in identification of conserved miRs where the sequence is well maintained across plant species. The targets for these miRs can also be easily predicted using Parallel Analysis of RNA End (PARE) sequencing, where miR and its target mRNA have often nearly perfect complementarily (Rhoades et al., 2002; Bonnet et al., 2004b; Jones-Rhoades and Bartel, 2004). The HTS data also provided a useful source to hunt for the non-conserved or species-specific miRs based on the criteria of miR annotation (Discussed in Section The Influence of Bioinformatics Approaches on microRNA Nomenclature and Annotation).
This HTS approach was initially used to visualize the repertoire of sRNAs in Arabidopsis (Rajagopalan et al., 2006; Fahlgren et al., 2007), followed by investigation on the rice miR expression profiles in drought and salt stress responses (Sunkar et al., 2008). Later, Liu and Zhang identified 67 arsenite-responsive miRs belonging to 26 miR families from Oryza sativa (Liu and Zhang, 2012). Solexa sequencing was also used to identify conserved and novel miRs in Glycine max libraries from water deficit and rust infections (Kulcheski et al., 2011), cold responsive miRs in trifoliate orange, Poncirus trifoliate, (Zhang et al., 2014a), drought and salinity responsive miRs in Gossypium hirsutum (Xie et al., 2015), heat stress induced miRs in Brassica napus (Yu et al., 2012), and salt stressed miRs in Raphanus sativus (Sun et al., 2015). Regulation of miRs in response to various abiotic stresses was studied in Arabidopsis, under drought, heat, salt, and metal ions such as copper (Cu), cadmium (Cd), sulfur (S) excess or deficiency, using sRNA NGS libraries. The search for most profound changes in miR expression patterns identified that miR319a/b, miR319b.2, and miR400 were responsive to most of the stresses under study (Barciszewska-Pacak et al., 2015).
Comparative profiles of miR expression during cold stress among Arabidopsis, Brachypodium, and Populus trichocarpa revealed that miR397 and miR169 are up-regulated. This indicated the presence of conserved cold responsive pathways in all the species. Whereas the differences in the pathways was highlighted by miR172 which was up-regulated in Arabidopsis and Brachypodium but not in poplar (Zhang et al., 2009a). Opposing patterns of miR regulation in different plant species during cold stress were observed for miR168 and miR171. The miRs are up-regulated in poplar (Lu et al., 2008) and Arabidopsis (Liu et al., 2008a) but down-regulated in rice (Lv et al., 2010). Likewise the HTS analysis of salt stressed sRNAome identified 211 conserved miRs and 162 novel miRs, belonging to 93 families between Populus trichocarpa and P. euphratica (Li et al., 2013). Using the approach of comparative miR profiling followed by experimental validation, our group identified 59 Osa-miRs that show tissue-preferential expression patterns and significantly supplemented 51 potential interactive nodes in these tissues (Mittal et al., 2013).
HTS technology has also played a crucial role in identification and characterization of the miR targets with PARE or Degradome sequencing. This involves sequencing of the entire pool of cleaved targets followed by mapping of the miR-guided cleavage sites (Ding et al., 2012). In Populus, 112 transcripts targeted by 51 identified miRs families were validated by using degradome sequencing (Li et al., 2013). These are several reports which used HTS of sRNA pools and degradome analysis to identify targets of stress induced miRs such as, in maize (Liu et al., 2014), tomato (Cao et al., 2014), Raphanus sativus (Wang et al., 2014), Populus (Chen et al., 2015), rice (Qin et al., 2015), Phaseolus vulgaris (Formey, 2015), and barley (Hackenberg et al., 2015).
It has been shown that plant miRs also act by inhibiting mRNA translation (Brodersen et al., 2008; Lanet et al., 2009), therefore such targets tend to get overlooked during degradome sequencing. The HTS techniques are also being employed for sequencing the whole transcriptome pools to identify the miR targets in Medicago (Cheung et al., 2006), Zea mays (Emrich et al., 2007), and Arabidopsis (Weber et al., 2007). The combined strategy of sRNAs and mRNAs (transcriptome) sequencing enabled the identification of new genes, involved in nitrate regulation and management of carbon and nitrogen metabolism in Arabidopsis. This study identified miR5640 and its target, AtPPC3, leading to the preposition that the responsive miR/target might be involved in modulating the carbon flux to assimilate nitrate into amino acids (Vidal et al., 2013).
The Influence of Bioinformatics Approaches on microRNA Nomenclature and Annotation
The in silico approaches have also played a dominant role in the identification of plant miRs and their targets. The advancement in molecular and computational approaches has not only resulted in the exponential growth in the discovery and study of sRNA biology but has also provided a deeper insight into the miR regulatory circuits. At the same time, they have been instrumental in defining and redefining the rules for annotating the miRs and their nomenclature.
A miR registry system was adopted in 2004 to facilitate a complete and searchable place for the published miRs and to provide a systematic rule so that the new miRs can be assigned with a distinctive name prior to publication of their discovery (Ambros et al., 2003; Griffiths-Jones, 2004). In miRBase the nomenclature of miRs starts with initial 3 letters signifying the organism, followed by a number which is simply a sequential numerical identifier based on sequence similarity, suffixed by “miR,” trailed by alphabet letters which denotes the family member (Figure 4). It was later enforced that sequences showing homology within organisms and mature identical sequences coming from two or more different organism should be assigned the same family names (Meyers et al., 2008). Sequences with no similarity to previously reported sequence were considered novel and assigned next number in the series (Griffiths-Jones, 2004). It is observed that in miRBase Medicago truncatula, mtr-miR2592 is the largest miR family with 66 members, while in rice; the largest family is seen for Osa-miR395 with 25 members. The occurrence of more than 1 mature sequence from same precursor is designated by an integer followed by a dot at the end (Griffiths-Jones, 2004; Meyers et al., 2008). With the accumulation of HTS data and the experimental validation that both miR and miR* of same precursor can be functional, it was decided to add a suffix of 3p and 5p at the end of the sequence to represent the presence of miR on 3′ or 5′ arm of stem loop precursor (Meyers et al., 2008).

Figure 4. Nomenclature schema of miRs.
The processing of biological information through bioinformatics tools and computational biology methods has now become crucial for elucidating complicated biological problems in genomics, proteomics, as well as in metabolomics. With the accumulation of huge sRNA sequencing datasets, it is almost impossible to analyze each and every sequence through direct experimental approaches. This has necessitated the role of bioinformatics tools and databases in analyzing and screening the huge data sets in a short time period, with minimum costs and without compromising on the specificity of analysis.
The primary criteria for annotation of plant miRs is the precise excision of a miR/miR* duplex from the stem of a single-stranded, stem-loop precursor. Computational algorithms use these criteria to predict the RNA secondary structure for the sequences identified from the genomic DNA, transcript or ESTs. Subsequently the annotation rules are followed to distinguish a miR from the sRNA pool. The first set of guidelines for miR annotation was based on specific expression and biogenesis criteria (Ambros et al., 2003). The expression criteria included the identification by cloning and/or detection by hybridization and phylogenetic conservation of the miR sequence. While the biogenesis criteria included the presence of a characteristic hairpin structured precursor transcript, conservation of the precursor secondary structure and increased accumulation of a precursor in absence or reduction in Dicer activity (Ambros et al., 2003).
The advancement in sequencing technologies provided with highly sensitive techniques for obtaining the complete small RNA profiles that could distinguish between fragments differing by a single base. This also provided an excellent medium to search for known and novel miR family members, their precursors, and modified versions. The bioinformatics based analysis of HTS datasets, made it feasible to predict the entire set of miRs present in a RNA sample. This was also utilized to retrieve the information on expression profiles, putative target transcripts, the miR isoforms, and sequence variants of miRs through differential expression profiling under various conditions (Moxon et al., 2008; Addo-Quaye et al., 2009; Yang and Li, 2011b; Neilsen et al., 2012). Dedicated web servers like isomiRex (Sablok et al., 2013) are available online for identification of the sequence variants using HTS data.
With the development in computational tools and the availability of genomic sequences the rules were further refined to include characteristics that are both necessary and sufficient for miR annotation. It was proposed that the prediction criteria should include that the miR and miR* are derived from opposite arm of same precursor such that they form a duplex with two nucleotide overhang at the 3′ end, base pairing of miR and miR* should have less than four mismatched bases, the asymmetric bulges are minimum in size and frequency specifically in miR/miR* duplex. sRNA-producing stem-loops that violate one of these criteria could still be annotated as miRs, provided that there is conclusive experimental evidence of precise miR/miR* excision (Meyers et al., 2008). In continuation to the guidelines set by Ambros et al. (2003) it was recognized that conservation of miRs, assessed using either bioinformatics or direct experimentation, was still a powerful indicator of their functional relevance though it need not be necessary for annotation as many plant miRs lack homologs in other species. It was proposed that identification of a target is not necessary for miR annotation as targets could not be predicted for many of the less-conserved miRs or the predicted targets lacked experimental confirmation.
It is being observed that increased coverage of deep-sequencing results have resulted in capturing sequences of ever-lower abundance. This has made the identification of miRs even more challenging. A number of recent publications have attempted use additional criteria based on patterns of mapped reads (Hendrix et al., 2010). The consensus set of guidelines that have started to emerge lay importance to the presence of multiple reads with consistent processing of the 5′-end of the mature sequence preferably from several independent experiments. The mapped reads should not overlap other annotated transcripts as they may represent fragments of mRNAs or other known RNA types.
Various tools were developed based on the annotation guidelines to analyze the HTS data sets. The major steps adopted by various available tools for prediction of novel miRs and their target identification are discussed in Figure 5. Basically the sequenced reads are selected, based on the average quality score appended with each base, and subjected to 3′ adapter trimming. This can be achieved by designing specific scripts (using languages such as PERL) or by using various available tools such as NGSQC Toolkit (Patel and Jain, 2012), FASTX-Toolkit (Gordon and Hannon, 2010), CLC Genomics Workbench (Matvienko)1 etc. Next the reads with length of 18–24 nucleotides are selected and aligned to the corresponding genome of the plant species under consideration using tools such as bowtie, soap, and bwa. The aligned reads are then used to filter out sequences mapping with other sRNAs such as, tRNA, rRNA, sRNA, snRNA, snoRNA, and known miRs. The remaining reads are used to retrieve the potential precursors from the reference genome and their secondary structure is predicted. Excellent softwares like Mfold (Zuker, 2003), RNAfold (Denman, 1993) etc. are freely available and have been useful in identifying the appropriately folded structures. Then these candidate precursors are evaluated on the basis of the annotation criteria (Meyers et al., 2008). The expression profiles of identified known and novel miRs from sequence pools are achieved by calculating the number of times a unique read occurred in the entire sRNA pool and normalized against total reads. Reads Per Million (RPM) for each sequence occurring in each sample is most common way to achieve the normalized expression of each sequence. RPM = (Actual read count/total number of reads in sample) × 1,000,000) (Motameny et al., 2010).
