 Na Liu
Na Liu Shougang Zhuang
Shougang Zhuang- 1Department of Nephrology, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, China
- 2Department of Medicine, Rhode Island Hospital and Alpert Medical School, Brown University, Providence, RI, USA
Histone deacetylases (HDACs) induce deacetylation of both histone and non-histone proteins and play a critical role in the modulation of physiological and pathological gene expression. Pharmacological inhibition of HDAC has been reported to attenuate progression of renal fibrogenesis in obstructed kidney and reduce cyst formation in polycystic kidney disease. HDAC inhibitors (HDACis) are also able to ameliorate renal lesions in diabetes nephropathy, lupus nephritis, aristolochic acid nephropathy, and transplant nephropathy. The beneficial effects of HDACis are associated with their anti-fibrosis, anti-inflammation, and immunosuppressant effects. In this review, we summarize recent advances on the treatment of various chronic kidney diseases with HDACis in pre-clinical models.
Introduction
Histone and non-histone protein acetylation has been widely studied in the field of cancer research (Kwon et al., 2009; Mahalingam et al., 2010; Selinger et al., 2011). Histone acetyltransferases (HATs) and histone deacetylases (HDACs) can mediate the acetylated/deacetylated states of histones (Bush and McKinsey, 2010). HATs induce acetylation of histones H3 and H4 on lysine amino groups (Spencer and Davie, 1999) whereas HDACs remove acetyl groups from the acetylated proteins. Acetylation of the ε-amino groups of lysine residues in nucleosomal histone tails by HATs is considered necessary for the chromatin structure to relax, allowing activation of transcriptional activators and initiation of gene induction. Inhibition of HDACs with HDAC inhibitors (HDACis) also enhances the deposition of acetylated histones H3 and H4, thereby modifying chromatin structure and regulating gene transcription (Turner, 1993; Van Lint et al., 1996). In addition, HDACs are able to catalyze deacetylation of many non-histone proteins, thus, they are also called lysine deacetylases to describe their functions more precisely (Glozak et al., 2005). HDACis have been reported to regulate gene transcription positively or negatively in a gene-specific manner (Marks et al., 2000).
HDACs are divided into four groups, mainly according to the homology of yeast HDACs. Class I HDACs (HDAC1, 2, 3, and 8) are critically connected with yeast RPD3 gene. Class II HDACs (HDAC4, 5, 6, 7, 9, and 10) are related to yeast Hda1 gene. Class III HDACs (SIRT1-7) are homologous to silent information regulator 2 (Sir2) and have no sequence similarity to class I and II HDACs; these Sir2 proteins, also called sirtuins are unaffected by known class I/II HDACis. Class IV (HDAC11) has conserved residues in its catalytic regions that are shared by both class I and II HDACs. Class I and II HDACs need Zn2+ for their enzymatic reaction. Class IV also has a Zn2+ based reaction mechanism. However, class III HDACs do not require Zn2+ for their catalysis, but strictly depend on the cofactor NAD+ (Pang and Zhuang, 2010).
Currently, the expression profiles and distribution of HDACs in the kidney have not been completely clarified. It has been documented that class I HDAC isoforms are expressed in the cortex of developmental kidney, renal fibroblasts, and renal tubular cells (Pang et al., 2011; Chen and El-Dahr, 2013; Tang et al., 2013). HDAC5 and 6 have been identified in the renal tubules (Marumo et al., 2008; Liu et al., 2012b). SIRT1 is expressed in both renal tubules (Zhou et al., 2013) and fibroblasts (Ponnusamy et al., 2014) and HDAC11 is expressed in renal tubules (Kim et al., 2013). HDACs have been shown to be involved in a variety of cellular functions such as proliferation, survival, differentiation, and immunological responses (Van Beneden et al., 2013).
Most functional roles of HDACs are revealed by application of HDACis, which are classified into four categories according to chemical structures: hydroxamates (e.g., vorinostat), cyclic peptides (e.g., romidepsin), aliphatic acids (e.g., phenylbutyrate), and benzamides (entinostat) (Miller et al., 2003; Marks et al., 2004; Dokmanovic and Marks, 2005; Ma et al., 2009). HDACis primarily target the zinc domains to exert the biological effects through cell cycle arrest, differentiation and apoptosis in a variety of tumor models (Acharya et al., 2005). Treatment of renal cell carcinoma with HDACis also resulted in tumor growth inhibition (Ramakrishnan and Pili, 2013). To date, two HDACis, vorinostat, and romideps, have been approved by the FDA to treat cutaneous and peripheral T cell lymphoma. Other 20 different HDACis have been tested in the clinic (West and Johnstone, 2014). The common side-effects of HDACis in humans include fatigue, nausea, and vomiting and resolve upon treatment withdrawal (Minucci and Pelicci, 2006).
Although the tumor has been the primary target for HDACis, HDAC inhibition has also shown beneficial effects in some non-neoplastic disorders. HDACis are effective in attenuating the pathogenesis of several forms of chronic kidney disease and improving renal function. In this article, we highlight the therapeutic application of HDACis in pre-clinical models of renal injury and discuss the mechanisms involved (Figure 1).
Figure 1. The mechanisms by which HDACIs attenuate chronic kidney diseases. HDACIs can protect against chronic kidney diseases through multiple mechanisms as indicated.