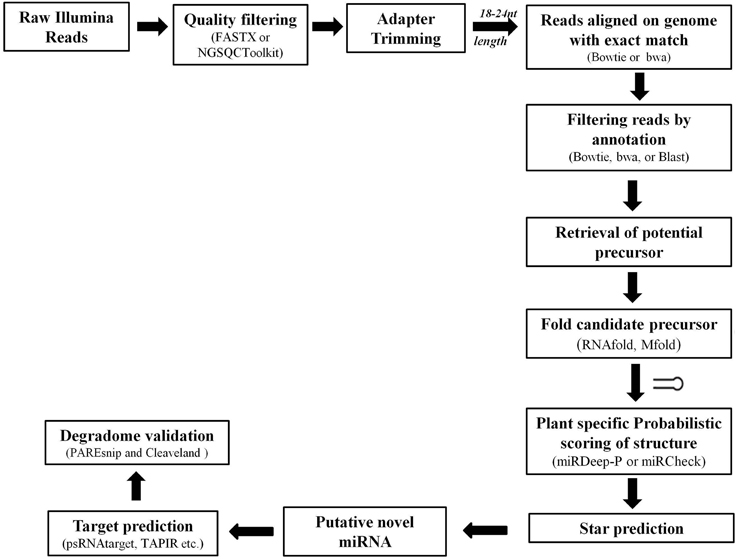
Figure 5. Pipeline showing major steps for miR identification from high-throughput sequencing data.
microRNA Repositories
The study of miR and their targets by analyzing the sRNA and transcriptome sequences is greatly facilitated by the availability of numerous freely accessible tools and databases, which can be used by experimental researchers without any specialization in bioinformatics. The various web-based tools and databases available for the prediction and analysis of plant miRs and their targets are listed in Tables 1, 2, respectively. Each of these is based on different algorithms and methodologies and has their respective strengths and shortcomings. However, the major limitation in most of these techniques is the requirement for a known sequence and the search for a conserved hairpin loop structure (Unver et al., 2009). To overcome these limitations, Kadri et al. (2009) developed the Hierarchical Hidden Markov Model (HHMM) that employs region-based structural information of pre-miRs without relying on phylogenetic conservation. It obtains the secondary structures on the basis of minimum free energy and then classifies the sequence with HHMM (Kadri et al., 2009). Some of the popularly used tools are discussed below.

Table 1. Major Plant databases providing information on the miR and their targets.
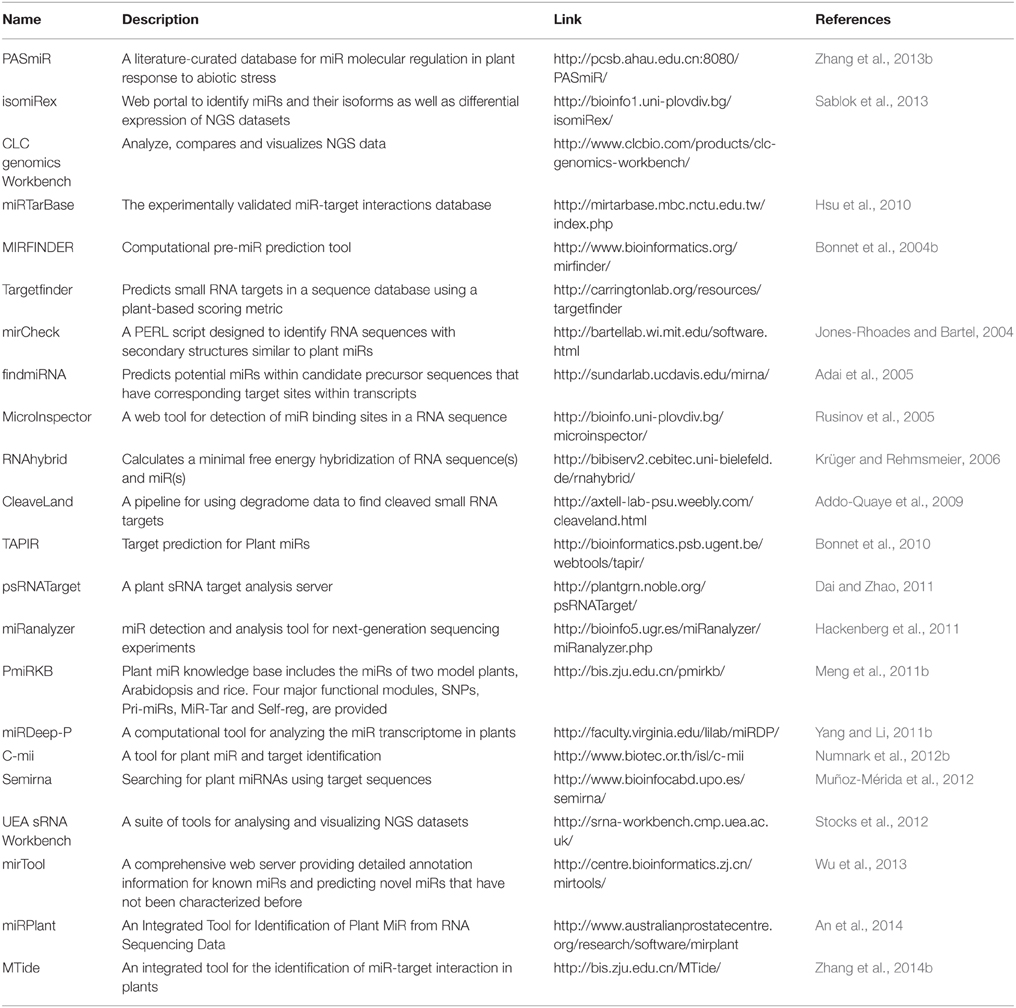
Table 2. Major tools for analyzing plant miRs and their targets.
miRcheck
This is an algorithm written in the form of a PERL script for identifying 20 mers having potential to encode plant miRs. The tool requires input of a putative hairpin sequences and their secondary structures. The presence of candidate 20 mer sequences is then searched within the hairpin to predict potential plant miR. This algorithm was first used for identifying conserved miRs in Arabidopsis and rice (Jones-Rhoades and Bartel, 2004).
UEA sRNA Workbench
It is a comprehensive tool for the complete analysis of sRNA sequencing data and provides the convenience of using the facilities provided by different tools in one place. Its Graphical User Interface (GUI) makes it easy to use for researchers, do not needs any prior knowledge of computer programming (Moxon et al., 2008). It can be downloaded and installed locally, and it also has a web-based facility of doing the same analysis in form of UEA sRNA toolkit which is freely accessible. Table 3 lists all the available tools at UEA sRNA Workbench.

Table 3. Tools available on UEA sRNA Workbench and their functions in analyzing the sRNA sequencing data.
TAPIR
This is an online web server for prediction of targets of plant miRs. It can characterize miR-targets duplexes with large loops which are usually not detectible by traditional target prediction tools. The prediction results are driven by a combination of two different algorithms. The first one is the fast and canonical FASTA local alignment program which cannot detect duplexes with large number of bulges and/or mismatches (Pearson, 2004) and second one is RNAhybrid (Krüger and Rehmsmeier, 2006) for detection of miR-mRNA duplexes (Bonnet et al., 2010). Though it is a good option for miR target prediction but is not preferred as the users face problem in analyzing large datasets on the online server.
CLC Genomics Workbench
It is a commercial software developed by QIAGEN that offers Quality Check (QC) and pre-processing of NGS data. Although it is a good tool for preprocessing of NGS data but it focuses more on other genomic areas such as de novo assembly and it doesn't provides the facility to process the sRNA data for miR and target identification. In relation to the sRNAs it has been majorly used in initial steps of quality filtering, adapter trimming and calculating abundances of sRNA libraries. It can also generate genome alignments by using standalone blast search. The workbench provides an interactive visualization to the differential expression and statistical analysis of RNA-Seq and sRNA data.
C-mii
It uses a homology-based approach for plant miR and target identification. The tool aligns known miRs from different plant species to the EST sequences of the query plant species using blast homology search. The aligned sequences are allowed to fold in to the characteristic hairpin loop structures to identify the putative miRs. The predicted miR sequences are further used for identifying perfect or nearly perfect complimentary sites on the input transcript sequences to identify the putative targets. The tool has a unique feature of predicting the secondary structures of the miR-target duplexes. The identified targets can be annotated further by searching their functions and Gene Ontologies (GO) (Numnark et al., 2012a). It provides user friendly GUI, and is easily downloadable hence it can be easily used for analyzing large datasets. However, the major limitation lies in the search and availability of homologous sequences, so it cannot be used to analyze the NGS datasets.
miRDeep-P
It is a collection of PERL scripts that are used for prediction of novel miRs from deep sequencing data. It was developed by incorporating the plant-miR specific criteria to miRDeep (Friedländer et al., 2008). Its pipeline utilizes bowtie for sequence alignments and RNAfold for secondary structure prediction of putative precursors. The remaining steps such as extracting potential precursor sequences and identification of putative novel miR is regulated by specific scripts (Yang and Li, 2011a). Although it is a specialized tool for identification of plant miRs, but does not has a GUI interface. So the user needs to work through command line for its execution, which warrants knowledge on PERL scripting.
CleaveLand
It is a general pipeline, available as a combination of PERL scripts, for detecting miR-cleaved target transcripts from degradome datasets (Addo-Quaye et al., 2009). It can be executed by a single command and requires input of degradome sequences, sRNAs, and an mRNA database to yield an output of cleaved targets. The pipeline runs in command mode and requires the co-installation of several dependencies such as PERL, R, samtools, bowtie, RNAplex etc.
ARMOUR
The accumulation of sequencing data has generated the need for a comprehensive and integrated database of miR:mRNA, expression profile information and target information. Our group has developed ARMOUR database (A Rice miRNA: mRNA Interaction Resource) that consolidates extensive datasets of rice miRs from various deep sequencing datasets for examining the expression changes with respect to their targets. Development of such interactomes for different plant species shall provide a valuable tool to biologists for selecting miRs for further functional studies.
Perspectives
miRs are an extensive class of endogenous, small regulators of gene expression in the numerous developmental and signaling pathways. There is ample evidence for the role of miRs in abiotic stress mediated genomic changes that result in attenuation of plant growth and development. The different experimental approaches have identified the intriguing expression profiles of miRs in distinctive tissues and/or stages of development. The regulation of miR expression also varies between the domesticated plant species and their wild relatives. Sequence-based profiling along with computational analysis has played a pivotal role in the identification of stress-responsive miRs, although these results require independent experimental validations. sRNA blot and RT-PCR analysis have played an equally important part in systematically confirming the profiling data. The identification of putative targets for these miRs has provided robust confirmation of their stress responsiveness. This has also enabled quantification of their effect on the genetic networks, such that many of the stress regulated miRs have emerged as potential candidates for improving plant performance under stress. However, so many efforts are still required for in-depth analysis of the miR modulation of each gene product induced by abiotic stress(es) and its interacting partners. This requires development of reliable and rigorous assays for firm characterization of the spatio-temporal regulation of these miRs under stress conditions. The potential of computational biology needs to be tapped for performing an extensive comparison of miR expression profiles among agriculturally important crops during environmental stress conditions to tap key target nodes that need to be modulated for improving crop tolerance to environmental stress. The development and integration of plant synthetic biology tools and approaches will add new functionalities and perspectives in the miR biology to make them relevant for genetic engineering programs for enhancing abiotic stress tolerance.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
There is a vast literature on miRs, so we offer our apologies to researchers whose work could not be cited here. The research in our lab is supported through different grants from the Department of Biotechnology (DBT), Government of India.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphys.2015.00286
Supplementary Table 1. List of all abiotic stress associated plant miRs.
Footnotes
1. ^Matvienko, M. CLC Genomics Workbench.
References
Adai, A., Johnson, C., Mlotshwa, S., Archer-Evans, S., Manocha, V., Vance, V., et al. (2005). Computational prediction of miRNAs in Arabidopsis thaliana. Genome Res. 15, 78–91. doi: 10.1101/gr.2908205
Addo-Quaye, C., Miller, W., and Axtell, M. J. (2009). CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 25, 130–131. doi: 10.1093/bioinformatics/btn604
Alam, M., Stuchbury, T., Naylor, R., and Rashid, M. (2004). Effect of salinity on growth of some modern rice cultivars. J. Agron. 3, 1–10. doi: 10.3923/ja.2004.1.10
Ambros, V., Bartel, B., Bartel, D. P., Burge, C. B., Carrington, J. C., Chen, X., et al. (2003). A uniform system for microRNA annotation. RNA 9, 277–279. doi: 10.1261/rna.2183803
An, J., Lai, J., Sajjanhar, A., Lehman, M. L., and Nelson, C. C. (2014). miRPlant: an integrated tool for identification of plant miRNA from RNA sequencing data. BMC Bioinformatics 15:275. doi: 10.1186/1471-2105-15-275
Apel, K., and Hirt, H. (2004). Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 55, 373–399. doi: 10.1146/annurev.arplant.55.031903.141701
Arenas-Huertero, C., Pérez, B., Rabanal, F., Blanco-Melo, D., De la Rosa, C., Estrada-Navarrete, G., et al. (2009). Conserved and novel miRNAs in the legume Phaseolus vulgaris in response to stress. Plant Mol. Biol. 70, 385–401. doi: 10.1007/s11103-009-9480-3
Aroca, R., Irigoyen, J. J., and Sánchez-DıìAz, M. (2001). Photosynthetic characteristics and protective mechanisms against oxidative stress during chilling and subsequent recovery in two maize varieties differing in chilling sensitivity. Plant Sci. 161, 719–726. doi: 10.1016/S0168-9452(01)00460-5
Aroca, R., Porcel, R., and Ruiz-Lozano, J. M. (2012). Regulation of root water uptake under abiotic stress conditions. J. Exp. Bot. 63, 43–57. doi: 10.1093/jxb/err266
Arteaga-Vázquez, M., Caballero-Pérez, J., and Vielle-Calzada, J.-P. (2006). A family of microRNAs present in plants and animals. Plant Cell Online 18, 3355–3369. doi: 10.1105/tpc.106.044420
Axtell, M. J., and Bowman, J. L. (2008). Evolution of plant microRNAs and their targets. Trends Plant Sci. 13, 343–349. doi: 10.1016/j.tplants.2008.03.009
Bailey-Serres, J., and Voesenek, L. (2008). Flooding stress: acclimations and genetic diversity. Annu. Rev. Plant Biol. 59, 313–339. doi: 10.1146/annurev.arplant.59.032607.092752
Barciszewska-Pacak, M., Milanowska, K., Knop, K., Bielewicz, D., Nuc, P., Plewka, P., et al. (2015). Arabidopsis microRNA expression regulation in a wide range of abiotic stress responses. Front. Plant Sci. 6:410. doi: 10.3389/fpls.2015.00410
Bartels, D., and Sunkar, R. (2005). Drought and salt tolerance in plants. Crit. Rev. Plant Sci. 24, 23–58. doi: 10.1080/07352680590910410
Baumberger, N., and Baulcombe, D. (2005). Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits microRNAs and short interfering RNAs. Proc. Natl. Acad. Sci. U.S.A. 102, 11928–11933. doi: 10.1073/pnas.0505461102
Berezikov, E., Cuppen, E., and Plasterk, R. H. (2006). Approaches to microRNA discovery. Nat. Genet. 38, S2–S7. doi: 10.1038/ng1794
Bonnet, E., He, Y., Billiau, K., and Van de Peer, Y. (2010). TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 26, 1566–1568. doi: 10.1093/bioinformatics/btq233
Bonnet, E., Wuyts, J., Rouzé, P., and Van de Peer, Y. (2004a). Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes. Proc. Natl. Acad. Sci. U.S.A. 101, 11511–11516. doi: 10.1073/pnas.0404025101
Bonnet, E., Wuyts, J., Rouzé, P., and Van de Peer, Y. (2004b). Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics 20, 2911–2917. doi: 10.1093/bioinformatics/bth374
Bréda, N., Granier, A., and Aussenac, G. (1995). Effects of thinning on soil and tree water relations, transpiration and growth in an oak forest (Quercus petraea (Matt.) Liebl.). Tree Physiol. 15, 295–306. doi: 10.1093/treephys/15.5.295
Brodersen, P., Sakvarelidze-Achard, L., Bruun-Rasmussen, M., Dunoyer, P., Yamamoto, Y. Y., Sieburth, L., et al. (2008). Widespread translational inhibition by plant miRNAs and siRNAs. Science 320, 1185–1190. doi: 10.1126/science.1159151
Burge, S. W., Daub, J., Eberhardt, R., Tate, J., Barquist, L., Nawrocki, E. P., et al. (2013). Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 41, D226–D232. doi: 10.1093/nar/gks1005
Cai, L., Rensing, C., Li, X., and Wang, G. (2009). Novel gene clusters involved in arsenite oxidation and resistance in two arsenite oxidizers: Achromobacter sp. SY8 and Pseudomonas sp. TS44. Appl. Microbiol. Biotechnol. 83, 715–725. doi: 10.1007/s00253-009-1929-4
Cao, X., Wu, Z., Jiang, F., Zhou, R., and Yang, Z. (2014). Identification of chilling stress-responsive tomato microRNAs and their target genes by high-throughput sequencing and degradome analysis. BMC Genomics 15:1130. doi: 10.1186/1471-2164-15-1130
Carra, A., Mica, E., Gambino, G., Pindo, M., Moser, C., Pè, M. E., et al. (2009). Cloning and characterization of small non−coding RNAs from grape. Plant J. 59, 750–763. doi: 10.1111/j.1365-313X.2009.03906.x
Carrington, J. C., and Ambros, V. (2003). Role of microRNAs in plant and animal development. Science 301, 336–338. doi: 10.1126/science.1085242
Chen, H.-M., Chen, L.-T., Patel, K., Li, Y.-H., Baulcombe, D. C., and Wu, S.-H. (2010). 22-Nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc. Natl. Acad. Sci. U.S.A. 107, 15269–15274. doi: 10.1073/pnas.1001738107
Chen, H., and Xiong, L. (2012). “Genome-wide transcriptional reprogramming under drought stress,” in Plant Responses to Drought Stress, ed R. Aroca (Berlin; Heidelberg: Springer), 273–289.
Chen, M., Bao, H., Wu, Q., and Wang, Y. (2015). Transcriptome-wide identification of miRNA targets under nitrogen deficiency in populus tomentosa using degradome sequencing. Int. J. Mol. Sci. 16, 13937–13958. doi: 10.3390/ijms160613937
Chen, X. (2008). “MicroRNA metabolism in plants,” in Rna Interference, ed P. J. Paddisom and P. K. Vogt (Berlin; Heidelberg: Springer), 117–136.
Chen, X. (2009). Small RNAs and their roles in plant development. Ann. Rev. Cell Dev. 25, 21–44. doi: 10.1146/annurev.cellbio.042308.113417
Cheung, F., Haas, B. J., Goldberg, S. M., May, G. D., Xiao, Y., and Town, C. D. (2006). Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology. BMC Genomics 7:272. doi: 10.1186/1471-2164-7-272
Chinnusamy, V., Zhu, J., and Zhu, J.-K. (2007). Cold stress regulation of gene expression in plants. Trends Plant Sci. 12, 444–451. doi: 10.1016/j.tplants.2007.07.002
Choi, D. H., Hwang, C. Y., and Cho, B. C. (2003). Comparison of virus- and bacterivory-induced bacterial mortality in the eutrophic Masan Bay, Korea. Aquat. Microb. Ecol. 30, 117–125. doi: 10.3354/ame030117
Covarrubias, A. A., and Reyes, J. L. (2010). Post-transcriptional gene regulation of salinity and drought responses by plant microRNAs. Plant Cell Environ. 33, 481–489. doi: 10.1111/j.1365-3040.2009.02048.x
Cramer, G. R., Urano, K., Delrot, S., Pezzotti, M., and Shinozaki, K. (2011). Effects of abiotic stress on plants: a systems biology perspective. BMC Plant Biol. 11:163. doi: 10.1186/1471-2229-11-163
Dai, X., and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. doi: 10.1093/nar/gkr319
Denman, R. (1993). Using RNAFOLD to predict the activity of small catalytic RNAs. Biotechniques 15, 1090–1095.
de Oliveira, A. B., Gomes-Filho, E., and Alencar, N. L. M. (2013). “Comparison between the water and salt stress effects on plant growth and development,” in Responses of Organisms to Water Stress, ed S. Akinci (INTECH Open Access Publisher).
Diédhiou, C., and Golldack, D. (2006). Salt-dependent regulation of chloride channel transcripts in rice. Plant Sci. 170, 793–800. doi: 10.1016/j.plantsci.2005.11.014
Ding, D., Zhang, L., Wang, H., Liu, Z., Zhang, Z., and Zheng, Y. (2009). Differential expression of miRNAs in response to salt stress in maize roots. Ann. Bot. 103, 29–38. doi: 10.1093/aob/mcn205
Ding, J., Zhou, S., and Guan, J. (2012). Finding microRNA targets in plants: current status and perspectives. Genomics Proteomics Bioinformatics 10, 264–275. doi: 10.1016/j.gpb.2012.09.003
Ding, Y., Chen, Z., and Zhu, C. (2011). Microarray-based analysis of cadmium-responsive microRNAs in rice (Oryza sativa). J. Exp. Bot. 62, 3563–3573. doi: 10.1093/jxb/err046
Dong, Z., Han, M.-H., and Fedoroff, N. (2008). The RNA-binding proteins HYL1 and SE promote accurate in vitro processing of pri-miRNA by DCL1. Proc. Natl. Acad. Sci. U.S.A. 105, 9970–9975. doi: 10.1073/pnas.0803356105
Dugas, D. V., and Bartel, B. (2004). MicroRNA regulation of gene expression in plants. Curr. Opin. Plant Biol. 7, 512–520. doi: 10.1016/j.pbi.2004.07.011
Duque, A. S., Farinha, A. P., Da Silva, A. B., De Almeida, A. M., Santos, D., Da Silva, J. M., et al. (2013). “Abiotic stress responses in plants: unraveling the complexity of genes and networks to survive,” in Abiotic Stress-Plant Responses and Applications in Agriculture, eds K. Vahdati and C. Leslie (INTECH Open Access Publisher).