HDACs in Renal Interstitial Fibrosis
Progression and development of renal injuries ultimately leads to renal interstitial fibrosis (Neilson, 2006; Wynn, 2008). Kidney fibrosis is characterized with activation and proliferation of renal interstitial fibroblasts as well as accumulation of extracellular matrix (ECM) components. During development of renal fibrosis, multiple cytokine, and growth factor signaling pathways are activated and involved in this process. Emerging evidence indicates that HDACs are also implicated in renal fibrogenesis. The mechanisms by which HDACs mediates renal fibrogenesis remain elusive, but may be associated with regulating the expression of inflammatory and profibrotic genes and activation of cell signaling pathways that mediate renal fibrosis. In the earlier studies, Pang et al. (2009) demonstrated that treatment with trichostatin A (TSA), a pan HDACi that can block both class I and class II HDACs, attenuates renal fibrosis in a murine model of unilateral ureteral obstruction (UUO). TSA treatment also significantly inhibits expression of α-SMA and fibronectin, two hallmarks of activated fibroblasts (Pang et al., 2009). Moreover, silencing of HDAC1 or HDAC2 using specific siRNA blocked renal fibroblast proliferation and reduced phosphorylation of STAT3 (signal transducer and activator of transcription 3), a signaling molecule associated with proliferation of renal fibroblasts and development of renal fibrosis (Pang et al., 2011). Recently, Manson et al. (2014) demonstrated that TSA treatment preserves the expression of Bmp-7 transcription and attenuates the pathogenesis of renal injury in obstructive nephropathy. As BMP-7 is a potent anti-fibrotic molecule, restoration of BMP-7 expression by TSA represents another mechanism by which HDACis protect against chronic kidney injury. In addition, Marumo et al. (Liu et al., 2013) showed that HDAC inhibition also alleviates renal fibrosis through suppression of inflammatory responses in the injured kidney.
Since non-selective blockade of class I and class II HDACs does not allow elucidation of their individual roles in renal fibrogenesis, Liu et al. (2013) further examined the effect of MS-275, a selective class I inhibitor, on UUO injury and renal interstitial fibroblast activation and proliferation. Administration of MS-275 inhibited both renal fibroblast activation and proliferation and attenuated progression of kidney fibrosis. MS-275 treatment also inhibited UUO-induced production of TGF-β 1 and phosphorylation of Smad3 and EGFR (epidermal growth factor receptor). These results suggest that specific blockade of only class I HDAC can inhibit renal fibrogenesis through a mechanism involved in the inactivation of the TGF-β 1/Smad3 and EGFR signaling pathways. Although there is no study thus far to compare the pharmacological effect of TSA and MS-275 on renal fibroblast activation and development of renal fibrosis, it has been reported that pre-treatment with either valproic acid, another inhibitor of class I or TSA attenuates glomerulosclerosis and tubulointerstitial fibrosis to the similar degree. Delayed administration of these two inhibitors also showed comparable effects on the inhibition of renal fibrosis (Van Beneden et al., 2013). Thus, it appears that class I HDACs play a pre-dominant role in regulating renal fibrogenesis. Class III HDACs have been implicated in the regulation of renal fibrosis. It is evident that specific inhibition of SIRT1/2 alleviates progression of renal fibrogenesis and reduces renal fibroblast activation (Ponnusamy et al., 2014). Mechanistic studies showed that blocking SIRT1/2 inhibits activation of epidermal growth factor receptor (EGFR) and platelet derived growth factor receptors (PDGFR), two growth factor receptors associated with renal fibrosis (Chen et al., 2011; Liu et al., 2012a).
Collectively, these studies indicate that HDACs contribute to renal fibroblast activation and fibrogenesis. Additional studies are needed to clarify the role of individual HDAC isoforms in mediating these processes and elucidate the mechanisms involved in a great detail.
HDACs in Polycystic Kidney Diseases
Autosomal dominant polycystic kidney disease (ADPKD) is very common hereditary kidney diseases in humans, affecting 1/500 in the United States (Gabow, 1993). In ADPKD patients, a large number of bilateral kidney cysts displace normal kidney parenchyma, leading to end-stage renal disease (ESRD). ADPKD is mainly caused by gene mutations in one of two genes: PKD1, which accounts for approximately 85–95% of the cases and PKD2, which affects about 5–15% of the cases (Peters and Sandkuijl, 1992). ADPKD is characterized by development of multiple bilateral renal cysts, and increased renal epithelial cell proliferation and fluid secretion. As the gene product of PKD2, polycystin-2 (PC2), either alone or in complex with the gene product of PKD1, polycystin-1 (PC1), functions as a calcium-permeable cation channel and regulates intracellular Ca2+ levels, alteration of signaling pathways regulated by calcium such as cAMP-dependent B-Raf and ERK (extracellular signal-regulated kinase) activation resulted in abnormal proliferation of tubule epithelial cells (Yamaguchi et al., 2006). In addition, activation of many other signaling pathways and transcription factors such as EGFR and p53 is also involved in the development and growth of polycystic kidneys (Harris and Torres, 2014).