Duursma, R. A., Kolari, P., Perämäki, M., Nikinmaa, E., Hari, P., Delzon, S., et al. (2008). Predicting the decline in daily maximum transpiration rate of two pine stands during drought based on constant minimum leaf water potential and plant hydraulic conductance. Tree Physiol. 28:265. doi: 10.1093/treephys/28.2.265
Eldem, V., Okay, S., and Ünver, T. (2013). Plant microRNAs: new players in functional genomics. Turkish J. Agri. Forestry 37, 1–21. doi: 10.3906/tar-1206-50
Emrich, S. J., Barbazuk, W. B., Li, L., and Schnable, P. S. (2007). Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res. 17, 69–73. doi: 10.1101/gr.5145806
Fahlgren, N., Howell, M. D., Kasschau, K. D., Chapman, E. J., Sullivan, C. M., Cumbie, J. S., et al. (2007). High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS ONE 2:e219. doi: 10.1371/journal.pone.0000219
Fang, Y., and Spector, D. L. (2007). Identification of nuclear dicing bodies containing proteins for microRNA biogenesis in living Arabidopsis plants. Curr. Biol. 17, 818–823. doi: 10.1016/j.cub.2007.04.005
Floyd, S. K., and Bowman, J. L. (2004). Gene regulation: ancient microRNA target sequences in plants. Nature 428, 485–486. doi: 10.1038/428485a
Folkes, L., Moxon, S., Woolfenden, H. C., Stocks, M. B., Szittya, G., Dalmay, T., et al. (2012). PAREsnip: a tool for rapid genome-wide discovery of small RNA/target interactions evidenced through degradome sequencing. Nucleic Acids Res. 40, e103–e103. doi: 10.1093/nar/gks277
Formey, D. (2015). “Genome-wide identification of the phaseolus vulgaris sRNAome using small RNA and degradome sequencing,” in Plant and Animal Genome XXIII Conference: Plant and Animal Genome (San Diego, CA).
Foyer, C., Descourvieres, P., and Kunert, K. (1994). Protection against oxygen radicals: an important defence mechanism studied in transgenic plants. Plant Cell Environ. 17, 507–523. doi: 10.1111/j.1365-3040.1994.tb00146.x
Foyer, C. H., and Noctor, G. (2005). Oxidant and antioxidant signalling in plants: a re−evaluation of the concept of oxidative stress in a physiological context. Plant Cell Environ. 28, 1056–1071. doi: 10.1111/j.1365-3040.2005.01327.x
Friedländer, M. R., Chen, W., Adamidi, C., Maaskola, J., Einspanier, R., Knespel, S., et al. (2008). Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 26, 407–415. doi: 10.1038/nbt1394
Fujii, H., Chiou, T.-J., Lin, S.-I., Aung, K., and Zhu, J.-K. (2005). A miRNA involved in phosphate-starvation response in Arabidopsis. Curr. Biol. 15, 2038–2043. doi: 10.1016/j.cub.2005.10.016
Fujioka, Y., Utsumi, M., Ohba, Y., and Watanabe, Y. (2007). Location of a possible miRNA processing site in SmD3/SmB nuclear bodies in Arabidopsis. Plant Cell Physiol. 48, 1243–1253. doi: 10.1093/pcp/pcm099
Garzon, R., Fabbri, M., Cimmino, A., Calin, G. A., and Croce, C. M. (2006). MicroRNA expression and function in cancer. Trends Mol. Med. 12, 580–587. doi: 10.1016/j.molmed.2006.10.006
Ghosh, D., and Xu, J. (2014). Abiotic stress responses in plant roots: a proteomics perspective. Front. Plant Sci. 5:6. doi: 10.3389/fpls.2014.00006
Giri, J. (2011). Glycinebetaine and abiotic stress tolerance in plants. Plant Signal. Behav. 6, 1746–1751. doi: 10.4161/psb.6.11.17801
Gleave, A. P., Ampomah-Dwamena, C., Berthold, S., Dejnoprat, S., Karunairetnam, S., Nain, B., et al. (2008). Identification and characterisation of primary microRNAs from apple (Malus domestica cv. Royal Gala) expressed sequence tags. Tree Genetics Genomes 4, 343–358. doi: 10.1007/s11295-007-0113-1
Gordon, A., and Hannon, G. (2010). Fastx-toolkit. FASTQ/A Short-reads Pre-processing Tools. Unpublished Available online at: http://hannonlab.cshl.edu/fastx_toolkit
Goswami, S., Kumar, R. R., and Rai, R. D. (2014). Heat-responsive microRNAs regulate the transcription factors and heat shock proteins in modulating thermo-stability of starch biosynthesis enzymes in wheat (‘Triticum aestivum’L.) under the heat stress. Aust. J. Crop Sci. 8, 697–705.
Grass, L. (1994). Effect of Heat Stress during Seed Development and Maturation on Wheat (Triticum durum) Seed Quality. Retrospective theses and dissertations, Digital Repository @ Iowa State University. Available online at: http://lib.dr.iastate.edu/
Griffiths-Jones, S. (2004). The microRNA registry. Nucleic Acids Res. 32, D109–D111. doi: 10.1093/nar/gkh023
Griffiths-Jones, S., Saini, H. K., van Dongen, S., and Enright, A. J. (2008). miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154–D158. doi: 10.1093/nar/gkm952
Guan, Q., Lu, X., Zeng, H., Zhang, Y., and Zhu, J. (2013). Heat stress induction of miR398 triggers a regulatory loop that is critical for thermotolerance in Arabidopsis. Plant J. 74, 840–851. doi: 10.1111/tpj.12169
Hackenberg, M., Gustafson, P., Langridge, P., and Shi, B. J. (2015). Differential expression of microRNAs and other small RNAs in barley between water and drought conditions. Plant Biotechnol. J. 13, 2–13. doi: 10.1111/pbi.12220
Hackenberg, M., Rodríguez-Ezpeleta, N., and Aransay, A. M. (2011). miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res. 39, W132–W138. doi: 10.1093/nar/gkr247
Hakim, L. (2013). Variability and correlation of agronomic characters of Mungbean germplasm and their utilization for variety improvement program. Indones. J. Agric. Sci. 9, 24–28.
Han, M.-H., Goud, S., Song, L., and Fedoroff, N. (2004). The Arabidopsis double-stranded RNA-binding protein HYL1 plays a role in microRNA-mediated gene regulation. Proc. Natl. Acad. Sci. U.S.A. 101, 1093–1098. doi: 10.1073/pnas.0307969100
Hasanuzzaman, M., Nahar, K., Alam, M. M., Roychowdhury, R., and Fujita, M. (2013a). Physiological, biochemical, and molecular mechanisms of heat stress tolerance in plants. Int. J. Mol. Sci. 14, 9643–9684. doi: 10.3390/ijms14059643
Hasanuzzaman, M., Nahar, K., and Fujita, M. (2013b). “Extreme temperature responses, oxidative stress and antioxidant defense in plants,” in Abiotic StressŮPlant Responses and Applications in Agriculture, ed Kourosh Vahdati (INTECH Open Access Publisher). Available online at: http://www.intechopen.com/books/abiotic-stress-plant-responses-and-applications-in-agriculture/extreme-temperature-responses-oxidative-stress-and-antioxidant-defense-in-plants
Hasanuzzaman, M., Nahar, K., and Fujita, M. (2013c). “Plant response to salt stress and role of exogenous protectants to mitigate salt-induced damages,” in Ecophysiology and Responses of Plants Under Salt Stress (New York, NY: Springer), 25–87.
Hedhly, A. (2011). Sensitivity of flowering plant gametophytes to temperature fluctuations. Environ. Exp. Bot. 74, 9–16. doi: 10.1016/j.envexpbot.2011.03.016
Hendrix, D., Levine, M., and Shi, W. (2010). Method miRTRAP, a computational method for the systematic identification of miRNAs from high throughput sequencing data. Genome Biol. 11:R39. doi: 10.1186/gb-2010-11-4-r39
Hiraguri, A., Itoh, R., Kondo, N., Nomura, Y., Aizawa, D., Murai, Y., et al. (2005). Specific interactions between Dicer-like proteins and HYL1/DRB-family dsRNA-binding proteins in Arabidopsis thaliana. Plant Mol. Biol. 57, 173–188. doi: 10.1007/s11103-004-6853-5
Hsu, S.-D., Lin, F.-M., Wu, W.-Y., Liang, C., Huang, W.-C., Chan, W.-L., et al. (2010). miRTarBase: a database curates experimentally validated microRNA–target interactions. Nucleic Acids Res. 42, D78–D85. doi: 10.1093/nar/gkq1107
Jagadeeswaran, G., Zheng, Y., Sumathipala, N., Jiang, H., Arrese, E. L., Soulages, J. L., et al. (2010). Deep sequencing of small RNA libraries reveals dynamic regulation of conserved and novel microRNAs and microRNA-stars during silkworm development. BMC Genomics 11:52. doi: 10.1186/1471-2164-11-52
Jia, L., Zhang, D., Qi, X., Ma, B., Xiang, Z., and He, N. (2014). Identification of the conserved and novel miRNAs in Mulberry by high-throughput sequencing. PLoS ONE 9:e104409. doi: 10.1371/journal.pone.0104409
Jia, X., Wang, W.-X., Ren, L., Chen, Q.-J., Mendu, V., Willcut, B., et al. (2009). Differential and dynamic regulation of miR398 in response to ABA and salt stress in Populus tremula and Arabidopsis thaliana. Plant Mol. Biol. 71, 51–59. doi: 10.1007/s11103-009-9508-8
Jin, W., Li, N., Zhang, B., Wu, F., Li, W., Guo, A., et al. (2008). Identification and verification of microRNA in wheat (Triticum aestivum). J. Plant Res. 121, 351–355. doi: 10.1007/s10265-007-0139-3
Jones-Rhoades, M. W., and Bartel, D. P. (2004). Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 14, 787–799. doi: 10.1016/j.molcel.2004.05.027
Jones-Rhoades, M. W., Bartel, D. P., and Bartel, B. (2006). MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 57, 19–53. doi: 10.1146/annurev.arplant.57.032905.105218
Joseph, E. A., and Mohanan, K. (2013). A study on the effect of salinity stress on the growth and yield of some native rice cultivars of Kerala state of India. Agric. Fish 2, 141–150. doi: 10.11648/j.aff.20130203.14
Jung, H. J., and Kang, H. (2007). Expression and functional analyses of microRNA417 in Arabidopsis thaliana under stress conditions. Plant Physiol. Biochem. 45, 805–811. doi: 10.1016/j.plaphy.2007.07.015
Kacperska, A. (2004). Sensor types in signal transduction pathways in plant cells responding to abiotic stressors: do they depend on stress intensity? Physiol. Plant 122, 159–168. doi: 10.1111/j.0031-9317.2004.00388.x
Kadri, S., Hinman, V., and Benos, P. V. (2009). HHMMiR: efficient de novo prediction of microRNAs using hierarchical hidden Markov models. BMC Bioinformatics 10:S35. doi: 10.1186/1471-2105-10-S1-S35
Kaiser, W. M. (1987). Effects of water deficit on photosynthetic capacity. Physiol. Plant 71, 142–149. doi: 10.1111/j.1399-3054.1987.tb04631.x
Kang, K., Lee, K., Park, S., Kim, Y. S., and Back, K. (2010). Enhanced production of melatonin by ectopic overexpression of human serotonin N−acetyltransferase plays a role in cold resistance in transgenic rice seedlings. J. Pineal Res. 49, 176–182. doi: 10.1111/j.1600-079x.2010.00783.x
Kantar, M., Lucas, S. J., and Budak, H. (2011). miRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta 233, 471–484. doi: 10.1007/s00425-010-1309-4
Kantar, M., Unver, T., and Budak, H. (2010). Regulation of barley miRNAs upon dehydration stress correlated with target gene expression. Funct. Integr. Genomics 10, 493–507. doi: 10.1007/s10142-010-0181-4
Khan, Y., Yadav, A., Bonthala, V. S., Muthamilarasan, M., Yadav, C. B., and Prasad, M. (2014). Comprehensive genome-wide identification and expression profiling of foxtail millet [Setaria italica (L.)] miRNAs in response to abiotic stress and development of miRNA database. Plant Cell Tiss. Organ Cult. 118, 279–292. doi: 10.1007/s11240-014-0480-x
Khraiwesh, B., Zhu, J. K., and Zhu, J. (2012). Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta 1819, 137–148. doi: 10.1016/j.bbagrm.2011.05.001
Kim, S., Yang, J.-Y., Xu, J., Jang, I.-C., Prigge, M. J., and Chua, N.-H. (2008). Two cap-binding proteins CBP20 and CBP80 are involved in processing primary MicroRNAs. Plant Cell Physiol. 49, 1634–1644. doi: 10.1093/pcp/pcn146
Kim, Y. J., Zheng, B., Yu, Y., Won, S. Y., Mo, B., and Chen, X. (2011). The role of Mediator in small and long noncoding RNA production in Arabidopsis thaliana. EMBO J. 30, 814–822. doi: 10.1038/emboj.2011.3
Kozomara, A., and Griffiths-Jones, S. (2010). miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39, D152–D157. doi: 10.1093/nar/gkq1027
Kozomara, A., and Griffiths-Jones, S. (2014). miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73. doi: 10.1093/nar/gkt1181
Krüger, J., and Rehmsmeier, M. (2006). RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 34, W451–W454. doi: 10.1093/nar/gkl243
Kulcheski, F. R., de Oliveira, L. F., Molina, L. G., Almerão, M. P., Rodrigues, F. A., Marcolino, J., et al. (2011). Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genomics 12:307. doi: 10.1186/1471-2164-12-307
Kurihara, Y., Takashi, Y., and Watanabe, Y. (2006). The interaction between DCL1 and HYL1 is important for efficient and precise processing of pri-miRNA in plant microRNA biogenesis. RNA 12, 206–212. doi: 10.1261/rna.2146906
Kurihara, Y., and Watanabe, Y. (2004). Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. U.S.A. 101, 12753–12758. doi: 10.1073/pnas.0403115101
Lagos-Quintana, M., Rauhut, R., Lendeckel, W., and Tuschl, T. (2001). Identification of novel genes coding for small expressed RNAs. Science 294, 853–858. doi: 10.1126/science.1064921
Lanet, E., Delannoy, E., Sormani, R., Floris, M., Brodersen, P., Crété, P., et al. (2009). Biochemical evidence for translational repression by Arabidopsis microRNAs. Plant Cell Online 21, 1762–1768. doi: 10.1105/tpc.108.063412
Lau, N. C., Lim, L. P., Weinstein, E. G., and Bartel, D. P. (2001). An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294, 858–862. doi: 10.1126/science.1065062
Laufs, P., Peaucelle, A., Morin, H., and Traas, J. (2004). MicroRNA regulation of the CUC genes is required for boundary size control in Arabidopsis meristems. Development 131, 4311–4322. doi: 10.1242/dev.01320
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854. doi: 10.1016/0092-8674(93)90529-Y
Lee, Y., Kim, M., Han, J., Yeom, K. H., Lee, S., Baek, S. H., et al. (2004). MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060. doi: 10.1038/sj.emboj.7600385
Li, B., Duan, H., Li, J., Deng, X. W., Yin, W., and Xia, X. (2013). Global identification of miRNAs and targets in Populus euphratica under salt stress. Plant Mol. Biol. 81, 525–539. doi: 10.1007/s11103-013-0010-y
Li, J., Yang, Z., Yu, B., Liu, J., and Chen, X. (2005a). Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr. Biol. 15, 1501–1507. doi: 10.1016/j.cub.2005.07.029
Li, T., Li, H., Zhang, Y.-X., and Liu, J.-Y. (2011). Identification and analysis of seven H2O2-responsive miRNAs and 32 new miRNAs in the seedlings of rice (Oryza sativa L. ssp. indica). Nucleic Acids Res. 39, 2821–2833. doi: 10.1093/nar/gkq1047
Li, W.-X., Oono, Y., Zhu, J., He, X.-J., Wu, J.-M., Iida, K., et al. (2008). The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell Online 20, 2238–2251. doi: 10.1105/tpc.108.059444
Li, Y., Li, W., and Jin, Y.-X. (2005b). Computational identification of novel family members of microRNA genes in Arabidopsis thaliana and Oryza sativa. Acta Biochim. Biophys. Sin. 37, 75–87. doi: 10.1093/abbs/37.2.75
Lima, J. C., Arenhart, R. A., Margis-Pinheiro, M., and Margis, R. (2011). Aluminum triggers broad changes in microRNA expression in rice roots. Genet. Mol. Res. 10, 2817–2832. doi: 10.4238/2011.November.10.4
Liu, H.-H., Tian, X., Li, Y.-J., Wu, C.-A., and Zheng, C.-C. (2008a). Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 14, 836–843. doi: 10.1261/rna.895308
Liu, H., Qin, C., Chen, Z., Zuo, T., Yang, X., Zhou, H., et al. (2014). Identification of miRNAs and their target genes in developing maize ears by combined small RNA and degradome sequencing. BMC Genomics 15:25. doi: 10.1186/1471-2164-15-25
Liu, Q., and Zhang, H. (2012). Molecular identification and analysis of arsenite stress-responsive miRNAs in rice. J. Agric. Food Chem. 60, 6524–6536. doi: 10.1021/jf300724t
Liu, Q., Zhang, Y.-C., Wang, C.-Y., Luo, Y.-C., Huang, Q.-J., Chen, S.-Y., et al. (2009). Expression analysis of phytohormone-regulated microRNAs in rice, implying their regulation roles in plant hormone signaling. FEBS Lett. 583, 723–728. doi: 10.1016/j.febslet.2009.01.020
Liu, X., Hua, X., Guo, J., Qi, D., Wang, L., Liu, Z., et al. (2008b). Enhanced tolerance to drought stress in transgenic tobacco plants overexpressing VTE1 for increased tocopherol production from Arabidopsis thaliana. Biotechnol. Lett. 30, 1275–1280. doi: 10.1007/s10529-008-9672-y
Llave, C., Kasschau, K. D., Rector, M. A., and Carrington, J. C. (2002). Endogenous and silencing-associated small RNAs in plants. Plant Cell Online 14, 1605–1619. doi: 10.1105/tpc.003210
Lobbes, D., Rallapalli, G., Schmidt, D. D., Martin, C., and Clarke, J. (2006). SERRATE: a new player on the plant microRNA scene. EMBO Rep. 7, 1052–1058. doi: 10.1038/sj.embor.7400806
Lu, C., and Fedoroff, N. (2000). A mutation in the Arabidopsis HYL1 gene encoding a dsRNA binding protein affects responses to abscisic acid, auxin, and cytokinin. Plant Cell 12, 2351–2365. doi: 10.1105/tpc.12.12.2351
Lu, S., Sun, Y. H., and Chiang, V. L. (2008). Stress−responsive microRNAs in Populus. Plant J. 55, 131–151. doi: 10.1111/j.1365-313X.2008.03497.x
Lu, S., Sun, Y.-H., Shi, R., Clark, C., Li, L., and Chiang, V. L. (2005). Novel and mechanical stress–responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell Online 17, 2186–2203. doi: 10.1105/tpc.105.033456
Lukasik, A., Pietrykowska, H., Paczek, L., Szweykowska-Kulinska, Z., and Zielenkiewicz, P. (2013). High-throughput sequencing identification of novel and conserved miRNAs in the Brassica oleracea leaves. BMC Genomics 14:801. doi: 10.1186/1471-2164-14-801
Lv, D. K., Bai, X., Li, Y., Ding, X. D., Ge, Y., Cai, H., et al. (2010). Profiling of cold-stress-responsive miRNAs in rice by microarrays. Gene 459, 39–47. doi: 10.1016/j.gene.2010.03.011
Machida, S., Chen, H.-Y., and Adam Yuan, Y. (2011). Molecular insights into miRNA processing by Arabidopsis thaliana SERRATE. Nucleic Acids Res. 39, 7828–7836. doi: 10.1093/nar/gkr428
Mahajan, S., and Tuteja, N. (2005). Cold, salinity and drought stresses: an overview. Arch. Biochem. Biophys. 444, 139–158. doi: 10.1016/j.abb.2005.10.018
Mahmood, A., Latif, T., and Khan, M. A. (2009). Effect of salinity on growth, yield and yield components in basmati rice germplasm. Pak. J. Bot. 41, 3035–3045.