Emerging evidence has revealed the regulatory role of HDACs in the pathogenesis of polycystic kidneys. Xie et al., showed (Xia et al., 2010) that histone deacetylase 5 (HDAC5) is a target of polycystin-dependent fluid stress sensing in renal epithelial cells in mice. Stimulation of polarized epithelial monolayers with fluid flow induced phosphorylation and nuclear export of HDAC5 whereas dwonregulation of HDAC5 or treatment with TSA reduced cyst formation in Pkd2−/− mouse embryos. Cao et al. (2009) demonstrated that TSA treatment can affect both body curvature and laterality, two pathological changes associated with cyst formation in zebrafish and block cyst formation in pkd2 knockdown animals. Treatment with valproic acid (VPA), a class I specific HDACi, also delays the development of cyst production and improves renal function in a mouse ADPKD model. In addition, Fan et al. showed that administration of TSA in pregnant mice prevented cyst formation in Pkd1 mutant embryonic kidneys (Fan et al., 2012). TSA treatment can ameliorate p53-induced repression of the PKD1 expression, Chang et al. (2006) and Thivierge et al. (2006). As EGFR activation and nuclear translocation of β-catenin are essential for ADPKD, the role of HDAC6 in regulating these biological responses was examined. HDAC6 inhibition blocks EGF-induced β-catenin nuclear localization, leading to inhibition of epithelial cell proliferation and promotion of EGFR degradation (Li et al., 2008). These studies suggest that class I/II HDAC activation is essential for PKD development and that HDACis may be possible drug treatments for PKD.
A recent study further reveals that SIRT is also involved in the pathogenesis of ADPKD (Zhou et al., 2013). SIRT1 upregulation was observed in embryonic and post-natal Pkd1-mutant mouse renal epithelial cells and tissues whereas double conditional knockouts of PKD1 and SIRT1 as well as inhibition of SIRT1 with a pan-sirtuin inhibitor (nicotinamide) or a SIRT1-specific inhibitor (EX-527) resulted in delayed renal cyst formation. Silence or inhibition of SIRT1 also reduced renal epithelial cell proliferation, but potentiated apoptosis. Further studies show that SIRT1 mediates cystic epithelial cell proliferation through altering retinoblastoma (RB) protein acetylation/phosphorylation and promotes their survival via p53 deacetylation. This study elucidates a functional role of SIRT1 in regulating ADPKD and provides a molecular basis for using SIRT1 inhibitors to interfere with cyst formation (Zhou et al., 2013).
HDACs in Diabetic Nephropathy
Diabetic nephropathy (DN) is characterized by ECM protein accumulation in glomerular mesangium and tubulointerstitium with thickening of glomerular and tubular basement membranes, ultimately progressing to glomerulosclerosis and tubulo-interstitial fibrosis (Mauer et al., 1984). The earliest finding of renal involvement in DN is glomerular hypertrophy, which is caused by glomerular hyper-filtration. Although targeting diverse signaling pathways has been reported to attenuate the pathogenesis of DN, two animal studies have demonstrated the inhibitory effect of HDACis on DN. Gilbert et al. showed that vorinostat administration resulted in attenuation of renal hypertrophy in rats (Gilbert et al., 2011). Advani et al. demonstrated that vorinostat was effective in decreasing albuminuria and mesangial matrix accumulation in streptozotocin–wild-type mice (Advani et al., 2011). In vitro, treatment with VPA and SK-7041, two class I-selective HDACis, can also reduce expression of ECM components in renal epithelial cells (NRK52-E) (Noh et al., 2009). In addition, HDAC2 activity was upregulated in the kidneys of strotozotocin (STZ) induced diabetic rats and db/db mice. Treatment with N-acetylcysteine, an antioxidant, decreased TGF-β 1 mediated activation of HDAC2 in NRK52-E cells (Noh et al., 2009). These data suggest that HDACs are required for the development of DN and that reactive oxygen species may play an essential role in mediating TGF-β 1-induced activation of HDAC2.
EGFR activation has been shown to be implicated in the DN (Chen et al., 2014; Zhang et al., 2014). To understand whether EGFR expression is associated with the HDAC activity, Gilbert et al. (2011) further investigated the effect of vorinostat on the expression of EGFR in the early stage of diabetes. They found that daily treatment with vorinostat in diabetic rats for 4 weeks remarkably reduced EGFR expression and subsequently inhibited kidney growth and glomerular hypertrophy. In cultured rat proximal tubule cells, treatment with vorinostat also decreased EGFR expression, concomitant with cellular proliferation inhibition. Therefore, HDACs may regulate early DN through activation of the EGFR signaling pathway.
Podocyte damage accelerates the development of DN, characterized by loss of cytoskeleton protein integrity, such as nephrin. An early study showed that miR-29a is a potent regulator that inhibits fibrotic matrix expression in high glucose–stressed renal proximal tubule cells (Du et al., 2010). A recent study (Lin et al., 2014) indicated that miR-29a is protective against diabetes-induced podocyte damage, glomerular fibrosis and inflammation, and renal dysfunction. However, HDAC4-dependent H3K9 hypoacetylation counteracts miR-29a transcription in high glucose–stressed podocytes, suggesting that HDAC4 may be an important mediator in diabetic podocytopathy. Indeed, stimulation of podocytes with high glucose, advanced glycation end products, or transforming growth factor-β can increase HDAC4 expression and specific silencing of HDAC4 reduces podocyte injury in streptozotocin-induced diabetic rats and diabetic db/db mice (Wang et al., 2014). Further studies showed that the protective effect of HDAC4 inhibition is associated with prevention of autophagy defects and suppression of renal inflammation (Wang et al., 2014). Therefore, HDAC4 is a critical epigenetic mediator in the pathogenesis of DN, and specific inhibition of HDAC4 could serve as a therapeutic approach for DN and related renal diseases.