Mallory, A. C., and Vaucheret, H. (2006). Functions of microRNAs and related small RNAs in plants. Nat. Genet. 38(Suppl.), S31–S36. doi: 10.1038/ng0706-850b
Manavella, P. A., Hagmann, J., Ott, F., Laubinger, S., Franz, M., Macek, B., et al. (2012a). Fast-forward genetics identifies plant CPL phosphatases as regulators of miRNA processing factor HYL1. Cell 151, 859–870. doi: 10.1016/j.cell.2012.09.039
Manavella, P. A., Koenig, D., and Weigel, D. (2012b). Plant secondary siRNA production determined by microRNA-duplex structure. Proc. Natl. Acad. Sci. U.S.A. 109, 2461–2466. doi: 10.1073/pnas.1200169109
Matsui, T., Omasa, K., and Horie, T. (2000). High temperature at flowering inhibits swelling of pollen grains, a driving force for thecae dehiscence in rice (Oryza sativa L.). Plant Prod. Sci. 3, 430–434. doi: 10.1626/pps.3.430
Meng, X. Z., Zheng, T. S., Chen, X., Wang, J. B., Zhang, W. H., Pan, S. H., et al. (2011a). microRNA expression alteration after arsenic trioxide treatment in HepG−2 cells. J. Gastroenterol. Hepatol. 26, 186–193. doi: 10.1111/j.1440-1746.2010.06317.x
Meng, Y., Gou, L., Chen, D., Mao, C., Jin, Y., Wu, P., et al. (2011b). PmiRKB: a plant microRNA knowledge base. Nucleic Acids Res. 39, D181–D187. doi: 10.1093/nar/gkq721
Meyers, B. C., Axtell, M. J., Bartel, B., Bartel, D. P., Baulcombe, D., Bowman, J. L., et al. (2008). Criteria for annotation of plant MicroRNAs. Plant Cell 20, 3186–3190. doi: 10.1105/tpc.108.064311
Millar, A. A., and Gubler, F. (2005). The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell Online 17, 705–721. doi: 10.1105/tpc.104.027920
Mishra, N. S., and Mukherjee, S. K. (2007). A peep into the plant miRNA world. Open Plant Sci. J. 1, 1–9. doi: 10.2174/1874294700701010001
Mittal, D., Mukherjee, S. K., Vasudevan, M., and Mishra, N. S. (2013). Identification of tissue−preferential expression patterns of rice miRNAs. J. Cell. Biochem. 114, 2071–2081. doi: 10.1002/jcb.24552
Mittler, R. (2002). Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 7, 405–410. doi: 10.1016/S1360-1385(02)02312-9
Mittler, R., Vanderauwera, S., Gollery, M., and Van Breusegem, F. (2004). Reactive oxygen gene network of plants. Trends Plant Sci. 9, 490–498. doi: 10.1016/j.tplants.2004.08.009
Mohorianu, I., Stocks, M. B., Wood, J., Dalmay, T., and Moulton, V. (2013). CoLIde: a bioinformatics tool for CO-expression based small RNA L oci Ide ntification using high-throughput sequencing data. RNA Biol. 10, 1221–1230. doi: 10.4161/rna.25538
Mondal, T. K., and Ganie, S. A. (2014). Identification and characterization of salt responsive miRNA-SSR markers in rice (Oryza sativa). Gene 535, 204–209. doi: 10.1016/j.gene.2013.11.033
Motameny, S., Wolters, S., Nürnberg, P., and Schumacher, B. (2010). Next generation sequencing of miRNAs–strategies, resources and methods. Genes 1, 70–84. doi: 10.3390/genes1010070
Moxon, S., Schwach, F., Dalmay, T., Maclean, D., Studholme, D. J., and Moulton, V. (2008). A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics 24, 2252–2253. doi: 10.1093/bioinformatics/btn428
Muñoz-Mérida, A., Perkins, J. R., Viguera, E., Thode, G., Bejarano, E. R., and Pérez-Pulido, A. J. (2012). Semirna: searching for plant miRNAs using target sequences. Omics 16, 168–177. doi: 10.1089/omi.2011.0115
Nakano, M., Nobuta, K., Vemaraju, K., Tej, S. S., Skogen, J. W., and Meyers, B. C. (2006). Plant MPSS databases: signature-based transcriptional resources for analyses of mRNA and small RNA. Nucleic Acids Res. 34, D731–D735. doi: 10.1093/nar/gkj077
Nakashima, K., Ito, Y., and Yamaguchi-Shinozaki, K. (2009). Transcriptional regulatory networks in response to abiotic stresses in Arabidopsis and grasses. Plant Physiol. 149, 88–95. doi: 10.1104/pp.108.129791
Naqvi, A. R., Sarwat, M., Hasan, S., and Roychodhury, N. (2012). Biogenesis, functions and fate of plant microRNAs. J. Cell. Physiol. 227, 3163–3168. doi: 10.1002/jcp.24052
Neilsen, C. T., Goodall, G. J., and Bracken, C. P. (2012). IsomiRs–the overlooked repertoire in the dynamic microRNAome. Trends Genet. 28, 544–549. doi: 10.1016/j.tig.2012.07.005
Nobel, P. S., and Cui, M. (1992). Hydraulic conductances of the soil, the root-soil air gap, and the root: changes for desert succulents in drying soil. J. Exp. Bot. 43, 319–326. doi: 10.1093/jxb/43.3.319
North, G. B., and Nobel, P. S. (1997). Drought-induced changes in soil contact and hydraulic conductivity for roots of Opuntia ficus-indica with and without rhizosheaths. Plant Soil 191, 249–258. doi: 10.1023/A:1004213728734
Nozawa, M., Miura, S., and Nei, M. (2010). Origins and evolution of microRNA genes in Drosophila species. Genome Biol. Evol. 2, 180–189. doi: 10.1093/gbe/evq009
Numnark, S., Mhuantong, W., Ingsriswang, S., and Wichadakul, D. (2012a). C-mii: a tool for plant miRNA and target identification. BMC Genomics 13:S16. doi: 10.1186/1471-2164-13-S7-S16
Numnark, S., Mhuantong, W., Ingsriswang, S., and Wichadakul, D. (2012b). C-mii: a tool for plant miRNA and target identification. BMC Genomics 13(Suppl. 7):S16. doi: 10.1186/1471-2164-13-S7-S16
Olena, A. F., and Patton, J. G. (2010). Genomic organization of microRNAs. J. Cell. Physiol. 222, 540–545. doi: 10.1002/jcp.21993
Oosterhuis, D. M. (2013). Global Warming and Cotton Productivity. Vol. 29. Cartagena de India: ON ADVISOR.
Ori, N., Cohen, A. R., Etzioni, A., Brand, A., Yanai, O., Shleizer, S., et al. (2007). Regulation of LANCEOLATE by miR319 is required for compound-leaf development in tomato. Nat. Genet. 39, 787–791. doi: 10.1038/ng2036
Palatnik, J. F., Allen, E., Wu, X., Schommer, C., Schwab, R., Carrington, J. C., et al. (2003). Control of leaf morphogenesis by microRNAs. Nature 425, 257–263. doi: 10.1038/nature01958
Pandey, B., Gupta, O. P., Pandey, D. M., Sharma, I., and Sharma, P. (2013). Identification of new stress-induced microRNA and their targets in wheat using computational approach. Plant Signal. Behav 8:e23932. doi: 10.4161/psb.23932
Park, M. Y., Wu, G., Gonzalez-Sulser, A., Vaucheret, H., and Poethig, R. S. (2005). Nuclear processing and export of microRNAs in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 102, 3691–3696. doi: 10.1073/pnas.0405570102
Park, W., Li, J., Song, R., Messing, J., and Chen, X. (2002). CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr. Biol. 12, 1484–1495. doi: 10.1016/S0960-9822(02)01017-5
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7:e30619. doi: 10.1371/journal.pone.0030619
Pearson, W. (2004). Finding protein and nucleotide similarities with FASTA. Curr. Protoc Bioinformatics Chapter 3:Unit3.9. doi: 10.1002/0471250953.bi0309s04
Perata, P., Armstrong, W., and Voesenek, L. A. (2011). Plants and flooding stress. New Phytol. 190, 269–273. doi: 10.1111/j.1469-8137.2011.03702.x
Petrov, V., Hille, J., Mueller-Roeber, B., and Gechev, T. S. (2015). ROS-mediated abiotic stress-induced programmed cell death in plants. Front. Plant Sci. 6:69. doi: 10.3389/fpls.2015.00069
Pfalz, J., Liebers, M., Hirth, M., Grübler, B., Holtzegel, U., Schröter, Y., et al. (2012). Environmental control of plant nuclear gene expression by chloroplast redox signals. Front. Plant Sci. 3:257. doi: 10.3389/fpls.2012.00257
Pierik, R., and Testerink, C. (2014). The art of being flexible: how to escape from shade, salt, and drought. Plant Physiol. 166, 5–22. doi: 10.1104/pp.114.239160
Prasad, P., Boote, K., Allen, L., Sheehy, J., and Thomas, J. (2006). Species, ecotype and cultivar differences in spikelet fertility and harvest index of rice in response to high temperature stress. Field Crops Res. 95, 398–411. doi: 10.1016/j.fcr.2005.04.008
Prasad, P. V., Craufurd, P., and Summerfield, R. (2000). Effect of high air and soil temperature on dry matter production, pod yield and yield components of groundnut. Plant Soil 222, 231–239. doi: 10.1023/A:1004793220787
Qi, Y., Denli, A. M., and Hannon, G. J. (2005). Biochemical specialization within Arabidopsis RNA silencing pathways. Mol. Cell 19, 421–428. doi: 10.1016/j.molcel.2005.06.014
Qin, H., Chen, F., Huan, X., Machida, S., Song, J., and Yuan, Y. A. (2010). Structure of the Arabidopsis thaliana DCL4 DUF283 domain reveals a noncanonical double-stranded RNA-binding fold for protein–protein interaction. RNA 16, 474–481. doi: 10.1261/rna.1965310
Qin, J., Ma, X., Tang, Z., and Meng, Y. (2015). Construction of regulatory networks mediated by small RNAs responsive to abiotic stresses in rice (Oryza sativa). Comput. Biol. Chem. 58, 69–80. doi: 10.1016/j.compbiolchem.2015.05.006
Rajagopalan, R., Vaucheret, H., Trejo, J., and Bartel, D. P. (2006). A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 20, 3407–3425. doi: 10.1101/gad.1476406
Rasia, R. M., Mateos, J., Bologna, N. G., Burdisso, P., Imbert, L., Palatnik, J. F., et al. (2010). Structure and RNA interactions of the plant MicroRNA processing-associated protein HYL1. Biochemistry 49, 8237–8239. doi: 10.1021/bi100672x
Reinhart, B. J., Weinstein, E. G., Rhoades, M. W., Bartel, B., and Bartel, D. P. (2002). MicroRNAs in plants. Genes Dev. 16, 1616–1626. doi: 10.1101/gad.1004402
Rejeb, I. B., Pastor, V., and Mauch-Mani, B. (2014). Plant responses to simultaneous biotic and abiotic stress: molecular mechanisms. Plants 3, 458–475. doi: 10.3390/plants3040458
Ren, G., Xie, M., Dou, Y., Zhang, S., Zhang, C., and Yu, B. (2012). Regulation of miRNA abundance by RNA binding protein TOUGH in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 109, 12817–12821. doi: 10.1073/pnas.1204915109
Reyes, J. L., and Chua, N. H. (2007). ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 49, 592–606. doi: 10.1111/j.1365-313X.2006.02980.x
Rhoades, M. W., Reinhart, B. J., Lim, L. P., Burge, C. B., Bartel, B., and Bartel, D. P. (2002). Prediction of plant microRNA targets. Cell 110, 513–520. doi: 10.1016/S0092-8674(02)00863-2
Rodríguez, M., Canales, E., and Borrás-Hidalgo, O. (2005). Molecular aspects of abiotic stress in plants. Biotecnol. Apli. 22, 1–10.
Rosewick, N., Durkin, K., Momont, M., Takeda, H., Caiment, F., Cleuter, Y., et al. (2013). ST105 Deep sequencing reveals abundant Pol III retroviral microRNA cluster in Bovine Leukemia Virus-induced leukemia. J. Acq. Imm. Def. Syndr. 62, 66. doi: 10.1097/01.qai.0000429267.82844.b6
Rossi, M., Trupiano, D., Tamburro, M., Ripabelli, G., Montagnoli, A., Chiatante, D., et al. (2015). MicroRNAs expression patterns in the response of poplar woody root to bending stress. Planta 242, 339–351. doi: 10.1007/s00425-015-2311-7
Roychoudhury, A., and Chakraborty, M. (2013). Biochemical and molecular basis of varietal difference in plant salt tolerance. Ann. Rev. Res. Biol. 3, 422–454.
Rubio-Somoza, I., and Weigel, D. (2011). MicroRNA networks and developmental plasticity in plants. Trends Plant Sci. 16, 258–264. doi: 10.1016/j.tplants.2011.03.001
Rusinov, V., Baev, V., Minkov, I. N., and Tabler, M. (2005). MicroInspector: a web tool for detection of miRNA binding sites in an RNA sequence. Nucleic Acids Res. 33, W696–W700. doi: 10.1093/nar/gki364
Sablok, G., Milev, I., Minkov, G., Minkov, I., Varotto, C., Yahubyan, G., et al. (2013). isomiRex: web-based identification of microRNAs, isomiR variations and differential expression using next-generation sequencing datasets. FEBS Lett. 587, 2629–2634. doi: 10.1016/j.febslet.2013.06.047
Sanan-Mishra, N., Kumar, V., Sopory, S. K., and Mukherjee, S. K. (2009). Cloning and validation of novel miRNA from basmati rice indicates cross talk between abiotic and biotic stresses. Mol. Genetics Genomics 282, 463–474. doi: 10.1007/s00438-009-0478-y
Sanan-Mishra, N., Varanasi, S. P., and Mukherjee, S. K. (2013). Micro-regulators of auxin action. Plant Cell Rep. 32, 733–740. doi: 10.1007/s00299-013-1425-2
Schauer, S. E., Jacobsen, S. E., Meinke, D. W., and Ray, A. (2002). DICER-LIKE1: blind men and elephants in Arabidopsis development. Trends Plant Sci. 7, 487–491. doi: 10.1016/S1360-1385(02)02355-5
Shanker, A., and Venkateswarlu, B. (eds.). (2011). “Abiotic stress response in plants-physiological, biochemical and genetic perspectives,” in Agricultural and Biological Sciences (InTech).
Sharma, N., Tripathi, A., and Sanan-Mishra, N. (2015). Profiling the expression domains of a rice-specific microRNA under stress. Front. Plant Sci. 6:333. doi: 10.3389/fpls.2015.00333
Shi, Z.-M., Wang, J., Yan, Z., You, Y.-P., Li, C.-Y., Qian, X., et al. (2012). MiR-128 inhibits tumor growth and angiogenesis by targeting p70S6K1. PLoS ONE 7:e32709. doi: 10.1371/journal.pone.0032709
Slezak-Prochazka, I., Kluiver, J., de Jong, D., Kortman, G., Halsema, N., Poppema, S., et al. (2013). Cellular localization and processing of primary transcripts of exonic microRNAs. PLoS ONE 8:e76647. doi: 10.1371/journal.pone.0076647
Sodha, N., and Karan, R. (2015). Omics study for abiotic stress responses in plants. Adv. Plants Agric. Res. 2, 00037. doi: 10.15406/apar.2015.02.00037
Song, C., Wang, C., Zhang, C., Korir, N. K., Yu, H., Ma, Z., et al. (2010). Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genomics 11:431. doi: 10.1186/1471-2164-11-431
Song, L., Han, M.-H., Lesicka, J., and Fedoroff, N. (2007). Arabidopsis primary microRNA processing proteins HYL1 and DCL1 define a nuclear body distinct from the Cajal body. Proc. Natl. Acad. Sci. U.S.A. 104, 5437–5442. doi: 10.1073/pnas.0701061104
Stocks, M. B., Moxon, S., Mapleson, D., Woolfenden, H. C., Mohorianu, I., Folkes, L., et al. (2012). The UEA sRNA workbench: a suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 28, 2059–2061. doi: 10.1093/bioinformatics/bts311
Sun, X., Dong, B., Yin, L., Zhang, R., Du, W., Liu, D., et al. (2013). PMTED: a plant microRNA target expression database. BMC Bioinformatics 14:174. doi: 10.1186/1471-2105-14-174
Sun, X., Xu, L., Wang, Y., Yu, R., Zhu, X., Luo, X., et al. (2015). Identification of novel and salt-responsive miRNAs to explore miRNA-mediated regulatory network of salt stress response in radish (Raphanus sativus L.). BMC Genomics 16:197. doi: 10.1186/s12864-015-1416-5
Sunkar, R., Chinnusamy, V., Zhu, J., and Zhu, J.-K. (2007). Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 12, 301–309. doi: 10.1016/j.tplants.2007.05.001
Sunkar, R., Girke, T., Jain, P. K., and Zhu, J.-K. (2005). Cloning and characterization of microRNAs from rice. Plant Cell Online 17, 1397–1411. doi: 10.1105/tpc.105.031682
Sunkar, R., and Jagadeeswaran, G. (2008). In silico identification of conserved microRNAs in large number of diverse plant species. BMC Plant Biol. 8:37. doi: 10.1186/1471-2229-8-37
Sunkar, R., Kapoor, A., and Zhu, J.-K. (2006). Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell Online 18, 2051–2065. doi: 10.1105/tpc.106.041673
Sunkar, R., Zhou, X., Zheng, Y., Zhang, W., and Zhu, J. K. (2008). Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 8:25. doi: 10.1186/1471-2229-8-25
Sunkar, R., and Zhu, J. K. (2004). Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16, 2001–2019. doi: 10.1105/tpc.104.022830
Suzuki, K., Tsukaguchi, T., Takeda, H., and Egawa, Y. (2001). Decrease of pollen stainability of green bean at high temperatures and relationship to heat tolerance. J. Am. Soc. Horticul. Sci. 126, 571–574.
Thakur, P., Kumar, S., Malik, J. A., Berger, J. D., and Nayyar, H. (2010). Cold stress effects on reproductive development in grain crops: an overview. Environ. Exp. Bot. 67, 429–443. doi: 10.1016/j.envexpbot.2009.09.004
Thiebaut, F., Rojas, C. A., Almeida, K. L., Grativol, C., Domiciano, G. C., Lamb, C. R., et al. (2012). Regulation of miR319 during cold stress in sugarcane. Plant Cell Environ. 35, 502–512. doi: 10.1111/j.1365-3040.2011.02430.x
Trindade, I., Capitão, C., Dalmay, T., Fevereiro, M. P., and Santos, D. M. (2010). miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta 231, 705–716. doi: 10.1007/s00425-009-1078-0
Unver, T., Namuth-Covert, D. M., and Budak, H. (2009). Review of current methodological approaches for characterizing microRNAs in plants. Int. J. Plant Genomics 2009:262463. doi: 10.1155/2009/262463
Upadhyaya, H., and Panda, S. K. (2013). Abiotic stress responses in tea [Camellia sinensis L (O) Kuntze]: an overview. Rev. Agricul. Sci. 1, 1–10. doi: 10.7831/ras.1.1
Vazquez, F. (2006). Arabidopsis endogenous small RNAs: highways and byways. Trends Plant Sci. 11, 460–468. doi: 10.1016/j.tplants.2006.07.006
Vidal, E. A., Moyano, T. C., Krouk, G., Katari, M. S., Tanurdzic, M., McCombie, W. R., et al. (2013). Integrated RNA-seq and sRNA-seq analysis identifies novel nitrate-responsive genes in Arabidopsis thaliana roots. BMC Genomics 14:701. doi: 10.1186/1471-2164-14-701
Voinnet, O. (2009). Origin, biogenesis, and activity of plant microRNAs. Cell 136, 669–687. doi: 10.1016/j.cell.2009.01.046
Wahid, A., and Close, T. (2007). Expression of dehydrins under heat stress and their relationship with water relations of sugarcane leaves. Biol. Plant. 51, 104–109. doi: 10.1007/s10535-007-0021-0
Wahid, F., Shehzad, A., Khan, T., and Kim, Y. Y. (2010). MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim. Biophys. Acta Mol. Cell Res. 1803, 1231–1243. doi: 10.1016/j.bbamcr.2010.06.013
Wan, P., Wu, J., Zhou, Y., Xiao, J., Feng, J., Zhao, W., et al. (2011). Computational analysis of drought stress-associated miRNAs and miRNA co-regulation network in Physcomitrella patens. Genomics Proteomics Bioinformatics 9, 37–44. doi: 10.1016/S1672-0229(11)60006-5
Wang, L., Li, P., and Brutnell, T. P. (2010). Exploring plant transcriptomes using ultra high-throughput sequencing. Brief. Funct. Genomics 9, 118–128. doi: 10.1093/bfgp/elp057
Wang, R., Xu, L., Zhu, X., Zhai, L., Wang, Y., Yu, R., et al. (2014). Transcriptome-wide characterization of novel and heat-stress-responsive microRNAs in radish (Raphanus Sativus L.) using next-generation sequencing. Plant Mol. Biol. Rep. 33, 867–880. doi: 10.1007/s11105-014-0786-1
Wang, X.-J., Reyes, J. L., Chua, N.-H., and Gaasterland, T. (2004). Prediction and identification of Arabidopsis thaliana microRNAs and their mRNA targets. Genome Biol. 5:R65. doi: 10.1186/gb-2004-5-9-r65
Wang, X., Zhang, J., Li, F., Gu, J., He, T., Zhang, X., et al. (2005). MicroRNA identification based on sequence and structure alignment. Bioinformatics 21, 3610–3614. doi: 10.1093/bioinformatics/bti562
Weber, A. P., Weber, K. L., Carr, K., Wilkerson, C., and Ohlrogge, J. B. (2007). Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiol. 144, 32–42. doi: 10.1104/pp.107.096677
Weber, M. J. (2005). New human and mouse microRNA genes found by homology search. FEBS J. 272, 59–73. doi: 10.1111/j.1432-1033.2004.04389.x
Wu, J., Liu, Q., Wang, X., Zheng, J., Wang, T., You, M., et al. (2013). mirTools 2.0 for non-coding RNA discovery, profiling, and functional annotation based on high-throughput sequencing. RNA Biol. 10, 1087–1092. doi: 10.4161/rna.25193