While application of HDACis is effective in the attenuation of DN, the combination of HDACis with other inhibitors might have additive or synergistic effects. Although such studies have not been performed in the DN, the combination of an ACE inhibitor with a HDACi has been reported to provide a better renal protection in a mouse model of HIV-associated nephropathy (Zhong et al., 2013). These two inhibitors can affect several important pathways involved in kidney inflammation and fibrosis, such as NF-κ B, interleukin-1, TGF-β, mitogen-activated protein kinase, and apoptosis signaling (Zhong et al., 2013). Thus, examination of the therapeutic effect of HDACis in the treatment of DN in combination with other drugs is warranted.
HDACs in Lupus Nephritis
Systemic lupus erythematosus (SLE) is a very common autoimmune disease (Alderaan et al., 2015). Two studies have examined the role of HDACs in the pathogenesis of SLE in the MRL-lpr/lpr murine model of lupus. Mishra et al. (2003) demonstrated a remarkable reduction in proteinuria, glomerulonephritis, and spleen weight after treatment with TSA. TSA was also effective in the downregulation of IL-12, IFN-γ, IL-6, and IL-10 expression levels in splenocytes of this model. Regna et al. showed that HDAC inhibition with ITF2357, a specific inhibitor of class I and II HDAC, reduces sera and urinary markers of lupus, and suppresses expression of several inflammatory cytokines (IL-1β, TNF-α, IL-6, and IFN-γ) and improves kidney histopathology (Regna et al., 2014). These data suggest that class I and II HDACs contribute to the development of lupus and application of HDACis may have therapeutic benefits in the treatment of SLE.
Aristolochic Acid Nephropathy
Aristolochic acid nephropathy is a progressive renal interstitial fibrosis, frequently associated with urothelial malignancies (Debelle et al., 2008). Recently, Novitskaya et al. (2014) examined the effect of a HDACi, 4-(phenylthio)butanoic acid (PTBA) analog methyl-4-(phenylthio)butanoate (M4PTB), on the kidney injury induced by aristolochic acid. They found that treatment with M4PTB promotes renal recovery and reduces renal fibrosis after aristolochic acid injury in mice. These beneficial effects are associated with increased renal tubular cell proliferation and decreased G2/M arrest of regenerating renal tubular epithelial cells. Furthermore, M4PTB treatment decreased peritubular macrophage infiltration and expression of macrophage chemokines such as CX3Cl1 and CCL2. Since an increased number of renal epithelial cell arrested at G2/M phase of cell cycle represents a maladaptive repair process that leads to renal fibrosis (Yang et al., 2010), class I/II HDACs may contribute to the development of CKD after aristolochic acid injury.
HDACs in Transplant Kidney Injury
Calcineurin inhibitors (CNIs) decrease the rate of acute rejection in renal transplantation patients, but side effects such as nephrotoxicity, neurotoxicity, and diabetogenicity (Shapiro et al., 1994), limit application of these drugs. Furthermore, CNIs are less effective in preventing chronic allograft rejection and the promotion of tolerance. Therefore, it is essential to search for novel and safer immunosuppressants with different mechanisms. Histone deacetylases (HDACs) are known to mediate transcription of genes that trigger immunological responses (Johnstone, 2002; Remiszewski, 2002). Studies examining the effect of HDACis on kidney injury after transplantation suggest that HDACis can initiate immunosuppression and prolong graft survival (Takahashi et al., 1996; Mishra et al., 2001). Edens et al. showed that HDACis were able to induce antigen-specific energy in lymphocytes (Edens et al., 2006). Tao et al. reported that HDAC inhibition improved the generation and function of regulatory T cells (Tao and Hancock, 2007). Moreover, Kinugasa et al. demonstrated that FR276457 had a remarkable immunosuppressive effect in a heterotopic cardiac transplant rat model (Kinugasa et al., 2008) and was able to prolong the median survival time (MST) in transplanted grafts in a canine renal transplant model. The combination of FR276457 with tacrolimus can also prevent allograft rejection. Histopathological analysis indicated that FR276457 suppressed mononuclear cell infiltration and vasculitis. Therefore, HDAC inhibition may prolong the MST in transplanted grafts when administered alone or combined with other immunosuppressive agents.
Conclusion and Future Directions
Numerous studies have shown that treatment with HDACis is able to inhibit activation and proliferation of cultured renal interstitial fibroblasts and attenuate renal fibrosis in animal models. HDAC inhibition is also beneficial for other chronic kidney diseases caused by the diverse etiologies, as listed in Table 1. Given the large number of distinct HDACs, most studies in this field are currently conducted by using pan-HDACis or class-specific HDACis. Thus, there is a need to identify specific functions of each HDAC and to develop small molecule inhibitors that can selectively modulate the activities of individual HDAC isoforms. In addition, it is necessary to clarify the profile of HDAC-modulated proteins in the setting of renal fibrosis and other kidney diseases by using some novel techniques such as proteomics to globally analyze protein lysine acetylation in response to HDAC inhibition. Although numerous clinical trials for application of HDACIs in tumors have been reported, there is no clinical trial thus far to test the therapeutic effects of those inhibitors in patients with CKD. Therefore, further investigation of the mechanisms, efficacy and toxicity of HDACis in the pre-clinical model of CKD will be helpful for initiating clinical trials to assess the feasibility of HDACis in the treatment of this disease in the future.
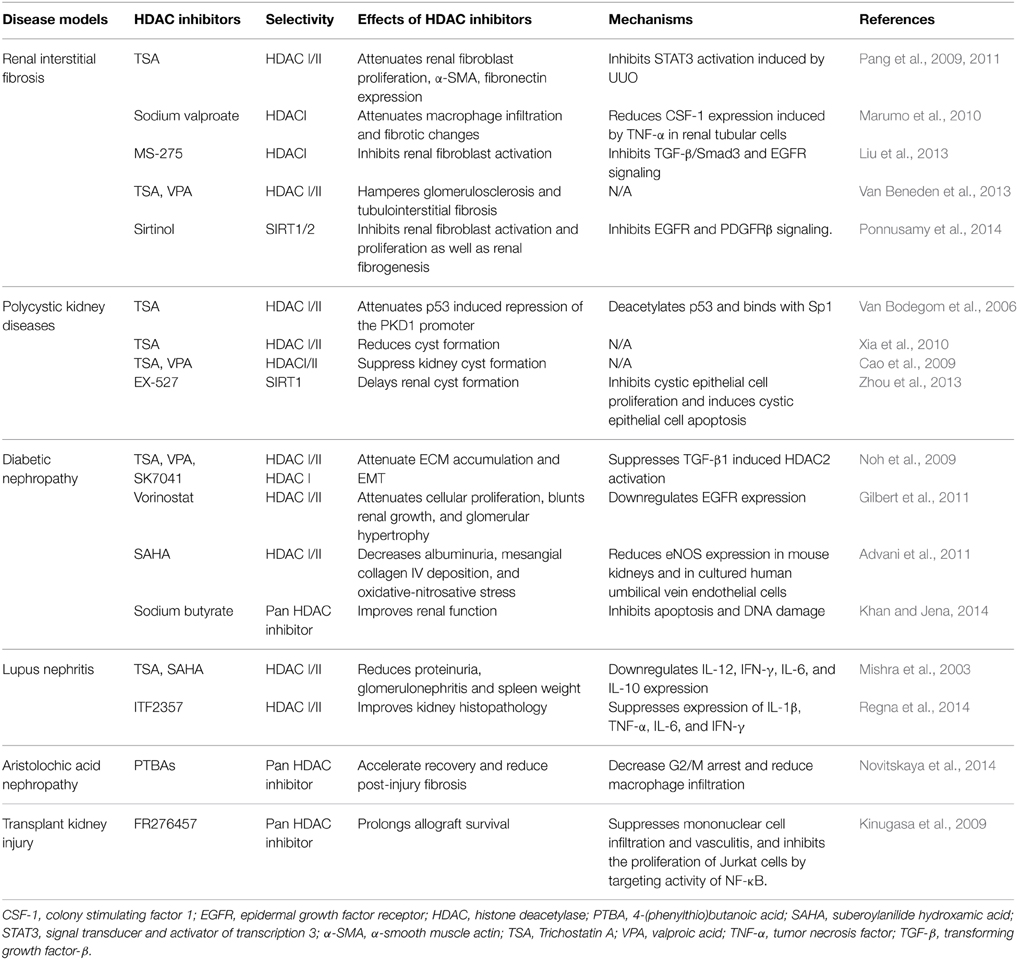
Table 1. Effects of HDAC inhibitors on experimental kidney disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. George Bayliss for critically reading and editing this manuscript. This study was supported by the National Nature Science Foundation of China Grants (81270778 and 81470920 to SZ, 81200492 and 81470991 to NL), the Shanghai Scientific Committee of China (13PJ1406900 to NL), Key Discipline Construction Project of Pudong Health Bureau of Shanghai (PWZx2014-06 to SZ), and US National Institutes of Health (2R01DK08506505A1 to SZ).
References
Acharya, M. R., Sparreboom, A., Venitz, J., and Figg, W. D. (2005). Rational development of histone deacetylase inhibitors as anticancer agents: a review. Mol. Pharmacol. 68, 917–932. doi: 10.1124/mol.105.014167
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Advani, A., Huang, Q., Thai, K., Advani, S. L., White, K. E., Kelly, D. J., et al. (2011). Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am. J. Pathol. 178, 2205–2214. doi: 10.1016/j.ajpath.2011.01.044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alderaan, K., Sekicki, V., Magder, L. S., and Petri, M. (2015). Risk factors for cataracts in systemic lupus erythematosus (SLE). Rheumatol. Int. 35, 701–708. doi: 10.1007/s00296-014-3129-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bush, E. W., and McKinsey, T. A. (2010). Protein acetylation in the cardiorenal axis: the promise of histone deacetylase inhibitors. Circ. Res. 106, 272–284. doi: 10.1161/CIRCRESAHA.109.209338
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cao, Y., Semanchik, N., Lee, S. H., Somlo, S., Barbano, P. E., Coifman, R., et al. (2009). Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc. Natl. Acad. Sci. U.S.A. 106, 21819–21824. doi: 10.1073/pnas.0911987106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chang, M. Y., Parker, E., Ibrahim, S., Shortland, J. R., Nahas, M. E., Haylor, J. L., et al. (2006). Haploinsufficiency of Pkd2 is associated with increased tubular cell proliferation and interstitial fibrosis in two murine Pkd2 models. Nephrol. Dial. Transplant 21, 2078–2084. doi: 10.1093/ndt/gfl150
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, J., Chen, J. K., and Harris, R. C. (2014). EGF receptor deletion in podocytes attenuates diabetic nephropathy. J. Am. Soc. Nephrol. doi: 10.1681/ASN.2014020192. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, S., and El-Dahr, S. S. (2013). Histone deacetylases in kidney development: implications for disease and therapy. Pediatr. Nephrol. 28, 689–698. doi: 10.1007/s00467-012-2223-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, Y. T., Chang, F. C., Wu, C. F., Chou, Y. H., Hsu, H. L., Chiang, W. C., et al. (2011). Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 80, 1170–1181. doi: 10.1038/ki.2011.208
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Debelle, F. D., Vanherweghem, J. L., and Nortier, J. L. (2008). Aristolochic acid nephropathy: a worldwide problem. Kidney Int. 74, 158–169. doi: 10.1038/ki.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dokmanovic, M., and Marks, P. A. (2005). Prospects: histone deacetylase inhibitors. J. Cell. Biochem. 96, 293–304. doi: 10.1002/jcb.20532
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Du, B., Ma, L. M., Huang, M. B., Zhou, H., Huang, H. L., Shao, P., et al. (2010). High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett. 584, 811–816. doi: 10.1016/j.febslet.2009.12.053
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Edens, R. E., Dagtas, S., and Gilbert, K. M. (2006). Histone deacetylase inhibitors induce antigen specific anergy in lymphocytes: a comparative study. Int. Immunopharmacol. 6, 1673–1681. doi: 10.1016/j.intimp.2006.07.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fan, L. X., Li, X., Magenheimer, B., Calvet, J. P., and Li, X. (2012). Inhibition of histone deacetylases targets the transcription regulator Id2 to attenuate cystic epithelial cell proliferation. Kidney Int. 81, 76–85. doi: 10.1038/ki.2011.296
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gilbert, R. E., Huang, Q., Thai, K., Advani, S. L., Lee, K., Yuen, D. A., et al. (2011). Histone deacetylase inhibition attenuates diabetes-associated kidney growth: potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 79, 1312–1321. doi: 10.1038/ki.2011.39