Wu, S. C., and Zhang, Y. (2010). Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 11, 607–620. doi: 10.1038/nrm2950
Xie, F. L., Huang, S. Q., Guo, K., Xiang, A. L., Zhu, Y. Y., Nie, L., et al. (2007). Computational identification of novel microRNAs and targets in Brassica napus. FEBS Lett. 581, 1464–1474. doi: 10.1016/j.febslet.2007.02.074
Xie, F., Wang, Q., Sun, R., and Zhang, B. (2015). Deep sequencing reveals important roles of microRNAs in response to drought and salinity stress in cotton. J. Exp. Bot. 66, 789–804. doi: 10.1093/jxb/eru437
Xie, Z., Allen, E., Fahlgren, N., Calamar, A., Givan, S. A., and Carrington, J. C. (2005a). Expression of Arabidopsis MIRNA genes. Plant Physiol. 138, 2145–2154. doi: 10.1104/pp.105.062943
Xie, Z., Allen, E., Wilken, A., and Carrington, J. C. (2005b). DICER-LIKE 4 functions in trans-acting small interfering RNA biogenesis and vegetative phase change in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 102, 12984–12989. doi: 10.1073/pnas.0506426102
Yang, J.-H., and Qu, L.-H. (2012). DeepBase: annotation and discovery of microRNAs and other noncoding RNAs from deep-sequencing data. Methods Mol. Biol. 822, 233–248. doi: 10.1007/978-1-61779-427-8_16
Yang, L., Liu, Z., Lu, F., Dong, A., and Huang, H. (2006). SERRATE is a novel nuclear regulator in primary microRNA processing in Arabidopsis. Plant J. 47, 841–850. doi: 10.1111/j.1365-313X.2006.02835.x
Yang, S. W., Chen, H.-Y., Yang, J., Machida, S., Chua, N.-H., and Yuan, Y. A. (2010). Structure of Arabidopsis HYPONASTIC LEAVES1 and its molecular implications for miRNA processing. Structure 18, 594–605. doi: 10.1016/j.str.2010.02.006
Yang, X., and Li, L. (2011a). miRDeep-P: a computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 27, 2614–2615. doi: 10.1093/bioinformatics/btr430
Yang, X., and Li, L. (2011b). miRDeep-P: a computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 27, 2614–2615. doi: 10.1093/bioinformatics/btr430
Yi, X., Zhang, Z., Ling, Y., Xu, W., and Su, Z. (2015). PNRD: a plant non-coding RNA database. Nucleic Acids Res. 43, D982–D989. doi: 10.1093/nar/gku1162
Yin, Z., Li, C., Han, X., and Shen, F. (2008). Identification of conserved microRNAs and their target genes in tomato (Lycopersicon esculentum). Gene 414, 60–66. doi: 10.1016/j.gene.2008.02.007
Yin, Z., Li, Y., Yu, J., Liu, Y., Li, C., Han, X., et al. (2012). Difference in miRNA expression profiles between two cotton cultivars with distinct salt sensitivity. Mol. Biol. Rep. 39, 4961–4970. doi: 10.1007/s11033-011-1292-2
Yu, B., Bi, L., Zheng, B., Ji, L., Chevalier, D., Agarwal, M., et al. (2008). The FHA domain proteins DAWDLE in Arabidopsis and SNIP1 in humans act in small RNA biogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 10073–10078. doi: 10.1073/pnas.0804218105
Yu, J., Matsuura, T., Yoshikawa, Y., Islam, M. N., and Hori, M. (2005). In situ analysis of performance degradation of a PEMFC under nonsaturated humidification. Electrochem. Solid State Lett. 8, A156–A158. doi: 10.1149/1.1854781
Yu, X., Wang, H., Lu, Y., de Ruiter, M., Cariaso, M., Prins, M., et al. (2012). Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. J. Exp. Bot. 63, 1025–1038. doi: 10.1093/jxb/err337
Zhang, B. H., Pan, X. P., Wang, Q. L., Cobb, G. P., and Anderson, T. A. (2005). Identification and characterization of new plant microRNAs using EST analysis. Cell Res. 15, 336–360. doi: 10.1038/sj.cr.7290302
Zhang, B., Pan, X., and Anderson, T. A. (2006a). Identification of 188 conserved maize microRNAs and their targets. FEBS Lett. 580, 3753–3762. doi: 10.1016/j.febslet.2006.05.063
Zhang, B., Pan, X., Cannon, C. H., Cobb, G. P., and Anderson, T. A. (2006b). Conservation and divergence of plant microRNA genes. Plant J. 46, 243–259. doi: 10.1111/j.1365-313X.2006.02697.x
Zhang, B., Pan, X., and Stellwag, E. J. (2008a). Identification of soybean microRNAs and their targets. Planta 229, 161–182. doi: 10.1007/s00425-008-0818-x
Zhan, X., Wang, B., Li, H., Liu, R., Kalia, R. K., Zhu, J.-K., et al. (2012). Arabidopsis proline-rich protein important for development and abiotic stress tolerance is involved in microRNA biogenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 18198–18203. doi: 10.1073/pnas.1216199109
Zhang, J., Xu, Y., Huan, Q., and Chong, K. (2009a). Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics 10:449. doi: 10.1186/1471-2164-10-449
Zhang, J., Zeng, R., Chen, J., Liu, X., and Liao, Q. (2008b). Identification of conserved microRNAs and their targets from Solanum lycopersicum Mill. Gene 423, 1–7. doi: 10.1016/j.gene.2008.05.023
Zhang, L., Chia, J.-M., Kumari, S., Stein, J. C., Liu, Z., Narechania, A., et al. (2009b). A genome-wide characterization of microRNA genes in maize. PLoS Genet. 5:e1000716. doi: 10.1371/journal.pgen.1000716
Zhang, S., Xie, M., Ren, G., and Yu, B. (2013a). CDC5, a DNA binding protein, positively regulates posttranscriptional processing and/or transcription of primary microRNA transcripts. Proc. Natl. Acad. Sci. U.S.A. 110, 17588–17593. doi: 10.1073/pnas.1310644110
Zhang, S., Yue, Y., Sheng, L., Wu, Y., Fan, G., Li, A., et al. (2013b). PASmiR: a literature-curated database for miRNA molecular regulation in plant response to abiotic stress. BMC Plant Biol. 13:33. doi: 10.1186/1471-2229-13-33
Zhang, X.-N., Li, X., and Liu, J.-H. (2014a). Identification of conserved and novel cold-responsive microRNAs in trifoliate orange (Poncirus trifoliata (L.) Raf.) using high-throughput sequencing. Plant Mol. Biol. Rep. 32, 328–341. doi: 10.1007/s11105-013-0649-1
Zhang, Y., Yu, M., Yu, H., Han, J., Song, C., Ma, R., et al. (2012). Computational identification of microRNAs in peach expressed sequence tags and validation of their precise sequences by miR-RACE. Mol. Biol. Rep. 39, 1975–1987. doi: 10.1007/s11033-011-0944-6
Zhang, Z., Jiang, L., Wang, J., and Chen, M. (2014b). MTide: an integrated tool for the identification of miRNA-target interaction in plants. Bioinformatics btu633.
Zhang, Z., Yu, J., Li, D., Liu, F., Zhou, X., Wang, T., et al. (2010). PMRD: plant microRNA database. Nucleic Acids Res. 38, D806–D813. doi: 10.1093/nar/gkp818
Zhao, B., Ge, L., Liang, R., Li, W., Ruan, K., Lin, H., et al. (2009). Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol. Biol. 10:29. doi: 10.1186/1471-2199-10-29
Zhao, B., Liang, R., Ge, L., Li, W., Xiao, H., Lin, H., et al. (2007). Identification of drought-induced microRNAs in rice. Biochem. Biophys. Res. Commun. 354, 585–590. doi: 10.1016/j.bbrc.2007.01.022
Zheng, F., Liao, Y.-J., Cai, M.-Y., Liu, Y.-H., Liu, T.-H., Chen, S.-P., et al. (2012). The putative tumour suppressor microRNA-124 modulates hepatocellular carcinoma cell aggressiveness by repressing ROCK2 and EZH2. Gut 61, 278–289. doi: 10.1136/gut.2011.239145
Zhong, R., and Ye, Z.-H. (2004). Amphivasal vascular bundle 1, a gain-of-function mutation of the IFL1/REV gene, is associated with alterations in the polarity of leaves, stems and carpels. Plant Cell Physiol. 45, 369–385. doi: 10.1093/pcp/pch051
Zhou, L., Liu, Y., Liu, Z., Kong, D., Duan, M., and Luo, L. (2010). Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J. Exp. Bot. 61, 4157–4168. doi: 10.1093/jxb/erq237
Zhou, X., Wang, G., Sutoh, K., Zhu, J.-K., and Zhang, W. (2008). Identification of cold-inducible microRNAs in plants by transcriptome analysis. Biochim. Biophys. Acta. Gene Regul. Mechan.1779, 780–788. doi: 10.1016/j.bbagrm.2008.04.005
Zhou, X., Wang, G., and Zhang, W. (2007). UV-B responsive microRNA genes in Arabidopsis thaliana. Mol. Syst. Biol. 3, 103. doi: 10.1038/msb4100143
Zinn, K. E., Tunc-Ozdemir, M., and Harper, J. F. (2010). Temperature stress and plant sexual reproduction: uncovering the weakest links. J. Exp. Bot. 61, 1959–1968. doi: 10.1093/jxb/erq053
Keywords: microRNA, abiotic stress, high-throughput sequencing, microarray, bioinformatic approached, degradome, NGS
Citation: Tripathi A, Goswami K and Sanan-Mishra N (2015) Role of bioinformatics in establishing microRNAs as modulators of abiotic stress responses: the new revolution. Front. Physiol. 6:286. doi: 10.3389/fphys.2015.00286
Received: 05 June 2015; Accepted: 28 September 2015;
Published: 26 October 2015.
Edited by:
Manoj Prasad, National Institute of Plant Genome Research, IndiaReviewed by:
Debasis Chattopadhyay, National Institute of Plant Genome Research, IndiaAmit Katiyar, All India Institute of Medical Sciences, India
Copyright © 2015 Tripathi, Goswami and Sanan-Mishra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neeti Sanan-Mishra, bmVldGlAaWNnZWIucmVzLmlu
†These authors have contributed equally to this work.