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Glozak, M. A., Sengupta, N., Zhang, X., and Seto, E. (2005). Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23. doi: 10.1016/j.gene.2005.09.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Harris, P. C., and Torres, V. E. (2014). Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Invest. 124, 2315–2324. doi: 10.1172/JCI72272
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johnstone, R. W. (2002). Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 1, 287–299. doi: 10.1038/nrd772
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khan, S., and Jena, G. (2014). Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-β1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem. Toxicol. 73, 127–139. doi: 10.1016/j.fct.2014.08.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, J. I., Jung, K. J., Jang, H. S., and Park, K. M. (2013). Gender-specific role of HDAC11 in kidney ischemia- and reperfusion-induced PAI-1 expression and injury. Am. J. Physiol. Renal Physiol. 305, F61–F70. doi: 10.1152/ajprenal.00015.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kinugasa, F., Nagatomi, I., Nakanishi, T., Noto, T., Mori, H., Matsuoka, H., et al. (2009). Effect of the immunosuppressant histone deacetylase inhibitor FR276457 in a canine renal transplant model. Transpl. Immunol. 21, 198–202. doi: 10.1016/j.trim.2009.04.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kinugasa, F., Yamada, T., Noto, T., Matsuoka, H., Mori, H., Sudo, Y., et al. (2008). Effect of a new immunosuppressant histon deacetylase (HDAC) inhibitor FR276457 in a rat cardiac transplant model. Biol. Pharm. Bull. 31, 1723–1726. doi: 10.1248/bpb.31.1723
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kwon, H. K., Ahn, S. H., Park, S. H., Park, J. H., Park, J. W., Kim, H. M., et al. (2009). A novel gamma-lactam-based histone deacetylase inhibitor potently inhibits the growth of human breast and renal cancer cells. Biol. Pharm. Bull. 32, 1723–1727. doi: 10.1248/bpb.32.1723
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, Y., Zhang, X., Polakiewicz, R. D., Yao, T. P., and Comb, M. J. (2008). HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J. Biol. Chem. 283, 12686–12690. doi: 10.1074/jbc.C700185200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, C. L., Lee, P. H., Hsu, Y. C., Lei, C. C., Ko, J. Y., Chuang, P. C., et al. (2014). MicroRNA-29a promotion of nephrin acetylation ameliorates hyperglycemia-induced podocyte dysfunction. J. Am. Soc. Nephrol. 25, 1698–1709. doi: 10.1681/ASN.2013050527
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, N., Guo, J. K., Pang, M., Tolbert, E., Ponnusamy, M., Gong, R., et al. (2012a). Genetic or pharmacologic blockade of EGFR inhibits renal fibrosis. J. Am. Soc. Nephrol. 23, 854–867. doi: 10.1681/ASN.2011050493
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, N., He, S., Ma, L., Ponnusamy, M., Tang, J., Tolbert, E., et al. (2013). Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS ONE 8:e54001. doi: 10.1371/journal.pone.0054001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, W., Fan, L. X., Zhou, X., Sweeney, W. E. Jr., Avner, E. D., and Li, X. (2012b). HDAC6 regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation in renal epithelial cells. PLoS ONE 7:e49418. doi: 10.1371/journal.pone.0049418
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ma, X., Ezzeldin, H. H., and Diasio, R. B. (2009). Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs 69, 1911–1934. doi: 10.2165/11315680-000000000-00000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mahalingam, D., Medina, E. C., Esquivel, J. A. 2nd., Espitia, C. M., Smith, S., Oberheu, K., et al. (2010). Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin. Cancer Res. 16, 141–153. doi: 10.1158/1078-0432.CCR-09-1385
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Manson, S. R., Song, J. B., Hruska, K. A., and Austin, P. F. (2014). HDAC dependent transcriptional repression of Bmp-7 potentiates TGF-beta mediated renal fibrosis in obstructive uropathy. J. Urol. 191, 242–252. doi: 10.1016/j.juro.2013.06.110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marks, P. A., Richon, V. M., Miller, T., and Kelly, W. K. (2004). Histone deacetylase inhibitors. Adv. Cancer Res. 91, 137–168. doi: 10.1016/S0065-230X(04)91004-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marks, P. A., Richon, V. M., and Rifkind, R. A. (2000). Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 92, 1210–1216. doi: 10.1093/jnci/92.15.1210
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marumo, T., Hishikawa, K., Yoshikawa, M., and Fujita, T. (2008). Epigenetic regulation of BMP7 in the regenerative response to ischemia. J. Am. Soc. Nephrol. 19, 1311–1320. doi: 10.1681/ASN.2007091040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marumo, T., Hishikawa, K., Yoshikawa, M., Hirahashi, J., Kawachi, S., and Fujita, T. (2010). Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am. J. Physiol. Renal Physiol. 298:F133–F141. doi: 10.1152/ajprenal.00400.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mauer, S. M., Steffes, M. W., Ellis, E. N., Sutherland, D. E., Brown, D. M., and Goetz, F. C. (1984). Structural-functional relationships in diabetic nephropathy. J. Clin. Invest. 74, 1143–1155. doi: 10.1172/JCI111523
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miller, T. A., Witter, D. J., and Belvedere, S. (2003). Histone deacetylase inhibitors. J. Med. Chem. 46, 5097–5116. doi: 10.1021/jm0303094
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Minucci, S., and Pelicci, P. G. (2006). Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 6, 38–51. doi: 10.1038/nrc1779
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mishra, N., Brown, D. R., Olorenshaw, I. M., and Kammer, G. M. (2001). Trichostatin A reverses skewed expression of CD154, interleukin-10, and interferon-gamma gene and protein expression in lupus T cells. Proc. Natl. Acad. Sci. U.S.A. 98, 2628–2633. doi: 10.1073/pnas.051507098
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mishra, N., Reilly, C. M., Brown, D. R., Ruiz, P., and Gilkeson, G. S. (2003). Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Invest. 111, 539–552. doi: 10.1172/JCI16153
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neilson, E. G. (2006). Mechanisms of disease: fibroblasts–a new look at an old problem. Nat. Clin. Pract. Nephrol. 2, 101–108. doi: 10.1038/ncpneph0093
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Noh, H., Oh, E. Y., Seo, J. Y., Yu, M. R., Kim, Y. O., Ha, H., et al. (2009). Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am. J. Physiol. Renal Physiol. 297, F729–F739. doi: 10.1152/ajprenal.00086.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Novitskaya, T., McDermott, L., Zhang, K. X., Chiba, T., Paueksakon, P., Hukriede, N. A., et al. (2014). A PTBA small molecule enhances recovery and reduces postinjury fibrosis after aristolochic acid-induced kidney injury. Am. J. Physiol. Renal Physiol. 306, F496–F504. doi: 10.1152/ajprenal.00534.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pang, M., Kothapally, J., Mao, H., Tolbert, E., Ponnusamy, M., Chin, Y. E., et al. (2009). Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am. J. Physiol. Renal Physiol. 297, F996–F1005. doi: 10.1152/ajprenal.00282.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pang, M., Ma, L., Liu, N., Ponnusamy, M., Zhao, T. C., Yan, H., et al. (2011). Histone deacetylase 1/2 mediates proliferation of renal interstitial fibroblasts and expression of cell cycle proteins. J. Cell. Biochem. 112, 2138–2148. doi: 10.1002/jcb.23135
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pang, M., and Zhuang, S. (2010). Histone deacetylase: a potential therapeutic target for fibrotic disorders. J. Pharmacol. Exp. Ther. 335, 266–272. doi: 10.1124/jpet.110.168385
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Peters, D. J., and Sandkuijl, L. A. (1992). Genetic heterogeneity of polycystic kidney disease in Europe. Contrib. Nephrol. 97, 128–139.
Ponnusamy, M., Zhou, X., Yan, Y., Tang, J., Tolbert, E., Zhao, T. C., et al. (2014). Blocking sirtuin 1 and 2 inhibits renal interstitial fibroblast activation and attenuates renal interstitial fibrosis in obstructive nephropathy. J. Pharmacol. Exp. Ther. 350, 243–256. doi: 10.1124/jpet.113.212076
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramakrishnan, S., and Pili, R. (2013). Histone deacetylase inhibitors and epigenetic modifications as a novel strategy in renal cell carcinoma. Cancer J. 19, 333–340. doi: 10.1097/PPO.0b013e3182a09e07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Regna, N. L., Chafin, C. B., Hammond, S. E., Puthiyaveetil, A. G., Caudell, D. L., and Reilly, C. M. (2014). Class I and II histone deacetylase inhibition by ITF2357 reduces SLE pathogenesis in vivo. Clin. Immunol. 151, 29–42. doi: 10.1016/j.clim.2014.01.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Remiszewski, S. W. (2002). Recent advances in the discovery of small molecule histone deacetylase inhibitors. Curr. Opin. Drug Discov. Devel. 5, 487–499.
Selinger, C. I., Cooper, W. A., Al-Sohaily, S., Mladenova, D. N., Pangon, L., Kennedy, C. W., et al. (2011). Loss of special AT-rich binding protein 1 expression is a marker of poor survival in lung cancer. J. Thorac. Oncol. 6, 1179–1189. doi: 10.1097/JTO.0b013e31821b4ce0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shapiro, R., Jordan, M., Scantlebury, V. P., Vivas, C., Gritsch, H. A., Rao, A. S., et al. (1994). Renal transplantation at the University of Pittsburgh: the impact of FK506. Clin. Transpl. 8, 229–236.
Spencer, V. A., and Davie, J. R. (1999). Role of covalent modifications of histones in regulating gene expression. Gene 240, 1–12.
Takahashi, I., Miyaji, H., Yoshida, T., Sato, S., and Mizukami, T. (1996). Selective inhibition of IL-2 gene expression by trichostatin A, a potent inhibitor of mammalian histone deacetylase. J. Antibiot. 49, 453–457.
Tang, J., Yan, Y., Zhao, T. C., Bayliss, G., Yan, H., and Zhuang, S. (2013). Class I histone deacetylase activity is required for proliferation of renal epithelial cells. Am. J. Physiol. Renal Physiol. 305, F244–F254. doi: 10.1152/ajprenal.00126.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tao, R., and Hancock, W. W. (2007). Regulating regulatory T cells to achieve transplant tolerance. HBPD INT 6, 348–357.
Thivierge, C., Kurbegovic, A., Couillard, M., Guillaume, R., Cote, O., and Trudel, M. (2006). Overexpression of PKD1 causes polycystic kidney disease. Mol. Cell. Biol. 26, 1538–1548. doi: 10.1128/MCB.26.4.1538-1548.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Beneden, K., Geers, C., Pauwels, M., Mannaerts, I., Wissing, K. M., Van Den Branden, C., et al. (2013). Comparison of trichostatin A and valproic acid treatment regimens in a mouse model of kidney fibrosis. Toxicol. Appl. Pharmacol. 271, 276–284. doi: 10.1016/j.taap.2013.05.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Bodegom, D., Saifudeen, Z., Dipp, S., Puri, S., Magenheimer, B. S., Calvet, J. P., et al. (2006). The polycystic kidney disease-1 gene is a target for p53-mediated transcriptional repression. J. Biol. Chem. 281, 31234–31244.
Van Lint, C., Emiliani, S., and Verdin, E. (1996). The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 5, 245–253.
Wang, X., Liu, J., Zhen, J., Zhang, C., Wan, Q., Liu, G., et al. (2014). Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 86, 712–725. doi: 10.1038/ki.2014.111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
West, A. C., and Johnstone, R. W. (2014). New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 124, 30–39. doi: 10.1172/JCI69738
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wynn, T. A. (2008). Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210. doi: 10.1002/path.2277
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xia, S., Li, X., Johnson, T., Seidel, C., Wallace, D. P., and Li, R. (2010). Polycystin-dependent fluid flow sensing targets histone deacetylase 5 to prevent the development of renal cysts. Development 137, 1075–1084. doi: 10.1242/dev.049437
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamaguchi, T., Hempson, S. J., Reif, G. A., Hedge, A. M., and Wallace, D. P. (2006). Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J. Am. Soc. Nephrol. 17, 178–187. doi: 10.1681/ASN.2005060645
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, L., Besschetnova, T. Y., Brooks, C. R., Shah, J. V., and Bonventre, J. V. (2010). Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 16, 535–543, 531p following 143. doi: 10.1038/nm.2144
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, M. Z., Wang, Y., Paueksakon, P., and Harris, R. C. (2014). Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes 63, 2063–2072. doi: 10.2337/db13-1279
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhong, Y., Chen, E. Y., Liu, R., Chuang, P. Y., Mallipattu, S. K., Tan, C. M., et al. (2013). Renoprotective effect of combined inhibition of angiotensin-converting enzyme and histone deacetylase. J. Am. Soc. Nephrol. 24, 801–811. doi: 10.1681/ASN.2012060590
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, X., Fan, L. X., Sweeney, W. E. Jr., Denu, J. M., Avner, E. D., and Li, X. (2013). Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J. Clin. Invest. 123, 3084–3098. doi: 10.1172/JCI64401
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: histone deacetylases, chronic kidney diseases, renal fibrosis, renal fibroblasts
Citation: Liu N and Zhuang S (2015) Treatment of chronic kidney diseases with histone deacetylase inhibitors. Front. Physiol. 6:121. doi: 10.3389/fphys.2015.00121
Received: 20 December 2014; Paper pending published: 09 February 2015;
Accepted: 02 April 2015; Published: 28 April 2015.
Edited by:
Hui Y. Lan, The Chinese University of Hong Kong, ChinaReviewed by:
Agnieszka Swiatecka-Urban, University of Pittsburgh School of Medicine, USAFiona McDonald, University of Otago, Dunedin, New Zealand
Copyright © 2015 Liu and Zhuang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shougang Zhuang, Department of Nephrology, Shanghai East Hospital, Tongji University School of Medicine, Shanghai 200120, China;
Department of Medicine, Rhode Island Hospital, Middle House 301, 593 Eddy Street, Providence, RI 02903, USA,c3podWFuZ0BsaWZlc3Bhbi5vcmc=